
Submitted:
27 November 2023
Posted:
27 November 2023
You are already at the latest version
Abstract
With the term neuroinflammation has defined the typical inflammatory response of the brain closely related to the onset of many neurological diseases. Neuroinflammation is well known, but its mechanisms and pathways are not entirely comprehended. Currently, some progress has been achieved through many efforts and research. Consequently, new cellular and molecular mecha-nisms, diverse from conventional ones, are emerging. In listing some of those that will be the sub-ject of our description and discussion, essential are the important role of peripheral and infiltrated monocytes and clonotypic cells, alterations in gut/brain axis, dysregulation of the apelinergic sys-tem, as well as changes in the endothelial glycocalyx of blood-brain barrier, variation in expres-sion of some genes and levels of the encoding molecules by microRNAs (miRNAs), or other epige-netic factors and distinctive transcriptional factors, as well as the role of autophagy, ferroptosis, sex differences and modifications in the circadian cycle. Such mentioned mechanisms can add significant pieces in understanding the complex etiological puzzle of neuroinflammation. In addi-tion, they could represent biomarkers and targets of neurodegenerative diseases, in increase in our old populations.
Keywords:
Neuroinflammation
; neurodegenerative diseases
; emerging mechanisms.
1. Introduction
The term neuroinflammation indicates the representative pathological condition induced in the brain by several (local and systemic) triggering factors (e.g., infections, trauma, ischemia, toxins, alterations of microbiota-brain axis, etc.) and driving factors (e.g., genetic, vascular and brain factors: for example, alterations in the expression of neurotrophins and components of the endothelial glycocalyx and/or endothelium) [1,2]. Neuroinflammation has evocated by typical immune cells residing in the brain, and well-known to have a main role in central nervous system (CNS) homeostasis, as well as in contributing to the development of neurodegenerative diseases (ND), constituting their typical hallmark [3,4,5]. However, recent evidence suggests that both peripheral and infiltrating monocytes, as well as cells of clonotypic immunity, constitute other crucial actors of neuroinflammation [5,6]. Nevertheless, such evidence needs to be supported by larger studies, even if the growing data obtained in recent years appear promising and demonstrate the crucial contribution of this immune component in brain health and disease [3,4,5,6].
Other unconventional mechanisms related to the onset of neuroinflammation have recently emerged in the literature. Among these, it includes the altered relationship of gut microbiota with CNS, known as gut-brain microbiota (MGB) axis [7,8]. Of note, also is the autophagy, although the related mechanisms remain indefinable, and further investigation is necessary [9]. Interestingly, also is the contribution of the ferroptosis, a new form of cell death, caused by iron-dependent lipid peroxidation [10,11,12], and associated with the pathogenesis of many diseases, such as ND. Such link has led to suppose that iron homeostasis, oxidative stress, and neuroinflammation are involved in controlling the evocation of ferroptosis and neuronal health [10,11,12]. In such process, the apelinergic system mediated by ELA/APJ signaling, has also been recently documented to participate in this regulation [13]. Another nonclassical mechanism related to onset of neuroinflammation appears to be the modified expression of neurotrophins, such as BDNF [14].
Furthermore, it is also emerging that neuroinflammation also is the result of the modulation in expression of genes encoding immune and injury’s molecules. In such process, miRNAs [15] and epigenetic factors [16], including A-to-I RNA editing, M6A RNA methylation, and alternative splicing [15,16,17] have been recently revealed to have a fundamental role. Finally, circadian rhythm disorders have recently been also discovered to impact both the onset and development of neuroinflammation, trough the activation of glial cells and peripheral immune responses [18].
In line of such emerging evidence, insights have been achieved in identifying the mechanisms related to the complex neuroinflammation, although the complex puzzle is not complete and further studies are needed. In this review article, we try to shed light on the above and others by integrating recent clinical studies and experimental observations.
2. Recent evidence on peripheral and infiltrating monocytes and clonotypic immune cells in neuroinflammation and their sex- and gender-mediated modulation
Monocytes and clonotypic immune cells represent key actors of neuroinflammation, as recently underlined (see Figure 1) [18,19,20,21]. Such cells physiologically show a double function: a) first facilitate immune surveillance and confer resistance against neurotropic viruses [18,19,20,21], and b) second, maintain CNS homeostasis and integrity, by stimulating neurogenesis, and improving cognitive function. Thus, in ND, they appear deregulated; precisely, the recent data evidence a deregulation of clonotypic immunity. Clonotypic cells commonly appears to have altered levels and functions, and consequently mediate abnormal immune responses [4]. Likewise, monocytes are frequently modified, both peripherally and centrally, in quantity and quality with altered profiles and phenotypes. Data from recent studies in both human and animal models report that monocytes, different subsets of lymphocytes and their mediators can mediate both defensive or noxious mechanisms in ND, disturbing both their progression and risk of death. Noteworthy, in fact, is the crosstalk between peripheral and CNS immune cells, significantly linked to the survival of ND patients, since changes in peripheral macrophages can diminish inflammation at the periphery along nerves and in CNS [18,19,20,21]. In this complex context, it has been demonstrated that extracellular vehicles (EVs), putative vehicles for misfolded proteins and mediators of inflammation between cells, have a crucial role [6]. The following is evidence from the literature on the two cellular actors in neuroinflammation.
2.1. Monocytes in neuroinflammation
The participation of monocytes in the evocation of neuroinflammation has recently emerged from both clinical studies and experimental observations [21,22,23,24,25]. Accordingly, degenerative conditions in the mouse brain have been revealed to provoke infiltration of peripheral immune cells [21,22,23,24,25]. Accordingly, a study in a in vivo model of Parkinson's disease (PD) has discovered infiltration of peripheral blood monocytes into the brain, which can evolve in neurodegeneration [26]. During cerebrovascular disease, it has been also observed infiltration of peripheral blood monocytes into the brain [27]. In addition, monocytes from patients with multiple sclerosis have been found to secrete some anti-neurodegenerative mediators [28]. In addition, in vitro studies have reported that high levels of chemokine CXCL12 from monocytes induce endothelial cells (EC) activation, thereby facilitating lymphocyte transmigration, and validating the crucial role of monocytes in the infiltration of immune cells in the brain [29]. Another study has established that brain leukocyte infiltration origins from peripheral inflammation [30]. Furthermore, circulating Ly-6C+ myeloid precursors have been observed to migrate into CNS and have a pathogenic role in individuals affected by autoimmune demyelinating disease [31]. Moreover, monocytes have been also demonstrated to phagocyte surplus brain proteins [32], such as amyloid-β peptide [33].
Differently, other animal models, such as the murine stroke model, have evidenced a neuroprotective function of monocytes [34]. Precisely, they provide bioactive substances to brain cells [35,36]. In contrast, other investigations have reported the toxic action towards neuronal cells, of monocytes, by releasing saturated fatty acids, which can cause diverse pathologies, such as autoimmune disorders [21,22,23,24,25]. Thus, monocytes through different mechanisms, actively contribute to the development of neuroinflammation, although some require further investigation.
2.2 Clonotypic immune cells in neuroinflammation and ND
The involvement of clonotypic cells in neuroinflammation has only been theorized in previous research. However, recent evidence has largely established the key role of both T and B cell subsets and demonstrated their infiltration into the CNS, by determining different effects depending on different subsets [37,38]. Therefore, CD4+ CD25+ regulatory T cells (Tregs) and CD4+ T helper (Th)2 cells have a neuroprotective effect. CD4+ Th1, CD4+ Th17, cytotoxic CD8+, and Natural killer (NK) cells and effector T lymphocytes (Teffs) mediate an accelerate progression of neuroinflammation, which evolve in a exacerbated/accelerated neurodegeneration and an increased mortality’s risk [39,40,41,42]. Furthermore, they show different circulating levels. Precisely, circulating Th17 cells have shown higher levels in subjects with Mild Cognitive Impairment (MCI) respect to cognitively normal subjects or those with non-Alzheimer’s MCI [42]. In contrast, circulating Th1 cells have been demonstrated to have higher levels in subjects with AD [43]. Th2 cells and Th2-associated molecules have been observed to have lower levels in AD subjects [43]. Other studies have reported higher levels of circulating Th17 populations in individuals with AD [44,45]. Tregs cells have also been discovered to have lower levels in patients with AD [46]. They are tanti-inflammatory populations with opposite effects respect to Th17 cells. Fu et al. have described low levels of Tregs cells in AD subjects [47]. Contrarily, another investigation has observed no difference in Tregs levels between MCI, AD, and healthy subjects, although Treg levels are associated positively with total tau and pTau181 in AD subjects [48].
In patients with amyotrophic lateral sclerosis (ALS), a chronic, progressive disease characterized by the gradual degeneration of upper and lower motor neurons, different T subsets with diverse functions have been detected, related to the stage of the disease. Precisely, T cells have been revealed to be in augment and evocate survival of motoneuron cells (MNs) in superoxide dismutase (SOD)1 mutant mice, through a protective neuroinflammatory response, likely mediated by Interleukin-4 (IL-4). Motor impairment is, while, accompanied by a decline in the functions of regulatory T cells, which impede the microglia activation in SOD1 mutant mice [49]. Thus, neuroprotective functions of the immune cells may occur in the early stage of the disease, although other studies are imperative to approve this finding. Moreover, disease progression is related to several alterations in the immune system, such as the acquisition of an inflammatory phenotype of microglia cells [50], thymic involution [51], augmented levels of pro-inflammatory cytokines [52], and leucocyte CNS infiltration [53].
Insufficient evidence with contradictory data is available on the function of B lymphocytes, plasma cells, and antibodies in neuroinflammation and ND [54,55] and their contribution in AD pathogenesis, needs further investigations. They could also make clear the diverse and obscure points of neuroinflammation and ND [56]. Furthermore, clinical trials on AD and other ND have failed to provide promising results [57], because of diverse causes, including the relevant role of sex/gender dimorphism (which also justifies the differences observed in the onset, progression, and hallmarks of neuroinflammation in the various ND), which will be described in the following section [58].
2.3. Considerations on immune cells infiltrating the CNS and new evidence on the migration of immune cells outside the CNS.
The mechanisms related to the infiltration of peripheral immune cells into the CNS in neuroinflammation and ND have been gaining a great interest in recent years, by leading to the development of numerous immunomodulatory therapies to regulate immune cell infiltration at BBB, the choroid plexus (ChP) epithelium, and glial barrier. For instance, the therapy with Natalizumab, a drug that blocks adhesion and trafficking of leukocytes across BBB, has been used for almost two decades to treat MS [59,60].
Furthermore, it has been recently evidenced that crucial CNS immune cell populations, i.e., dendritic cells (DC), T cells, B cells and other myeloid cell populations, may migrate out of the CNS and mediate signals from the CNS to peripheral lymphatics [61]. Such has been supported by recent evidence that demonstrates the involvement of meningeal lymphatic system not only in fluid homeostatic CNS functions, but also in allowing immune cell migration, and facilitating DC migration from the CNS to the meningeal borders and draining cervical lymph nodes [61]. Thus, they can mediate immunity in the lymph nodes and consequently modulate disease onset and/or progression [61].
However, significant work is needed to elucidate the function of each CNS-associated lymphatic region in overall CNS immunity. The results obtained could facilitate the development of new therapeutic strategies to target lymphatic-leukocyte interactions in the treatment of CNS diseases. Diverse lymphatic-leukocyte pathways have been recently studied in both the peripheral and CNS lymphatic systems. However, other studies are needed to comprehend their therapeutic capacity in several homeostatic and neuropathological conditions.
