
Submitted:
24 November 2023
Posted:
28 November 2023
You are already at the latest version
Abstract
Dravet syndrome (DS), also known as severe myoclonic epilepsy of infancy, is a rare and drug-resistant form of developmental and epileptic encephalopathies, which is both debilitating and challenging to manage, typically arising during the first year of life, with seizures often triggered by fever, infections, or vaccinations. It is characterized by frequent and prolonged seizures, developmental delays, and various other neurological and behavioral impairments. Most cases result from pathogenic mutations in the sodium voltage-gated channel alpha subunit 1 (SCN1A) gene, which encodes a critical voltage-gated sodium channel subunit involved in neuronal excitability. Precision medicine offers significant potential for improving DS diagnosis and treatment. Early genetic testing enables timely and accurate diagnosis. Advances in our understanding of DS's underlying genetic mechanisms and neurobiology have enabled the development of targeted therapies, such as gene therapy, offering more effective and less invasive treatment options for patients with DS. Targeted and gene therapies provide hope for more effective and personalized treatments. However, research into novel approaches remains in its early stages, and their clinical application remains to be seen. This review addresses the current understanding of clinical DS features, genetic involvement in DS development, and outcomes of novel DS therapies.
Keywords:
Dravet Syndrome
; developmental epileptic encephalopathy
; genetics
; targeted therapy
; epilepsy syndrome
1. Introduction
1.1. Epileptic (EEs) and Developmental Encephalopathies (DEs)
The International League Against Epilepsy (ILAE) defines an EE as “the epileptiform activity itself contributes to severe cognitive and behavioral impairments beyond that expected from the underlying pathology alone (such as a cortical malformation)” [1]. EEs are conditions that can worsen over time, be seen along a spectrum of severity, and across all epilepsies, and occur at any age, although most first appear in infancy and childhood [1,2]. The EE Triad includes seizures (refractory), epileptiform activity on electroencephalography (EEG), and adverse effects on development, cognition, and often behavior [3]. Therefore, if patients have no epileptiform activity, they do not have an EE. Importantly, if the treatments can ameliorate the epileptiform discharges of a patient with EE, its developmental consequences can also be improved [4]. DEs are pre-existing developmental delays or intellectual disabilities due to a non-progressive brain state, and the degree of disability may become more apparent with brain maturation. Patients with DEs have a higher risk of epilepsy than the general population, suggesting that developmental delays are the direct result of the underlying cause of their epilepsy. Genetic causes are also frequent in this population [3]. Due to several connections and overlaps between EEs and DEs, the term developmental and epileptic encephalopathies (DEEs) was coined in the formal 2017 classification revision.
1.2. DEEs
DEEs encompass a group of severe neurological disorders that combine EEs and DEs, leading to a complex clinical presentation involving seizures, developmental delays, and cognitive impairments [4]. These conditions are often associated with a high disease burden, including significant morbidity, mortality, and reduced quality of life [5].
Various factors contribute to developing DEEs, including genetic mutations, structural brain abnormalities, and metabolic disorders. In neonatal onset DEEs, nearly 60% of cases are attributable to specific causes, compared to approximately 50% of later onset DEEs [5]. Genetic mutations cause a significant proportion of DEEs, with many cases involving genes associated with synaptic function, ion channels, and neurotransmitter signaling. Advances in genetic testing, such as whole exome sequencing and targeted gene panels, have improved our understanding of the genetic landscape underlying DEEs [6]. DEEs are characterized by several key features, including variable age of onset, etiologies, seizure types, and EEG patterns, which may change as age advances; drug-resistant seizures; developmental delays and intellectual disability (ID); co-occurring motor, speech, and behavioral impairments; and a high risk of complications. Their outcomes are very poor even though seizures cease, and sometimes early death. In addition, various epilepsy syndromes fall within the DEE category (Figure 1). However, a key component of the concept is that the amelioration of the epileptiform activity may potentially improve the developmental consequences of the disorder [3,4,7].

Table 1.
Various epilepsy syndromes fall within the DEE category.
| DEE | Syndrome or Known Name | Phenotype MIM Number |
Genes | Genetic Loci |
|---|---|---|---|---|
| DEE1 | LISX2; XLAG;Hydranencephaly with abnormal genitalia | 300215 | ARX | Xp21.3 |
| Proud syndrome | 300004 | ARX | Xp21.3 | |
| PRTS; MRX36; MRXS1 | 309510 | ARX | Xp21.3 | |
| XLID29; MRX29;MRX32MRX33;MRX29;MRX38;MRX43;MRX52;MRX54;MRX76; MRX87 | 300419 | ARX | Xp21.3 | |
| DEE2 | EIEE2; ISSX2 | 300672 | CDKL5 | Xp22.13 |
| DEE3 | EIEE3 | 609302 | SLC25A22 | 11p15 |
| DEE4 | EIEE4 | 612164 | STXBP1 | 9q34 |
| DEE5 | EIEE5 | 613477 | SPTAN1 | 9q34 |
| DEE6A | EIEE6; SMEI;DS | 607208 | SCN1A | 2q24.3 |
| DEE6B | Phenotypes caused by heterozygous mutation in the SCN1A gene, not DS | 619317 | SCN1A | 2q24.3 |
| DEE7 | EIEE7 | 613720 | KCNQ2 | 20q13 |
| DEE8 | EIEE8; Hyperkplexia and epilepsy | 300607 | ARHGEF9 | Xq11.1 |
| DEE9 | EIEE9; EFMR; Juberg-Hellman syndrome | 300088 | PCDH19 | Xq22.1 |
| DEE10 | EIEE10; MCSZ | 613402 | PNKP | 19q13 |
| DEE11 | EIEE11 | 613721 | SCN2A | 2q24 |
| DEE12 | EIEE12 | 613722 | PLCB1 | 20p12.3 |
| DEE13 | EIEE13 | 614558 | SCN8A | 12q13 |
| DEE14 | EIEE14 | 614959 | KCNT1 | 9q34 |
| DEE15 | EIEE15 | 615006 | ST3GAL3 | 1p34 |
| DEE16 | EIEE16 | 615338 | TBC1D24 | 16p13 |
| DEE17 | EIEE17 | 615473 | GNAO1 | 16q13 |
| DEE18 | EIEE18 | 615476 | SZT2 | 1p34 |
| DEE19 | EIEE19 | 615744 | GABRA1 | 5q34 |
| DEE20 | EIEE20; GPIBD4MCAHS2 | 300868 | PIGA | Xp22 |
| DEE21 | EIEE21 | 615833 | NECAP1 | 12p13 |
| DEE22 | EIEE22; CDGIIm | 300896 | SLC35A2 | Xp11 |
| DEE23 | EIEE23 | 615859 | DOCK7 | 1p31 |
| DEE24 | EIEE24 | 615871 | HCN1 | 5p12 |
| DEE25 | EIEE25 | 615905 | SLC13A5 | 17p13 |
| DEE26 | EIEE26 | 616056 | KCNB1 | 20q13 |
| DEE27 | EIEE27 | 616139 | GRIN2B | 12p12 |
| DEE28 | EIEE28 | 616211 | WWOX | 16q23 |
| DEE29 | EIEE29 | 616339 | AARS | 16q22 |
| DEE30 | EIEE30 | 616341 | SIK1 | 21q22 |
| DEE31A | EIEE31 | 616346 | DNM1 | 9q34 |
| DEE31B | - | 620352 | DNM1 | 9q34 |
| DEE32 | EIEE32 | 616366 | KCNA2 | 1p13 |
| DEE33 | EIEE33 | 616409 | EEF1A2 | 20q13 |
| DEE34 | EIEE34 | 616645 | SLC12A5 | 20q12 |
| DEE35 | EIEE35 | 616647 | ITPA | 20p13 |
| DEE36 | EIEE36; CDG1s | 300884 | ALG13 | Xq23 |
| DEE37 | EIEE37 | 616981 | FRRS1L | 9q31 |
| DEE38 | EIEE38; GPIBD23 | 617020 | ARV1 | 1q42 |
| DEE39 | EIEE39; AGC1 DEFICIENCY | 612949 | SLC25A12 | 2q31 |
| DEE40 | EIEE40 | 617065 | GUF1 | 4p12 |
| DEE41 | EIEE41 | 617105 | SLC1A2 | 11p13 |
| DEE42 | EIEE42 | 617106 | CACNA1A | 19p13 |
| DEE43 | EIEE43 | 617113 | GABRB3 | 15q11 |
| DEE44 | EIEE44 | 617132 | UBA5 | 3q22 |
| DEE45 | EIEE45 | 617153 | GABRB1 | 4p13 |
| DEE46 | EIEE46 | 617162 | GRIN2D | 19q13 |
| DEE47 | EIEE47 | 617166 | FGF12 | 3q28 |
| DEE48 | EIEE48 | 617276 | AP3B2 | 15q25 |
| DEE49 | EIEE49 | 617281 | DENND5A | 11p15 |
| DEE50 | EIEE50; CDG1Z | 616457 | CAD | 2p23 |
| DEE51 | EIEE51 | 617339 | MDH2 | 7q11 |
| DEE52 | EIEE52 | 617350 | SCN1B | 19q13 |
| DEE53 | EIEE53 | 617389 | SYNJ1 | 21q22 |
| DEE54 | EIEE54 | 617391 | HNRNPU | 1q44. |
| DEE55 | EIEE55; GPIBD14 | 617599 | PIGP | 21q22 |
| DEE56 | EIEE56 | 617665 | YWHAG | 7q11 |
| DEE57 | EIEE57 | 617771 | KCNT2 | 1q31 |
| DEE58 | EIEE58 | 617830 | NTRK2 | 9q21 |
| DEE59 | EIEE59 | 617904 | GABBR2 | 9q22. |
| DEE60 | EIEE60 | 617929 | CNPY3 | 6p21 |
| DEE61 | EIEE61 | 617933 | ADAM22 | 7q21 |
| DEE62 | EIEE62 | 617938 | SCN3A | 2q24 |
| DEE63 | EIEE63 | 617976 | CPLX1 | 4p16 |
| DEE64 | EIEE64 | 618004 | RHOBTB2 | 8p21 |
| DEE65 | EIEE65 | 618008 | CYFIP2 | 5q33 |
| DEE66 | EIEE66 | 618067 | PACS2 | 14q32 |
| DEE67 | EIEE67 | 618141 | CUX2 | 12q23 |
| DEE68 | EIEE68 | 618201 | TRAK1 | 3p25 |
| DEE69 | EIEE69 | 618285 | CACNA1E | 1q25 |
| DEE70 | EIEE70 | 618298 | PHACTR1 | 6p24 |
| DEE71 | EIEE71; Glutaminase deficiency with neonatal epileptic encephalopathy | 618328 | GLS | 2q32 |
| DEE72 | EIEE72 | 618374 | NEUROD2 | 17q12 |
| DEE73 | EIEE73 | 618379 | RNF13 | 3q25 |
| DEE74 | EIEE74 | 618396 | GABRG2 | 5q34 |
| DEE75 | EIEE75 | 618437 | PARS2 | 1p32 |
| DEE76 | EIEE76; DECAM | 618468 | ACTL6B | 7q22. |
| DEE77 | EIEE77; GPIBD19 | 618548 | PIGQ | 16p13 |
| DEE78 | EIEE78 | 618557 | GABRA2 | 4p13 |
| DEE79 | EIEE79 | 618559 | GABRA5 | 15q11 |
| DEE80 | EIEE80; GPIBD20 | 618580 | PIGB | 15q21. |
| DEE81 | EIEE81 | 618663 | DMXL2 | 15q21 |
| DEE82 | EIEE82; GOT2 deficiency | 618721 | GOT2 | 16q21 |
| DEE83 | EIEE83; Barakat-Perenthaler syndrome | 618744 | UGP2 | 2p14 |
| DEE84 | EIEE84; Jamuar syndrome | 618792 | UGDH | 4p14 |
| DEE85 | EIEE85 | 301044 | SMC1A | Xp11 |
| DEE86 | EIEE86 | 618910 | DALRD3 | 3p21 |
| DEE87 | EIEE87 | 618916 | CDK19 | 6q21. |
| DEE88 | EIEE88 | 618959 | MDH1 | 2p15 |
| DEE89 | - | 619124 | GAD1 | 2q31 |
| DEE90 | - | 301058 | FGF13 | Xq26 |
| DEE91 | IECEE1 | 617711 | PPP3CA | 4q24 |
| DEE92 | IECEE2 | 617829 | GABRB2 | 5q34 |
| DEE93 | - | 618012 | ATP6V1A | 3q13 |
| DEE94 | EEOC | 615369 | CHD2 | 15q26 |
| DEE95 | GPIBD18 | 618143 | PIGS | 17q11 |
| DEE96 | - | 619340 | NSF | 17q21 |
| DEE97 | - | 619561 | iCELF2 | 10p14. |
| DEE98 | - | 619605 | ATP1A2 | 1q23 |
| DEE99 | 619606 | ATP1A3 | 19q13. | |
| DEE100 | - | 619777 | FBXO28 | 1q42 |
| DEE101 | - | 619814 | GRIN1 | 9q34 |
| DEE102 | - | 619881 | SLC38A3 | 3p21 |
| DEE103 | - | 619913 | KCNC2 | 12q21 |
| DEE104 | - | 619970 | ATP6V0A1 | 17q21 |
| DEE105 | - | 619983 | HID1 | 17q25. |
| DEE106 | - | 620028 | UFSP2 | 4q35 |
| DEE107 | - | 620033 | NAPB | 20p11 |
| DEE108 | - | 620115 | MAST3 | 19p13 |
| DEE109 | - | 620145 | FZR1 | 19p13 |
| DEE110 | - | 620149 | CACNA2D1 | 7q21 |
| DEE111 | - | 62054 | DEPDC5 | 22q12.2-q12.3 |
| DEE112 | - | 620537 | KCNH5 | 14q23.2 |
ACC: agenesis of the corpus callosum; AGC1 deficiency: aspartate-glutamate carrier 1 deficiency; CDG: congenital disorder of glycosylation; DECAM: developmental delay, epileptic encephalopathy, cerebral atrophy, and abnormal myelination; DS: Dravet syndrome; EEOC: epileptic encephalopathy, childhood-onset; EFMR: epilepsy and mental retardation restricted to females; GOT2 deficiency: mitochondrial glutamate oxaloacetate transaminase deficiency; GPIBD: glycophosphatidylinositol biosynthesis defect; IECEE: epileptic encephalopathy, infantile or early childhood; ISSX2: infantile spasm syndrome, X-linked 2; LISX2: lissencephaly, X-linked, 2; MCSZ: microcephaly, seizures, and developmental delay; MCAHS: multiple congenital anomalies-hypotonia-seizures syndrome; MRX: mental retardation, X-linked; MRXS1: mental retardation, X-linked, syndromic 1; PRTS: Partington syndrome; SMEI: severe myoclonic epilepsy of infancy; XLAG: X-linked lissencephaly with ambiguous genitalia; XLID29: intellectual developmental disorder, X-linked 29; -: no available data. (Adopted and modified from https://omim.org/entry/308350; accessed on 11/21/2023).
