
Submitted:
29 November 2023
Posted:
30 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Severe cases of SARS-CoV-2 infection are characterized by an immune response that leads to the overproduction of pro-inflammatory cytokines, resulting in damage to the lungs and other organs. This remarkable increase in cytokines and other inflammatory molecules is primary caused by viral proteins, in particular ORF8, a unique accessory protein specific to SARS-CoV-2. In addition to its role in cytokine induction, ORF8 is attributed with various other functions, including its ability to evade type I interferon responses. Despite the evidence, the mechanisms through which ORF8 exerts these functions remains a subject of controversy. In this mini review, we discuss the multifaceted roles of ORF8 as a modulator of cytokine response, focusing on type I interferon and IL-6. We also discuss data from our studies indicating that ORF8 augments production of IL-6 induced by Poly(I:C) in human embryonic kidney (HEK)-293 and monocyte-derived dendritic cells (mono-DCs).
Keywords:
SARS CoV-2
; COVID-19
; ORF8
; cytokine storm
1. Introduction
SARS-CoV-2 and ORF8
Severe acute respiratory syndrome (SARS) coronavirus (CoV) type 2 (SARS-CoV-2), the causative agent of coronavirus infectious disease 2019 (COVID-19), is a member of the β-coronavirus family, along with SARS-CoV and middle east respiratory syndrome (MERS) virus [1,2,3,4]. In the majority of healthy individuals, SARS-CoV-2 infection is asymptomatic or causes mild to moderate illness with symptoms as fever, headache, cough and breathlessness. However, severe cases develop acute respiratory distress syndrome (ARDS) and acute lung injury, leading to morbidity and mortality [2,3]. It is well established that the severity of COVID-19 correlates with viral replication and hyper-responsiveness of the host immune system. The latter frequently involves an excessive production of cytokines by the host, referred to as ‘cytokine storm’. This exacerbated cytokine response can lead to a multi-organ failure and, in some cases, to a fatal outcome [5,6,7]. COVID-19 patients with severe disease have an exacerbated inflammatory response, evidenced by high levels of inflammatory markers (C-reactive protein, ferritin, D-dimer) in the blood, an increased neutrophil-to-lymphocyte ratio, high serum levels of pro-inflammatory chemokines and cytokines, such as interleukins IL-2, IL-6, IL-10 and IFN-γ, and low serum levels of type I and III interferons (IFNs) [8,9,10,11,12,13,14].
The SARS-CoV-2 genome is a positive-strand RNA that comprises 14 open reading frames (ORFs). ORF1a and ORF1b, the largest ORFs, are translated into polyproteins pp1a and pp1ab, which are cleaved to produce non-structural proteins Nsp1 to Nsp6. The other ORFs encode the structural proteins: spike (S), membrane (M), envelope (E) and nucleocapsid (N), and the accessory proteins ORF3a, ORF4, ORF6, ORF7a, ORF7b, ORF8, ORF9b and ORF10 [15]. The accessory proteins of coronaviruses are not considered to be primarily required for viral replication in vitro. However, those proteins serve important functions during virus infection in vivo, contributing to immune evasion, cytokine induction and enhanced virulence [16,17,18].
SARS-CoV-2 ORF8 is poorly conserved among other human coronaviruses, but shares 95% amino acid sequence similarity with Bat-RaTG13-CoV, indicating its origin from Bat-RaTG13-CoV ORF8 [19,20]. The SARS-CoV-2 ORF8 is particularly relevant due to its high susceptibility to deletions and to point mutations, and it is associated with disease’s progression and outcome [21,22,23,24,25,26]. Notably, a mutation at position 84 that changes leucine to serine (ORF8 L84S) was the most frequent mutation in the first six months of the pandemic and underwent significant selection pressure [26,27]. This mutation has been related to mild disease outcome [25], similarly to SARS-CoV-2 strains that lack the ORF8 gene [28], indicating that ORF8 is a virulence factor.
ORF8 is abundantly secreted both in vitro and in vivo, and it is highly immunogenic [29,30,47,48,49]. Among the accessory proteins of SARS-CoV-2, ORF8 has the largest protein interactome network [30], and several studies performed in the past few years have reported several biological properties of this protein, including immune evasion and signaling activation.
Here we summarize research on SARS-CoV-2 ORF8 as a modulator of the cytokine response, in particular the pathways linked to type I IFN (IFN-α and IFN-β) and IL-6. ORF8 structure, evolution, roles in adaptive immunity and overall functions have been extensively reviewed elsewhere [31,32,33,34,35,36]. We focus on ORF8’s capacity to modulate type I IFN and other cytokine responses, discussing the existing data and introduce new findings from our laboratory, indicating that the ORF8 L84S variant augments the levels of IL-6 and IFN-β induced by polyinosinic:polycytidylic acid (poly(I:C)) in human embryonic kidney (HEK)-293 and monocyte-derived dendritic cells (mono-DCs).
Cytokine Responses to SARS-CoV-2
Reduced innate antiviral defenses associated with exuberant inflammatory responses are hallmark features of COVID-19 [8,9,10,11,12,13]. Clinical and experimental data indicate an exacerbated production of pro-inflammatory cytokines, in particular IL-6, associated with a deficient production of type I and III IFNs in infected individuals, cells and animal models [11,26,37]
Innate antiviral immunity is triggered by the recognition of viral pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs), leading to production of type I IFNs and pro-inflammatory cytokines. These cytokines function to inhibit viral replication and to regulate induction of adaptive immunity [38]. Type I IFNs constitute one of the first lines of defense against viruses. IFN production leads to induction of several IFN-stimulated genes (ISGs), via the janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, that encode proteins functioning to restrict viral replication in infected and neighboring cells [39,40].
In RNA viruses, including SARS-CoV, SARS-CoV-2 and MERS virus, the double-strand RNA (dsRNA) generated during genome replication and transcription is sensed in the endosome by Toll-like receptors (TLRs) 3 and 7, or in the cytoplasm by the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), RIG-I and/or melanoma differentiation-associated protein (MDA5) [41]. RIG-I and MDA5 interact with the adapter mitochondrial antiviral signaling (MAVS) protein, recruiting two IκB kinase (IKK)-related kinases, TANK-binding kinase 1 (TBK1) and inducible IκB kinase (IKKi). These kinases phosphorylate interferon regulatory factor (IRF) 3 and 7, leading to their dimerization and translocation to the nucleus, resulting in the activation of IFN-α/β expression.