2.4. The sex/gender dimorphism: an important modifier of immune system and physiology of brain, and a crucial differential driver in diseases
With term sex indicates diverse biological and physiological features of male and female individuals, while the term gender regards, the social and cultural differences, between men and women [58]. Biological, socioeconomic, and cultural differences impact the health and diseases of individuals. Therefore, the sexual dimorphism between women and men mainly fixes differences in the anatomy and physiology of the body systems, while gender indicates the socially created norms for imposing and specifying roles, relationships, among the individuals of a specific society and time. Accordingly, sexual biological differences characterize the morphology of the nervous system between the sexes, as evidenced by studies conducted in human and animal models [58]. Particularly, amygdalae are larger in males than in females. In addition, in regard memory formation, the dimorphism modulates emotional memories in the female amygdala involve the left region (visually predominant, positive, and negative emotions), while in males they involve the right region of the brain (negative emotional responses). Furthermore, prefrontal cortical regions show higher expression of estrogen receptors. Such could elucidate the differences in decision making between the two sexes. Structural neuroimaging investigations have evidenced in males decreased levels of overall cortical thickness and augmented rate of cortical thickness decline. In females, a greater white matter volume has been, while, observed. Moreover, humans also have sex variations in neurotransmitter systems, including adrenergic, serotonergic, cholinergic systems, corticosterone, benzodiazepine, and cholecystokinin, factors largely associated with episodic memory. Males have higher levels serotonin than females, which may influence the disease states evocated by serotonin dysfunction [58]. Such can result in differences in the learning process, as detected during stress and the subsequent conditioning (enhanced in males but inhibited in females). Such modulation appears to be related to sex hormones, as evidenced by studies in human and animal models of cognitive decline, neurodegenerative and psychiatric diseases. Exactly, sex hormones impact the permeability of BBB, assumed to be one of the key pathophysiological hallmarks of ND. Such discoveries could have larger consequences in these disease processes; however, further research is needed for verifying and validating this relationship [58].
Sexual dimorphism also modulates the immune system of both sexes; females show stronger responses to pathogens in both innate and adaptive immunity than males. Such influences the recovery from infections and improves vaccine efficacy [62]. However, stronger immune responses simultaneously dispose females to a higher risk for autoimmune diseases, even if the mechanisms related to these alterations are not yet fully defined. Furthermore, the association between the biological effect of sex chromosomes and sex hormones on the immune system is well understood, including the quantity and function of immune cells, as well as the levels of cytokines or other circulating immune mediators [58].
Therefore, sex represents a significant modifier of physiology and disease via genetic, epigenetic, and hormonal regulations, as emerged from sex/gender medicine, born in the late of 1990s to detect changes between diseases and their determinants in men and women [58]. Women and men have been displayed to vary in the mechanisms and pathways of the pathophysiology of their diseases, as well as in the clinical manifestation, prognosis, and outcomes. Such aspect is studying about the major ND [58,63], and can modify their traditional criteria and guidelines, that appear today inadequate between the two sexes, and particularly for women. In women, the diagnosis is more challenging due to lower sensitivity and specificity of traditional diagnostic tests. This has resulted in researching new protocols. However, to date, few, and scattered records, have been achieved, and are of little application in medical practice. Accordingly, clinicians and researchers show a little attention on sex and gender in health planning and medical practice.
Therefore, increased efforts are needed to include sex and gender in modern medical research, clinical trials, and clinical practice; in addition, there needs to be a greater appreciation by medical societies and institutions regarding the sex and gender differences that characterize and differentiate women and men and their related diseases. Finally, various gender equity measures need to be promoted at all levels by biomedical and pharmaceutical organizations for the development of different biomarker panels and the most appropriate therapies for both sexes.
3. Changes in the MGB axis and neuroinflammation
The relationship of gut microbiota and CNS is known as MGB axis (see Figure 1), first discovered, and proposed in 2012 [64], and evocated by neuroanatomical brain structures, and intestinal nerves located in the intestinal wall [64]. It can transmit signals to the brain by the vagus nerve, with a response mediated by descending branch, which in turn regulates intestinal activities. In addition, the neuroendocrine axis, represented by the hypothalamic–pituitary–adrenal (HPA) axis, is another component of MGB axis [65]. The HPA controls the variations in the composition and function of the gut microbiome. HPA dysfunctions have been shown to have a crucial role in the pathogenesis of neuropsychiatric diseases. Specifically, HPA mediates the activation of inflammatory signaling pathways resulting in the release of inflammatory mediators, such as tumor necrosis factor α (TNF-α), interferon-γ (IFN-γ) and interleukin 6 (IL-6) [66]. In turn, these mediators may damage BBB integrity and contribute to brain diseases through the systemic circulation and by damaging the gut mucosal barrier. In addition, the induced HPA inflammatory response also modulates the secretion of glucocorticoids [67], which in turn stimulates gut function and the production of pro-inflammatory factors [68]. This vicious cycle also causes the activation of enteric immune cells, such as Th17 and NK cells, which can translocate into the brain and cause neuroinflammation [69]. Neuroinflammation, in turn, also modifies the gut microbial composition, which further stimulates enteric immune cells and microbiota-derived metabolites, which act as regulators in this bidirectional through inflammatory signals. Because of the immunoregulatory function of gut microbiota, alterations in the commensal microbiota, termed as dysbiosis, have recently attracted increasing interest for its pathogenetic role in immune-mediated diseases.
Dysbiosis is described by the variations in host–microbe interactions and associates with low-grade inflammation, metabolic syndrome, gastrointestinal tract infections and inflammatory bowel disease, as well as autoimmune diseases such as systemic lupus erythematosus, systemic sclerosis, Sjogren’s syndrome, antiphospholipid antibody syndrome, multiple sclerosis (MS) and myasthenia gravis (MG). MG is a neuromuscular autoimmune disease caused by immune-mediated destruction of the neuromuscular junction (NMJ). Although antibodies against neuromuscular junction components are documented, the MG pathogenesis remains unclear, and it is probably multifactorial [70]. Based on emerging evidence, it has recently been proposed that perturbations in human microbiota may contribute to MG pathogenesis and clinical course. Accordingly, some products derived from commensal microflora have been exhibited to have anti-inflammatory effects, while other have been revealed to possess pro-inflammatory properties. In addition, MG patients compared with age-matched controls showed a distinctive composition of the oral and gut microbiota, with a typical increase in Streptococcus and Bacteroides and a reduction in Clostridia, as well as a reduction of short-chain fatty acid. Moreover, restoration of gut microbiota disruption was evidenced after probiotic administration, followed by improvement of symptoms in MG cases [7].
This evidence emphasizes that MGB axis maintains host health, and changes in the composition of the gut microbiota and the MGB axis has a key role in the onset of MG and some ND, such as PD and AD. However, the related molecular and cellular mechanisms are complex and remain unclear. Behavioral phenotypes can be transmitted from humans to animals through transplantation/translocation of the gut microbiota, indicating that the latter may be an important regulator of ND [7]. However, further research is required to determine whether the findings in animals can be extended to humans and to elucidate the relevant mechanisms by which the gut microbiota regulates neuroinflammation and ND. Such investigations could contribute to the development of new microbiota-based strategies for diagnosis, treatment, and clinical management of neuroinflammation and ND.
4. Autophagy
Autophagy is a physiologically regulated and evolutionarily conserved process, involving the degradation of cytoplasmic proteins and other macromolecules within the lysosome in multicellular organisms [71]. Neuroinflammation and ND are categorized by protein aggregation and alterations in autophagy have been associated with these disorders, as established by several studies [72,73,74]. However, the mechanisms related to autophagy during neuroinflammation, and ND, remain less undoubtedly understood [75,76,77]. Drugs, such as cocaine [78], or other toxic substances of exogenous or endogenous nature, pathogens, can evoke autophagic cell death in astrocytes, consequently contributing to the pathogenesis of neurodegeneration [79]. Recently, it has been also disclosed a close relationship between glia maturation factor (GMF), autophagy-related proteins, and the NLRP3 inflammasome in AD patients [80]. A shift of microglia from M1 to M2 after autophagy induction has also been detected [81]. However, further investigations are needed, which could provide basic understanding on the etiology of ND or neuroinflammation-associated disorders.
5. Ferroptosis in neuroinflammation
Ferroptosis, introduced in 2012 by Brent R. Stockwell, is a new form of regulated cell death (RCD) characterized by the accumulation of lethal amounts of iron- and lipid-dependent reactive oxygen species [82]. Ferroptosis displays morphological and biochemical differences from other conventional RCD forms, mostly through characteristic mitochondrial shrinkage that differentiate it from other RCD [82]. Severe peroxidation of membranes containing polyunsaturated fatty acids (PUFAs) is the main driving force behind ferroptosis, not only regulated by lipid and iron metabolism, but also by amino acid metabolism and signaling transduction. The critical steps involved in ferroptosis comprise the accumulation of intracellular free iron, glutathione depletion, and peroxidation of PUFA-rich membranes [82,83].
Ferroptosis is gradually documented as a crucial factor in the pathogenesis of several diseases, such as ND [82,83,84,85]. Recent studies have designated the complicated relations between iron metabolism and disorders associated with neuroinflammation. Emerging evidence suggests that iron homeostasis, oxidative stress, and subsequent neuroinflammation contribute to the regulation of ferroptosis and neuronal health [82,83,84,85]. However, the precise molecular mechanisms underlying the involvement of ferroptosis in the pathological processes of neurodegeneration and its impact on neuronal dysfunction remain incompletely understood. Nevertheless, it has recently been revealed that ferroptosis is probably regulated by ELA/APJ signaling of the apelinergic system [13]. ELA is a peptide hormone belonging to the adipokine group and a component of the apelinergic system, discovered in 2013-2014 [13]. Given its high homology with apelin, it binds to the APJ receptor and probably mediates similar effects. Regulation mediated by ELA/APJ signaling could be a promising strategy for the treatment of NDs, such as stroke [13]. On the other hand, a recent study in mouse models of middle cerebral artery occlusion (MCAO) showed the protective role of the ELA-APJ axis in ischemic stroke, after treatment with ELA-32 [widely quoted in 13]. A reduction in cerebral ischemic lesion and an improvement in neurobehavioral and cognitive deficits was detected. Furthermore, ELA-32 administration has been disclosed to improve neuronal ferroptosis, accompanied by reduced iron deposition, decreased mitochondrial damage, improved lipid peroxidation, and glutathione reduction. These findings have suggested that the ELA-APJ axis attenuates neuronal ferroptosis after ischemic stroke and that ELA-32 peptide may be a putative therapeutic pathway for ischemic stroke [widely quoted in 13]. However, further data are to provide other/novel strategies to modulate the onset of neuroinflammation and ND, such as stroke.
