
Submitted:
30 November 2023
Posted:
30 November 2023
You are already at the latest version
Abstract
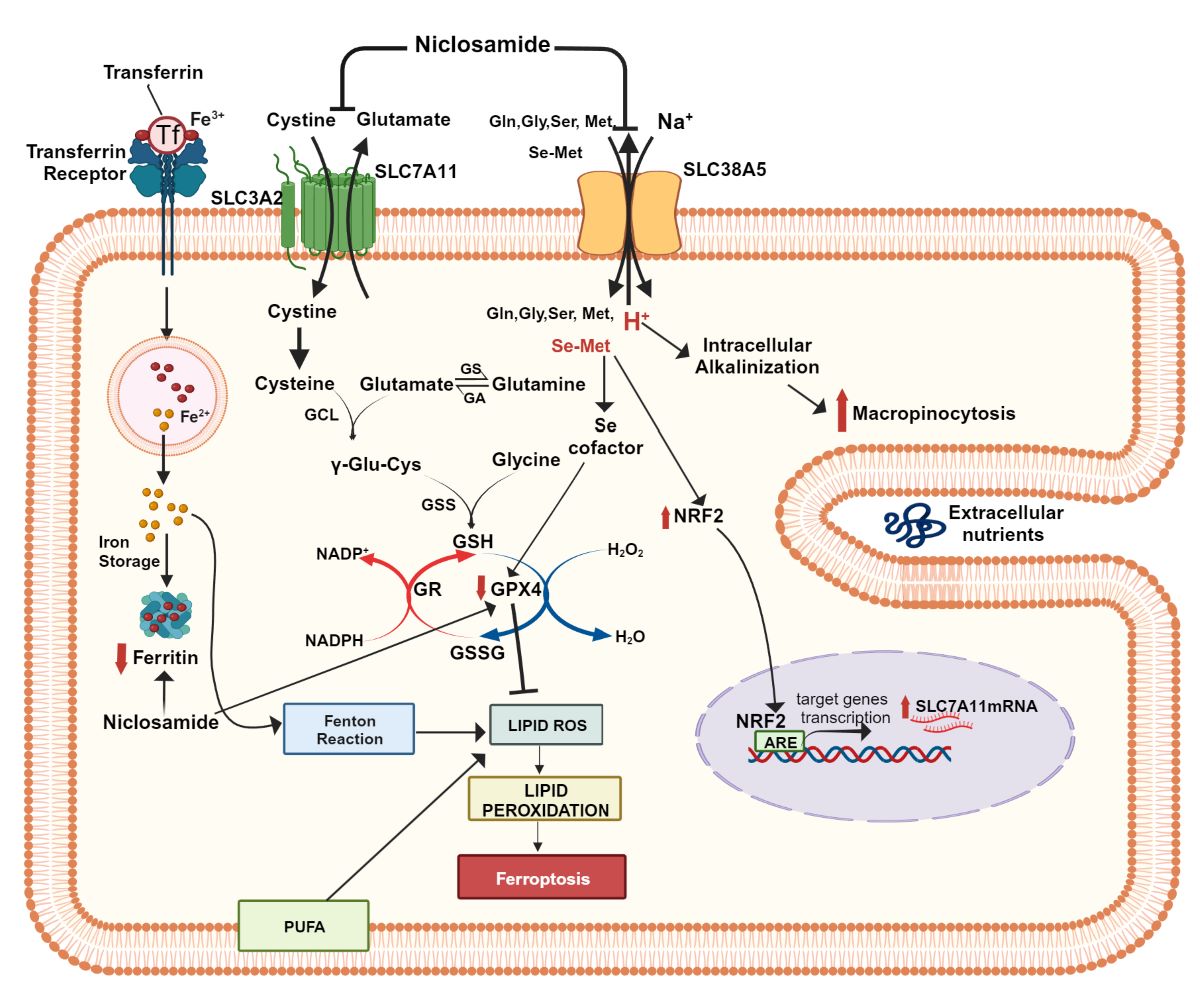
The amino acid transporters SLC38A5 and SLC7A11 are upregulated in triple-negative breast cancer (TNBC). SLC38A5 transports glutamine, methionine, glycine and serine, and therefore activates mTOR signaling and induces epigenetic modifications. SLC7A11 transports cystine and increases cellular levels of glutathione, which protects against oxidative stress and lipid peroxidation via glutathione peroxidase, a seleno (Se)-enzyme. The primary source of Se is dietary Se-methionine (Se-Met). Since SLC38A5 transports methionine, we examined its role in Se-Met uptake in TNBC cells. We found that SLC38A5 interacts with methionine and Se-Met with comparable affinity. We also examined the influence of Se-Met on Nrf2 in TNBC cells. Se-Met activated Nrf2 and induced expression of Nrf2-target genes, including SLC7A11. Our previous work discovered niclosamide, an antiparasitic drug, as a potent inhibitor of SLC38A5. Here we found SLC7A11 to be inhibited by niclosamide with an IC50 value in the range of 0.1-0.2 M. In addition to the direct inhibition of SLC38A5 and SLC7A11, pretreatment of TNBC cells with niclosamide reduced the expression of both transporters. Niclosamide decreased glutathione levels, inhibited proliferation, suppressed GPX4 expression, increased lipid peroxidation, and induced ferroptosis in TNBC cells. It also significantly reduced the growth of the TNBC cell line MB231 in mouse xenografts.
Keywords:
SLC38A5
; SLC7A11
; TNBC cells
; seleno-methionine
; glutathione
; glutathione peroxidase
; Nrf2
; ferroptosis
; lipid peroxidation
; niclosamide
1. Introduction
Iron is an obligatory element for survival of all cells. It is involved in almost each and every biological process such as energy production, anabolism, catabolism, oxygen delivery, and metabolism/disposal of drugs/xenobiotics. Iron exists in two different valency states: ferrous (Fe2+) and ferric (Fe3+). It can be found in cells either in a free form (labile iron) or bound to proteins such as ferritin and transferrin. Two most important biologically active forms of iron are the iron-sulfur cluster (Fe-S) and heme. The transition between the two valency states is either enzyme-catalyzed (ferroxidases and ferrireductases) or simply the result of electron transfer between iron and electron acceptors such as oxygen. The labile free iron in the ferrous form is highly oxidative with reactivity towards hydrogen peroxide and lipid hydroperoxides via Fenton reaction resulting in the generation of hydroxyl and lipid alkoxyl radicals, which are extremely detrimental to cells promoting a unique form of cell death known as ferroptosis [1-3].
Since many of the biological processes requiring iron are essential for cell proliferation, cancer cells are obligatorily programed to accumulate iron at levels more than that found in normal cells; in other words, cancer cells are “addicted” to iron [4,5]. This poses a conundrum: how can cancer cells accumulate excess iron to support their growth and proliferation without risking themselves to ferroptotic cell death? Cancer cells overcome this problem by enhancing the efficacy of their antioxidant machinery. The most important component of this machinery is the glutathione (GSH)/glutathione peroxidase (GPX)/glutathione reductase (GR) system which involves the thiol-containing tripeptide glutathione (γ-glutamylcysteinylglycine), the selenium-containing enzyme GPX (particularly GPX4) and the reducing power NADPH. Cancer cells induce the cystine transporter SLC7A11 to increase the cellular levels of GSH [6,7], upregulate hexose monophosphate shunt and malic enzyme to increase the production of NADPH [8,9], and increase the expression of GPX4 [10], which collectively detoxify hydroxyl and lipid alkoxyl radicals to protect the cells from ferroptosis despite the presence of excessive iron within the cells.
We undertook the present investigation to determine the potential crosstalk between two specific amino acid transporters in triple-negative breast cancer (TNBC) in enhancing the antioxidant machinery and to identify an effective pharmacologic strategy to interfere with this pathway as a plausible anticancer approach. SLC38A5 (also known as SN2 or SNAT5) is a transporter that mediates Na+-dependent influx of glutamine, methionine, serine and glycine into cells in exchange for intracellular H+ [11,12]. It is upregulated in TNBC [13] and pancreatic cancer [14]. In addition to mediating the uptake of amino acids, SLC38A5 also promotes macropinocytosis, a special form of nutrient uptake in cancer cells [13,15,16]. Based on the amino acid selectivity of this transporter, we recently proposed that the transporter is uniquely suitable to influence one-carbon metabolism and mTOR signaling [17]. In the present study with TNBC cells, we explored the possibility that SLC38A5 could transport seleno-methionine (Se-Met), the most predominant dietary source of selenium. Since the selenoenzyme GPX4 is a critical component of the antioxidant machinery in TNBC cells, the possible involvement of SLC38A5 in the delivery of selenium into cancer cells in the form of Se-Met could represent a novel function of the transporter in promoting cancer growth. Previous work in our lab has shown that Se-Met is an activator of Nrf2, an important transcription factor related to the antioxidant machinery [18]. The targets for Nrf2 include the cystine transporter SLC7A11 and the glutamate-cysteine ligase, the first enzyme in the synthesis of glutathione [19]. Similar to SLC38A5, SLC7A11 is also upregulated in TNBC [20,21]. Therefore, we hypothesized that there could be a functional coupling between SLC38A5 and SLC7A11 via Se-Met. We tested this hypothesis in the present study using TNBC cells. In addition, we have identified niclosamide, an FDA-approved antiparasitic drug, as a potent inhibitor of the function and expression of SLC38A5 and SLC7A11, consequently inducing oxidative stress, lipid peroxidation, and ferroptotic cell death in TNBC cells, and suppression of growth of a TNBC cell line into tumors in mouse xenografts. Niclosamide is known to elicit anticancer effects via multiple mechanisms in several cancer types [22-24]. The findings of the present study provide yet another novel, hitherto unknown, pharmacologic mechanism for the anticancer efficacy of this drug.
2. Materials and Methods
2.1. Materials
[2,3-3H]-L-Serine (specific radioactivity, 15 Ci/mmol) was purchased from Moravek, Inc. (Brea, CA, USA). [3H]-Glutamate (specific radioactivity 50.8 Ci/mmol) was purchased from PerkinElmer Corp (Waltham, MA, USA). Niclosamide, monomethyl fumarate, methionine, seleno-methionine and buthionine sulphoximine (BSO) were purchased from Millipore-Sigma (St. Louis, MO, USA).
2.2. Cell lines and culture conditions
We used three breast cancer cell lines (all representing triple-negative breast cancer). All media contained 10% fetal bovine serum. Cell cultures were tested every month for mycoplasma using a commercially available detection kit (cat. no. G238; Applied Biological Materials, Inc. Richmond, British Columbia, Canada). All cell lines used in the present study were mycoplasma-free. Two of the breast cancer cell lines were from ATCC: MDA-MB231 (cat. no. CRM-HTB-26) and MDA-MB453 (cat. no. HTB-131) cells were cultured in Leibovitz’s L-15 medium. One patient-derived TNBC cell line, identified as TXBR-100, was provided by the TTUHSC Cancer Center. This cell line was cultured in a special medium consisting of Dulbecco’s modified Eagle’s medium and Ham’s F12 medium, in a 1:1 ratio, supplemented with 20 ng/mL EGF, 0.01 mg/mL insulin, 500 ng/mL hydrocortisone, and 100 ng/mL cholera toxin.
2.3. Uptake measurement
Uptake of radiolabeled amino acids was used to monitor the transport function of SLC38A5. Since SLC38A5 is a Na+-coupled transporter with the involvement of H+ movement in the opposite direction, the uptake assays were done at pH 8.5 to create an outwardly directed H+-gradient across the plasma membrane to maximize the uptake activity. As there are several Na+-coupled amino acid transporters for serine that was used as the substrate in most of the experiments in the present study, we cannot specifically monitor the function of SLC38A5 by using Na+- containing buffer. However, unlike other Na+-coupled transporters, SLC38A5 is tolerant to Li+ (i.e., SLC38A5 functions when Na+ is replaced with Li+). Therefore, we used an uptake buffer with LiCl in place of NaCl. The composition of the uptake buffer was 25 mM Tris/Hepes, pH 8.5, containing 140 mM LiCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4 and 5 mM glucose. Serine that was used as the substrate to monitor the transport function of SLC38A5 is also substrate for SLC7A5 (LAT1), which is a Na+-independent amino acid transporter; therefore, uptake of serine via this transporter will contribute to the total uptake measured in the LiCl-containing buffer, thus confounding the interpretation of SLC38A5-specific uptake. Therefore, we needed to suppress SLC7A5-mediated serine uptake while measuring the transport activity of SLC38A5. This was done by including 5 mM tryptophan in the uptake buffer to compete with and block the transport of serine mediated by SLC7A5; SLC38A5 does not transport tryptophan and therefore SLC38A5-mediated uptake will not be affected by tryptophan. To determine the contribution of diffusion to the total uptake of serine, the same uptake buffer but with LiCl replaced isosmotically with N-methyl-D-glucamine chloride (NMDGCl) was used. As such, the uptake was measured in two buffers: (i) LiCl-buffer, pH 8.5 with 5 mM tryptophan; (ii) NMDGCl-buffer, pH 8.5 with 5 mM tryptophan. The uptake in NMDGCl-buffer was subtracted from the uptake in LiCl-buffer to determine the transport activity of SLC38A5 [15].