Additionally, MAVS recruits TANK1 by tumor necrosis factor receptor associated factor 6 (TRAF6), and activates the NF-κB signaling pathway, leading to cytokine production. Alternatively, sensing of viral RNAs by most TLRs, except for TLR3, involves the adaptor myeloid differentiation primary response gene 88 (MyD88). After TLR engagement, MyD88 forms a complex with IL-1 receptor-associated kinase (IRAK) family members, including IRAK1, IRAK2 and IRAK4, referred to as the Myddosome. IRAK1 associates with the RING-domain E3 ubiquitin ligase TRAF6, leading to activation of TAK1 followed by the activation of the NF-κB and mitogen-activated protein kinase (MAPK) pathways and the production of pro-inflammatory cytokines [42,43].
TLRs localized in endosomes (TLR3, TLR7, TLR8 and TLR9) activate NF-κB and IRF7. While TLR7, TLR8 and TLR9 use MyD88, TLR3 uses the adaptor TIR-domain-containing adaptor-inducing interferon-β (TRIF), and TLR4 uses both MyD88 and TRIF adaptors. TRIF binds to TRAF3, which then recruits the IKK-related kinases TBK1 and IKKε, activating IRF3 that mediates transcription of type I IFNs. Additionally, TRIF interacts with TRAF6 and promotes the activation of NF-κB and MAPKs [42,43].
Notably, recent studies have shown that RNA virus infection activates the cytoplasmic DNA sensor cGAS/STING by directly recognizing viral components or by sensing cellular DNA released from mitochondria or nuclei during cellular stress [44]. Accordingly, Neufeldt et al (2022) showed that SARS-CoV-2 directs a cGAS-STING-mediated, NF-κB-driven inflammatory immune response in human epithelial cells that likely contributes to inflammatory responses seen in patients [45].
The viral antagonism of host innate immune responses is critical for virus replication and often determines the outcome of infection. Several SARS-CoV-2 proteins can block innate immunity, mainly IFN induction or signaling pathways. These proteins include Nsp1, Nsp3, Nsp12, Nsp13, Nsp14, Nsp15, M, N, S, ORF3b, ORF6 and ORF8 [11,46,47,48].
Among the accessory proteins, SARS-CoV-2 ORF8 is particularly remarkable. ORF8 is a poorly conserved accessory protein among human coronaviruses [36], which is highly expressed, secreted from infected cells [49,50,51,52] and very immunogenic in SARS-CoV-2-infected individuals [29,46,47,48,54,56,57]. ORF8 has the largest protein interactome network of all the accessory proteins, and several of its biological functions are still being investigated. ORF8 interacts with the major histocompatibility complex class I (MHC-I), targeting it for degradation at lysosomes and causing SARS-CoV-2-infected cells to be less resistant to lysis by cytotoxic T-cells [54]. ORF8 has also been implicated in the escape of humoral immune responses. It binds to monocytes causing a decrease in the levels of CD16 and reduction in the ability to mediate antibody-dependent cellular cytotoxicity (ADCC) [55]. Additionally, its interaction with the human complement components C3/C3b and their metabolites leads to complement inhibitory activity [56]. In particular, the roles of ORF8 in modulation of type I IFN and other cytokine responses are discussed.
SARS-CoV-2 ORF8 as a Type I IFN Antagonist
Several studies investigated the effect of SARS-CoV-2 ORF8 on type I IFN production. Li et al (2020) showed that ORF8 inhibits IFN-β- and NF-κB-responsive promoters induced by Sendai virus in a luciferase reporter system in HEK-293 cells. Moreover, ORF8 also inhibited IFN induction via the IFN-stimulated response element (ISRE) after treatment with IFN-β, indicating that the protein is able to inhibit both IFN induction and signalling [57]. Additionally, Rashid et al (2021) showed that both ORF8 84L and ORF8 L84S induce endoplasmic reticulum stress by activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1) pathways. At the same time, ORF8 exhibited IFN antagonist function, as demonstrated by a decrease in IRF3 nuclear translocation, IFN-β production and expression of ISG15 and ISG56 after poly(I:C) transfection of HEK-293 cells [58]. Further evidence suggests that ORF8 causes deamidation of IRF3 via cellular CTP synthetase 1 (CTPS1), resulting in loss of binding to IRF3-responsive promoters and reduced IFN expression [59].
Similar inhibitory activities of ORF8 84L and ORF8 L84S were also shown in another study in which both variants inhibited production of IFN-β, MDA5, RIG-I, ISG15, ISG56, IRF3 and other IFN-related genes after poly(I:C) transfection in HeLa cells. In this work, ORF8 was shown to interact with heat shock protein 90 β family member 1 (HSP90B1), inhibiting its IFN inducer function [60]. Moreover, there are evidence indicating that ORF8 affects the IFN-β pathway in a cell type specific manner, since ORF8 84L inhibited expression of ISGs responsive to IFN-β by poly(I:C) stimulation, such as OAS3 and IFITM1, in HEK-293 but not A549 cells [61]. However, while ORF8 did not inhibit IFN-β signaling in A549 cells, it showed antagonism of the IFN-γ pathway [61].
