Submitted:
28 November 2023
Posted:
30 November 2023
You are already at the latest version
Abstract
Cadmium (Cd) is a pervasive toxic metal, present in most food types, cigarette smoke, and contaminated air. It has no nutritional or physiological value, but most cells in the body will assimilate Cd, as its charge and ionic radius are similar to the essential metals, iron, zinc, and calcium (Fe, Zn, Ca), which are required for cellular metabolism and functional integrity. The kidney is regarded as the principal site of Cd toxicity as it preferentially accumulates in the proximal tubular epithelium here. As these cells die due to toxic accumulation, Cd complexed with metallothionein are released into tubular lumen and excreted. Excretion of Cd, signifying current renal cell toxicity, is used as a measure of long-term Cd exposure. Approximately 10% of the global population are now living with diabetes and over 80% of these are overweight or obese. This association has fueled an intense search for any exogenous chemicals and life-style factors that could induce excessive weight gain. Whilst epidemiological studies have clearly linked diabetes to Cd exposure, this appears to be independent of obesity. Indeed, obesity may enhance diabetogenic effects of Cd. This review highlights Cd exposure sources and levels associated with diabetes type 2 along with the pathophysiologic mechanisms by which Cd disrupt glucose metabolism. Special emphasis is on roles of the liver and kidney in glucose homeostasis, heme-glucose cross-talk, and the involvement of heme oxygenase-1 and -2 (HO-1 and HO-2). From heme degradation, both HO-1 and HO-2 release Fe, carbon monoxide and a precursor substrate for producing a cytoprotective biochemical, bilirubin, whilst HO-2 appears to have also anti-diabetic and anti-obese actions.
Keywords:
bilirubin
; cadmium
; diabetes type 2
; glucose homeostasis
; gluconeogenesis
; glycolysis
; heme
; heme oxygenase
; hyperglycemia
; mitochondria
; obesity.
1. Introduction
Cadmium (Cd) is a redox inert metal, present in relatively low levels in the Earth’s crust and most surface soils [1,2,3]. It is primary present as a sulphide such as in greenockite and zinc ore, sphalerite [2]. Hence, mining, smelting, and refining of zinc ores yield Cd as the main byproduct. It has been widely used in many industrial processes because of its metallic and anti-corrosive properties [1,2]. The realization in the 1940s that the condition referred to as “itai-itai” disease in the Jinzu river basin of Japan was due to excessive exposure to Cd through consumption of rice grown on paddy soils contaminated with the discharge from zinc mining, brought into focus the real threat to health posed by this metal [4,5,6].
Because of its notoriously high toxicity, the worldwide production and industrial applications of Cd have significantly declined. However, the use of Cd-contaminated phosphate fertilizers persists, and adds substantial amount of Cd to the food chain in most parts of the world [7,8,9,10]. Cd, like all other metals, is nonbiodegradable, and consequently, it persists indefinitely in the environment. Because of its efficient soil-to-plant transference, Cd can accumulate in vegetation even the levels of Cd in soils are very low [6,11].
The kidney and bone were described as the organs severely affected in “itai-itai” disease patients [4,5]. Current evidence, however, suggests that effects of Cd exposure extend beyond kidney and bone, and that Cd even at very low levels can adversely impact the functions of most organs of the body. Many pathologic conditions, including hypertension, diabetes type 2 (DM), chronic kidney disease (CKD), osteoporosis, non-alcoholic fatty liver, infertility, and various types of cancer have been linked to environmental Cd exposure in the general populations of many countries [12,13,14].
The living organism does not synthesize or break down metals, and transporter systems and pathways have evolved, consequently, to acquire from exogenous sources all metals such as iron, zinc, manganese, calcium, copper, and cobalt (Fe, Zn, Mn, Ca, Cu, Co) [15,16,17,18,19]. These multiple transporters systems, in turn, provide Cd entry routes into most cells in the body. Similarly, through these metal transporters, Cd reaches the inner membrane of the mitochondria, where it affects the synthesis of adenosine triphosphate (ATP), inhibits the electron transport chain, and promotes the formation of reactive oxygen species (ROS) with resultant oxidative stress conditions [20,21,22,23,24].
The organs with high metabolic activities and high energy demands, like kidneys, ovaries, and testes, are particularly sensitive to Cd-induced mitochondrial dysfunction, a central mechanism by which Cd affects most cells in the body.
The present review has its focus on the impact of environmental Cd exposure on the prevalence of metabolic derangement, pre-diabetes and diabetes, defined as fasting plasma glucose ≥ 110 mg/dL and 126 mg/dL, respectively. The global prevalence of diabetes has reached epidemic proportions, and the epidemic is most frequently attributed to the concurrently increasing prevalence of obesity. Environmental Cd exposure is unrecognized and ignored contributing factor. Intriguingly, intracellular levels of the natural cyclic AMP antagonist prostaglandyl-inositol cyclic phosphate (cyclic PIP) and cyclic adenosine monophosphate have been postulated to be the biochemicals that trigger insulin resistance, a dominant feature of DM [25,26].
Diabetes together with its complication, diabetic kidney disease (DKD) is the leading cause of kidney failure worldwide. Given that the global incidence of diabetes and DKD, and their treatment costs continue to rise, developing strategies to prevent diabetes and DKD is of global importance.
Current evidence suggests that environmental exposure to even low levels of Cd promotes the development and progression of DKD [27,28], and that people with DM are highly susceptible to the nephrotoxicity of Cd [29,30,31,32]. A no-observed-adverse-effect level of kidney Cd accumulation is as little as 0.01 µg/g creatinine [33,34]. A dietary exposure guideline of 0.83 μg per kg body weight per day (58 µg/day for a 70-kg person), and a nephrotoxicity threshold level of 5.24 µg/g creatinine [35,36] are not protective of human health.
2. Exposure Sources, Dosimetry and Health Risk Assessment
2.1. Dietary exposure to cadmium
For non-smoking and non-occupationally exposed populations, diet is the major environmental sources of Cd exposure. This exposure source is indicated by levels of Cd and various contaminants, and food additives in the human diet reported in total diet study (TDS), a food safety monitoring program, conducted by food authority agencies [37,38]. TDS involves collection of samples of foodstuffs from supermarkets and retail stores for quantitation of various food additives, pesticide residues, contaminants, and nutrients, and is thus known also as the “market basket survey”. The level of exposure is computed based on the concentration of a given contaminant in collected food samples and an amount of the food item consumed per day. Typically, the median and the 90th percentile concentration levels of a contaminant are used, to approximate the exposure levels among average and high consumers, respectively [39,40].
TDS data indicated that dietary Cd exposure levels varied widely among populations, but foods that are frequently consumed in large quantities, such as rice, potatoes, wheat, and leafy salad vegetables are consistently identified as the major sources of Cd [14]. It is noteworthy that rice is a staple food for over half of the world’s population, and in some regions, rice contributes to more than 50% of the total Cd intake, detailed below.
Dietary Cd exposure levels among women in two Cd-polluted areas of Japan were 55.7 µg/d (1.03 µg/kg body weight) and 48.7 µg/d (0.86 µg/kg bw/d) [6]. Rice and its products constituted 40–50% of these Cd exposures. Differences between the two groups with respect to dietary Cd exposure were attributed mostly to Cd levels in rice consumed in the two locations. Cd was found in all plant foods investigated, but especially in spinach, Japanese parsley, garland chrysanthemum, Japanese mustard spinach, belvedere fruit, shiitake mushrooms, and seaweed. Important animal sources of Cd included shellfish, salted squid guts, scallops, oysters, and freshwater clams. Chocolate and tea leaves contained also high-Cd levels, but the metal was undetectable in brewed tea.
In comparison, the mean dietary Cd exposure in the general Japanese population was 0.35 μg/kg bw/day, ranging between 0.25 and 0.45 μg/kg bw/day [41]. Respective contribution of exposure to Cd in rice and its products, green vegetables, cereals, and seeds plus potatoes were 38%, 17%, and 11%.
Dietary Cd exposure in China was 32.7 μg/day, with rice and vegetables being the main sources [42]. In Mongolia, potatoes were the main source of Cd, which contributed to 24% of total Cd exposure [42]. Nori, peanuts, squid, cuttlefish and mushrooms had notably high Cd levels [43,44].
Dietary Cd exposure in South Korea was 12.6 μg/day [45]. Cereals and vegetables, beverages, fruits and nuts, and dairy products (milk included) were the main dietary sources. Cereals, oil seeds and fruits, and vegetables had relatively high Cd concentrations. A Cd exposure of 22 μg/day was reported in another Korean study [46]. Given average rice consumption of 587-611 g/day, rice was the major contributor (40.3%) to total Cd exposure, followed by squid (11.8%), eel (11.0%), crab (8.6%), shellfish (3.6%), kimchi (Korean cabbage; 3.5%) and seaweed (3.5%).
2.2. The intestinal absorption of cadmium
As detailed above, dietary Cd exposure is inevitable for most people, given the presence of the metal in staples and nearly all food types. In all likelihoods, Cd in the gut gains an entry into the portal blood system through transporters for calcium, zinc, manganese, iron, copper and cobalt (Ca, Zn, Mn, Fe, Cu, and Co) [47,48,49]. Examples of such metal transporters are members of the Zrt- and Irt-related protein (ZIP) of the zinc transporter family and the Ca2+-selective channel TRPV6 [50,51,52,53]. Furthermore, Cd complexed with the metal binding protein, metallothionine (MT) and phytochelatin (PC) as CdMT and CdPC can be absorbed through transcytosis, and endocytosis, mediated by the human neutrophil gelatinase-associated lipocalin (hNGAL) [54,55,56].
Similarly, through the transporters for essential metals, Cd can enter most cells in the body, including hepatocytes [57], kidney tubular epithelial cells [58,59,60,61,62,63,64], adipocytes [65], insulin producing pancreatic β-cells [66], ovaries [67], testes [68], and erythrocytes [69,70,71]. However, no physiologic mechanism exists to eliminate Cd. Consequently, virtually all of the acquired metal is retained and the cellular levels of Cd increased with age (duration of exposure).
The body burden of is essentially determined by absorption rate. In theory, the absorption rate of Cd will increase when the body content of Fe, Zn, or Ca is low, and when diets are deficient in these nutritionally essential metals.
Table 1 provides Cd accumulation levels in various tissues, recorded in Australian and Japanese autopsy studies.
Preferential Cd accumulation in the kidney cortex and in female gender were apparent from both Australian and Japanese autopsy studies [58,59]. The mean liver Cd in Australian women was 1.74-fold higher than men, and after adjustment for inhalation exposure, women had higher kidney cortical Cd content than did men [58]. In comparison, the mean liver Cd in women living in a non-Cd contaminated location of Japan was 1.6-fold higher than men [59].
Fractionally, the difference between men and women in kidney cortex content is smaller than the difference in hepatic content. A plausible interpretation is that women have lower iron stores, and adjustments to increase intestinal iron absorption lead to increased absorption and liver uptake of Cd from dietary exposure. Redistribution of hepatic Cd to the kidney may be sufficient to cause a higher kidney content of Cd as well, but not so great as to obscure the dietary origin of the increased Cd burden.
2.3. Blood and plasma (serum) cadmium
Cd enters erythrocytes through the chloride/bicarbonate anion exchanger ([Cl−/HCO3−], AE1, SLC4A1) [68,69,70], and iron transporters that were responsible also for erythrocytic uptake of lead (Pb) and Zn [72,73,74]. Cd may induce the erythrocyte membrane morphological change [75], leading to premature hemolysis in the reticuloendothelial system, and thereby shorten cellular lifespan [76]. Cd may induce eryptosis, erythrocytic suicidal cell death, which is the mechanism by which injured red blood cells are removed [77,78].
Most of the circulating Cd is bound to hemoglobin in red blood cells [79]. Less than 10% of Cd the circulation is in the plasma, where it is associated with histidine and thiols (-SH) of peptides and proteins, such as pre-albumin, albumin, α2-macroglobulin, and immunoglobulins G and A [80,81,82]. Examples of the non-protein plasma thiols that interact with Cd are glutathione (GSH), cysteine, cysteinylglycine, homocysteine, and γ-glutamylcysteine [83,84]. The total concentrations of these non-protein thiols are in the low µM range (12–20 µM). In comparison, albumin thiol is more abundant (0.6 mM), implying a significant role of albumin in the transport and delivery of Cd to cells throughout the body [85].
The estimated half-life of blood Cd varied from 75 to 128 days [86]. Because the mean life-span of erythrocytes is 120 days, blood Cd is used as an indicator of recent exposure to the metal. In an epidemiologic investigation, a significant correlation was observed between blood and urine, which suggested that a blood Cd level may reflect partially long-term exposure [87].
In theory, plasma Cd is more predictive of tissue toxic injury than erythrocytic Cd because plasma Cd is readily exchangeable with other metals in target tissues. However, use of plasma Cd in exposure assessment is limited because of the high cost involved in its quantification as plasma Cd is in a nano molar (nM) range. Presently, the distribution of Cd in whole blood and plasma is not known. The relationship between blood and plasma Cd at varying exposure levels could provide a method to predict plasma Cd from whole blood/erythrocyte Cd data.
2.4. Excretion of cadmium siginfies kidney tubular cell injury and death
The kidney is an organ, where most Cd accumulates. This is because kidney tubular epithelial cells are equipped with protein internalization mechanisms that enable reabsorption of virtually all proteins in the ultrafiltrate [88,89,90,91]. These protein retrieval systems are additional to various metal transporters expressed in the apical and basolateral membranes of tubular cells [14]. These multiple metal transporters and protein re-absorptive pathways provide Cd several entry routes into tubular cells along with proteins to which it is bound such as albumin, transferrin receptor [91].
Experimental studies in rats using microinjection technique found that 70–90% of Cd was taken up in the S1-segment of the proximal tubule [92,93], where the megalin/cubillin receptor-mediated endocytosis is involved. The neutrophil gelatinase-associated lipocalin (NGAL)/lipocalin-2 receptor system has also been implicated in reabsorption of Cd-protein complexes in the distal tubule and the collecting duct [94,95,96].
The multiple entry routes of Cd, but there is no exit route means that most acquired Cd is retained in kidneys, and is released to tubular lumen due to cell death from Cd toxicity [14,97]. Thus, the excretion of Cd signifies tubular cell injury and death induced by a cumulative burden of Cd [14,97].
Figure 1 shows the mitochondrion as the toxicity target of Cd.
2.5. Is urinary β2M indicative of tubulopathy ?
β2M protein with a molecular weight of 11,800 Da, is synthesized and shed by all nucleated cells [98]. β2M is filtered freely by glomeruli, and is reabsorbed almost completely by proximal tubular cells [99]. Increased β2M excretion has been used to document tubular dysfunction [100,101] and was described as a key feature of Cd nephropathy. However, there are attributes of β2M excretion that compromise its utility for such purposes. First, β2M production rises in response to many inflammatory and neoplastic conditions [102]. Second, if reabsorption rates of β2M per nephron remain constant as its production rates change, excretion will vary directly with its production. Third, if the production and reabsorption per nephron remain constant as nephrons are lost, the excretion of β2M will rise.
The increased β2M excretion due to Cd-induced nephron loss has been revealed in a dose-response analysis, where β2M excretion of 100–299, 300–999, and ≥ 1000 μg/g creatinine were associated with 4.7-fold, 6.2-fold and 10.5-fold increases in the likelihood of eGFR ≤ 60 mL/min/1.73 m2 [103].
A threshold of toxicity is defined as the highest dose that does not produce an adverse effect in the most sensitive organ (endpoint) [104]. A rise of β2M excretion above 300 µg/g creatinine (tubular proteinuria) is the manifestation of severe toxicity of Cd in kidneys, and its use as an endpoint to determine an exposure guideline is inappropriate.
