
Submitted:
27 November 2023
Posted:
29 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID-19 pandemic caused by the virus SARS-CoV-2 has been the greatest global threat to human health in the last three years. The most widely used methodologies for the diagnosis of COVID-19 are quantitative reverse transcription polymerase chain reaction (RT-qPCR), used to detect the viral genome, and rapid antigen tests (RATs), also known as lateral flow tests, that employ specific antibodies to detect the viral spike protein. While PCR is considered the gold standard test in terms of specificity and sensitivity, it is time-consuming and requires specialized instrumentation operated by skilled personnel. In contrast, RATs can be used in-home or at point-of care but are less sensitive leading to a higher rate of false negative results. An alternative methodology for the detection of virus is the use of electrochemical biosensors that employ disposable strip electrodes to which binding of viral components elicits a change in electrical signal. Laser-scribed graphene (LSG) electrodes are attractive candidates for use as biosensors electrodes suitable for SARS-COV-2 detection due to their low-cost, patternability and ease of fabrication. In this work, we describe the development of a biosensor for COVID-19 detection that exploits a split-ester bond ligase system (termed ‘EsterLigase’) for immobilization of a virus-specific nanobody to maintain the out-of-plane orientation of the probe to ensure the efficacy of the probe-target recognition process. An anti-spike VHH E nanobody, genetically fused with the EsterLigase domain, was used as the specific probe for the spike receptor-binding domain (SP-RBD) protein as the target. The recognition between the two was measured by the change in the charge transfer resistance, determined by fitting the electrochemical impedance spectroscopy (EIS) spectra. The developed LSG-based biosensor achieved a linear detection range for the SP-RBD from 15 pM to 150 nM with a sensitivity of 0.0866 [log(M)]-1 and a limit of detection (LOD) of 7.68 pM.
Keywords:
Laser-scribed graphene
; biosensor
; nanobody
; electrochemistry
; COVID-19
1. Introduction
In December 2019, a respiratory infectious disease later called COVID-19 was reported in Wuhan, China [1]. Caused by a novel coronavirus, SARS-CoV-2, its rapid spread via aerosol transmission has resulted in one of the largest pandemics in human history [2]. Fast and accurate diagnostic methods have proven indispensable in tracking spread of the virus during the pandemic and in the isolation of infected cases to mitigate the spread of virus and its impact on societies. Reverse transcription polymerase chain reaction (RT-PCR) tests were developed rapidly following the publication of the genome sequence of SARS-CoV-2. RT-PCR remains the most sensitive diagnostic method for COVID-19 with a limit of detection as low as 3.9 and 3.6 copies of genomic RNA [3]. A drawback of RT-PCR tests however is the requirement for specialized equipment and skilled operators limiting the execution of tests to laboratory environments for RNA extraction after transportation from point-of testing. However, it can only be conducted in a laboratory and requires highly skilled operators. As a result, a PCR test usually takes from 3 hours to 24 hours to be completed.
Serological antibody tests using enzyme-linked immunoassay (ELISA) are another option for COVID-19 diagnosis. These are commonly used as supplementary tests for negative RT-PCR cases. ELISA employs recombinant spike protein and nucleocapsid protein as the probes to detect antibodies of COVID-19 IgG/IgM. Zhang et al. used anti-human IgG-HRP (horseradish peroxidase) conjugated monoclonal antibody and anti-human IgM for IgG/IgM test [4]. The presence of IgM in the sample indicates a recent infection by COVID-19. The paper-based ELISA is a fast, cheap and portable diagnostic method that can be used for point-of-care tests [5]. However, this technology does not provide a quantitative analysis. The detection of antibodies is also not suitable for diagnosing the COVID-19 infection at an early stage, because both IgM and IgG can only be detected after 5 days of infection [4]. The sensitivity of ELISA for the combined IgM/IgG test is 87.3% which is relatively low compared to RT-PCT tests. [6] Another lateral flow technology for COVID detection is the rapid antigen test (RAT) methodology that is currently widely used for rapid COVID tests at home [7]. The mechanism of RAT is that a specimen analyte, containing SARS-CoV-2 nucleocapsid (N) antigen suspended in an assay buffer, is deposited in the sample well. The specimen sample diffuses to the conjugate pad where nucleocapsid (N) antigen binds with the AuNPs-modified anti-nucleocapsid protein antibody. This process results in a colour change of the test line due to the presence of AuNPs. The RAT test is quick and simple, however does not provide a quantitative result and is of limited sensitivity (CT value of 25 for the detection of COVID-19 [7]).
As RT-PCR, ELISA-based and RAT-based detection methods all have their inherent limitations, alternative methodologies continue to be developed for portable, rapid, accurate and sensitive detection of COVID-19 and similar infectious diseases. Biosensors based on field-effect transistors (FET)-based are an attractive option in clinical diagnosis, point-of-care testing and on-site detection [8]. Seo et al. developed a field-effect transistor (FET) biosensor for the rapid detection of COVID-19 virus where SARS-CoV-2 spike antibody (IgG) was anchored on the graphene channel as a probe for the detection of whole virus [9]. The LoD of this FET device was reported as 1 fg/mL for spike protein and 2.42×102 copies/mL for whole virus in clinical samples. Electrochemical biosensors are also attractive candidates for point-of-care COVID-19 tests since they are cost-efficient and easy to operate. Yakoh et al. reported a paper-based analytical device for diagnosing COVID-19 ePAD [10]. The working electrode was prepared from graphene oxide, followed by attachment of SARS-CoV-2 antibodies (both IgG and IgM) on the graphene oxide through EDC/NHS chemistry. The SARS-CoV-2 spike protein containing receptor-binding domain (SP-RBD) was captured by SARS-CoV-2 antibodies on the ePAD, and the signal was measured by means of square-wave voltammetry (SWV). This sensor provided a linear detection range toward the spike protein between 1–1000 ng/mL with a LOD of 0.11 ng/mL.
Laser Scribed Graphene (LSG) is made by direct laser scribing on a polymer substrate, such as polyimide (PI), leading to C-O, C-N and C=O bonds breaking and re-forming, with a release of CO2/NO2/H2O gases. The remaining aromatic carbons form graphene [11]. Compared to graphene FET sensor [9] and other disposable, paper-based electrochemical sensors [10,12,13], fabrication of laser-scribed graphene electrodes is simple and time- and cost-efficient, and the electrodes can be easily patterned; thus, the process is amenable to mass production. LSG electrodes have been employed in a number of applications, including supercapacitor [14], hydrogen evolution [15] and biosensor [16,17,18]. Beduk et al. reported a gold-coated LSG electrode for SARS-CoV-2 S1 spike protein detection where the LSG electrode surface was modified by electrochemically deposited AuNPs [19]. SARS-CoV-2 spike protein antibody was anchored onto the AuNPs. The biorecognition event was evaluated by differential pulse voltammetry (DPV) measurements in potassium ferri/ferrocyanide solution where the bulky spike protein bound to the electrode surface hindered the electron transfer process leading to a decrease in DPV current peak. The dynamic range was found to be in 10 - 75 ng/ml range, with LoD of 2.9 ng/ml.
Nanobodies are recombinant proteins that contain the variable domain of antibodies comprising a single heavy chain only, typically produced by camels, lammas and other members of the Family Cammelidae [20]. These single-chain antibody fragments are advantageous as they are highly stable with strong affinity for their cognate antigen. To the best of our knowledge, the only known biosensor using nanobody receptor for SARS-CoV-2 detection is an organic electrochemical transistor (OECT) reported by Guo et al. [21] which has not yet been applied to LSG-based biosensors. In their design of the nanobody-based OECT sensor, they employed a nanobody-SpyCatcher fusion protein as a probe for COVID-19 and MERS antigens detection. In the OECT-gate functionalization process, the 7-methoxycoumarin-4-acetic acid (MCA)-modified SpyTag peptide was immobilized on the 1,6-hexanedithiol self-assembly monolayer through a flexible linker on top of a gold gate electrode. The SpyCatcher domain could specifically bind to SpyTag peptide, through forming an isopeptide bond. The SpyCatcher domain was then fused with a spike-specific nanobody through a 12-nm-long flexible linker. This SpyTag/SpyCatcher system helped to control the orientation of the nanobody during immobilization and improved the sensor performance. Their devices provided fast readout, within 10 mins of incubation with clinic nasopharyngeal swab and saliva samples. The limit of detection for SARS-CoV-2 S1 protein (in universal transport medium) was 1.453 pg/mL.