6. The close link of endothelial dysfunction with neuroinflammation and ND
Another determinant condition, contributing to neuroinflammation and ND, is the endothelial dysfunction, characterized by typical cellular and molecular mechanisms, including changes in the glycocalyx [86,87]. Endothelium represents an important constituent of the brain; precisely its cells, EC, are the fundamental components of the neurovascular unit (NVU), composed by EC arranged with neurons, glial cells, and other vascular elements [86,87]. NUV contributes to maintenance of CNS homeostasis, physiological neurotransmission and neuronal survival [62]. Furthermore, EC and glial cells, such as microglia cells, contribute to create the BBB, and to provide both nutrients and toxins. Therefore, any disfunction of the NVU, and BBB, determines the onset of neuroinflammation, which in turn causes age-associated cognitive deficits and, consequently, onset of ND [86,87,88]. Usually, NVU impairment is evocated by the altered clearance of amyloid-β peptide and its consequent accumulation in the brain, which triggers neuroinflammation through the release of toxic small molecules and inflammatory products, which cross the damaged BBB [62]. Cardiovascular and cerebrovascular diseases (i.e., macro-infarcts, lacunes, microbleeds- atherosclerosis, arteriolosclerosis and cerebral amyloid angiopathy (CAA) can also evocate NVU destruction via a direct action [62,86]. Particularly, microvascular diseases have been also demonstrated to impact NVU by triggering variations in the physiological brain oxygenation [62,86,87,88]. In turn, these modifications determine a reduced blood flow and the onset of subsequent hypoxia, which results not associated with infarction, although chronic hypoxia-ischemia is recognized to cause chronic damage to the NVU. Thus, NVU dysfunction, similarly to endothelium dysfunction for CVD, is the key determinant of neuroinflammation and ND onset [62,86,87,88]. Moreover, this dysfunction cotemporally evocates BBB dysfunction, related to many ND, such as stroke, MS, AD and PD. This accompanies an augmented passage of neuroinflammatory molecules to the brain [86,87,88], which may influence the onset of neurodegeneration. However, BBB breakdown is primarily associated with the aging process, and particularly with vascular aging. In addition, it appears to originate from the hippocamp level. This condition also happens in subjects without cognitive impairment, and more rapidity in old age and particularly with concomitant MCI [86,87,88]. During regular aging, the damage of BBB impacts the CA1 region and the dentate gyrus, but not the CA3 region [86,87,88]. In addition, hippocampal BBB distribution has been discovered to be a condition that temporally precedes the onset of hippocampal atrophy [86,87,88]. By analyzing cerebrospinal fluid from MCI cases than cognitively normal persons, augmented damage biomarkers (i.e., platelet-derived growth factor receptor (PDGFR)-β) of pericytes have been detected, suggesting an early role of pericytes than other cell types in the BBB breakdown [86,87,88].
In case of AD, a reduced presence of tight junctions in brain EC has been assessed, with EC showing an abnormal morphology. In addition, the typical Tau deposition also affects the diameter of blood vessels, which appear small often not containing red blood cells [86,87,88]. This determines an anomalous angiogenesis in AD cases, likely due to altered function and quantity of trophic factors. Accordingly, patients with AD have increased levels of VEGF, the stimulator of endothelial proliferation and a neuroprotective factor, both in serum and temporal cortex and hypothalamus [86,87,88]. This accompanies a decreased expression and levels of both VEGFR-1 and VEGFR-2. Some studies have attributed to VEGF itself, the cause of such decrease in levels of two receptors. Precisely, VEGF seems to mediate such effect through a ligand-mediated endocytosis mechanism [86,87,88]. Thus, VEGF in AD has a role of antagonist versus its receptors, by resulting in an altered angiogenesis. Moreover, the in vitro Aβ accumulation has been demonstrated to be able to reduce the mRNA levels of VEGFR-1 and VEGFR-2 resulting in increased VEGF reactivity [86,87,88].
Likewise, in ALS patients, alterations in NVU and angiogenic factors, including VEGF, have also recently been observed. Accordingly, animal and human experimental investigations on SOD-1 mutant mice and ALS patients, respectively, have shown reduced levels of tight junctions, such as ZO-1, and BBB breakdown, which precede motor neuron death. Such suggests that vascular damage represents an early pathogenetic ALS mechanism [62,86,87,88,89]. In addition, metalloproteinase-MMP-2 and MMP-9 high levels have been detected in the peripheral blood samples from ALS cases. Nicaise and colleagues have reported variations in the composition of vascular NVU elements in the early ALS stage [62]. In similar manner, in a SOD-1 mice model, a blood-spinal cord barrier (BSCB) dysfunction has been observed and characterized by the ex-erythrocyte extravasation, neurotoxic hemoglobin accumulation and NUV injury via iron-dependent oxidative stress [62]. Moreover, the alterations in NVU appears to be not only structural but also functional, as confirmed by investigation in G93A SOD1 mice, showing a downregulation of Glut-1 and CD146 expression early and late in the disease [62]. Elevated levels of VEGF, particularly VEGF-A, have been also quantified in the blood and CSF from ALS patients than healthy controls [62], possibly due to a compensatory mechanism. In SOD-1 mutant mice, VEGF-A has been displayed to exercise neuroprotective effects, by dropping MN cell death via the activation of the PI3K-Akt pathway [62]; and this may result in a delay of disease onset [62,86,87,88,89]. However, other studies have shown reduced levels of VEGF and its receptor VEGFR in ALS, and exclusively in subjects homozygous for certain haplotypes, given by three polymorphisms in their genes (−2578C/A, −1154 G/A, and −634G/C) [62]. The reason has been attributed to the destabilization of VEGF mRNA induced by SOD1 protein [62]. In addition, many studies have revealed a neuroprotective role of VEGF in ALS. For example, studies in cell cultures have reported a protective role of VEGF in the NSC-34 motor neuron cell line [62].
This evidence globally suggests that the treatment of neuroinflammation and ND, or better of inflammation of the entire cardiovascular system, could improve the health status of cerebrovascular system, and represent the best avenue for the development of potential strategies for getting better the blood flow at the cerebral microvascular level, by protecting BBB and NVU. Preserving the integrity, permeability, and function of BBB and NVU could stop or delay the progression of neuroinflammation and ND. For achieving this goal, it is imperative to identify all the pathways involved in the pathophysiology of these diseases, and particularly those related to BBB and NVU dysfunction. Surely, this objective can be realized by performing multiple omics investigations, offering the opportunity of acquiring major, relevant, and new data. Accordingly, such studies are encouraged.
7. miRNAs and epigenetic factors in neuroinflammation and ND
Recent evidence reports that modulation of genes related to neuroinflammation can mitigate it. Factors mediating such role, are microRNAs and other epigenetic factors, that are universal regulators of differentiation, activation, and polarization of all the cells of human body, including immune and neuronal cells. Such evidence has led to focus on them the recent investigations, which demonstrate different expression levels of both factors in microglia, both in normal and inflamed CNS, suggesting their role in brain health and neuroinflammation-associated disorders [90]. Precisely, low levels of mature micro-RNAs have been discovered in human temporal lobes from cases with epilepsy and in the hippocamp of patients with sclerosis [91,92]. Among these, microRNA-155 has been revealed to negatively regulate BBB function during the progression of neuroinflammation in neurodegeneration [93]. The miR-195 has been detected to inhibit autophagy after peripheral nerve damage [94]. In contrast, microRNA-188-3p, in a upregulated state, limits the neuroinflammation and improves memory in AD patients AD [95]. miR-137 mediates attenuation of a-beta-induced neurotoxicity in Neuro2a cells [96]. The miR-124 expression has been displayed to modify the promoter DNA methylation and microglial functions [97]. Notably, microRNA-30e has also been shown to control neuroinflammation modulating NLRP3 in an MPTP-induced PD model [98]. MicroRNA-129-5p has been demonstrated to exacerbate neuroinflammation and BBB injury [99]. Similarly, miR-17-92 has been detected to trigger the differentiation of neurons during neuroinflammatory conditions [100]. MicroRNA-139 has been related to the AD pathogenesis via cannabinoids receptors [101]. Thus, microRNAs represent good therapeutic targets for the development of novel anti-neuroinflammatory AD treatments [90].
Moreover, post-transcriptional RNA changes represent epigenetic factors, able to modulate mRNA coding properties, stability, translatability expanding the genome’s coding capacity. They have been reported to influence neuroinflammation. Especially, A-to-I RNA editing, m6A RNA methylation, and alternative splicing (AS), demonstrated to impact neuronal cell life cycle, alterations in neuron cell mechanisms, have been shown to contribute significantly to affect neuroinflammation and age-related neurodegeneration [102]. A-to-I RNA editing is a post-transcriptional mechanism, that modulates the double-stranded (ds) RNA structures; it is catalyzed by the adenosinedeaminases family of RNA acting enzymes (ADAR), and consists in the deamination of specific adenosine (A) into inosine (I), by altering both coding and non-coding transcripts [102]. Three ADAR enzymes are detected in mammalian cells: ADAR1, ADAR2 and ADAR3. They show a high expression and activity in the brain, regulating neurodevelopment, brain function, and physiological brain aging [widely quoted in 102]. Consequently, brain appears to be vulnerable to ADAR actions and RNA editing dysregulation, potentially starting CNS disorders, such as glioblastoma, epilepsy, and ND [widely quoted in 102]. A recent study has discovered that ADAR1 and ADAR2 expression is negatively associated with age. Indeed, a low expression has been detected in older adults. ADAR3 has been, while, observed to overexpressed in older individuals, and particularly in males [102].
Concerning, N6-methyladenosine (m6A), a dynamic and reversible post-transcriptional alteration, adding a methyl group to the N6position in selected adenosines of each type of RNA [102], have been reported an altered expression both in mouse and human brains with aging. Concerning ND, anomalous m6A changes have been detected in AD, PD, and ALS. Moreover, epigenetic factor represents the most predominant epigenetic factor in the brain. It finely regulates many processes regarding brain function and development [102]. ND, including mainly AD, PD, ALS, frontotemporal dementia (FTD) and familial dysautonomia (FD), have been assessed to have AS alterations [102].
The key function of post-transcriptional RNA modifications in brain aging and neurodegeneration emphasizes the possibility to perform interventions, able to reduce or inhibit these processes; among these, antisense oligonucleotides (ASOs) can modify their expression. Recent studies are testing ASOs to eliminate causative splicing defects in PD, AD, FTD and ALS [widely quoted in 102].
The cumulative evidence about the contribution and the serious impact of A-to-I RNA editing, m6A RNA methylation, and alternative splicing on brain aging process, neuroinflammation, and ND, emphasizes the necessity to execute investigations on the relationship between these processes, how these mechanisms may impact each other to modulate them simultaneously.
8. Transcriptional factors and related pathways: focus on NF-kB (nuclear factor kappa-light-chain enhancer of activated B cells) and related pathways
Other modulating factors of neuroinflammation are transcriptional factors, which can activate an inflammatory network in the brain; they are all linked to the NF-κB pathway, an ancient signaling pathway specialized in host defense [103]. The NF-κB pathway is a cytoplasmatic seno, constitute by a protein’s complex, comprising Rel family proteins-RelA/p65, c-Rel and RelB- and NF-κB components-p50/p105 and p52/p100, and commonly inhibited by binding to IκB proteins (i.e., IκBα, IκBβ, IκBγ, IκBδ, IκBϵ, IκBζ and Bcl3) and via the action of many signaling pathways and negative feedback loops regulating in diverse mechanisms at various levels of the signaling cascades. Its activation can be induced by immune insults and external and internal danger signals, such as oxidative and genotoxic stress and tissue injury [103]. In addition, its induction is linked to Toll-like receptors (TLRs) and inflammasome [104,105,106], as well as several upstream kinase cascades via canonical or non-canonical pathways. IKKα/β and NIK are the most important upstream kinases. IKKγ are generally referred to a nuclear factor-kappa B Essential Modulator (NEMO), an important regulatory component of the IKK complex linked upstream to genotoxic signals and IL-1 and TNF receptor mediated signaling [104,105,106]. NF-κB complex activation, playing the crucial role of pleiotropic mediator of gene expression, determines its translocation into the nucleus and the expression of target genes, encoding various molecules, such as pro-inflammatory cytokines, chemokines, adhesion molecules, eicosanoids, growth factors, metallo-proteinases, nitric oxide, etc. [103]. NF-κB signaling has been reported to be one of the major pathways stimulating neuroinflammation [103,107].