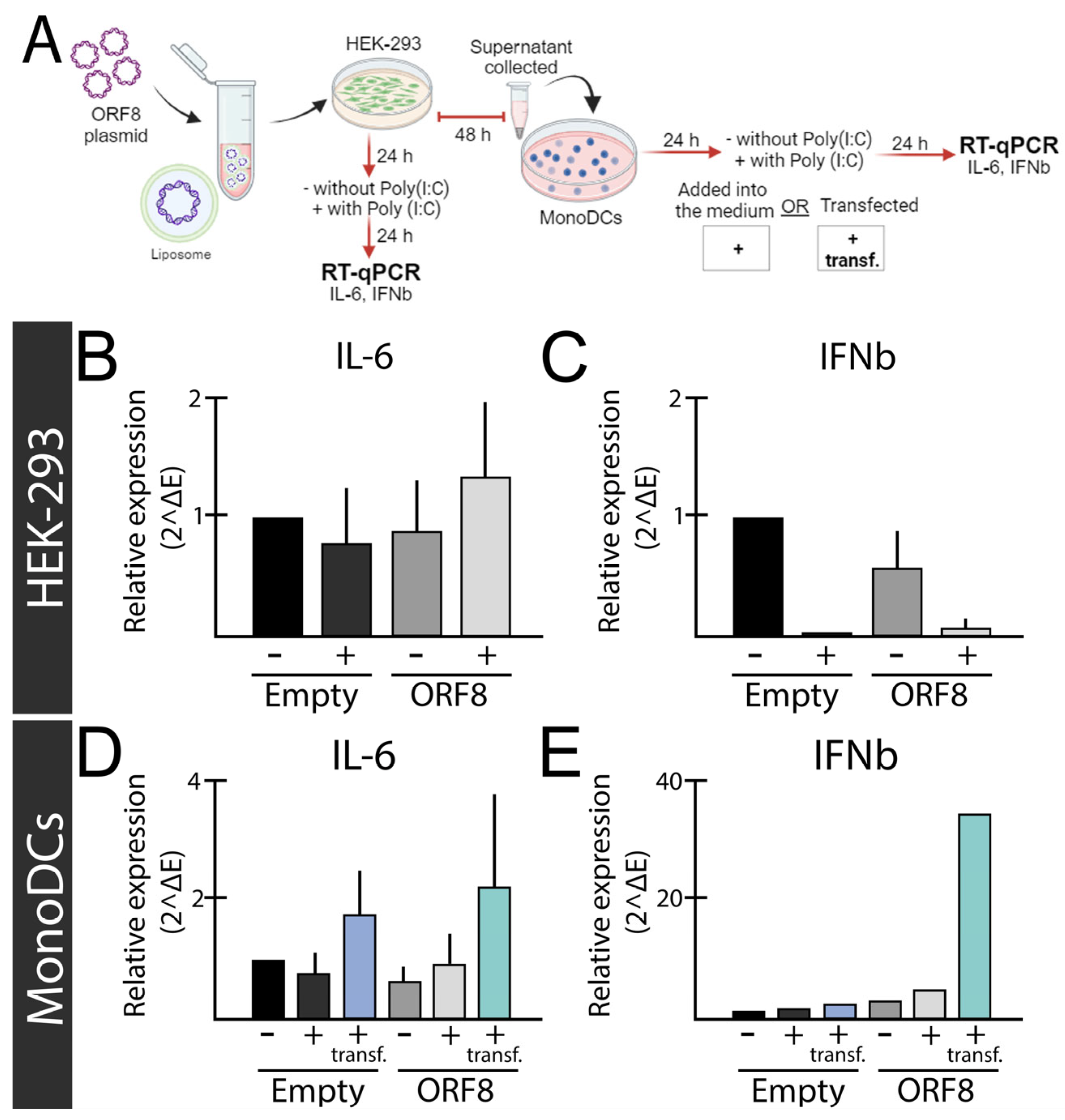
Contrary to the previous findings, ORF8 neither inhibited activation of the IFN-β promoter by the RIG-I caspase recruitment domains (CARDs) [11,43] or Sendai virus [11], nor ISRE activation by IFN-β in HEK-293 cells. Noteworthy, in experiments performed in our laboratory, we observed that ORF8 L84S upon poly(I:C) stimulation in HEK-293 inhibits IFN-β , while the same effect was not observed monocyte-derived dendritic cells (mono-DCs) (Figure 1C,E). In fact, our results indicate that ORF8 L84S can augment type I IFN-β expression by transfected poly(I:C) in mono-DCs (Figure 1E), as further discussed in this review. Therefore, our analysis corroborates the previous results that ORF8 inhibits IFN-β expression by poly(I:C) stimulation in HEK-293 cells, but not in mono DCs.
Overall, although several studies have pointed a role of ORF8 in IFN-β antagonism, there are clearly inconsistences and the reasons for these discrepancies may be explained by differences in cell types, assay methods, assay timing after IFN-β stimulation or amino acid variations in ORF8. Therefore, further experiments are necessary to clearly stablish the possible role of ORF8 in the type I IFN antagonistic activity, and to identify which conditions it may happens. Arguably, if ORF8 has this activity it may not be its primary function, as compared to other SARS-CoV-2 proteins, such as ORF6, which is a well-stablished potent IFN-β antagonist [46].
Figure 1.
Relative expression levels of IL-6 and IFN-B genes after ORF8 expression. (A) Experimental design used for the experiment. (B,C) HEK-293 cells were transfected with ORF8 or empty plasmid. To induce immune response, Poly(I:C) (2 μg/ml) was added into the media (+ med.) after 24 hours of transfection. The relative expression of IL-6 (B) and IFNb (C) were evaluated after 48 h of transfection. (D,E) Supernant of HEK-293 cells transfected with ORF8 and empty vector were added onto mono-DCs, either alone (-) or in combination with Poly(I:C) (2 μg/ml) in the media (+ med.) or transfected (+ transf.). After 48 hours, cells were subjected to real time-quantitative PCR (rt-qPCR) to evaluate IL-6 (D) and IFNb (E) levels.
Figure 1.
Relative expression levels of IL-6 and IFN-B genes after ORF8 expression. (A) Experimental design used for the experiment. (B,C) HEK-293 cells were transfected with ORF8 or empty plasmid. To induce immune response, Poly(I:C) (2 μg/ml) was added into the media (+ med.) after 24 hours of transfection. The relative expression of IL-6 (B) and IFNb (C) were evaluated after 48 h of transfection. (D,E) Supernant of HEK-293 cells transfected with ORF8 and empty vector were added onto mono-DCs, either alone (-) or in combination with Poly(I:C) (2 μg/ml) in the media (+ med.) or transfected (+ transf.). After 48 hours, cells were subjected to real time-quantitative PCR (rt-qPCR) to evaluate IL-6 (D) and IFNb (E) levels.
SARS-CoV-2 ORF8 as a Pro-Inflammatory Virokine
In addition to its role in immune evasion, plenty of evidence suggests that ORF8 acts as a virokine inducing pro-inflammatory responses. As mentioned, ORF8 is secreted from infected cells [49,50,51,52] and various studies, described below, indicate that the extracellular protein is able to activate inflammatory cytokines. This idea matches the fact that the presence of ORF8 in the plasma of infected individuals is conversely associated with survival [62,63].
Several studies have shown modulation of pathways linked to cytokine expression by ORF8 in vitro. Interactome and effectome analyses indicate that ORF8 interacts with the transforming growth factor (TGF)-β1-latent TGF-β binding protein 1 (LTBP1) complex, potentially dysregulating the TGF-β signaling pathway [63]. ORF8 was also shown to interact with the PRR nucleotide oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) in CD14+/CD16+ monocytes, inducing a cytokine response. According to the proposed model, ORF8 enters monocytes through a non-receptor-mediated process and binds NLRP3 intracellularly, a process that needs to be further investigated [64].