In this study, a disposable, single-use electrochemical biosensor for the detection of SARS-CoV-2 spike protein was developed. The disposable laser-scribed graphene electrodes were produced and modified by immobilizing SARS-CoV-2 spike protein utilizing a split-ester bond ligase system [22]. Firstly, an ester bond peptide tag (E-Tag) was modified with a C-terminal pyrene and immobilized on the graphene electrode. A spike-specific VHH E nanobody [23] genetically fused with a C-terminal ester bond ligase (EsterLigase) was subsequently captured and covalently ligated to the electrode via a spontaneous autocatalytic ester bond formed between the E-Tag and EsterLigase. The presence of the SARS-CoV-2 spike protein containing receptor-binding domain (SP-RBD) was then measured by electrochemical impedance spectroscopy (EIS) in a buffer solution containing ferri/ferrocyanide (Fe[(CN)6]3-/4-) redox couple. A linear detection range from 15 pM to 150 nM, a LoD of 7.68 pM and excellent selectivity towards other potential viral proteins were demonstrated.
2. Materials and Methods
2.1. Materials and Chemicals
Potassium ferricyanide (III), potassium ferrocyanide (II) trihydrate and phosphate buffered saline (PBS) tablets were purchased from Sigma-Aldrich. 4-(Pyren-1yl) butanoic acid (PBA) and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from AK Scientific Inc. SP-RBD, Influenza H3N2 and H1N1 were purchased from Thermo Fisher Scientific. The chemicals and materials used for EsterLigase-VHH E production pyrene-E-Tag synthesis is provided in Supporting Information S1.
2.2. EsterLigase-VHH E production
The split-Ester bond ligation system was identified and developed using previously described protocols [24]. Briefly, an intramolecular ester bond-containing domain was identified in a cell surface adhesin from Gemella bergeriae (ATCC 700627). The domain (residues 732-882, Uniprot U2Q1B9) was split at residue Asp 862 to produce a N-terminal EsterLigase and a C-terminal E-Tag that when mixed covalently ligate together through a spontaneous ‘intramolecular’ ester bond as the domain reassembles.
The VHH E nanobody [23] EsterLigase fusion protein (Figure S1 A) was synthesized as a gBlock (GenScript), cloned into the expression vector pProExHta with In-Fusion® cloning and transformed into chemically competent Stellar™ E. coli cells (Clontech). A single colony was cultured overnight in 10 mL of 2×YT medium at 30°C and transferred to 2 L baffled culture flasks containing 1 L of 2×YT medium supplemented to 0.1 mg/mL ampicillin. Cultures were grown at 30° with shaking to an OD600 of ~0.8 before induction with 0.3 mM isopropyl-β-D-1-thiogalctopyranoside (IPTG). The culture was transferred to 18°C and incubated for 16 h before harvesting by centrifugation. The cell pellet was re-suspended in 20 mL lysis buffer (50 mM Tris.Cl pH 8.0, 500 mM NaCl, 10 mM imidazole, 0.5 mM TCEP) supplemented with complete protease inhibitor cocktail mini tablets (EDTA free; Roche), and transferred to 50 mL falcon tubes and stored at -20°C.
Cell pellets were lysed using a cell disruptor (Constant Cell Disruption Systems) at 124 kPa and the lysate clarified by centrifugation at 30,000 ×g for 20 min at 4°C. The soluble protein was applied to a 5 mL IMAC column (HiTrap), bound VHH E nanobody EsterLigase protein washed with wash buffer (50 mM Tris.Cl pH 8.0, 300 mM NaCl, 20 mM imidazole) and eluted in a linear gradient with elution buffer (50 mM Tris.Cl pH 8.0, 300 mM NaCl, 500 mM imidazole). The N-terminal vector derived polyhistidine tag was cleaved from VHH E nanobody EsterLigase protein with a 1:100 ratio of recombinant Tobacco Etch Virus (rTEV) protease to recombinant protein, and concurrently dialyzed against 1 L buffer (10 mM Tris-Cl [pH 8.0], 100 mM NaCl, 1 mM β-mercaptoethanol) at 4°C for 16 h. Cleaved VHH E nanobody EsterLigase protein was separated from the rTEV-His6 protease by subtractive IMAC. The unbound protein containing cleaved VHH E nanobody EsterLigase protein was collected and concentrated using a 30-kDa-molecular-weight-cutoff (MWCO) protein concentrator (VivaScience) and purified by size exclusion chromatography on a Superdex S200 10/300 column (GE Healthcare). Purified VHH E nanobody EsterLigase eluted as a single peak and was judged to be approximately >99% pure, as inferred by SDS-PAGE analysis.
2.3. Synthesis of pyrene-E-Tag
The E-Tag peptide (Figure S1B) was prepared by Fmoc solid-phase peptide synthesis (Fmoc-SPPS) [25], with coupling reactions performed under microwave irradiation for 5 min at 70 oC using a Biotage Biotage® Initiator+ Alstra™ (Uppsala, Sweden). All other chemical manipulations were undertaken at room temperature. Briefly, the peptide was prepared as the corresponding C-terminal amide derivative using Fmoc-Rink amide linker with TentaGel S NH2 resin (0.23 mmol/g, Rapp Polymere, Tubingen, Germany). The E-Tag compliment peptide sequence was modified to contain a GSGSGAKG spacer sequence at its C-terminus. The Lysine residue of this sequence was incorporated with orthogonal side chain protection as Fmoc-Lys(Dde)-OH. Upon completion of the linear peptide sequence, the N-terminal Fmoc group was exchanged for Boc protection on-resin by treatment with Boc2O in DMF [26], followed by removal of the Dde protecting group from the Nε amino group of the most C-terminal lysine with a 2% hydrazine solution in DMF [27]. To the free amino group was coupled 4-(pyren-1-yl)butanoic acid (5 equiv., AK Scientific, Union City, CA, USA) with HATU (4.8 equiv.) for 30 min at rt. Upon resin cleavage the crude peptide was purified by RP-HPLC to afford the title compound in 25.6 % overall yield (based on resin loading) in 95.5% purity (Figure S2). The identity of the pyrene-E-Tag was confirmed by mass spectrometry (Figure S3).
2.4. Fabrication of LSG electrodes
The laser-scribed graphene (LSG) electrodes on the polyimide (PI) sheet were used as sensing strips for the impedimetric detection of SP-RBD. The LSG electrodes were fabricated using a Universal Laser system (laser cutter VLS3.50) equipped with a CO2 laser. The fabrication method and optimized laser parameters were previously reported by Zhu et al. [17]. In short, the wavelength of the laser was set to 10.6 μm, the optimized lens substrate distance was 5.1 cm, the laser speed was 0.45 cm/s, the power was 2.7 W and pulses per inch (PPI) was 1000. The laser scribing process was conducted in a vacuum chamber.
Each LSG sensing strip contained an LSG working electrode (WE), an LSG counter electrode (CE) and an LSG electrode with Ag paste painted and dried in an oven at 60 °C for 30 mins as the reference electrode (RE). The diameter of the working electrode was 2 mm.
The fabricated LSG electrodes were characterized by scanning electron microscopy (SEM) using a Philip ML30S FEG Scanning Electron Microscope. The LSG electrodes were sputter-coated with 20 nm gold before imaging.