Recent studies have evidenced the beneficial effect of dietary supplementation with anti-inflammatory compounds on cognitive decline, neuroinflammation and oxidative stress, acting on NF-kB pathway in AD-like animal models [as extensively cited in 108, 109]. Precisely, curcumin, krill oil, chicoric acid, plasmalogens, lycopene, tryptophan-related dipeptides, hesperidin, and selenium peptides have been tested, despite their heterogeneity, and have shown beneficial effects on cognitive deficits and lipopolysaccharide (LPS)-induced neuroinflammatory responses in rodents, by affecting NF-κB pathway [107,108,109]. Overall, dietary interventions could represent positive factors in contradicting AD, or other ND, by acting on neuroprotection and immune regulation. For example, the treatment with metformin, an antidiabetic drug, has demonstrated anti-inflammatory effects via many mechanisms, revealing its potential as therapeutic target for neuroinflammation.
However, as evidenced in such review, the mechanisms involved in neuroinflammation are various and complex, and numerous molecules are combined in a network and consequently can modify each other. For example, metformin significantly prevents nuclear translocation of p65, but the pretreatment with compound C, an AMPK inhibitor, eliminates this effect, while silencing HMGB1 abolish the NF-κB activation. SIRT1 deacetylates FoxO, increasing its transcriptional activity. mTOR in dendritic cells regulates FoxO1 through AKT. Interactions between the various molecules need to be further explored to clarify their specific mechanisms and provide more guidance for the treatment of neuroinflammation [110].
Based on the evidence described above, mTOR and AKT pathways, as well as JAK-STAT, and PPARγ, and Notch pathways constitute other crucial pathways in neuroinflammation [9,111,112,113,114,115,116]. They represent highly conserved signaling hubs that coordinate neuronal activity and brain development and participate in neuroinflammation. Accordingly, hyperactivation of JAK/STAT, mTOR, and inhibition of PPARγ and AKT signaling have been associated with various neuro-complications, including neuroinflammation, apoptosis, and oxidative stress [113,114,115,116]. Remarkably, target modulators have also been described to act during acute and chronic neurological deficits. For example, natural products, such as osthole, an important ingredient of traditional Chinese medicinal plants often found in various plants of the Apiaceae family, have been shown to target these pathways [1117]. Precisely, osthole induces neurogenesis and neuronal function via the stimulation of Notch, BDNF/Trk, and P13k/Akt signaling pathways. This upregulates the expression of various proteins, such as BDNF, TrkB, CREB, Nrf-2, P13k, and Akt, and inhibits MAPK/NF-κB-mediated transcription of genes involved in the production of inflammatory cytokines and the NLRP-3 inflammasome. Thus, modulation of Notch, BDNF/Trk, MAPK/NF-κB, and P13k/Akt signaling pathways by osthole confers protection against neuroinflammation and ND [117].
Evidence described above suggests the neuroprotective potential of several compounds and natural products as possible therapeutic agents for neuroinflammation and NDs. However, a limitation of some of such substances is their low bioavailability and solubility in water. Furthermore, the use of innovative nanotechnology or the incorporation of a more polar group would be advantageous to increase the bioactivity and physicochemical properties of such compounds or natural products, such as osthole. To this end, liposomes, microspheres, nanoparticles, transferosomes, ectosomes, lipid-based systems, etc. have been developed for the modified delivery of various herbal drugs. For example, osthole-loaded nanoemulsion has been reported to effectively target the brain and have beneficial effects in the treatment of AD. Therefore, the development of potential nanocarriers such as liposomes, microspheres, and nano-emulsions could improve the bioavailability of such compounds [108,109]. However, further studies are needed to evaluate the real therapeutic effect of such compounds on neuroinflammation.
9. Circadian cycle and neuroinflammation
Another determinant of neuroinflammation is the alteration of circadian cycle/rhythm, a fundamental process of life developed during the long-term evolution of organisms. It has diverse functions: maintaining the proliferation, migration, and activation of cells, and particularly of immune cells [118,119]. Studies have shown that circadian rhythm disorders impact the onset and development of neuroinflammation by activation of glial cells and peripheral immune responses [119,120,121]. Animal models exposed to nightshifts or night light have shown to have significant levels of activated microglia and proinflammatory cytokines in brain [122,123]. Sleep deprivation has been also demonstrated to activate the transcriptional factor NF-κB and intensify the release of IL-1β and TNF-α in the hippocampus, resulting in neuronal injury [124]. Studies have also recognized that mRNA levels of IL-1β and TNF-α in brain tissue of experimental animals evocate significant variations in circadian rhythm, dependable by alterations in the sleep–wake cycle [125]. Inhibition of such cytokines has been reported to decrease spontaneous nonrapid eye movement sleep in experimental animals [126,127], confirming that pro-inflammatory cytokines mediate effects on circadian cycle and neuroimmune function. In addition, alterations in circadian system influence microglial activation and their phenotypes [122,123,124]. Accordingly, studies have shown that under conditions of light exposure, the inflammatory activity of rat microglia is enhanced, with a significant increase of TNF-α, IL-1β, and IL-6 [122,123,124,128].
The circadian cycle also shows a serious role in the regulation of the peripheral immune system [129]. Most peripheral immune cells, including innate and adaptive cells, have their own molecular clock and display significant rhythmic differences during recruitment and activation processes [130]. Studies have established a regulation of the bone marrow chemokine CXCL12 on the hypothalamic sympathetic–parasympathetic nervous system in a circadian manner, resulting in periodic fluctuations in CXCL12 levels and CXCR4 receptor activation to maintain daily rhythmic variations in neutrophil quantities in the bone marrow blood reserve [131,132]. In macrophages, the circadian cycle impacts their pattern recognition receptors signaling pathways, inflammatory mediators, and phagocytic activity [133]. In such mechanism, it has been discovered that Krüppel-like factor 4 (KLF4), whose expression is time-regulated, regulates macrophage phenotype and rhythmic expression of inflammatory factors. Its expression (that is of KLF4) is downregulated in aging macrophages, and the disruption of macrophages circadian rhythms after inhibition of KLF4 gene expression further confirms the close relationship between the expression circadian rhythms and the disruption of the innate immune state [134]. Some studies have proposed that the REV-ERBα clock gene mediates the expression of the PI3K/Akt signaling pathway and is involved in the regulation of the diurnal rhythm of macrophage polarization [135]. REV-ERBα could also represent a potential target for regulating circadian rhythms and inflammatory response. Similarly, adaptive immune responses also display rhythmicity. Studies have revealed that the strength of the immune response varies significantly during the different hours of the day. For example, more CD8+ T cells are produced in midday than night in response to antigen immunization, and the rhythmic response dissolves by knocking out the Bmal1 gene in T cells, further validating the relevance of circadian rhythms in modulating adaptive immune responses [136].
10. Chronic low-grade inflammation and neurodegenerative diseases
In the previous paragraphs, several molecular aspects related to neuroinflammation in ND, particularly during dementia and AD were described. Although numerous research and pathophysiological hypotheses on potential inflammatory processes during ND have been proposed, currently the precise nature and temporal characteristics of the relationship between neuroinflammation and ND remain largely unknown. Clinical and preclinical studies have described how systemic chronic inflammation (SCI) should be considered as a potential driver for the onset of the neurodegenerative process associated with cognitive impairment [137,138]. Several studies have proposed the concept of chronic low-grade inflammation as potentially causal in the etiopathogenesis of dementia and other ARDs of the elderly individual, and the term inflammaging has been coined for this phenomenon [139,140]. Specifically, inflammaging refers to the presence of chronic low-grade systemic inflammation that occurs during aging in the absence of overt infection (the so-called sterile inflammation). Clinical and epidemiological studies have shown that this process is a relevant risk factor for morbidity and mortality in the elderly [139,140]. In particular, the presence of SCI leads to an increased risk of metabolic diseases (e.g., hypertension, diabetes, dyslipidaemia) but also osteoporosis, cancer, and cardiovascular, neurodegenerative, and autoimmune diseases [137].
SCI involves several cytokines and transcription factors that regulate chronic inflammation at the tissue and causal levels for different ARDs. Among the cytokines involved IL-6 is probably the one most associated with a robust chronic inflammatory response that characterizes different ARDs [141]; other inflammatory cytokines that participate in the inflammatory process during ARD are IL-1β and TNF-α [141]. In turn, cytokines interact with specific tissue surface receptors that regulate the inflammatory cascade by regulating transcriptional processes. The two main protein transcription factors associated with SCI are: the STAT (signal transducer and activator of transcription) and NF-κB [103]. These proteins cascade regulate a series of genes that code for the formation of inflammatory cytokines.
Over the last decade the role of low-grade SCI in periodontal disease (PeD) has been suggested as a potential risk factor for overall dementia and particularly AD. Several authors have described the presence of significantly elevated antibody levels toward specific oral cavity opportunistic pathogens causing PeD in subjects with AD but also MCI compared with control subjects without cognitive impairment [143]. Regarding specific oral pathogens, the one most implicated in the link between dementia and PeD appears to be porphyromonas gingivalis [144], but significantly elevated levels of oral microbial load of other pathogens such as fusobacterium nucleatum and treponema denticola have been described in subjects with AD and MCI compared with control subjects [144]. Data from a recent national US retrospective cohort study showed that periodontal pathogens increase the risk of AD incidence and mortality [145]. In addition, data from a recent meta-analysis showed that the risk of cognitive disorder in individuals with PeD increases as the severity of PeD increases, and this risk appears to be greater in the female sex [146]. There are at least two main mechanisms by which PeD can cause cognitive disorders. The first involves the presence of an increased cerebral inflammatory state caused by the SCI process originating from oral pathogens; the second involves a direct action of periodontal bacteria on the CNS that cross the BBB, cause its breakdown with subsequent, potential triggering of the pre-existing neurodegenerative process [143,144,145,146].
In addition to increased risk of dementia, some studies have suggested that PeD may increase the risk of PD []; however, data from a recent meta-analysis revealed no association between PeD and increased risk of PD [147]. In conclusion, PeD is associated with an increased risk of overall dementia, AD, and MCI, and this appears to be due to low-grade SCI sustained by the oral pathogens that cause PeD. However, prospective data on large population cohorts are needed to confirm the role of PeD as a risk factor for AD, dementia, and possibly other neurodegenerative diseases. If confirmed such data will have major implications for the treatment and prevention of cognitive disorders.
11. Conclusions
With this review, we have provided an overview of the new mechanisms associated with the relationship between neuroinflammation and subsequent onset of ND (see Figure 2A and B). This latter offers the possibility to hypothesize and develop new treatments and to identify diagnostic and prognostic biomarker profiles for neuroinflammation and ND. The latter could include the assessment of transmigration and activation levels of monocytes, as well as the levels, activation, and quantification of clonotypic cells and their mediators, and the evaluation of expression of NF-kB, and other transcriptional factors (i.e., ERG factor), and the related pathways [148,149]. The concomitant assessment of microRNAs and epigenetic factors involved in the regulation of these mechanisms could be further helpful. Furthermore, the targeting of autophagy and ferroptosis is gaining more and more interest, as it contributes to the modulation of neuroinflammation and the onset of ND, as well as to endothelial-related BBB dysfunction. Taking all together, the complex pathophysiology and pathogenesis of neuroinflammation and ND might appear clearer, as well as all pathways and cells. This could facilitate the identification of biomarkers and targets and, consequently, the management of NDs. Therefore, further efforts and investigations are needed. Advances in omics methodologies, artificial intelligence, machine learning, advanced biological techniques, metagenomics, and meta-transcriptomics are currently important in neuroscientific research and could be used to achieve this goal, similarly, large-scale randomized controlled trials are needed. Such studies would pave the way for next-generation treatment strategy capable of modulating neuroinflammation during ND.