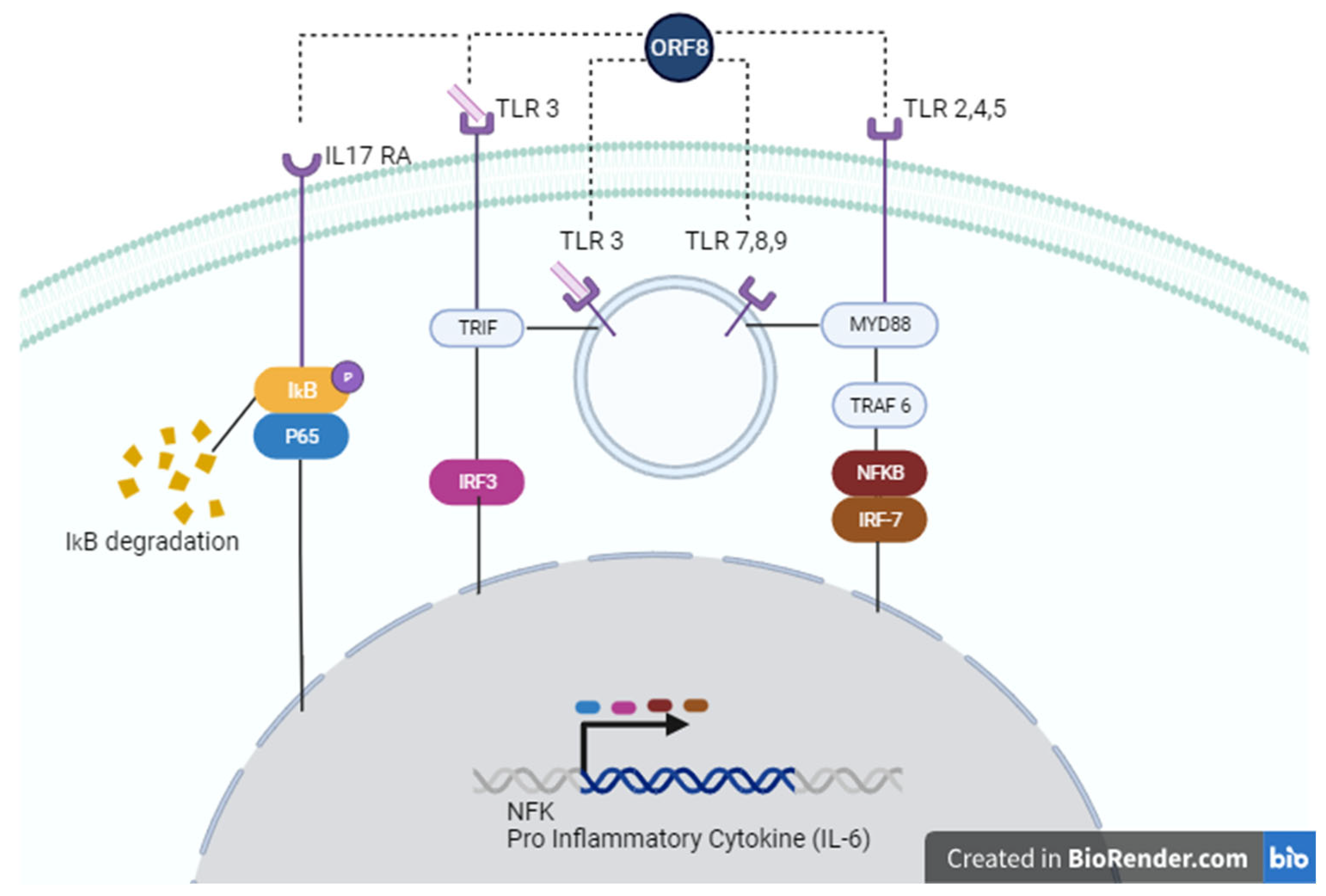
Two subsequent studies indicate that ORF8 has the ability to mimic the pro-inflammatory human IL-17 cytokine and binds to RAW 264.7 murine macrophages, as well as human CD14+ THP-1 and U973 monocytes [49,65]. Similar to IL-17, ORF8 binds to human hIL-17RA/C complex and induces their heterodimerization and subsequent activation of downstream inflammatory responses. These responses include phosphorylation of p65 and IκΒα as well as degradation of IκΒα, and subsequent activation of the NF-κB signaling pathway (Figure 3) [49]. However, despite the similarities, signaling induced by ORF8 appears to have some differences with the IL-17 pathway. In fact, ORF8 induces expression of genes coding for pro-inflammatory factors of the IL-17 receptor pathway, including CCL20, CXCL1, CXCL2 and IL-6, in THP-1 and U937 cells, but also causes overexpression of genes not known to be induced by IL-17, such as COL17A1, MMP10 and SERPINB2 [49]. Notably, variations in ORF8 appear to affect its ability to induce cytokine expression, given that the ORF8 L84S variant, and to a lesser extent variants V62L and S24L, showed reduction in expression of CCL20, CXCL1 and IL-6 compared to wild-type ORF8 (84L), suggesting that these variants are associated with an attenuated inflammation phenotype [49].
In addition to the effects of mutations on ORF8 function, glycosylation appears to affect protein activity. Accordingly, determination of the X-ray crystallographic structure of ORF8 indicates that four pairs of disulphide bonds and glycosylation at residue N78 are essential for stabilizing protein structure, and that glycosylation regulates binding to monocytes [66], which appears to be required for cytokine induction [49].
It was demonstrated that secretion of glycosylated ORF8 occurs by the conventional way, through the Golgi apparatus, while unglycosylated ORF8 is secreted via unconventional pathways. However, only the unglycosylated ORF8, due to a mutation at the N78 residue, was able to bind to the IL-17RA receptor and induce cytokine expression in macrophages or mice in one study [51]. Conversely, in another study, glycosylated ORF8 (purified from HEK-293 culture supernatants) induced inflammation, whereas the unglycosylated form (produced in E. coli) did not. However, in the same work, ORF8 induced high levels of cytokine synthesis even when protein glycosylation levels were reduced by Brefeldin A or Monensin treatment [49]. Therefore, further studies are required to clarify the role of glycosylated versus unglycosylated forms of ORF8 regarding differential functions and relevance during infection, especially since glycosylated ORF8 seems to be predominantly secreted in supernatants of SARS-CoV-2-infected cells and sera of COVID-19 patients [64].