2.5. Functionalization of LSG electrodes
A stock solution (500 μM) of 4-(pyren-1yl) butanoic acid (PBA) was made by dissolving 4.3 mg of BPA in 30 ml of ethanol with sonication. This PBA solution was diluted 100 times with ethanol and then 10 times with PBS to obtain 500 nM of PBA in a mixture of PBS and ethanol (9:1 vol:vol). A 500 nM solution of the pyrene-E-Tag linker was prepared in PBS buffer. To maintain the space between pyrene-E-Tag linkers and EsterLigase-nanobody, 10 μL of 500 nM PBA solution, 10 μL of 500 nM pyrene-E-Tag linker PBS solution, and 80 μL PBS buffer were mixed, providing a final solution containing 50 nM pyrene-E-Tag linker and 50 nM PBA. The LSG working electrode was first incubated with 20 μL of 50 nM pyrene-E-Tag linker/PBA (1:1 mol/mol, 50 nM each) mixture for 3 hours at room temperature and then washed with PBS buffer to remove the unbound pyrene-E-Tag linker and PBA (referreferred here as “LSG/PBA-pyrene-E-Tag linker”). The LSG/PBA-pyrene-E-Tag linker was incubated with 20 μL of 2.5 μM EsterLigase-nanobody in 50 mM HEPES solution containing 20 vol% glycerol overnight at 4 °C in the fridge; the resulting modified electrode is referred here as “LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody”. Glycerol was used as it was shown that it can facilitate the ligation reaction by stabilizing nanobody structure and preventing aggregation [28]. The electrode was washed with PBS buffer 3 times to remove the unreacted nanobody before the detection of SP-RBD.
2.6. SP-RBD detection by electrochemical impedance spectroscopy (EIS)
The LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody was incubated with 20 μL of SP-RBD of various concentrations in PBS solution for 30 mins at room temperature. After incubation with SP-RBD, LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody was then washed with PBS buffer thoroughly. The EIS was carried out using PalmSens 4. The EIS measurements were performed in PBS buffer, containing 5 mM Fe[(CN)6]3-/4- (1:1 mol:mol) as a redox couple, with a bias potential of 0.14 V (vs. Ag reference electrode) and an amplitude of 10 mV in a frequency range from 100 kHz to 0.1 Hz.
2.7. Fabrication of low-dimensional graphene and AFM characterization of surface modification
To construct a flat 2D graphene surface for AFM measurements, a graphene oxide (GO) film was prepared on 200 nm SiO2/Si substrate using an electrospraying technique. The solution consisted of 5 mg/mL graphene oxide in ethanol/propylene carbonate with a ratio of 4:3:3 (GO: ethanol: propylene carbonate). The flow rate, potential and height for the electrospraying of GO wet film were 90 μL/h, 3.3 kV and 15 mm, respectively. The GO film was then treated with vacuum-flash assisted evaporation for 10 minutes and heated up to 120°C for 30 minutes to remove all solution on the surface. After the preparation of GO film, a vapour phase reduction was carried out for 8 h at 100 ºC, where a mixture solution of HI/AcOH (1:1) was used as a reducing agent. The rGO film was annealed at 100 °C for 2 h to stabilize the binding between rGO film and SiO2/Si substrate.
The 2D graphene surface was then modified by first treating the surface with 20 μL of 2.5 nM pyrene-E-Tag linker in PBS solution for 3 h at room temperature. After washing with PBS buffer, the graphene surface was incubated with 20 μL of 5 μM EsterLigase-nanobody in HEPES solution containing 20 vol % glycerol and stored overnight at 4 °C in the fridge. 20 μL of 1.5 nM SP-RBD was added to the surface and waited for 30 mins incubation before AFM measurements. The AFM images were taken on separate graphene chips for each step of the modification process.
The AFM imaging of the graphene surface was conducted on a Cypher ES AFM (Asylum Research) in tapping mode using a silicon tip. The resonance frequency of the tip was 150 kHz. The samples were transferred to a 15 mm-diameter AFM specimen disc using double sided tape.
2.8. FT-IR characterization of LSG electrode surface modification with pyrene-E-Tag linker
The Attenuated total reflectance (ATR)-FTIR characterization was carried out by an FTIR Bruker Vertex 70 spectrometer. The bare LSG electrode was modified with 20 μL of 250 nM pyrene-E-Tag linker in PBS solution for 3 h and then washed with PBS buffer for 3 times before FT-IR measurement.
3. Results and discussion
3.1. Design of LSG-based electrochemical biosensor for the detection of SARS-CoV-2 spike protein RBD
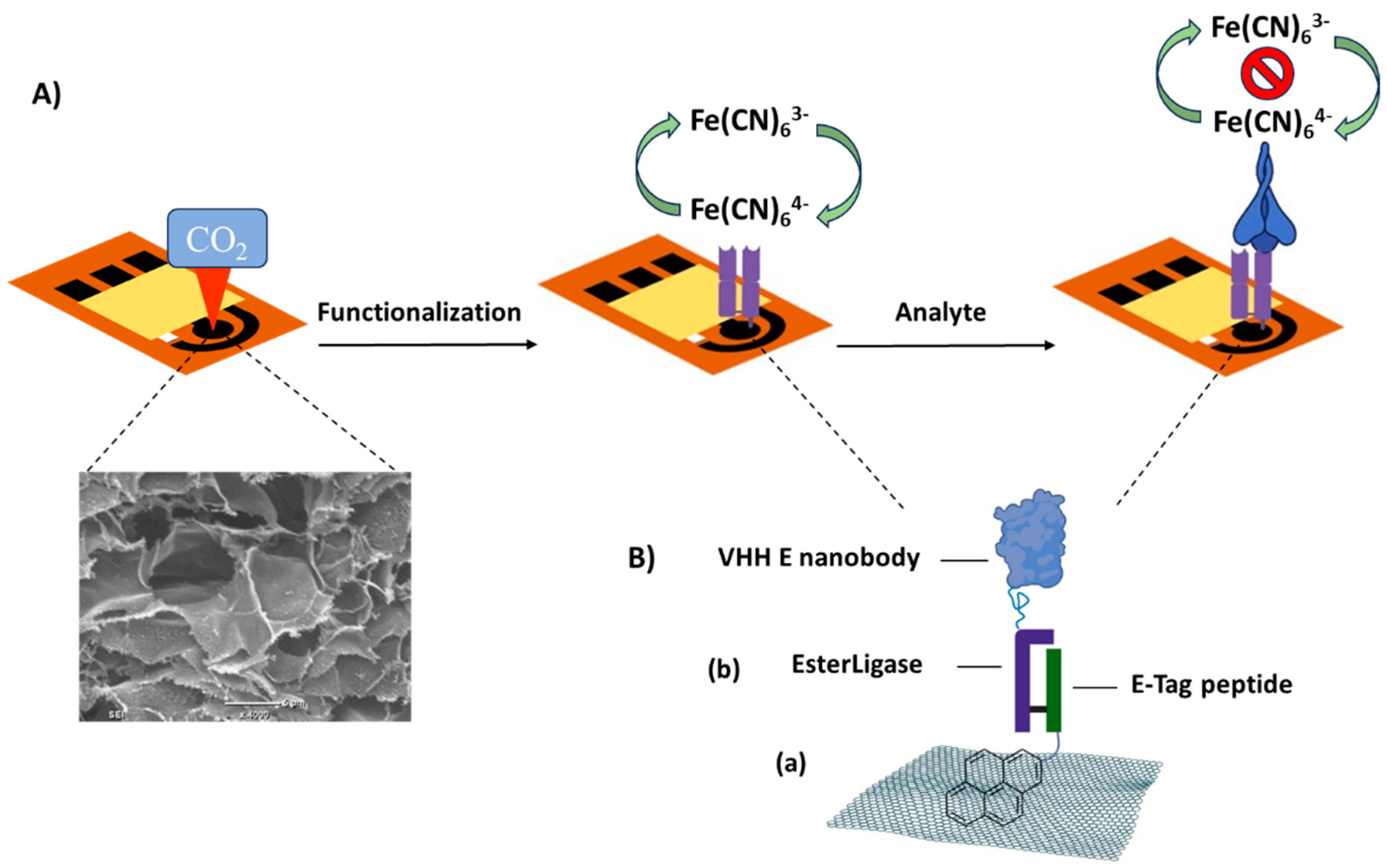
A schematic of the LSG-based biosensor design for the detection of SP-RBD is presented in Figure 1A. The pyrene-E-Tag linker and PBA spacer were assembled onto the laser-scribed graphene (LSG) surface via п-п stacking [29]. The pyrene-E-Tag linker can be specifically recognized by EsterLigase that was fused with the anti-spike VHH E nanobody. EsterLigase can spontaneously form an easter bond with E-Tag peptide, formed between Thr of the EsterLigase and Gln of E-tag side chains [22], thereby modifying the working electrode with the nanobody and preserving the vertical orientation of the nanobody. The anti-spike VHH E nanobody, one of the variable domains of heavy-chain-only antibodies (VHHs) derived from llama [23], acted as the bio-recognition element for the detection of SP-RBD (Figure 1B). The biorecognition between the target and the nanobody is similar to antigen and antibody recognition; however, the nanobody is much smaller than an antibody [23]. The binding between the pyrene-E-Tag linker and the nanobody was validated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Figure S4). The result indicated the formation of a larger species after mixing the anti-spike VHH E nanobody with the pyrene-E-Tag linker, confirming the successful binding of EsterLigase-nanobody to pyrene-E-Tag linker.