2.6. Health risk assessment of cadmium exposure
2.7.1 Exposure guideline
The Joint FAO/WHO Expert Committee on Food Additives and Contaminants (JECFA) used tubular proteinuria, defined as a rise of urinary excretion of β2-microglobulin (β2M) above 300 µg/g creatinine, to indicate the nephrotoxicity of dietary Cd exposure [35]. Based solely on this endpoint, a tolerable monthly intake (TMI) of Cd was found to be 25 μg per kg body weight per month, equivalent to 0.83 μg per kg body weight per day [35]. A Cd excretion of 5.24 μg/g creatinine was suggested to be a nephrotoxicity threshold value [35].
The European Food Safety Authority (EFSA) employed the same endpoint, but a Cd excretion of 1 μg/g creatinine was designated as the toxicity threshold after inclusion of an uncertainty factor (safety margin) [102]. A dietary exposure of Cd at 0.36 μg/kg body weight per day for 50 years was viewed as an acceptable Cd ingestion level or reference dose (RfD) [109].
2.7.2 Population data
In a risk analysis of Chinese population data, a dietary exposure level of 16.8 µg/day for a 60-kg person (0.28 μg/kg body weight per day) was suggested to be a tolerable intake level, when tubular proteinuria (β2M) was an endpoint [110]. A corresponding threshold level of Cd excretion was 3.07 μg/g creatinine.
In risk analysis of Thai population data, nephron loss was used as an endpoint, and Cd excretion level that is likely to produce a negligible adverse effect, termed benchmark dose limit or the NOAEL equivalent was 0.01 µg/g creatinine [33].
3. Cadmium, Obesity and Diseases with High Prevalence
Numerous population studies have linked diseases with high prevalence, such as DM and CKD, to lifelong exposure to environmental Cd. In the present review, however, we highlight data from the U.S. general population, recorded in the National Health and Nutrition Examination Survey (NHANES). The U.S. NHANES provides data on levels exposure to more than 200 chemicals, experienced by the representative of U.S. general population [118]. Urinary and blood Cd levels were quantified using standardized methodology that enables the comparison of data across NHANES cycles [118].
Table 2 provides evidence that Cd exposure, even at low levels, may increase the prevalence of pre-diabetes, diabetes, CKD and liver disease in the representative U.S. population [119,120,121,122,123,124,125,126,127,128].
As data in Table 2 indicate, low environmental Cd exposure in the U.S. has been linked to CKD and liver disease in additional to prediabetes and diabetes.
Increases in the risks of prediabetes and diabetes among NHANES 1988-1994 participants were associated with urinary Cd levels of 1–2 µg/g creatinine [119]. An in-creased risk of prediabetes among NHANES 2005-2010 was associated with urinary Cd levels ≥ 0.7 µg/g creatinine after adjustment for covariates [120]. Obesity was associated with prediabetes in both men and women, and there was evidence that obesity may enhance diabetogenic effects of Cd among men [121].
3.1. Dietary exposure: The U.S. experience
TDS data indicate average dietary Cd exposure in the U.S. was 4.63 μg/d [130]. This figure was computed from levels of Cd found in 260 food items in the 2006–2013 market basket survey together with 24-h dietary recall data from 12,523 participants in NHANES 2007–2012, aged 2 years and older. A dietary assessment of U.S. women (n = 1002, mean age 63.4) reported mean dietary Cd exposure of 10.4 μg/day, and mean Cd excretion of 0.62 μg/g creatinine [131].
Cereals and bread, leafy vegetables, potatoes, legumes and nuts, stem/root vegetables, and fruits contributed to 34%, 20%, 11%, 7%, and 6% of total intake, respectively. Foods that contain relatively high Cd levels are spaghetti, bread, potatoes, and potato chips which contributed the most to total Cd intake, followed by lettuce, spinach, tomatoes, and beer. Lettuce was a main Cd source for White people and Black people. Tortillas and rice were the main Cd sources for Hispanic Americans and Asians plus other ethnicities [130].
3.2. Cadmium and its inverse relationship with obesity
Studies from the U.S., and other countries consistently observed inverse relationships of urinary and blood Cd levels with various measurements of obesity. The reason for this phenomenon has never been investigated and largely ignored. However, it at least indicates that the diabetogenicity of Cd is unrelated to obesity, and Cd exposure at least accounts for diabetes among lean subjects.
3.2.1. Children and adolescents
Urinary Cd was associated with a 54% reduction in risk of obesity in children and adolescents enrolled in NHANES 1999-2011; an inverse association between urinary Cd and obesity was stronger in a younger (6-12 years) than the older age group (13-19 years) [132]. Urinary Cd levels inversely associated with height and body mass index (BMI) in Flemish children, aged 14-15 years [133].
3.2.2. Adults
Urinary Cd inversely associated with central obesity among participants of NHANES 1999-2002 [134]. Among NHANES 2003-2010 participants, their blood Cd levels inversely associated with BMI [135]. In another analysis of data from NHANES 2001–2014 participants aged 20-80 years (n = 3,982), urinary Cd levels were not associated with the risk of metabolic syndrome, but they were associated with a decreased risk of abdominal obesity [136]. In a meta-analysis of data from 11 cross-sectional studies, Cd exposure was not associated with an in-creased risk of metabolic syndrome but it was associated with dyslipidemia, especially in Asian population [137].
Similarly, an inverse association between blood Cd and BMI was seen in non-smokers in the Canadian Health Survey 2007-2011 [138]. An inverse relationship between Cd exposure and various measurements of obesity was seen in both men and women in a study of the indigenous population of Northern Québec, Canada, where obesity was highly prevalent [139].
An inverse association between blood Cd and BMI was noted in a group of Korean men, 40–70 years of age [140]. This Korean population study observed an inverse correlation between fasting blood glucose and urinary Cd excretion levels, and a 1.81-fold increase in risk of diabetes among men who had urinary Cd > 2 μg/g creatinine.
In a Thai population study, increases in risk of diabetes in men and women were not associated with obesity/overweight, but were associated with blood Cd and Pb levels above median values of 0.3 µg/L and 2.12 µg/dL for Cd and Pb, respectively [141].
In a Chinese study, urinary Cd excretion ≥ 2.95 µg/g creatinine were associated with reduced risk of weight gain and obesity [142]. In a study of non-occupationally exposed residents of Shanghai, the median urinary Cd excretion was 0.77 μg/g creatinine and higher urinary Cd levels were associated with lower BMI values [143].
3.3. The U.S. population risk analysis of Cd-associated diabetes
The geometric mean, the 50th, 75th, 90th, and 95th percentile values for urinary Cd levels in the representative U.S. general population were 0.210, 0.208, 0.412, 0.678, and 0.949 µg/g creatinine, and the corresponding values for blood Cd were 0.304, 0.300, 0.500, 1.10, and 1.60 µg/L, respectively [144]. Although the U.S. population mean Cd excretion was less than 0.5 µg/g creatinine, 2.5%, 7.1%, and 16% of non-smoking women (aged ≥ 20 years) were found to have Cd excretion levels > 1, > 0.7, and > 0.5 μg/g creatinine, respectively [145]. Given that Cd excretion > 0.5 μg/g creatinine were found in 16% of non-smoking U.S. women [145], the proportion of people, especially women, who were at risk of Cd-associated adverse effects is not negligible
Rish analysis of data from 4,530 adults enrolled in NHANES 1999–2006 showed that mean Cd excretion levels of 0.198 and 0.365 μg/g creatinine were associated with the likelihood that the prevalence of diabetes to be less than 5% and 10%, respectively [114]. These Cd excretion levels were 3.78% and 6.97% of the nephrotoxicity threshold level determined from the β2M endpoint [35].
The Cd excretion of 0.198 and 0.365 μg/g creatinine associated with 5% and 10% prevalence of diabetes were in ranges with the 50th and 75th percentiles of Cd excretion. Thus, the proportion of the U.S. adults at-risk of Cd-associated diabetes was substantial. Similarly, Cd excretion was inversely associated with bone mineral density, and low environmental Cd exposure in the U.S. accounted for 16% of osteoporosis cases, aged 50-79 years [117].
4. Cadmium, the Liver, Kidney and Diabetes Type 2
The kidney is the only organ other than the liver that produces and releases glucose into the circulation [146,147,148,149]. The liver and kidney are directly involved in blood glucose control. The kidney contributes to 20-25% of plasma glucose after an overnight fast, and it releases into the circulation 60% of plasma glucose in the postprandial period [148]. In diabetes type 1, there is an impairment in renal release of glucose [148]
The kidney is also responsible for filtration, and reabsorption of glucose. In normal health, an approximate of 160 to 180 gm of glucose is retrieved each day [146,147,148,149]. The sodium glucose co-transporter 2 (SGLT2) and SGLT1 mediate 90% and 10% of the tubular reabsorption of glucose, respectively [149]. Increased renal expression of these glucose transporters has been implicated in an elevation of renal threshold for glucose excretion in diabetes type 2 patients [147].
Loss of tubular gluconeogenesis and a switch to glycolysis are known pathologic features of CKD [149]. In clinical trials, SGLT2 inhibitors were effective to attenuate the deterioration of kidney function in CKD patients [150].
Cd-associated GFR reductions, albuminuria and hypertension were more severe in those who had diabetes [30,32,34,128,141]. These results are replicated in experimental studies [151,152]. In cross-sectional and prospective cohort studies of 231 diabetic patients in the Netherlands, both Cd and active smoking were associated with a progressive decline in eGFR [27,28]. Collectively, these findings support the premise that exposure to even low levels of environmental Cd promote the development and progression of DKD.
As a summary, environmental sources, entry routes of Cd and its toxicity targets are depicted in Figure 3.
In the presence of multiple Cd entry routes, the proximal tubule accumulates most Cd acquired (Section 2.4). In effect, Cd amount in kidney cortex, as µg/g wet tissue weight, is the highest (Table 1). For instance, respective mean Cd in lung, liver and kidney cortex of Australians, aged 2−70 years (mean 39.9) were 0.12, 0.99, and 20.5 μg/g wet tissue weight, while the mean urinary Cd was 0.62 μg/L, range; 0.05−2.88 μg/L [58].
In a study of kidney transplant donors, a Cd excretion of 0.42 μg/g creatinine corresponded to kidney cortical Cd of 25 μg/g wet kidney weight [153]. In female kidney transplant donors, the mean values for Cd excretion, blood Cd and kidney cortical Cd were 0.34 μg/g creatinine, 0.54 μg/L and 17.1 μg/g kidney wet weight, respectively [154]. The corresponding figures in men were 0.23 μg/g creatinine, 0.46 μg/L and 12.5 μg/g, all of which were lower than in women [154].
The rates of Cd accumulation found in Australian autopsy study were 3–5 µg/g wet tissue weight for each 10-year increase in age, reaching 25.9 µg/g wet tissue weight in 50 years [58]. After adjustment for age and inhalational exposure, the rate of Cd accumulation in kidneys was higher in females than males [58].
Similarly, the rate of Cd accumulation found in non-smoking Swedish kidney transplant donors was 3.9 μg/g kidney wet weight for every 10-year increase in age. Non-smoking women with low body iron stores had an Cd accumulation rate of 4.5 μg/g kidney wet weight in 10 years [155].
5. Cadmium and Diabetes: Experimental Studies
This section provides a summary of findings from experimental studies attempted to shed light on how Cd causes diabetes. However, most experimental studies examined Cd-induced diabetes along with the impacts of high-fat diet in the belief that obesity is a major contributing factor.
Like obesity, ample epidemiologic and experimental data suggest, as a mitochondrial toxicant, Cd can cause oxidative stress, inflammation [156,157,158], disrupt intermediary metabolism, and insulin resistance in many tissues, including insulin-dependent and non-dependent types [159,160,161].
Also, Cd may have an indirect effect on diabetes through induction of hyperuricemia. An association between prevalence of hyperuricemia and Cd exposure has been noted in Chinese [162], the U.S. [163], and Korean [164,165] population studies. Pancreatic β-cell death and a reduction in glucose-stimulated insulin secretion have been demonstrated in mice with hyperuricemia due to uricase deficiency [166,167].
5.1. Cadmium and pancreatic β cells
In cell culture, pancreatic β cells progressively accumulated Cd from medium containing Cd in nanomolar concentrations similar to human plasma Cd levels. An effect of Cd on insulin secretion occurred at the onset of cell death [168].
In another study using the human β cell line (the INS-1), Cd concentration ten-fold below the level causing cell death produced no effects on mitochondrial function, assessed with the energy charge and ATP synthesis [169]. This Cd concentration, however, induced mitochondrial morphological change toward circularity, indicative of fission. The increased circularity suggested mitochondrial adaptive response to low-level Cd.
Thus, a sublethal Cd dose caused mitochondria to undergo morphological adaptative change as the mechanism to offset an effect of Cd on energy output and insulin secretion [169]. If cellular Cd influx continues, impairment of this organelle may contribute to cellular dysfunction and decreased viability of β-cells.
Through mathematical modeling of oral glucose tolerance test data, an effect of Cd on the sensitivity of pancreatic β cells to glucose has also been demonstrated. Perinatal exposure to low-level Cd in mother’s milk reduced pancreatic β-cell sensitivity to glucose stimulation [170].
In rats, fasting plasma glucose was increased 12 weeks after Cd treatment, at which time pancreatic islets from the Cd-treated group showed less glucose-stimulated insulin release than islets from saline-treated control animals [171]. At this stage, Cd accumulation in isolated islets was 5 times higher than in pancreatic parenchyma but 30% lower than in renal cortex [171]. These relative pancreatic and renal Cd accumulation levels paralleled human data (Table 1).
5.2. Cadmium and “metal stressed” fat cells
In a Swiss autopsy study, Cd accumulation in omentum visceral and abdominal subcutaneous fat tissues [were quantified [172]. Cd in the same range found in those postmortem fat tissue samples disrupted cellular zinc homeostasis and caused an increase in expression of various pro-inflammatory cytokines in the adipose-derived human mesenchymal stem cells [172].
In a Spanish cohort study, Cd levels in fat tissues were higher in those with lower BMI values [173]. This observation is an additional to insulin resistance and higher plasma insulin levels in smokers with adipose tissue Cd levels in the middle tertile, compared to those with adipose tissue Cd levels in the bottom tertile [174].
In Cd-treated mice, abnormal differentiation of the adipocyte was evident from its small size, and a reduced secretion of adiponectin [175,176]. In Cd-treated rats, subcutaneous fat tissue accumulated more Cd than did abdominal and retroperitoneal adipose tissues, and all three fat tissue types had reduced adiponectin and leptin transcript levels [177].
6. Cadmium, Metal Stress Response, and Deranged Glucose Metabolism
This section focuses on the universal cellular stress response involving the induction of heme oxygenase-1 (HO-1) and Cd-induced hyperglycemia. Figure 4 provides metabolic pathways for glucose, heme and glutathione, which are central to the understanding of these topics.
Glucose can be metabolized via glycolysis to pyruvate. Pyruvate can be metabolized via three possible routes; pyruvate dehydrogenase (PDH) and oxidative phosphorylation, via pyruvate carboxylase (PC) to replenish the intermediates of tricarboxylic acid (TCA) cycle for heme biosynthesis, and via lactate dehydrogenase (LDH) to lactate.
Glucose can be generated from precursor substrates such as lactate and alanine via gluconeogenesis. Surplus glucose is stored as glycogen via glycogenesis. Glucose in glycogen can be retrieved via glycogenolysis.
Glucose can be metabolized via the pentose phosphate pathway to generate NADPH (H+) required for the degradation of heme, catalyzed by HO-1 and HO-2. NADPH (H+) is also required in the regeneration of GSH from GSSG, a product of ROS action.
The enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 4 (PFKFB4) regulates glycolysis through controlling levels of F-2,6-P2. This metabolite intermediate of glucose, F-2,6-P2 acts as an activator (+) of glycolysis and an inhibitor (−) of gluconeogenesis.
6.1. Cellular response to stress
Activation and repression of the HO-1 gene are universal cellular stress responses that are required for cell survival under the influence of environmental stressors of various forms. Every nucleated cell in the body must synthesize heme, and the de novo heme biosynthesis supplies heme that can rapidly be catabolized to release Fe, carbon monoxide, and a precursor substrate (biliverdin IXα) to produce bilirubin [178,179,180,181,182].
However, unlike endogenous (physiologic) HO-1 activators, the HO-1 induction by Cd does not result in the formation of bilirubin [178]. The rapid and massive HO-1 expression in response to Cd, leads to a transient increase in the intracellular heme concentration. This results in the stimulation of gluconeogenesis, and a shift to dominant glycolysis, a known pathologic feature of CKD [149].
The finding that Cd induces HO-1 expression without a concomitant bilirubin formation is of significance in Cd research. It explains the pervasiveness of Cd toxicity as it increases cellular oxidative stress and lowers cellular antioxidant capacity at the same time. This knowledge comes from a methodology breakthrough in measuring bilirubin in cells, where it is produced [178]
6.2. How does Cd activate HO-1 expression?
Cd induces expression of HO-1 by mechanisms different from those used by physiologic HO-1 activators that include prostaglandin D2 (PGD2) [183,184,185].
PGD2 is a major cyclooxygenase mediator, synthesized by activated mast cells and other immune cells, and is implicated in allergic disorders [185]. In a study of a cell culture model of human retinal epithelial cells, PGD2 was found to activate the HO-1-gene promoter through D-prostanoid 2 (DP2) receptor in an enhancer manner [183].
In comparison, Cd activates the HO-1 promoter via the Cd response element (CdRE), and Maf recognition antioxidant response element (MARE), also known as stress response element (StRE) [186]. Cd also suppresses lysosomal degradation of Nrf2 [187 153] and causes nuclear export of the HO-1 gene repressor Bach1, which allows transactivation of the HO-1 gene by the Nrf2/small Maf complex [188].
6.3. Cadmium-induced hyperglycemia
The ability of Cd to induce hyperglycemia was first demonstrated in neonatal rats [189] and then in adult rats [190,191]. The liver of Cd-exposed neonatal rats had a reduced glycogen, and an enhanced gluconeogenesis, evident from activity of enzymes in gluconeogenesis; pyruvate carboxylase, phosphoenolpyruvate carboxy kinase, fructose-1,6-biphosphatase, and glucose-6-phosphatase [189]. Hyperglycemia in Cd-exposed rats developed long before the onset of kidney toxicity [192].
In rats exposed to Cd via intraperitoneal injection for 45 days, depletion of hepatic and renal glycogen was noted along with enhanced activity of the rate-limiting enzymes in gluconeogenesis [193]. These changes remained 4 weeks after exposure cessation.
In another study, the effects of Cd on hepatic glucose metabolism remained one year after Cd treatment was discontinued [194]. The persistent Cd effects can be expected because most Cd is retained by cells, which provide ample of opportunity for Cd to exert toxicity. A study in rats showed Cd was excreted only when cell die [195].
Female preponderance effect of Cd has been investigated in rats exposed to Cd in drinking water for 3 months. The increment of plasma insulin levels in response to fasting and glucose stimulation was found in female rats only [196]. The increased plasma insulin levels were attributable to impaired hepatic extraction of insulin.
Cd exposure in utero has been investigated in rats, where dams were exposed to Cd for a period of 21 days before mating, 21 days during gestation, and 21 days during lactation [197]. The effects of Cd exposure on the metabolism of glucose and lipids were examined at 21, 26, and 60 days of age. Collective data indicated changes in glucose homeostasis in pubs born from Cd-exposed dams that may increase susceptibility to development of diabetes [197]. Effects of early life exposure to Cd on the development of diabetes later in life have been reviewed by Saedi et al. [198].
6.4. The molecular basis for deranged cellular glucose metabolism after Cd exposure
Therapeutic effects of metformin, an anti-diabetic drug, were evaluated, using male Wistar rats given Cd in drinking water (32.5 ppm) alone or Cd plus metformin (200 mg/kg/day) [199]. Cd caused hyperinsulinemia, insulin resistance, adipocyte dysfunction, loss of hepatic insulin sensitivity. Increased lipid accumulation was also seen in various tissues, while glycogen in liver, heart, and renal cortex were diminished, but was increased in the muscle. Metformin showed a limited therapeutic efficiency on Cd-induced glucose tolerance and lipid accumulation.
Current evidence suggests that metformin has the ability to stimulate synthesis of natural cyclic AMP antagonist prostaglandylinositol cyclic phosphate (cyclic PIP) [26]. The increment of cyclic PIP appeared to account for most of the beneficial effects of metformin; the reduction of blood glucose levels, the inhibition of cAMP synthesis, gluconeogenesis and an increase in sensitivity to insulin [26]. It has been postulated that insulin resistance is due to an imbalance action of cyclic PIP and cAMP [25].
6.5. The physiologic heme catabolism
In concert with NADPH-cytochrome P450 reductase, HO-1 and HO-2 catalyze heme degradation with the resultant release of Fe, CO, and biliverdin IXα [200,201,202,203]. Biliverdin IXα is converted to bilirubin almost instantly by biliverdin reductase. The bulk of Fe release by HO-1 and HO-2 is reutilized in the synthesis of hemoglobin and other hemoproteins, including nitric oxide synthase, various enzymes of the mitochondrial respiratory chain and the cytochrome P450 super family [182,204]. This makes heme degradation by HO-1 and HO-2 indispensable. Two other products of heme degradation, namely CO and bilirubin, are known for their anti-inflammatory and antioxidant properties [178,179,180,181,182].
Because CO is produced exclusively by HO-1 and HO-2, an exhaled CO can serve as a biomarker for heme degradation. In healthy individuals, levels of exhaled CO increase with increasing blood glucose and both exhaled CO and blood glucose levels return to their respective baseline values 40 minutes after glucose administration. These data suggest that levels of HO activity may influence blood glucose levels [205]. The relationship between exhaled CO and blood glucose has been observed as well in diabetics. As expected, the levels of exhaled CO are greater in diabetic subjects, compared to non-diabetic controls [205]. The elevated CO exhaled in diabetic subjects is attributable to HO-1 induction in response to rising oxidative (high-glucose) stress, reflected by elevated levels of damage proteins such as glycosylated haemoglobin. The exhaled CO-blood glucose correlation implies that exhaled CO can be used in monitoring disease progression in diabetes patients [205].
In the Goto-Kakizaki rats, a model for hyperglycemia and insulin resistance without obesity [206], induction of HO-1 causes a reduction in fasting blood glucose levels and prevents a rise in blood glucose in post absorptive state [207]. In an obese mouse model of diabetes, induction of HO-1 prevents weight gain, decreases visceral and subcutaneous fat content, improves both insulin sensitivity and glucose tolerance [208].
6.6. Heme oxygenase enzymes
HO-1 and HO-2 are products of two different genes [194]. The promoter of the human HO-1 gene is unique because it contains the GT repeats, not found in rodent or murine species [200,201,202]. The genetic polymorphisms, such as long GT repeats, are associated with an elevated risk for various diseases, type 2 diabetes included [209,210].
Expression of the HO-1 gene is regulated by a cascade of transcription factors such as CLOCK, Bmal and Per, that generate day-night cyclical expression of the genes involved in energy metabolism [211,212,213,214]. Disruption of the diurnal cycle caused obesity in mice [215].
Expression of the HO-1 gene is regulated also by heme (its own substrate), the levels of glucose, oxygen, and shear stress [200,201,202,216]. HO-1 is a component of innate immune responses [217 206].
The catalytic domains of HO-1 and HO-2 are highly homologous, sharing 93% of their amino acid sequences. HO-2, however, contains an additional domain, which has Cys-Pro dipeptide motifs that allows binding of heme and interacting with other proteins that include Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways [218,219,220] and PFKFB4, the key regulator of glycolysis. This HO-2 domain accounts for the its biologic roles that are distinct from those of HO-1, such as protection against ischemic acute kidney injury [221] and anti-diabetic properties, detailed below.
HO-2 deficiency causes neither lethality nor infertility. The HO-2 knockout mice reproduce offsprings that undergo normal development to adulthood, but develops the symptomatic spectrum of human type-2 diabetes; hyperglycemia, increased fat deposition, insulin resistance and hypertension with aging [222,223,224]. Normal fertility and normal development in HO-2 knockout mice suggests that HO-1 could compensate for heme degradation of HO-However, HO-1 could not compensate for anti-diabetogenic function of HO-2, thereby suggesting such function is unique to HO-2.
6.7. Maintenance of blood glucose by HO-1, HO-2 and PFKFB4
Data from protein microarray connected HO-2 with glycolysis through its interaction with 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) [224]. In liver, PFKFB4 is the key regulator of glycolysis [225] and HO-2 deficiency causes persistent hyperglycemia due to an impaired ability to suppress glucose production.
PFKFB4 expression is regulated by the hepatocyte nuclear factor-6 (Hnf-6) [226] and Hnf6-knockout caused diabetes in mice [227]. PFKFB4 protein phosphorylation, mediated by the cAMP dependent protein kinase A (PKA) decreased fructose 2,6 bisphosphate (F-2,6-P2) level in the liver, thereby increasing gluconeogenesis with concomitantly reducing glycolysis [227]. Figure 5 depicts, the homeostasis of blood glucose requires coordinated expression of HO-1, HO-2 and PFKFB4.
Both HO-1 and HO-2 are required to prevent a fall or a rise in blood glucose levels during fasting and post absorptive periods, respectively. In fasting state, HO-1 up-regulation concurrent with PFKFB4 down-regulation results in enhanced glucose production with minimal use of glucose. In the post-absorptive state, HO-1 down regulation concurrent with HO-2 plus PFKFB4 up-regulation results in suppressed glucose production and increased use of glucose.
HO-2 is required for the up-regulation of PFKFB4. Failure in any of these (HO-1, HO-2 and PFKFB4) can result in hyperglycemia due to over production of glucose in fasting state in combination with impaired ability to suppress its production.
Hepatic HO-1 protein expression in HO-2 deficient mice were 35%-40% lower than the wild-type mouse [223,228]. A marked reduction in HO-1 expression could render the hepatocyte to oxidative damage. However, the repression of the HO-1 gene expression is a necessary metabolic adaptation to safeguard the cellular redox state. This could be achieved by utilizing NADPH (H+) for regenerating GSH from GSSG rather than for heme catabolism (Figure 4). GSH recycling is a mechanism for maintenance of cell redox state. It is central to cell function integrity.
7. Conclusion
An inverse relationship between various measurements of obesity and Cd exposure means that the diabetogenic action of Cd is independently of adiposity. Like obesity, Cd induces oxidative stress, chronic systemic inflammation, and insulin resistance in many tissues, including insulin-dependent and non-dependent types. Of principal concern, research data show that obesity may enhance the diabetogenic effects of Cd, and that the risk of diabetes was higher in obese persons, compared to the non-obese with the same overall Cd burden.
Cd-induced HO-1 expression does not yield bilirubin formation because Cd activation of the HO-1 gene is through the mechanisms different from those of physiologic HO-1 gene activators. The instantaneously massive increase in HO-1 expression induced through such mechanism raises intracellular concentration of heme, a stimulator of gluconeogenesis, and persistent hyperglycemia ensues as cellular Cd influx continues. Metformin is ineffective to prevent the expression of diabetic symptoms induced by Cd.
Cd increases the risk of both CKD and diabetes type 2, and promotes the progression of DKD, the perfect storm to health care system. Because CKD and diabetes are highly prevalent worldwide, even small increases in risks of these diseases yield large number of cases that are preventable by early minimization of exposure.
Current dietary exposure guidelines at 0.83 µg/kg body weight/day (58 µg/day for a 70-kg person), and a nephrotoxicity threshold of Cd at 5.24 µg/g creatinine are not protective of human health. Adverse effects observed at Cd excretion below 5.24 µg/g creatinine such as eGFR decline is concerning. The Cd-induced nephron loss, evident from a fall of eGFR to below 60 mL/min/1.73m2 is irreversible.
Minimization of Cd contamination of the food chains, and its levels in food crop are essentially preventive public measures. Sufficient intake of Zn and maintenance of an adequate body Fe stores are additional logical interventions. Induced HO-1 expression with a concomitant increase in bilirubin production may be a complementary measure to mitigate harmful effects of inevitable exposure to Cd. Further research dissecting the molecular basis for a renoprotection of HO-2 and its anti-obese and anti-diabetogenic properties are imperative.
Author Contributions
Conceptualization, S.S.; writing—original draft preparation, S.S.; writing—review and editing, S.S. The author has agreed to the published version of the manuscript
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported with resources from the Centre for Kidney Disease Research, Translational Research Institute, and the Department of Kidney and Transplant Services, Princess Alexandra Hospital.
Conflicts of Interest
The author declares no conflict of interest.