2. Dravet Syndrome (DS)
DS, also known as severe myoclonic epilepsy of infancy, epilepsy with polymorphic seizures, and polymorphic epilepsy of infancy, is a severe drug-resistant form of DEE that is both debilitating and challenging to manage [8]. It is a rare, genetic epileptic disorder typically arising during the first year of life, with seizures often triggered by fever, infections, or vaccinations [9]. Over 80% of cases result from pathogenic mutations in the sodium voltage-gated channel alpha subunit 1 (SCN1A) gene, which encodes a critical voltage-gated sodium channel subunit involved in neuronal excitability [10]. While DS's exact pathophysiological mechanisms remain incompletely understood, its underlying genetic basis contributes to the refractory nature of seizures and the diversity of associated developmental and cognitive impairments [11]. Patients may experience frequent and prolonged seizures, developmental delays, intellectual disabilities, motor impairments, and behavioral challenges [12]. DS substantially burdens affected individuals and their families with a broad spectrum of clinical features and multi-systemic comorbidities [13]. Patients with DS have a significantly increased risk of premature mortality, with sudden unexpected death in epilepsy (SUDEP) being the leading cause.
With the promising results of gene therapy for SCN1A-mutated experimental models, prompt identification and accurate definition of DS have become crucial to optimize patients' outcomes. However, DS typically presents with a broad range of phenotypes, while molecular identification may miss 20% of the patients with DS but without an SCN1A mutation. A recent report found that traditional DS phenotypic spectrums and diagnostic criteria may limit the accurate diagnosis of patients [14]. For example, they found the age of onset to extend to 19 months of life instead of the previously established 12 months. In addition, fever-related seizures were less prevalent than previously thought. In contrast, tonic-clonic seizures were the most common seizure form; hemiclonic seizures were previously believed to be the most common form in DS [14].
Precision medicine, a medical approach that tailors treatment to patients individual characteristics, has the potential to significantly improve DS management. Several gene therapy approaches have been proposed and investigated in recent years, including antisense oligonucleotides (ASOs) and adeno-associated virus (AAV)-mediated gene replacement [15,16]. In addition, pharmacological approaches addressing the imbalance between excitatory and inhibitory neurotransmissions caused by SCN1A dysfunction have been investigated, such as stiripentol, fenfluramine, and cannabidiol (CBD) [8]. As more treatment options tailored to DS become available, we aim to comprehensively review the current understanding of clinical DS features, genetic involvement in DS development, and outcomes of novel DS therapies.
2.1. Epidemiology
Data indicate that DS has a global incidence of approximately 6.5 per 100,000 live births [9,17,18,19]. However, it may vary substantially across ethnicities and geographical regions. For example, in the United States (US), a population-based study found that the incidence of DS was 1 per 15,700 live births, with six of the eight identified cases having a de novo SCN1A missense mutation. This finding indicates that DS due to an SCN1A mutation is twice as common in the US as previously thought [19]. Data from Denmark also indicated an incidence of DS of 1 per 22,000 live births, with 15 of the 17 patients having an SCN1A mutation [20]. In Germany, the 10-year prevalence of DS was estimated to be 4.7 per 100,000 people [21]. The estimated prevalence is 1/20,000–1/30,000 in France [22] and 1/ 45, 700 children aged under 18 years in Sweden [23]. DS with a confirmed SCN1A mutation has an incidence of at least 1/40, 900 births in the UK [24], and 1/ 15, 500 live births in Scotland [18]. While rare, DS accounts for approximately 2%–5% of all childhood epilepsies [25]. Males and females are equally affected [26]. Nevertheless, more extensive population-based studies are needed to accurately determine the prevalence and distribution of DS across different populations.
2.2. Clinical Presentation of DS
2.2.1. Neurodevelopment in DS Starts before the Age of Two Years and Continues to Worsen
Most children with DS show neurodevelopmental delay, often having delays in gross-motor, fine-motor, language, and psychomotor function, leading to global neurodevelopmental delays that majorly impacts affected children [27]. Generally, affected children are almost normal at baseline. Motor delays slowly appear in them before the age of six years and rapidly worsen with age [24,28,29,30,31,32]. While gross-motor milestone achievement is diverse in DS, a report assessing children with DS aged over two years showed that approximately half had significant delays in independent sitting and walking [31]. These results are consistent with our experiences and several studies, despites differences in different domains, irrelevant to the different courses of their seizure histories [30,32]. Crouch gait, an abnormal walking pattern, is mainly caused by spasticity characterized by excessive knee and hip flexion and ankle dorsiflexion during the stance phase [33]. One study that performed formal gait analysis on patients with DS aged 2–34 years found crouch gait in 88% of adolescents aged >13 years, 50% of children aged ≥6 years, and no children aged <6 years [34]. Affected children are reported to frequently show neurological impairments, such as ataxia, extrapyramidal signs, myoclonus [31], and tremor [11]. Mounting evidence shows that fine-motor skills are even more affected than gross motor skills in affected children [28,30]. Verheyen et al. reported that 100% of children with DS developed gross-motor delay, while 77% developed fine-motor delay [30,32]. While language may be less affected in some patients, their social abilities are usually maintained [35].
A significant delay in psychomotor development is one characteristic of DS, primarily moderate to severe [32,36,37,38]. Poorer cognitive outcomes are reportedly associated with higher frequencies of seizures [32], sttus epilepticus (SE), and interictal EEG abnormalities in one year old [24], and the early appearance of myoclonus, absence seizures [39], and motor dysfunction, including ataxia, dyskinesia, hypotonia, and spasticity [24]. Pathogenic SCN1A mutations have also been reported to be associated with poor cognitive outcomes [40]. Many children with DS experience a decline in their intelligence quotient (IQ) [41]. One study showed that ID was typically apparent in children with DS by age 18-60 months [42]. Another study involving 21 children with DS showed a decline in IQ over time, with no child having a normal IQ beyond the age of six years. These children also showed signs suggestive of attention-deficit hyperactivity disorder (ADHD) [43]. Nabbout et al reported that IQ was generally normal before age two but declined after three years, and ADHD was also prevalent [40]. A quarter of children with DS show autistic behavior with poor eye contact and stereotypes [44].The global decline in DS may be due to a combination of factors, including seizure effects, the underlying genetic defect, and medication effects. Seizures can damage the brain, and the SCN1A gene mutation can lead to abnormal brain development. While seizures may lessen in the third phase, behavioral, communication, and developmental manifestations may persist from infancy to adulthood [31]. Medications for treating seizures, (such as sodium channel blockers: (e.g., carbamazepine, eslicarbazepine, lamotrigine, oxcarbazepine, phenytoin, and vigabatrin) can also have cognitive side effects [45]. Whether early and proper control of seizures in children with DS can stop their development of psychological disorders, such as ADHD and autistic-like behavior deserves further study.
2.2.2. Triggering Factors
DS is a severe form of DEE that typically begins during the first year of life. In children, common triggers include fever, infection, sun exposure, flashing lights, high ambient temperature, visual patterns, hot-water baths, intense physical exercise, eating, and overexcitement [11,46,47]. Compared to children with DS, adultswith DS may be much less susceptible to seizures induced by hyperthermia and other triggers that induced seizures in childhood. However, seizures tend to exacerbate in older children and adults with the use of sodium channel blockers [42].
2.2.3. Seizure Patterns
Initially, otherwise healthy children may experience frequent, prolonged hemiclonic or generalized seizures [11]. Patients can suffer from recurring febrile and afebrile seizures that impact different body parts as they grow. Various seizure forms can emerge between the ages of one and four years, including myoclonic, atypical absence, focal, and generalized tonic-clonic seizures. Focal seizures might progress to focal motor or bilateral convulsive seizures. However, tonic seizures are uncommon in individuals with DS [12,48].
In a Chinese retrospective study, 77% of children with DS and SCN1A mutations had seizure onset before seven months, with 72% experiencing febrile seizures lasting >15 minutes and 67% having multiple febrile seizures within a day [49]. Li et al. found that the age of onset extended to 19 months of life and that tonic-clonic seizure was the most common seizure form [14]. Another report found that seizure onset under seven months; frequent, prolonged seizures; partial, focal, and myoclonic seizures; and hot water induced seizure significantly correlated with DS [50].
Convulsive and non-convulsive SE frequently occurs in DS. Non-convulsive status epilepticus (NCSE), also called "obtundation status epilepticus," is reported to be about 40% of patients with DS in several series can be life-threatening [41,51]. Moverover, NCSE can present subtle symptoms that make it difficult to recognize [11,52]. As epilepsy progresses, children with DS often experience a decrease in seizure frequency and intensity as they transition into adolescence and adulthood. While fever sensitivity remains, its effects lessen over time. Myoclonic, atypical absence, focal seizures with altered awareness, and SE become less frequent in adults [53]. In adulthood, the most common seizure type is generalized tonic-clonic, which may have a focal origin and primarily occur during sleep. Tonic vibratory or clonic movements may occur in asymmetrical or bilateral patterns [11]. In a study of five patients with DS who transitioned into adulthood, their EEG recording showed that three had focal seizures, with or without progression into bilateral convulsive seizures. However, obtundation and tonic events occurred in the other two patients [53]. In a report followed 53 children with DS for up to 14 years, seizure frequency was weekly in more than two-thirds of cases, while monthly and daily frequencies were observed in 13% each [54]. Several studies have shown that the frequency and intensity of seizures may gradually decrease during adolescence and into adulthood but vary among individuals [38,55,56,57,58,59]. In addition to age-related maturation effects, whether the roles of sex-related hormones contribute to the evolution of seizures in DS needs further investigations.
2.2.4. EEG Evolution in DS
During the first year of life, EEGs are typically normal for patients with DS. As the disease evolves, the EEG may remain normal or become slower while sleep patterns generally remain intact. Common observations include generalized spike-and-wave patterns, polyspike waves, and multifocal spikes. However, few studies have comprehensively examined the development of EEG findings in DS over time [11]. In an EEG study of 22 children with DS during the first five years after diagnosis, all children had a normal EEG background at onset, but 27% showed background slowing after six months. At the onset of seizures, epileptiform discharges were present in 27% of the children, increasing to 64% by the five-year follow-up. In addition, while almost 57% of the children had multifocal epileptiform after five years, approximately 30% and 14% had focal epileptiform discharges and generalized epileptiform, respectively. The rate of photoparoxysmal response increased to 41% from a baseline of 9% after five years [60].
Another report showed that 81% of children with DS had abnormal EEG results; 25% had an abnormal EEG at the time of seizure onset. Generalized spike-wave discharges were the most common epileptiform pattern [61]. Other reports confirmed these findings and showed that epileptiform discharges were present in most patients, with multifocal epileptiform discharges being the most common [62]. Notably, Genton et al. described varying EEG backgrounds and epileptiform activity patterns among 24 patients with DS, with photosensitivity and pattern sensitivity typically disappearing by 20 years of age [53]. The diameter of the human optic nerve varies across developmental stages. It is small at birth, and enlarges during childhood, and remains constant in adulthood [63]. The growth of the optic nerve is generally correlated with the evolution of photosensitivity and pattern sensitivity in DS, suggesting gene therapy for DS should be given before adulthood.
2.2.5. Neuroimaging in DS
Structural brain imaging using magnetic resonance imaging (MRI) is mostly normal in DS; however, abnormalities have been reported in some cases. A study reviewing 120 Italian children with DS found cortical development malformations in four [64]. In another study, among 18 children with DS who underwent MRI after age three, seven showed hippocampal sclerosis or a loss of grey-white distinction in the temporal lobe [65]. A comparative study of nine patients (3 adults and 6 children) with DS and SCN1A mutations and nine seizure-free controls showed that patients with DS had globally reduced grey and white matter volumes. However, total intracranial volume has been reported to decrease significantly with age [66]. Another study showed that some patients with DS had cerebral or cerebellar atrophy, or hippocampal sclerosis [67].
Figure 1.
Comorbidities in individuals with DS. TSH: thyroid-stimulating hormone; IGF-1: insulin-like growth factor 1; SUDEP: sudden unexpected death in epilepsy.
Figure 1.
Comorbidities in individuals with DS. TSH: thyroid-stimulating hormone; IGF-1: insulin-like growth factor 1; SUDEP: sudden unexpected death in epilepsy.