Figure 1.
(A) Scheme of LSG-based SARS-CoV-2 spike RBD protein biosensor for the detection of SP-RBD. (B) Surface modification of the LSG electrode: (a) the immobilisation of pyrene-modified E-Tag peptide to the LSG surface via п- п stacking. (b) The conjugation between EsterLigase and E-Tag through the formation of an ester bond. The mechanism of conjugation was exploited by Paul et al. [22].
Figure 1.
(A) Scheme of LSG-based SARS-CoV-2 spike RBD protein biosensor for the detection of SP-RBD. (B) Surface modification of the LSG electrode: (a) the immobilisation of pyrene-modified E-Tag peptide to the LSG surface via п- п stacking. (b) The conjugation between EsterLigase and E-Tag through the formation of an ester bond. The mechanism of conjugation was exploited by Paul et al. [22].
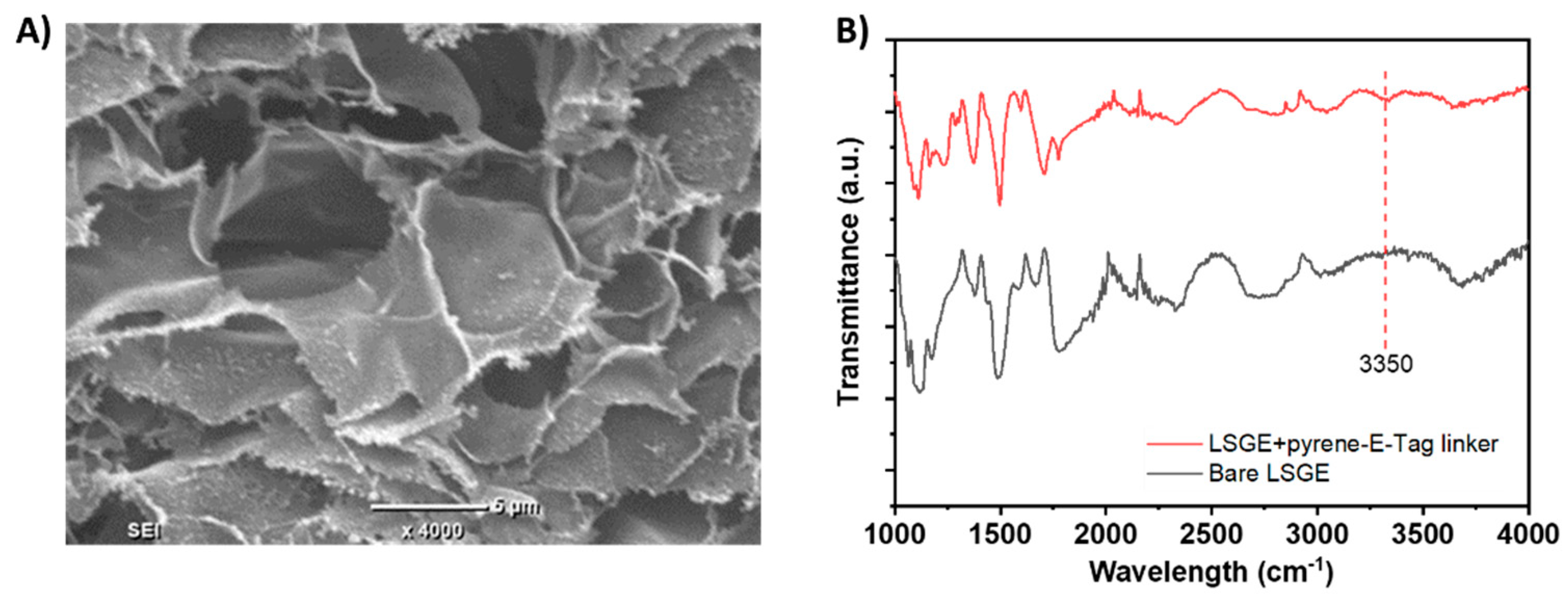
The morphology of the bare LSG electrodes was characterized by SEM imaging (Figure 2A). The highly porous structure of the LSG electrode can be observed, which indicates a high surface area for surface functionalization [17]. FTIR spectrum of the LSG electrode before and after the modification with the pyrene-E-Tag linker (Figure 2B) confirms the successful attachment of pyrene-E-Tag linker on the LSG surface. A new peak around 3350 cm-1, corresponding to the N-H stretch from the secondary amine within pyrene-E-Tag linker, was observed after the modification.
Figure 2.
(A) SEM image for bare LSG electrode. (B) FTIR spectra of bare LSG electrode and LSG electrode functionalized with pyrene-E-Tag linker.
Figure 2.
(A) SEM image for bare LSG electrode. (B) FTIR spectra of bare LSG electrode and LSG electrode functionalized with pyrene-E-Tag linker.