The heterodimeric amino acid transporter xCT/4F2hc is responsible for the activity of the amino acid transport system known as SLC7A11. It is a Na+-independent system that mediates the cellular entry of cystine in exchange for intracellular glutamate under physiological conditions. However, we routinely measure the activity of this transporter by cellular uptake of [3H]-glutamate under Na+-free conditions. Under these conditions, SLC7A11 mediates the cellular entry of [3H]-glutamate in exchange for intracellular unlabeled glutamate. The primary reason for not using radiolabeled cystine directly in uptake measurement of SLC7A11 transport activity includes difficulties such as insolubility of cystine, the availability of radiolabeled cystine from most commercial sources only in 35S-form with its relatively low half-life and the inability to determine the relative amounts of reduced and oxidized forms of cystine in the stock solution. Transport activity with L-[3H]-glutamate was measured using a Na+-free uptake buffer (25 mM Hepes/Tris, 140 mM N-methyl-D-glucamine chloride, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, and 5 mM glucose, pH 7.5). Non-carrier-mediated uptake (i.e, diffusional component) was determined by measuring the uptake of [3H]-glutamate in the presence of excess unlabeled glutamate (5 mM). The transport activity of SLC7A11 was calculated by subtracting the diffusional component from total uptake [25,26].
Cells were seeded in 24-well culture plates (2 × 105 cells/well) with the culture medium and were allowed to grow to confluency, which normally took 2 or 3 days depending on the cell line. Confluent cultures were used for uptake measurements. On the day of uptake measurement, the culture plates were kept in a water bath at 37 °C. The medium was aspirated, and the cells were washed with uptake buffers. The uptake medium (250 μl) containing corresponding labeled amino acid as the tracer along with unlabeled glutamate, methionine, Se-Met (different concentrations for a dose-response study) was added to the cells. Following incubation for 15 or 30 min, the medium was removed, and the cells were washed three times with ice-cold uptake buffer. The cells were then lysed in 1% sodium dodecyl sulfate/0.2 N NaOH and used for measurement of radioactivity.
2.4. RT-PCR
Total RNA was extracted from cells using TRIzol Reagent (ThermoFisher Scientific, Waltham, MA, USA), and the RNA was reverse-transcribed using a high-capacity cDNA reverse transcription kit (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. PCR and quantitative PCR were performed with Takara Taq Hot Start Version (TaKaRa Biotechnology, Shiga, Japan) or Power SYBR Green PCR master mix (Bio-Rad, Hercules, CA, USA). Primer sequences are shown in Supplemental Table 1. The relative mRNA expression was determined by the 2−∆∆Ct method. 18S was used as a housekeeping gene for normalization.
2.5. Protein isolation and western blot
Cells and tumor tissues were lysed in Pierce™ RIPA buffer (ThermoFisher Scientific, Waltham, MA, U.S.A.) supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific, Waltham, MA, USA). Homogenates were centrifuged, and supernatants were used for protein measurement via Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific, Waltham, MA, U.S.A.). Western blot samples were prepared in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, USA). They were loaded onto a SDS–PAGE gel and transferred onto a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, U.S.A.). The membrane was blocked, and antibodies diluted in 5% nonfat dry milk (Bio-Rad Laboratories, Hercules, CA, USA) or in 5% bovine serum albumin (Irvine Scientific, Santa Ana, CA, U.S.A.) were used. Protein bands were visualized using Pierce™ ECL Western Blotting Substrate (ThermoFisher Scientific, Waltham, MA, USA) and developed on the autoradiography film (Santa Cruz, Dallas, TX, USA.). Primary antibodies were purchased either from Cell Signaling (Danvers, MA, USA) [anti-GPX4 (#52455), anti-FTH (#4393), anti-HSP60 (#12165)] or from Abcam (Waltham, MA, USA) [Nrf2 (#ab62352)]. Secondary antibody Horseradish peroxidase-conjugated goat anti-rabbit (#1706515) was purchased from Bio-Rad Laboratories (Hercules, CA, USA). For quantification of protein levels by the densitometric analysis, the experiment was carried out in triplicate and the data were collected from the resultant three Western blots.
2.6. Ferroptosis assay
Cells were cultured onto a 25-mm glass coverslip until they reach 60-70% confluency (~ 48 h) at 37 oC and 5% CO2. At the time of the experiment, cells were washed with NaCl buffer, pH 7.5 and then incubated with 1 µM of LipiRadical Green (a lipid radical detection reagent, FDV-0042, Funakoshi, Tokyo, Japan) in NaCl buffer, pH 7.5 for 20 min and then washed with NaCl buffer, pH 7.5. To analyze the effects of modifiers of lipid peroxidation, the cells were co-treated with the modifiers and LipiRadical Green together for 20 min and then washed. The glass coverslip containing the cells was placed into a chamber and then transferred onto a stage of an inverted microscope. The fluorescence imaging were observed using a Nikon laser-scanning confocal microscopy, with a 60x objective, at 488 nm. The images represent a maximum projection intensity derived from a Z-stack. The fluorescence quantification was performed by measuring the corrected total cell fluorescence (CTCF) using Image J and the following formula; CTCF = (integrated density) – (area of cell of interest) x (mean fluorescence of background).
2.7. Glutathione and lipid peroxidation assay
Control and niclosamide-treated cells were used for measurement of cellular glutathione levels as instructed in the manufacturer’s protocol (GSH-Glo assay, Promega, Madison, WI, USA). The levels of malondialdehyde were measured using lipid peroxidation kit [Lipid Peroxidation (MDA) Assay Kit (MAK085), Millipore-sigma, St. Louis, MO, USA].
2.8. Colony formation assay
We performed the colony-formation (clonogenic) assay with different doses of niclosamide on two different TNBC cell lines. Initial seeding was done with 500 cells/well and culture was continued for 10 days with culture medium replaced with fresh medium with freshly prepared niclosamide every other day. At the end of the 10-day time period, the medium was removed and the colonies were fixed with ice-cold methanol/acetone and then stained with Giemsa stain. After examination, lysis buffer added (1% sodium dodecyl sulfate/0.2 N NaOH) and incubated in shaker to extract the Giemsa stain and quantified using plate reader.
2.9. MTT assay
Cells were seeded in 96-well plates; after 24 h, niclosamide treatment was initiated. Cells were then cultured for 72 h with fresh medium containing freshly prepared niclosamide supplied every 24 h. Cells were washed with phosphate-buffered saline twice followed by MTT reagent (ATCC). Treatment and lysis of the cells were done as per the manufacturer's instructions. Absorbance of the lysate was measured at 550 nm.
2.10. Mouse xenograft experiments
Female athymic nude mice (4-week-old) were purchased from Jackson Laboratories and housed under standard conditions. MD-MB231 cells were injected into lower mammary fat pad (5 × 106 cells). All cells were suspended in serum-free media and Matrigel (1:1 ratio), with 100 µl of suspension being injected to each mouse. Mice were treated by daily intraperitoneal injection of niclosamide (4 mg/kg/day) and vehicle (dimethylsulfoxide) control. The treatment began when the tumor size was 100-150 mm3. Tumor size was measured biweekly with a caliper, with tumor volume calculated using the formula (width2 × length)/2. Tumors were allowed to grow for 7 weeks; mice were then euthanized via isoflurane injection and tumors harvested. The animal study protocol was approved by the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee (IACUC protocol number 18005) and the experiments were conducted in the same institution. RNA and protein were prepared from the tumor tissue for qPCR and western blotting.
2.11. Homology modeling and docking studies
SLC38A5 homology modeling and docking studies performed as previously published [15] to determine the theoretical values for the binding energies for the interaction of methionine and seleno-methionine with SLC38A5. Since the cryo-EM crystal structure of human SLC38A5 is not known, we used the structures of closely related transporters as templates for our purpose [15]. With regard to structural modeling of the cystine/glutamate antiporter, we have information on the cryo-EM crystal structure of human transporter-chaperone complex SLC7A11/SLC3A2 (PDB: 7P9V) [27]. This structure of the in-vivo functional heterodimer was used for docking studies to determine the binding energies for methionine and selenomethionine. In the case of both transporters, the docking simulations were conducted using AutoDock/Vina in conjunction with the USCF Chimera program [28,29]. A grid with dimensions of 30 x 30 x 30 (Å3) was employed, focusing on the cavity within the structures of the two proteins where ligand binding was observed.
2.12. Statistics
Uptake experiments were routinely done in triplicates, and each experiment was repeated at least thrice using independent cell cultures. Statistical analysis was performed with a two- tailed, paired Student’s t-test for single comparison and a p-value < 0.05 was considered statistically significant. Data are given as means ± S.E. For quantification of fluorescence signals in image analysis related to ferroptosis, ANOVA followed by Dunn’s test was used to determine the significance of difference among the different groups.
3. Results
3.1. Expression patterns for SLC38 gene family members in breast cancer
The SLC38 gene family consists of 11 members, and the best characterized among them in terms of transport function are five members, all belonging to two subclasses: system A (SLC38A1, SLC38A2, and SLC38A4) and system N (SLC38A3 and SLC38A5) [30]. There is evidence for a role for two of these transporters in TNBC: SLC38A2 [31] and SLC38A5 [13, 15]. Using the transcriptomic data from publicly available datasets, we analyzed the expression of the remaining three members in breast cancer (Supplemental Fig. S1). This analysis showed that SLC38A1 is upregulated in estrogen receptor-positive luminal-type breast cancer. In contrast to SLC38A1, SLC38A3 is downregulated in this type whereas SLC38A4 remains unaltered. None of the three is altered in HER2-positive breast cancer. In TNBC, SLC38A1 remains unaltered, SLC38A3 is upregulated, and SLC38A4 is downregulated.
3.2. Interaction of seleno-methionine (Se-Met) with human SLC38A5
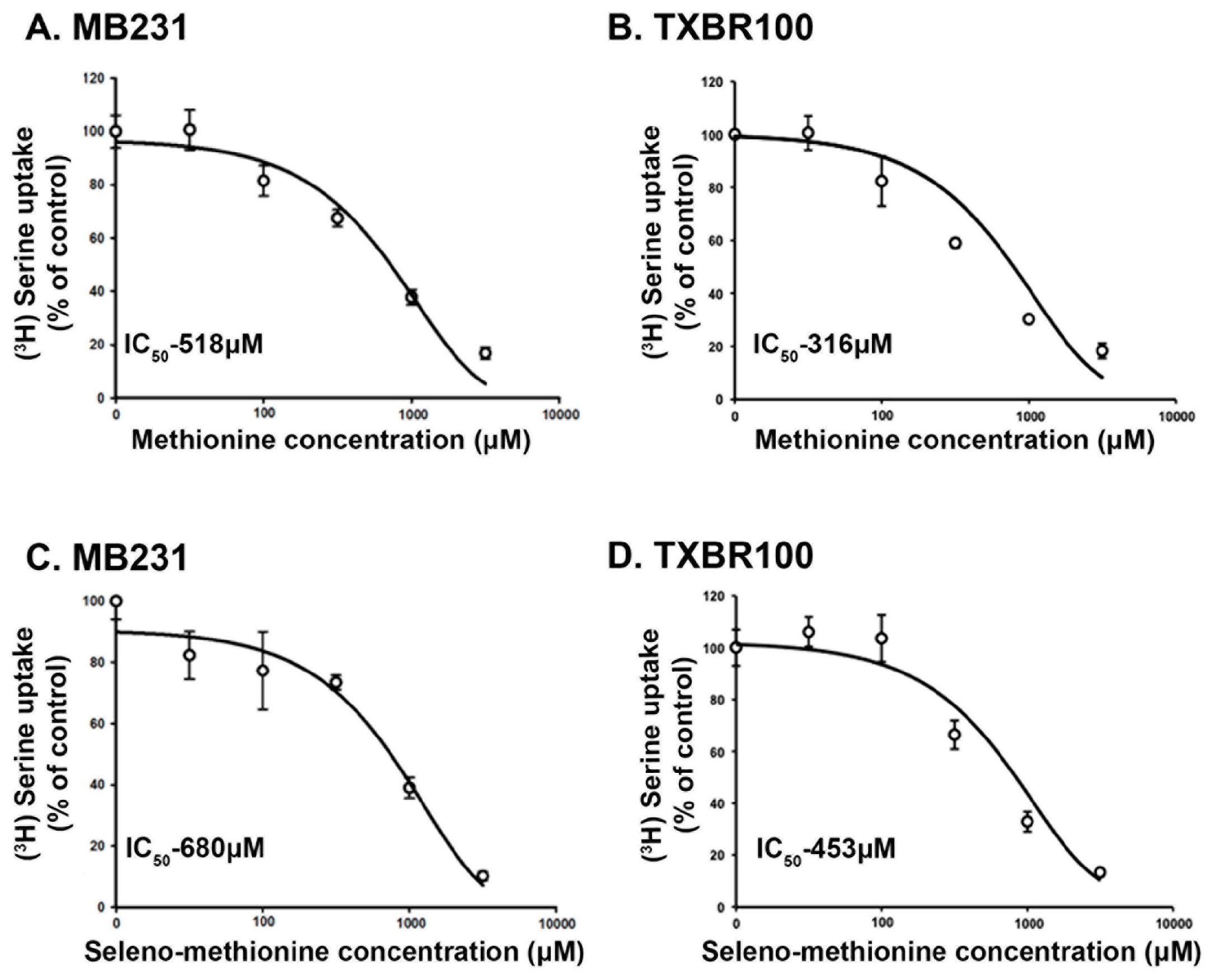
There is no information in published literature on transport systems available for Se-Met in mammalian cells. However, it is very likely that any transport system that is capable of transporting methionine might transport Se-Met because the only structural difference between methionine and Se-Met is the replacement of sulfur in methionine with Se. Therefore, we hypothesized that SLC38A5 that transports methionine would be able to recognize Se-Met as a substrate. Since radiolabeled Se-Met is not available from any commercial source, we decided to address this issue by monitoring the ability of Se-Met to compete with serine for uptake that is mediated by SLC38A5. Serine uptake was measured in two different TNBC cell lines (MB231 and TXBR100). The uptake conditions were designed such that only SLC38A5-mediated uptake of serine was measured as described in the Methods section. The abilities of methionine and Se-Met as competitors of serine uptake was compared in a dose-response experiment in both cell lines (Fig. 1). Methionine as well as Se-Met competed with serine for SLC38A5-mediated transport. In MB231 cells, the IC50 values for the inhibition of serine uptake by methionine and Se-Met were 518 μM and 680 μM, respectively (Fig. 1A, C). A similar inhibitory potency was observed in the TXBR100 cell line also (IC50 values: 316 μM for methionine and 453 μM for Se-Met) (Fig. 1B, D). Since the concentration of serine in these uptake experiments was only 5 μM, very small compared to its Kt value for SLC38A5-mediated transport (200 - 1300 μM) [11, 32], the IC50 values for methionine and Se-Met can be approximated to their corresponding Kt values.
