
Submitted:
30 November 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
Malaria parasite harbors an essential prokaryotic-like organelle characterized by multiple mem-branes, akin to a plastid. This organelle, known as the apicoplast, evolved from secondary en-dosymbiosis. In this event, a photosynthetic alga was engulfed by a eukaryotic host and subsequently transformed into a symbiotic entity from its free-living origin. As a result, the apicoplast transformed into a remnant structure housing a circular genome, enveloped by four membranes. Concurrently, the host cell relinquished significant metabolic pathways, and a substantial portion of the apicoplast genome migrated to the host cell, establishing a symbiotic relationship. This unique symbiotic bond designates the apicoplast as a constrained environment for diverse metabolic pathways, vital for parasite survival. Notably, the non-mevalonate biosynthesis (MEP/DOXP) pathway is identified as pivotal for the parasite's viability across parasitic stages. In this study, we conducted a comparative analysis of Plasmodium falciparum MEP pathway enzymes, juxtaposing them with their counterparts in other apicomplexans, plants, and pathogenic microorganisms to uncover the conserved traits of these enzymes. Additionally, predictive modeling was employed to shed light on the protein structures, with specific emphasis on elucidating the active site characteristics and examining their binding affinities with various substrates and inhibitors. This article presents a comprehensive in-silico analysis of MEP pathway enzymes, showcasing their potential as prospective targets for innovative antimalarial drugs. The elucidation of these enzymes' intricate functions provides a foundation for the development of advanced therapeutic strategies in the battle against malaria.
Keywords:
Malaria
; Apicoplast
; Plasmodium falciparum
; Isoprenoid.
1. Introduction
Malaria is a mosquito-borne infectious disease and Plasmodium falciparum is the etiological agent of human malaria, one of the most widespread diseases in tropical and subtropical regions [1]. Drug resistance is one of the biggest problems in controlling the disease, which leads to the need to discover new antimalarial compounds. The malaria parasite belongs to the phylum apicomplexan and contains a multimembrane vital organelle called apicoplast. It originated from the process of secondary endosymbiosis however certain aspects of apicoplast biology remain enigmatic. Recent studies indicate that apicomplexans share ancestral ties with diatoms and kelps and apicoplast originated from the acquisition of a red algal endosymbiont [2]. Questions persist regarding apicoplast division and maintenance, how the organelle preserves its degenerative genome amid the rapidly shifting parasite stages, and the mechanisms underpinning transport across its multifaceted membranes. Apicoplast biogenesis and housekeeping processer were also exploited as a drug target leading to the delayed death of the parasite [3].
Apicoplast houses various metabolic pathways and isoprenoid biosynthesis makes it indispensable for the parasite. Two primary routes to synthesized isoprene units: the mevalonate (MVA) pathway as in human host or the Mevalonate independent (MEP/DOXP) pathway in prokaryotes (Figure 1). Studies conducted over the past decade have firmly established the essentiality of the MEP pathway within the apicoplast during the erythrocytic stages of the malaria parasite [4,5]. These investigations encompassed the comprehensive characterization of various enzymes intrinsic to the pathway, substrate labeling experiments, and the quantification of pathway metabolites [6,7,8]. Fosmidomycin (Fos) was proposed as a promising inhibitor of MEP, however, it failed in clinical studies. MEP pathway inhibition with Fos and its rescue with an external supply of Isopentenyl pyrophosphate (IPP) also established the apicoplast as the sole source of isoprenoids [9]. This rescue experiment was further exploited to indirectly identify drug molecules exclusive to the apicoplast [10]. A forward genetic screen employed to study the link between febrile temperature rise and artemisinin resistance leads to the identification of the upregulation of genes involved the in MEP pathway [11]. The anaerobic environment in apicoplast is an essential feature for the optimal functionality of key enzymes involved in the MEP pathway and it was achieved by the ferredoxin redox system [12,13] and could be explored as a potential drug target. Intriguingly, ATP molecules required for the pathway enzymes are exclusively generated via glycolysis instead of mitochondrial respiration also supports the need for an anaerobic environment in apicoplast [14].
Isoprenoids are significantly important for protein prenylation, vesicular trafficking, cell wall construction, mitochondrial ubiquinone, and heme A biosynthesis [15,16,17,18]. Isoprenoid alcohol, Dolichol’s involvement in post-translational modification also supports the MEP pathway’s essentiality [19]. Although these functions are performed in the parasite cytosol, a recent gene knockout study revealed a novel arm of isoprenoid metabolism required for apicoplast biosynthesis [20]. MEP Pathway inhibition could initiate the isoprenoids scavenge from the host cell [21] however long-term external supply is not feasible. Putative transporters, such as the triose phosphate transporter (TPT) and pyruvate phosphate transporter (PPT), are believed to be present on the apicoplast membrane. Yet, the intricacies of transport across these membranes remain elusive. For instance, the knockout of TPT resulted in the immediate demise of the parasite, indicating a failure to acquire dihydroxyacetone phosphate (DHAP), an MEP pathway precursor [22]. Indeed, studies corroborate the apicoplast's confinement as the exclusive site for the MEP pathway in malaria parasites. Nevertheless, the export mechanism of Isopentenyl pyrophosphate (IPP) units from the apicoplast continues to elude understanding.
The enzymatic cascade of the MEP pathway conserved across prokaryotes, plants, and diverse organisms, has been deciphered through sequence-based analyses, facilitating the mapping of these enzymes within malaria parasites. In malaria parasites, these proteins are expressed in the nuclear genome and are targeted to the apicoplast through Nuclear-encoded apicoplast-targeted (NEAT) sequences, marked by sequence length and polymorphism. The presence of conserved domains and signature motifs offers valuable insights into the functional attributes of these enzymes. In-silico analyses have further unveiled the presence of various functional domains, suggesting supplementary roles contributing to parasite survival. Accumulated evidence underscores the exclusive and indispensable nature of the MEP pathway for parasite viability, thereby elevating the pursuit of enzyme inhibitors as a promising therapeutic strategy. This study encompasses a comprehensive comparative analysis of the pathway's enzymes, meticulously detailing critical conserved domains and yielding structural insights pivotal for the design of novel inhibitory agents.
2. Results and Discussion
CD, Signature motif, MSA, Phylogenetic analysis and Structural insights
Initial sequence analysis was undertaken to discern the conserved domains and distinctive signature motifs within the MEP pathway enzymes of the malaria parasite. This analysis was juxtaposed against analogous enzymes from other parasites, prokaryotes, and plants. Notably, in malaria parasites and plants, the MEP pathway exhibits a compartmentalized distribution, whereby the pertinent enzymes are encoded within the nuclear genome. These enzymes possess bipartite leader sequences that facilitate their targeted translocation to specific organelles, with the apicoplast serving as the subcellular destination in malaria parasites. The identification of function-specific conserved domains and motifs within these enzymes stands as compelling evidence of their catalytic role (Table 1). This compartmentalization bears substantive significance, as the apicoplast furnishes an optimal reducing milieu that is paramount for the enzymatic activities integral to the pathway.
2.1. DXS (DXP Synthase) Enzyme
The MEP pathway commences with the synthesis of 1-deoxy-D-xylulose-5-phosphate (DXP/DOXP) through an acyloin condensation process involving pyruvate and glyceraldehyde 3-phosphate (GA3P). This crucial step is enzymatically catalyzed by DXS (EC 4.1.3.37) in the presence of thymine diphosphate (ThDP). Notably, this rate-limiting step assumes a pivotal role in controlling the flux of the pathway [23,24]. Transcriptomic analyses of P. falciparum indicate an upregulation of the DXS enzyme during the trophozoite stages, juxtaposed with a downregulation evident in the schizont stages (Supp data) [25]. Comprehensive analysis of the conserved domains within the DXS enzyme unearthed three characteristic thiamine-dependent domains: TPP (thiamine pyrophosphate binding domain; 412-551), PYR (pyrimidine binding domain; 828-994), and transketolase C domain (1064-1191) (Figure 2a). The functionality of these domains was well-established for pyruvate decarboxylating [26] and ThDP-dependent transketolase enzymes, preceding the characterization of the DXS enzyme [26,27]. Intriguingly, the N-terminal region of these domains exhibited significant variation in malaria parasites, prokaryotes, and plants, largely attributed to the influence of NEAT sequences. This region, characterized by an abundance of basic amino acid residues and susceptibility to post-targeting cleavage, suggests its pivotal role in apicoplast targeting. Moreover, the presence of repeated asparagine residues contributes to the elongation of the P. falciparum DXS enzyme. Notably, a conserved signature motif pattern involving glycine and histidine as central residues was discerned in all DXS enzymes (Table 1).