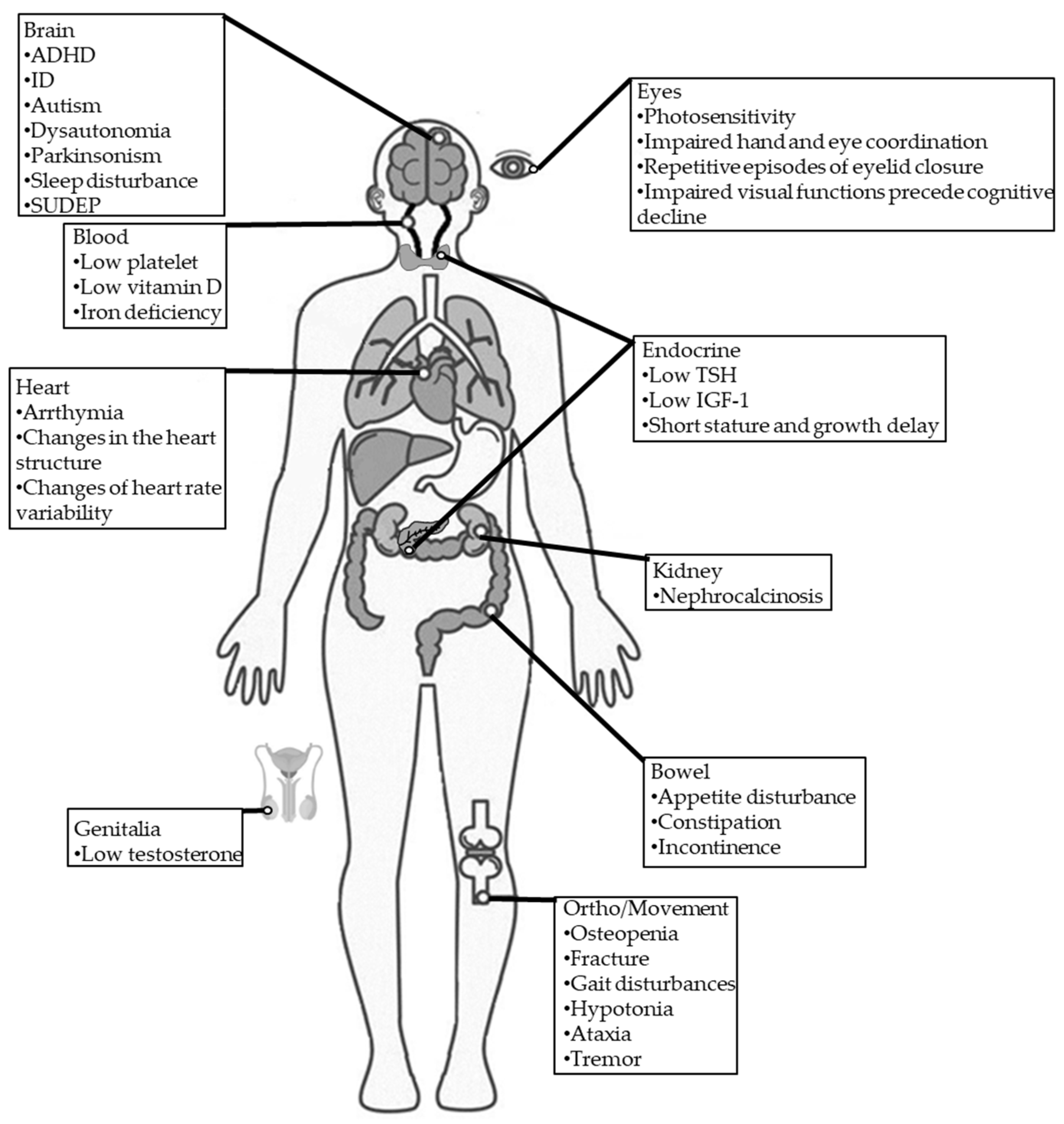
2.2.6. DS Comorbidities
DS is often associated with various comorbidities, such as blood, cardiac, digestive, endocrine, infection, neurodegenerative, orthopedic, and sleep impairments (Figure 1) [13].
Psychomotor
Blood
Approximately one-third of patients with DS were reported to have low platelets, vitamin D deficiency, and iron deficiency [13].
Cardiac
Digestive
Gastrointestinal problems are commonly reported symptoms in DS [13]. A cross-section study found that more than one third of patients with DS had dysphariga-related symptoms, such as impaied chewing, impaired swallowing, choking and drooling; more than 40% of pateints with DS had behavioral symptoms, such as picky eating and distracted during meals, and more than half of patients with DS had gastrointestinal symptoms, such as constipatio, and loss of appetite [79]. Stiripentol, a very effective AED against seizures in DS, was reported to be linked to worsening gastrointestinal symptoms in patients with DS [80].
Endocrine
Endocrine abnormalities associated with DS are very rare. Only one retrospective study involving 68 children with DS showed decreases in mean height and weight that were insensitive to age, sex, family history andanti-epileptic drugs (AEDs). Furthermore, they also found that some had low thyroid-stimulating hormone (TSH), insulin-like growth factor 1(IGF-1), and testosterone levels, supporting that endocrine dysfunction is related to DS [81]. Delayed and precocious puberty were also reported in patients with DS due to SCN1A mutation [13].
Infection
Chronic infectious diseases such as bronchitis, pneumonia, and otitis media have been frequently reported in patients with DS [13].
Neurodegenerative
Dysautonomia (autonomic dysfunction) of the heart may induce critical arrhythmia, which is a risk factor associated with sudden death in several heart diseases, with numerous experimental models supporting this association [82]. In addition, dysautonomia-induced overheating may potentially trigger seizures in patients with DS [1]. However, the underlying cause of dysautonomia is unclear. In another study of 12 adults with DS, Fasano et al. identified Parkinsonism features in 91%. Two patients experienced significant improvement in their Parkinsonian symptoms after receiving levodopa treatment [83].
Orthopedic/Movement
Individuals with drug-resistant epilepsy (DRE) often experience osteopenia, leading to elevated fracture risk [84]. However, osteopenia risk has not been studied in patients with DS. The reported incidence of ataxia(62%) [85], crouch gait (40%) [85], and hemiparesis (4.8%) [85] are common in patients with DS.
Sleep
Eyes
Photosensitivity is common in patients with DS [13]. It may be noticed when a flash of light induces eyelid myoclonic jerks, myoclonia, absences or generalized tonic–clonic seizures by eye closure, watching television, or fixation on patterns [52]. One study involving 12 patients with DS found that their hand and eye coordination were significantly impaired [35]. Importantly, impaired visual functions were found to precede cognitive decline in patients with DS, and visual functions were found to continually deteriorate in patients with DS, specifically those involving a specific visuo-motor dorsal pathway [89]. Besides visual impairment, eyelid closure may also be involved in DS. A case series reported that pateints with DS presented repetitive episodes of eyelid closure, which was proposed to be an early motor trait of DS [90].
2.2.7. SUDEP and Mortality
The incidence of SUDEP is significantly higher in patients with DS than in the general epilepsy population, with an estimated incidence of 9.32 per 1000-person-years [91].The risk of mortality is notably high in patients with DS. While mortality risk is higher for patients with DS than the general population in all age groups, with a reported mortality rate of 3%–21%, the highest mortality rate is observed during childhood. Previous studies have shown that the most common causes of death in patients with DS are SE, SUDEP, and drowning/injury during a seizure event [11]. Current evidence indicates that SUDEP risk is 15 times higher in DS than in other epilepsies [26]. The exact etiology of SUDEP in DS remains unclear. However, it has been hypothesized that hyper-stimulation of parasympathetic activity after generalized seizure increases the risks of severe bradycardia and electrical heart failure. A previous animal study showed that SCN1A-mutant mice had brain-induced bradycardia after a seizure [92]. Other studies have proposed that SCN1A haploinsufficiency contributes to ventricular dysfunction and arrhythmia in mice with a SCN1A mutation [93,94]. Because prolonged seizures without appropriate intervention may lead to SE or increase the risk of SUDEP, the detection of seizures is considerably important. While the core features of DS remain consistent across both children and adults, adults often represent a diagnostic challenge for DS [42] since they usually have different clinical features (Table 2).
3. DS Diagnosis and Long-Term Prognosis
3.1. Diagnostic Criteria
A large dutch cohort study revealed that the time for making a corrective diagnosis of DS is frequently delayed, even as late as three-years of age [95]. With the disease progression and exposure to inappropriate AEDs, patients with DS may experience seizure exacerbation and adverse neuro-development. Therefore, the diagnostic criteria for DS are critical and essential. While the diagnostic criteria for DS have evolved over time, DS remains a clinical diagnosis and should be suspected in previously healthy infants presenting with drug-resistant seizures before the first year of life, typically accompanied by fever and associated with neurodevelopmental regression. The ILAE diagnostic criteria for DS are summarized as follows [9]:
- Seizure onset in a previously healthy infant within the first year of life [96].
- A history of febrile and afebrile seizures [97].
- Seizures resistant to conventional AEDs [98].
- Prolonged, recurrent febrile and afebrile seizures [97].
- Appearance of multiple seizure types, including febrile or afebrile generalized/unilateral clonic/tonic seizures, myoclonic, and atypical absence. Focal seizures progress to focal motor or bilateral convulsive seizures [99].
- Children show cognitive and behavioral impairments from the second year of life [97].
- At the onset of the first seizures, neither parents nor physicians notice any developmental delay, which insidiously appears during the second year of life [9].
- A family history of epilepsy. SCN1A gene mutationswere initially identified in cases of genetic epilepsy with febrile seizures plus (GEFS+). A family history of epilepsy or febrile seizures has been noted in 15%–35% of DS cases, with affected family members often showing GEFS+. DS has been documented in identical twins, infrequently in non-identical twins, and occasionally among multiple siblings within a single family. A family history of febrile seizures and epilepsy in individuals with DS and de novo SCN1A mutations implies a polygenic mode of inheritance. Additional modifier genes, such as sodium voltage-gated channel alpha subunit 9 (SCN9A), may contribute to DS manifestations [100,101]. Abnormal EEG findings are characterized by generalized or multifocal epileptiform discharges [9].
- Seizures with photosensitivity are more challanging to control in DS [9].
- Psychomotor slowing followed by crouch gait, pyramidal signs, or interictal myoclonus [9].
- Recent consensus strongly recommended genetic testing for patients with suspected DS [9].
3.2. Genetic Testing in DS
Genetic testing for DS is vital for several reasons, including early and accurate diagnosis, tailored treatment strategies, and genetic counseling for families [100]. Early DS diagnosis is crucial since it has been shown that early recognition and intervention can significantly improve patient outcomes and quality of life [102]. In addition to guiding treatment, genetic testing for DS is essential for providing accurate genetic counseling to families. Since DS is an autosomal dominant disorder with a 50% chance of inheritance for each offspring of an affected parent [48], identifying its genetic basis allows families to make informed decisions about family planning and prenatal testing. Moreover, identifying the specific SCN1A mutation can help predict the severity of the disease and provide prognostic information since some SCN1A mutations have been associated with more severe phenotypes [10].
4. The Role of SCN1A in DS
4.1. SCN1A Mutations in DS
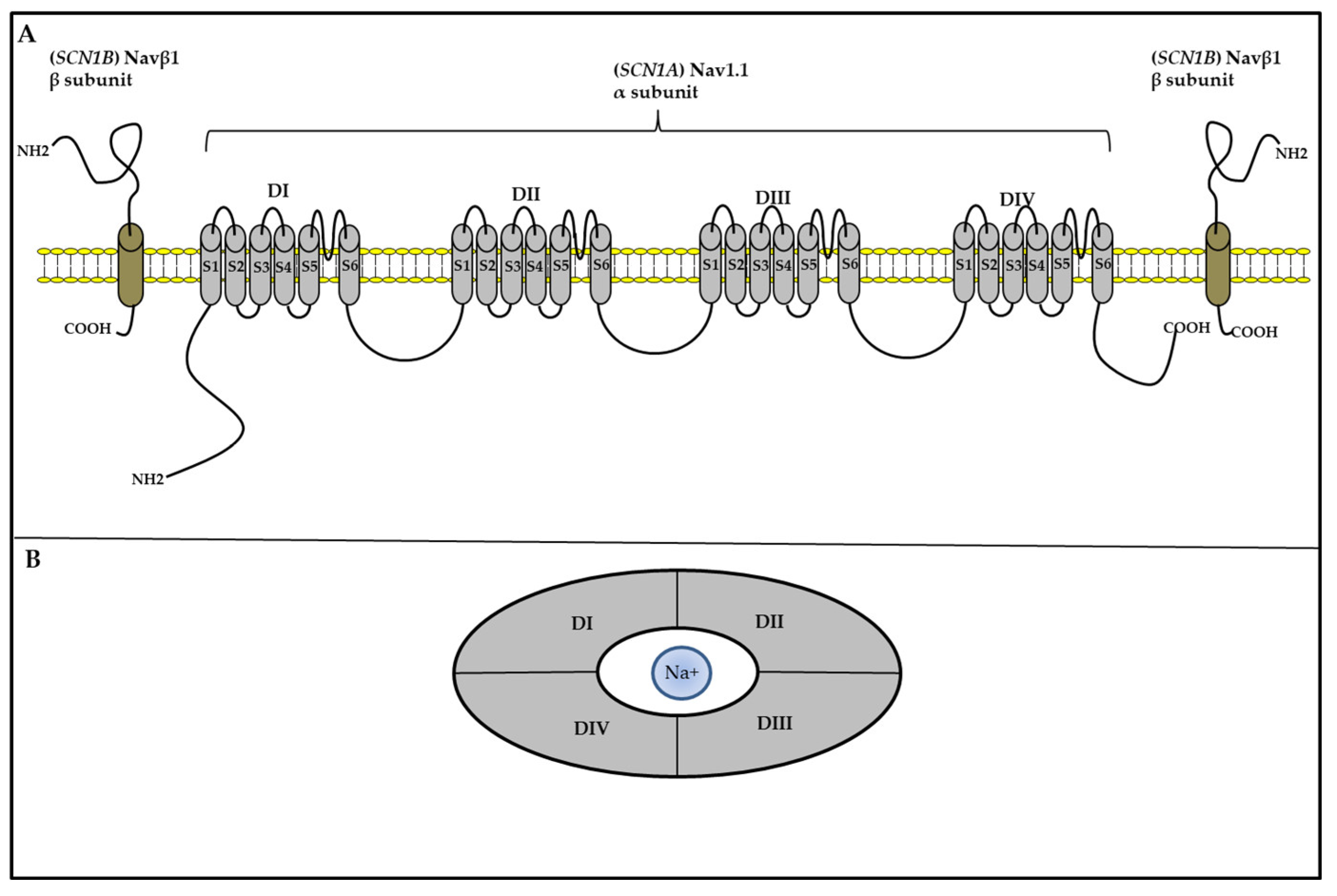
The voltage-gated sodium channel (VGSC) family comprises nine isoforms (Nav1.1–Nav1.9). Structurally, these VGSCs contain one of nine large α-subunits, which in humans are encoded bynine distinct genes (SCN1A-5A, and SCN7A-10A), and one of four ß-subunits (Navβ1–Navβ4), which in human are encoded by four distinct genes genes (SCN1B-4B) [103]. The α-subunits have all the machinery functions for channel cell surface expression, ion conduction, voltage sensing and gating, and inactivation. The β-subunits are accessories to α subunits, and help regulate gating, kinetics, and channel surface density [104].
SCN1A mutations account for nearly 80% of DS cases [49], and more than 2584 mutations have been identified to date (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php). The SCN1A gene is located on chromosome 2q24.3, and contains 27 exons. It encodes the sodium voltage-gated channel alpha subunit 1 (Nav1.1) with 2,009 amino acids (~229 kDa) [105]. The Nav1.1 channel comprises four repeated domains (DI-DIV), each containing six α-helical transmembrane segments (S1-S6) spanning the cell membrane; S4 is a voltage sensor, and S5 and S6 form the pores, that allow ions to pass through (Figure 1) [106].