Author Contributions
Conceptualization C.R.B.; literature search C.R.B and R.M.; data curation, C.R.B. and R.M..; writing—original draft preparation, C.R.B and R.M..; writing—review and editing C.R.B. and R.M.; figure production, C.R.B.; supervision, C.R.B. and R.M. Both co-authors have read and agreed to the submitted version of the manuscript.
References
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: the devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation — a common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Chasaide, C.N.; A Lynch, M. The role of the immune system in driving neuroinflammation. Brain Neurosci. Adv. 2020, 4. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2016, 17, 49–59. [Google Scholar] [CrossRef]
- Carata, E.; Muci, M.; Di Giulio, S.; Mariano, S.; Panzarini, E. Looking to the Future of the Role of Macrophages and Extracellular Vesicles in Neuroinflammation in ALS. Int. J. Mol. Sci. 2023, 24, 11251. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Iacono, S.; Balistreri, C.R. The Role of Human Microbiota in Myasthenia Gravis: A Narrative Review. Neurol. Int. 2023, 15, 392–404. [Google Scholar] [CrossRef]
- Celorrio, M.; Shumilov, K.; Friess, S.H. Gut microbial regulation of innate and adaptive immunity after traumatic brain injury. Neural Regen. Res. 2023, 19, 272–276. [Google Scholar] [CrossRef]
- Sanghai, N.; Tranmer, G.K. Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View. Cells 2023, 12, 2318. [Google Scholar] [CrossRef]
- Li, L.; Guo, L.; Gao, R.; Yao, M.; Qu, X.; Sun, G.; Fu, Q.; Hu, C.; Han, G. Ferroptosis: a new regulatory mechanism in neuropathic pain. Front. Aging Neurosci. 2023, 15, 1206851. [Google Scholar] [CrossRef]
- Massaccesi, L.; Balistreri, C.R. Biomarkers of Oxidative Stress in Acute and Chronic Diseases. Antioxidants 2022, 11, 1766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-B.; Jia, X.; Cao, Q.; Chen, Y.-T.; Tong, J.; Lu, G.-D.; Li, D.-J.; Han, T.; Zhuang, C.-L.; Wang, P. Ferroptosis-Regulated Cell Death as a Therapeutic Strategy for Neurodegenerative Diseases: Current Status and Future Prospects. ACS Chem. Neurosci. 2023, 14, 2995–3012. [Google Scholar] [CrossRef] [PubMed]
- Monastero, R.; Magro, D.; Venezia, M.; Pisano, C.; Balistreri, C.R. A promising therapeutic peptide and preventive/diagnostic biomarker for age-related diseases: The Elabela/Apela/Toddler peptide. Ageing Res. Rev. 2023, 91, 102076. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Iacono, S.; Ragonese, P.; Aridon, P.; Salemi, G.; Balistreri, C.R. A Brief Overview on BDNF-Trk Pathway in the Nervous System: A Potential Biomarker or Possible Target in Treatment of Multiple Sclerosis? Front. Neurol. 2022, 13, 917527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, A.; Gu, R.; Tong, Y.; Cheng, J. Role and regulatory mechanism of microRNA mediated neuroinflammation in neuronal system diseases. Front. Immunol. 2023, 14, 1238930. [Google Scholar] [CrossRef] [PubMed]
- Tregub, P.P.; Ibrahimli, I.; Averchuk, A.S.; Salmina, A.B.; Litvitskiy, P.F.; Manasova, Z.S.; Popova, I.A. The Role of microRNAs in Epigenetic Regulation of Signaling Pathways in Neurological Pathologies. Int. J. Mol. Sci. 2023, 24, 12899. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, V.; La Rosa, P.; Guida, E.; Colopi, A.; Caratelli, S.; De Paolis, F.; Gallo, A.; Cenciarelli, C.; Sconocchia, G.; Dolci, S.; et al. Contribution of A-to-I RNA editing, M6A RNA Methylation, and Alternative Splicing to physiological brain aging and neurodegenerative diseases. Mech. Ageing Dev. 2023, 212, 111807. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Chen, G. Circadian cycle and neuroinflammation. Open Life Sci. 2023, 18, 20220712. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.L.; Groenendijk, L.; Overdevest, R.; Fowke, T.M.; Annida, R.; Mocellin, O.; de Vries, H.E.; Wevers, N.R. Human BBB-on-a-chip reveals barrier disruption, endothelial inflammation, and T cell migration under neuroinflammatory conditions. Front. Mol. Neurosci. 2023, 16, 1250123. [Google Scholar] [CrossRef]
- Bruno, M.; Bonomi, C.G.; Ricci, F.; Di Donna, M.G.; Mercuri, N.B.; Koch, G.; Martorana, A.; Motta, C. Blood–brain barrier permeability is associated with different neuroinflammatory profiles in Alzheimer's disease. Eur. J. Neurol. 2023, 31. [Google Scholar] [CrossRef]
- Lauritsen, J.; Romero-Ramos, M. The systemic immune response in Parkinson’s disease: focus on the peripheral immune component. Trends Neurosci. 2023, 46, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Miró-Mur, F.; Pérez-De-Puig, I.; Ferrer-Ferrer, M.; Urra, X.; Justicia, C.; Chamorro, A.; Planas, A.M. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain, Behav. Immun. 2016, 53, 18–33. [CrossRef]
- Jones, K.A.; Maltby, S.; Plank, M.W.; Kluge, M.; Nilsson, M.; Foster, P.S.; Walker, F.R. Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain, Behav. Immun. 2018, 67, 299–307. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Li, Y.; Jiao, Y.; Doran, S.J.; Khan, N.; Henry, R.J.; Brunner, K.; Loane, D.J.; Faden, A.I.; Szeto, G.L.; Wu, J. The brain-bone marrow axis and its implications for chronic traumatic brain injury. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Côté, M.; Poirier, A.-A.; Aubé, B.; Jobin, C.; Lacroix, S.; Soulet, D. Partial depletion of the proinflammatory monocyte population is neuroprotective in the myenteric plexus but not in the basal ganglia in a MPTP mouse model of Parkinson’s disease. Brain Behav. Immun. 2015, 46, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Veenstra, M.; Gaskill, P.J.; Morgello, S.; Calderon, T.M.; Berman, J.W. Monocytes Mediate HIV Neuropathogenesis: Mechanisms that Contribute to HIV Associated Neurocognitive Disorders. Curr. HIV Res. 2014, 12, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Molnarfi, N., Benkhoucha. Interferonbeta induces hepatocyte growth factor in monocytes of multiple sclerosis patients. PLoS ONE 2012, 7, e49882. [Google Scholar] [CrossRef] [PubMed]
- Man, S.; Tucky, B.; Cotleur, A.; Drazba, J.; Takeshita, Y.; Ransohoff, R.M.; Yu, H.; Lee, H.; Cazanave, S.C.; Warren, A.D.; et al. CXCL12-Induced Monocyte-Endothelial Interactions Promote Lymphocyte Transmigration Across an in Vitro Blood-Brain Barrier. Sci. Transl. Med. 2012, 4, 119ra14. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Strazielle, N.; Ghersi-Egea, J.-F. Brain leukocyte infiltration initiated by peripheral inflammation or experimental autoimmune encephalomyelitis occurs through pathways connected to the CSF-filled compartments of the forebrain and midbrain. J. Neuroinflamm. 2012, 9, 187–187. [Google Scholar] [CrossRef]
- Vojdani, A., Lambert. The role of Th17 in neuroimmune disorders: target for CAM therapy. Part II. Evid. Based Complement. Alternat. Med. 2011, 984965. [Google Scholar] [CrossRef]
- Kadiu, I.; Glanzer, J.G.; Kipnis, J.; Gendelman, H.E.; Thomas, M.P. Mononuclear phagocytes in the pathogenesis of neurodegenerative diseases. Neurotox. Res. 2005, 8, 25–50. [Google Scholar] [CrossRef]
- Cashman, J.R.; Gagliardi, S.; Lanier, M.; Ghirmai, S.; Abel, K.J.; Fiala, M. Curcumins promote monocytic gene expression related to beta-amyloid and superoxide dismutase clearance. Neurodegener. Dis. 2012, 10, 274–276. [Google Scholar] [CrossRef]
- Kohman, R.A. Aging microglia: relevance to cognition and neural plasticity. Meth.Mol. Biol 2012, 934, 193–218. [Google Scholar] [CrossRef]
- Böttger, D.; Ullrich, C.; Humpel, C. Monocytes deliver bioactive nerve growth factor through a brain capillary endothelial cell-monolayer in vitro and counteract degeneration of cholinergic neurons. Brain Res. 2010, 1312, 108–119. [Google Scholar] [CrossRef]
- Karlmark, K.; Tacke, F.; Dunay, I. Monocytes in health and disease — Minireview. Eur. J. Microbiol. Immunol. 2012, 2, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Di Stefano, V.; Iacono, S.; Lupica, A.; Brighina, F.; Monastero, R.; Balistreri, C.R. Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis. Brain Sci. 2022, 12, 1412. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; Park, J.; Hall, C.K.; Lin, C.-C.J.; Chen, M.; von Maydell, D.; Kruskop, J.M.; Kang, B.; Choi, Y.; Prokopenko, D.; et al. Infiltrating CD8+ T cells exacerbate Alzheimer’s disease pathology in a 3D human neuroimmune axis model. Nat. Neurosci. 2023, 26, 1489–1504. [Google Scholar] [CrossRef]
- Kimura, K.; Nishigori, R.; Hamatani, M.; Sawamura, M.; Ashida, S.; Fujii, C.; Takata, M.; Lin, Y.; Sato, W.; Okamoto, T. Resident Memory-like CD8+ T Cells Are Involved in Chronic Inflammatory and Neurodegenerative Diseases in the CNS. Neurol.-Neuroimmunol. Neuroinflamm. 2024, 11, e200172. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Yang, Y.-T.; Tansey, M.G. Central and peripheral innate and adaptive immunity in Parkinson’s disease. Sci. Transl. Med. 2023, 15, eadk3225. [Google Scholar] [CrossRef]
- Jia, Z.; Guo, M.; Ge, X.; Chen, F.; Lei, P. IL-33/ST2 Axis: A Potential Therapeutic Target in Neurodegenerative Diseases. Biomolecules 2023, 13, 1494. [Google Scholar] [CrossRef]
- Lutshumba, J.; Wilcock, D.M.; Monson, N.L.; Stowe, A.M. Sex-based differences in effector cells of the adaptive immune system during Alzheimer's disease and related dementias. Neurobiol. Dis. 2023, 184, 106202. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, J.; Qu, Q.; Zhao, X.; Zhang, J. JKAP, Th1 cells, and Th17 cells are dysregulated and inter-correlated, among them JKAP and Th17 cells relate to cognitive impairment progression in Alzheimer’s disease patients. Ir. J. Med Sci. (1971-) 2021, 191, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wei, B.; Li, L.; Wang, B.; Sun, M. Th17 cells and inflammation in neurological disorders: Possible mechanisms of action. Front. Immunol. 2022, 13, 932152. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, T.J.; Taha, L.; Spitzer, P.; Hellstern, J.; Herrmann, M.; Kornhuber, J.; Maler, J.M. Imbalance of Circulating Th17 and Regulatory T Cells in Alzheimer’s Disease: A Case Control Study. Front. Immunol. 2018, 9, 1213. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Sheikhi, A.; Jafarzadeh, Z.; Nemati, M. Differential roles of regulatory T cells in Alzheimer's disease. Cell. Immunol. 2023, 393, 104778. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Duan, J.; Mo, J.; Xiao, H.; Huang, Y.; Chen, W.; Xiang, S.; Yang, F.; Chen, Y.; Xu, S. Mild Cognitive Impairment Patients Have Higher Regulatory T-Cell Proportions Compared with Alzheimer's Disease-Related Dementia Patients. Front. Aging Neurosci. 2021, 12. [Google Scholar] [CrossRef]
- Oberstein, T.J.; Taha, L.; Spitzer, P.; Hellstern, J.; Herrmann, M.; Kornhuber, J.; Maler, J.M. Imbalance of Circulating Th17 and Regulatory T Cells in Alzheimer’s Disease: A Case Control Study. Front. Immunol. 2018, 9, 1213. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Seksenyan, A.; Ron-Harel, N.; Azoulay, D.; Cahalon, L.; Cardon, M.; Rogeri, P.; Ko, M.K.; Weil, M.; Bulvik, S.; Rechavi, G.; et al. Thymic involution, a co-morbidity factor in amyotrophic lateral sclerosis. J. Cell. Mol. Med. 2009, 14, 2470–2482. [Google Scholar] [CrossRef]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 1–11. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Liao, B.; Henkel, J.S.; Appel, S.H. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol. Dis. 2012, 48, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; He, L.; Ren, M.; Lu, Y.; Meng, H.; Yin, D.; Chen, S.; Zhou, Q. Paraneoplastic Amyotrophic Lateral Sclerosis: Case Series and Literature Review. Brain Sci. 2022, 12, 1053. [Google Scholar] [CrossRef] [PubMed]