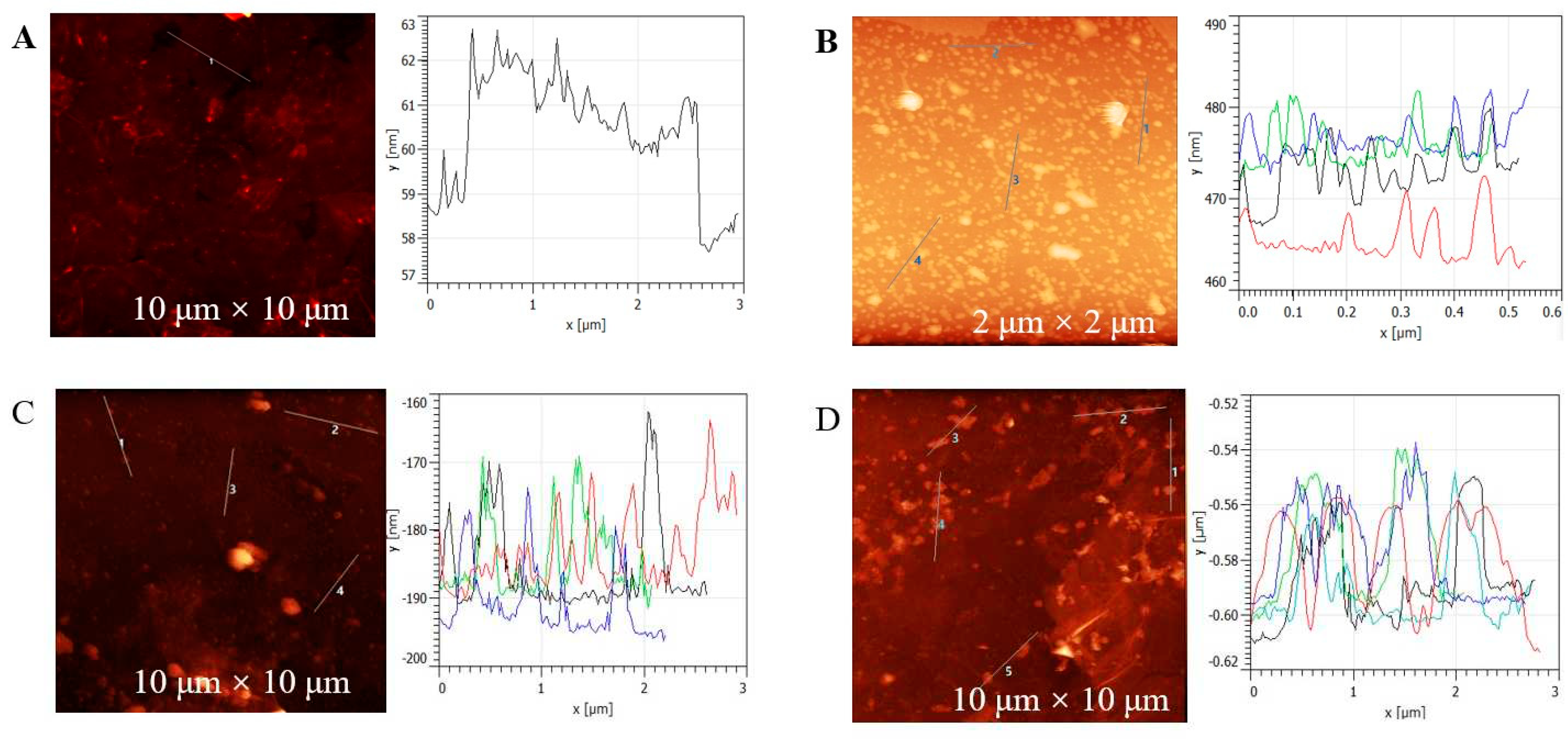
AFM was employed to characterise the functionalisation of the working electrode. Since the porous LSG was not ideal for AFM studies, a low-dimensional graphene surface was fabricated for AFM measurements. The bare graphene layer had a thickness of 3 ± 0.5 nm (Figure 3A), while the pyrene-E-Tag linker functionalized graphene had a thickness of ~5 nm (Figure 3B). After the conjugation of EsterLigase-nanobody, the thickness increased to 10-15 nm, where the thickness of EsterLigase-nanobody was ca. 5-10 nm, calculated by subtracting the thickness of pyrene-E-Tag linker (Figure 3C). The thickness of SP-RBD was estimated as 25-30 nm in Figures 3D. The increment in thickness from 3 nm for bare graphene to ~40 nm after all steps of modification indicates a successful functionalization of the graphene surface.
Figure 3.
AFM images for each step of the graphene surface modification: (A) Bare graphene surface. Graphene surface modified with (B) pyrene-E-Tag linker, (C) pyrene-E-Tag linker/EsterLigase-nanobody, and (D) after incubation with 1.5 nM of SP-RBD for 30 mins.
Figure 3.
AFM images for each step of the graphene surface modification: (A) Bare graphene surface. Graphene surface modified with (B) pyrene-E-Tag linker, (C) pyrene-E-Tag linker/EsterLigase-nanobody, and (D) after incubation with 1.5 nM of SP-RBD for 30 mins.
3.2. Feasibility of SARS-CoV-2 sensing strip to detect SP-RBD protein
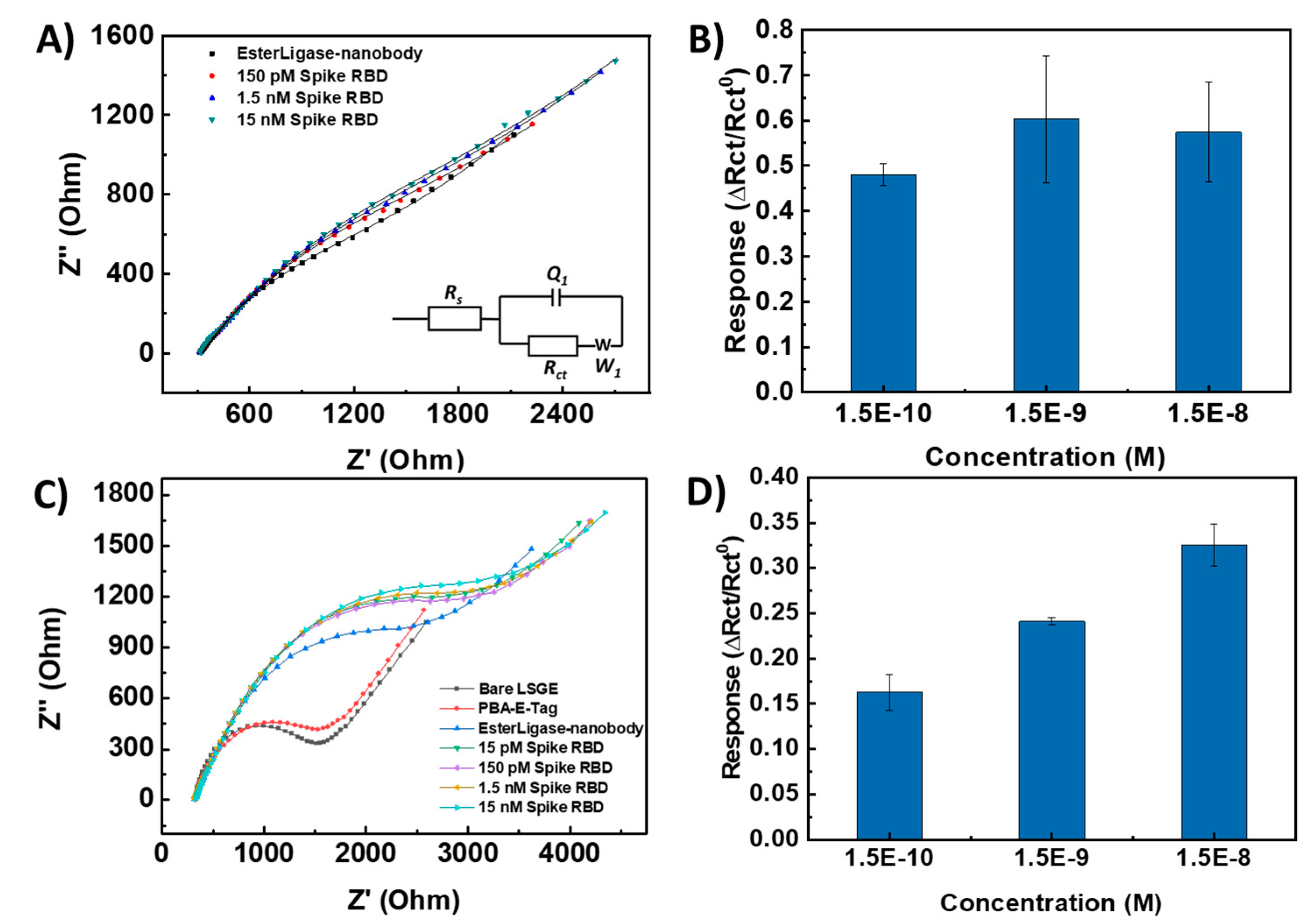
After confirming the successful functionalization of the EsterLigase-nanobody onto the LSG electrode, the binding between the anti-spike VHH E nanobody and SP-RBD was investigated by EIS, as shown in Figure 4. The Randle’s equivalent circuit model was used to fit the spectra (Figure 4A inset). It consisted of solution resistance (Rs), charge transfer resistance (Rct), constant phase element (Q1) and Warburg resistance (W1). Normalized change in charge transfer resistance (∆Rct/Rct0) before and after the detection of SP-RBD was used as the sensor response, where Rct0 represents the charge transfer resistance of LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody. After the detection of SP-RBD, an increase in Rct was observed due to the blockage of Fe[(CN)6]3-/4- to access the LSG/PBA-E-Tag /EsterLigase-nanobody.