Similar to other VGSCs, the Nav1.1 channel maintains the balance between neuronal excitation and inhibition, shaping complex neural networks that underlie cognition, perception, and behavior [107]. NaV1.1 channel is predominantly expressed in the central nervous system, particularly in inhibitory GABAergic interneurons, where it contributes to regulating neuronal excitability and fine-tuning synaptic transmission [106]. Dysfunction of the NaV1.1 channel has been implicated in various neurological disorders, including epilepsy, migraine, and autism spectrum disorders [108]
Mutations in the SCN1A gene were initially identified in cases of GEFS+. While most mutations develop due to de-novo truncating or missense changes, a family history of epilepsy or febrile seizures has been noted in 15%–35% of DS cases, with affected family members often exhibiting GEFS+. DS has been documented in identical twins, infrequently in non-identical twins, and occasionally among multiple siblings within a single family. The presence of unaffected or mildly-affected parents can be explained by the possibility of mosaic mutations, whether somatic or germline; previous reports suggested that mosaicism is present in 7% of DS families [109,110]. A family history of febrile seizures and epilepsy in individuals with DS and de novo SCN1A mutations implies a polygenic mode of inheritance. Additional modifier genes, such as sodium voltage-gated channel α subunit 9 (SCN9A), may contribute to the clinical presentations of DS [100,101]. The Nav1.1 channel activation depends on activating the fourth transmembrane segment (S4) of the four repeated domains (DI-DIV). On the other hand, the loop between DIII and DIV interacts with loops between S5 and S6 to inactivate the channel. SCN1A mutations affect the function of NaV1.1 differently based on the involved amino acids [111]. In return, there are obvious genotype-phenotype correlations in DS syndrome, and the disease severity can be closely correlated with the extent of mutations. Although GEFS+ is associated with less severe seizures, patients with GEFS+ and SCN1A mutations (nearly 10-15% of the patients) are characterized by hyperthermia-induced severe seizures [112]. In a previous retrospective study, patients with DS exhibited higher frequencies of truncating mutations and greater alterations in amino acid polarity compared to patients with GEFS+, supporting the involvement of SCN1A mutations in the clinical severity of DS [113]. However, it should be noted that the relationship between SCN1A mutations and phenotype has not been fully elucidated yet. Other factors like genetic background may affect DS's clinical severity. As the expression of voltage-gated sodium channels changes during the developmental process, it is expected that the SCN1A expression may play a role in the developmental onset and susceptibility to febrile seizures [114,115].
Figure 2.
The typical structure of a voltage-gated Nav channel. (A) The VGSC contains an α and β subunit, which are transmembrane proteins.The α subunit contains four homologous domains (DI-IV). Each domain comprises six transmembrane regions (S1–S6). S1–S4 forms the voltage-sensing domain, and S5 and S6 comprise the pore-forming domain. The β-subunits have a single transmembrane segment, a short intracellular domain with a single loop. (B) Top view from the extracellular face and side view of the Nav channel. The four domains of the α subunit form an Na+ -permeable pore lined by the re-entrant S5–S6 pore-loop segments.
Figure 2.
The typical structure of a voltage-gated Nav channel. (A) The VGSC contains an α and β subunit, which are transmembrane proteins.The α subunit contains four homologous domains (DI-IV). Each domain comprises six transmembrane regions (S1–S6). S1–S4 forms the voltage-sensing domain, and S5 and S6 comprise the pore-forming domain. The β-subunits have a single transmembrane segment, a short intracellular domain with a single loop. (B) Top view from the extracellular face and side view of the Nav channel. The four domains of the α subunit form an Na+ -permeable pore lined by the re-entrant S5–S6 pore-loop segments.
4.2. SCN1A-Mediated Hyperexcitability in DS: The Sodium Channel Interneuronopathy
Functional studies have shown that SCN1A mutants function differently, including loss-of-function (LOF) and gain-of-function (GOF). Among the VGSC subtypes, Nav1.1is the most important VGSC subtype for initiating action potentials in bipolar GABAergic inhibitory interneurons, which have crucial inhibitory activity [116]. Consequently, SCN1A LOF variants may cause dysfunction in those inhibitory interneurons and alter neuronal excitability by reducing inhibition, leading to a predisposition to seizures [117]. AEDs such as valproic acid (VPA), which can elevate gamma aminobutyric acid (GABA)levels by inhibiting transaminase, may be effective. However, sodium channel blockers may aggravate seizure attacks [118]. GOF reflects a small and non-inactivating current becoming a large and activating one, leading to neuron hyperexcitability [119]. SCN1A GOF-mutant-associated epilepsy may be controlled by the appropriate use of adequate AEDs that can inhibit sodium channels, such as phenytoin and carbamazepine. Modifying the gating properties of the SCN1A mutant can lead to an imbalance in excitatory to inhibitory neurotransmission in neural circuits, causesing both SCN1A-associated epilepsies and co-morbidities [102].
In addition, SCN1A haploinsufficiency decreases the availability of functional NaV1.1 channels on the cell membrane, impairing their ability to release the inhibitory neurotransmitter GABA effectively [102]. The reduced inhibitory input onto excitatory neurons disrupts the balance between excitation and inhibition in the neuronal network, resulting in hyperexcitability [120]. This heightened excitability renders the brain more susceptible to seizures, often triggered by external factors such as fever, stress, or sensory stimulation [11]. Several experimental models confirmed the involvement of SCN1A LOF in hyperexcitability and seizures [117,121]. In an SCN1A knock-out model, there was increased susceptibility to hyperthermia-induced seizures, which was found to arise from the reduced sodium currents in the inhibitory GABAergic interneurons. It was also noted that there is impaired inhibitory input to postsynaptic cells [120].
The hypothesis of dysregulated inhibitory interneurons in DS is further supported by the NaV1.1 expression distribution pattern. Three major classes of interneurons, distinguished by their molecular markers and functional properties, have been extensively studied: parvalbumin (PV), somatostatin (SST), and serotonin receptor 3a (5-HT3aR) expressing interneurons [122]. PV interneurons are fast-spiking cells that predominantly target the perisomatic region of excitatory neurons [123]. They play a crucial role in regulating the temporal aspects of neural activity, such as generating gamma oscillations, which are essential for cognitive processes like attention and memory [124]. Previous reports found that the NaV1.1 expression is more predominant in the PV+ interneurons of the neocortex, hippocampus, and cerebellum, supporting the involvement of PV+ GABAergic neurons in SCN1A-related epilepsy [111]. Besides, recent evidence highlighted that, in DS models, deletion of Nav1.1 in PV+ interneurons led to autistic-like social interaction deficits and pro-epileptic effects [125].
4.3. Interneuronopathy and Cognition, Developmental Impairment, and Comorbidities
4.3.1. Role of Interneuronopathy in Cognition and Development
When evaluating the impact of SCN1A mutations on cognition in DS, it is crucial to understand Nav1.1 function in the healthy and developing brain. Research from animal models indicates that SCN1A mutations in DS impair the function of GABAergic interneurons, particularly PV+ interneurons. These cells are essential for the spatiotemporal regulation of neural network activity during cognitive processes. Thus, even a slight impairment in their function may lead to significant cognitive and behavioral disturbances, contributing to severe cognitive impairments in individuals with DS [102].
Interneurons are more diverse and complex than initially thought, serving roles beyond simply regulating excitability. They can be classified based on their target cells' dendritic or perisomatic domain, with the latter being characterized by the expression of PV. Perisomatic-targeting PV+ interneurons are essential for regulating neural network synchrony and temporal patterns, as demonstrated by their role in gamma and theta oscillations [123,126]. These oscillations are vital for cognitive processes and require the proper function of PV+ fast-spiking interneurons, which are believed to be primarily affected by DS [111]. Gamma oscillations are a type of brain rhythm associated with cognitive functions such as sensory processing, memory, and attention. PV+ interneurons play a significant role in generating cortical gamma oscillations. In animal models of DS, disrupted gamma oscillations have been observed, which may contribute to the cognitive impairments observed in affected individuals [127].
Theta oscillations, which play a significant role in cognitive function and provide a temporal framework for neural network activity, also rely on PV+ interneurons. In the hippocampus, PV+ interneurons help generate theta oscillations, and blocking their synaptic transmission directly impairs spatial working memory [128,129]. Furthermore, PV+ GABAergic neurons in distal network structures are required for proper hippocampal and cortical oscillations. Disruptions in these oscillations can result in severe spatial memory impairment, exemplifying the importance of oscillatory network activity in shaping pyramidal cell output during cognitive processing. Consequently, any dysfunction in interneurons, as seen in DS, can significantly affect cognitive processes [130].
Interneurons can have a developmental role in patients with SCN1A mutations. In mice, for example, SCN1A expression coincides with the neurophysiological maturation of fast-spiking interneurons and the evolution of spatial cognition. PV+ fast-spiking interneurons undergo a drop in action potential breadth and an increase in firing frequency during the second and third postnatal weeks, changing them from slow to fast signalling cells [131]. The discovery that interneurons in SCN1A-mutant mice do not develop the narrow action potential width characteristic of mature cells suggests that SCN1A expression is important in this metamorphosis [120].
5. Novel Pharmacological Approaches for DS Management: What Is the Role of Precision Medicine?
5.1. Standard DS Management
Seizures are known to lead to ID, and SE may cause numerous hospitalizations and death. Since seizures are frequently pharmacoresistant in patients with DS [132], control of convulsive seizures should be prioritized over nonconvulsive seizures, given their greater impact on quality of life and higher association with SUDEP [133]. Several AEDs and surgical options are currently available for DS. Complete seizure control is typically unachievable in patients with DS, and the management goal should focus on reducing the risk of extended seizures and SE [54]. Measures to reduce seizure triggers are also recommended, including allowing the child to nap if tired, avoidance of high ambient temperature, prophylactic antipyretics for vaccination and illness, prophylactic benzodiazepines or antibiotics for febrile illness, and avoiding overexertion or flashing lights [42].
Most patients with DS require at least two AEDs to achieve seizure control. Both clobazam and VPA are widely recommended first-line options for patients with DS [54]. VPA, a branched-chain fatty acid, can be administered intravenously, orally, or rectally and is generally well tolerated. VPA’s mechanisms of action include affecting the GABAergic system, inhibiting α-ketoglutarate dehydrogenase, GABA transaminase, and succinate semialdehyde dehydrogenase, and enhancing glutamate decarboxylase to increase GABA levels in plasma and several brain regions. Eventually, VPA can stimulate GABA receptors to interfere with sodium channels, modulate potassium and calcium conductance, affect serotoninergic and dopaminergic transmission, and alter cerebral metabolism [134]. All make VPA a very effective AED against a range of generalized and focal seizure types including tonic-clonic, myoclonic, tonic, partial, and absence seizures [135]. The starting VPA dose is recommended between 10 and 15 mg/kg/day, divided into two or three doses, followed by gradual increases to target doses in the range of 25 to 60 mg/kg/day based on patients’ tolerability and clinical responses. Despite several advantages, VPA is uncommonly used as monotherapy in patients with DS. Three studies reported responder rates of 23-52% [136,137,138]. However, the literature and personal experience suggest that polypharmacy, instead of VPA alone, may be sufficient to control seizures in patients with DS. Notably, VPA may increase serum concentrations of lamotrigine, phenobarbital, the metabolite 10,11-epoxide of carbamazepine, ethosuximide and rufinamide because VPA can inhibit cytochrome P450s, uridine glucuronyl transferases and epoxide hydrolase. Common side effects of VPA include abdominal cramps, diarrhea, impaired coagulation, nausea, neutropenia, vomiting, and weight gain. VPA may be associated with hepatotoxicity, pancreatitis, teratogenicity, and endocrine disturbance, such as increased total testosterone levels, menstrual abnormalities, obesity, polycystic ovary syndrome, and teratogenicity [134].
Clobazam, (7-chloro-1-methyl-5-phenyl-1,5-benzodiazepine), first synthesized in 1966, has been marketed as an anxiolytic agent since 1975 and as an AED since 1984 [139,140]. Clobazam is a benzodiazepine, and has good oral tolerability, safety, and broadspectrum anti-seizure effects with weak sedative and amnesic effects than other benzodiazepines [141]. Due to its excellent effects and safety, clobazam is now approved as an adjunctive treatment for epilepsy in more than 100 countries [142]. The anti-seizure activity of clobazam may be mediated through its GABA-A receptor agonist activity and its active metabolite, N-desmethyl-clobazam (norclobazam) [141].
The double-blind, placebo controlled, efficacy and safety CONTAIN (ClObazam in patieNTs with LennoxGAstaut SyNdrome) trial showed that physicians and caregivers assessments indicated clobazam significantly improved weekly drop seizure rates in Lennox Gastaut Syndrome (LGS) [143]. Another study involving patients with LGS receiving adjunctive clobazam showed sustained seizure-free and significant seizure improvements at stable dosages through three years of therapy [144]. These findings support the excellent anti-seizure effects of clobazam. Since AEDs can only partially control DS-related seizures [132], clinical trials using valproate, stiripentol and clobazame only achieved 71%-89% responder rate in reducing seizure frequency in patients with DS [136,145]. Consistent with these findings, treating DS with a triple therapy of stiripentol, valproate and clobazam resulted in about twofold increases in clobazam and 5-7- fold increases in norclobazam levels but no change in plasma valproate levels [146]. Therefore, valproate, stiripentol and clobazame are regarded as the “gold standard treatment.”
The initial clobazam dose is 0.2 mg/kg/day, divided into twice daily doses, followed by gradual increases to reach a target dose of 0.3-1.0 mg/kg/day, up to a maximum of 2 mg/kg/ day. It is recommended that clobazam dose escalation should not exceed weekly since clobazam and norclobazam for concentrations in patients’ serum may reach a steady state after 5 and 9 days, respectively [147,148]. Adverse effects include upper respiratory infections, constipation, drooling, lethargy, somnolence, pyrexia, and respiratory depression, especially at high doses or with concomitant opioids [143,144].