- Plantone, D.; Pardini, M.; Locci, S.; Nobili, F.; De Stefano, N. B Lymphocytes in Alzheimer’s Disease—A Comprehensive Review. J. Alzheimer's Dis. 2022, 88, 1241–1262. [Google Scholar] [CrossRef] [PubMed]
- Ashford, M.T.; Zhu, D.; Bride, J.; McLean, E.; Aaronson, A.; Conti, C.; Cypress, C.; Griffin, P.; Ross, R.; Duncan, T. Understanding Online Registry Facilitators and Barriers Experienced by Black Brain Health Registry Participants: The Community Engaged Digital Alzheimer’s Research (CEDAR) Study. J. Prev. Alzheimer's Dis. 2023, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Asher, S.; Priefer, R. Alzheimer's disease failed clinical trials. Life Sci. 2022, 306, 120861. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R. New frontiers in ageing and longevity: Sex and gender medicine. Mech. Ageing Dev. 2023, 214, 111850. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, S.; Cocozza, G.; Bernardini, G.; Savage, J.; Raspa, M.; Aronica, E.; Tremblay, M.-E.; Ransohoff, R.M.; Santoni, A.; Limatola, C. Blocking immune cell infiltration of the central nervous system to tame Neuroinflammation in Amyotrophic lateral sclerosis. Brain Behav. Immun. 2022, 105, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rekik, A.; Aissi, M.; Rekik, I.; Mhiri, M.; Frih, M.A. Brain atrophy patterns in multiple sclerosis patients treated with natalizumab and its clinical correlates. Brain Behav. 2022, 12, e2573. [Google Scholar] [CrossRef] [PubMed]
- Laaker, C.; Baenen, C.; Kovács, K.G.; Sandor, M.; Fabry, Z. Immune cells as messengers from the CNS to the periphery: the role of the meningeal lymphatic system in immune cell migration from the CNS. Front. Immunol. 2023, 14, 1233908. [Google Scholar] [CrossRef]
- Schirò, G.; Balistreri, C.R. The close link between brain vascular pathological conditions and neurodegenerative diseases: Focus on some examples and potential treatments. Vasc. Pharmacol. 2021, 142, 106951. [Google Scholar] [CrossRef]
- Nicoletti, A.; Baschi, R.; Cicero, C.E.; Iacono, S.; Re, V.L.; Luca, A.; Schirò, G.; Monastero, R. Sex and gender differences in Alzheimer’s disease, Parkinson’s disease, and Amyotrophic Lateral Sclerosis: A narrative review. Mech. Ageing Dev. 2023, 212, 111821. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.-W.; Gao, X.-B. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678.e23. [Google Scholar] [CrossRef] [PubMed]
- Agirman, G.; Yu, K.B.; Hsiao, E.Y. Signaling inflammation across the gut-brain axis. Science 2021, 374, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- McCarville, J.L.; Chen, G.Y.; Cuevas, V.D.; Troha, K.; Ayres, J.S. Microbiota Metabolites in Health and Disease. Annu. Rev. Immunol. 2020, 38, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zeng, B.; Zeng, L.; Du, X.; Li, B.; Huo, R.; Liu, L.; Wang, H.; Dong, M.; Pan, J.; et al. Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl. Psychiatry 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- O'Riordan, K.J.; Collins, M.K.; Moloney, G.M.; Knox, E.G.; Aburto, M.R.; Fülling, C.; Morley, S.J.; Clarke, G.; Schellekens, H.; Cryan, J.F. Short chain fatty acids: Microbial metabolites for gut-brain axis signalling. Mol. Cell. Endocrinol. 2022, 546, 111572. [Google Scholar] [CrossRef]
- Thibaut, M.M.; Bindels, L.B. Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol. Med. 2022, 28, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Angstwurm, K.; Mergenthaler, P.; Kohler, S.; Schönenberger, S.; Bösel, J.; Neumann, U.; Vidal, A.; Huttner, H.B.; Gerner, S.T.; et al. Myasthenic crisis demanding mechanical ventilation. Neurology 2020, 94, E299–E313. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Zhang, Q.; Fu, C.; Jiang, W.; Xue, J.; Liu, S.; Meng, Q.; Ai, L.; Zhi, X.; et al. Autophagy in Disease Onset and Progression. Aging Dis. 2023. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Qu, X.; Sun, G.; Fu, Q.; Han, G. Stress/cell death pathways, neuroinflammation, and neuropathic pain. Immunol. Rev. 2023, 321, 33–51. [Google Scholar] [CrossRef]
- Qu, L.; Li, Y.; Liu, F.; Fang, Y.; He, J.; Ma, J.; Xu, T.; Wang, L.; Lei, P.; Dong, H.; et al. Microbiota-Gut-Brain Axis Dysregulation in Alzheimer's Disease: Multi-Pathway Effects and Therapeutic Potential. Aging Dis. 2023. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Xing, Z.; Peng, C.; Li, D. Autophagy in Neuroinflammation: A Focus on Epigenetic Regulation. Aging Dis. 2024, 15, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-W.; Zhu, X.-X.; Tang, D.-S.; Lu, J.-H. Targeting autophagy in Alzheimer’s disease: Animal models and mechanisms. Zool. Res. 2023, 44, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Nechushtai, L.; Frenkel, D.; Pinkas-Kramarski, R. Autophagy in Parkinson’s Disease. Biomolecules 2023, 13, 1435. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Gökmen, .R.; Uysal, H.; Köksoy, S.; Bilge, U.; Manguoğlu, A.E. Autophagy dysregulation plays a crucial role in regulatory T-cell loss and neuroinflammation in amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler. Front. Degener. 2023, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Niu, F.; Tom, E.; Liao, K.; Periyasamy, P.; Buch, S. Cocaine Mediated Neuroinflammation: Role of Dysregulated Autophagy in Pericytes. Mol. Neurobiol. 2018, 56, 3576–3590. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Thangaraj, A.; Chivero, E.T.; Guo, M.-L.; Periyasamy, P.; Buch, S. Role of Dysregulated Autophagy in HIV Tat, Cocaine, and cART Mediated NLRP3 Activation in Microglia. J. Neuroimmune Pharmacol. 2023, 18, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Elkhoely, A.; Estfanous, R.S.; Alrobaian, M.; Borg, H.M.; Kabel, A.M. Repositioning itraconazole for amelioration of bleomycin-induced pulmonary fibrosis: Targeting HMGB1/TLR4 Axis, NLRP3 inflammasome/NF-κB signaling, and autophagy. Life Sci. 2023, 313, 121288. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, X.; Zhang, H.; Lin, X.; Chen, X.; Zhang, Y.; Lin, X.; Huang, L.; Zhuge, Q. Dimethyl itaconate inhibits LPS-induced microglia inflammation and inflammasome-mediated pyroptosis via inducing autophagy and regulating the Nrf-2/HO-1 signaling pathway. Mol. Med. Rep. 2021, 24, 1–14. [Google Scholar] [CrossRef]
- Li, S.; Huang, P.; Lai, F.; Zhang, T.; Guan, J.; Wan, H.; He, Y. Mechanisms of Ferritinophagy and Ferroptosis in Diseases. Mol. Neurobiol. 2023, 61, 1605–1626. [Google Scholar] [CrossRef]
- Li, L.; Guo, L.; Gao, R.; Yao, M.; Qu, X.; Sun, G.; Fu, Q.; Hu, C.; Han, G. Ferroptosis: a new regulatory mechanism in neuropathic pain. Front. Aging Neurosci. 2023, 15, 1206851. [Google Scholar] [CrossRef] [PubMed]
- Chavoshinezhad, S.; Beirami, E.; Izadpanah, E.; Feligioni, M.; Hassanzadeh, K. Molecular mechanism and potential therapeutic targets of necroptosis and ferroptosis in Alzheimer's disease. Biomed. Pharmacother. 2023, 168, 115656. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; John, U.; Bano, N.; Khan, S.; Bhat, S.A. Oxytosis/Ferroptosis in Neurodegeneration: the Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol. Neurobiol. 2023, 61, 1507–1526. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R. Vascular ageing and the related complications in the brain: New insights on related mechanisms and their translational applications. Mech. Ageing Dev. 2021, 196, 111469. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Giarratana, R.M.; Scola, L.; Balistreri, C.R. The close link between the fetal programming imprinting and neurodegeneration in adulthood: The key role of “hemogenic endothelium” programming. Mech. Ageing Dev. 2021, 195, 111461. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R. Promising Strategies for Preserving Adult Endothelium Health and Reversing Its Dysfunction: From Liquid Biopsy to New Omics Technologies and Noninvasive Circulating Biomarkers. Int. J. Mol. Sci. 2022, 23, 7548. [Google Scholar] [CrossRef]
- Olivieri, F.; Pompilio, G.; Balistreri, C.R. Endothelial progenitor cells in ageing. Mech. Ageing Dev. 2016, 159, 1–3. [Google Scholar] [CrossRef]
- Tregub, P.P.; Ibrahimli, I.; Averchuk, A.S.; Salmina, A.B.; Litvitskiy, P.F.; Manasova, Z.S.; Popova, I.A. The Role of microRNAs in Epigenetic Regulation of Signaling Pathways in Neurological Pathologies. Int. J. Mol. Sci. 2023, 24, 12899. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, M.; Conti, G.O. Environment and Neurodegenerative Diseases: An Update on miRNA Role. MicroRNA 2017, 6, 157–165. [Google Scholar] [CrossRef]
- Zhang, J.; Li, A.; Gu, R.; Tong, Y.; Cheng, J. Role and regulatory mechanism of microRNA mediated neuroinflammation in neuronal system diseases. Front. Immunol. 2023, 14, 1238930. [Google Scholar] [CrossRef]
- Rastegar-Moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. Roles of the miR-155 in Neuroinflammation and Neurological Disorders: A Potent Biological and Therapeutic Target. Cell. Mol. Neurobiol. 2022, 43, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, H.; Xie, W.; Wei, N.; Liu, M. MicroRNA-195 triggers neuroinflammation in Parkinson's disease in a Rho-associated kinase 1-dependent manner. Mol. Med. Rep. 2019, 19, 5153–5161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.; Teng, Z.; Tang, Y.-P.; Chen, C. Synaptic and Cognitive Improvements by Inhibition of 2-AG Metabolism Are through Upregulation of MicroRNA-188-3p in a Mouse Model of Alzheimer's Disease. J. Neurosci. 2014, 34, 14919–14933. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Tan, J.; Zhang, J. miR-137 attenuates Aβ-induced neurotoxicity through inactivation of NF-κB pathway by targeting TNFAIP1 in Neuro2a cells. Biochem. Biophys. Res. Commun. 2017, 490, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, Z.; Dong, J.; Bi, H.; Wang, B.; Du, K.; Zhang, C.; Chen, L. lncRNA XIST inhibition promotes M2 polarization of microglial and aggravates the spinal cord injury via regulating miR-124–3p / IRF1 axis. Heliyon 2023, 9, e17852. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, H.; Ma, J.; Luo, S.; Chen, S.; Gu, Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum. Cell 2017, 31, 106–115. [Google Scholar] [CrossRef]
- Singh, V.; Kushwaha, S.; Ansari, J.A.; Gangopadhyay, S.; Mishra, S.K.; Dey, R.K.; Giri, A.K.; Patnaik, S.; Ghosh, D. MicroRNA-129-5p-regulated microglial expression of the surface receptor CD200R1 controls neuroinflammation. J. Biol. Chem. 2021, 298, 101521. [Google Scholar] [CrossRef]
- Nasirishargh, A.; Kumar, P.; Ramasubramanian, L.; Clark, K.; Hao, D.; Lazar, S.V.; Wang, A. Exosomal microRNAs from mesenchymal stem/stromal cells: Biology and applications in neuroprotection. World J. Stem Cells 2021, 13, 776–794. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Bao, J.; Su, J.; Huang, W. MicroRNA-139 modulates Alzheimer’s-associated pathogenesis in SAMP8 mice by targeting cannabinoid receptor type 2. Evolution 2017, 16. [Google Scholar] [CrossRef]