SARS-CoV-2 ORF8 as a Potential Cytokine Modulator Through TLRs
Intriguingly, a recent study by Ponde et al (2023) challenges previous findings in myeloid cells [48,55]. Using unglycosylated ORF8 produced in bacterial cells, they show that IL-17RA and IL-17RC are not required for ORF8 signaling in macrophages and monocytes. Instead, it was demonstrated that signaling by ORF8 depends on the TLR/IL-1 family adaptor MyD88, indicating that it occurs though TLR recognition (Figure 3) [67]. The authors argue that the co-immunoprecipitation and ligation assays performed in previous studies do not prove a direct interaction between the IL-17 receptor and ORF8 [48,55]. Therefore, additional experiments may be required to convincingly demonstrate binding to and signaling though the IL-17 receptor by ORF8.
Notably, we observed that secreted ORF8 L84S expressed in HEK-293 cells augments expression of IL-6 in HEK-293 cells and mono-DCs as measured by RT-qPCR (Figure 1B,D) and enzyme-linked immunosorbent assay (ELISA) (Figure 2). As mentioned earlier, we also observed induction of IFN-β in mono-DCs (Figure 1E), but not in HEK-293 cells (Figure 1C) in the presence of poly(I:C), while ORF8 alone (without poly(I:C) stimulation) did not induce cytokine expression in any cells (Figure 1).
In agreement with our findings, as reported in by Kriplani et al (2021, preprint) ORF8 L84S variant, but not ORF8 L84, induces IL-6 production in primary monocyte-derived macrophages (MDMs) in the presence of poly(I:C). However, in this work both ORF8 84L and L84S combined with poly(I:C) neither induced nor inhibited IFN-β production. This observation appears to be at odds to our findings and may be explained by differences in cell types. Although we did not test ORF8 L84 in our assays, these findings suggest that ORF8 L84S can induce IL-6 production in mono-DCs and MDMs, likely in a sequence specific manner. We believe that the most plausible explanation for these findings is that ORF8 acts through on one or more cellular PRRs which sense dsRNA (poly(I:C)), such as TLR3, RIG-I or MDA5 [68], increasing IFN-β and IL6.
TLR3 localizes to both the plasma membrane and the endosome in HEK-293 cells [64,65] but is exclusively intracellular in immature dendritic cells (iDCs) [70]. Coupled with our observation that activation of IL-6 occurs in HEK-293 cells with media supplemented poly(I:C) but only upon transfection in mono-DCs (Figure 1B,D), we hypothesize that ORF8 is acting through TLR3 (Figure 3).
In line with this idea, the work by Ponde et al (2023) demonstrated that ORF8 signaling depends on MyD88, the canonical adaptor for inflammatory signaling downstream of several TRL family members (TLR2, TLR4, TLR5, TLR7, TLR8 and TLR9), except TRL3, which uses the adaptor TRIF (Figure 3). However, recent evidence indicates that TLR3 might nonetheless use, at least in certain circumstances, the MyD88-mediated pathway [71,72].
Interestingly, it is not uncommon for viral proteins to enhance poly(I:C)- and viral dsRNA-induced TLR3 signaling, as exemplified by polymerases from two genotypes of hepatitis C virus and three viral capsid proteins (1bD21, 2a.m26–30, B-cp, Rcp and H-cp183). These proteins have the common property to bind dsRNA [72]. It is possible that viral proteins released by cell lysis or secreted from infected cells bind to RNA and/or cell surface or intracellular receptors after being taken up by cells, subsequently activating innate immune responses. Based on the available evidence including our new findings, we believe that it is necessary to investigate whether TLR3 or other PRRs are required for ORF8-activated innate immune signaling.
Figure 2.
IL-6 levels induced by ORF8 measured by enzyme-linked immunosorbent assay (ELISA) assay. (A) Experimental design used for the experiment. (B) Supernant of HEK-293 cells transfected with ORF8 and empty vector were added onto mono-DCs, either alone (-) or in combination with Poly(I:C) (2 μg/ml) in the media (+ med.) or transfected (+ transf.). After 48 hours, the supernatants were collected, and subjected to ELISA for IL-6. The bars represent the mean value normalized by the control (empty supernatant).
Figure 2.
IL-6 levels induced by ORF8 measured by enzyme-linked immunosorbent assay (ELISA) assay. (A) Experimental design used for the experiment. (B) Supernant of HEK-293 cells transfected with ORF8 and empty vector were added onto mono-DCs, either alone (-) or in combination with Poly(I:C) (2 μg/ml) in the media (+ med.) or transfected (+ transf.). After 48 hours, the supernatants were collected, and subjected to ELISA for IL-6. The bars represent the mean value normalized by the control (empty supernatant).