References
- Järup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.G. Natural sources of metals to the environment. Hum. Ecol. Risk Assess. 2010, 6, 945–963. [Google Scholar] [CrossRef]
- ATSDR (Agency for Toxic Substances and Disease Registry). Toxicological Profile for Cadmium; Department of Health and Humans Services, Public Health Service, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2012. [Google Scholar]
- Aoshima, K. Epidemiology of renal tubular dysfunction in the inhabitants of a cadmium-polluted area in the Jinzu River basin in Toyama Prefecture. Tohoku J. Exp. Med. 1987, 152, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Aoshima, K.; Oguma, E.; Sasaki, S.; Miyamoto, K.; Hosoi, Y.; Katoh, T.; Kayama, F. Latest status of cadmium accumulation and its effects on kidneys, bone, and erythropoiesis in inhabitants of the formerly cadmium-polluted Jinzu River Basin in Toyama, Japan, after restoration of rice paddies. Int. Arch. Occup. Environ. Health 2010, 83, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Oguma, E.; Sasaki, S.; Miyamoto, K.; Hosoi, Y.; Ono, A.; Kayama, F. Exposure assessment of cadmium in female farmers in cadmium-polluted areas in Northern Japan. Toxics 2020, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Zarcinas, B.A.; Pongsakul, P.; McLaughlin, M.J.; Cozens, G. Heavy metals in soils and crops in Southeast Asia. 2. Thailand. Environ. Geochem. Health 2004, 26, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Verbeeck, M.; Salaets, P.; Smolders, E. Trace element concentrations in mineral phosphate fertilizers used in Europe: A balanced survey. Sci. Total Environ. 2020, 712, 136419. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Zhou, S.; Zhou, Y.; Jia, Z.; Guo, T.; Wang, J. Cadmium pollution of soil-rice ecosystems in rice cultivation dominated regions in China: A review. Environ. Pollut. 2021, 280, 116965. [Google Scholar] [CrossRef] [PubMed]
- McDowell, R.W.; Gray, C.W. Do soil cadmium concentrations decline after phosphate fertiliser application is stopped: A comparison of long-term pasture trials in New Zealand? Sci. Total Environ. 2022, 804, 150047. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Duan, Z.; Zhang, L.; Sun, D.; Li, X. The status and research progress of cadmium pollution in rice- (Oryza sativa L.) and wheat- (Triticum aestivum L.) cropping systems in China: A critical review. Toxics 2022, 10, 794. [Google Scholar] [CrossRef]
- Satarug, S.; C. Gobe, G.; A. Vesey, D.; Phelps, K.R. Cadmium and lead exposure, nephrotoxicity, and mortality. Toxics 2020, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Gobe, G.C.; Vesey, D.A. Multiple targets of toxicity in environmental exposure to low-dose cadmium. Toxics 2022, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Phelps, K.R. Estimation of health risks associated with dietary cadmium exposure. Arch. Toxicol. 2023, 97, 329–358. [Google Scholar] [CrossRef] [PubMed]
- Nishito, Y.; Kambe, T. (2018) Absorption mechanisms of iron, copper, and zinc: an overview. J. Nutr. Sci. Vitaminol. (Tokyo) 2018, 64, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Frazer, D.M.; Anderson, G.J. Iron homeostasis: transport, metabolism, and regulation. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Knez, M.; Graham, R.D.; Welch, R.M.; Stangoulis, J.C. New perspectives on the regulation of iron absorption via cellular zinc concentrations in humans. Crit. Rev. Food Sci. Nutr. 2017, 57, 2128–2143. [Google Scholar] [CrossRef] [PubMed]
- Kondaiah, P.; Yaduvanshi, P.S.; Sharp, P.A.; Pullakhandam, R. ; Iron and zinc homeostasis and interactions: Does enteric zinc excretion cross-talk with intestinal iron absorption? Nutrients 2019, 11, 1885. [Google Scholar] [CrossRef] [PubMed]
- Kondaiah, P.; Palika, R.; Mashurabad, P.; Singh Yaduvanshi, P.; Sharp, P.; Pullakhandam, R. Effect of zinc depletion/repletion on intestinal iron absorption and iron status in rats. J. Nutr. Biochem. 2021, 97, 108800. [Google Scholar] [CrossRef] [PubMed]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 2010, 198, 49–55. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-induced cytotoxicity: Effects on mitochondrial electron transport chain. Front. Cell Dev. Biol. 2020, 8, 604377. [Google Scholar] [CrossRef]
- Lee, W.K.; Thévenod, F. Cell organelles as targets of mammalian cadmium toxicity. Arch. Toxicol. 2020, 94, 1017–1049. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K.; Garrick, M.D. Iron and cadmium entry into renal mitochondria: Physiological and toxicological implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.N.; Colone, M.; Gambioli, R.; Stringaro, A.; Unfer, V. Oxidative stress and male fertility: Role of antioxidants and inositols. Antioxidants 2021, 10, 1283. [Google Scholar] [CrossRef] [PubMed]
- Wasner, H.K. Metformin’s mechanism of action is stimulation of the biosynthesis of the natural cyclic AMP antagonist prostaglandylinositol cyclic phosphate (cyclic PIP). Int. J. Mol. Sci. 2022, 23, 2200. [Google Scholar] [CrossRef] [PubMed]
- Wasner, H.K. Insulin resistance develops due to an imbalance in the synthesis of cyclic AMP and the natural cyclic AMP antagonist prostaglandylinositol cyclic phosphate (cyclic PIP). Stresses 2023, 3, 762–767. [Google Scholar] [CrossRef]
- Hagedoorn, I.J.M.; Gant, C.M.; Huizen, S.V.; Maatman, R.G.H.J.; Navis, G.; Bakker, S.J.L.; Laverman, G.D. Lifestyle-related exposure to cadmium and lead is associated with diabetic kidney disease. J. Clin. Med. 2020, 9, 2432. [Google Scholar] [CrossRef]
- Oosterwijk, M.M.; Hagedoorn, I.J.M.; Maatman, R.G.H.J.; Bakker, S.J.L.; Navis, G.; Laverman, G.D. Cadmium, active smoking and renal function deterioration in patients with type 2 diabetes. Nephrol. Dial. Transplant 2023, 38, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Haswell-Elkins, M.; Satarug, S.; O’Rourke, P.; Moore, M.; Ng, J.; McGrath, V.; Walmby, M. Striking association between urinary cadmium level and albuminuria among Torres Strait Islander people with diabetes. Environ. Res. 2008, 106, 379–383. [Google Scholar] [CrossRef]
- Barregard, L.; Bergström, G.; Fagerberg, B. Cadmium, type 2 diabetes, and kidney damage in a cohort of middle-aged women. Environ. Res. 2014, 135, 311–316. [Google Scholar] [CrossRef]
- Satarug, S.; Yimthiang, S.; Pouyfung, P.; Khamphaya, T.; Vesey, D.A. Cadmium-induced tubular dysfunction in type 2 diabetes: A population-based cross-sectional study. Toxics 2023, 11, 390. [Google Scholar] [CrossRef]
- Yimthiang, S.; Vesey, D.A.; Pouyfung, P.; Khamphaya, T.; Gobe, G.C.; Satarug, S. Chronic kidney disease induced by cadmium and diabetes: A quantitative case-control study. Int. J. Mol. Sci. 2023, 24, 9050. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Đorđević, A.B.; Yimthiang, S.; Vesey, D.A.; Gobe, G.C. The NOAEL equivalent of environmental cadmium exposure associated with GFR reduction and chronic kidney disease. Toxics 2022, 10, 614. [Google Scholar] [CrossRef] [PubMed]
- Yimthiang, S.; Pouyfung, P.; Khamphaya, T.; Vesey, D.A.; Gobe, G.C.; Satarug, S. Evidence linking cadmium exposure and β2-microglobulin to increased risk of hypertension in diabetes type 2. Toxics 2023, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- JECFA. Evaluation of certain Food Additives and Contaminants. In Proceedings of the Seventy-Third Meeting of the Joint FAO/WHO Expert Committee on Food Additives, Geneva, Switzerland, 8–17 June 2010; Food and Agriculture Organization of the United Nations: Rome, Italy; World Health Organization: Geneva, Switzerland, 2010; Available online: https://apps.who.int/iris/handle/10665/44521 (accessed on 9 November 2023).
- Wong, C.; Roberts, S.M.; Saab, I.N. Review of regulatory reference values and background levels for heavy metals in the human diet. Regul. Toxicol. Pharmacol. 2022, 130, 105122. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.K.; Bolger, P.M.; Carrington, C.D. Update of US FDA’s total diet study food list and diets. J. Expo. Sci. Environ. Epidemiol. 2007, 17, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Callan, A.; Hinwood, A.; Devine, A. Metals in commonly eaten groceries in Western Australia: A market basket survey and dietary assessment. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2014, 31, 1968–1981. [Google Scholar] [CrossRef]
- Sand, S.; Becker, W. Assessment of dietary cadmium exposure in Sweden and population health concern including scenario analysis. Food Chem. Toxicol. 2012, 50, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.A.; Lindtner, O.; Blume, K.; Heinemeyer, G.; Schneider, K. Cadmium exposure from food: the German LExUKon project. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2014, 31, 1038–1051. [Google Scholar] [CrossRef]
- Watanabe, T.; Kataoka, Y.; Hayashi, K.; Matsuda, R.; Uneyama, C. Dietary exposure of the Japanese general population to elements: total diet study 2013–2018. Food Saf (Tokyo) 2022, 10, 83–101. [Google Scholar] [CrossRef]
- Wei, J.; Gao, J.; Cen, K. Levels of eight heavy metals and health risk assessment considering food consumption by China’s residents based on the 5th China total diet study. Sci. Total Environ. 2019, 689, 1141–1148. [Google Scholar] [CrossRef]
- Xiao, G.; Liu, Y.; Dong, K.F.; Lu, J. Regional characteristics of cadmium intake in adult residents from the 4th and 5th Chinese total diet study. Environ. Sci. Pollut. Res. Int. 2020, 27, 3850–3857. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Wang, H.; Cui, L.; Zhang, Z.; Guo, J.; Liu, S.; Cui, W. Contamination and health risk assessment of lead, arsenic, cadmium, and aluminum from a total diet study of Jilin Province, China. Food Sci. Nutr. 2020, 8, 5631–5640. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.A.; Kwon, H.J.; Ha, M.; Kim, H.; Oh, S.Y.; Kim, J.S.; Lee, S.A.; Park, J.D.; Hong, Y.S.; Sohn, S.J.; et al. Korean research project on the integrated exposure assessment of hazardous substances for food safety. Environ. Health Toxicol. 2015, 30, e2015004. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.; Woo, H.D.; Kim, D.W.; Choi, I.J.; Kim, Y.I.; Kim, J. Association between dietary cadmium intake and early gastric cancer risk in a Korean population: A case-control study. Eur. J. Nutr. 2019, 58, 3255–3266. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.N.; Liu, Z.; Wang, B.; Miller, M.L.; Afton, S.E.; Soleimani, M.; Nebert, D.W. Oral cadmium in mice carrying 5 versus 2 copies of the Slc39a8 gene: Comparison of uptake, distribution, metal content, and toxicity. Int. J. Toxicol. 2014, 33, 14–20. [Google Scholar] [CrossRef]
- Fujishiro, H.; Himeno, S. New insights into the roles of ZIP8, a cadmium and manganese transporter, and its relation to human diseases. Biol. Pharm. Bull. 2019, 42, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Fels, J.; Lee, W.K.; Zarbock, R. Channels, transporters and receptors for cadmium and cadmium complexes in eukaryotic cells: myths and facts. Biometals 2019, 32, 469–489. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Danko, T.; Bergeron, M.J.; Balazs, B.; Suzuki, Y.; Zsembery, A.; Hediger, M.A. Heavy metal cations permeate the TRPV6 epithelial cation channel. Cell Calcium 2011, 49, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Montalbetti, N.; Franz, M.C.; Graeter, S.; Simonin, A.; Hediger, M.A. Human TRPV5 and TRPV6: key players in cadmium and zinc toxicity. Cell Calcium 2013, 54, 276–286. [Google Scholar] [CrossRef]
- Jorge-Nebert, L.F.; Gálvez-Peralta, M.; Landero Figueroa, J.; Somarathna, M.; Hojyo, S.; Fukada, T.; Nebert, D.W. Comparing gene expression during cadmium uptake and distribution: untreated versus oral Cd-treated wild-type and ZIP14 knockout mice. Toxicol. Sci. 2015, 143, 26–35. [Google Scholar] [CrossRef]
- Fujishiro, H.; Hamao, S.; Tanaka, R.; Kambe, T.; Himeno, S. Concentration-dependent roles of DMT1 and ZIP14 in cadmium absorption in Caco-2 cells. J. Toxicol. Sci. 2017, 42, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; el Belbasi, H.I.; Min, K.S.; Onosaka, S.; Okada, Y.; Matsumoto, Y.; Mutoh, N.; Tanaka, K. Fate of cadmium bound to phytochelatin in rats. Res. Commun. Chem. Pathol. Pharmacol. 1993, 82, 357–365. [Google Scholar] [PubMed]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Thévenod, F. Expression and function of the lipocalin-2 (24p3/NGAL) receptor in rodent and human intestinal epithelia. PLoS One 2013, 8, e71586. [Google Scholar] [CrossRef] [PubMed]
- Langelueddecke, C.; Lee, W.K.; Thévenod, F. Differential transcytosis and toxicity of the hNGAL receptor ligands cadmium-metallothionein and cadmium-phytochelatin in colon-like Caco-2 cells: implications for in vivo cadmium toxicity. Toxicol. Lett. 2014, 226, 228–235. [Google Scholar] [CrossRef]
- DelRaso, N.J.; Foy, B.D.; Gearhart, J.M. , Frazier, J.M. Cadmium uptake kinetics in rat hepatocytes: correction for albumin binding. Toxicol. Sci. 2003, 72, 19–30. [Google Scholar] [CrossRef]
- Satarug, S.; Baker, J.R.; Reilly, P.E.; Moore, M.R.; Williams, D.J. Cadmium levels in the lung, liver, kidney cortex, and urine samples from Australians without occupational exposure to metals. Arch. Environ. Health 2002, 57, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Uetani, M.; Kobayashi, E.; Suwazono, Y.; Honda, R.; Nishijo, M.; Nakagawa, H.; Kido, T.; Nogawa, K. Tissue cadmium (Cd) concentrations of people living in a Cd polluted area, Japan. Biometals 2006, 19, 521–525. [Google Scholar] [CrossRef]