- Tassinari, V.; La Rosa, P.; Guida, E.; Colopi, A.; Caratelli, S.; De Paolis, F.; Gallo, A.; Cenciarelli, C.; Sconocchia, G.; Dolci, S.; et al. Contribution of A-to-I RNA editing, M6A RNA Methylation, and Alternative Splicing to physiological brain aging and neurodegenerative diseases. Mech. Ageing Dev. 2023, 212, 111807. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. NF-κB pathway activators as potential ageing biomarkers: targets for new therapeutic strategies. Immun. Ageing 2013, 10, 24–16. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Colonna-Romano, G.; Lio, D.; Candore, G.; Caruso, C. TLR4 Polymorphisms and Ageing: Implications for the Pathophysiology of Age-Related Diseases. J. Clin. Immunol. 2009, 29, 406–415. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.-I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Nampoothiri, M.; Gurram, P.C.; Manandhar, S.; Satarker, S.; Mudgal, J.; Arora, D. Dopaminergic Signaling as a Plausible Modulator of Astrocytic Toll-Like Receptor 4: A Crosstalk between Neuroinflammation and Cognition. CNS Neurol. Disord.-Drug Targets 2023, 22, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Sivamaruthi, B.S.; Raghani, N.; Chorawala, M.; Bhattacharya, S.; Prajapati, B.G.; Elossaily, G.M.; Chaiyasut, C. NF-κB Pathway and Its Inhibitors: A Promising Frontier in the Management of Alzheimer’s Disease. Biomedicines 2023, 11, 2587. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Chen, Y.; Ye, B.; Guo, W.; Wang, D.; He, J. Natural products for the treatment of neurodegenerative diseases. Phytomedicine 2023, 121, 155101. [Google Scholar] [CrossRef] [PubMed]
- Decandia, D.; Gelfo, F.; Landolfo, E.; Balsamo, F.; Petrosini, L.; Cutuli, D. Dietary Protection against Cognitive Impairment, Neuroinflammation and Oxidative Stress in Alzheimer’s Disease Animal Models of Lipopolysaccharide-Induced Inflammation. Int. J. Mol. Sci. 2023, 24, 5921. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R. Anti-Inflamm-Ageing and/or Anti-Age-Related Disease Emerging Treatments: A Historical Alchemy or Revolutionary Effective Procedures? Mediat. Inflamm. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Dias-Carvalho, A.; Sá, S.I.; Carvalho, F.; Fernandes, E.; Costa, V.M. Inflammation as common link to progressive neurological diseases. Arch. Toxicol. 2023, 98, 95–119. [Google Scholar] [CrossRef]
- Koole, L.; Martinez-Martinez, P.; van Amelsvoort, T.; Evelo, C.T.; Ehrhart, F. Interactive neuroinflammation pathways and transcriptomics-based identification of drugs and chemical compounds for schizophrenia. World J. Biol. Psychiatry 2023, 25, 116–129. [Google Scholar] [CrossRef]
- Lashgari, N.-A.; Roudsari, N.M.; Shamsnia, H.S.; Shayan, M.; Momtaz, S.; Abdolghaffari, A.H. TLR/mTOR inflammatory signaling pathway: novel insight for the treatment of schizophrenia. Can. J. Physiol. Pharmacol. 2024, 102, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Sarapultsev, A.; Gusev, E.; Komelkova, M.; Utepova, I.; Luo, S.; Hu, D. JAK-STAT signaling in inflammation and stress-related diseases: implications for therapeutic interventions. Mol. Biomed. 2023, 4, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Roy, S.; Zehravi, M.; Paul, S.; Sutradhar, H.; Yaidikar, L.; Kumar, B.R.; Dogiparthi, L.K.; Prema, S.; Nainu, F.; et al. Polyphenols Targeting MAP Kinase Signaling Pathway in Neurological Diseases: Understanding Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 2023, 1–21. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, X.; Zhang, R.; Wang, Z.; Gan, J.; Gao, Q.; Yang, L.; Xu, P.; Jiang, X. Inflammatory signaling pathways in the treatment of Alzheimer's disease with inhibitors, natural products and metabolites (Review). Int. J. Mol. Med. 2023, 52, 1–42. [Google Scholar] [CrossRef]
- Singh, L.; Bhatti, R. Signaling Pathways Involved in the Neuroprotective Effect of Osthole: Evidence and Mechanisms. Mol. Neurobiol. 2023, 61, 1100–1118. [Google Scholar] [CrossRef]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef]
- Chen, R.; Routh, B.N.; Gaudet, A.D.; Fonken, L.K. Circadian Regulation of the Neuroimmune Environment Across the Lifespan: From Brain Development to Aging. J. Biol. Rhythm. 2023, 38, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, J.; Chen, G. Circadian cycle and neuroinflammation. Open Life Sci. 2023, 18, 20220712. [Google Scholar] [CrossRef]
- Ahmad, F.; Sachdeva, P.; Sarkar, J.; Izhaar, R. Circadian dysfunction and Alzheimer's disease – An updated review. Aging Med. 2022, 6, 71–81. [Google Scholar] [CrossRef]
- Meng, D.; Yang, M.; Zhang, H.; Zhang, L.; Song, H.; Liu, Y.; Zeng, Y.; Yang, B.; Wang, X.; Chen, Y.; et al. Microglia activation mediates circadian rhythm disruption-induced cognitive impairment in mice. J. Neuroimmunol. 2023, 379, 578102. [Google Scholar] [CrossRef]
- Srinivasan, M.; Walker, C. Circadian Clock, Glucocorticoids and NF-κB Signaling in Neuroinflammation- Implicating Glucocorticoid Induced Leucine Zipper as a Molecular Link. ASN Neuro 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Aho, V.; Ollila, H.M.; Rantanen, V.; Kronholm, E.; Surakka, I.; van Leeuwen, W.M.A.; Lehto, M.; Matikainen, S.; Ripatti, S.; Härmä, M.; et al. Partial Sleep Restriction Activates Immune Response-Related Gene Expression Pathways: Experimental and Epidemiological Studies in Humans. PLoS ONE 2013, 8, e77184. [Google Scholar] [CrossRef]
- Taishi, P.; Chen, Z.; Obál, F., Jr.; Hansen, M.K.; Zhang, J.; Fang, J.; Krueger, J.M. Sleep-Associated Changes in Interleukin-1β mRNA in the Brain. J. Interf. Cytokine Res. 1998, 18, 793–798. [Google Scholar] [CrossRef]
- Szentirmai,.; Kapás, L. Sleep and body temperature in TNFα knockout mice: The effects of sleep deprivation, β3-AR stimulation and exogenous TNFα. Brain Behav. Immun. 2019, 81, 260–271. [CrossRef]
- Nguyen, J.; Gibbons, C.M.; Dykstra-Aiello, C.; Ellingsen, R.; Koh, K.M.S.; Taishi, P.; Krueger, J.M. Interleukin-1 receptor accessory proteins are required for normal homeostatic responses to sleep deprivation. J. Appl. Physiol. 2019, 127, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.E.; Miyanishi, K.; Takeda, H.; Islam, A.; Matsuoka, N.; Kubo, M.; Matsumoto, S.; Kunieda, T.; Nomoto, M.; Yano, H.; et al. Phagocytic elimination of synapses by microglia during sleep. Glia 2020, 68, 44–59. [Google Scholar] [CrossRef]
- Klarica, M.; Radoš, M.; Orešković, D. The Movement of Cerebrospinal Fluid and Its Relationship with Substances Behavior in Cerebrospinal and Interstitial Fluid. Neuroscience 2019, 414, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Hand, L.E.; Gray, K.J.; Dickson, S.H.; Simpkins, D.A.; Ray, D.W.; Konkel, J.E.; Hepworth, M.R.; Gibbs, J.E. Regulatory T cells confer a circadian signature on inflammatory arthritis. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- García-García, A.; Korn, C.; García-Fernández, M.; Domingues, O.; Villadiego, J.; Martín-Pérez, D.; Isern, J.; Bejarano-García, J.A.; Zimmer, J.; Pérez-Simón, J.A.; et al. Dual cholinergic signals regulate daily migration of hematopoietic stem cells and leukocytes. Blood 2019, 133, 224–236. [Google Scholar] [CrossRef]
- Adrover, J.M.; del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossío, I. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 50, 390–402.e10. [Google Scholar] [CrossRef]
- O'Siorain, J.R.; Curtis, A.M. Circadian Control of Redox Reactions in the Macrophage Inflammatory Response. Antioxidants Redox Signal. 2022, 37, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Tsai, C.; Litichevskiy, L.; Shipony, Z.; Iweka, C.A.; Schneider, K.M.; Chuluun, B.; Heller, H.C.; Menon, V.; Thaiss, C.A.; et al. Aging disrupts circadian gene regulation and function in macrophages. Nat. Immunol. 2021, 23, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jin, X.; Xu, F.; Wang, S.; Liu, L.; Li, X.; Lin, H.; Du, M. Circadian rhythm-associated Rev-erbα modulates polarization of decidual macrophage via the PI3K/Akt signaling pathway. Am. J. Reprod. Immunol. 2021, 86, e13436. [Google Scholar] [CrossRef] [PubMed]
- Nobis, C.C.; Laramée, G.D.; Kervezee, L.; De Sousa, D.M.; Labrecque, N.; Cermakian, N. The circadian clock of CD8 T cells modulates their early response to vaccination and the rhythmicity of related signaling pathways. Proc. Natl. Acad. Sci. 2019, 116, 20077–20086. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Monastero, R.; Caruso, C.; Vasto, S. Alzheimer’s disease and infections, where we stand and where we go. Immun. Ageing 2014, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Proctor, M.J.; McMillan, D.C.; Horgan, P.G.; Fletcher, C.D.; Talwar, D.; Morrison, D.S. Systemic Inflammation Predicts All-Cause Mortality: A Glasgow Inflammation Outcome Study. PLOS ONE 2015, 10, e0116206. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in Aging and Chronic Disease: A Magnificent Pathway. Journals Gerontol. Ser. A 2006, 61, 575–584. [Google Scholar] [CrossRef]
- Wright, T.M.; A Feghali, C. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–26. [Google Scholar] [CrossRef]
- Parra-Torres, V.; Melgar-Rodríguez, S.; Muñoz-Manríquez, C.; Sanhueza, B.; Cafferata, E.A.; Paula-Lima, A.C.; Díaz-Zúñiga, J. Periodontal bacteria in the brain—Implication for Alzheimer's disease: A systematic review. Oral Dis. 2021, 29, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dashper, S.G.; Zhao, R. Association Between Oral Bacteria and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer's Dis. 2023, 91, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Hossain, S.; El-Hajj, Z.W.; Weiss, J.; Zonderman, A.B. Clinical and Bacterial Markers of Periodontitis and Their Association with Incident All-Cause and Alzheimer’s Disease Dementia in a Large National Survey. J. Alzheimer's Dis. 2020, 75, 157–172. [CrossRef]
- Larvin, H.; Gao, C.; Kang, J.; Aggarwal, V.R.; Pavitt, S.; Wu, J. The impact of study factors in the association of periodontal disease and cognitive disorders: systematic review and meta-analysis. J. Am. Geriatr. Soc. 2023, 52, afad015. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jin, Y.; Li, K.; Qiu, H.; Jiang, Z.; Zhu, J.; Chen, S.; Xie, W.; Chen, G.; Yang, D. Is There an Association Between Parkinson’s Disease and Periodontitis? A Systematic Review and Meta-Analysis. J. Park. Dis. 2023, 13, 1107–1125. [Google Scholar] [CrossRef]
- Yazdanpanahi, N.; Etemadifar, M.; Kardi, M.-T.; Shams, E.; Shahbazi, S. Investigation of ERG Gene Expression in Iranian Patients with Multiple Sclerosis. Immunol. Investig. 2018, 47, 351–359. [Google Scholar] [CrossRef]
- Pisano, C.; Terriaca, S.; Scioli, M.G.; Nardi, P.; Altieri, C.; Orlandi, A.; Ruvolo, G.; Balistreri, C.R. The Endothelial Transcription Factor ERG Mediates a Differential Role in the Aneurysmatic Ascending Aorta with Bicuspid or Tricuspid Aorta Valve: A Preliminary Study. Int. J. Mol. Sci. 2022, 23, 10848. [Google Scholar] [CrossRef]
Figure 1.