The necessity of the use of 1-pyrene butyric acid (PBA) to construct a successful sensing platform was investigated. For LSG/pyrene-E-Tag linker/EsterLigase-nanobody sensors where PBA was not used, the semicircle of the Nyquist plot (representing the charge transfer process on the LSG surface [30]) is not obvious (Figure 4A), with the ∆Rct/Rct0 signal presented in Figure 4B. The signal only slightly increased from 0.48 for 150 pM SP-RBD to 0.65 for 15 nM SP-RBD. The large semicircles indicate a slow charge transfer kinetics due to a high blockage of highly dense EsterLigase-nanobody on the LSG surface. The standard deviations (error bars) after detection of SP-RBD on the LSG/pyrene-E-Tag linker/EsterLigase-nanobody electrode was large (Figure 4B), indicating the poor repeatability of the sensor response from independent sensor strips. To improve the repeatability, PBA, which also contains a pyrene group for self-assembling onto the LSG surface through п-п stacking, was mixed with the pyrene-E-Tag linker (1:1 mol:mol, 50 nM each) in order to space out the EsterLigase-nanobodies on the LSG surface and to improve their orientation. When a mixture of PBA and pyrene-E-Tag linker (1:1 mol:mol, 50 nM each) was used for the LSG surface modification, the semicircle in the Nyquist plot of EIS (Figure 4C) is more obvious, which indicates a stronger and faster charge transfer reaction of Fe[(CN)6]3-/4- on the LSG surface. The sensor responses, ∆Rct/Rct0, were from 0.16 to 0.31 after the detection of 150 pM to 15 nM SP-RBD (Figure 4D). The value of sensing response (∆Rct/Rct0) of LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody was smaller (∆Rct/Rct0=0.31) after the detection of 15 nM SP-RBD, compared with LSG/pyrene-E-Tag linker/EsterLigase-nanobody without PBA (∆Rct/Rct0=0.65). We suggest these results from the smaller amount of anti-spike VHH E nanobodies on the LSG surface when the mixed pyrene-E-Tag linker and PBA monolayer was assembled on the surface.
Figure 4.
(A) Nyquist plot of LSG/pyrene-E-Tag linker/EsterLigase-nanobody electrode after the detection of SP-RBD in concentrations from 150 pM to 15 nM. (B) Normalised response, ∆Rct/Rct0, of LSG/pyrene-E-Tag linker/EsterLigase-nanobody for the detection of SP-RBD from 150 pM to 15 nM. (C) Nyquist plot of LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody electrode after the detection of SP-RBD detection from 150 pM to 15 nM. (D) ∆Rct/Rct0 of LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody after the detection of SP-RBD protein from 150 pM to 15 nM. The sensitivity has been improved after using PBA for modification. (Error bars were obtained from the standard deviations of 3 independent experiments.)
Figure 4.
(A) Nyquist plot of LSG/pyrene-E-Tag linker/EsterLigase-nanobody electrode after the detection of SP-RBD in concentrations from 150 pM to 15 nM. (B) Normalised response, ∆Rct/Rct0, of LSG/pyrene-E-Tag linker/EsterLigase-nanobody for the detection of SP-RBD from 150 pM to 15 nM. (C) Nyquist plot of LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody electrode after the detection of SP-RBD detection from 150 pM to 15 nM. (D) ∆Rct/Rct0 of LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody after the detection of SP-RBD protein from 150 pM to 15 nM. The sensitivity has been improved after using PBA for modification. (Error bars were obtained from the standard deviations of 3 independent experiments.)
The control experiments were carried out by functionalizing LSG working electrode in the absence of either the pyrene-E-Tag linker or EsterLigase-nanobody, named LSG/PBA-pyrene-E-Tag linker and LSG/PBA/EsterLigase-nanobody. The LSG/PBA-pyrene-E-Tag linker and LSG/PBA/EsterLigase-nanobody electrode strips were applied for the detection of 1.5 nM SP-RBD, as shown in Figures 5A and B. The LSG/PBA-pyrene-E-Tag linker electrode strip had a ∆Rct/Rct0 of 0.0080±0.0004 (Figure 5C), which was negligible when compared with the ∆Rct/Rct0 of 0.240±0.008 obtained from LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody, indicating a specificity between EsterLigase-nanobody and SP-RBD. A ∆Rct/Rct0 of 0.0058±0.0029 was obtained from the LSG/PBA/EsterLigase-nanobody, which was much smaller than that from LSG/PBA-pyrene-E-Tag linker/SpyCatcer-nanobody, again demonstrating the specific binding between pyrene-E-Tag linker and EsterLigase-nanobody. The Rct increased after the incubation of LSG/PBA with EsterLigase-nanobody (Figure 5B), indicating some non-specific adsorption of EsterLigase-nanobody on the LSG electrode. However, the sensor response was negligible, demonstrating the pyrene-E-Tag linker’s role in facilitating the orientation of EsterLigase-nanobody for the binding of SP-RBD and the importance of the EsterLigase-nanobody orientation on the sensor’s surface.
3.2. Sensing performance of the SARS-CoV-2 sensing strip
The sensing responses of the sensor to 5 different concentrations of SP- RBD from 15 pM to 150 nM were measured on three independent sensors. A control sensor was functionalized with PBA only instead of the mixture of PBA and pyrene-E-Tag linker and exposed to 1.5 nM of SP-RBD. The obtained dose-response curve, shown in Figure 5D, indicates a linear detection range between 15 pM to 150 nM for SP-RBD, with a LoD of 7.68 pM (3 x S/N).
To investigate the specificity of the developed LSG/PBA-pyrene-E-Tag linker/ EsterLigase-nanobody sensing strip towards the detection of SP-RBD, the sensor strip was exposed to H1N1 and H3N2 influenza spike proteins as interfering targets. The results (Figure 5E) suggest that there is no binding between the influenza spike protein and EsterLigase-nanobody, with ∆Rct/Rct0 of 0.120±0.003 and 0.120±0.016 (n=3) after exposure to 1.5 nM of either H1N1 or H3N2 spike proteins, respectively. These values are much smaller than the response to 1.5 nM of SP-RBD (∆Rct/Rct0 = 0.240±0.008) and similar to the change in the signal when the sensor surface is not functionalized with the probe at all.
Figure 5.
Nyquist plot of (A) LSG/PBA/EsterLigase-nanobody and (B) LSG/ LSG/PBA-pyrene-E-Tag linker after exposure to 1.5 nM SP-RBD. (C) Normalized responses (∆Rct/Rct0) from (A) and (B). ∆Rct/Rct0 to 1.5 nM SP-RBD as a positive given for comparison. (D) Calibration curve of the LSG/PBA-pyrene-E-Tag linker/ EsterLigase-nanobody sensor (∆Rct/Rct0) for the detection of SARS-CoV-2 SP- RBD at different concentrations from 15 pM to 150 nM (n= 3) and the control LSG electrode strip with the modification with only PBA after the detection of 1.5 nM of SP-RBD. (E) LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody sensor responses, ∆Rct/Rct0, after exposure to H1N1 and H3N2 influenza spike proteins (n=3), as controls. ∆Rct/Rct0 to 1.5 nM SP-RBD as a positive given for comparison. (Error bars were obtained from the standard deviations of 3 independent experiments.)
Figure 5.