Figure 1.
Dose-response relationship for the inhibition of SLC38A5-mediated serine uptake by methionine and selenomethionine in TNBC cell lines MB231 and TXBR100. The uptake of [3H]-serine that was mediated specifically by SLC38A5 was monitored under the conditions detailed in the Methods section. Uptake in the absence of methionine or selenomethionine was taken as 100%, and the uptake in the presence of methionine or selenomethionine was determined as the percent of this control uptake. The IC50 values (concentration of methionine or selenomethionine at which the inhibition was 50% of the control uptake) were calculated using the Sigma plot program.
Figure 1.
Dose-response relationship for the inhibition of SLC38A5-mediated serine uptake by methionine and selenomethionine in TNBC cell lines MB231 and TXBR100. The uptake of [3H]-serine that was mediated specifically by SLC38A5 was monitored under the conditions detailed in the Methods section. Uptake in the absence of methionine or selenomethionine was taken as 100%, and the uptake in the presence of methionine or selenomethionine was determined as the percent of this control uptake. The IC50 values (concentration of methionine or selenomethionine at which the inhibition was 50% of the control uptake) were calculated using the Sigma plot program.
We also analyzed the interaction of methionine and Se-Met with human SLC38A5 using a theoretical approach. We modeled the interaction of these two amino acids with the transporter by molecular docking as we described in our previous publication where we used a similar approach to identify the FDA-approved drug niclosamide as a ligand for the transporter [15]. With this approach, we determined that methionine interacted with SLC38A5 with a binding energy of −4.8 kcal/mol and that Se-Met also interacted with the transporter with a similar binding energy (−4.9 kcal/mol). These binding energies translate into corresponding dissociation constants (KD) of 295 μM for methionine and 250 μM for Se-Met. As the dissociation constant is an approximate equivalent of Kt values, these theoretically determined values are in the same range as the experimentally determined values shown in Fig. 1. In addition to the similar binding affinities between methionine and Se-Met for SLC38A5, the amino acid residues in SLC38A5 that are responsible for the binding of methionine and Se-Met are also the same (Y142, L194, T197, S198, F267, A268, E274 and T349). However, the interaction of Se-Met showed participation of two additional amino acid residues (H134 and N135).
3.3. Potentiation of Nrf2 and its anti-oxidant signaling in TNBC cells by Se-Met
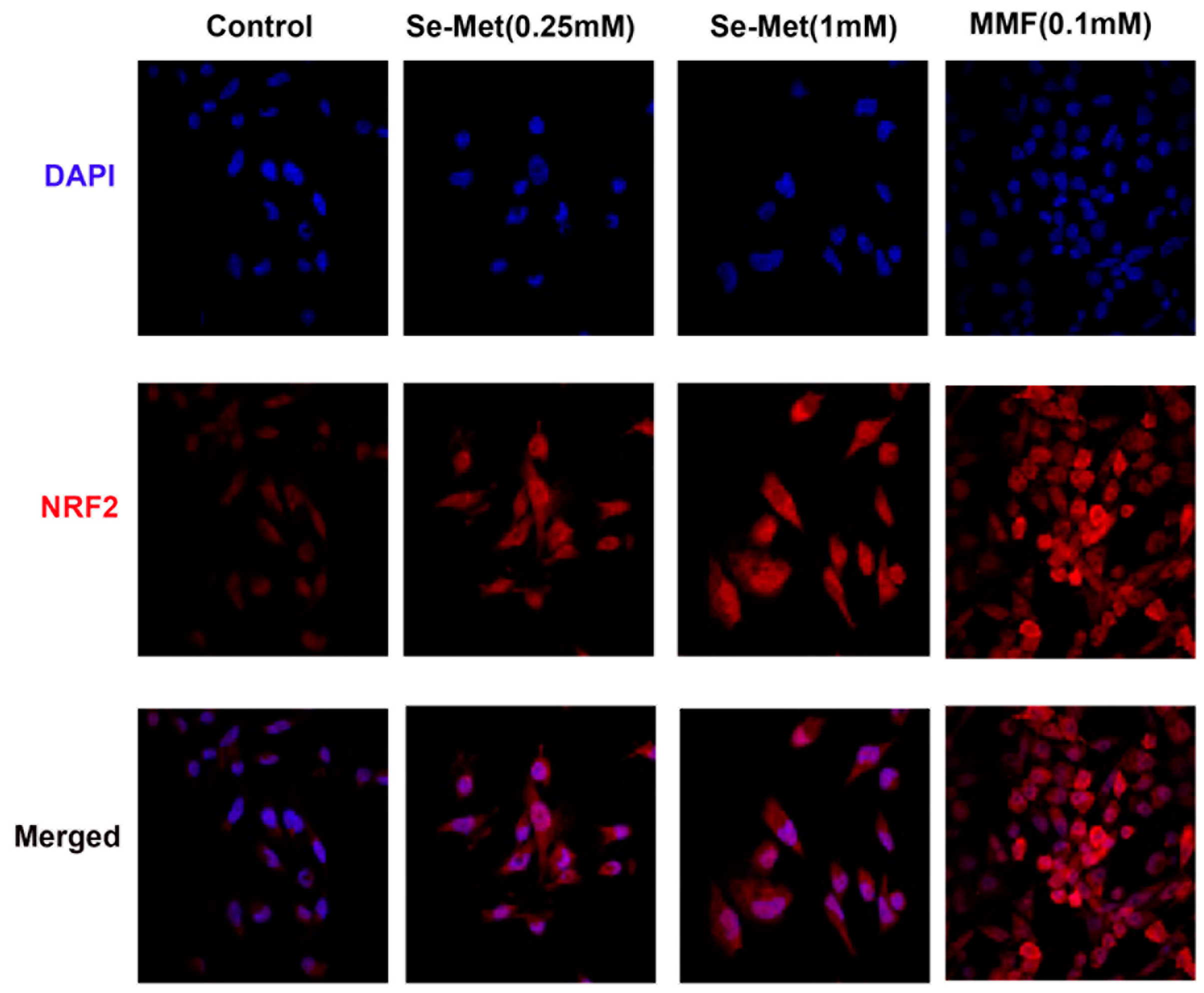
Se-Met is the primary dietary source of selenium [33] and the sole biological function of this micronutrient is its involvement in the catalytic activities of selenoproteins such as glutathione peroxidases which are components of cellular antioxidant machinery [34]. However, we recently reported that Se-Met also potentiates the antioxidant machinery in mammalian cells by a second mechanism by enhancing the expression and activity of Nrf2, an important antioxidant transcription factor [35]. Since TNBC cells have robust expression of SLC38A5 and also since SLC38A5 appears to mediate the cellular entry of Se-Met, we asked if Se-Met would potentiate the antioxidant machinery in TNBC cells by enhancing Nrf2 signaling. To address this question, we exposed two different TNBC cell lines (MB231 and TXBR100) to Se-Met (250 μM and 1 mM) for 24 h and then examined the expression and subcellular localization of Nrf2 by immunostaining. These studies showed that exposure of the cells to Se-Met increased cellular levels as well as nuclear localization of Nrf2 (Fig. 2). The effect was noticeable even at 250 μM. In these experiments, we used monomethylfumarate (0.1 mM) as a positive control because this compound is a known activator of Nrf2 signaling [36]. Monomethylfumarate increased the cellular levels and the nuclear localization of Nrf2 in MB231 cells (Fig. 2). Similar results were obtained with TXBR100 cells (Supplemental Fig. S2).
Figure 2.
Influence of Se-Met on the expression levels of Nrf2 protein in the TNBC cell line MB231. The cells were treated with Se-Met at 0.25 mM or 1 mM for 24 h and then the cells were fixed and Nrf2 protein levels monitored by immunofluorescence. Monomethylfumarate (MMF) was used at 0.1 mM as a positive control for induction of Nrf2 protein. DAPI was used as a marker for nucleus.
Figure 2.
Influence of Se-Met on the expression levels of Nrf2 protein in the TNBC cell line MB231. The cells were treated with Se-Met at 0.25 mM or 1 mM for 24 h and then the cells were fixed and Nrf2 protein levels monitored by immunofluorescence. Monomethylfumarate (MMF) was used at 0.1 mM as a positive control for induction of Nrf2 protein. DAPI was used as a marker for nucleus.
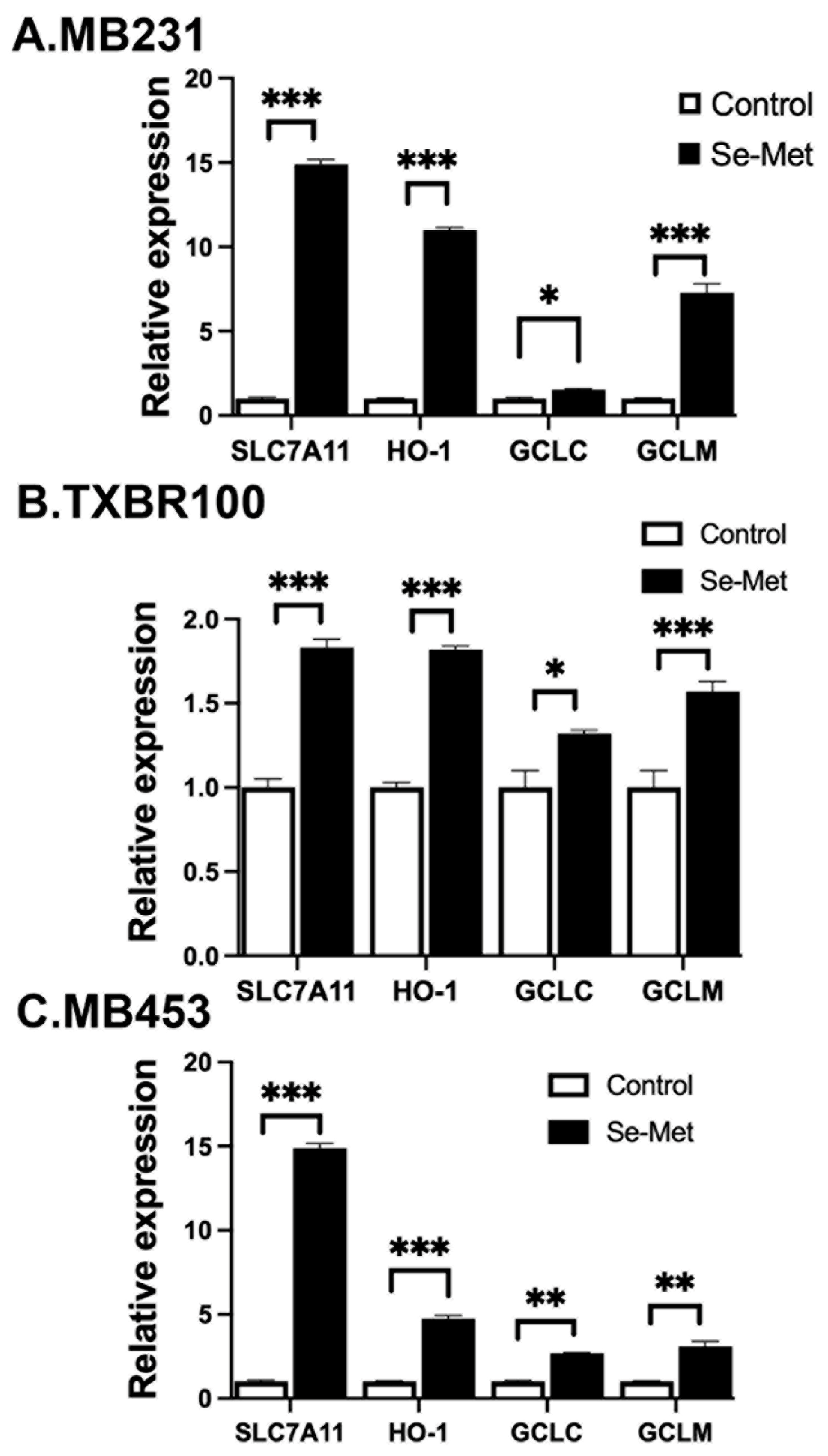
Nrf2 is a transcription factor and its antioxidant activity is evident from the biological activities of its transcriptional targets, which include the cystine transporter SLC7A11, the heme-degrading enzyme heme oxygenase-1 (HO-1), and the components of the glutathione synthetic pathway (the catalytic and the modulatory subunits of glutamate-cysteine ligase (GCLC and GCLM) that catalyzes the first step in the synthesis of glutathione) [37]. To determine if the observed increase in cellular and nuclear levels of Nrf2 seen in Se-Met-exposed TNBC cells is accompanied with a parallel increase in the expression of the above-mentioned Nrf2 target genes, we compared the levels of mRNA for SLC7A11, HO-1, GCLC and GCLM in control and Se-Met-treated MB231, TXBR100 and MB453 cells by real-time PCR (Fig. 3). We found a significant increase in mRNA levels for all four targets in response to Se-Met treatment in all three TNBC cell lines.