Multiple sequence alignment (MSA) disclosed a similarity range of 30 to 92%, primarily concentrated within the CD and signature motif regions, while the rest of the sequence exhibited length polymorphism (Supp data). The conserved N-terminal TPP binding domain is distinguished by the presence of highly conserved residues -GGH- (underlined in red; Figure 2b) at its commencement, followed by -LNDN- (underlined in blue; Figure 2b) where the histidine residue in -GGH- is critically important for the catalytic activity. The PYR binding domain spans from -IAEQ- to -DGATH-, with an invariant glutamate residue specifically associated with transketolase activity. Notably, a signature motif was identified within this domain (underlined in brown; Figure 2b,). These residues actively contribute to synthase activity and are fairly conserved across diverse organisms. Variability was predominantly observed in the unordered loop connecting the three domains.
The phylogenetic tree of DXS enzymes closely aligned with the CD and signature motif analysis. Within this context, all Plasmodium parasites clustered in the same clade, while P. cynomolgi, P. knowlesi, P. inui, and P. vivax branched apart from rodent parasites such as P. yoelii, P. berghei, P. chabaudi, and P. vinckei. The phylogenetic tree construction employed the sequence from M. avium as an outgroup. Notably, other apicomplexan parasites formed a separate clade distinct from Plasmodium parasites. Meanwhile, T. gondii exhibited proximity to A. thaliana, while plants and prokaryotes displayed clustering originating from a shared clade (Figure 2c).
2.1.1. PfDXS Enzyme’s Structural Insight
Similar to other ThDP-dependent enzymes, the DXS enzyme undertakes two consecutive half-reactions. The initial step involves the attack of an activated ThDP ylide on the first substrate, GA3P. Subsequently, the second step presents three potential mechanisms: (i) a classical ping-pong mechanism; (ii) an ordered sequential kinetic mechanism; or (iii) an alternate random sequential mechanism [28]. Notably, in Plasmodium, this step is executed through an alternate random sequential kinetic mechanism, a distinctive observation for ThDP-dependent enzymes. Structural elucidation highlights the interaction of TPP DXS domain residues with Mg2+ ions, a critical factor governing both the structural and functional attributes of the DXS enzyme [29].
The three-dimensional structure of the PfDXS enzyme was derived from the AlphaFold server [30,31]. Structural analysis revealed that PfDXS exists as a dimer, with each subunit harboring an active site. This stands in contrast to other ThDP-dependent enzymes, where the active site is located at the dimer interface [29,32]. A distinct aspect of DXS enzyme functionality lies in its requirement for ternary complex formation for activity [33]. However, initial template structure unavailability resulted in an unresolved stretch of approximately 300 residues, leading to high predicted aligned error (PAE). Conversely, the TPP and PYR binding domains exhibited well-resolved structures. Despite the conserved arrangement of the domains, a distinguishing feature in PfDXS is the presence of a stretch of repeated asparagine residues in place of the unstructured linker region, which forms a central loop in the protein structure. This region displayed high evolutionary variability in both length and sequence, observed across all known DXS proteins (Figure 2d). Intriguingly, truncation of this unstructured loop did not induce alterations in enzymatic activity or biophysical properties [34].
Active site residues were conserved in various DXS enzymes except malaria parasite, where NEAT sequences induced length polymorphism. Mutational studies revealed the substrate binding residues, His (458, 489, 941), Ser 522, Glu 879, Tyr 902, Arg 930, and Asp 937 in PfDXS, positioned at the interaction site for the TPP and PYR binding domains. Mutational studies also supported the essentiality of Arg 985 however it could be replaced with lysine without having structural alteration [35].
2.1.2. PfDXS Enzyme’s Inhibitors Studies
DXS enzyme’s indispensable role and involvement in pathway regulation makes it a potential target for specific inhibitor development, so understanding the binding activity and catalytic mechanism was pivotal. Therefore, the protein structure was subjected to docking studies with its substrates. TPP, interacting with Ser 465, Gln 490, Gly 627, Asp 628, Asn 655, Val 660, and Lys 751 residues conserved across various organisms, underscores key interactions (Figure 2e; Supp data). Leveraging the transketolase domain's susceptibility to Pyrimidinone derivatives, ketoclomazone was docked against the enzyme, engaging with pivotal residues Asn 657 and Lys 751 (Figure S1). Other inhibitors, such as ß-fluropyruvate and MAP (Methylacetylphosphonate), engaged with residues Ser 514, Thr 940 (Figure 2f) and Lys 751, Ser 752, Gln 659 (Figure 2g, Figure S2 and Figure S3) respectively, aligning with the ThDP domain, representing a competitive inhibition. Additionally features distinguishing DXS from other ThDP-dependent enzymes also present an avenue for the development of novel inhibitors.
2.2. IspC (1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase) Enzyme
IspC, also known as DXR (EC 1.1.1.267), assumes the role of catalyzing the second step within the pathway, converting DOXP to 2-C-methyl-D-erythritol-4-phosphate (MEP) through utilization of the NADPH pro-S hydride. This homo-dimeric class B dehydrogenase enzyme, IspC, features three conserved domains as evidenced by CD analysis: DXP reductoisomerase (80-212), DXP reductoisomerase catalytic domain (227-323), and DXP reductoisomerase C-terminal (357-475) (Figure 3a). Although no distinct signature motif has been identified in any IspC enzymes, a lower pI value is attributed to the prevalence of negatively charged amino acid residues such as Asp and Glu, in contrast to the positively charged Lys and Arg residues. The mRNA expression dynamics of the PfIspC enzyme revealed an immediate post-invasion downregulation, followed by a discernible upregulation in late trophozoite stages, subsequently attenuating prior to the release of merozoites (Supp data) [25].
Upon subjecting the IspC enzyme to MSA, a significant level of identity within the conserved domain regions emerged, ranging from 35 to 93% (Supp data). The DXP reductoisomerase domain constitutes the NADPH binding site, serving as an energy source (underlined in red; Figure 3b). The DXP reductoisomerase catalytic domain governs the enzyme's active site and oscillates between open and closed conformations (underlined in blue; Figure 3b), while the DXP reductoisomerase C-terminal forms a left-handed super-helix to confer stability (underlined in brown; Figure 3b). The phylogenetic tree depicting the IspC enzyme's relationships mirrors the architecture observed in the DXS enzyme, except for T. gondii, which diverges from the prokaryotic and plant clades (Figure 3C). This phylogenetic tree construction utilized the sequence from M. avium as an outgroup.
2.2.1. PfIspC Enzyme’s Structural Insight
The three-dimensional structure of PfIspC was retrieved and analyzed for its binding affinity for its substrates and inhibitors. IspC is a bisubstrate enzyme where the dinucleotide binding fold at the N-terminal domain is specific for NADPH binding and this interaction induces conformational changes to facilitate the binding of DOXP. The central catalytic domain accommodates the binding of divalent cations (Mn2+ or Mg2+) as well as the phosphate group of DOXP. A linker region between the catalytic and C-terminal domain bridges the entire monomer, forms a V-shape architecture, and provides structural support (Figure 3d). Remarkably, the enzyme’s active site exists in two confirmations: open and closed. In the open state DOXP ingress and bind whereas in the closed state, a flap covers the active site to initiate the catalytic function [36]. In PfIspC, DOXP and NADPH molecules interact within the active site defined by conserved residues Lys (205 and 312), Asp 231, Glu (233 and 315), Ser (232, 269, 270, and 306), and Asn 311 (Figure 3e; Supp data). Additionally, conserved residues Asp 231, Glu 233, and Glu 315 binding with metal ligand, Mg2+, are pivotal for catalytic reaction. Interestingly interaction of Mg2+ in the closed confirmation is contingent on the redox milieu prevalent in the apicoplast.