- Barregard, L.; Fabricius-Lagging, E.; Lundh, T.; Mölne, J.; Wallin, M.; Olausson, M.; Modigh, C.; Sallsten, G. Cadmium, mercury, and lead in kidney cortex of living kidney donors: Impact of different exposure sources. Environ. Res. 2010, 110, 47–54. [Google Scholar] [CrossRef]
- Barregard, L.; Sallsten, G.; Lundh, T.; Mölne, J. Low-level exposure to lead, cadmium and mercury, and histopathological findings in kidney biopsies. Environ. Res. 2022, 211, 113119. [Google Scholar] [CrossRef]
- Ajjimaporn, A.; Botsford, T.; Garrett, S.H.; Sens, M.A.; Zhou, X.D.; Dunlevy, J.R.; Sens, D.A.; Somji, S. ZIP8 expression in human proximal tubule cells, human urothelial cells transformed by Cd+2 and As+3 and in specimens of normal human urothelium and urothelial cancer. Cancer Cell Int. 2012, 12, 16. [Google Scholar] [CrossRef]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Wolff, N.A.; Lee, W.K.; Thévenod, F. Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J. Biol. Chem. 2012, 287, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K.; Garrick, M.D. Iron and cadmium entry into renal mitochondria: physiological and toxicological implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Bellido, I.; Gómez-Peña, C.; Pérez-Carrascosa, F.M.; Vrhovnik, P.; Mustieles, V.; Echeverría, R.; Fiket, Ž.; Pérez-Díaz, C.; Barrios-Rodríguez, R.; Jiménez-Moleón, J.J.; et al. Adipose tissue cadmium concentrations as a potential risk factor for insulin resistance and future type 2 diabetes mellitus in GraMo adult cohort. Sci. Total. Environ. 2021, 780, 146359. [Google Scholar] [CrossRef] [PubMed]
- El Muayed, M.; Raja, M.R.; Zhang, X.; MacRenaris, K.W.; Bhatt, S.; Chen, X.; Urbanek, M.; O’Halloran, T.V.; Lowe, W.L., Jr. Accumulation of cadmium in insulin-producing β cells. Islets 2012, 4, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.; Zsolnai, B.; Paksy, K.; Náray, M.; Ungváry, G. Age dependent accumulation of cadmium in the human ovary. Reprod. Toxicol. 1993, 7, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Oldereid, N.B.; Thomassen, Y.; Attramadal, A.; Olaisen, B.; Purvis, K. Concentrations of lead, cadmium and zinc in the tissues of reproductive organs of men. J. Reprod. Fertil. 1993, 99, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.; Garay, R.; Alda. , J.O. Cadmium uptake through the anion exchanger in human red blood cells. J. Physiol. 1991, 443, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Satchwell, T.J.; Toye, A.M. Anion exchanger 1 in red blood cells and kidney: Band 3’s in a pod. Biochem. Cell Biol. 2011, 89, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.D.; Boron, W.F. The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol. Rev. 2013, 93, 803–959. [Google Scholar] [CrossRef]
- Savigni, D.L.; Morgan, E.H. Transport mechanisms for iron and other transition metals in rat and rabbit erythroid cells. J. Physiol. 1998, 508, 837–850. [Google Scholar] [CrossRef]
- Simons, T.J. The role of anion transport in the passive movement of lead across the human red cell membrane. J. Physiol. 1986, 378, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Simons, T.J. Lead transport and binding by human erythrocytes in vitro. Pflugers Arch. 1993, 423, 307–313. [Google Scholar] [CrossRef]
- Demchenkov, E.L.; Nagdalian, A.A.; Budkevich, R.O.; Oboturova, N.P.; Okolelova, A.I. Usage of atomic force microscopy for detection of the damaging effect of CdCl2 on red blood cells membrane. Ecotoxicol. Environ. Saf. 2021, 208, 111683. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Oguma, E.; Kayama, F. Cadmium induces anemia through interdependent progress of hemolysis, body iron accumulation, and insufficient erythropoietin production in rats. Toxicol. Sci. 2011, 122, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Abed, M.; Lang, E.; Föller, M. Oxidative stress and suicidal erythrocyte death. Antioxid. Redox Signal. 2014, 21, 138–153. [Google Scholar] [CrossRef]
- Lang, E.; Lang, F. Mechanisms and pathophysiological significance of eryptosis, the suicidal erythrocyte death. Semin. Cell Dev. Biol. 2015, 39, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A. , Friberg, L., 1957. The distribution of cadmium in blood after repeated exposure. Scand. J. Clin. Lab. Invest. 9, 67-70. [CrossRef]
- Scott, B.J.; Bradwell, A.R. Identification of the serum binding proteins for iron, zinc, cadmium, nickel, and calcium. Clin. Chem. 1983, 29, 629–633. [Google Scholar] [CrossRef]
- Horn, N.M.; Thomas, A.L. Interactions between the histidine stimulation of cadmium and zinc influx into human erythrocytes. J. Physiol. 1996, 496, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.A.; Sarpong-Kumankomah, S.; Nehzati, S.; George, G.N.; Gailer, J. Remarkable differences in the biochemical fate of Cd2+, Hg2+, CH3Hg+ and thimerosal in red blood cell lysate. Metallomics 2017, 9, 1060. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.T.; Keir, J.L.; Boshart, S.J.; Lobanov, V.P.; Ruhland, A.M.; Bahl, N.; Gailer, J. Mobilization of Cd from human serum albumin by small molecular weight thiols. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 958, 16–21. [Google Scholar] [CrossRef]
- Sagmeister, P.; Gibson, M.A.; McDade, K.H.; Gailer, J. Physiologically relevant plasma d,l-homocysteine concentrations mobilize Cd from human serum albumin. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1027, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Turell, L.; Radi, R.; Alvarez, B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013, 65, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Järup, L.; Rogenfelt, A.; Elinder, C.G.; Nogawa, K.; Kjellström, T. Biological half-time of cadmium in the blood of workers after cessation of exposure. Scand. J. Work Environ. Health 1983, 9, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.V.; Newcomb, P.A. Cadmium blood and urine concentrations as measures of exposure: NHANES 1999-2010. J. Expo. Sci. Environ. Epidemiol. 2014, 24, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, B.A.; Sandoval, R.M.; Yadav, S.P.S.; Wagner, M.C. Albumin uptake and processing by the proximal tubule: Physiological, pathological, and therapeutic implications. Physiol. Rev. 2022, 102, 1625–1667. [Google Scholar] [CrossRef] [PubMed]
- Eshbach, M.L.; Weisz, O.A. Receptor-mediated endocytosis in the proximal tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Herbrechter, R.; Schlabs, C.; Pethe, A.; Lee, W.K.; Wolff, N.A.; Roussa, E. Role of the SLC22A17/lipocalin-2 receptor in renal endocytosis of proteins/metalloproteins: a focus on iron- and cadmium-binding proteins. Am. J. Physiol. Renal Physiol. 2023, 325, F564–F577. [Google Scholar] [CrossRef]
- Barbier, O.; Jacquillet, G.; Tauc, M.; Poujeol, P.; Cougnon, M. Acute study of interaction among cadmium, and zinc transport along the rat nephron in vivo. Am. J. Physiol. Ren. Physiol. 2004, 287, F1067–F1075. [Google Scholar] [CrossRef]
- Wang, Y.; Zalups, R.K.; Barfuss, D.W. Potential mechanisms involved in the absorptive transport of cadmium in isolated perfused rabbit renal proximal tubules. Toxicol. Lett. 2010, 193, 61–68. [Google Scholar] [CrossRef]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Wolff, N.A.; Lee, W.K.; Thévenod, F. Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J. Biol. Chem. 2012, 287, 159–169. [Google Scholar] [CrossRef]
- Fels, J.; Scharner, B.; Zarbock, R.; Zavala-Guevara, I.P.; Lee, W.K.; Barbier, O.C.; Thévenod, F. Cadmium complexed with β2-microglubulin, albumin and lipocalin-2 rather than metallothionein cause megalin:cubilin dependent toxicity of the renal proximal tubule. Int. J. Mol. Sci. 2019, 20, 2379. [Google Scholar] [CrossRef] [PubMed]
- Zavala-Guevara, I.P.; Ortega-Romero, M.S.; Narváez-Morales, J.; Jacobo-Estrada, T.L.; Lee, W.K.; Arreola-Mendoza, L.; Thévenod, F.; Barbier, O.C. Increased endocytosis of cadmium-metallothionein through the 24p3 receptor in an in vivo model with reduced proximal tubular activity. Int. J. Mol. Sci. 2021, 22, 7262. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Ruangyuttikarn, W.; Nishijo, M.; Gobe, G.C.; Phelps, K.R. The source and pathophysiologic significance of excreted cadmium. Toxics 2019, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, C.P.; Chen, S.S.; Ng, Y.H.; Roumelioti, M.E.; Shaffi, K.; Singh, P.P.; Tzamaloukas, A.H. Rediscovering beta-2 microglobulin as a biomarker across the spectrum of kidney diseases. Front. Med. 2017, 4, 73. [Google Scholar] [CrossRef]
- Gauthier, C.; Nguyen-Simonnet, H.; Vincent, C.; Revillard, J.-P.; Pellet, M.V. Renal tubular absorption of beta 2 microglobulin. Kidney Int. 1984, 26, 170–175. [Google Scholar] [CrossRef]
- Portman, R.J.; Kissane, J.M.; Robson, A.M. Use of B2-microglobulin to diagnose tubulo-interstitial renal lesions in children. Kidney Int. 1986, 30, 91–98. [Google Scholar] [CrossRef]
- Peterson, P.A.; Evrin, P.-E.; Berggard, I. Differentiation of glomerular, tubular, and normal proteinuria: Determination of urinary excretion of B2-microglobulin, albumin, and total protein. J. Clin. Investig. 1969, 48, 1189–1198. [Google Scholar] [CrossRef]
- Forman, D.T. Beta-2 microglobulin—An immunogenetic marker of inflammatory and malignant origin. Ann. Clin. Lab. Sci. 1982, 12, 447–451. [Google Scholar] [PubMed]
- Satarug, S.; Vesey, D.A.; Nishijo, M.; Ruangyuttikarn, W.; Gobe, G.C. The inverse association of glomerular function and urinary β2-MG excretion and its implications for cadmium health risk assessment. Environ. Res. 2019, 173, 40–47. [Google Scholar] [CrossRef]
- Moffett, D.B.; Mumtaz, M.M.; Sullivan, D.W., Jr.; Whittaker, M.H. Chapter 13, General Considerations of Dose-Effect and Dose-Response Relationships. In Handbook on the Toxicology of Metals, 5th ed.; Volume I: General Considerations; Nordberg, G., Costa, M., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 299–317. [Google Scholar]
- Roels, H.A.; Lauwerys, R.R.; Buchet, J.P.; Bernard, A.M.; Vos, A.; Oversteyns, M. Health significance of cadmium induced renal dysfunction: a five year follow up. Br. J. Ind. Med. 1989, 46, 755–764. [Google Scholar] [CrossRef]
- Misra, R.R.; Hochadel, J.F.; Smith, G.T.; Cook, J.C.; Waalkes, M.P.; Wink, D.A. Evidence that nitric oxide enhances cadmium toxicity by displacing the metal from metallothionein. Chem Res Toxicol. 1996, 9, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Baker, J.R.; Reilly, P.E.; Esumi, H.; Moore, M.R. Evidence for a synergistic interaction between cadmium and endotoxin toxicity and for nitric oxide and cadmium displacement of metals in the kidney. Nitric Oxide 2000, 4, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Phelps, K.R. Chapter 14, Cadmium exposure and toxicity. In: Bagchi M, Bagchi D (eds) Metal Toxicology, 2021. CRC Press.
- EFSA. European Food Safety Agency, Statement on tolerable weekly intake for cadmium. EFSA J. 2011, 9, 1975. [CrossRef]
- Qing, Y.; Yang, J.; Zhu, Y.; Li, Y.; Zheng, W.; Wu, M.; He, G. Dose-response evaluation of urinary cadmium and kidney injury biomarkers in Chinese residents and dietary limit standards. Environ. Health 2021, 20, 75. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Yimthiang, S.; Buha Đorđević, A. Health risk in a geographic area of Thailand with endemic cadmium contamination: Focus on albuminuria. Toxics 2023, 11, 68. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Khamphaya, T.; Pouyfung, P.; Gobe, G.C.; Yimthiang, S. Estimation of the cadmium nephrotoxicity threshold from loss of glomerular filtration rate and albuminuria. Toxics 2023, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Đorđević, A.B. The validity of benchmark dose limit analysis for estimating permissible accumulation of cadmium. Int. J. Environ. Res. Public Health 2022, 19, 1569. [Google Scholar] [CrossRef]
- Shi, P.; Yan, H.; Fan, X.; Xi, S. A benchmark dose analysis for urinary cadmium and type 2 diabetes mellitus. Environ. Pollut. 2021, 273, 116519. [Google Scholar] [CrossRef]
- Leconte, S.; Rousselle, C.; Bodin, L.; Clinard, F.; Carne, G. Refinement of health-based guidance values for cadmium in the French population based on modelling. Toxicol. Lett. 2021, 340, 43–51. [Google Scholar] [CrossRef]
- Ougier, E.; Fiore, K.; Rousselle, C.; Assunção, R.; Martins, C.; Buekers, J. Burden of osteoporosis and costs associated with human biomonitored cadmium exposure in three European countries: France, Spain and Belgium. Int. J. Hyg. Environ. Health 2021, 234, 113747. [Google Scholar] [CrossRef] [PubMed]
- Pouillot, R.; Santillana Farakos, S.; Van Doren, J.M. Modeling the risk of low bone mass and osteoporosis as a function of urinary cadmium in U.S adults aged 50-79 years. Environ. Res. 2022, 212(Pt B), 113315. [CrossRef]