Figure 3.
Influence of Se-Met on the expression of Nrf2 target genes in three different TNBC cell lines (MB231, TXBR100, and MB453). Cells were treated with Se-Met (1 mM) for 16 h, following which RNA was prepared from the cells for use in qRT-PCR. Cells cultured under identical conditions but in the absence of Se-Met were used as control. The mRNA levels for SLC7A11, heme oxygenase-1 (HO-1, catalytic subunit of glutamate-cysteine ligase (GCLC) and modulatory subunit of glutamate-cysteine ligase (GCLM) were monitored by quantitative PCR as described in the Methods section. For each gene, the mRNA level in control (i.e., cultured in the absence of Se-Met) cells was taken as 1. Data are given as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Figure 3.
Influence of Se-Met on the expression of Nrf2 target genes in three different TNBC cell lines (MB231, TXBR100, and MB453). Cells were treated with Se-Met (1 mM) for 16 h, following which RNA was prepared from the cells for use in qRT-PCR. Cells cultured under identical conditions but in the absence of Se-Met were used as control. The mRNA levels for SLC7A11, heme oxygenase-1 (HO-1, catalytic subunit of glutamate-cysteine ligase (GCLC) and modulatory subunit of glutamate-cysteine ligase (GCLM) were monitored by quantitative PCR as described in the Methods section. For each gene, the mRNA level in control (i.e., cultured in the absence of Se-Met) cells was taken as 1. Data are given as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
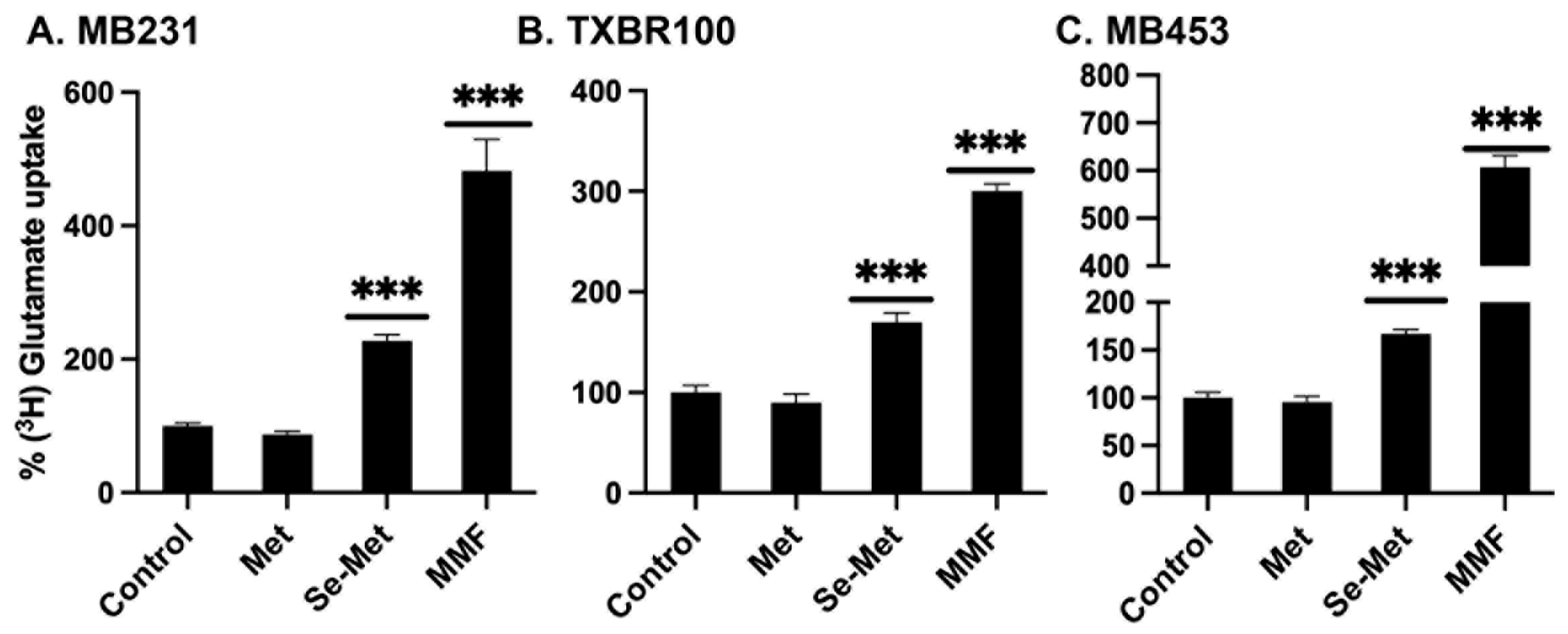
To corroborate the data on the Se-Met-induced changes in mRNA levels with functional data, we selected SLC7A11 for examination. We measured the transport activity of SLC7A11 in control cells and in cells treated with Se-Met. We used monomethylfumarate as a positive control. In addition, we compared the effects of Se-Met with that of methionine to determine if the observed effects are specific for Se-Met. The transport activity of SLC7A11, monitored as glutamate uptake as detailed in the Methods section, increased substantially in Se-Met-treated cells (all three TNBC cell lines: MB231, TXBR100 and MB453) (Fig. 4). The positive control monomethylfumarate also elicited the stimulatory effect on SLC7A11 transport activity. Importantly, methionine did not have any effect, indicating that the potentiating effect on Nrf2 signaling in TNBC cells is specific for Se-Met and that the effect is not seen with methionine.
Figure 4.
Influence of methionine (Met), selenomethionine (Se-Met) and monomethylfumarate (MMF) on the transport activity of SLC7A11 in three different TNBC cell lines (MB231, TXBR100, and MB453). The cells were treated with Met, Se-Met or MMF at 1 mM for 16 h and then the cells were used to monitor SLC7A11 transport activity using [3H]-glutamate as the tracer. Unlabeled glutamate was present at 5 μM. The SLC7A11-specific uptake was determined using the experimental conditions detailed in the Methods section. Uptake in control cells (i.e., cultured in the absence of Met, Se-Met and MMF) was taken as 100% and the uptake for treated cells was calculated as the percent of this control uptake. Data are given as mean ± S.E. ***, p < 0.001.
Figure 4.
Influence of methionine (Met), selenomethionine (Se-Met) and monomethylfumarate (MMF) on the transport activity of SLC7A11 in three different TNBC cell lines (MB231, TXBR100, and MB453). The cells were treated with Met, Se-Met or MMF at 1 mM for 16 h and then the cells were used to monitor SLC7A11 transport activity using [3H]-glutamate as the tracer. Unlabeled glutamate was present at 5 μM. The SLC7A11-specific uptake was determined using the experimental conditions detailed in the Methods section. Uptake in control cells (i.e., cultured in the absence of Met, Se-Met and MMF) was taken as 100% and the uptake for treated cells was calculated as the percent of this control uptake. Data are given as mean ± S.E. ***, p < 0.001.
3.4. Identification of niclosamide as a potent blocker of SLC7A11
We reported recently that the FDA-approved antihelminthic drug niclosamide is a potent inhibitor of SLC38A5 and SLC38A5-mediated macropinocytosis [15]. This study involved first a theoretical molecular docking approach, then followed by actual experimentation in mammalian cells. We followed a similar strategy to determine if niclosamide has any effect on SLC7A11. First, we analyzed the binding of niclosamide with human SLC7A11 using the recently elucidated cryo-EM structure of this transporter (Methods section). We modeled the binding of niclosamide with SLC7A11 and compared it with the binding of erastin, the most potent blocker of SLC7A11 known to date in the published literature [38]. The results, shown in Supplemental Fig. S3, demonstrate that both compounds interacted with the transporter even though the amino acid residues involved in the binding are different for both compounds as could be expected based on the differences in their chemical structures. The binding-energy obtained from this approach was −8.0 kcal/mol for niclosamide; the corresponding value for erastin was −7.3 kcal/mol. We also did the docking for the influx substrate cystine and the exchange substrate glutamate for the transporter; the binding-energies were −4.8 and −4.7 kcal/mol, respectively. The theoretical KD values derived from these binding-energy values are: 1.3 μM for niclosamide, 4.3 μM for erastin, 300 μM for cystine, and 350 μM for glutamate.
Figure 5.
Direct effect of niclosamide on SLC7A11 transport activity (A-C) and the correlation between theoretically calculated and experimentally determined dissociation constants for various substrates and inhibitors (D). The transport activity of SLC7A11 was measured in three different TNBC cell lines (MB231, TXBR100, and MB453) as detailed in the Methods section. Niclosamide was present only during uptake. The dissociation constants for the substrates and inhibitors were determined in the present study or taken from published reports (see the text). For A-C, data are given as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. .
Figure 5.
Direct effect of niclosamide on SLC7A11 transport activity (A-C) and the correlation between theoretically calculated and experimentally determined dissociation constants for various substrates and inhibitors (D). The transport activity of SLC7A11 was measured in three different TNBC cell lines (MB231, TXBR100, and MB453) as detailed in the Methods section. Niclosamide was present only during uptake. The dissociation constants for the substrates and inhibitors were determined in the present study or taken from published reports (see the text). For A-C, data are given as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. .