Previous reports showed that clobazam and VPA led to significant seizure reduction in patients with DS when combined with stiripentol [137,149]. Stiripentol is an AED specifically developed to treat DS. It was granted orphan drug designation in the European Union in 2001 and the US in 2008. Stiripentol acts as a positive allosteric modulator of the γ-aminobutyric acid type A (GABAA) receptor, enhancing GABA's inhibitory effect on neuronal activity [150]. The efficacy of stiripentol in DS was demonstrated in a pivotal clinical trial, which showed that 71% of the patients in the stiripentol group had a ≥50% reduction in seizure frequency during the two-month treatment period compared to only 5% in the placebo group [149]. Stiripentol or topiramate should be combined with the clobazam and VPA regimen, especially when the first-line regimen fails to achieve adequate control. Clonazepam, levetiracetam, zonisamide, ethosuximide, and phenobarbital may be used in cases with inadequate control on the first and second lines [42]. A high-fat, low-carbohydrate ketogenic diet for one year was reported to achieve a ≥75% reduction in seizure frequency and severity in nearly 80% of children with DS [151]. The initial stiripentol dose varies in different regions. The starting dose is 20 mg/kg/day up to a maximum of 1000mg/day in Japan, 50 mg/kg/day up to a maximum of 3000mg/day in the USA, and 20-50 mg/kg/day, divided into two to three daily doses, in Eruope [152,153].
While histopathological results may help uncover the genetic pathophysiology of DS-related epilepsy, studies on histopathological outcomes in DS are scarce [154]. Surgical options for these patients are limited and generally considered a last resort when pharmacological and dietary interventions have proven insufficient. A case series While surgical interventions for other forms of epilepsy, such as resective surgery or the implantation of a vagus nerve stimulator, are effective, their applicability to DS is not as well-established due to its genetic and generalized nature [7]. Vagus nerve stimulation (VNS) has been used as a treatment option for patients with DRE, including a small subset of patients with DS. A few case reports and small-scale studies have reported varying degrees of success with VNS in patients with DS [155]. However, corpus callosotomy use has been documented in patients with DS, but with mixed results [156]. There is limited evidence for the effectiveness of deep brain stimulation in DS [157].
Despite the plethora of available AEDs and trials of surgical options, the management of DS remains suboptimal since patients with DS may respond differently to various therapies. Therefore, personalized management is crucial to optimize treatment outcomes and ensure the best possible quality of life for individuals with DS.
5.2. Novel Pharmacological Approaches
5.2.1. CBD
CBD is a non-psychoactive compound found in the cannabis plant that has gained significant attention for its potential therapeutic effects in various neurological disorders, including DS. The results of CBD treatment for DS have been promising, as demonstrated by two pivotal clinical trials. Based on their findings, the US Food and Drug Administration (FDA) approved CBD for DS in 2018.
In the first randomized, double-blind, placebo-controlled trial, 120 children and young adults with DS were enrolled and treated with either CBD or a placebo in addition to their existing AEDs. Its results showed a significant reduction in the median frequency of convulsive seizures per month in the CBD group compared to the placebo group. The median reduction in seizure frequency was 38.9% in the CBD group compared to 13.3% in the placebo group. Moreover, 5% of the patients in the CBD group became seizure-free during the trial, compared to none in the placebo group. Regarding safety, some adverse events were reported in both the CBD and placebo groups. The most common side effects in the CBD group included diarrhea, vomiting, fatigue, pyrexia, somnolence, and abnormal liver function tests. However, most of these side effects were considered mild to moderate in severity [158]. The other double-blind trial showed that CBD reduced the seizure frequency by 45.7%–48.7% compared to 26.9% in the placebo group [159]. An open-label extension of the trial showed a median reduction in seizure frequency of 44% after a 48-week follow-up period [160].
5.2.2. Atypical Sodium Channel Blocker
Phenotypic variability and a wide range of SCN1A mutations in DS have led studies to investigate various treatment options. One such option is atypical sodium channel blockers, which target the neuronal voltage-gated sodium channels. These channels, particularly NaV1.1 encoded by SCN1A, are impaired by SCN1A mutations in patients with DS, leading to increased neuronal excitability and seizures. GS967 is one atypical sodium channel blocker that has been evaluated for DS. In a previous experimental model study, GS967 reduced seizure frequency and premature death in DS mice, attributed to the inhibition of sustained sodium currents and subsequent inhibition of synchronous repetitive action potential [161].
5.2.3. Fenfluramine
Fenfluramine was initially used as an appetite suppressant in the 1960s but was withdrawn from the market due to cardiovascular valvular side effects [162]. However, due to its inhibition of the serotonin (5-hydroxytryptamine [5-HT]) transporter, activation of multiple 5-HT receptors, enhancement of 5-HT release into the synapse [163], positive modulation of the sigma-1 receptor [164], and some promising results, low dose fenfluramine was allowed for use in children with intractable epilepsy [162]. Furthermore, these results also support it as a potential treatment for DS [165,166]. The efficacy of low-dose fenfluramine in DS was demonstrated in a randomized, double-blind, placebo-controlled trial by Lagae et al. This trial enrolled 119 children and adolescents with DS experiencing drug-resistant seizures who were randomized to receive either fenfluramine or a placebo in addition to their existing AEDs. Its results showed a significant reduction in the monthly seizure frequency in the fenfluramine group (62.3%) compared to the placebo group (13.2%). Additionally, 54% of the patients in the fenfluramine group experienced a reduced seizure frequency of ≥50%, compared to only 5% in the placebo group [167]. This trial's positive results led the FDA to approve fenfluramine in 2020 for treating seizures associated with DS in patients aged ≥2 years. Additionally, patients given fenfluramine had significantly longer seizure-free periods [167,168,169]. Fenfluramine use is reported to significantly reduce SE episodes [170,171,172,173]. Moreover, fenfluramine may reduce SUDEP mortality rates in patients with DS [174]. Fenfluramine is generally well tolerated. Its common adverse effects include upper respiratory tract infection, decreased weight, diarrhea, fatigue, lethargy, and pyrexia. Fenfluramine is contraindicated in patients with aortic or mitral valvular heart disease and pulmonary arterial hypertension. The prescribing physicians must conduct periodic echocardiograms in patients taking fenfluramine to monitor their risk for heart disease and cardiac death [175,176]. If patients are co-administered other serotonergic drugs, clinicians should be aware of the possibility of serotonin syndrome [175,176]. If patients also take VPA, clinicians should keep in mind that fenfluramine may exacerbate VPA-induced thrombocytopenia [177]. Fenfluramine is taken twice daily. However, an initial dose of 0.2 mg/kg/day is recommended, increasing to 0.4 mg/kg/day after seven days; 0.4 mg/kg/day is the recommended maintenance dose in patients taking stiripentol, which can increase plasma fenfluramine concentrations. If patients do not take stiripentol, a further increase to the suggested maintenance dose of 0.7 mg/kg/day can occur after another seven days [175].
5.2.4. Other Agents
Other agents currently being studied in patients with DS include soticlestat (a cholesterol 24-hydroxylase inhibitor) and medications targeting the serotonin pathway, such as clemizole and lorcaserin [8]. In addition, tiagabine, which inhibits GABA reuptake into presynaptic neurons, has emerged as a possible treatment for DS. Tiagabine can increase GABA availability and enhance its inhibitory effect on neuronal activity. While tiagabine has been approved for the adjunctive treatment of partial seizures in adults and children aged ≥12 years, its role in DS has not been well-established. A synergism between tiagabine and clonazepam was evident in DS models, with reduced side effects for single drugs [178]. However, it is important to note that the evidence supporting tiagabine use in DS is limited and not based on rigorous clinical trials.
5.3. Disease-Modifying Therapies
5.3.1. ASO Therapy
ASO involves using small, synthetic RNA molecules to modulate the expression of specific genes. In the context of DS, ASO therapy has been investigated as a potential treatment option due to its genetic nature (i.e., mutations in the SCN1A gene encoding the voltage-gated sodium channel NaV1.1). Preclinical studies have shown that ASO therapy significantly reduced electrographic seizures and mortality in DS and improved behavioral outcomes in the treated mice, whose brain NaV1.1 levels increased and became comparable to wild-type mice [15]. The preliminary results of the phase I/IIa MONARCH trial showed that ASO therapy was well-tolerable and reduced convulsive seizure frequency (median reductions of 17%–37%) [179]. While these findings are encouraging, further studies are needed to optimize the design and delivery of ASOs and assess this therapeutic approach's long-term safety and efficacy in human patients with DS.
5.3.2. AAV-9 Based Gene Therapy
AAV-9 delivers functional copies of genes directly into target cells, compensating for the effects of genetic mutations. ETX-101 is a gene therapy using AAV-9 as a delivery method, which contains an engineered transcription factor governed by a regulatory element specific to GABAergic cells. Its purpose is to increase the transcription and subsequent translation of the SCN1A gene in inhibitory interneurons. This therapy has been examined in a mouse model of DS, in which it decreased the frequency and intensity of seizures during postnatal days 26–28. Additionally, it reduced mortality rates up to 470 days after treatment [16].
6. Expert Perspectives and Future Directions
Precision medicine, an emerging approach that tailors medical treatment to an individual's unique genetic, environmental, and lifestyle factors, has the potential to revolutionize DS management. A significant advance in understanding DS is the identification of the SCN1A gene as its primary cause in most cases. This discovery has motivated research into developing gene therapies that could potentially target and correct the underlying genetic defects. Targeted gene therapies can potentially provide a more effective and long-lasting solution for the management of patients with DS since theose therapies address its root cause rather than only managing its symptoms. Several studies have shown that certain medications may be more effective in individuals with specific genetic variants, paving the way for tailored pharmacological interventions in DS. However, gene therapy for DS is still in its early stages, and further research is needed to determine its safety, efficacy, and feasibility in clinical settings.
Genetic testing plays a critical role in implementing precision medicine for DS management. Early and accurate DS diagnosis through genetic testing allows healthcare providers to promptly implement appropriate interventions, improving the overall prognosis and quality of life for affected individuals. Moreover, genetic testing can identify carriers of SCN1A mutations among family members, enabling informed reproductive choices and early intervention strategies for at-risk offspring. As our understanding of the genetic underpinnings of DS expands, the role of genetic testing in guiding precision medicine approaches will continue to grow.
7. Conclusions
Precision medicine has significant potential for improving the diagnosis and treatment of DS. Early genetic testing enables timely and accurate diagnosis. Advances in our understanding of DS's underlying genetic mechanisms and neurobiology have enabled the development of targeted therapies, such as gene therapy or optogenetics, that might offer more effective and less invasive treatment options for patients with DS. Targeted therapies and gene therapy provide hope for more effective and personalized treatments. However, research into these therapeutic approaches is still in its early stages, and their clinical application remains to be seen.
Author Contributions
Conceptualization, H.-C.F.; investigation, K.-L.C., L.-C.L.; resources, K.-L.C., L.-C.L., and I.-C.C.; data curation, K.-L.C., L.-C.L. and I.-C.C.; writing—original draft preparation, H.-C.F.; writing—review and editing, I.-C.C., and C.-M.C.; supervision, I.-C.C., and C.-M.C.; project administration, K.-L.C., L.-C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Tungs’ Taichung metroharbor hospital, grant number TTMHH-R1120001; TTMHH-C1120007.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable because this review did not report any new data.