- Calafat, A.M. The U.S. national health and nutrition examination survey and human exposure to environmental chemicals. Int. J. Hyg. Environ. Health 2012, 215, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Il’yasova, D.; Ivanova, A. Urinary cadmium, impaired fasting glucose, and diabetes in the NHANES III. Diabetes Care 2003, 26, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Wallia, A.; Allen, N.B.; Badon, S.; El Muayed, M. Association between urinary cadmium levels and prediabetes in the NHANES 2005–2010 population. Int. J. Hyg. Environ. Health 2014, 217, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhi, X.; Xu, M.; Li, B.; Zhang, Z. Gender-specific differences of interaction between cadmium exposure and obesity on prediabetes in the NHANES 2007-2012 population. Endocrine 2018, 61, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hyder, O.; Chung, M.; Cosgrove, D.; Herman, J.M.; Li, Z.; Firoozmand, A.; Gurakar, A.; Koteish, A.; Pawlik, T.M. Cadmium exposure and liver disease among US adults. J. Gastrointest. Surg. 2013, 17, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Min, J.Y.; Min, K.B. Association between cadmium exposure and liver function in adults in the United States: A cross-sectional study. J. Prev. Med. Public Health 2021, 54, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Weng, Z.; Liang, J.; Liu, Q.; Zhang, X.; Xu, J.; Xu, C.; Gu, A. Association between urinary cadmium concentrations and liver function in adolescents. Environ. Sci. Pollut. Res. Int. 2022, 29, 39768–39776. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.M.; Costanzi, S.; Naticchia, A.; Sturniolo, A.; Gambaro, G. Low level exposure to cadmium increases the risk of chronic kidney disease: Analysis of the NHANES 1999–2006. BMC Public Health 2010, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.J.; Wang, J.J.; Mao, J.H.; Shu, Q.; Du, L.Z. Relationships of cadmium, lead, and mercury levels with albuminuria in US adults: Results from the National Health and Nutrition Examination Survey Database, 2009–2012. Am. J. Epidemiol. 2019, 188, 1281–1287. [Google Scholar] [CrossRef]
- Lin, Y.S.; Ho, W.C.; Caffrey, J.L.; Sonawane, B. Low serum zinc is associated with elevated risk of cadmium nephrotoxicity. Environ. Res. 2014, 134, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, J.M.; Ricardo, A.C.; Persky, V.; Turyk, M. Associations between blood cadmium concentration and kidney function in the U.S. population: Impact of sex, diabetes and hypertension. Environ. Res. 2018, 169, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Oh, S.; Kang, H.; Kim, S.; Lee, G.; Li, L.; Kim, C.T.; An, J.N.; Oh, Y.K.; Lim, C.S.; et al. Environment-wide association study of CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Melough, M.M.; Vance, T.M.; Noh, H.; Koo, S.I.; Chun, O.K. Dietary cadmium intake and sources in the US. Nutrients 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, S.M.; Adams, S.V.; Shafer, M.; Meliker, J.R.; Li, W.; Luo, J.; Neuhouser, M.L.; Newcomb, P.A. Urinary cadmium and estimated dietary cadmium in the Women's Health Initiative. J. Expo. Sci. Environ. Epidemiol. 2016, 26, 303–308. [Google Scholar] [CrossRef]
- Shao, W.; Liu, Q.; He, X.; Liu, H.; Gu, A.; Jiang, Z. Association between level of urinary trace heavy metals and obesity among children aged 6-19 years: NHANES 1999–2011. Environ. Sci. Pollut. Res. Int. 2017, 24, 11573–11581. [Google Scholar] [CrossRef] [PubMed]
- Dhooge, W.; Hond, E.D.; Koppen, G.; Bruckers, L.; Nelen, V.; Van De Mieroop, E.; Bilau, M.; Croes, K.; Baeyens, W.; Schoeters, G.; et al. Internal exposure to pollutants and body size in Flemish adolescents and adults: Associations and dose-response relationships. Environ. Int. 2010, 36, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Padilla, M.A.; Elobeid, M.; Ruden, D.M.; Allison, D.B. An examination of the association of selected toxic metals with total and central obesity indices: NHANES 99-02. Int. J. Environ. Res. Public Health 2010, 7, 3332–3347. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.B. Effect of pregnancy on the levels of blood cadmium, lead, and mercury for females aged 17-39 years old: Data from National Health and Nutrition Examination Survey 2003–2010. J. Toxicol. Environ. Health A 2013, 76, 58–69. [Google Scholar] [CrossRef]
- Noor, N.; Zong, G.; Seely, E.W.; Weisskopf, M.; James-Todd, T. Urinary cadmium concentrations and metabolic syndrome in U.S. adults: The National Health and Nutrition Examination Survey 2001-2014. Environ Int. 2018, 21, 349–356. [Google Scholar] [CrossRef]
- Lu, L.; Li, Y.; Chen, C.; Zhang, Y.; Guo, W.; Zhang, S.; Kahe, K. Associations of cadmium exposure with risk of metabolic syndrome and its individual components: a meta-analysis. J. Expo. Sci. Environ. Epidemiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Garner, R.; Levallois, P. Cadmium levels and sources of exposure among Canadian adults. Health Rep. 2016, 27, 10–18. [Google Scholar] [PubMed]
- Akbar, L.; Zuk, A.M.; Martin, I.D.; Liberda, E.N.; Tsuji, L.J.S. Potential obesogenic effect of a complex contaminant mixture on Cree First Nations adults of Northern Québec, Canada. Environ. Res. 2021, 192, 110478. [Google Scholar] [CrossRef]
- Son, H.S.; Kim, S.G.; Suh, B.S.; Park, D.U.; Kim, D.S.; Yu, S.D.; Hong, Y.S.; Park, J.D.; Lee, B.K.; Moon, J.D.; et al. Association of cadmium with diabetes in middle-aged residents of abandoned metal mines: The first health effect surveillance for residents in abandoned metal mines. Ann. Occup. Environ. Med. 2015, 27, 20. [Google Scholar] [CrossRef] [PubMed]
- Yimthiang, S.; Pouyfung, P.; Khamphaya, T.; Kuraeiad, S.; Wongrith, P.; Vesey, D.A.; Gobe, G.C.; Satarug, S. Effects of environmental exposure to cadmium and lead on the risks of diabetes and kidney dysfunction. Int. J. Environ. Res. Public Health 2022, 19, 2259. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Wang, N.; Chen, Y.; Chen, C.; Han, B.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Lu, M.; et al. Blood cadmium in Chinese adults and its relationships with diabetes and obesity. Environ. Sci. Pollut. Res. Int. 2016, 23, 18714–18723. [Google Scholar] [CrossRef]
- Feng, X.; Zhou, R.; Jiang, Q.; Wang, Y.; Chen, C. Analysis of cadmium accumulation in community adults and its correlation with low-grade albuminuria. Sci. Total Environ. 2022, 834, 155210. [Google Scholar] [CrossRef] [PubMed]
- Crinnion, W.J. The CDC fourth national report on human exposure to environmental chemicals: what it tells us about our toxic burden and how it assists environmental medicine physicians. Altern. Med. Rev. 2010, 15, 101–109. [Google Scholar]
- Mortensen, M.E. , Wong, L.Y., Osterloh, J.D. Smoking status and urine cadmium above levels associated with subclinical renal effects in U.S. adults without chronic kidney disease. Int. J. Hyg. Environ. Health 2011, 214, 305–310. [Google Scholar] [CrossRef]
- Triplitt, C.L. Examining the mechanisms of glucose regulation. Am. J. Manag. Care. 2012, 18, S4–S10. [Google Scholar]
- Wilding, J.P. The role of the kidneys in glucose homeostasis in type 2 diabetes: clinical implications and therapeutic significance through sodium glucose co-transporter 2 inhibitors. Metabolism 2014, 63, 1228–1237. [Google Scholar] [CrossRef]
- Alsahli, M.; Gerich, J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef]
- Dalga, D.; Verissimo, T.; de Seigneux, S. Gluconeogenesis in the kidney: in health and in chronic kidney disease. Clin. Kidney J. 2023, 16, 1249–1257. [Google Scholar] [CrossRef]
- Duo, Y.; Gao, J.; Yuan, T.; Zhao, W. Effect of sodium-glucose cotransporter 2 inhibitors on the rate of decline in kidney function: A systematic review and meta-analysis. J. Diabetes 2023, 15, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.A.; Nisa, Z.U.; Mehmood, A.; Anjum, M.S.; Shahzad, K. Metal-induced nephrotoxicity to diabetic and non-diabetic Wistar rats. Environ. Sci. Pollut. Res. Int. 2019, 26, 31111–31118. [Google Scholar] [CrossRef]
- Riaz, M.A.; Nisa, Z.U.; Anjum, M.S.; Butt, H.; Mehmood, A.; Riaz, A.; Akhtar, A.B.T. Assessment of metals induced histopathological and gene expression changes in different organs of non-diabetic and diabetic rats. Sci. Rep. 2020, 10, 5897. [Google Scholar] [CrossRef] [PubMed]
- Akerstrom, M.; Barregard, L.; Lundh, T.; Sallsten, G. The relationship between cadmium in kidney and cadmium in urine and blood in an environmentally exposed population. Toxicol. Appl. Pharmacol. 2013, 268, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Wallin, M.; Sallsten, G.; Lundh, T.; Barregard, L. Low-level cadmium exposure and effects on kidney function. Occup. Environ. Med. 2014, 71, 848–854. [Google Scholar] [CrossRef]
- Barregard, L.; Fabricius-Lagging, E.; Lundh, T.; Mölne, J.; Wallin, M.; Olausson, M.; Modigh, C.; Sallsten, G. Cadmium, mercury, and lead in kidney cortex of living kidney donors: Impact of different exposure sources. Environ. Res. 2010, 110, 47–54. [Google Scholar] [CrossRef]
- Jacquet, A.; Ounnas, F.; Lénon, M.; Arnaud, J.; Demeilliers, C.; Moulis, J.M. Chronic exposure to low-level cadmium in diabetes: Role of oxidative stress and comparison with polychlorinated biphenyls. Curr. Drug Targets 2016, 17, 1385–1413. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C. Mitigation of cadmium toxicity through modulation of the frontline cellular stress response. Stresses 2022, 2, 355–372. [Google Scholar] [CrossRef]
- Moulis, J.M. Cellular dynamics of transition metal exchange on proteins: A challenge but a bonanza for coordination chemistry. Biomolecules 2020, 10, 1584. [Google Scholar] [CrossRef] [PubMed]
- Buha, A.; Đukić-Ćosić, D.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Moulis, J.M.; Goumenou, M.; Wallace, D. Emerging links between cadmium exposure and insulin resistance: Human, animal, and cell study data. Toxics 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Waalkes, M.P.; Flores Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Moroni-González, D.; Díaz, A.; Eduardo, B.; Samuel, T. Oral subacute exposure to cadmium LOAEL dose induces insulin resistance and impairment of the hormonal and metabolic liver-adipose axis in Wistar rats. Biol. Trace Elem. Res. 2022, 200, 4370–4384. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, N.; Chen, C.; Nie, X.; Han, B.; Li, Q.; Zhu, C.; Chen, Y.; Xia, F.; Chen, Y.; et al. Cadmium exposure and its association with serum uric acid and hyperuricemia. Sci. Rep. 2017, 7, 550. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Li, S.; Zhou, Y.; Sun, D. Association between low-level blood cadmium exposure and hyperuricemia in the American general population: A cross-sectional study. Biol. Trace Elem. Res. 2022, 200, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Kim, Y.; Lihm, H.; Kang, J. Associations between blood lead, cadmium, and mercury levels with hyperuricemia in the Korean general population: A retrospective analysis of population-based nationally representative data. Int. J. Rheum. Dis. 2019, 22, 1435–1444. [Google Scholar] [CrossRef]
- Park, J. , Kim, Y. Associations of blood heavy metals with uric acid in the Korean general population: analysis of data from the 2016–2017 Korean national health and nutrition examination survey. Biol Trace Elem Res. 2021, 199, 102–112. [Google Scholar] [CrossRef]
- Lu, J.; He, Y.; Cui, L.; Xing, X.; Liu, Z.; Li, X.; Zhang, H.; Li, H.; Sun, W.; Ji, A.; et al. Hyperuricemia predisposes to the onset of diabetes via promoting pancreatic β-cell death in uricase-deficient male mice. Diabetes 2020, 69, 1149–1163. [Google Scholar] [CrossRef]
- Ghasemi, A. Uric acid-induced pancreatic β-cell dysfunction. BMC Endocr. Disord. 2021, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Moulis, J.M.; Nahoui-Zarouri, I.; Lénon, M.; Cottet-Rousselle, C. Low-level cadmium doses do not jeopardize the insulin secretion pathway of β-cell models until the onset of cell death. J. Trace Elem. Med. Biol. 2021, 68, 126834. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, A.; Cottet-Rousselle, C.; Arnaud, J.; Julien Saint Amand, K.; Ben Messaoud, R.; Lénon, M.; Demeilliers, C.; Moulis, J.M. Mitochondrial morphology and function of the pancreatic β-cells INS-1 model upon chronic exposure to sub-lethal cadmium doses. Toxics 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Rocca, A.; Fanchon, E.; Moulis, J.-M. Theoretical modeling of oral glucose tolerance tests guides the interpretation of the impact of perinatal cadmium exposure on the offspring’s glucose homeostasis. Toxics 2020, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.; Olsen, A.; Nguyen, J.; Wong, W.; El Muayed, M.; Edwards, J. Pancreatic islets accumulate cadmium in a rodent model of cadmium-induced hyperglycemia. Int. J. Mol. Sci. 2020, 22, 360. [Google Scholar] [CrossRef] [PubMed]
- Gasser, M.; Lenglet, S.; Bararpour, N.; Sajic, T.; Wiskott, K.; Augsburger, M.; Fracasso, T.; Gilardi, F.; Thomas, A. Cadmium acute exposure induces metabolic and transcriptomic perturbations in human mature adipocytes. Toxicol. 2022, 470, 153153. [Google Scholar] [CrossRef] [PubMed]
- Echeverría, R.; Vrhovnik, P.; Salcedo-Bellido, I.; Iribarne-Durán, L.M.; Fiket, Ž.; Dolenec, M.; Martin-Olmedo, P.; Olea, N.; Arrebola, J.P. Levels and determinants of adipose tissue cadmium concentrations in an adult cohort from Southern Spain. Sci. Total. Environ. 2019, 670, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Bellido, I.; Gómez-Peña, C.; Pérez-Carrascosa, F.M.; Vrhovnik, P.; Mustieles, V.; Echeverría, R.; Fiket, Ž.; Pérez-Díaz, C.; Barrios-Rodríguez, R.; Jiménez-Moleón, J.J.; et al. ; Adipose tissue cadmium concentrations as a potential risk factor for insulin resistance and future type 2 diabetes mellitus in GraMo adult cohort. Sci. Total Environ. 2021, 780, 146359. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Sugimoto, H.; Furuichi, R.; Kadota, Y.; Inoue, M.; Setsu, K.; Suzuki, S.; Sato, M. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicol. 2010, 267, 20–26. [Google Scholar] [CrossRef]