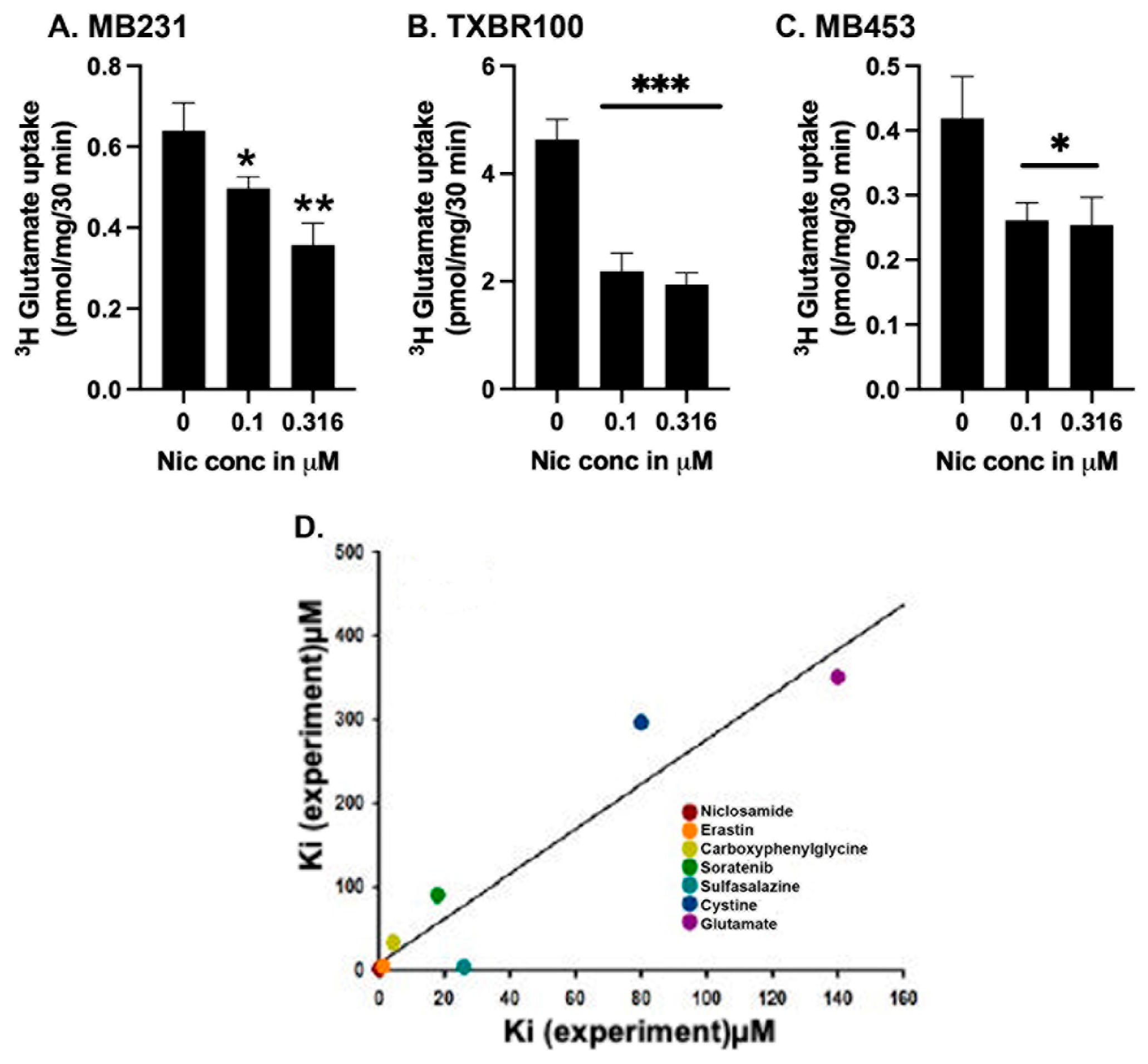
We then examined experimentally the ability of niclosamide to inhibit SLC7A11 transport activity in TNBC cells by using glutamate uptake as the readout (Fig. 5 A-C). In three different TNBC cell lines, niclosamide inhibited the uptake with an IC50 value of ~0.3 μM, an affinity at least 3 to 4 times greater than the experimental value known for erastin [38,39]. Because of the significant difference between the experimental (~0.3 μM) and theoretical (1.3 μM) values for niclosamide affinity, we compared the experimental and theoretical values for the affinities of the two substrates (cystine and glutamate) and five known inhibitors of this transporter (erastin, sulfasalazine, sorafenib, S-4-carboxy phenylglycine along with niclosamide) to see if a similar trend exists for the other substrates and inhibitors as well (Fig. 5D). The experimental and theoretical values correlated well for all of them (r2 = 0.9; p<0.001), thereby providing credibility to the potential of this theoretical approach to discover new inhibitors as we demonstrated with niclosamide.
3.5. Influence of niclosamide on Nrf2 expression in TNBC cells
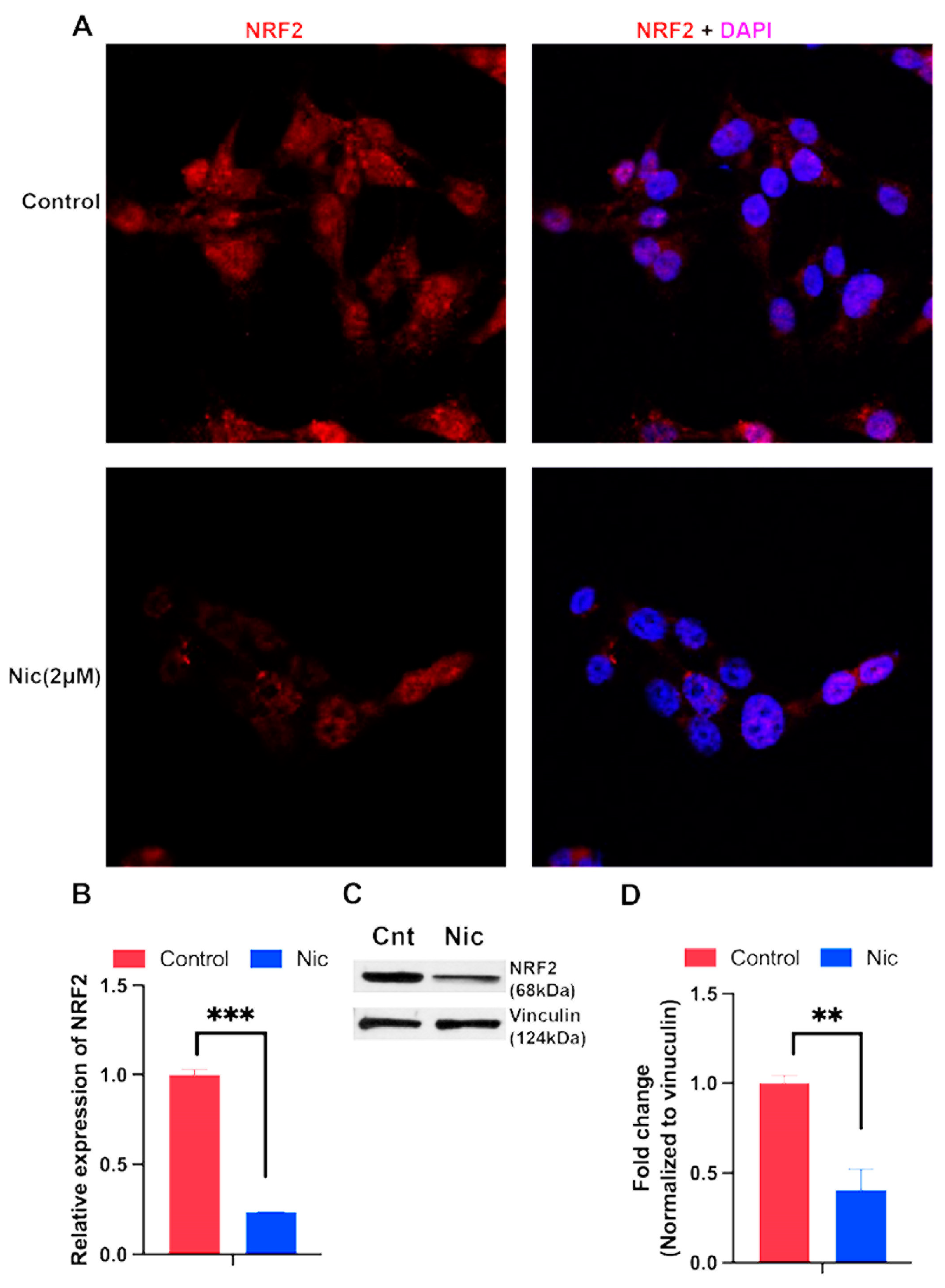
Niclosamide targets multiple signaling pathways in mammalian cells; these pathways include those involving Wnt, STAT3, mTOR, and NOTCH [40-42]. Since we found in the present study that niclosamide blocks the transport function of the cystine transporter SLC7A11, a key component of the cellular antioxidant machinery, we asked if treatment of TNBC cells with this drug would have any impact on the expression of Nrf2, a key transcription factor that protects the cells against oxidative stress. Treatment of MB231 cells with niclosamide (1-2.5 μM) for a short time (4 h) did not have any noticeable effect on Nrf2 levels or its subcellular localization as assessed by immunostaining. However, when the treatment time was extended to 24 h, niclosamide (2 μM) decreased the levels of Nrf2 signals within the nucleus (Fig. 6A). We then assessed the effects of the drug on Nrf2 mRNA levels by qRT-PCR (Fig. 6B) and protein levels by western blot (Fig. 6C). Treatment of MB231 cells with 2 μM niclosamide for 24 h significantly decreased the Nrf2 mRNA and protein levels. In TXBR100 cells however, 24-h treatment with niclosamide (2 μM) did not alter the levels of Nrf2 mRNA but significantly decreased protein level (Supplement Fig S4).
Figure 6.
Influence of niclosamide on Nrf2 mRNA and protein expression in MB231 cells. The cells were treated with niclosamide (2 μM) for 24 h and then immunofluorescence was used to localize the Nrf2 protein in cells (A). RNA and protein lysates were prepared from control and treated cells to monitor Nrf2 mRNA (B) and protein (C, D) levels. mRNA levels were determined by qRT-PCR. Protein levels were determined by western blot. For B and D, data are given as mean ± S.E. **, p < 0.01; ***, p < 0.001.
Figure 6.
Influence of niclosamide on Nrf2 mRNA and protein expression in MB231 cells. The cells were treated with niclosamide (2 μM) for 24 h and then immunofluorescence was used to localize the Nrf2 protein in cells (A). RNA and protein lysates were prepared from control and treated cells to monitor Nrf2 mRNA (B) and protein (C, D) levels. mRNA levels were determined by qRT-PCR. Protein levels were determined by western blot. For B and D, data are given as mean ± S.E. **, p < 0.01; ***, p < 0.001.
3.6. Induction of ferroptosis by niclosamide in TNBC cells
The data presented thus far above show that SLC38A5, which is upregulated in TNBC, could provide selenium to cancer cells via mediation of cellular uptake of Se-Met and that Se-Met potentiates Nrf2 signaling with resultant increase in expression and function of SLC7A11 and glutathione-synthesizing machinery. The data also show that the FDA-approved drug niclosamide blocks the transport function of SLC38A5 and SLC7A11 and also decreases the cellular levels of the antioxidant transcription factor Nrf2. This suggests that niclosamide is capable of inducing oxidative stress in cancer cells by decreasing cellular levels of glutathione. Cancer cells are “addicted” to iron as iron is needed for multiple biochemical pathways that are closely associated with cell proliferation and growth [43-45]. But excess free “labile” iron is detrimental to cells because of its ability to generate reactive oxygen species via Fenton reaction and cause breakdown of polyunsaturated fatty acids present in the form of phospholipids in biological membranes, a process known as lipid peroxidation. This pathway leads to a unique form of cell death, called ferroptosis. As such, cancer cells need to accumulate excess iron to support cell proliferation but at the same time have to find ways to protect themselves from ferroptotic cell death. They manage to do this by upregulating their antioxidant machinery. Since our studies have now shown that niclosamide interferes with this protective mechanism against ferroptosis, we asked whether niclosamide renders TNBC cells susceptible to ferroptotic cell death. We addressed this question with two different TNBC cells (MB231 and TXBR100). Ferroptosis was monitored using a fluorescent probe that is selective for byproducts of lipid peroxidation as described in the Methods section. We found that niclosamide at low micromolar concentrations (5 μM) induced ferroptosis in both cell lines (Fig. 7).
Figure 7.
Induction of ferroptosis by niclosamide (5 μM) in MB231 cells (treatment for 30 min) and TXBR100 cells (treatment for 30 min, 2 h and 4 h). Data are given as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001, all compared to their respective controls (no exposure to niclosamide).
Figure 7.
Induction of ferroptosis by niclosamide (5 μM) in MB231 cells (treatment for 30 min) and TXBR100 cells (treatment for 30 min, 2 h and 4 h). Data are given as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001, all compared to their respective controls (no exposure to niclosamide).
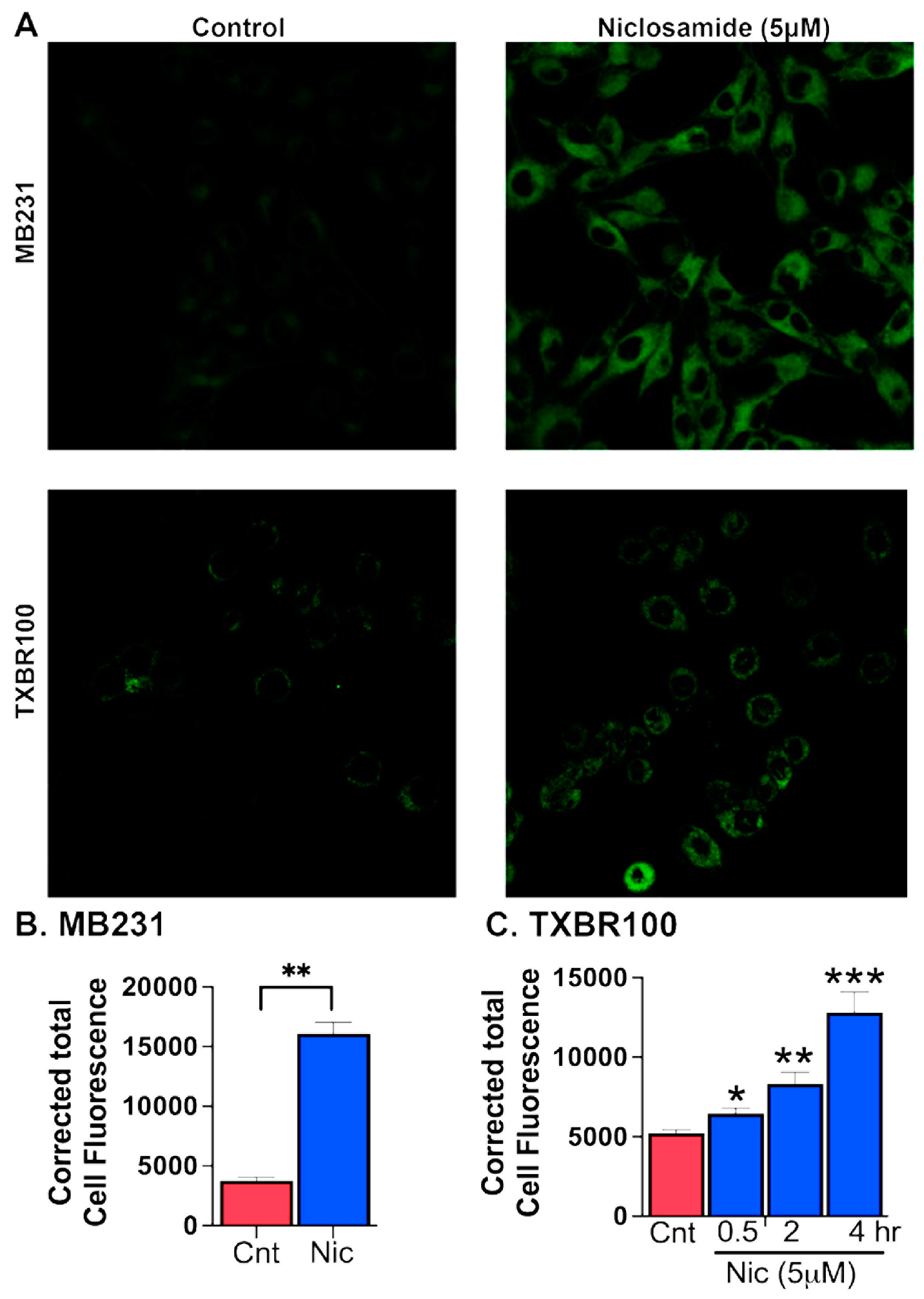
However, we noted a significant difference between MB231 cells and TXBR100 cells, the former being more sensitive to niclosamide-induced ferroptosis than the latter. In MB231 cells, a 30-min exposure to the drug was sufficient to induce marked ferroptosis whereas there was a much lower magnitude of ferroptosis induction in TXBR100cells under identical experimental conditions. However, when the exposure time to the drug was increased from 30 min to 2 h and 4 h, we saw a much higher magnitude of ferroptosis induction even in TXBR100 cells.