Nyquist plot of (A) LSG/PBA/EsterLigase-nanobody and (B) LSG/ LSG/PBA-pyrene-E-Tag linker after exposure to 1.5 nM SP-RBD. (C) Normalized responses (∆Rct/Rct0) from (A) and (B). ∆Rct/Rct0 to 1.5 nM SP-RBD as a positive given for comparison. (D) Calibration curve of the LSG/PBA-pyrene-E-Tag linker/ EsterLigase-nanobody sensor (∆Rct/Rct0) for the detection of SARS-CoV-2 SP- RBD at different concentrations from 15 pM to 150 nM (n= 3) and the control LSG electrode strip with the modification with only PBA after the detection of 1.5 nM of SP-RBD. (E) LSG/PBA-pyrene-E-Tag linker/EsterLigase-nanobody sensor responses, ∆Rct/Rct0, after exposure to H1N1 and H3N2 influenza spike proteins (n=3), as controls. ∆Rct/Rct0 to 1.5 nM SP-RBD as a positive given for comparison. (Error bars were obtained from the standard deviations of 3 independent experiments.)
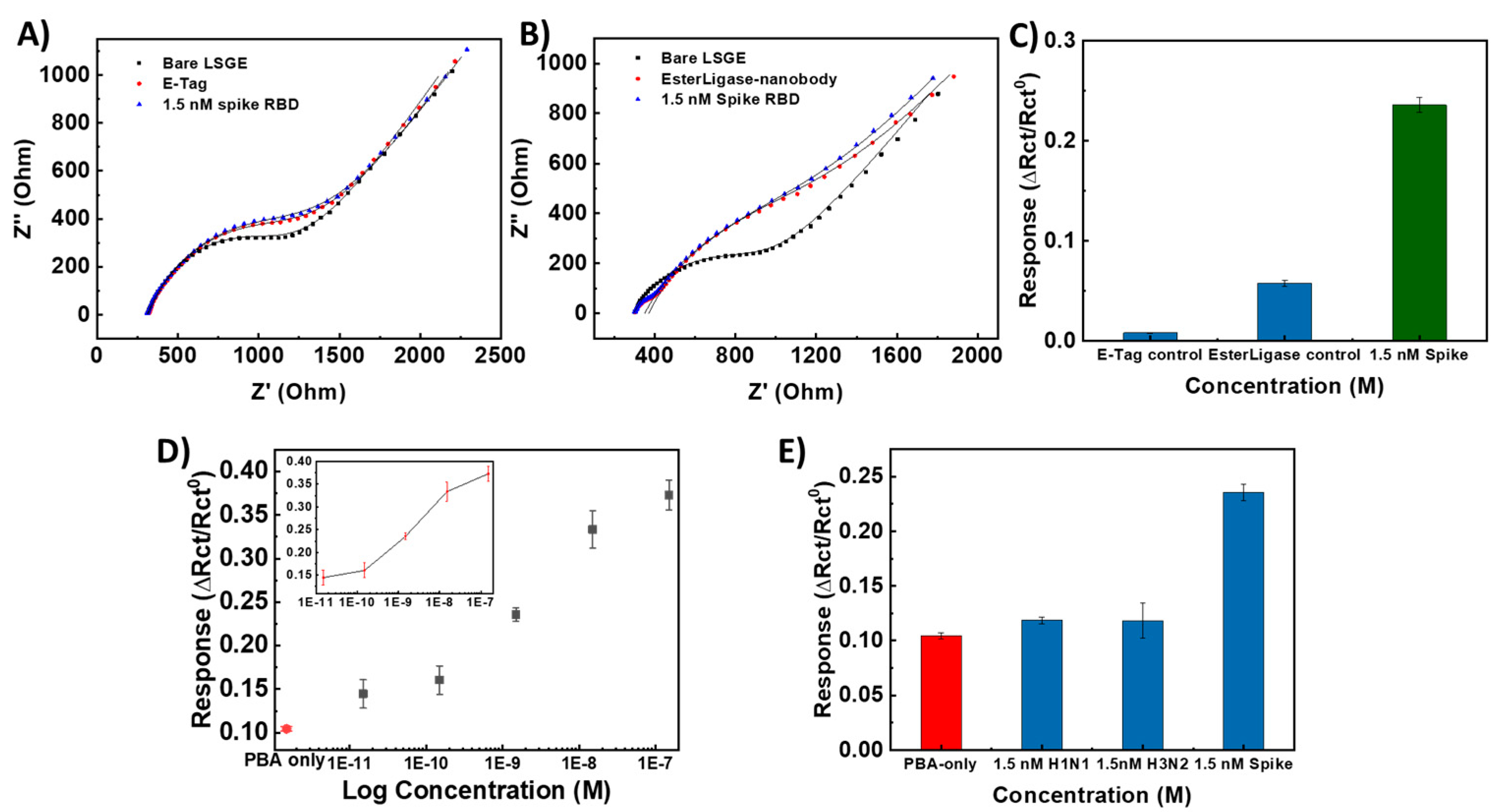
Table 1 summarizes the sensing responses from comparable electrochemical biosensors for SARS-CoV-2 detection. Compared to the reported sensors, such as LSG/AuNS electrodes that require 1h for target incubation [19], our design can provide a faster readout after 30 min of incubation and ca. 3 mins for EIS measurement, with a lower LoD of 7.68 pM, compared to 116 nM [19]. The LSG electrode strip in this work is portable and disposable, which could be applied for point-of-care detection [13,19] and be convenient to use. Our disposable sensing strips platform has the potential for portable, point-of-care diagnostic applications.

Table 1.
Comparison with various electrochemical biosensor.
| Technique | Analyte | Linear range | LoD | Ref. |
| μPAD, EIS | IgG | 10-1000 ng/mL | 0.4 pg/mL | [12] |
| AuNPs modified Paper electrodes | N-protein | 585 - 5.854 × 107 copies/μL | 6.9 copies/μL | [13] |
| LSG/AuNS electrods,DPV | S-protein RBD | 5-500 ng/mL | 2.9 ng/mL | [19] |
| Co-TNTs | S-protein RBD | 14-1400 nM | 0.7 nM | [31] |
| 3D nanoprinting rGO, EIS | S-protein RBD | 1.0 fM-1 nM | 16.9 fM | [32] |
| LSG, EIS | S-protein RBD | 15 pM-150 nM | 7.68 pM | This work |
4. Conclusions
This study describes a disposable, portable and electrochemical LSG-based sensor based on a bio-recognition system with pyrene-E-Tag linker and EsterLigase-nanobody for the detection of SP-RBD. The developed sensor provides a linear detection range from 15 pM to 150 nM towards the detection of SP-RBD, with a LoD of 7.68 pM and excellent selectivity towards other potential viral proteins. The developed sensor can provide the rapid readout within 30 min. This developed sensor has the potential for rapid, sensitive, selective and point-of-care diagnosis of SARS-CoV-2.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, R.W., B.Z. and J.T.-S.; methodology, R.W., B.Z., P.Y., Y.L., A.J.C., C.S.; validation, formal analysis, investigation, R.W.; writing—original draft preparation, R.W and B.Z.; writing—review and editing, P.Y., A.J.C., J.T., J.T.-S.; supervision, B.Z., and J.T.-S.; project administration, B. Z., J.T.-S.; funding acquisition, J.T.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Authors would like to than Health Research Council (HRC) for the financial support, grant number 3723813.
Data Availability Statement
Data is available upon requirement.