- Kawakami, T.; Nishiyama, K.; Kadota, Y.; Sato, M.; Inoue, M.; Suzuki, S. Cadmium modulates adipocyte functions in metallothionein-null mice. Toxicol. Appl. Pharmacol. 2013, 272, 625–636. [Google Scholar] [CrossRef]
- Attia, S.M.; Das, S.C.; Varadharajan, K.; Al-Naemi, H.A. White adipose tissue as a target for cadmium toxicity. Front. Pharmacol. 2022, 13, 1010817. [Google Scholar] [CrossRef]
- Takeda, T.A.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Hortmann, M.; Oelze, M.; Opitz, B.; Steven, S.; Schell, R.; Knorr, M.; Karbach, S.; Schuhmacher, S.; Wenzel, P.; et al. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell Cardiol. 2010, 49, 186–195. [Google Scholar] [CrossRef]
- Durante, W. Targeting heme oxygenase-1 in the arterial response to injury and disease. Antioxidants (Basel) 2020, 9, 829. [Google Scholar] [CrossRef]
- Vitek, L.; Hinds, T.D. Jr.; Stec, D.E.; Tiribelli, C. The physiology of bilirubin: health and disease equilibrium. Trends Mol Med. 2023, 29, 315–328. [Google Scholar] [CrossRef]
- Nath KA, Singh RD, Croatt AJ, Adams CM. Heme proteins and kidney injury: Beyond rhabdomyolysis. Kidney360, 2022; 3, 1969-1979. [CrossRef]
- Satarug, S.; Wisedpanichkij, R.; Takeda, K.; Li, B.; Na-Bangchang, K.; Moore, M.R. , Shibahara, S. Prostaglandin D2 induces heme oxygenase-1 mRNA expression through the DP2 receptor. Biochem. Biophys. Res. Commun. 2008, 377, 878–883. [Google Scholar] [CrossRef]
- Boonprasert, K.; Satarug, S.; Morais, C.; Gobe, G.C.; Johnson, D.W.; Na-Bangchang, K.; Vesey, D.A. The stress response of human proximal tubule cells to cadmium involves up-regulation of haemoxygenase 1 and metallothionein but not cyto-chrome P450 enzymes. Toxicol. Lett. 2016, 249, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Spik, I.; Brénuchon, C.; Angéli, V.; Staumont, D.; Fleury, S.; Capron, M.; Trottein, F.; Dombrowicz, D. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J. Immunol. 2005, 174, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ishizawa, S.; Sato, M.; Yoshida, T.; Shibahara, S. Identification of a cis-acting element that is responsible for cadmium-mediated induction of the human heme oxygenase gene. J. Biol. Chem. 1994, 269, 22858–22867. [Google Scholar] [CrossRef]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef]
- Suzuki, H.; Tashiro, S.; Sun, J.; Doi, H.; Satomi, S.; Igarashi, K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J. Biol. Chem. 2003, 278, 49246–49253. [Google Scholar] [CrossRef] [PubMed]
- Merali, Z.; Singhal, R.L. Diabetogenic effects of chronic oral cadmium administration to neonatal rats. Br. J. Pharmacol. 1980, 69, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.R.; Early, J.L.; Nonavinakere, V.K.; Mallory, Z. Effect of cadmium on blood glucose level in the rat. Toxicol Lett. 1990, 54, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.R.; Soliman, M.R.; Early, J.L. 2nd. Acute effects of cadmium and selenium on glucose output from rat liver hepatocytes using various gluconeogenic precursors. Toxicol. 1990, 65, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Prozialeck, W.C. Cadmium, diabetes and chronic kidney disease. Toxicol. Appl. Pharmacol. 2009, 238, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Merali, Z.; Kacew, S.; Singhal, R.L. Response of hepatic carbohydrate and cyclic AMP metabolism to cadmium treatment in rats. Can. J. Physiol. Pharmacol. 1975, 53, 174–184. [Google Scholar] [CrossRef]
- Wang, B.; Luo, Q.; Shao, C.; Li, X.; Li, F.; Liu, Y.; Sun, L.; Li. Y.; Cai, L. The late and persistent pathogenic effects of cadmium at very low levels on the kidney of rats. Dose Response 2013, 11, 60–81. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Hayakawa, K.; Miyaji, K.; Ishikawa, H.; Hata, M. Cadmium nephrotoxicity and evacuation from the body in a rat modeled subchronic intoxication. Int. J. Urol. 2003, 10, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, A.; Arnaud, J.; Hininger-Favier, I.; Hazane-Puch, F.; Couturier, K.; Lénon, M.; Lamarche, F.; Ounnas, F.; Fontaine, E.; Moulis, J.M.; Demeilliers, C. Impact of chronic and low cadmium exposure of rats: sex specific disruption of glucose metabolism. Chemosphere 2018, 207, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, A.; Barbeau, D.; Arnaud, J.; Hijazi, S.; Hazane-Puch, F.; Lamarche, F.; Quiclet, C.; Couturier, K.; Fontaine, E.; Moulis, J.M.; Demeilliers, C. Impact of maternal low-level cadmium exposure on glucose and lipid metabolism of the litter at different ages after weaning. Chemosphere 2019, 219, 109–121. [Google Scholar] [CrossRef]
- Saedi, S.; Watson, S.E.; Young, J.L.; Tan, Y.; Wintergerst, K.A.; Cai, L. Does maternal low-dose cadmium exposure increase the risk of offspring to develop metabolic syndrome and/or type 2 diabetes? Life Sci. 2023, 315, 121385. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Ortega, V.E.; Brambila, E.; Flores-Hernández, J.Á.; Díaz, A.; Peña-Rosas, U.; Moroni-González, D.; Aburto-Luna, V.; Treviño, S. The NOAEL Metformin dose is ineffective against metabolic disruption induced by chronic cadmium exposure in Wistar rats. Toxics 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Yoshizawa, M.; Suzuki, H.; Takeda, K.; Meguro, K.; Endo, K. Functional analysis of cDNAs for two types of human heme oxygenase and evidence for their separate regulation. J. Biochem. (Tokyo) 1993, 113, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S. The heme oxygenase dilemma in cellular homeostasis: new insights for the feedback regulation of heme catabolism. Tohoku J. Exp. Med. 2003, 200, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Han, F.; Li, B.; Takeda, K. Hypoxia and heme oxygenases: Oxygen sensing and regulation of expression. Antiox. Redox Signal. 2007, 9, 2209–2225. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on heme oxygenase-2: focus on cellular protection and oxygen response. Oxid. Med. Cell Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef] [PubMed]
- Schultz, I.J.; Chen, C.; Paw, B.H.; Hamza, I. Iron, porphyrin trafficking in heme biogenesis. J. Biol. Chem. 2010, 285, 26753–26759. [Google Scholar] [CrossRef] [PubMed]
- Paredi, P.; Biernacki, W.; Invernizzi, G.; Kharitonov, S.A.; Barnes, P.J. Exhaled carbon monoxide levels elevated in diabetes and correlated with glucose concentration in blood: a new test for monitoring the disease? Chest 1999, 116, 1007–1011. [Google Scholar] [CrossRef]
- Goto, Y.; Kakizaki, M.; Masaki, N. Production of spontaneous diabetic rats by repetition of selective breeding. Tohoku J. Exp. Med. 1976, 119, 85–90. [Google Scholar] [CrossRef]
- Ndisang, J.F.; Lane, N.; Jadhav, A. Upregulation of the heme oxygenase system ameliorates postprandial and fasting hyperglycemia in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1029–E1041. [Google Scholar] [CrossRef]
- Li, M.; Kim, D.H.; Tsenovoy, P.L.; Peterson, S.J.; Rezzani, R.; Rodella, L.F.; Aronow, W.S.; Ikehara, S.; Abraham, N.G. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008, 57, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Song, F.; Li, X.; Rong, S.; Yang, W.; Wang, D.; Xu, J.; Fu, J.; Zhao, Y.; Liu, L. Association between heme oxy-genase-1 gene promoter polymorphisms and type 2 diabetes mellitus: a HuGE review and meta-analysis. Am. J. Epidemiol. 2010, 172, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.L.; Sun, L.; Wang, Y.X.; Sun, B.H.; Li, Y.F.; Jin, Y.L. Association between HO-1 gene promoter polymorphisms and diseases (Review). Mol. Med. Rep. 2022, 25, 29. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, B.; Emmett, M.J.; Damle, M.; Sun, Z.; Feng, D.; Armour, S.M.; Remsberg, J.R.; Jager, J.; Soccio, R.E.; Steger, D.J.; Lazar, M.A. Gene regulation. Discrete functions of nuclear receptor Rev-erbα couple metabolism to the clock. Science 2015, 348, 1488–1492. [Google Scholar] [CrossRef]
- Everett, L.J.; Lazar, M.A. Nuclear receptor Rev-erbα: up, down, and all around. Trends Endocrinol Metab. 2014, 25, 586–592. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef]
- Medina, M.V.; Sapochnik, D.; Garcia Solá, M.; Coso, O. Regulation of the expression of heme oxygenase-1: signal transduction, gene promoter activation, and beyond. Antioxid. Redox Signal. 2020, 32, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: the circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef]
- Wu, N.; Yin, L.; Hanniman, E.A.; Joshi, S; Lazar, M. A. Negative feedback maintenance of heme homeostasis by its re-ceptor, Rev-erb alpha. Genes Dev. 2009, 23, 2201–2209. [Google Scholar] [CrossRef]
- Igarashi, K.; Watanabe-Matsui, M. Wearing red for signaling: The Heme-Bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J. Exp. Med. 2014, 232, 229–253. [Google Scholar] [CrossRef]
- Hanna, D.A. , Moore, C.M.; Liu, L.; Yuan, X.; Dominic, I.M.; Fleischhacker, A.S.; Hamza, I.; Ragsdale. S.W.; Reddi, A.R. Heme oxygenase-2 (HO-2) binds and buffers labile ferric heme in human embryonic kidney cells. J. Biol. Chem. 2022, 298, 101549. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Carter, E.L.; Ragsdale, S.W. Redox regulation of heme oxygenase-2 and the transcription factor, Rev-Erb, through heme regulatory motifs. Antioxid. Redox Signal. 2018, 29, 1841–1857. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Gunawan, A.L.; Kochert, B.A. , Liu, L.; Wales, T.E.; Borowy, M.C.; Engen, J.R.; Ragsdale, S.W. The heme-regulatory motifs of heme oxygenase-2 contribute to the transfer of heme to the catalytic site for degradation. J. Biol. Chem. 2020, 295, 5177–5191. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Garovic, V.D. , Grande, J.P., Croatt, A.J., Ackerman, A.W.; Farrugia, G.; Katusic, Z.S.; Belcher, J.D.; Vercellotti, G.M. Heme oxygenase-2 protects against ischemic acute kidney injury: influence of age and sex. Am. J. Physiol. Renal Physiol, 2019; 317, F695-F704. [Google Scholar] [CrossRef]
- Goodman, A.I.; Chander, P.N.; Rezzani, R.; Schwartzman, M.L.; Regan, R.F.; Rodella, L.; Turkseven, S.; Lianos, E.A.; Dennery, P.A.; Abraham, N.G. (2006) Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J. Am. Soc. Nephrol. 2006. 17, 1073–1081. [CrossRef]
- Sodhi, K.; Inoue, K.; Gotlinger, K.H.; Canestraro, M.; Vanella, L.; Kim, D.H.; Manthati, V.L.; Koduru, S.R.; Falck, J.R.; Schwartzman, M.L.; Abraham, N.G. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J. Pharmacol. Exp. Therapeu. 2009, 331, 906–916. [Google Scholar] [CrossRef]
- Li, B.; Takeda, K.; Ishikawa, K.; Yoshizawa, M.; Sato, M.; Shibahara, S.; Furuyama, K. Coordinated expression of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 4 and heme oxygenase 2: Evidence for a regulatory link between glycolysis and heme catabolism. Tohoku J. Exp. Med. 2012, 228, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Okar, D.A.; Manzano, A.; Navarro-Sabatè, A.; Riera, L.; Bartrons, R.; Lange, A.J. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2, 6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004, 381, 561–579. [Google Scholar] [CrossRef]
- Lannoy, V.J.; Decaux, J.F.; Pierreux, C.E.; Lemaigre, F.P.; Rousseau, G.G. Liver glucokinase gene expression is controlled by the onecut transcription factor hepatocyte nuclear factor-6. Diabetologia 2002, 45, 1136–1141. [Google Scholar] [CrossRef]
- Han, F.; Takeda, K.; Ishikawa, K.; Ono, M.; Date, F.; Yokoyama, S.; Furuyama, K.; Shinozawa, Y.; Urade, Y.; Shibahara, S. Induction of lipocalin-type prostaglandin D synthase in mouse heart under hypoxemia. Biochem. Biophys. Res. Commun. 2009, 385, 449–453. [Google Scholar] [CrossRef]
Figure 1.
Cadmium-induced kidney tubular cell death. Cd uses the Zn carrier metallothionein (MT) and transporters of Ca and Fe, metal coupling unit (MCU) and the mammalian transporter of metals DMT1 to reach the mitochondrial inner membrane [14]. There, Cd reduces ATP output and promotes reactive oxygen species (ROS) formation. Extensive damage causes a release of mitochondrial DNA (mtDNA). The DNA-sensing mechanism (cGAS-STING) and nuclear factor-kappaB (NF-кB) signaling pathways are activated, proinflammatory cytokines are released, and cell death ensues.
Figure 1.
Cadmium-induced kidney tubular cell death. Cd uses the Zn carrier metallothionein (MT) and transporters of Ca and Fe, metal coupling unit (MCU) and the mammalian transporter of metals DMT1 to reach the mitochondrial inner membrane [14]. There, Cd reduces ATP output and promotes reactive oxygen species (ROS) formation. Extensive damage causes a release of mitochondrial DNA (mtDNA). The DNA-sensing mechanism (cGAS-STING) and nuclear factor-kappaB (NF-кB) signaling pathways are activated, proinflammatory cytokines are released, and cell death ensues.
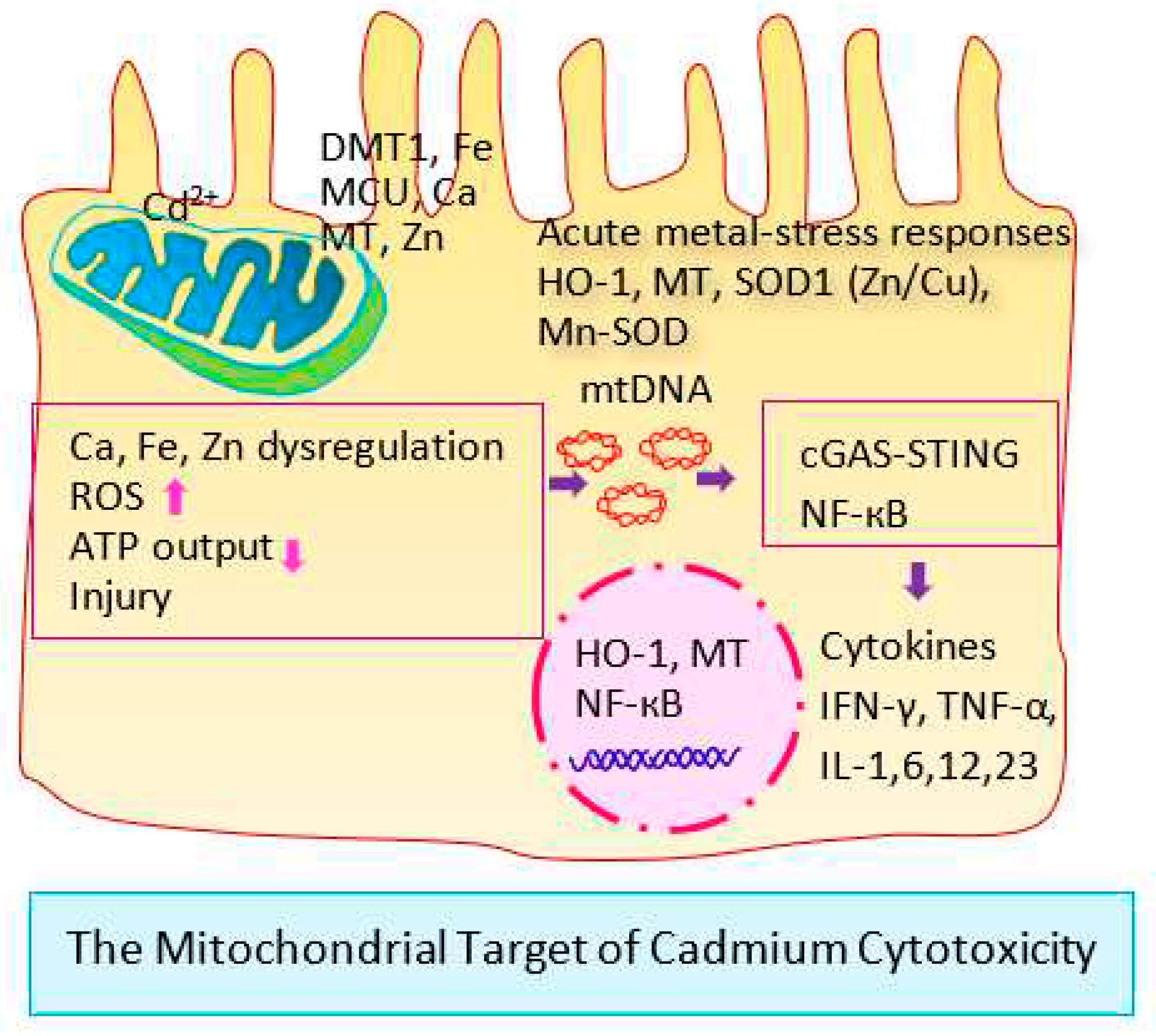
Figure 2.
Protein re-absorption by the kidney proximal tubular cells (PTCs). (a) Reabsorption of albumin and β2-microglobulin (β2M) in lumen through transcytosis and receptor-mediated endocytosis (RME). (b) Cd-induced RME dysfunction compromises reabsorption, and increases excretion of albumin and β2M.
Figure 2.
Protein re-absorption by the kidney proximal tubular cells (PTCs). (a) Reabsorption of albumin and β2-microglobulin (β2M) in lumen through transcytosis and receptor-mediated endocytosis (RME). (b) Cd-induced RME dysfunction compromises reabsorption, and increases excretion of albumin and β2M.
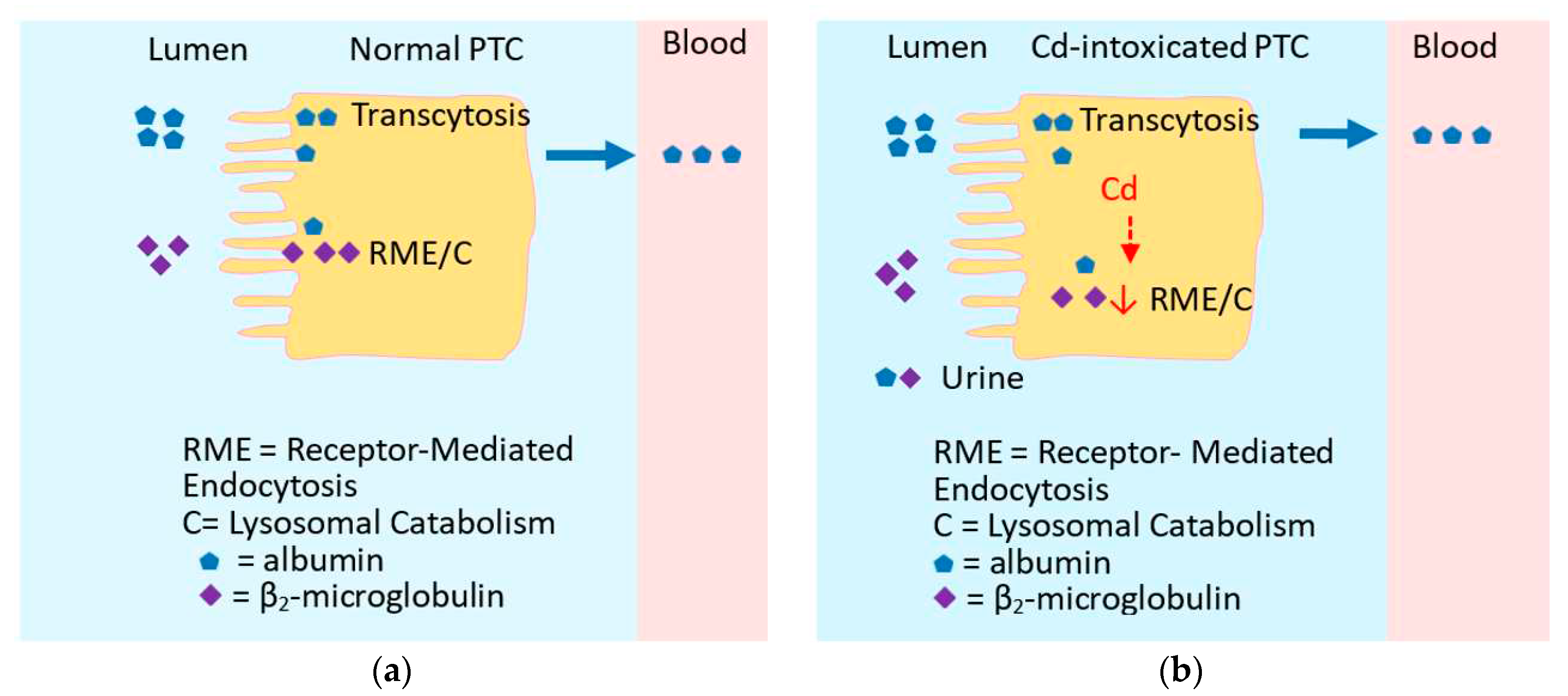
Figure 3.