3.7. Biochemical consequences of niclosamide treatment in TNBC cells relevant to ferroptosis induction
To understand the molecular basis of the observed induction of ferroptotic cell death by niclosamide, we monitored the levels of glutathione and the lipid peroxidation marker malondialdehyde in control and niclosamide-treated TNBC cells. MB231 cells and TXBR100 cells were treated with 1 or 2 μM niclosamide for 24 h and then cell lysates were prepared for determination of glutathione and malondialdehyde levels. In the case of glutathione, we used buthionine sulfoximine as a positive control; this compound is a widely used potent inhibitor of glutathione synthesis. In both cell lines, treatment with niclosamide led to a marked decrease in cellular levels of glutathione (Fig. 8 A, B). As expected, buthionine sulfoximine (100 μM) caused ~90% inhibition in glutathione in both cell lines. In comparison, the decrease in glutathione levels was 60-70% when the cells were treated with 2 μM niclosamide. Niclosamide also led to a significant increase in the cellular content of malondialdehyde (Fig. 8C). These data showing a decrease in glutathione and an increase in lipid peroxidation with niclosamide corroborate with the findings that niclosamide blocks the transport activity of SLC38A5 and SLC7A11 and also suppresses Nrf2 signaling with the resultant decrease in cysteine supply and glutathione synthesis.
Figure 8.
Induction of oxidative stress and lipid peroxidation and suppression of SLC38A5/SLC7A11 expression by niclosamide in TNBC cells. MB231 and TXBR100 cells were treated with niclosamide (2 μM) for 24 h and then RNA and protein lysates were prepared from untreated and treated cells. The levels of glutathione (A, B) and malondialdehyde (MDA) (C) were measured using commercially available assay kits. The levels of mRNAs for SLC38A5 and SLC7A11 were monitored by qRT-PCR with 18S mRNA as the internal control (D, E). The protein levels of glutathione peroxidase 4 (GPX4) and ferritin heavy chain (FTH1) were analyzed by western blot (F) and the protein bands quantified by densitometry (G).
Figure 8.
Induction of oxidative stress and lipid peroxidation and suppression of SLC38A5/SLC7A11 expression by niclosamide in TNBC cells. MB231 and TXBR100 cells were treated with niclosamide (2 μM) for 24 h and then RNA and protein lysates were prepared from untreated and treated cells. The levels of glutathione (A, B) and malondialdehyde (MDA) (C) were measured using commercially available assay kits. The levels of mRNAs for SLC38A5 and SLC7A11 were monitored by qRT-PCR with 18S mRNA as the internal control (D, E). The protein levels of glutathione peroxidase 4 (GPX4) and ferritin heavy chain (FTH1) were analyzed by western blot (F) and the protein bands quantified by densitometry (G).
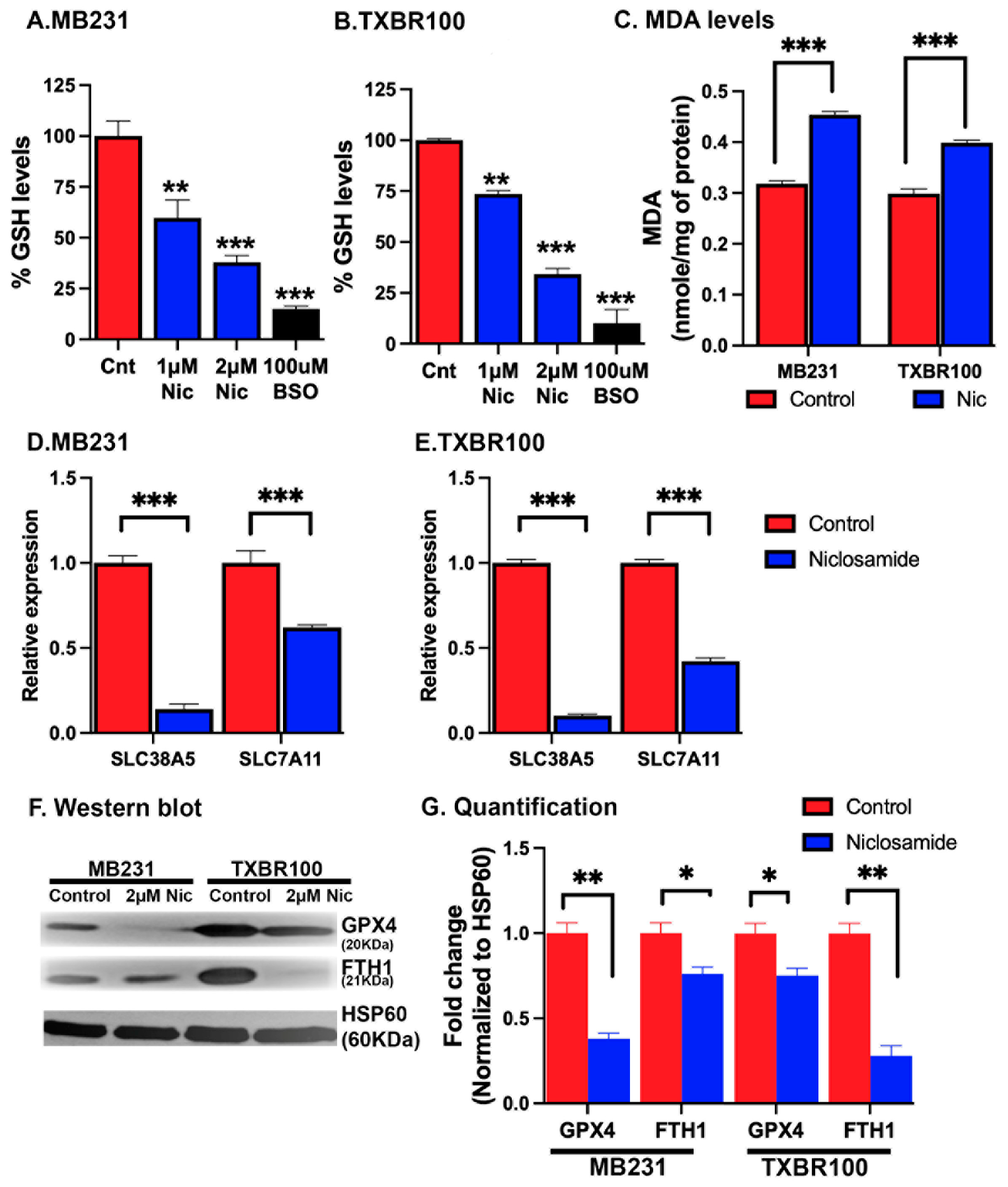
Since niclosamide interferes with multiple signaling pathways [40-42], we asked if the drug is capable of affecting the expression of SLC38A5 and SLC7A11 in addition to its ability to block the function of these two transporters by its direction interaction with the transporters. To address this question, we monitored the levels of mRNA for these two transporters in control and niclosamide-treated TNBC cells. We found that niclosamide decreased the levels of SLC38A5 mRNA and SLC7A11 mRNA to a marked extent in both cell lines (Fig. 8 D, E). The effect on SLC38A5 mRNA was much more in magnitude than the effect on SLC7A11 mRNA. These data show that niclosamide elicits a negative impact on these two transporters by two distinct mechanisms: (i) it directly interacts with the transporters and blocks their transport function, and (ii) it also suppresses the expression of these two transporters.
Since niclosamide treatment increased lipid peroxidation and ferroptosis, we monitored the impact of the drug exposure on the cellular levels of glutathione peroxidase 4 (the enzyme that removes hydrogen peroxide and lipid peroxides) and ferritin (the storage protein for iron that reduces the cellular levels of free “labile” iron). In MB231 cells as well as in TXBR100 cells, niclosamide treatment decreased glutathione peroxidase 4 protein levels (Fig. 8F, G) Ferritin levels also were lower in niclosamide-treated cells than in control cells, but the effect was much more pronounced in TXBR100 cells than in MB231 cells (Fig. 8F). The decrease in glutathione peroxidase 4 in the treated cells is expected to increase lipid peroxide-dependent membrane damage and the decrease in ferritin levels is expected to increase “labile” free iron with resultant increase in the generation of toxic hydroxyl radicals that promote lipid peroxidation. Together, these two changes lead to ferroptosis in niclosamide-treated cells.
3.8. Effect of niclosamide on colony formation and cell proliferation in TNBC cells
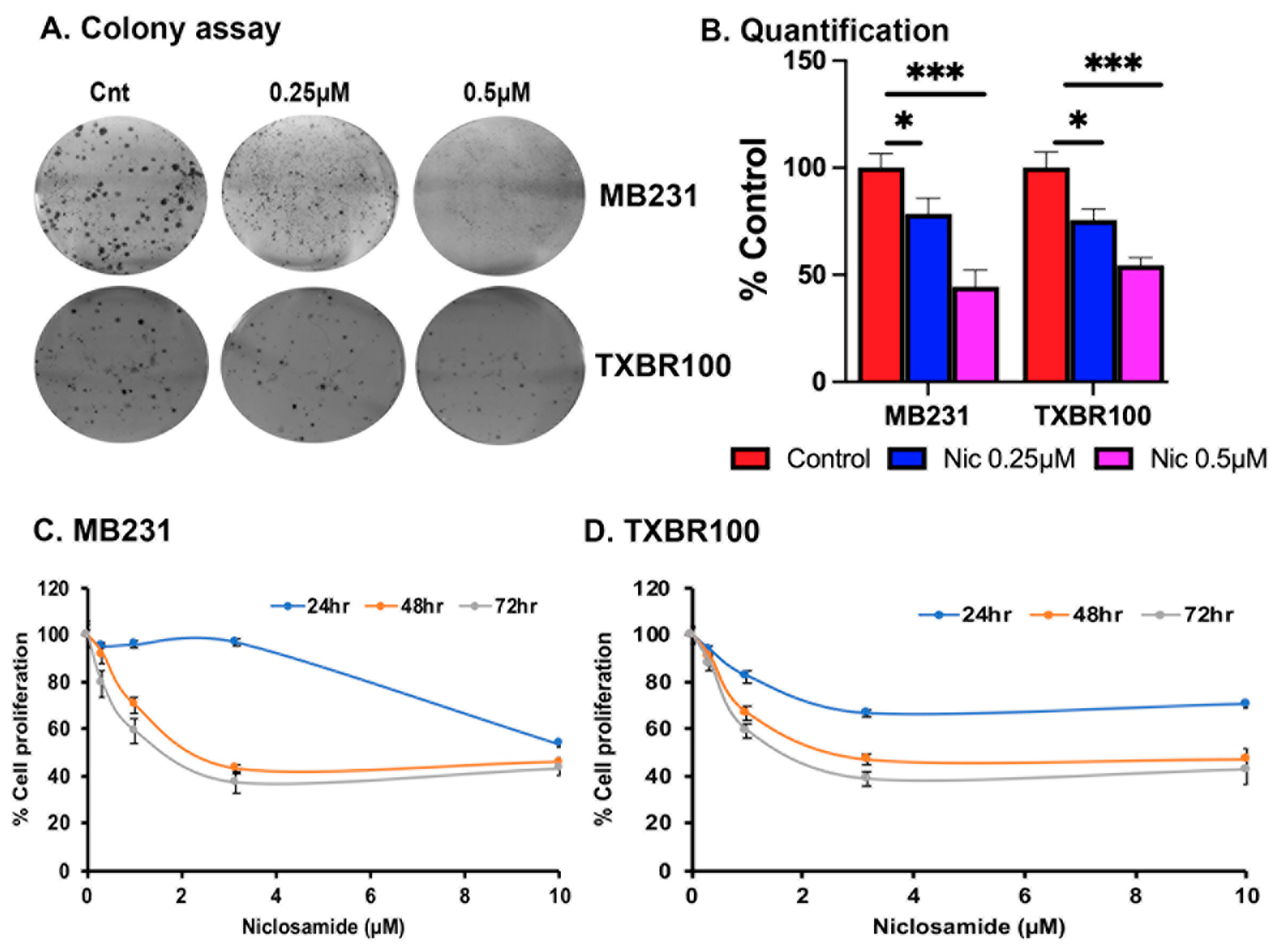
We assessed the impact of niclosamide treatment on the proliferation and colony-formation ability of TNBC cells (MB231 and TXBR100) in vitro. Exposure of the cells to niclosamide had a drastic inhibitory effect on colony formation (>50% decrease at 0.5 μM) and cell proliferation (>50% decrease at 2 μM) (Fig. 9). Even with 0.25 μM niclosamide, a significant inhibitory effect was noticeable in the colony-formation assay, though the magnitude of the effect was lower at this concentration compared to the effect observed with 0.5 μM niclosamide.