2.2.2. PfIspC Enzyme’s Inhibitors Studies
Initially identified Fos and its analogs were monosubstrate competitive inhibitors functional against DOXP whereas uncompetitive to NADPH thus required NADPH binding on active site to facilitate inhibitors binding. Fos sensitivity varied within different developmental stages of Plasmodium, demonstrating maximal efficacy during erythrocytic stages due to higher uptake compared to liver stages [37]. These inhibitors were ineffective against the conversion between open and close confirmation and suffered from low bioavailability, limited adsorption, shorter half-life, and malaria recrudescence [38]. To address this concern, diverse analogs of Fos have been synthesized by different research groups, achieving success in restraining the growth of chloroquine-resistant P. falciparum Dd2 strain [39,40]. Structural analysis reveals Fos’s affinity to conserved residues within the catalytic domain, forming interactions with NADPH and the DOXP substrate (Figure 3f and Figure S4). An alternative approach rather than designing and synthesizing Fos analogs could be employed i.e., a DNA aptamer designed against IspC had higher specificity against PfIspC compared to E. coli IspC [41].
2.3. IspD [2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (YgbP)] Enzyme
The enzyme IspD (EC 2.7.7.60) spearheads the cytidylation process, playing a pivotal role in the third step of the pathway, with its activity dependent on divalent cations such as Mg2+ or Mn2+. Its catalytic mechanism involves the utilization of nucleotide derivatives, cytosine-5′-triphosphate (CTP) and 2-C-methyl-D-erythritol-4-phosphate (MEP) as substrates, to yield 4-diphosphocytidyl-2C-methyl-D-erythritol (CDP-ME) and pyrophosphate as a byproduct. In the analysis of transcriptomic data, a constitutive expression profile was discerned for the PfIspD enzyme, exhibiting a marginal upregulation specifically during the trophozoite stage (Supp data) [25].
CD analysis revealed that the IspD enzyme belongs to the Glycosyl transferase family A (GT-A) domain, also known as the CDP-ME synthase domain (512-677) (Figure 4a), where Rossmann fold architecture enables direct nucleotide derivative binding. Sequence analysis unveils two distinctive conserved signature motifs: GXG (194-260) and [IVT]-[LIVMC]-[IVT]-[HS]-D-[SGAV]-[AV]-R (510-593) (Figure 4b). Notably, the GXG motif, rich in glycine, and located towards the N-terminal, offers a suitable orientation for adjacent conserved signature motif substrate binding (Table 1). Catalytic activity of the IspD enzyme adheres to an associative mechanism, characterized by nucleophilic attack on the α-phosphate of CTP by the 4-phosphate of MEP, leading to a negatively charged penta-coordinate transition state. The collapse of this transition state culminates in the release of pyrophosphate and CDP-ME molecules. Within this process, two lysine residues, 215 and 662, play a critical role in stabilizing the pentavalent transition state. MSA highlights the conservation of these fundamental basic residues within the IspD active site across all apicomplexan parasites with sequence identity spanning between 20 to 78%, attributed to the sequence length variation caused by NEAT sequences (Figure 4b; Supp data).Top of Form Phylogenetic tree analysis positions the IspD enzyme of malaria parasites within a shared clade, while T. gondii aligns closer to the prokaryotes and plant clade (Figure 4c). This phylogenetic tree construction utilized the sequence from T. gondii as an outgroup.
2.3.1. PfIspD Enzyme’s Structural Insight
The X-ray crystal structure of the IspD protein has been elucidated in several pathogens and plants, serving as a template for predicting the structure in P. falciparum. Notably, the crystal structure of the bifunctional enzyme IspDF has also been resolved in the food-borne pathogen Campylobacter jejuni [42]. While both enzymes are involved in distinct steps of the MEP pathway, they are encoded as a single transcript and assemble into a complex quaternary structure, comprising a monofunctional IspD dimer and a monofunctional IspF trimer. An implied physical association between IspDF and the IspE enzyme further suggests a coordinated interplay for the precise execution of the pathway [43]. However, electrophoresis on native gel and size exclusion chromatography did not confirm the formation of such assemblies for the enzymes [44].
Despite the presence of NEAT sequences leading to sequence length variability, critical residues like Thr 549 and Arg 574 in the IspD enzyme are conserved across various organisms, playing a pivotal role in interacting with the MEP substrate. Moreover, several other residues forming the catalytic site, including His 515, Asp 516, Ala 518, Arg 519, Lys 551, Thr 582, Pro 583, and Glu 584, remain conserved across the IspD enzyme sequences (Figure 4d; Supp data). The catalytic activity of the IspD enzyme has also been observed in P. vivax field isolates, with novel inhibitors being identified using a substrate-dependent assay [7].
In the PfIspD enzyme, the conserved residues discerned through MSA and CDD analysis collectively form a binding cavity. Upon molecular docking, substrate molecules MEP and CTP interact with conserved residues such as Lys 610, Thr 612, Asn (618, 620, 621), Asp 635, and Ser 638, as well as Lys 464, Phe 578, Ala 580, Asn (618, 620), Ser (637, 638), and Lys 662, respectively (Figure 4e,f). Notably, the binding of negatively charged substrates heavily relies on interaction with positively charged catalytic residues, an intricate interplay essential for substrate recognition and enzymatic activity.
2.3.2. PfIspD Enzyme’s Inhibitors Studies
The catalytic site of the IspD enzyme exhibits a pronounced affinity towards the phosphate groups within its substrates, which offers an avenue for designing diverse analogs to explore their in-vitro impact on parasite growth. Past efforts in in-silico screening assays have led to the identification of inhibitory molecules, subsequently prompting the development of in-vitro assays to assess the efficacy of these compounds [7].
L-erythritol-4-phosphate (E4P) was recognized as an initial inhibitor of IspD in Brucella abortus, albeit with a modest IC50 of 1.36 mM [45]. Subsequently, a synthetic compound named 7-hydroxy [1,2,4] triazolo [1,5-a] pyrimidine (Azolopyrimidine) emerged as an IspD inhibitor for A. thaliana, showing inhibition at the nanomolar level [46]. In docking analysis, we observed Azolopyrimidine interact with amino acid residues Glu 368, Lys 381, Asn (463, 465, 618), and Ile 616 of the PfIspD (Figure 4g). Another noteworthy inhibitor, MMV008138 competes with the CTP substrate as established earlier [47,48], however, found effective against P. vivax IspD only at lower CTP concentrations [49]. Furthermore, its activity does not extend to the liver stages of P. yoelii or the sexual stages of P. falciparum [50]. In asexual stages, MMV008138 exhibited a discernible impact on late trophozoite and early schizont stages, whereas its influence on the unchanged ring stage is attributed to either a limited need for isoprenoids or an ample supply thereof for parasite survival [51]. Detailed interaction studies unveiled the binding of MMV008138 with conserved residues Ile 614, Asn 615, and Lys 662 within the catalytic site of PfIspD (Figure S5). The congruence in the catalytic site features of IspC and IspD enzymes prompted an investigation into the effects of Fos on the PfIspD enzyme. Notably, Fos exhibited binding affinity with amino acid residues Ala 580, Asn (618, 620), Asp 635, and Ser 637-638 h, Figure S6–S9).