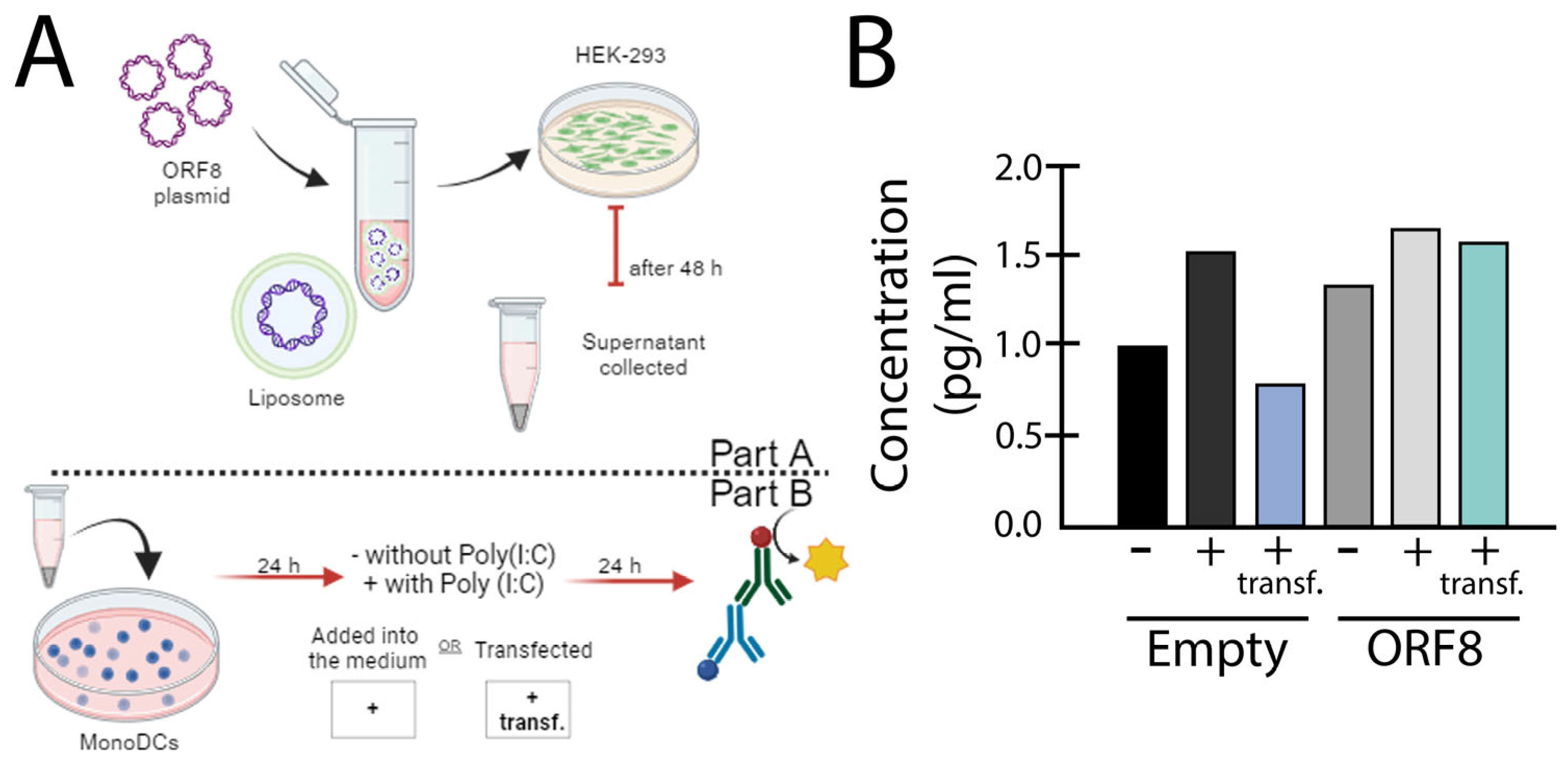
Figure 3.
Possible signaling pathways activated by ORF8. ORF8 accessory protein can act through IL-17 RA (left), TLR 3 (left, center), TLR 7,8,9 (center) or TLR 2,4,5 (right). In the IL-17 RA cascate, ORF8 leads to IkB/P65 phosphorylation, causing degradation of IκΒα, and subsequent activation of the NF-κB signaling pathway. Recognition of ORF8 by TLRs activates TRIF and/or MyD88 pathways, enhancing gene transcription of NF-κB genes or pro-inflammatory cytokines, such as IL-6.
Figure 3.
Possible signaling pathways activated by ORF8. ORF8 accessory protein can act through IL-17 RA (left), TLR 3 (left, center), TLR 7,8,9 (center) or TLR 2,4,5 (right). In the IL-17 RA cascate, ORF8 leads to IkB/P65 phosphorylation, causing degradation of IκΒα, and subsequent activation of the NF-κB signaling pathway. Recognition of ORF8 by TLRs activates TRIF and/or MyD88 pathways, enhancing gene transcription of NF-κB genes or pro-inflammatory cytokines, such as IL-6.
Concluding Remarks
A great amount of work has been done to understand the mechanisms that activate cytokine storm during SARS-CoV-2 infection, since the identification of the virus causative of COVID-19. There is plenty evidence that the viral accessory protein ORF8 is an important player in this regard. ORF8 has the capability to induce inflammatory responses, yet conversely, several studies indicate that it inhibits type I Interferon. However, it's worth noting that not all studies have consistently supported this inhibition effect. These two activities diverge from one to another and the precise mechanisms by which ORF8 functions are still not known. It is possible that ORF8 acts as an anti- and pro-inflammatory factor in different phases of infections: at early and late times, respectively, and more studies need to be done to unveil this question. One important question is if the release of secreted ORF8, activating inflammation has any benefit for the virus or it is just a consequence of viral replication contributing to immune pathology associated with viral infection.
We commented here about the current data providing substantial evidence that ORF8 is a virokine; however at this point there are a number of unanswered questions mainly regarding the receptors and mechanisms involved in signaling induction by ORF8 (Figure 3). In addition, we provide data showing that ORF8 increases cytokine expression in presence of poly(I:C) suggesting that it acts through a cellular receptor that senses dsRNA. It is possible that ORF8 acts through one or more cellular receptors (Figure 3) and more studies are important to answer this question, since it is a virulence factor that contributes to immunopathology and can be a target for antiviral therapy.
Author Contributions
Conceptualization, MCCS; methodology, MIM, GC, ILM, GBL; formal analysis, MCCS, SDS, AHK, EP, MN; investigation, MCCS, EP, MN; data curation, MCCS, GC, MIM; writing—original draft preparation, MCCS, writing—review and editing, EP and MN; supervision, project administration, and funding acquisition, MCCS. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Federal University of ABC (UFABC), São Bernardo do Campo, São Paulo, Brazil.