Acknowledgments
We would like to express our sincere gratitude to Hsiu Ching Tsai (Department of Nursing, Tungs’ Taichung metro harbor hospital, Taichung, Taiwan) for collecting and analysis data. We would like to thank I-Ching Chou (Department of Pediatrics, Children’s medical center, China Medical University Hospital, Taichung, Taiwan), Shyi-Jou Chen (Department of Pediatrics, Tri-Service general hospital, Taipei, Taiwan), Wang-Tso Lee (Department of Pediatric Neurology, National Taiwan University Children’s Hospital), Kuang-Lin Lin and Huei-Shyong Wang (Division of Pediatric Neurology, Chang Gung Children’s Hospital at Linkou, Taoyuan, Taiwan; College of Medicine, Chang Gung University, Taoyuan, Taiwan) for helpful suggestions and comments on the MS.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAV | adeno-associated virus |
| ACC | agenesis of the corpus callosum |
| ADHD | attention deficit hyperactivity disorder |
| AGC1 | aspartate-glutamate carrier 1 |
| ASO | antisense oligonucleotides |
| AEDs | antiepileptic drugs |
| CBD | cannabidiol |
| CDG | congenital disorder of glycosylation |
| CONTAIN | ClObazam in patieNTs with LennoxGAstaut SyNdrome |
| DEs | developmental encephalopathies |
| DEE | developmental and epileptic encephalopathies |
| DECAM | developmental delay, epileptic encephalopathy, cerebral atrophy, and abnormal myelination |
| DRE | drug-resistant epilepsy |
| DS | Dravet syndrome |
| EEs | epileptic encephalopathies |
| EEG | electroencephalography |
| EFMR | epilepsy and mental retardation restricted to females |
| EEOC | epileptic encephalopathy, childhood-onset |
| FDA | Food and Drug Administration |
| GABA | γ-aminobutyric acid |
| GEFS+ | genetic epilepsy with febrile seizures plus |
| GOF | gain-of-function |
| GOT2 | glutamate oxaloacetate transaminase |
| GPIBD | glycophosphatidylinositol biosynthesis defect |
| IECEE | epileptic encephalopathy, infantile or early childhood |
| IGF-1 | insulin-like growth factor 1 |
| ILAE | International League Against Epilepsy |
| ISSX2 | infantile spasm syndrome, X-linked 2 |
| IQ | intelligence quotient |
| LGS | Lennox Gastaut syndrome |
| LISX2 | lissencephaly, X-linked, 2 |
| LOF | loss-of-function |
| MCAHS | multiple congenital anomalies-hypotonia-seizures syndrome |
| MCSZ | microcephaly, seizures, and developmental delay |
| MRX | mental retardation, X-linked |
| MRXS1 | mental retardation, X-linked, syndromic 1 |
| MRI | magnetic resonance imaging |
| NCSE | non-convulsive status epilepticus |
| PV | parvalbumin |
| PRTS | Partington syndrome |
| SCN1A | voltage-gated channel alpha subunit 1 |
| SCN9A | voltage-gated channel α subunit 9 |
| SE | status epilepticus |
| SMEI | severe myoclonic epilepsy of infancy |
| SST | somatostatin |
| SUDEP | sudden unexpected death in epilepsy |
| TSH | thyroid-stimulating hormone |
| VGSC | voltage-gated sodium channel |
| VNS | vagus nerve stimulation |
| VPA | valproic acid |
| XLAG | X-linked lissencephaly with ambiguous genitalia |
| XLID29 | intellectual developmental disorder, X-linked 29 |
| 5-HT | 5-hydroxytryptamine |
| 5-HT3aR | serotonin receptor 3a |
References
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef]
- Engel, J., Jr.; International League Against, E. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001, 42, 796–803. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Liao, J. Deciphering the concepts behind "Epileptic encephalopathy" and "Developmental and epileptic encephalopathy". Eur. J. Paediatr. Neurol. 2020, 24, 11–14. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Palmer, E.E.; Howell, K.; Scheffer, I.E. Natural History Studies and Clinical Trial Readiness for Genetic Developmental and Epileptic Encephalopathies. Neurotherapeutics 2021, 18, 1432–1444. [Google Scholar] [CrossRef]
- Symonds, J.D.; McTague, A. Epilepsy and developmental disorders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 2020, 24, 15–23. [Google Scholar] [CrossRef]
- Khan, S.; Al Baradie, R. Epileptic encephalopathies: An overview. Epilepsy Res. Treat. 2012, 2012, 403592. [Google Scholar] [CrossRef]
- Sullivan, J.; Wirrell, E.C. Dravet Syndrome as an Example of Precision Medicine in Epilepsy. Epilepsy Curr. 2023, 23, 4–7. [Google Scholar] [CrossRef]
- Zuberi, S.M.; Wirrell, E.; Yozawitz, E.; Wilmshurst, J.M.; Specchio, N.; Riney, K.; Pressler, R.; Auvin, S.; Samia, P.; Hirsch, E.; et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1349–1397. [Google Scholar] [CrossRef]
- Cetica, V.; Chiari, S.; Mei, D.; Parrini, E.; Grisotto, L.; Marini, C.; Pucatti, D.; Ferrari, A.; Sicca, F.; Specchio, N.; et al. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology 2017, 88, 1037–1044. [Google Scholar] [CrossRef]
- Connolly, M.B. Dravet Syndrome: Diagnosis and Long-Term Course. Can. J. Neurol. Sci. 2016, 43 (Suppl 3), S3–8. [Google Scholar] [CrossRef]
- Anwar, A.; Saleem, S.; Patel, U.K.; Arumaithurai, K.; Malik, P. Dravet Syndrome: An Overview. Cureus 2019, 11, e5006. [Google Scholar] [CrossRef]
- Villas, N.; Meskis, M.A.; Goodliffe, S. Dravet syndrome: Characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. 2017, 74, 81–86. [Google Scholar] [CrossRef]
- Li, W.; Schneider, A.L.; Scheffer, I.E. Defining Dravet syndrome: An essential pre-requisite for precision medicine trials. Epilepsia 2021, 62, 2205–2217. [Google Scholar] [CrossRef]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena; Aznarez, I.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Tanenhaus, A.; Stowe, T.; Young, A.; McLaughlin, J.; Aeran, R.; Lin, I.W.; Li, J.; Hosur, R.; Chen, M.; Leedy, J.; et al. Cell-Selective Adeno-Associated Virus-Mediated SCN1A Gene Regulation Therapy Rescues Mortality and Seizure Phenotypes in a Dravet Syndrome Mouse Model and Is Well Tolerated in Nonhuman Primates. Hum. Gene Ther. 2022, 33, 579–597. [Google Scholar] [CrossRef]
- Symonds, J.D.; Elliott, K.S.; Shetty, J.; Armstrong, M.; Brunklaus, A.; Cutcutache, I.; Diver, L.A.; Dorris, L.; Gardiner, S.; Jollands, A.; et al. Early childhood epilepsies: Epidemiology, classification, aetiology, and socio-economic determinants. Brain 2021, 144, 2879–2891. [Google Scholar] [CrossRef]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O'Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef]
- Wu, Y.W.; Sullivan, J.; McDaniel, S.S.; Meisler, M.H.; Walsh, E.M.; Li, S.X.; Kuzniewicz, M.W. Incidence of Dravet Syndrome in a US Population. Pediatrics 2015, 136, e1310–e1315. [Google Scholar] [CrossRef]
- Bayat, A.; Hjalgrim, H.; Moller, R.S. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22,000: A population-based study from 2004 to 2009. Epilepsia 2015, 56, e36–e39. [Google Scholar] [CrossRef]
- Schubert-Bast, S.; Kay, L.; Simon, A.; Wyatt, G.; Holland, R.; Rosenow, F.; Strzelczyk, A. Epidemiology, healthcare resource use, and mortality in patients with probable Dravet syndrome: A population-based study on German health insurance data. Epilepsy Behav. 2022, 126, 108442. [Google Scholar] [CrossRef] [PubMed]
- Yakoub, M.; Dulac, O.; Jambaque, I.; Chiron, C.; Plouin, P. Early diagnosis of severe myoclonic epilepsy in infancy. Brain Dev. 1992, 14, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Rosander, C.; Hallbook, T. Dravet syndrome in Sweden: A population-based study. Dev. Med. Child. Neurol. 2015, 57, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Ellis, R.; Reavey, E.; Forbes, G.H.; Zuberi, S.M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012, 135, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Perez-Palma, E.; Ghanty, I.; Xinge, J.; Brilstra, E.; Ceulemans, B.; Chemaly, N.; de Lange, I.; Depienne, C.; Guerrini, R.; et al. Development and Validation of a Prediction Model for Early Diagnosis of SCN1A-Related Epilepsies. Neurology 2022, 98, e1163–e1174. [Google Scholar] [CrossRef] [PubMed]
- Skluzacek, J.V.; Watts, K.P.; Parsy, O.; Wical, B.; Camfield, P. Dravet syndrome and parent associations: The IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia 2011, 52 (Suppl 2), 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bluvstein, J.; Wenniger, S. Two Perspectives on Dravet Syndrome: Viewpoints from the Clinician and the Caregiver. Neurol. Ther. 2023, 12, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, D.; Chieffo, D.; Siracusano, R.; Waure, C.; Brogna, C.; Ranalli, D.; Contaldo, I.; Tortorella, G.; Dravet, C.; Mercuri, E.; et al. Cognitive decline in Dravet syndrome: Is there a cerebellar role? Epilepsy Res. 2013, 106, 211–221. [Google Scholar] [CrossRef]
- Ragona, F.; Brazzo, D.; De Giorgi, I.; Morbi, M.; Freri, E.; Teutonico, F.; Gennaro, E.; Zara, F.; Binelli, S.; Veggiotti, P.; et al. Dravet syndrome: Early clinical manifestations and cognitive outcome in 37 Italian patients. Brain Dev. 2010, 32, 71–77. [Google Scholar] [CrossRef]
- Riva, D.; Vago, C.; Pantaleoni, C.; Bulgheroni, S.; Mantegazza, M.; Franceschetti, S. Progressive neurocognitive decline in two children with Dravet syndrome, de novo SCN1A truncations and different epileptic phenotypes. Am. J. Med. Genet. A 2009, 149A, 2339–2345. [Google Scholar] [CrossRef]
- Verheyen, K.; Verbecque, E.; Ceulemans, B.; Schoonjans, A.S.; Van De Walle, P.; Hallemans, A. Motor development in children with Dravet syndrome. Dev. Med. Child. Neurol. 2019, 61, 950–956. [Google Scholar] [CrossRef]
- Wolff, M.; Casse-Perrot, C.; Dravet, C. Severe myoclonic epilepsy of infants (Dravet syndrome): Natural history and neuropsychological findings. Epilepsia 2006, 47 Suppl. 2, 45–48. [Google Scholar] [CrossRef]
- Kedem, P.; Scher, D.M. Evaluation and management of crouch gait. Curr. Opin. Pediatr. 2016, 28, 55–59. [Google Scholar] [CrossRef]
- Rodda, J.M.; Scheffer, I.E.; McMahon, J.M.; Berkovic, S.F.; Graham, H.K. Progressive gait deterioration in adolescents with Dravet syndrome. Arch. Neurol. 2012, 69, 873–878. [Google Scholar] [CrossRef]
- Chieffo, D.; Battaglia, D.; Lettori, D.; Del Re, M.; Brogna, C.; Dravet, C.; Mercuri, E.; Guzzetta, F. Neuropsychological development in children with Dravet syndrome. Epilepsy Res. 2011, 95, 86–93. [Google Scholar] [CrossRef]
- Guzzetta, F. Cognitive and behavioral characteristics of children with Dravet syndrome: An overview. Epilepsia 2011, 52 Suppl. 2, 35–38. [Google Scholar] [CrossRef]
- Jansson, J.S.; Hallbook, T.; Reilly, C. Intellectual functioning and behavior in Dravet syndrome: A systematic review. Epilepsy Behav. 2020, 108, 107079. [Google Scholar] [CrossRef]
- Takayama, R.; Fujiwara, T.; Shigematsu, H.; Imai, K.; Takahashi, Y.; Yamakawa, K.; Inoue, Y. Long-term course of Dravet syndrome: A study from an epilepsy center in Japan. Epilepsia 2014, 55, 528–538. [Google Scholar] [CrossRef]
- Ragona, F. Cognitive development in children with Dravet syndrome. Epilepsia 2011, 52 (Suppl 2), 39–43. [Google Scholar] [CrossRef]
- Nabbout, R.; Chemaly, N.; Chipaux, M.; Barcia, G.; Bouis, C.; Dubouch, C.; Leunen, D.; Jambaque, I.; Dulac, O.; Dellatolas, G.; et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J. Rare Dis. 2013, 8, 176. [Google Scholar] [CrossRef]
- Dravet, C.; Bureau, M.; Dalla Bernardina, B.; Guerrini, R. Severe myoclonic epilepsy in infancy (Dravet syndrome) 30 years later. Epilepsia 2011, 52 (Suppl 2), 1–2. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Laux, L.; Donner, E.; Jette, N.; Knupp, K.; Meskis, M.A.; Miller, I.; Sullivan, J.; Welborn, M.; Berg, A.T. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations from a North American Consensus Panel. Pediatr. Neurol. 2017, 68, 18–34.e13. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.; Laguitton, V.; Viellard, M.; Lepine, A.; Chabrol, B.; Dravet, C.; Milh, M. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav. 2014, 31, 143–148. [Google Scholar] [CrossRef]
- Gataullina, S.; Dulac, O. From genotype to phenotype in Dravet disease. Seizure 2017, 44, 58–64. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.M.; Berg, A.T.; Hood, V.; Knupp, K.G.; Koh, S.; Laux, L.; Meskis, M.A.; Miller, I.; Perry, M.S.; Scheffer, I.E.; et al. Dravet syndrome: A quick transition guide for the adult neurologist. Epilepsy Res. 2021, 177, 106743. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Hung, P.L.; Fan, P.C.; Lin, K.L.; Hsu, T.R.; Chou, I.J.; Ho, C.S.; Chou, I.C.; Lin, W.S.; Lee, I.C.; et al. Clinical spectrum and the comorbidities of Dravet syndrome in Taiwan and the possible molecular mechanisms. Sci. Rep. 2021, 11, 20242. [Google Scholar] [CrossRef] [PubMed]
- Dravet, C.; Oguni, H. Dravet syndrome (severe myoclonic epilepsy in infancy). Handb. Clin. Neurol. 2013, 111, 627–633. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Y.; Sun, H.; Liu, X.; Yang, X.; Xiong, H.; Jiang, Y.; Bao, X.; Wang, S.; Yang, Z.; et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev. 2014, 36, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Hattori, J.; Ouchida, M.; Ono, J.; Miyake, S.; Maniwa, S.; Mimaki, N.; Ohtsuka, Y.; Ohmori, I. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia 2008, 49, 626–633. [Google Scholar] [CrossRef]
- Oguni, H.; Hayashi, K.; Awaya, Y.; Fukuyama, Y.; Osawa, M. Severe myoclonic epilepsy in infants--a review based on the Tokyo Women's Medical University series of 84 cases. Brain Dev. 2001, 23, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Bureau, M.; Dalla Bernardina, B. Electroencephalographic characteristics of Dravet syndrome. Epilepsia 2011, 52 Suppl. 2, 13–23. [Google Scholar] [CrossRef]