(by Biorender software): Important drivers, i.e., alterations in the MGB axis, infections, ischemia contribute of the activation and infiltration of circulating monocytes in the brain and their specific secretion of inflammation mediators. These latter influence the activation of other resident immune cells thus leading to neuroinflammation and neurodegeneration. Similarly, clonotype cells influence neuroinflammation.
Figure 1.
(by Biorender software): Important drivers, i.e., alterations in the MGB axis, infections, ischemia contribute of the activation and infiltration of circulating monocytes in the brain and their specific secretion of inflammation mediators. These latter influence the activation of other resident immune cells thus leading to neuroinflammation and neurodegeneration. Similarly, clonotype cells influence neuroinflammation.
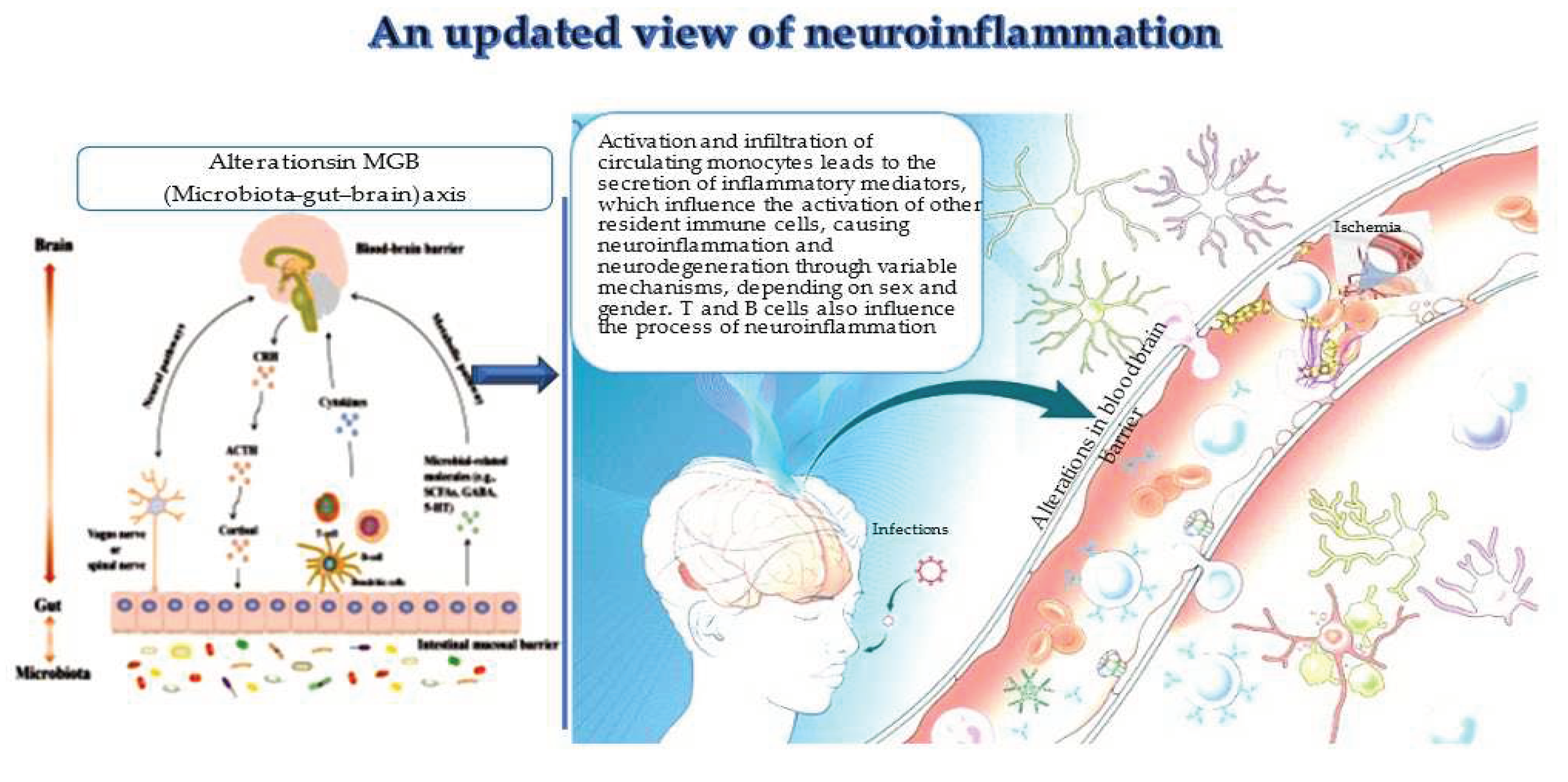
Figure 2.
A and B (by Biorender software): Model detailing the novel mechanisms described in the text involved in neuroinflammation and its relation to the onset of ND. In A, the unconventional mechanisms that determine the activation of NF-kB pathway through canonical or non-canonical signaling. In B, it is shown how activation of the NF-kB pathway can activate a network of different signaling pathways.
Figure 2.
A and B (by Biorender software): Model detailing the novel mechanisms described in the text involved in neuroinflammation and its relation to the onset of ND. In A, the unconventional mechanisms that determine the activation of NF-kB pathway through canonical or non-canonical signaling. In B, it is shown how activation of the NF-kB pathway can activate a network of different signaling pathways.
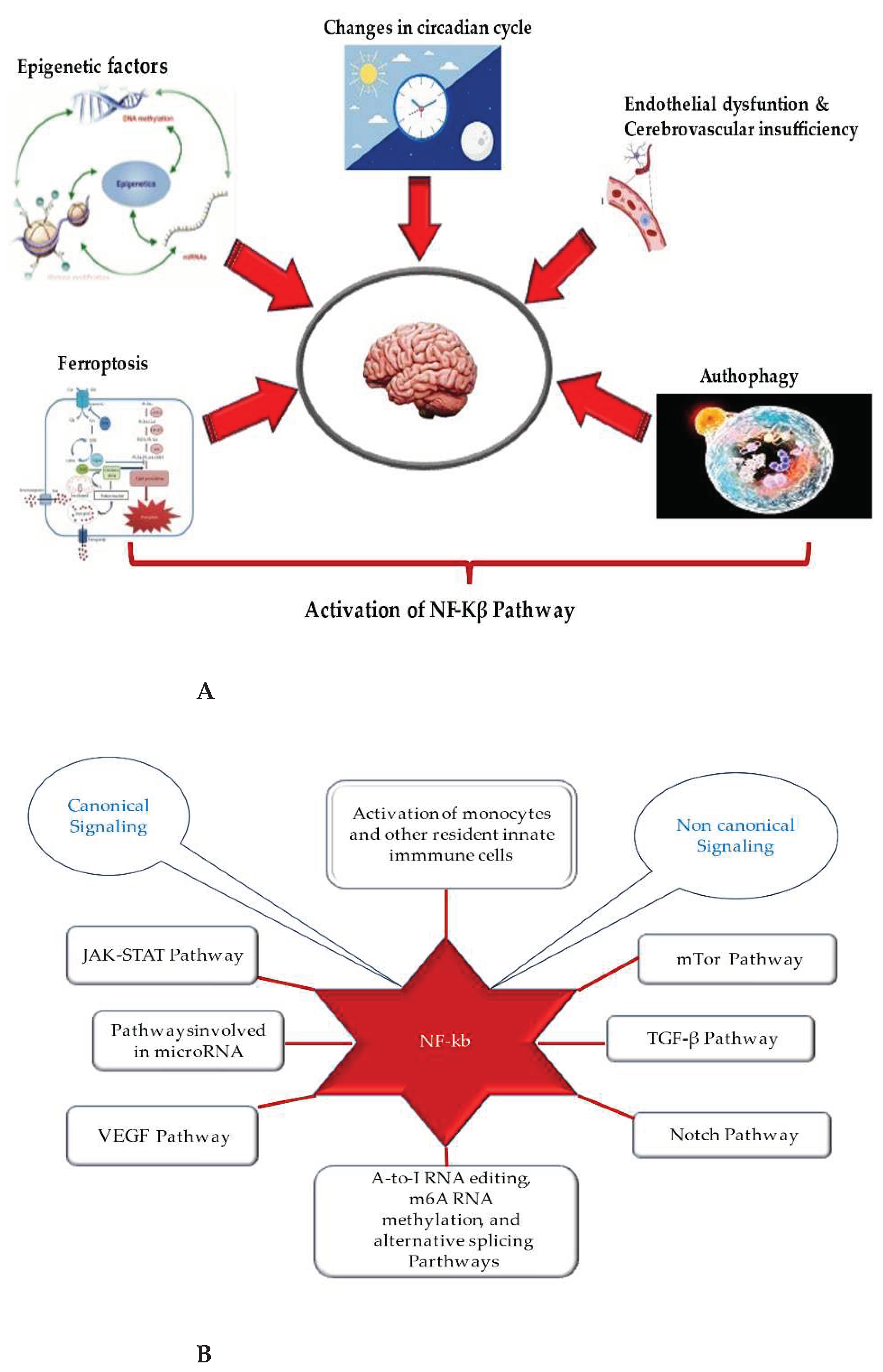
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