2.4. IspE [4-(cytidine-5-diphospho)-2-C-methyl-D-erythritol kinase (CMK)] Enzyme
IspE enzyme (EC 2.7.1.148) is ATP-dependent, and its primary function involves catalyzing the conversion of CDPME to 4-diphosphocytidyl–2C-methyl-D-erythritol-2-phosphate (CDP-ME-2P), resulting in the release of a single ADP molecule, with the divalent cation Mg2+ playing a pivotal role in this process. The enzyme follows a characteristic pattern of containing NEAT sequences, which are consistently observed in other malaria parasites and plants [52,53]. A constitutively expressed pattern was discerned for the PfIspE enzyme across diverse stages of the parasite's life cycle. Nevertheless, noticeable strain-to-strain variation in expression levels was also identified (Supp data) [25].
In a comprehensive CD analysis, the IspE enzyme reveals the presence of a functional domain associated with the GHMP (Galactose, Homoserine, Mevalonate, and Phosphomevalonate) kinase super-family (92-516). This domain comprises two distinct segments: the N-terminal ribosomal protein S5 domain (133-352), which forms a structural framework and provides a binding site for the phosphate group, and the C-terminal region (359-503), which acts as the substrate binding site (Figure 5a). Sequence-based comparisons indicate a high degree of conservation within the C-terminal region across the Plasmodium genus and prokaryotes. This conservation extends to the structural features, substrate specificity, and catalytic mechanisms of the enzyme. Notably, no signature motif has been identified in IspE proteins (Table 1).
MSA analysis highlights the presence of three conserved sequence motifs, namely -NIIIKVL- (255-261; underlined in red), -GVGGGSS- (289-295; underlined in red), and -MSGSGSS- (467-473; underlined in blue). These motifs are strategically positioned in proximity to each other at the domain-domain interface, collectively forming the enzyme's catalytic center. This arrangement facilitates the binding of the CDP-ME substrate and ATP, essential for the enzymatic activity (Figure 5b). The average sequence similarity score derived from MSA ranges from 21% to 92%, underscoring the presence of amino acid conservation throughout the sequence, particularly in regions associated with secondary structure and the active sites (Supp data). Although enzymes from the GHMP Kinase family participate in the Mevalonate (MVA) pathway in the human host, sequence alignment analysis does not reveal significant amino acid identity between these enzymes (data not shown). It is also noteworthy that the IspE enzyme is enriched with hydrophobic residues, which may contribute to its lower solubility and pose challenges for its study as a recombinant protein.
A phylogenetic tree provides compelling evidence of the independent evolutionary trajectory of Plasmodium IspE enzymes with P. cynomolgi, P. knowlesi, P. inui, and P. vivax forming a well-defined cluster. In contrast, rodent parasites such as P. yoelii, P. berghei, P. chabaudi, and P. vinckei constitute a separate lineage, distinct from the group. Notably, P. falciparum branches off into a separate evolutionary branch (Figure 5c). This unique clustering pattern sets them apart from other apicomplexans, plants, and algae. Such insights into the evolutionary relationships of IspE enzymes contribute to a deeper understanding of the genetic diversity and adaptation strategies within the Plasmodium genus, shedding light on the complex dynamics of these parasites and their interactions with various host organisms. This phylogenetic tree construction utilized the sequence from T. thermophilus as an outgroup.
2.4.1. PfIspE Enzyme’s Structural Insight
The structural elucidation of PfIspE was achieved through computational modeling, providing valuable insights into its structural and functional characteristics, particularly pertaining to the active site. PfIspE is monomeric in nature and exhibits a distinct two-domain fold characteristic of the GHMP kinase superfamily. These two domains consist of an ATP-binding domain and a substrate-binding domain. The active site of the enzyme is nestled within a deep cleft formed between these two domains. In this configuration, the N-terminal domain adopts a conformation conducive to phosphate binding, facilitating nucleotide (ATP) interaction, while the C-terminal domain serves as the substrate-binding region (Figure 5d; Supp data).
Detailed interaction studies have illuminated the specific binding interactions between substrate CDP-ME and PfIspE, facilitated by residues including Asn 141, Pro 285, Phe 287, Ser (288, 374, 470), Gly (289, 291), Val 290, and Tyr 378 (Figure 5e). Additionally, the substrate ATP has been found to bind to PfIspE with high affinity, involving key residues such as Phe 287, Ser (288, 294, 328, 374), Gly (289, 292), Asp 329, and Leu 372 (Figure 5f and Figure S10).
2.4.2. PfIspE Enzyme’s Inhibitors Studies
Employing a structure-based design approach, several inhibitors have been identified, primarily targeting the substrate rather than the enzyme itself. These inhibitors interact with hydrophobic residues within the cytidine binding pocket of PfIspE. Notably, substitutions with hydrophilic residues within this binding pocket deactivate the inhibitors. Given the relatively small size of the binding pocket in IspE, inhibitors designed for GHMP kinases were also screened against PfIspE. The docking studies revealed the binding of compound ethyl {3-[4-amino-5-{3-[(cyclopropyl sulfonyl)amino] prop-1-yn-1-yl}-2-oxopyrimidin-1(2H)-yl] oxetan-3-yl}acetate with CDM binding pocket residues Lys 139, Asn 141, Phe 287, Gly 293, Ser (294, 374, 470), Asp 329, and Tyr 378, elucidating the binding mechanisms and its potential as anti-malarial agents (Figure 5g and Figure S11).
2.5. IspF [2C-Methyl-D-erythritol-2, 4-cyclodiphosphate synthase (ygbB)] Enzyme
IspF (EC 4.6.1.12), an enzyme vital in the MEP pathway, plays a pivotal role in a distinctive cyclization reaction. This reaction involves the replacement of CMP (cytidine monophosphate) with two phosphate groups from CDP-ME-2P, ultimately yielding cyclic diphosphate MECP. Similar to IspD, this enzymatic conversion is contingent upon the binding of divalent cations, specifically Mg2+ or Zn2+. The exceptional cyclization reaction catalyzed by IspF, coupled with its indispensability in various pathogens, has garnered considerable attention as a potential target for inhibiting the MEP pathway and developing novel anti-infective agents. Evidence supporting IspF's role in feedback regulation within the MEP pathway has been substantiated through intermediate analysis. Specifically, the presence of MEP as a potential modulator significantly enhances the enzymatic activity of IspF [54]. As discussed earlier, the bifunctional IspDF enzyme catalyzes two steps of the MEP pathway concurrently, however, its role in enhanced metabolic flux is not reported. The transcriptomic data indicates an upregulation of the PfIspF enzyme during the trophozoite stages, concomitant with a downregulation observed in the schizont stages. (Supp data) [25].
In comparison to other enzymes in the MEP pathway, IspF is relatively small. Its CD analysis reveals the presence of a distinctive MECDP synthase domain (64-236) (Figure 6a). Functionally, IspF exists as a homotrimer, with each subunit harboring an active site that collectively forms a cleft between pairs of subunits. MSA indicates a high degree of sequence identity within the MECPD synthase domain (underlined in red; Figure 6b), with an overall similarity index ranging from 27 to 92% (Supp data). Additionally, a signature motif, S-[DN]-[GA]-D-LIVAP]-[LIVAG]-x-H-[STAC]-x(2)-[DNT]-[SAG]-x(2)-[SGA], characteristic of the IspF enzyme, has been identified (Table 1).
Phylogenetic analysis was conducted for the IspF enzyme using sequences obtained from various organisms, including plants. The dendrogram illustrates the clustering of Plasmodium IspF enzymes among P. cynomolgi, P. vivax, P. knowlesi, and P. inui, while rodent parasites such as P. yoelii, P. berghei, and P. chabaudi form a distinct lineage. P. vinckei and P. falciparum, on the other hand, form a separate branch (Figure 6c). Other apicomplexan parasites cluster in proximity to plants and prokaryotes. This phylogenetic analysis utilized the sequence from M. avium as an outgroup and reaffirms the independent evolution of Plasmodium IspF enzymes, as they group distinctly apart from other apicomplexan parasites, plants, and algae.