Figure 9.
Effects of niclosamide on colony formation (A, B) and cell proliferation (C, D). *, p < 0.05; ***, p < 0.001.
Figure 9.
Effects of niclosamide on colony formation (A, B) and cell proliferation (C, D). *, p < 0.05; ***, p < 0.001.
3.9. Effect of niclosamide on the growth of a TNBC cell line into tumor when xenografted in mice
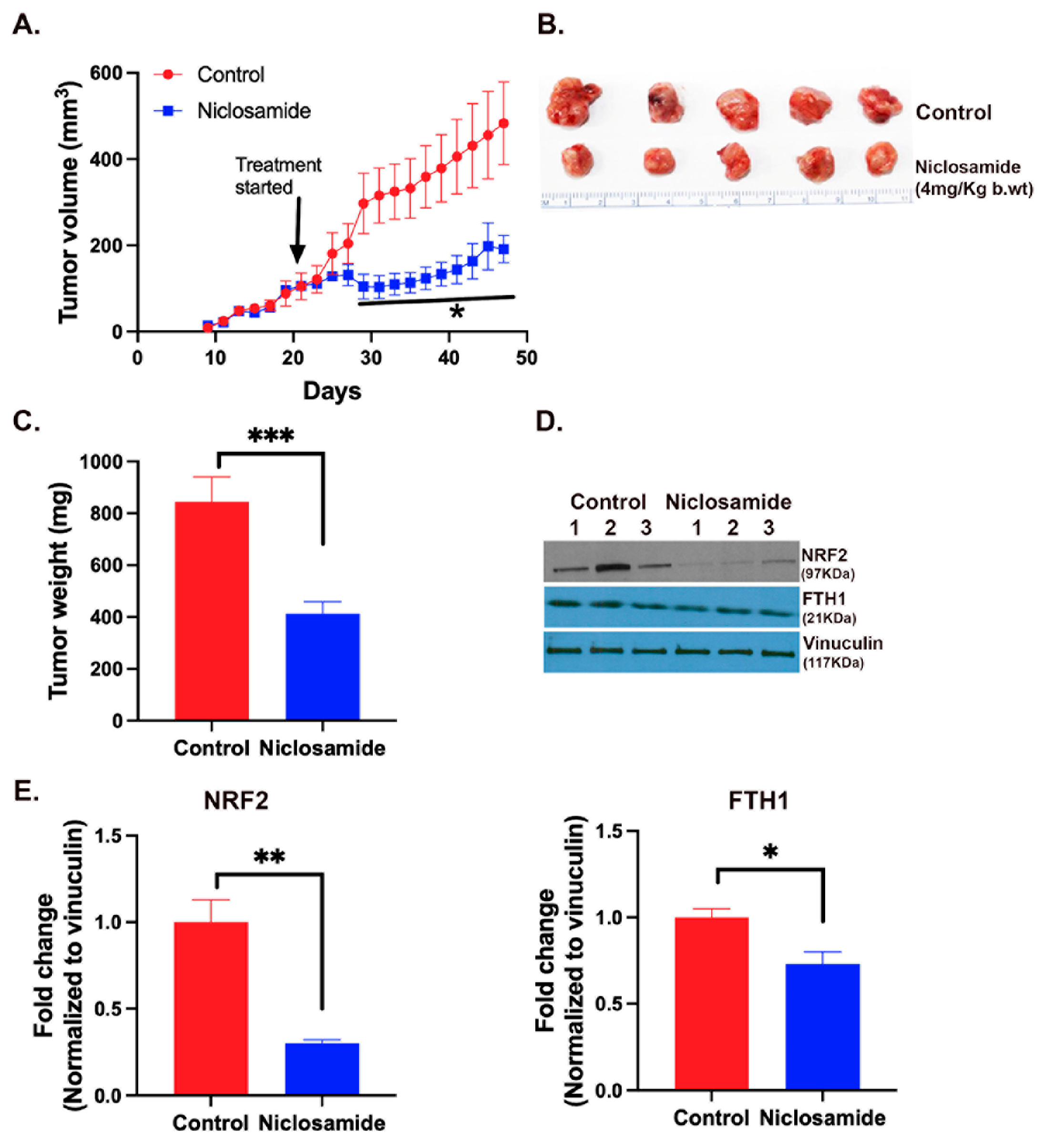
The in vitro experiments described above suggested a potent tumor-suppressive activity of niclosamide by the ability of the drug to induce oxidative stress and iron-induced ferroptosis via blockade of the expression and function of SLC38A5 and SLC7A11. Therefore, we examined the anticancer efficacy of this drug in vivo using the mouse xenograft model with the TNBC cell line MB231. We initiated the drug administration when the tumors grew to a size of ~100-150 mm3. The drug was given daily by intraperitoneal injection at a dose of 4 mg/kg. We found a drastic reduction in the growth of the tumors in drug-exposed mice (Fig. 10A-C). At the end of the experimental period, the tumors were harvested and prepared for qRT-PCR and western blot. The levels of mRNAs for Nrf2, SLC7A11 and SLC38A5 were markedly decreased in niclosamide-exposed tumors in comparison with control tumors (Supplemental Fig. S5). Western blot analysis showed that Nrf2 protein levels were also markedly decreased in niclosamide-exposed tumors in comparison with control tumors (Fig. 10 D, E). The protein levels of the ferritin heavy chain also decreased (Fig. 10 D, F) even though the magnitude of the decrease was smaller than that for Nrf2. These findings provide convincing evidence for the in vivo efficacy of niclosamide as an anticancer drug for TNBC and corroborative in vivo evidence for the molecular aspects of the drug’s anticancer effects which were observed in vitro.
Figure 10.
Tumor-suppressive efficacy of niclosamide in vivo in mouse xenograft. MB231 cells were injected into mammary fat pad of nude mice and the tumors were allowed to grow to an approximate size of 100-150 mm3. Niclosamide administration began at this point in one group of mice while the other group left untreated. The drug was given intraperitoneally daily (4 mg/kg) and the treatment continued for 4 weeks. Tumor volume during the experimental period (A) and tumor weights at the end of the experimental period (B, C) are given. Tumor tissues were prepared for western blot to monitor the protein levels of Nrf2 and ferritin heavy chain (FTH1) (D, E). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Figure 10.
Tumor-suppressive efficacy of niclosamide in vivo in mouse xenograft. MB231 cells were injected into mammary fat pad of nude mice and the tumors were allowed to grow to an approximate size of 100-150 mm3. Niclosamide administration began at this point in one group of mice while the other group left untreated. The drug was given intraperitoneally daily (4 mg/kg) and the treatment continued for 4 weeks. Tumor volume during the experimental period (A) and tumor weights at the end of the experimental period (B, C) are given. Tumor tissues were prepared for western blot to monitor the protein levels of Nrf2 and ferritin heavy chain (FTH1) (D, E). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
4. Discussion
Among the five members of the SLC38 gene family which have been characterized in detail as plasma membrane amino acid transporters, SLC38A5 and SLC38A3 are upregulated in TNBC. SLC38A2 and SLC38A4 are downregulated and SLC38A1 remains unaltered. We have recently shown that SLC38A5 functions as a tumor promoter in TNBC [13,15]. SLC38A2 has also been shown to promote TNBC [31] even though its expression is downregulated [13]. We have no information on the potential role of SLC38A3 which is upregulated and that of SLC38A4 which is downregulated. SLC38A3 and SLC38A5 possess almost identical functional features [30] and therefore it is likely that this transporter also promotes the growth and proliferation of TNBC cells. On the other hand, SLC38A4 has unique functional features such as the ability to transport not only neutral amino acids but also cationic amino acids [46,47]. It is also an imprinted gene [48] and has been shown to function as a tumor suppressor in some cancers [49]. Even though the downregulation of the transporter in TNBC may suggest a similar role in this cancer, it has not yet been determined experimentally.
Previous studies from our lab have uncovered an unconventional function of SLC38A5 [15]. Its transport function as an amino acid-dependent Na+/H+ exchanger couples amino acid entry into cells via this transporter to intracellular alkalinization, which promotes macropinocytosis. Since SLC38A3 functions in an identical manner in terms of transport modality, we have postulated that this transporter might also promote macropinocytosis even though it is only speculative at this time and has not yet been validated experimentally [16]. In the present study, we have uncovered another important functional feature of SLC38A5 in TNBC. It plays a role in selenium nutrition in TNBC cells by its ability to deliver Se-Met into cells. Selenium is obligatory for the antioxidant function in mammalian cells and therefore SLC38A5-mediated delivery of selenium ought to be an essential feature of this transporter as a tumor promoter in TNBC. In addition to the role in selenium nutrition, SLC38A5-mediated delivery of Se-Met plays a role in the control of Nrf2 signaling. Since SLC7A11 is an important target of Nrf2-mediated transcriptional activity, this suggests a functional coupling between SLC38A5 and SLC7A11 with Se-Met as an intermediate. SLC7A11 has already been shown to function as a tumor promoter in TNBC [20,21]. Therefore, the ability of SLC38A5 to potentiate Nrf2 signaling with resultant increase in SLC7A11 expression is important, underscoring the tumor-promoter role of SLC38A5.
Based on the data presented in this paper, we conclude that there are two aspects relating to the antioxidant function of SLC38A5 in TNBC cells. First, the function of the transporter has a direct positive effect on the antioxidant machinery of tumor cells by maintaining the optimal selenium nutrition via delivery of Se-Met. This micronutrient is obligatory for the function of glutathione peroxidases. Second, the SLC38A5-mediated delivery of Se-Met into tumor cells potentiates the activity of the transcription factor Nrf2, which not only increases the ability of the tumor cells to synthesize glutathione by upregulating the expression of GCLC and GCLM to promote the first step in the glutathione synthetic pathway but also induces the expression of the transporter SLC7A11 which provides cysteine (in the form of cystine), the rate-limiting amino acid for glutathione synthesis in tumor cells. Thus, Se-Met enables the functional coupling between SLC38A5 and SLC7A11, and this crosstalk between the two transporters forms an integral part of the antioxidant machinery in tumor cells.
The present study has also explored the impact of the FDA-approved drug niclosamide on the antioxidant machinery of TNBC cells. We have already shown that niclosamide is a potent inhibitor of SLC38A5 [15]. Therefore, we could speculate that the ability of TNBC cells to acquire Se-Met via SLC38A5 would be impaired when exposed to niclosamide, thus decreasing cellular levels of Se and hence the catalytic activity of glutathione peroxidases. The experiments described in the present study have discovered another important action of niclosamide that is related to the antioxidant machinery of TNBC cells. This drug is also a potent inhibitor of SLC7A11. In fact, the potency of inhibition observed identifies niclosamide as the most potent inhibitor of SLC7A11 known to date. This discovery has profound implications for the anticancer potential of niclosamide because SLC7A11 is considered as one of the promising drug targets for the treatment of not only TNBC but also other cancers. Interestingly, the ability of niclosamide to interfere with the antioxidant machinery of TNBC cells does not stop with its direct effect as an inhibitor of SLC38A5 and SLC7A11. The drug also suppresses the expression of both transporters. We have not explored the signaling pathway that is responsible for this effect, but niclosamide is known to elicit its pharmacological effects by suppressing multiple signaling pathways, including Wnt, STAT3, mTOR, etc. Additional studies are needed to tease out which of these pathways affected by niclosamide is responsible for the suppression of SLC38A5 and SLC7A11 in TNBC cells when exposed to the drug. It is possible that a single pathway may not be involved in the niclosamide-dependent regulation of SLC38A5 and SLC7A11. As multiple signaling pathways are affected by niclosamide, it is feasible that more than one signaling mechanisms participate in the suppression of SLC38A5 and SLC7A11.