Institutional Review Board Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
Not applicable
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhou, P.; Yang, X. Lou; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, K.; Antognini, D.; Combes, A.; Paden, M.; Zakhary, B.; Ogino, M.; Maclaren, G.; Brodie, D. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. The Lancet 2020, 395, 497–506. [Google Scholar]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a Novel Coronavirus Causing Severe Pneumonia in Human: A Descriptive Study. Chin Med J (Engl) 2020, 133, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Saksena, S. Illuminating the Immunopathology of SARS-CoV-2. [CrossRef]
- Merad, M.; Martin, J.C. Pathological Inflammation in Patients with COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Luo, R.; Zhang, M.; Wang, Y.; Song, T.; Tao, T.; Li, Z.; Jin, L.; Zheng, H.; Chen, W.; et al. A Cross-Talk between Epithelium and Endothelium Mediates Human Alveolar–Capillary Injury during SARS-CoV-2 Infection. Cell Death Dis 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The Cytokine Storm and COVID-19. J Med Virol 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev 2020. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and Evasion of Type I Interferon Responses by SARS-CoV-2. Nat Commun 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Merani, S.; Pawelec, G.; Kuchel, G.A.; McElhaney, J.E. Impact of Aging and Cytomegalovirus on Immunological Response to Influenza Vaccination and Infection. Front Immunol 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients from Wuhan, China. Intensive Care Med 2020, 46, 846–848. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. The Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat Rev Microbiol 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 Genome-Encoded Proteins. Sci China Life Sci 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front Immunol 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Rashid, F.; Xie, Z.; Suleman, M.; Shah, A.; Khan, S.; Luo, S. Roles and Functions of SARS-CoV-2 Proteins in Host Immune Evasion. Front Immunol 2022, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. The Lancet 2020. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F. Evolutionary Dynamics of the SARS-CoV-2 ORF8 Accessory Gene. Infection, Genetics and Evolution 2020, 85, 104525. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zheng, X.; Zhu, J.; Ding, R.; Jin, Y.; Zhang, W.; Yang, H.Y.; Zheng, Y.; Li, X.; Duan, G. Extended ORF8 Gene Region Is Valuable in the Epidemiological Investigation of Severe Acute Respiratory Syndrome-Similar Coronavirus. Journal of Infectious Diseases 2020, 222, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant Analysis of SARS-Cov-2 Genomes. Bull World Health Organ 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Badua, C.L.D.C.; Baldo, K.A.T.; Medina, P.M.B. Genomic and Proteomic Mutation Landscapes of SARS-CoV-2. J Med Virol 2021, 93, 1702–1721. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-CoV-2. Gene Rep 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different Mutations in SARS-CoV-2 Associate with Severe and Mild Outcome. Int J Antimicrob Agents 2021, 57, 0–4. [Google Scholar] [CrossRef] [PubMed]
- Ceraolo, C.; Giorgi, F.M. Genomic Variance of the 2019-NCoV Coronavirus. J Med Virol 2020. [CrossRef] [PubMed]
- Alkhansa, A.; Lakkis, G.; El Zein, L. Mutational Analysis of SARS-CoV-2 ORF8 during Six Months of COVID-19 Pandemic. Gene Rep 2021, 23, 101024. [Google Scholar] [CrossRef] [PubMed]
- Young, B.E.; Fong, S.W.; Chan, Y.H.; Mak, T.M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study. The Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Hachim, A.; Kavian, N.; Cohen, C.A.; Chin, A.W.H.; Chu, D.K.W.; Mok, C.K.P.; Tsang, O.T.Y.; Yeung, Y.C.; Perera, R.A.P.M.; Poon, L.L.M.; et al. ORF8 and ORF3b Antibodies Are Accurate Serological Markers of Early and Late SARS-CoV-2 Infection. Nat Immunol 2020, 21, 1293–1301. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V. V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Vinjamuri, S.; Li, L.; Bouvier, M. SARS-CoV-2 ORF8: One Protein, Seemingly One Structure, and Many Functions. Front Immunol 2022, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Laprise, F.; Liang, C. SARS-CoV-2 ORF8: A Rapidly Evolving Immune and Viral Modulator in COVID-19. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.; Ghosh, S.; Attrish, D.; Choudhury, P.P.; Seyran, M.; Pizzol, D.; Adadi, P.; Abd El Aziz, T.M.; Soares, A.; Kandimalla, R. ; et al. A Unique View of SARS-CoV-2 through the Lens of ORF8 Protein (Preprint). bioRxiv, 2020; 2020.08.25.267328. [Google Scholar]
- Mohammad, S.; Bouchama, A.; Alharbi, B.M.; Rashid, M.; Khatlani, T.S.; Gaber, N.S.; Malik, S.S. Sars-cov-2 Orf8 and Sars-cov Orf8ab: Genomic Divergence and Functional Convergence. Pathogens 2020, 9, 1–26. [Google Scholar] [CrossRef]
- Valcarcel, A.; Bensussen, A.; Álvarez-Buylla, E.R.; Díaz, J. Structural Analysis of SARS-CoV-2 ORF8 Protein: Pathogenic and Therapeutic Implications. Front Genet 2021, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L. Lost in Deletion: The Enigmatic ORF8 Protein of SARS-CoV-2. Biochem Biophys Res Commun 2021, 538, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C. ; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients.
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at Age 50: Past, Current and Future Impact on Biomedicine. Nat Rev Drug Discov 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; García-Sastre, A. Viruses and the Type I Interferon Antiviral System: Induction and Evasion. Int Rev Immunol 2002, 21, 305–337. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I Interferons (α/β) in Immunity and Autoimmunity. Annu Rev Immunol 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Islamuddin, M.; Mustfa, S.A.; Ullah, S.N.M.N.; Omer, U.; Kato, K.; Parveen, S. Innate Immune Response and Inflammasome Activation During SARS-CoV-2 Infection. Inflammation 2022, 45, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signaling Pathways. Front Immunol 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aluri, J.; Cooper, M.A.; Schuettpelz, L.G. Toll-like Receptor Signaling in the Establishment and Function of the Immune System. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Amurri, L.; Horvat, B.; Iampietro, M. Interplay between RNA Viruses and CGAS/STING Axis in Innate Immunity. Front Cell Infect Microbiol 2023, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 Infection Induces a pro-Inflammatory Cytokine Response through CGAS-STING and NF-ΚB. Commun Biol 2022, 5. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.K.; Lam, J.Y.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Cai, J.P.; Jin, D.Y.; To, K.K.W.; Chan, J.F.W.; et al. SARS-CoV-2 Nsp13, Nsp14, Nsp15 and Orf6 Function as Potent Interferon Antagonists. Emerg Microbes Infect 2020, 9, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and Nucleocapsid Proteins of SARS-CoV-2 Inhibit Type I Interferon Signaling Pathway. Virus Res 2020, 286, 198074. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.E.; Song, Y.J. Coordinated Regulation of Interferon and Inflammasome Signaling Pathways by SARS-CoV-2 Proteins. Journal of Microbiology 2022, 60, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xia, T.; Shin, W.J.; Yu, K.M.; Jung, W.; Herrmann, A.; Foo, S.S.; Chen, W.; Zhang, P.; Lee, J.S.; et al. Viral Mimicry of Interleukin-17A by SARS-CoV-2 ORF8. mBio 2022, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Imahashi, N.; Ohno, M.; Ode, H.; Nakata, Y.; Kubota, M.; Sugimoto, A.; Imahashi, M.; Yokomaku, Y.; Iwatani, Y. SARS-CoV-2 Accessory Protein ORF8 Is Secreted Extracellularly as a Glycoprotein Homodimer. Journal of Biological Chemistry 2022, 298, 101724. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Fu, B.; Xiong, Y.; Xing, N.; Xue, W.; Guo, D.; Zaky, M.; Pavani, K.; Kunec, D.; Trimpert, J.; et al. Unconventional Secretion of Unglycosylated ORF8 Is Critical for the Cytokine Storm during SARS-CoV-2 Infection. PLoS Pathog 2023, 19, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kriplani, N.; Clohisey, S.; Fonseca, S.; Fletcher, S.; Lee, H.-M.; Ashworth, J.; Kurian, D.; Lycett, S.J.; Tait-Burkard, C.; Baillie, J.K. ; et al. Secreted SARS-CoV-2 ORF8 Modulates the Cytokine Expression Profile of Human Macrophages. bioRxiv, 2021; 2021.08.13.456266. [Google Scholar]
- Protein, S.S.-. Crossm Accurate Diagnosis of COVID-19 by a Novel Immunogenic. 2020, 11, 1–13.