2.5.1. PfIspF Enzyme’s Structural Insight
Substrate-enzyme complex of IspF has been elucidated in P. falciparum (PDB Id: 4C81) [55], providing insight into the functional configuration of the enzyme (Figure 6d) that consists of a hydrophobic cavity at the core of the trimer with affinity towards IPP, DMAPP, geranyl diphosphate (GPP), and farnesyl diphosphate (FPP) however, none of them inhibits IspF [54]. The substrate alignment and its binding in the active site are stabilized by the divalent cation Zn2+ or Mg2+. Alpha fold servers predicted the protein structure with unstructured sequence forming a loop in the NEAT region, essential for the enzyme’s apicoplast targeting.
Substrate-based docking analysis revealed the interaction of CDP-ME-2P with amino acid residue Asp (71, 127), Ile (72, 138), His (73, 115), Gln 95, and Ser (114, 116) (Figure 6e; Supp data). A high apolar residue presence makes IspF an attractive drug target however its potential as a putative drug target is unexploited as fewer inhibitors were reported. Various anthranilate cytidine complexes were tried against E. coli IspF where fluorescent diammonium 5 ′ -O-{[({[2-({[5-(Dimethylamino) naphthalene-1- yl] sulfonyl} amino) ethyl] oxy} phosphinato) oxy] phosphinato} cytidine found to be inhibitive [56].
2.5.2. PfIspF Enzyme’s Inhibitors Studies
High-throughput screening led to the identification of non-cytidine-like thiazolopyrimidine derivatives as IspF inhibitors in Mycobacterium and P. falciparum [57]. In our docking analysis aryl bis sulfonamide interacts with conserved residues Ser (114, 116), His 115, Asp 127, and Asn 149 of PfIspF enzyme (Figure 6f and Figure S12) whereas non-cytidine-like thiazolopyrimidine derivative, Ethyl (2Z)-2-(3,5-Dibromo-4-hydroxybenzylidene)-7-methyl-3-oxo-5-(2-thienyl)-2,3-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimi-dine-6-carboxylate interact with Gln 95, Ser 114, His 115, Phe 142 and Asp 144 (Figure 6g and Figure S13).
2.6. IspG [4-Hydroxy-3-methyl-2-(E)-butenyl-4-diphosphate synthase (gcpE)] Enzyme
IspG enzyme plays a crucial role in the penultimate step of the MEP pathway, where its activity hinges on iron-sulfur clusters. IspG orchestrates the reduction of MECP through a multi-step enzymatic reaction, ultimately converting it into 4-hydroxy-3-methyl-2-(E)-butenyl-4-diphosphate (HMBDP). Electrons required in this catalytic process for MECP substrate binding are furnished by the ferredoxin/ferredoxin reductase/NADPH system through [4Fe-4S] clusters [58]. This electron transfer process is pivotal for the formation of a double bond, which, in turn, converts the cyclic form of the MECP molecule into the aliphatic HMBDP and results oxidized [4Fe-4S] cluster recharged by a ferredoxin molecule. In transcriptomic datasets, the PfIspG enzyme exhibits downregulation throughout the invasion process until the ring stages, followed by an upswing in expression levels during the transition from ring to trophozoite stages. Subsequently, a decline in expression is observed during the schizont stages (Supp data) [25].
CD analysis indicates that the IspG enzyme belongs to the GcpE superfamily (Figure 7a), encompassing two conserved domains. The first domain is the TIM (Triosephosphate isomerase) barrel domain (responsible for MECP substrate binding; 112-403, underlined in red), while the second domain is the C-terminal domain (responsible for housing [4Fe-4S] clusters; 716-817). While no distinctive signature motifs were identified, the presence of conserved cysteine residues toward the C-terminal domain is a noteworthy feature of this protein (underlined in blue). MSA underscores a similarity index ranging from 30 to 97%, with the highest identity observed in the TIM barrel and C-terminal domain regions (Figure 7b; Supp data). In phylogenetic tree analysis, malaria parasites were found to cluster together in a similar clade, suggesting a common evolutionary lineage, while T. gondii was situated within the clade associated with A. thaliana, signifying a divergence in evolutionary history (Figure 7i). This phylogenetic tree construction utilized the sequence from A. aeolicus as an outgroup.
2.6.1. PfIspG Enzyme’s Structural Insight
The structural analysis of IspG reveals a clear division into two distinct regions: the core region and the cap region. The core region adopts a TIM barrel structure, providing the substrate binding site and serving as the enzyme's catalytic center. In contrast, the cap region functions as a flap that can cover this core region, and it contains conserved cysteine residues positioned at specific distances from each other.
In this study, the structural model of PfIspG was obtained through the AlphaFold platform, followed by rigorous refinement and evaluation of protein structure quality using various online tools before further analysis. The resulting PfIspG structure exhibits a remarkable level of conservation within the conserved domain. The TIM barrel structure within the core region comprises ten beta-sheets and nine alpha-helices arranged alternately. It encompasses several conserved residues, including four arginine residues (Arg 169, 219, 268, and 377), Gln 140, Asp 200, Ser 314, and His 344 located in the beta barrel, as well as Met 142, Asn 221, Asn 273, Lys 316, Thr 348, Glu 349, and Ser 379 located in the loops connecting beta strands to alpha helices. Notably, the seventh and eighth helices of the TIM barrel contain conserved hydrophobic residues. The C-terminal domain, on the other hand, features three alpha-helices and five beta-sheets, along with three cysteine residues (Cys 732, 735, and 766) arranged in a loop and Glu 773 situated within an alpha-helix (Figure 7c).
This refined PfIspG structure was subjected to docking simulations with the MECP substrate, revealing robust interactions with conserved residues such as Arg (169, 219, 377), Thr (272, 348), Ser 314, Met 315, and Tyr 327 located on the beta-sheet of the TIM barrel domain (Figure 7d and Figure S14). Additionally, the [4Fe-4S] cluster displayed binding affinity with three cysteine residues (Cys 732, 735, and 766) and Glu 773 within the C-terminal region (Figure 7e). It's worth noting that IspG enzyme activity is critically reliant on [4Fe-4S] clusters, which are synthesized by the Suf system in the apicoplast and are subsequently transferred directly to these cysteine residues [58]. This interaction between the [4Fe-4S] cluster and the cysteine residues brings the cap and core regions into proximity, facilitating the transfer of electrons from the [4Fe-4S] cluster to the MECP substrate.
2.6.2. PfIspG Enzyme’s Inhibitors Studies
Furthermore, this study delves into the inhibitory effects of various molecules on PfIspG, including known IspG inhibitors from prokaryotes, as well as the identification of novel inhibitory compounds for PfIspG through high-throughput screening and in silico methods. Several diphosphate compounds containing functional groups like alkyne, carboxylate, pyridine, and imidazole moieties were docked against the PfIspG enzyme where compounds without triple bonds were significantly less active. Fos, an inhibitor of other pathway enzymes showed interaction with Arg (169, 219, 377), Ser 314, and Thr (272, 348) amino acid residues of PfIspG (Figure 7 and Figure S15). A strong binding affinity of atorvastatin with amino acid residue Ile 106, Tyr (116, 546), Lys (117, 136, 399, 438), Leu 400, Asp 454 was observed (Figure 7g) whereas Prop-2-yn-1-yl trihydrozen di phosphate interacts with Arg (169, 218, 267, 376), Thr 348 (Figure 7h and Figure S16). Alkynes has higher binding affinity for iron atom in the [4Fe-4S] cluster and form ferracyclopropene structure which raises the question of specificity concerning the mammalian iron-sulfur cluster, however, the coordination of the [4Fe-4S] cluster in IspG is very specific where three iron atoms coordinate with cysteine sulfur atoms and the fourth one binding a glutamate carboxylate. The latter is then replaced by the hydroxyl group of MECP before the catalytic reaction activation [59].