When the expression and function of SLC38A5 and SLC7A11 are impaired, one would expect decreased levels of glutathione and increased levels lipid peroxidation in niclosamide-treated cells. This is indeed supported by the results of the experiments described in the present study. Niclosamide treatment decreases glutathione levels and increases malondialdehyde levels in TNBC cells. In addition, niclosamide also decreases the levels of glutathione peroxidase 4 (GPX4) and ferritin (H chain). The exact mechanisms involved in this process remain to be investigated. It is known that glutathione peroxidase mRNA stability is influenced by the selenium nutritional status of the cells [50]. Selenium deficiency decreases the stability of GPX mRNAs via their 3’-untranslated regions. Therefore, we speculate that since the expression and function of SLC38A5 are drastically suppressed by niclosamide in TNBC cells, it would cause selenium deficiency due to impaired Se-Met delivery, which would then be expected to decrease the stability of GPX4 mRNA and hence its protein levels. Alternatively, any of the signaling pathways affected by niclosamide could also mediate the effect. The same is true for the decrease in ferritin levels. More work is needed to deduce the mechanisms involved in these effects.
The observed decrease in GPX4 and ferritin is directly related to the potentiating effect of niclosamide on ferroptosis in TNBC cells. The decrease in GPX4, coupled with the decrease in glutathione levels, would enhance lipid peroxidation, which is further supported by the observed increase in malondialdehyde levels. Ferritin sequesters iron and decreases the cellular levels of labile iron, thus protecting the cells from the potential prooxidant activity of free iron. Therefore, the decrease in ferritin levels in niclosamide-treated cells would be expected to increase the levels of labile iron, consequently enhancing lipid peroxidation and hence ferroptosis. Since tumor cells are known to be “addicted” to iron to promote their survival and proliferation, these cells potentiate their antioxidant machinery to protect themselves from ferroptosis. Our studies show that niclosamide effectively interferes with this protective mechanism, thus making the tumor cells susceptible to iron-induced cell death. This offers a novel anticancer mechanism for niclosamide.
The anticancer efficacy of niclosamide has been demonstrated in several cancers, both in vitro using cultured cells and in vivo using mouse xenografts [20-24]. The results of the present study offer further supportive evidence for the potential of niclosamide as an anticancer drug. A major hindrance in successful use of this drug for cancer treatment appears to be the low bioavailability when the drug is given orally [51]. This does not necessarily negate the therapeutic potential of this drug. Studies are ongoing to increase the bioavailability of niclosamide either by changing the formulation or by using a prodrug approach. In the former, nanoformulations could enhance the bioavailability; in the latter, niclosamide can be structurally modified such that the modified drug is recognized as a transportable substrate for intestinal nutrient transporters (e.g., peptide transporter), thus increasing the oral bioavailability of the drug. Therefore, niclosamide holds great potential for the treatment of cancers and our present study provides strong support for its use in the treatment of TNBC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplemental Figures Fig. S1 – S5; Supplemental Table 1.
Author Contributions
Conceptualization, M.M., S.S., Y.D.B. and V.G.; methodology, M.M. and S.S.; validation, S.S., G.D.N. and N.T.N.; molecular docking, V.J.M.; investigation, M.M., S.S., G.D.N. and S.R.S.; data curation, S.S. and V.G.; writing—original draft preparation, M.M., S.S. and V.G.; writing—review and editing, N.T.N. and Y.D.B.; supervision, S.S. and V.G.; project administration, V.G.; funding acquisition, V.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health, grant number CA277140 and the APC was funded by CA277140.
Institutional Review Board Statement
The animal study protocol was approved by the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee (IACUC protocol number 18005) and the Institutional Review Board (IRB). The animal experiments were all carried out at the Texas Tech University Health Sciences Center, Lubbock, TX.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data related to this study are given in the manuscript and in the Supplemental Materials section, which is freely available to the scientific community and the public.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gonciarz, R.L.; Collisson, E.A.; Renslo, A.R. Ferrous iron-dependent pharmacology. Trends Pharmacol. Sci. 2021, 42, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.Y.; Mina, E.; Roetto, A.; Porporato, P.E. Iron: An essential element of cancer metabolism. Cells 2020, 9, 2591. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Rather, G.M.; Pramono, A.A.; Szekely, Z.; Bertino, J.R.; Tedeschi, P.M. In cancer, all roads lead to NADPH. Pharmacol. Ther. 2021, 226, 107864. [Google Scholar] [CrossRef] [PubMed]
- Simmen, F.A.; Alhallak, I.; Simmen, R.C.M. Malic enzyme 1 (ME1) in the biology of cancer: it is not just intermediary metabolism. J. Mol. Endocrinol. 2020, 65, R77–R90. [Google Scholar] [CrossRef]
- Weaver, K.; Skouta, R. The selenoprotein glutathione peroxidase 4: From molecular mechanisms to novel therapeutic opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef]
- Nakanishi, T.; Sugawara, M.; Huang, W.; Martindale, R.G.; Leibach, F.H.; Ganapathy, M.E.; et al. Structure, function, and tissue expression pattern of human SN2, a subtype of the amino acid transport system N. Biochem. Biophys. Res. Commun. 2001, 281, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Kekuda, R.; Fei, Y.J.; Hatanaka, T.; Sugawara, M.; Martindale, R.G.; et al. Cloning and functional characterization of a new subtype of the amino acid transport system N. Am. J. Physiol. Cell Physiol. 2001, 281, C1757–C1768. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Sennoune, S.R.; Sharma, M.; Thangaraju, M.; Suresh, V.V.; Sniegowski, T.; et al. Expression and function of SLC38A5, an amino acid-coupled Na+/H+ exchanger, in triple-negative breast cancer and its relevance to macropinocytosis. Biochem. J. 2021, 478, 3957–3876. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, T.; Rajasekaran, D.; Sennoune, S.R.; Sukumaran, S.; Chen, F.; Fokar, M.; et al. Amino acid transporter SLC38A5 is a tumor promoter and a novel therapeutic target for pancreatic cancer. Sci. Rep. 2023, 13, 16863. [Google Scholar] [CrossRef] [PubMed]
- Sennoune, S.R.; Nandagopal, G.D.; Ramachandran, S.; Mathew, M.; Sivaprakasam, S.; Jaramillo-Martinez, V.; et al. Potent inhibition of micropinocytosis by niclosamide in cancer cells: A novel mechanism for the anticancer efficacy of the antihelminthic. Cancers (Basel) 2023, 15, 759. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Mathew, M.; Sivaprakasam, S.; Ramachandran, S.; Ganapathy, V. Unconventional functions of amino acid transporters: Role in micropinocytosis (SLC38A5/SLC38A3) and diet-induced obesity/metabolic syndrome (SLC6A19/SLC6A14/SLC6A6). Biomolecules 2022, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. SLC6A14 and SLC38A5 drive the glutaminolysis and serine-glycine-one-carbon pathways in cancer. Pharmaceuticals (Basel) 2021, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Ananth, S.; Miyauchi, S.; Thangaraju, M.; Jadeja, R.N.; Bartoli, M.; Ganapathy, V.; Martin, P.M. Selenomethionine (Se-Met) induces the cystine/glutamate exchanger SLC7A11 in cultured human retinal pigment epithelial (RPE) cells: Implications for antioxidant therapy in aging retina. Antioxidants (Basel) 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padro, M.; Daemen, A.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef]
- Hasegawa, M.; Takahashi, H.; Rajabi, H.; Alam, M.; Suzuki, Y.; Yin, L.; et al. Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget 2016, 7, 11756–11769. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, A.M.; Ye, J. The magic bullet: Niclosamide. Front. Oncol. 2022, 12, 1004978. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mook, Jr. R.A.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; Pacifico, T.; Luiz-Ferreira, A.; Monteleone, G.; Laudisi, F. Anthelmintic drug as emerging immune modulators in cancer. Int. J. Mol. Sci. 2023, 24, 6446. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, J.P.; Thangaraju, M.; Liu, K.; Ha, Y.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: role of cystine/glutamate exchanger. Biochem. J. 2009, 424, 243–52. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Kekuda, R.; Wang, H.; Prasad, P.D.; Mehta, P.; Huang, W.; et al. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2001, 42, 47–54. [Google Scholar] [PubMed]
- Parker, J.L.; Deme, J.C.; Kolokouris, D.; Kuteyi, G.; Biggin, P.C.; Lea, S.M.; Newstead, S. Molecular basis for redox control by the human cystine/glutamate antiporter system xc-. Nat. Commun. 2021, 12, 7147. [Google Scholar] [CrossRef]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; et al. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Broer, S. The SLC38 family of sodium-amino acid co-transporters. Pflugers Arch. 2014, 466, 155–172. [Google Scholar] [CrossRef]
- Morotti, M.; Zois, C.E.; El-Ansari, R.; Craze, M.L.; Rakha, E.A.; Fan, S.J.; et al. Increased expression of glutamine transporter SNAT2/SLC38A2 promotes glutamine dependence and oxidative stress resistance, and is associated with worse prognosis in triple-negative breast cancer. Br. J. Cancer 2021, 124, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Radzishevsky, I.; Odeh, M.; Bodner, O.; Zubedat, S.; Shaulov, L.; Litvak, M.; et al. Impairment of serine transport across the blood-brain barrier by deletion of Slc38a5 causes developmental delay and motor dysfunction. Proc. Natl. Acad. Sci. USA 2023, 120, e2302780120. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Sun, X.; Li, P.; Shen, X.; Fang, Y. Selenium in cereals: Insight into species of the element from total amount. Compr. Rev. Food Sci. Food Saf. 2021, 20, 2914–2940. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, J.K.; Power, R.; Toborek, M. Biological activity of selenium: Revisited. IUBMB Life 2016, 68, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ananth, S.; Miyauchi, S.; Thangaraju, M.; Jadeja, R.N.; Bartoli, M.; Ganapathy, V.; Martin, P.M. Selenomethionine (Se-Met) induces the cystine/glutamate exchanger SLC7A11 in cultured human retinal pigment epithelial (RPE) cells: Implications for antioxidant therapy in aging retina. Antioxidants (Basel) 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Promsote, W.; Makala, L.; Li, B.; Smith, S.B.; Singh, N.; Ganapathy, V.; et al. Monomethylfumarate induces γ-globin expression and fetal hemoglobin production in cultured human retinal pigment epithelial (RPE) and erythroid cells and in intact retina. Invest. Ophthalmol. Vis. Sci. 2014, 55, 5382–5393. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; et al. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [PubMed]
- Figuera-Losada, M.; Thomas, A.G.; Stathis, M.; Stockwell, B.R.; Rojas, C.; Slusher, B.S. Development of a primary microglia screening assay and its use to characterize inhibition of system xc- by erastin and its analogs. Biochem. Biophys. Rep. 2017, 9, 266–272. [Google Scholar] [CrossRef]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Repurposing of anthelminthics as anticancer drugs. Oncomedicine 2018, 3, 1–8. [Google Scholar] [CrossRef]
- Laudisi, F.; Maronek, M.; di Grazia, A.; Monteleone, G.; Stolfi, C. Repositioning of anthelmintic drugs for the treatment of cancers of the digestive system. Int. J. Mol. Sci. 2020, 21, 4957. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Jaramillo-Martinez, V.; Mathew, M.; Suresh, V.V.; Sivaprakasam, S.; Bhutia, Y.D.; Ganapathy, V. Sigma receptors: Novel regulators of iron/heme homeostasis and ferroptosis. Int. J. Mol. Sci. 2023, 24, 14672. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Nakanishi, T.; Fei, Y.J.; Martindale, R.G.; Ganapathy, M.E.; Leibach, F.H.; Ganapathy, V. Structure and function of ATA3, a new subtype of amino acid transport system A, primarily expressed in the liver and skeletal muscle. Biochim. Biophys. Acta 2000, 1509, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Huang, W.; Ling, R.; Prasad, P.D.; Sugawara, M.; Leibach, F.H.; Ganapathy, V. Evidence for the transport of neutral as well as cationic amino acids by ATA3, a novel and liver-specific subtype of amino acid transport system A. Biochim. Biophys. Acta 2001, 1510, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Dean, W.; Konfortova, G.; Kelsey, G. Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res. 2003, 13, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.H.; Wang, T.T.; Liu, X.N.; Zhu, X.T.; Dai, Y.Z.; et al. SLC38A4 functions as a tumour suppressor in hepatocellular carcinoma through modulating Wnt/β-catenin/MYC/HMGCS2 axis. Br. J. Cancer 2021, 125, 865–876. [Google Scholar] [CrossRef]
- Muller, C.; Wingler, K.; Brigelius-Flohe, R. 3’-UTRs of glutathione peroxidases differentially affect selenium-dependent mRNA stability and selenocysteine incorporation efficiency. Biol. Chem. 2003, 374, 11–18. [Google Scholar] [CrossRef]
- Barbosa, E.J.; Lobenberg, R.; Barros de Araujo, G.L.; Bou-Chacra, N.A. Niclosamide repositioning for treating cancer: Challenges and nano-based drug delivery opportunities. Eur. J. Pharm. Biopharm. 2019, 141, 58–69. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