- Genton, P.; Velizarova, R.; Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 2011, 52 (Suppl 2), 44–49. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H.; Fejerman, N. Dravet syndrome: A study of 53 patients. Epilepsy Res. 2006, 70 (Suppl 1), S231–238. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Kobayashi, K.; Yoshinaga, H.; Ohtsuka, Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 2010, 51, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52 (Suppl 2), 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.E.; Sadleir, L.G.; Harkin, L.A.; Vadlamudi, L.; McMahon, J.M.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F. Severe myoclonic epilepsy of infancy (Dravet syndrome): Recognition and diagnosis in adults. Neurology 2006, 67, 2224–2226. [Google Scholar] [CrossRef] [PubMed]
- Rilstone, J.J.; Coelho, F.M.; Minassian, B.A.; Andrade, D.M. Dravet syndrome: Seizure control and gait in adults with different SCN1A mutations. Epilepsia 2012, 53, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, A.; Zulfiqar-Ali, Q.; Marques, P.; Rong, M.; Andrade, D.M. A systematic review of adults with Dravet syndrome. Seizure 2021, 87, 39–45. [Google Scholar] [CrossRef]
- Specchio, N.; Balestri, M.; Trivisano, M.; Japaridze, N.; Striano, P.; Carotenuto, A.; Cappelletti, S.; Specchio, L.M.; Fusco, L.; Vigevano, F. Electroencephalographic features in dravet syndrome: Five-year follow-up study in 22 patients. J. Child. Neurol. 2012, 27, 439–444. [Google Scholar] [CrossRef]
- Korff, C.; Laux, L.; Kelley, K.; Goldstein, J.; Koh, S.; Nordli, D., Jr. Dravet syndrome (severe myoclonic epilepsy in infancy): A retrospective study of 16 patients. J. Child. Neurol. 2007, 22, 185–194. [Google Scholar] [CrossRef]
- Kim, S.H.; Nordli, D.R., Jr.; Berg, A.T.; Koh, S.; Laux, L. Ictal ontogeny in Dravet syndrome. Clin. Neurophysiol. 2015, 126, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Coleman-Belin, J.; Harris, A.; Chen, B.; Zhou, J.; Ciulla, T.; Verticchio, A.; Antman, G.; Chang, M.; Siesky, B. Aging Effects on Optic Nerve Neurodegeneration. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Barba, C.; Parrini, E.; Coras, R.; Galuppi, A.; Craiu, D.; Kluger, G.; Parmeggiani, A.; Pieper, T.; Schmitt-Mechelke, T.; Striano, P.; et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia 2014, 55, 1009–1019. [Google Scholar] [CrossRef]
- Gaily, E.; Anttonen, A.K.; Valanne, L.; Liukkonen, E.; Traskelin, A.L.; Polvi, A.; Lommi, M.; Muona, M.; Eriksson, K.; Lehesjoki, A.E. Dravet syndrome: New potential genetic modifiers, imaging abnormalities, and ictal findings. Epilepsia 2013, 54, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.; Garcia-Penton, L.; Canales-Rodriguez, E.J.; Lerma-Usabiaga, G.; Iturria-Medina, Y.; Roman, F.J.; Davidson, D.; Aleman-Gomez, Y.; Acha, J.; Carreiras, M. Brain morphometry of Dravet syndrome. Epilepsy Res. 2014, 108, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Bureau, M.G.P.; Dravet, C.; Delgado-Escueta, A.V.; Tassinari, C.A.; Thomas, P.; Wolff, P. Epileptic Syndromes in Infancy, Childhood and Adolescence, 5th ed.; John Libbey Eurotext: France, 2005. [Google Scholar]
- Berkvens, J.J.; Veugen, I.; Veendrick-Meekes, M.J.; Snoeijen-Schouwenaars, F.M.; Schelhaas, H.J.; Willemsen, M.H.; Tan, I.Y.; Aldenkamp, A.P. Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav. 2015, 47, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Boel, M.J. Behavioural and neuropsychological problems in refractory paediatric epilepsies. Eur. J. Paediatr. Neurol. 2004, 8, 291–297. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Li, B.M.; Li, Z.X.; Wang, J.; Liu, X.R.; Meng, H.; Tang, B.; Bian, W.J.; Shi, Y.W.; Liao, W.P. Few individuals with Lennox-Gastaut syndrome have autism spectrum disorder: A comparison with Dravet syndrome. J. Neurodev. Disord. 2018, 10, 10. [Google Scholar] [CrossRef]
- Kwong, A.K.; Fung, C.W.; Chan, S.Y.; Wong, V.C. Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome. PLoS ONE 2012, 7, e41802. [Google Scholar] [CrossRef]
- Li, B.M.; Liu, X.R.; Yi, Y.H.; Deng, Y.H.; Su, T.; Zou, X.; Liao, W.P. Autism in Dravet syndrome: Prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 2011, 21, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Ouss, L.; Leunen, D.; Laschet, J.; Chemaly, N.; Barcia, G.; Losito, E.M.; Aouidad, A.; Barrault, Z.; Desguerre, I.; Breuillard, D.; et al. Autism spectrum disorder and cognitive profile in children with Dravet syndrome: Delineation of a specific phenotype. Epilepsia Open 2019, 4, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Lagae, L.; Irwin, J.; Gibson, E.; Battersby, A. Caregiver impact and health service use in high and low severity Dravet syndrome: A multinational cohort study. Seizure 2019, 65, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60 (Suppl 3), S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Perulli, M.; Battista, A.; Sivo, S.; Turrini, I.; Musto, E.; Quintiliani, M.; Gambardella, M.L.; Contaldo, I.; Veredice, C.; Mercuri, E.M.; et al. Heart rate variability alterations in Dravet Syndrome: The role of status epilepticus and a possible association with mortality risk. Seizure 2022, 94, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Daverio, M.; Ciccone, O.; Boniver, C.; De Palma, L.; Corrado, D.; Vecchi, M. Supraventricular Tachycardia During Status Epilepticus in Dravet Syndrome: A Link Between Brain and Heart? Pediatr. Neurol. 2016, 56, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Ergul, Y.; Ekici, B.; Tatli, B.; Nisli, K.; Ozmen, M. QT and P wave dispersion and heart rate variability in patients with Dravet syndrome. Acta Neurol. Belg. 2013, 113, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Minderhoud, C.A.; Postma, A.; Jansen, F.E.; Verhoeven, J.S.; Schrijver, J.J.; Goudswaard, J.; Andreae, G.; Otte, W.M.; Braun, K.P.J.; Brilstra, E.H. Gastrointestinal and eating problems in SCN1A-related seizure disorders. Epilepsy Behav. 2023, 146, 109361. [Google Scholar] [CrossRef] [PubMed]
- Beck, V.C.; Isom, L.L.; Berg, A.T. Gastrointestinal Symptoms and Channelopathy-Associated Epilepsy. J. Pediatr. 2021, 237, 41–49 e41. [Google Scholar] [CrossRef]
- Eschbach, K.; Scarbro, S.; Juarez-Colunga, E.; Allen, V.; Hsu, S.; Knupp, K. Growth and endocrine function in children with Dravet syndrome. Seizure 2017, 52, 117–122. [Google Scholar] [CrossRef]
- Delogu, A.B.; Spinelli, A.; Battaglia, D.; Dravet, C.; De Nisco, A.; Saracino, A.; Romagnoli, C.; Lanza, G.A.; Crea, F. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia 2011, 52 (Suppl 2), 55–58. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Borlot, F.; Lang, A.E.; Andrade, D.M. Antecollis and levodopa-responsive parkinsonism are late features of Dravet syndrome. Neurology 2014, 82, 2250–2251. [Google Scholar] [CrossRef] [PubMed]
- Andress, D.L.; Ozuna, J.; Tirschwell, D.; Grande, L.; Johnson, M.; Jacobson, A.F.; Spain, W. Antiepileptic drug-induced bone loss in young male patients who have seizures. Arch. Neurol. 2002, 59, 781–786. [Google Scholar] [CrossRef]
- Bjurulf, B.; Reilly, C.; Sigurdsson, G.V.; Thunstrom, S.; Kolbjer, S.; Hallbook, T. Dravet syndrome in children-A population-based study. Epilepsy Res. 2022, 182, 106922. [Google Scholar] [CrossRef]
- Licheni, S.H.; McMahon, J.M.; Schneider, A.L.; Davey, M.J.; Scheffer, I.E. Sleep problems in Dravet syndrome: A modifiable comorbidity. Dev. Med. Child. Neurol. 2018, 60, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kalume, F.; Oakley, J.C.; Westenbroek, R.E.; Gile, J.; de la Iglesia, H.O.; Scheuer, T.; Catterall, W.A. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol. Dis. 2015, 77, 141–154. [Google Scholar] [CrossRef]
- Van Nuland, A.; Ivanenko, A.; Meskis, M.A.; Villas, N.; Knupp, K.G.; Berg, A.T. Sleep in Dravet syndrome: A parent-driven survey. Seizure 2021, 85, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci, D.; Chieffo, D.; Battaglia, D.; Brogna, C.; Contaldo, I.; De Clemente, V.; Losito, E.; Dravet, C.; Mercuri, E.; Guzzetta, F. A prospective longitudinal study on visuo-cognitive development in Dravet syndrome: Is there a "dorsal stream vulnerability"? Epilepsy Res. 2015, 109, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Sala-Coromina, J.; Raspall-Chaure, M.; Marce-Grau, A.; de la Ossa, A.M.; Macaya, A. Early-onset eyelid stereotypies are a frequent and distinctive feature in Dravet syndrome. Seizure 2021, 92, 155–157. [Google Scholar] [CrossRef]
- Cooper, M.S.; McIntosh, A.; Crompton, D.E.; McMahon, J.M.; Schneider, A.; Farrell, K.; Ganesan, V.; Gill, D.; Kivity, S.; Lerman-Sagie, T.; et al. Mortality in Dravet syndrome. Epilepsy Res. 2016, 128, 43–47. [Google Scholar] [CrossRef]
- Kearney, J. Sudden unexpected death in dravet syndrome. Epilepsy Curr. 2013, 13, 264–265. [Google Scholar] [CrossRef]
- Auerbach, D.S.; Jones, J.; Clawson, B.C.; Offord, J.; Lenk, G.M.; Ogiwara, I.; Yamakawa, K.; Meisler, M.H.; Parent, J.M.; Isom, L.L. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS ONE 2013, 8, e77843. [Google Scholar] [CrossRef]
- Cheah, C.S.; Westenbroek, R.E.; Roden, W.H.; Kalume, F.; Oakley, J.C.; Jansen, L.A.; Catterall, W.A. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels 2013, 7, 468–472. [Google Scholar] [CrossRef]
- de Lange, I.M.; Gunning, B.; Sonsma, A.C.M.; van Gemert, L.; van Kempen, M.; Verbeek, N.E.; Nicolai, J.; Knoers, N.; Koeleman, B.P.C.; Brilstra, E.H. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A-related seizure phenotypes. Epilepsia 2018, 59, 1154–1165. [Google Scholar] [CrossRef]
- Dravet, C.B.M.; Oguni, H.; Cokar, O.; Guerrini, R.; Wolf, P. Epileptic Syndromes in Infancy, Childhood and Adolescence, 6th ed.; John Libbey Eurotext: Arcueil, France, 2019. [Google Scholar]
- Dravet, C. Severe myoclonic epilepsy in infants and its related syndromes. Epilepsia 2000, 41 (Suppl 9), 7. [Google Scholar] [CrossRef]
- Ohki, T.; Watanabe, K.; Negoro, T.; Aso, K.; Haga, Y.; Kasai, K.; Kito, M.; Maeda, N. Severe myoclonic epilepsy in infancy: Evolution of seizures. Seizure 1997, 6, 219–224. [Google Scholar] [CrossRef]
- Ozmen, M.; Dilber, C.; Tatli, B.; Aydinli, N.; Caliskan, M.; Ekici, B. Severe myoclonic epilepsy of infancy (Dravet syndrome): Clinical and genetic features of nine Turkish patients. Ann. Indian. Acad. Neurol. 2011, 14, 178–181. [Google Scholar] [CrossRef]
- Depienne, C.; Trouillard, O.; Saint-Martin, C.; Gourfinkel-An, I.; Bouteiller, D.; Carpentier, W.; Keren, B.; Abert, B.; Gautier, A.; Baulac, S.; et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: Analysis of 333 patients. J. Med. Genet. 2009, 46, 183–191. [Google Scholar] [CrossRef]
- Singh, R.; Andermann, E.; Whitehouse, W.P.; Harvey, A.S.; Keene, D.L.; Seni, M.H.; Crossland, K.M.; Andermann, F.; Berkovic, S.F.; Scheffer, I.E. Severe myoclonic epilepsy of infancy: Extended spectrum of GEFS+? Epilepsia 2001, 42, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Dravet Syndrome: A Sodium Channel Interneuronopathy. Curr. Opin. Physiol. 2018, 2, 42–50. [Google Scholar] [CrossRef]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.M.; Isom, L.L. Voltage-gated sodium channel beta subunits: The power outside the pore in brain development and disease. Neuropharmacology 2018, 132, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Plummer, N.W.; Meisler, M.H. Evolution and diversity of mammalian sodium channel genes. Genomics 1999, 57, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Isom, L.L.; De Jongh, K.S.; Catterall, W.A. Auxiliary subunits of voltage-gated ion channels. Neuron 1994, 12, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Kalume, F.; Oakley, J.C. NaV1.1 channels and epilepsy. J. Physiol. 2010, 588, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, E.; Santorelli, F.M.; Bertini, E.; Buti, D.; Gaggero, R.; Gobbi, G.; Lini, M.; Granata, T.; Freri, E.; Parmeggiani, A.; et al. Somatic and germline mosaicisms in severe myoclonic epilepsy of infancy. Biochem. Biophys. Res. Commun. 2006, 341, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Mei, D.; Helen Cross, J.; Guerrini, R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia 2006, 47, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.C.; Morse, R.P.; Scott, R.C.; Holmes, G.L.; Lenck-Santini, P.P. SCN1A mutations in Dravet syndrome: Impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. 2012, 23, 177–186. [Google Scholar] [CrossRef]
- Gambardella, A.; Marini, C. Clinical spectrum of SCN1A mutations. Epilepsia 2009, 50 (Suppl 5), 20–23. [Google Scholar] [CrossRef]
- Zuberi, S.M.; Brunklaus, A.; Birch, R.; Reavey, E.; Duncan, J.; Forbes, G.H. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 2011, 76, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Beckh, S.; Noda, M.; Lubbert, H.; Numa, S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. 1989, 8, 3611–3616. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Ohmori, I.; Ouchida, M.; Ohno, Y.; Tsurumi, T.; Miki, T.; Wakamori, M.; Ishihara, S.; Yoshida, T.; Takizawa, A.; et al. A missense mutation of the gene encoding voltage-dependent sodium channel (Nav1.1) confers susceptibility to febrile seizures in rats. J. Neurosci. 2010, 30, 5744–5753. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.S.; Grunnet, M.; Bastlund, J.F. Therapeutic potential of Na(V)1.1 activators. Trends Pharmacol. Sci. 2014, 35, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.S.; Dutt, K.; Papale, L.A.; Dube, C.M.; Dutton, S.B.; de Haan, G.; Shankar, A.; Tufik, S.; Meisler, M.H.; Baram, T.Z.; et al. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 2010, 285, 9823–9834. [Google Scholar] [CrossRef]
- Hayashi, K.; Ueshima, S.; Ouchida, M.; Mashimo, T.; Nishiki, T.; Sendo, T.; Serikawa, T.; Matsui, H.; Ohmori, I. Therapy for hyperthermia-induced seizures in Scn1a mutant rats. Epilepsia 2011, 52, 1010–1017. [Google Scholar] [CrossRef]