2.7. IspH [4-Hydroxy-3-methyl-2-(E)-butenyl-4-diphosphate reductase (lytB)] Enzyme
IspH enzyme conducts the final step of the MEP pathway where it converts HMBDP to IPP and DMAPP, representing the ultimate product of the pathway. There are striking similarities between the functionality and requirement of the IspG and IspH enzymes i.e., contingent on the redox environment established within the apicoplast. The transcriptional profile of PfIspH enzyme mRNA undergoes a discernible downregulation during the initial developmental phases, with a subsequent upregulation in the trophozoite stages. A slight downregulation is noted during the transition from trophozoites to schizonts; however, a renewed upregulation is evident just prior to the release of merozoites (Supp data) [25].
CD analysis of IspH categorizes it within the LytB superfamily, delineating two distinct domains. One domain is specialized for the HMBDP substrate (spanning residues 220-535), while the other accommodates the crucial [3Fe-4S] clusters (Figure 8a). The binding of these [3Fe-4S] clusters play a pivotal role in inducing a closed conformation of the enzyme, a critical step within this pathway. Moreover, a signature motif was identified within the IspH protein of various organisms, adding to its distinctive features (Table 1).
In MSA, the similarity index exhibited variability ranging from 21% to 92%, with the most significant variations occurring towards the N-terminal region, primarily due to the presence of NEAT sequences (Supp data). Conversely, higher sequence identity was observed within the substrate-binding domain (underlined in red; Figure 8b), with a distinct presence of conserved cysteine residues towards the N-terminal region, facilitating the binding of [3Fe-4S] clusters (underlined in blue Figure 8b). Phylogenetic analysis, rooted from the plant IspH, delineated four separate clades. Interestingly, T. gondii IspH occupied an intermediate position between the clade encompassing the malaria parasite and prokaryotes, further reflecting the evolutionary diversity and distinctiveness of IspH enzymes within various organisms (Figure 8c). This phylogenetic tree construction utilized the sequence from A. protothecoides as an outgroup.
2.7.1. PfIspH Enzyme’s Structural Insight
Crystallographic structure of the IspH enzyme was resolved from A. aeolicus, E. coli, and P. falciparum [60], however, in PfIspH, the first 217 amino acids (NEAT) were not included in the construct utilized for the crystallization process. IspH enzyme folded structure resemble a three-leaf clover model and consist of three structurally similar domain without any sequence similarity. Each domain comprises four central parallel beta sheets surrounded by three or four alpha helices where the active site is buried in a hydrophobic cavity embedded between these three domains (Figure 8d). The [3Fe-4S] cluster essential for enzymatic reaction interacts with three conserved cysteine residues. Each domain contributes to the cluster coordination since these cysteines are located at the N-terminus of the first alpha helix of each of the three domains. Whereas the C-terminal loop contains a short beta-sheet that covers the active site pocket to protect the [3Fe-4S] cluster from the solvent. Like the IspG enzyme, the reaction takes place inside this close confirmation where the binding of a single sulfate ion in the substrate cavity is sufficient to induce these changes [60].
The docking analysis revealed the interaction of conserved cysteine residues (231, 315, and 413) with the [3Fe-4S] clusters (Figure 8e), whereas residues Cys 231, Gly 233, His 260, Thr 384, Ser 441, 485, Asn 443 interact with HMBDP substrate (Figure 8f). Here the binding of conserved Cys 231 is pivotal as it helps in the interaction between HMBDP substrate and [3Fe-4S] clusters. PfIspH enzyme’s active site is composed of high apolar amino acids that serve as a druggable pocket, along with it, an allosteric site having a high affinity towards substrate analog was also identified.
2.7.2. PfIspH Enzyme’s Inhibitors Studies
In silico screening was conducted utilizing thiol and amino derivatives of the HMBDP substrate to elucidate potential novel inhibitors for PfIspH. During this analysis, diphosphates incorporating alkyne and nitrile functional groups exhibited notable activity against the [3Fe-4S] cluster. In the subsequent docking analysis, the alkyne diphosphate derivative, namely (E)-4-mercapto-3-methyl but-2-enyl diphosphate, demonstrated a pronounced affinity for residues commonly associated with the HMBDP substrate (Figure 8g).
Furthermore, bisphosphate pyridine derivatives were systematically investigated as potential inhibitors. Among these, the Phenolphthalein diphosphate pyridine was observed to interact with key residues including Cys (231, 413), His 260, Ser (441, 442), and Asn 443 (Figure 8h and Figure S17). Notably, the binding affinity exhibited by these compounds with the conserved Cys residues implies their potential competitive binding with the [3Fe-4S] clusters.
3. Materials and Methods
3.1. Exploring Orthologous Sequences of MEP Pathway Enzymes
The protein sequences of MEP pathway enzymes from various organisms, including Apicomplexans, Prokaryotes, Chromalveolata, Chlorophyta, and Plants, were acquired from NCBI and PlasmoDB [25]. To unveil conserved amino acid residues, multiple sequence alignment was carried out using the CLUSTAL Omega tool [61]. This alignment allowed the identification of shared regions, conserved domains, and functional motifs across these sequences.
The presence of Nuclear encoded apicoplast targeted (NEAT) sequences was predicted through the application of PlasmoAP, a tool known for its accuracy in this regard [62]. PlasmoAP relies on the recognition of specific features, particularly the presence of basic amino acid residues and regions enriched in Lysine and Asparagine (KN) toward the N-terminal section of the proteins. It's worth mentioning that the NEAT region is prone to proteolytic cleavage, which occurs once the enzyme is properly targeted to the apicoplast. To further support this prediction, the presence of cleavage sites within the MEP pathway enzymes was assessed using the SignalP 4.1 server [63].
3.2. Conserved Domain Architecture and Signature Motifs
The MEP pathway in the malaria parasite exhibits prokaryotic origins that have been conserved within the eukaryotic host. To assess the preservation of molecular and cellular integrity in the enzymes participating in this pathway within the eukaryotic host, a comprehensive examination was conducted. This analysis encompassed the identification of conserved domains and signature motifs utilizing established tools and resources. Specifically, the Conserved Domain Search tool from NCBI [64], the InterPro server [65], and Prosite from ExPASy [66] were employed for this purpose.
3.3. Phylogenetic Tree Construction and Analysis
The evolutionary history of proteins integral to the MEP pathway in diverse pathogens and representative organisms was deduced using the Maximum Likelihood method, a computational approach implemented in MEGA X software [67,68]. Initially, the MSA generated through Clustal Omega was utilized to construct the phylogenetic tree. This process involved employing Neighbor-Join and BioNJ algorithms on a matrix of pairwise distances calculated using a JTT model. Subsequently, the tree topology with the highest log likelihood value was selected. To ensure robustness, a bootstrap consensus tree was established from 1000 replicates, providing a representation of the evolutionary relationships among the analyzed taxa [69]. It's important to note that any positions in the dataset containing gaps or missing data were excluded from the analysis. This resultant tree was scrutinized to discern the evolutionary positioning of the proteins under investigation, elucidating their evolutionary divergence across various Plasmodium species, as well as in relation to other apicomplexans, prokaryotes, and plants.
3.4. Protein Structure Prediction
Since crystal structures for the MEP pathway enzymes from Plasmodium parasites have not been experimentally resolved, predicting their 3D models was necessary. To accomplish this, we employed the HHpred server, a computational tool developed by Söding et al. in 2005 [70]. In this procedure, we submitted the protein sequences of P. falciparum to the HHpred online server. The selection of appropriate template structures was based on their sequence identity with the target protein and the availability of high-resolution structural data. Crystal structures corresponding to these templates were retrieved from the RPSC PDB server (https://www.rcsb.org).