Entry routes of cadmium and its toxicity targets. From the gut, Cd is delivered to liver via the portal blood system and is taken up by hepatocytes. The fraction of absorbed Cd not taken up by hepatocytes in the first pass reaches systemic circulation and is assimilated by cells of various organs throughout the body, including the heart, kidney, bone, thyroid gland, fat cells and pancreas.
Figure 3.
Entry routes of cadmium and its toxicity targets. From the gut, Cd is delivered to liver via the portal blood system and is taken up by hepatocytes. The fraction of absorbed Cd not taken up by hepatocytes in the first pass reaches systemic circulation and is assimilated by cells of various organs throughout the body, including the heart, kidney, bone, thyroid gland, fat cells and pancreas.
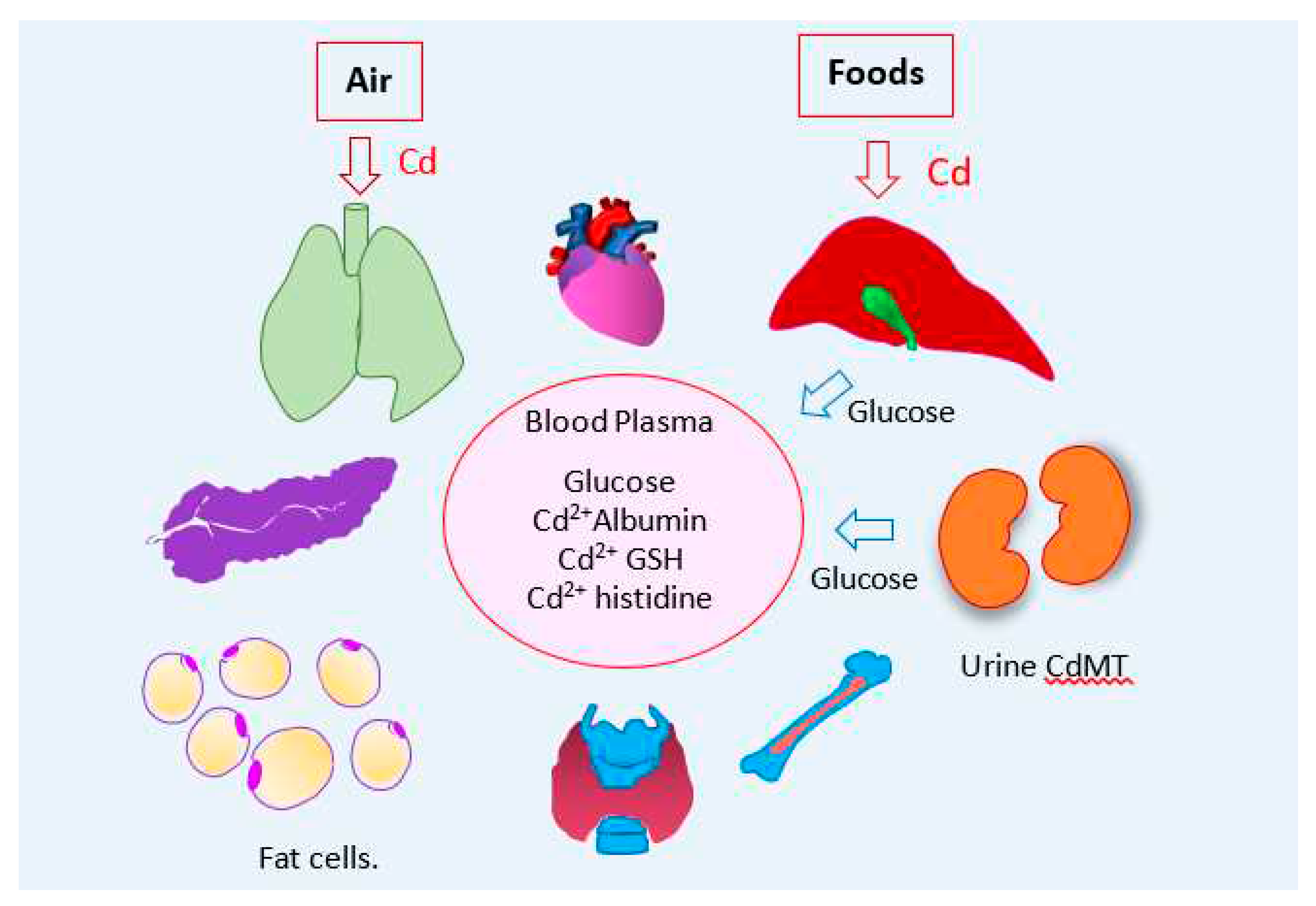
Figure 4.
Metabolic interrelationships of glucose, heme and glutathione. Abbreviations: ROS, reactive oxygen species; GSH, glutathione; GSSG, oxidized glutathione; HO-1, heme oxygenase-1; HO-2, heme oxygenase-2; CO, carbon monoxide; G-6-P; glucose-6-phosphate; F-1,6-P2, fructose 1,6 bisphosphate; F-2,6-P2, fructose 2,6 bisphosphate; TCA, tricarboxylic acid; F-6-P, fructose-6-phosphate; PFKFB4,6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 4.
Figure 4.
Metabolic interrelationships of glucose, heme and glutathione. Abbreviations: ROS, reactive oxygen species; GSH, glutathione; GSSG, oxidized glutathione; HO-1, heme oxygenase-1; HO-2, heme oxygenase-2; CO, carbon monoxide; G-6-P; glucose-6-phosphate; F-1,6-P2, fructose 1,6 bisphosphate; F-2,6-P2, fructose 2,6 bisphosphate; TCA, tricarboxylic acid; F-6-P, fructose-6-phosphate; PFKFB4,6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 4.
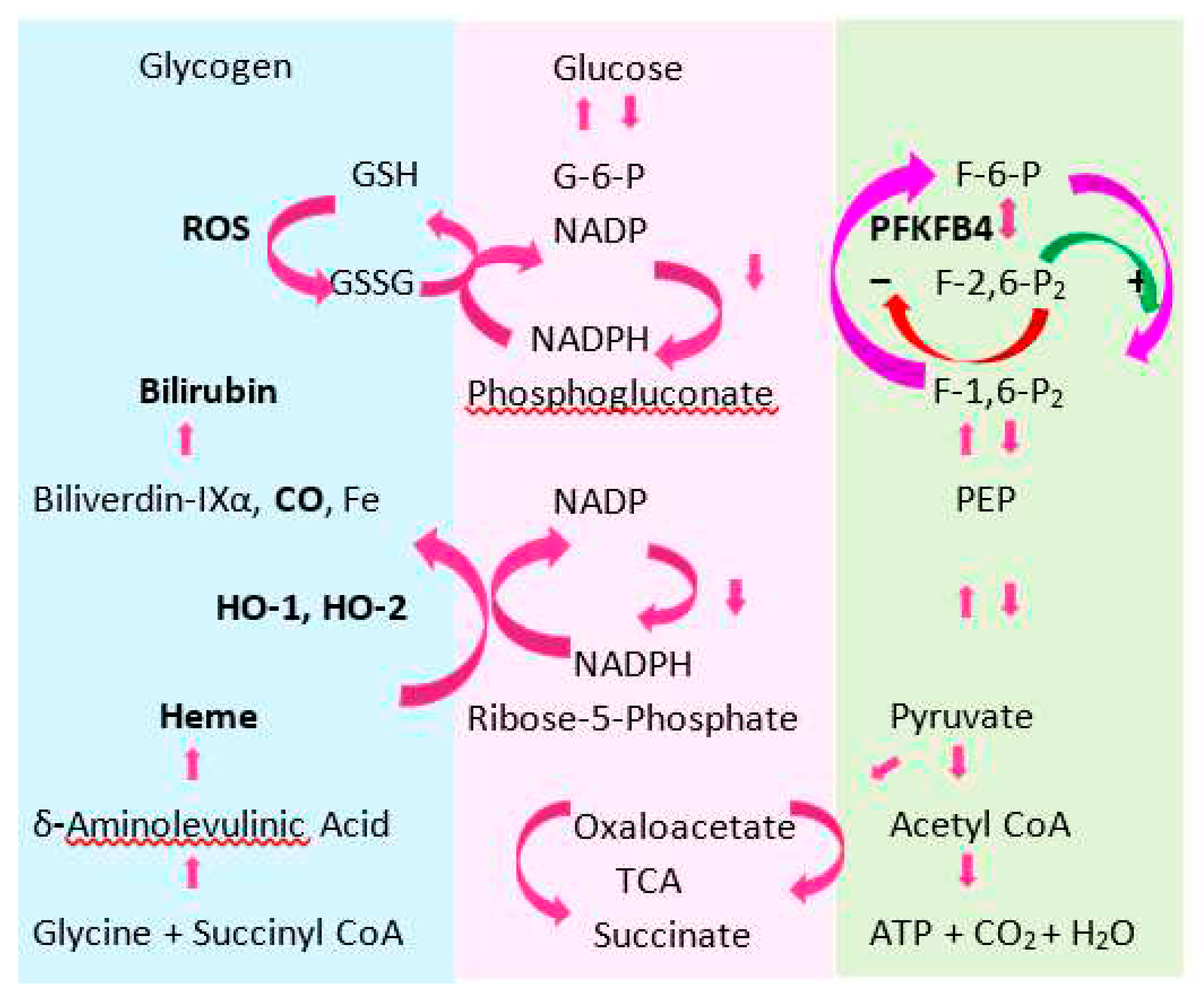
Figure 5.
Maintenance of plasma glucose through coordinated expression of HO-1, HO-2 and PFKFB4 in the liver (a) Changes in expression of HO-1 and PFKFB4 in the fasting state.; (b) Changes in expression of HO-1, HO-2 and PFKFB4 in the post absorptive period. Abbreviations: PFKFB4, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4; F-2,6-P2, fructose 2,6-biphosphate.
Figure 5.
Maintenance of plasma glucose through coordinated expression of HO-1, HO-2 and PFKFB4 in the liver (a) Changes in expression of HO-1 and PFKFB4 in the fasting state.; (b) Changes in expression of HO-1, HO-2 and PFKFB4 in the post absorptive period. Abbreviations: PFKFB4, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4; F-2,6-P2, fructose 2,6-biphosphate.


Table 1.
Gender- and organ-differentiated cadmium accumulation in humans.
| Tissues/Organs | Cd content in µg/g wet tissue weight | Country of origin/reference | |
|---|---|---|---|
| Males | Females | ||
| Lung | 0.11 ± 0.19 | 0.17 ± 0.35 | Australia, Satarug et al. [58] a |
| Liver | 0.78 ± 0.71 | 1.36 ± 0.96 | |
| Kidney cortex | 14.6 ± 12.4 | 18.1 ± 18.0 | |
| Liver | 7.9 (1.3−33.3) | 13.1 (3.1−106.4) | Japan, Uetani et al. 2006 [59] b |
| Kidney cortex | 72.1 (19.4−200) | 83.9 (3.9− 252.9) | |
| Kidney medulla | 18.3 (3.5−76.4) | 24.5 (4−105) | |
| Pancreas | 7.4 (3.0−25.9) | 10.5 (2.5−29.8) | |
| Thyroid | 10.6 (3.8−35) | 11.9 (3.9−56.4) | |
| Heart | 0.3 (0.1−0.5) | 0.4 (0.1−1.3) | |
| Muscle | 1.2 (0.3−3.2) | 2.2 (0.8−12.4) | |
| Aorta | 1.0 (0.4 −2.5) | 1.1 (0.3− 3) | |
| Bone | 0.4 (0.2−0.6) | 0.6 (0.2−1.6) | |
a Values were arithmetic mean ± standard deviation from 43 males and 18 females, aged 2–89 years (mean 38.5). b Values were geometric mean (lowest−highest Cd levels) from 36 males and 36 females, aged 60–91 years (mean 74).
Table 2.
Urinary and blood cadmium levels associated with increased risks of diseases with high prevalence.
Table 2.
Urinary and blood cadmium levels associated with increased risks of diseases with high prevalence.
| NHANES | Exposure and Risk Estimates | References |
|---|---|---|
| 1988−1994 n 8,722, ≥ 40 yrs |
Urinary Cd levels 1-2 μg/g creatinine were associated with prediabetes in (OR 1.48) and diabetes (OR 1.24). The respective OR values for prediabetes and diabetes rose to 2.05 and 1.45 at urine Cd > 2 µg/g creatinine. |
Schwartz et al. 2003 [119] |
| 2005−2010 n 2,398, ≥ 40 yrs |
Urinary Cd >1.4 µg/g creatinine in non-smokers were associated with pre-diabetes. | Wallia et al. 2014 [120] |
| 2007-2012 n 3,552, ≥ 20 yrs |
Urinary Cd quartile 4 was associated with prediabetes among men (OR 1.95). OR for prediabetes rose 3.4-fold in men with obesity and a high Cd exposure, compared to those with a normal weight and low Cd exposure. |
Jiang et al. 2018, [121] |
| 1988−1994 n 12,732, ≥ 20 yrs |
Urinary Cd levels ≥ 0.83 μg/g creatinine in women were associated with liver inflammation (OR 1.26). Urinary Cd ≥ 0.65 μg/g creatinine in men were associated with liver inflammation (OR 2.21), NAFLD (OR 1.30), and NASH (OR 1.95). |
Hyder et al. 2013 [122] |
| 1999−2015 n 11, 838, ≥ 20 yrs |
A 10-fold increment of urinary Cd was associated with elevated plasma levels of ALT (OR 1.36), and AST (OR 1.31). | Hong et al. 2021 [123] |
| 1999−2016 n 4411 adolescents |
Urinary Cd quartile 4 was associated with elevated plasma ALT (OR 1.40) and AST (OR 1.64). The effect was larger in boys than girls. |
Xu et al. 2022 [124] |
| 1999−2006 n 5,426, aged ≥ 20 yrs, |
Urinary Cd levels ≥ 1µg/L were associated with increased risk of albuminuria a (OR 1.41) and low GFR b (OR 1.48). | Ferraro et al. 2010 [125] |
| 2009−2012, n 2,926, aged ≥ 20 yrs |
Urinary Cd levels > 0.220 μg/L were associated with increased albumin excretion, compared with urinary Cd < 0.126 μg/L. Blood Cd levels > 0.349 μg/L associated with increased albumin excretion, compared with blood Cd < 0.243 μg/L. |
Zhu et al. 2019 [126] |
| 2011−2012 n 1,545, aged ≥ 20 yrs |
Blood Cd levels > 0.53 μg/L were associated with albuminuria (OR 2.04) and low GFR (OR 2.21). OR for albuminuria was increased to 3.38 in those with similar Cd exposure levels and serum Zn < 74 μg/dL. |
Lin et al. 2014 [127] |
| 2007−2012 n 12,577, aged ≥ 20 yrs |
Blood Cd levels > 0.61 μg/L were associated with low GFR (OR 1.80) and albuminuria (OR 1.60). GFR reduction associated with Cd was more pronounced in those with diabetes, hypertension, or both. |
Madrigal et al. 2019 [128] |
| 1999 − 2016, n 46,748, aged ≥ 20 yrs |
Of 262 chemicals tested, blood Cd was associated with all three kidney outcomes; low GFR, albuminuria, and low GFR plus albuminuria. | Lee et al. 2020 [129] |
NHANES, National Health and Nutrition Examination Survey; n, sample size; OR, odds ratio; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; ALT, alanine aminotransferase; AST, aspartate aminotransferase; a Albuminuria was defined as urinary albumin-to-creatinine ratio ≥ 30 mg/g creatinine; b Low GFR was defined as the estimated glomerular filtration rate < 60 ml/min/1.73 m2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