- The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. 2020.
- Beaudoin-Bussières, G.; Arduini, A.; Bourassa, C.; Medjahed, H.; Gendron-Lepage, G.; Richard, J.; Pan, Q.; Wang, Z.; Liang, C.; Finzi, A. SARS-CoV-2 Accessory Protein ORF8 Decreases Antibody-Dependent Cellular Cytotoxicity. Viruses 2022, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Dhyani, S.; Kumar, P.; Sharma, N.R.; Ganguly, S. SARS-CoV-2–Encoded ORF8 Protein Possesses Complement Inhibitory Properties. Journal of Biological Chemistry 2023, 299, 102930. [Google Scholar] [CrossRef]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and Nucleocapsid Proteins of SARS-CoV-2 Inhibit Type I Interferon Signaling Pathway. Virus Res 2020, 286. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Dzakah, E.E.; Wang, H.; Tang, S. The ORF8 Protein of SARS-CoV-2 Induced Endoplasmic Reticulum Stress and Mediated Immune Evasion by Antagonizing Production of Interferon Beta. Virus Res 2021, 296. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Wang, T.-Y.; Qin, C.; Espinosa, B.; Liu, Q.; Ekanayake, A.; Zhao, J.; Savas, A.C.; Zhang, S.; Zarinfar, M.; et al. Targeting CTP Synthetase 1 to Restore Interferon Induction and Impede Nucleotide Synthesis in SARS-CoV-2 Infection. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Z.; Yang, X.; Zhou, Y.; Gao, J.; Zhang, S.; Huang, S.; Cai, J.; Yu, J.; Zhao, W.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 ORF8 Protein Inhibits Type I Interferon Production by Targeting HSP90B1 Signaling. Front Cell Infect Microbiol 2022, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Subramanian, S.; Wu, L.; Bu, H.F.; Wang, X.; Du, C.; De Plaen, I.G.; Tan, X. Di SARS-CoV-2 ORF8 Forms Intracellular Aggregates and Inhibits IFNγ-Induced Antiviral Gene Expression in Human Lung Epithelial Cells. Front Immunol 2021, 12, 679482. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, M.; Suzuki, T.; Nakai, W.; Ono, C.; Matsuoka, S.; Iwatani, K.; Liu, Y.; Sakai, Y.; Nakagawa, A.; Tomii, K. et al. SARS-CoV-2 ORF8 Is a Viral Cytokine Regulating Immune Responses.
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A. ; et al. Multilevel Proteomics Reveals Host Perturbations by SARS-CoV-2 and SARS-CoV; Springer US, 2021; Vol. 594; ISBN 4158602103493.
- Wu, X.; Manske, M.K.; Ruan, G.; Nowakowski, K.E.; Abeykoon, J.P.; Tang, X.; Yu, Y.; Witter, T.L.; Taupin, V.; Paludo, J. ; et al. Secreted ORF8 Is a Pathogenic Cause of Severe Covid-19 and Potentially Targetable with Select NLRP3 Inhibitors. bioRxiv, 2021; 2021.12.02.470978. [Google Scholar]
- Lin, X.; Fu, B.; Yin, S.; Li, Z.; Liu, H.; Zhang, H.; Xing, N.; Wang, Y.; Xue, W.; Xiong, Y.; et al. ORF8 Contributes to Cytokine Storm during SARS-CoV-2 Infection by Activating IL-17 Pathway. iScience 2021, 24, 102293. [Google Scholar] [CrossRef]
- Wu, F.; Chen, X.; Ma, Y.; Wu, Y.; Li, R.; Huang, Y.; Zhang, R.; Zhou, Y.; Zhan, J.; Liu, S.; et al. Glycosylated, Lipid-Binding, CDR-Like Domains of SARS-CoV-2 ORF8 Indicate Unique Sites of Immune Regulation. Microbiol Spectr 2023, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ponde, N.O.; Shoger, K.E.; Khatun, M.S.; Sarkar, M.K.; Dey, I.; Taylor, T.C.; Cisney, R.N.; Arunkumar, S.P.; Gudjonsson, J.E.; Kolls, J.K.; et al. SARS-CoV-2 ORF8 Mediates Signals in Macrophages and Monocytes through MyD88 Independently of the IL-17 Receptor. The Journal of Immunology 2023, 211, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Hoose, S.; Schreiter, J.; Sawant, K. V.; Lamb, R.; Ranjith-Kumar, C.T.; Mills, J.; Mateo, L.S.; Jordan, J.L.; Kao, C.C. Secretion of the Human Toll-like Receptor 3 Ectodomain Is Affected by Single Nucleotide Polymorphisms and Regulated by Unc93b1. Journal of Biological Chemistry 2010, 285, 36635–36644. [Google Scholar] [CrossRef]
- Matsumoto, M.; Funami, K.; Tanabe, M.; Oshiumi, H.; Shingai, M.; Seto, Y.; Yamamoto, A.; Seya, T. Subcellular Localization of Toll-Like Receptor 3 in Human Dendritic Cells. The Journal of Immunology 2003, 171, 4934–4934. [Google Scholar] [CrossRef]
- Singh, A.; Basu, A.; Devkar, R. Investigation on the MyD88 Mediated TLR3 Signaling via Cell Surface in Breast Cancer. Annals of Oncology 2019, 30, iii46. [Google Scholar] [CrossRef]
- Lai, Y.; Yi, G.; Chen, A.; Bhardwaj, K.; Tragesser, B.J.; Valverde, R.A.; Zlotnick, A.; Mukhopadhyay, S.; Ranjith-Kumar, C.T.; Kao, C.C. Viral Double-Strand RNA-Binding Proteins Can Enhance Innate Immune Signaling by Toll-like Receptor 3. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