Subsequently, we employed the MODELLER 9v11 program, as developed by Eswar et al. in 2006 and based on principles outlined by Sali and Blundell in 1993 [71], to predict the protein structures. To assess the quality and reliability of these predicted structures, we calculated the Discrete Optimized Protein Energy (DOPE) score for each model. Among these structures, the one with the lowest DOPE score was chosen for further refinement. The selected structure underwent additional refinement processes, including energy minimization and molecular simulation, utilizing the GROMACS software package, developed by Van Der Spoel et al. in 2005 [72]. The overall procedure for structure refinement and the assessment of the refined structure's quality followed a method previously published by Saggu et al. in 2017 [4]. In addition, protein structures were retrieved from the AlphaFold server and underwent refinement before being utilized for various analyses [30,31].
3.5. Investigating Substrate Docking and In-Silico Drug Inhibition Strategies
To assess the binding affinity of MEP pathway enzymes toward their substrates, products, and potential inhibitors, we conducted molecular docking analyses. Initially, we identified the binding pockets within these enzymes, which are composed of conserved amino acid residues. This identification process was facilitated using the BioLip server, a tool developed by Yang et al. in 2013 [73]. Subsequently, based on the confidence score derived from the binding pocket identification, we carried out the docking simulations employing Molegro Virtual Docker, as established by Thomsen and Christensen in 2006 [74]. Binding energies for these interactions were calculated and compared with the enzyme-substrate and enzyme-inhibitor complex available in the database. Various inhibitors have lower binding energy with respect to the natural substrate, which shows that they can be used as potent inhibitors, whereas inhibitors with slightly higher binding energy can act as weak inhibitors (Table S1).
For this analysis, the chemical structures of the substrates and inhibitor molecules were obtained from the PubChem open chemistry and ChEMBL-NTD databases [75]. These chemical structures were then refined using LIGPREP v2.5, developed by Schrodinger LLC. Following these preparations, we performed docking simulations, generating protein-substrate and protein-inhibitor complexes. The resulting complexes were meticulously analyzed and further refined based on the protocol outlined in the previously published work [5].
4. Conclusion
In this study, a sequence and structure-based approach was employed to elucidate the mechanisms of action of the enzymes in the MEP pathway and to propose them as potential, yet untapped, drug targets. Crucial conserved residues, pivotal for the enzymatic processes, were identified, shedding light on the binding affinities of substrates through docking analyses. The conservation of pathway properties supports the hypothesis that inhibitory molecules could act effectively across various pathogens. Amino acid substitution analysis revealed that the enzyme's core domain is sensitive to mutational changes, offering a promising avenue to address drug resistance development issues. The sequential nature of the pathway presents an attractive target for combinable inhibitors, potentially leading to a higher rate of pathway blockage.
The insights gained from this study could serve as a foundational framework for the design or synthesis of inhibitory molecules with high specificity based on binding residue properties. However, it is essential to define critical parameters such as cellular uptake and the molecule's half-life before pursuing these compounds as potential therapeutics. Ultimately, this research has the potential to contribute significantly to the development of combination therapies targeting conserved protein architectures, not only for malaria but also for combating other infectious diseases prevalent in developing countries.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Multiple sequence alignment percent identity matrix of MEP’s enzymes and targeting of these enzymes in P. falciparum apicoplast is provided in the supplementary file. Full-scale images for the enzyme interaction with the substrates or inhibitors are added to avoid discrepancies about the interaction site. Binding energies and binding affinity calculated for various interactions is represented in Table S1.
Author Contributions
G.S.S and K.D equally contributed to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases. The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Data Availability Statement
Sequence annotations are provided in Table 1 to obtain the data from online servers; PlasmoDB or NCBI.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. World Malaria Report 2022.
- Janouškovec, J., Horák, A., Oborník, M., Lukeš, J., Keeling, P. J. A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc Natl Acad Sci U S A. 2010, 107, 10949-10954. https://doi.org/10.1073/pnas.1003335107. [CrossRef]
- Goodman, C.D., Su, V., McFadden, G.I. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 2007, 152(2), 181-191. https://doi.org/10.1016/j.molbiopara.2007.01.005. [CrossRef]
- Ralph, S.A., van Dooren, G.G., Waller, R.F., Crawford, M.J., Fraunholz, M.J., Foth, B.J., Tonkin, C.J., Roos, D.S., McFadden, G.I. Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol. 2004, 2, 203-216. https://doi.org/10.1038/nrmicro843. [CrossRef]
- Saggu, G.S., Garg, S., Pala, Z.R., Yadav, S.K., Kochar, S.K., Kochar, D.K., Saxena, V. Characterization of 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase (IspG) from Plasmodium vivax and it’s potential as an antimalarial drug target. Int J Biol Macromol. 2017, 96, 466-473. https://doi.org/10.1016/j.ijbiomac.2016.12.033. Epub 2016 Dec 20. [CrossRef]
- Couto, A.S., Kimura, E.A., Peres, V.J., Uhrig, M.L., Katzin, A.M. Active isoprenoid pathway in the intra-erythrocytic stages of Plasmodium falciparum presence of dolichols of 11 and 12 isoprene units. Biochem J. 1999, 341, 629-637.
- Zhang, B., Watts, K.M., Hodge, D., Kemp, L.M., Hunstad, D.A., Hicks, L.M., Odom, A.R. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry. 2011, 50, 3570-3577. https://doi.org/10.1021/bi200113y. Epub 2011 Apr 11. [CrossRef]
- Saggu, G.S., Garg, S., Pala, Z.R., Kochar, S.K., Saxena, V. Deciphering the role of IspD (2 C methyl D erythritol 4 phosphate cytidyltransferase) enzyme as a potential therapeutic drug target against Plasmodium vivax. Gene. 2018, 675, 240-253. https://doi.org/10.1016/j.gene.2018.06.084. Epub 2018 Jun 26. [CrossRef]
- Yeh, E., and DeRisi, J.L. Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum. PLoS Biol. 2011, 9, e1001138. https://doi.org/10.1371/journal.pbio.1001138. [CrossRef]
- Uddin, T., McFadden, G.I., Goodman, C.D. Validation of putative apicoplast-targeting drugs using a chemical supplementation assay in cultured human malaria parasites. Antimicrob Agents Chemother. 2018, 62(1), 10-1128. https://doi.org/10.1128/AAC.01161-17. [CrossRef]
- Zhang, M., Wang, C., Oberstaller, J., Thomas, P., Otto, T.D., Casandra, D., Adams, J.H. The apicoplast link to fever-survival and artemisinin-resistance in the malaria parasite. Nat Commun. 2021, 12(1), 4563. https://doi.org/10.1038/s41467-021-24814-1. [CrossRef]