- Rhodes, T.H.; Lossin, C.; Vanoye, C.G.; Wang, D.W.; George, A.L., Jr. Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc. Natl. Acad. Sci. U S A 2004, 101, 11147–11152. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Oakley, J.C.; Kalume, F.; Yu, F.H.; Scheuer, T.; Catterall, W.A. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. USA 2009, 106, 3994–3999. [Google Scholar] [CrossRef]
- Kepecs, A.; Fishell, G. Interneuron cell types are fit to function. Nature 2014, 505, 318–326. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kondo, S. Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J. Neurocytol. 2002, 31, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G.; Wang, X.J. Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.H.; Vaiana, M.; Nakuci, J.; Somarowthu, A.; Goff, K.M.; Goldstein, N.; Murthy, P.; Muldoon, S.F.; Goldberg, E.M. Interneuron Desynchronization Precedes Seizures in a Mouse Model of Dravet Syndrome. J. Neurosci. 2020, 40, 2764–2775. [Google Scholar] [CrossRef]
- Freund, T.F. Interneuron Diversity series: Rhythm and mood in perisomatic inhibition. Trends Neurosci. 2003, 26, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Kalume, F.; Westenbroek, R.E.; Cheah, C.S.; Yu, F.H.; Oakley, J.C.; Scheuer, T.; Catterall, W.A. Sudden unexpected death in a mouse model of Dravet syndrome. J. Clin. Invest. 2013, 123, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Caplan, J.B.; Madsen, J.R.; Schulze-Bonhage, A.; Aschenbrenner-Scheibe, R.; Newman, E.L.; Kahana, M.J. Human theta oscillations related to sensorimotor integration and spatial learning. J. Neurosci. 2003, 23, 4726–4736. [Google Scholar] [CrossRef]
- Cobb, S.R.; Buhl, E.H.; Halasy, K.; Paulsen, O.; Somogyi, P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 1995, 378, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.C.; Jiao, X.; Sinha, S.; Beck, K.D.; Servatius, R.J. Damage of GABAergic neurons in the medial septum impairs spatial working memory and extinction of active avoidance: Effects on proactive interference. Hippocampus 2011, 21, 835–846. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Jeong, H.Y.; Kruglikov, I.; Tremblay, R.; Lazarenko, R.M.; Rudy, B. Rapid developmental maturation of neocortical FS cell intrinsic excitability. Cereb. Cortex 2011, 21, 666–682. [Google Scholar] [CrossRef]
- Chiron, C.; Dulac, O. The pharmacologic treatment of Dravet syndrome. Epilepsia 2011, 52 Suppl. 2, 72–75. [Google Scholar] [CrossRef]
- Wirrell, E.C.; Hood, V.; Knupp, K.G.; Meskis, M.A.; Nabbout, R.; Scheffer, I.E.; Wilmshurst, J.; Sullivan, J. International consensus on diagnosis and management of Dravet syndrome. Epilepsia 2022, 63, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Lee, H.S.; Chang, K.P.; Lee, Y.Y.; Lai, H.C.; Hung, P.L.; Lee, H.F.; Chi, C.S. The Impact of Anti-Epileptic Drugs on Growth and Bone Metabolism. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W. Basic pharmacology of valproate: A review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 2002, 16, 669–694. [Google Scholar] [CrossRef]
- Dressler, A.; Trimmel-Schwahofer, P.; Reithofer, E.; Muhlebner, A.; Groppel, G.; Reiter-Fink, E.; Benninger, F.; Grassl, R.; Feucht, M. Efficacy and tolerability of the ketogenic diet in Dravet syndrome - Comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015, 109, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Ohtsuka, Y.; Oguni, H.; Tohyama, J.; Baba, H.; Fukushima, K.; Ohtani, H.; Takahashi, Y.; Ikeda, S. Stiripentol open study in Japanese patients with Dravet syndrome. Epilepsia 2009, 50, 2362–2368. [Google Scholar] [CrossRef]
- Shi, X.Y.; Tomonoh, Y.; Wang, W.Z.; Ishii, A.; Higurashi, N.; Kurahashi, H.; Kaneko, S.; Hirose, S.; Epilepsy Genetic Study Group, J. Efficacy of antiepileptic drugs for the treatment of Dravet syndrome with different genotypes. Brain Dev. 2016, 38, 40–46. [Google Scholar] [CrossRef]
- Freche, C. [Study of an anxiolytic, clobazam, in otorhinolaryngology in psychosomatic pharyngeal manifestations]. Sem. Hop. Ther. 1975, 51, 261–263. [Google Scholar] [PubMed]
- Group, C.C.C. Clobazam in treatment of refractory epilepsy: The Canadian experience. A retrospective study. Epilepsia 1991, 32, 407–416. [Google Scholar]
- Gauthier, A.C.; Mattson, R.H. Clobazam: A Safe, Efficacious, and Newly Rediscovered Therapeutic for Epilepsy. CNS Neurosci. Ther. 2015, 21, 543–548. [Google Scholar] [CrossRef]
- Pernea, M.; Sutcliffe, A.G. Clobazam and Its Use in Epilepsy. Pediatr. Rep. 2016, 8, 6516. [Google Scholar] [CrossRef]
- Ng, Y.T.; Conry, J.A.; Drummond, R.; Stolle, J.; Weinberg, M.A.; Investigators, O.V.S. Randomized, phase III study results of clobazam in Lennox-Gastaut syndrome. Neurology 2011, 77, 1473–1481. [Google Scholar] [CrossRef]
- Conry, J.A.; Ng, Y.T.; Kernitsky, L.; Mitchell, W.G.; Veidemanis, R.; Drummond, R.; Isojarvi, J.; Lee, D.; Paolicchi, J.M.; Investigators, O.V.S. Stable dosages of clobazam for Lennox-Gastaut syndrome are associated with sustained drop-seizure and total-seizure improvements over 3 years. Epilepsia 2014, 55, 558–567. [Google Scholar] [CrossRef]
- Wirrell, E.C.; Laux, L.; Franz, D.N.; Sullivan, J.; Saneto, R.P.; Morse, R.P.; Devinsky, O.; Chugani, H.; Hernandez, A.; Hamiwka, L.; et al. Stiripentol in Dravet syndrome: Results of a retrospective U.S. study. Epilepsia 2013, 54, 1595–1604. [Google Scholar] [CrossRef]
- Giraud, C.; Treluyer, J.M.; Rey, E.; Chiron, C.; Vincent, J.; Pons, G.; Tran, A. In vitro and in vivo inhibitory effect of stiripentol on clobazam metabolism. Drug Metab. Dispos. 2006, 34, 608–611. [Google Scholar] [CrossRef]
- Frisium (clobazam): Summary of product characteristics. Available online: https://www.medicines.org.uk/emc/product/1574/smpc#gref (accessed on 15 November 2021).
- Frisium (clobazam): Summary of product characteristics. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/203993s005lbl.pdf (accessed on 15 November 2021).
- Chiron, C.; Marchand, M.C.; Tran, A.; Rey, E.; d'Athis, P.; Vincent, J.; Dulac, O.; Pons, G. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet 2000, 356, 1638–1642. [Google Scholar] [CrossRef]
- Nickels, K.C.; Wirrell, E.C. Stiripentol in the Management of Epilepsy. CNS Drugs 2017, 31, 405–416. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Cersosimo, R.O.; Sakr, D.; Cresta, A.; Escobal, N.; Fejerman, N. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005, 46, 1539–1544. [Google Scholar] [CrossRef]
- Frampton, J.E. Stiripentol: A Review in Dravet Syndrome. Drugs 2019, 79, 1785–1796. [Google Scholar] [CrossRef]
- Yamada, M.; Suzuki, K.; Matsui, D.; Inoue, Y.; Ohtsuka, Y. Long-term safety and effectiveness of stiripentol in patients with Dravet syndrome: Interim report of a post-marketing surveillance study in Japan. Epilepsy Res. 2021, 170, 106535. [Google Scholar] [CrossRef]
- Na, J.H.; Shin, S.; Yang, D.; Kim, B.; Kim, H.D.; Kim, S.; Lee, J.S.; Choi, J.R.; Lee, S.T.; Kang, H.C. Targeted gene panel sequencing in early infantile onset developmental and epileptic encephalopathy. Brain Dev. 2020, 42, 438–448. [Google Scholar] [CrossRef]
- Dibue-Adjei, M.; Fischer, I.; Steiger, H.J.; Kamp, M.A. Efficacy of adjunctive vagus nerve stimulation in patients with Dravet syndrome: A meta-analysis of 68 patients. Seizure 2017, 50, 147–152. [Google Scholar] [CrossRef]
- Dlouhy, B.J.; Miller, B.; Jeong, A.; Bertrand, M.E.; Limbrick, D.D., Jr.; Smyth, M.D. Palliative epilepsy surgery in Dravet syndrome-case series and review of the literature. Childs Nerv. Syst. 2016, 32, 1703–1708. [Google Scholar] [CrossRef]
- Andrade, D.M.; Hamani, C.; Lozano, A.M.; Wennberg, R.A. Dravet syndrome and deep brain stimulation: Seizure control after 10 years of treatment. Epilepsia 2010, 51, 1314–1316. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Wright, S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med. 2017, 377, 699–700. [Google Scholar] [CrossRef]
- Miller, I.; Scheffer, I.E.; Gunning, B.; Sanchez-Carpintero, R.; Gil-Nagel, A.; Perry, M.S.; Saneto, R.P.; Checketts, D.; Dunayevich, E.; Knappertz, V.; et al. Dose-Ranging Effect of Adjunctive Oral Cannabidiol vs Placebo on Convulsive Seizure Frequency in Dravet Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 613–621. [Google Scholar] [CrossRef]
- Devinsky, O.; Nabbout, R.; Miller, I.; Laux, L.; Zolnowska, M.; Wright, S.; Roberts, C. Long-term cannabidiol treatment in patients with Dravet syndrome: An open-label extension trial. Epilepsia 2019, 60, 294–302. [Google Scholar] [CrossRef]
- Anderson, L.L.; Hawkins, N.A.; Thompson, C.H.; Kearney, J.A.; George, A.L., Jr. Unexpected Efficacy of a Novel Sodium Channel Modulator in Dravet Syndrome. Sci. Rep. 2017, 7, 1682. [Google Scholar] [CrossRef]
- Schoonjans, A.S.; Ceulemans, B. An Old Drug for a New Indication: Repurposing Fenfluramine from an Anorexigen to an Antiepileptic Drug. Clin. Pharmacol. Ther. 2019, 106, 929–932. [Google Scholar] [CrossRef]
- Schoonjans, A.S.; Ceulemans, B. A critical evaluation of fenfluramine hydrochloride for the treatment of Dravet syndrome. Expert. Rev. Neurother. 2022, 22, 351–364. [Google Scholar] [CrossRef]
- Martin, P.; de Witte, P.A.M.; Maurice, T.; Gammaitoni, A.; Farfel, G.; Galer, B. Fenfluramine acts as a positive modulator of sigma-1 receptors. Epilepsy Behav. 2020, 105, 106989. [Google Scholar] [CrossRef]
- Ceulemans, B.; Boel, M.; Leyssens, K.; Van Rossem, C.; Neels, P.; Jorens, P.G.; Lagae, L. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia 2012, 53, 1131–1139. [Google Scholar] [CrossRef]
- Ceulemans, B.; Schoonjans, A.S.; Marchau, F.; Paelinck, B.P.; Lagae, L. Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia 2016, 57, e129–e134. [Google Scholar] [CrossRef]
- Lagae, L.; Sullivan, J.; Knupp, K.; Laux, L.; Polster, T.; Nikanorova, M.; Devinsky, O.; Cross, J.H.; Guerrini, R.; Talwar, D.; et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: A randomised, double-blind, placebo-controlled trial. Lancet 2019, 394, 2243–2254. [Google Scholar] [CrossRef]
- Nabbout, R.; Mistry, A.; Zuberi, S.; Villeneuve, N.; Gil-Nagel, A.; Sanchez-Carpintero, R.; Stephani, U.; Laux, L.; Wirrell, E.; Knupp, K.; et al. Fenfluramine for Treatment-Resistant Seizures in Patients With Dravet Syndrome Receiving Stiripentol-Inclusive Regimens: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 300–308. [Google Scholar] [CrossRef]
- Sullivan, J.; Specchio, N.; Devinsky, O.; Auvin, S.; Perry, M.S.; Strzelczyk, A.; Gil-Nagel, A.; Dai, D.; Galer, B.S.; Gammaitoni, A.R. Fenfluramine significantly reduces day-to-day seizure burden by increasing number of seizure-free days and time between seizures in patients with Dravet syndrome: A time-to-event analysis. Epilepsia 2022, 63, 130–138. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Doccini, V.; Trivisano, M.; Darra, F.; Ragona, F.; Cossu, A.; Spolverato, S.; Battaglia, D.; Quintiliani, M.; et al. Efficacy and safety of Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: A real-world study. Epilepsia 2020, 61, 2405–2414. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Ferretti, A.; Trivisano, M.; Vigevano, F. Successful use of fenfluramine in nonconvulsive status epilepticus of Dravet syndrome. Epilepsia 2020, 61, 831–833. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Pringsheim, M.; Mayer, T.; Polster, T.; Klotz, K.A.; Muhle, H.; Alber, M.; Trollmann, R.; Spors, H.; Kluger, G.; et al. Efficacy, tolerability, and retention of fenfluramine for the treatment of seizures in patients with Dravet syndrome: Compassionate use program in Germany. Epilepsia 2021, 62, 2518–2527. [Google Scholar] [CrossRef]
- Trowbridge, S.; Poduri, A.; Olson, H. Early diagnosis and experimental treatment with fenfluramine via the Investigational New Drug mechanism in a boy with Dravet syndrome and recurrent status epilepticus. Epileptic Disord. 2021, 23, 954–956. [Google Scholar] [CrossRef]
- Cross, J.H.; Galer, B.S.; Gil-Nagel, A.; Devinsky, O.; Ceulemans, B.; Lagae, L.; Schoonjans, A.S.; Donner, E.; Wirrell, E.; Kothare, S.; et al. Impact of fenfluramine on the expected SUDEP mortality rates in patients with Dravet syndrome. Seizure 2021, 93, 154–159. [Google Scholar] [CrossRef]
- Fintepla: Prescribing information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212102s000lbl.pdf (accessed on 15 November 2021).
- Fintepla: Summary of product characteristics. Available online: https://www.medicines.org.uk/emc/product/11998/smpc#gref (accessed on 15 November 2021).
- Schoonjans, A.; Maes, P.; Ceulemans, B. Aggravation of valproic acid induced thrombocytopenia after the introduction of fenfluramine, a case report. Seizure 2021, 93, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Oakley, J.C.; Cho, A.R.; Cheah, C.S.; Scheuer, T.; Catterall, W.A. Synergistic GABA-enhancing therapy against seizures in a mouse model of Dravet syndrome. J. Pharmacol. Exp. Ther. 2013, 345, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Laux, L.R.C.; Knupp, K.G.; Schreiber, J.M.; Lallas, M.; Wirrell, E.; Devinsky, O.; Stutely, J.; Brathwaite, C.; Avendaño, J.; Parkerson, K.; Meena, M.; Wyant, N.; Ticho, B.; Sullivan, J. Positive Interim Safety, PK, and CSF Exposure Data from the Phase 1/2a MONARCH Study of STK-001, an Antisense Oligonucleotide (ASO), in Children and Adolescents with Dravet Syndrome (DS). In Proceedings of the American Epilepsy Society Annual meeting McCormick Place West, Chicago, IL, USA; 2021. [Google Scholar]
Table 2.
DS features in young children versus older previously undiagnosed children and adults.
| Features | Children | Older children & Adults |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