Acknowledgments
Authors would like to thank Health Research Council (HRC) for the financial support (3723813).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N Engl J Med 2020, 382, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, R.H.; Li, B.; Zheng, X.S.; Yang, X.L.; Hu, B.; Wang, Y.Y.; Xiao, G.F.; Yan, B.; Shi, Z.L.; Zhou, P. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect 2020, 9, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Huang, Z.; Pan, P.; Pan, Y.; Zuo, R.; Sun, Y.; Liu, X. A Nitrocellulose Paper-Based Multi-Well Plate for Point-of-Care ELISA. Micromachines (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Yan, M.; Li, H.; Liu, T.; Lin, C.; Huang, S.; Shen, C. Evaluation of Enzyme-Linked Immunoassay and Colloidal Gold-Immunochromatographic Assay Kit for Detection of Novel Coronavirus (SARS-Cov-2) Causing an Outbreak of Pneumonia (COVID-19). medRxiv, 2002; 2020.2002.2027.20028787. [Google Scholar] [CrossRef]
- Frew, E.; Roberts, D.; Barry, S.; Holden, M.; Restell Mand, A.; Mitsock, E.; Tan, E.; Yu, W.; Skog, J. A SARS-CoV-2 antigen rapid diagnostic test for resource limited settings. Sci. Rep. 2021, 11, 23009. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Wang, Q.; Xiao, M.; Zhong, D.; Sun, W.; Zhang, G.; Zhang, Z. Ultrasensitive Monolayer MoS2 Field-Effect Transistor Based DNA Sensors for Screening of Down Syndrome. Nano Lett. 2019, 19, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.; Lee, G.; Kim, M.J.; Baek, S.-H.; Choi, M.; Ku, K.B.; Lee, C.-S.; Jun, S.; Park, D.; Kim, H.G.; et al. Rapid Detection of COVID-19 Causative Virus (SARS-CoV-2) in Human Nasopharyngeal Swab Specimens Using Field-Effect Transistor-Based Biosensor. ACS Nano 2020, 14, 5135–5142. [Google Scholar] [CrossRef] [PubMed]
- Yakoh, A.; Pimpitak, U.; Rengpipat, S.; Hirankarn, N.; Chailapakul, O.; Chaiyo, S. Paper-based electrochemical biosensor for diagnosing COVID-19: Detection of SARS-CoV-2 antibodies and antigen. Biosens. Bioelectron. 2021, 176, 112912. [Google Scholar] [CrossRef] [PubMed]
- Beduk, T.; Ait Lahcen, A.; Tashkandi, N.; Salama, K.N. One-step electrosynthesized molecularly imprinted polymer on laser scribed graphene bisphenol a sensor. Sens. Actuators B Chem. 2020, 314, 128026. [Google Scholar] [CrossRef]
- Li, X.; Qin, Z.; Fu, H.; Li, T.; Peng, R.; Li, Z.; Rini, J.M.; Liu, X. Enhancing the performance of paper-based electrochemical impedance spectroscopy nanobiosensors: An experimental approach. Biosens. Bioelectron. 2021, 177, 112672. [Google Scholar] [CrossRef] [PubMed]
- Alafeef, M.; Dighe, K.; Moitra, P.; Pan, D. Rapid, Ultrasensitive, and Quantitative Detection of SARS-CoV-2 Using Antisense Oligonucleotides Directed Electrochemical Biosensor Chip. ACS Nano 2020, 14, 17028–17045. [Google Scholar] [CrossRef] [PubMed]
- El-Kady, M.F.; Strong, V.; Dubin, S.; Kaner, R.B. Laser Scribing of High-Performance and Flexible Graphene-Based Electrochemical Capacitors. Science 2012, 335, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Nayak, P.; Jiang, Q.; Kurra, N.; Wang, X.; Buttner, U.; Alshareef, H.N. Monolithic laser scribed graphene scaffolds with atomic layer deposited platinum for the hydrogen evolution reaction. J. Mater. Chem. A. 2017, 5, 20422–20427. [Google Scholar] [CrossRef]
- Xu, G.; Jarjes, Z.A.; Wang, H.-W.; Phillips, A.R.J.; Kilmartin, P.A.; Travas-Sejdic, J. Detection of Neurotransmitters by Three-Dimensional Laser-Scribed Graphene Grass Electrodes. ACS Appl. Mater. Interfaces 2018, 10, 42136–42145. [Google Scholar] [CrossRef]
- Chang, Z.; Zhu, B.; Liu, J.; Zhu, X.; Xu, M.; Travas-Sejdic, J. Electrochemical aptasensor for 17β-estradiol using disposable laser scribed graphene electrodes. Biosens. Bioelectron. 2021, 185, 113247. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Yu, L.; Beikzadeh, S.; Zhang, S.; Zhang, P.; Wang, L.; Travas-Sejdic, J. Disposable and portable gold nanoparticles modified - laser-scribed graphene sensing strips for electrochemical, non-enzymatic detection of glucose. Electrochim. Acta 2021, 378, 138132. [Google Scholar] [CrossRef]
- Beduk, T.; Beduk, D.; de Oliveira Filho, J.I.; Zihnioglu, F.; Cicek, C.; Sertoz, R.; Arda, B.; Goksel, T.; Turhan, K.; Salama, K.N.; Timur, S. Rapid Point-of-Care COVID-19 Diagnosis with a Gold-Nanoarchitecture-Assisted Laser-Scribed Graphene Biosensor. Anal. Chem. 2021, 93, 8585–8594. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.K.; Odongo, S.; Radwanska, M.; Magez, S. Nanobodies: A Review of Generation, Diagnostics and Therapeutics. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Wustoni, S.; Koklu, A.; Díaz-Galicia, E.; Moser, M.; Hama, A.; Alqahtani, A.A.; Ahmad, A.N.; Alhamlan, F.S.; Shuaib, M.; et al. Rapid single-molecule detection of COVID-19 and MERS antigens via nanobody-functionalized organic electrochemical transistors. Nat. Biomed. Eng. 2021, 5, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Young, P.G.; Yosaatmadja, Y.; Harris, P.W.R.; Leung, I.K.H.; Baker, E.N.; Squire, C.J. Harnessing ester bond chemistry for protein ligation. Chem comm 2017, 53, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Koenig, P.A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371. [Google Scholar] [CrossRef] [PubMed]
- Young, P.G.; Squire, C.J. Molecular Superglues: Discovery and Engineering Orthogonalization. Methods Mol Biol 2020, 2073, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.; He, J.; Yang, S.H.; Brimble, M.A.; Harris, P.W.R.; Cameron, A.J. Flow-Based Fmoc-SPPS Preparation and SAR Study of Cathelicidin-PY Reveals Selective Antimicrobial Activity. Molecules 2023, 28, 1993. [Google Scholar] [CrossRef] [PubMed]
- Nash, I.A.; Bycroft, B.W.; Chan, W.C. Dde — A selective primary amine protecting group: A facile solid phase synthetic approach to polyamine conjugates. Tetrahedron Lett. 1996, 37, 2625–2628. [Google Scholar] [CrossRef]
- Vagenende, V.; Yap, M.G.S.; Trout, B.L. Mechanisms of Protein Stabilization and Prevention of Protein Aggregation by Glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef] [PubMed]
- Fenzl, C.; Nayak, P.; Hirsch, T.; Wolfbeis, O.S.; Alshareef, H.N.; Baeumner, A.J. Laser-Scribed Graphene Electrodes for Aptamer-Based Biosensing. ACS Sens. 2017, 2, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Lazanas, A.C.; Prodromidis, M.I. Electrochemical Impedance Spectroscopy─A Tutorial. ACS meas. sci. au 2023, 3, 162–193. [Google Scholar] [CrossRef]
- Vadlamani, B.S.; Uppal, T.; Verma, S.C.; Misra, M. Functionalized TiO2 Nanotube-Based Electrochemical Biosensor for Rapid Detection of SARS-CoV-2. Sensors 2020, 20, 5871. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Hu, C.; Jahan, S.; Yuan, B.; Saleh, M.S.; Ju, E.; Gao, S.-J.; Panat, R. Sensing of COVID-19 Antibodies in Seconds via Aerosol Jet Nanoprinted Reduced-Graphene-Oxide-Coated 3D Electrodes. Adv Mater. 2021, 33, 2006647. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