- Swift, R.P., Rajaram, K., Elahi, R., Liu, H.B., Prigge, S.T. Roles of Ferredoxin-Dependent Proteins in the Apicoplast of Plasmodium falciparum Parasites. mBio. 2021, 13(1), e0302321-e0302321. https://doi.org/10.1128/mbio.03023-21. Epub 2022 Feb 15. [CrossRef]
- Saggu, G.S. Apicoplast Journey and Its Essentiality as a Compartment for Malaria Parasite Survival. Front Cell Infect Microbiol. 2022, 12, 881825. https://doi.org/10.3389/fcimb.2022.881825. [CrossRef]
- Painter, H.J., Morrisey, J.M., Mather, M.W., Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007, 446(7131), 88-91. https://doi.org/10.1038/nature05572. [CrossRef]
- Guggisberg, A.M., Park, J., Edwards, R.L., Kelly, M.L., Hodge, D.M., Tolia, N.H., Odom, A.R. A sugar phosphatase regulates the methylerythritol phosphate (MEP) pathway in malaria parasites. Nat Commun. 2014, 5(1), 4467. doi: 10.1038/ncomms5467. [CrossRef]
- Jomaa, H., Wiesner, J., Sanderbrand, S., Altincicek, B., Weidemeyer, C., Hintz, M., Turbachova, I., Eberl, M., Zeidler, J., Lichtenthaler, H.K. Soldati, D. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999, 285(5433), 1573-1576. https://doi.org/10.1126/science.285.5433.1573. [CrossRef]
- Van Dooren, G.G., Stimmler, L.M., McFadden, G.I. Metabolic maps and functions of the Plasmodium mitochondrion. FEMS Microbiol Rev. 2006, 30(4), 596-630. https://doi.org/10.1111/j.1574-6976.2006.00027.x. [CrossRef]
- Simão-Gurge, R.M., Wunderlich, G., Cricco, J.A., Cubillos, E.F.G., Doménech-Carbó, A., Cebrián-Torrejón, G., Katzin, A.M. Biosynthesis of heme O in intraerythrocytic stages of Plasmodium falciparum and potential inhibitors of this pathway. Sci Rep. 2019, 9(1), 19261. https://doi.org/10.1038/s41598-019-55506-y. [CrossRef]
- Bofill Verdaguer, I., Sussmann, R.A., Santiago, V.F., Palmisano, G., Moura, G.C., Mesquita, J.T., Crispim, M. Isoprenoid alcohols utilization by malaria parasites. Front Chem. 2022, 10, 1035548. https://doi.org/10.3389/fchem.2022.1035548. [CrossRef]
- Okada, M., Rajaram, K., Swift, R.P., Mixon, A., Maschek, J.A., Prigge, S.T., Sigala, P.A. Critical role for isoprenoids in apicoplast biogenesis by malaria parasites. Elife. 2022, 11, e73208. doi: 10.7554/eLife.73208. [CrossRef]
- Labaied, M., Jayabalasingham, B., Bano, N., Cha, S.J., Sandoval, J., Guan, G., Coppens, I. Plasmodium salvages cholesterol internalized by LDL and synthesized de novo in the liver. Cell Microbiol. 2011, 13, 569–586. DOI: 10.1111/j.1462-5822.2010.01555.x. [CrossRef]
- Banerjee, T., Jaijyan, D.K., Surolia, N., Singh, A.P., Surolia, A. Apicoplast triose phosphate transporter (TPT) gene knockout is lethal for Plasmodium. Mol Biochem Parasitol. 2012, 186(1), 44-50. https://doi.org/10.1016/j.molbiopara.2012.09.008. [CrossRef]
- Kuzuyama, T., Takagi, M., Kaneda, K. Studies on the nonmevalonate pathway: conversion of 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol to its 2-phospho derivative by 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase. Tetrahedron Lett. 2000, 41, 2925-2928. DOI: 10.1016/S0040-4039(00)00295-1. [CrossRef]
- Estévez, J.M., Cantero, A., Reindl, A., Reichler, S., León, P. 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem. 2001, 276(25), 22901-22909. DOI: 10.1074/jbc.M100854200. [CrossRef]
- Bahl, A., Brunk, B., Crabtree, J., Fraunholz, M.J., Gajria, B., Grant, G.R., Ginsburg, H., Gupta, D., Kissinger, J.C., and Labo, P. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003, 31, 212-215. https://doi.org/10.1093/nar/gkg081. [CrossRef]
- Bringer-Meyer, S., Sahm, H. Junctions of catabolic and anabolic pathways in Zymomonas mobilis: phosphoenolpyruvate carboxylase and malic enzyme. Appl Microbiol Biotechnol. 1989, 31, 529-536. https://doi.org/10.1007/bf00270789. [CrossRef]
- Sprenger, G.A., Schorken, U., Wiegert, T., Grolle, S., Graff, A.A.D., Taylor, S.V., Begley, T.P., Bringer-Meyer, S., Sahm, H. Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-D-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc Natl Acad Sci U S A. 1997, 94, 12857-12862. https://doi.org/10.1073/pnas.94.24.12857. [CrossRef]
- Brammer, L.A., Smith, J.M., Wade, H., Meyers, C.F. 1-Deoxy-D-xylulose-5-phosphate synthase catalyzes a novel random sequential mechanism. J Biol Chem. 2011, 286(42), 36522-36531. https://doi.org/10.1074/jbc.M111.259747. doi: 10.1074/jbc.M111.259747. [CrossRef]
- Xiang, S., Usunow, G., Lange, G., Busch, M., Tong, L. Crystal structure of 1-deoxy-D-xylulose 5-phosphate synthase, a crucial enzyme for isoprenoids biosynthesis. J Biol Chem. 2007, 282(4), 2676-2682. https://doi.org/10.1074/jbc.M610235200. [CrossRef]
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. Bridgland, A., Highly accurate protein structure prediction with AlphaFold. Nature. 2021, 596(7873), 583-589. https://doi.org/10.1038/s41586-021-03819-2. [CrossRef]
- Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., Yuan, D., Stroe, O., Wood, G., Laydon, A. Žídek, A., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, 439-444. https://doi.org/10.1093/nar/gkab1061. [CrossRef]
- Frank, R.A.W., Leeper, F.J., Luisi, B.F. Structure, mechanism and catalytic duality of thiamine-dependent enzymes. Cell Mol Life Sci. 2007, 64, 892-905. https://doi.org/10.1007/s00018-007-6423-5. [CrossRef]
- Eubanks, L.M., Poulter, C.D. Rhodobacter capsulatus 1-deoxy-D-xylulose 5-phosphate synthase: steady-state kinetics and substrate binding. Biochemistry. 2003, 42(4), 1140-1149. https://doi.org/10.1021/bi0205303. [CrossRef]
- Gierse, R.M., Reddem, E.R., Alhayek, A., Baitinger, D., Hamid, Z., Jakobi, H., Groves, M.R. Identification of a 1-deoxy-D-xylulose-5-phosphate synthase (DXS) mutant with improved crystallographic properties. Biochem Biophysical Res Commun. 2021, 539, 42-47. https://doi.org/10.1016/j.bbrc.2020.12.069. [CrossRef]
- Chen, P.Y.T., DeColli, A.A., Meyers, C.L.F., Drennan, C.L. X-ray crystallography–based structural elucidation of enzyme-bound intermediates along the 1-deoxy-d-xylulose 5-phosphate synthase reaction coordinate. J Biol Chem. 2019, 294(33), 12405-12414. https://doi.org/10.1074/jbc.RA119.009321. [CrossRef]
- Mac Sweeney, A., Lange, R., Fernandes, R.P., Schulz, H., Dale, G.E., Douangamath, A., Proteau, P.J. Oefner, C., The crystal structure of E. coli 1-deoxy-D-xylulose-5-phosphate reductoisomerase in a ternary complex with the antimalarial compound fosmidomycin and NADPH reveals a tight-binding closed enzyme conformation. J Mol Biol. 2005, 345(1), 115-127. https://doi.org/10.1016/j.jmb.2004.10.030. [CrossRef]
- Nair, S.C., Brooks, C.F., Goodman, C.D., Sturm, A., McFadden, G.I., Sundriyal, S., Anglin, J.L., Song, Y., Moreno, S.N.J. Apicoplast isoprenoid precursor synthesis and the molecular basis of fosmidomycin resistance in Toxoplasma gondii. J Exp Med. 2011, 208, 1547-1559. https://doi.org/10.1084/jem.20110039. [CrossRef]
- Wang, X., Edwards, R.L., Ball, H., Johnson, C., Haymond, A., Girma, M., Dowd, C.S. MEPicides: α, β-unsaturated fosmidomycin analogues as DXR inhibitors against malaria. J Med Chem. 2018, 61(19), 8847-8858. https://doi.org/10.1021/acs.jmedchem.8b01026. [CrossRef]
- Phillips, A.M.F., Nogueira, F., Murtinheira, F., Barros, M.T. Synthesis and antimalarial evaluation of prodrugs of novel fosmidomycin analogues. Bioorg Med Chem Lett. 2015, 25(10), 2112-2116. https://doi.org/10.1016/j.bmcl.2015.03.077. [CrossRef]
- Girma, M.B., Ball, H.S., Wang, X., Brothers, R.C., Jackson, E.R., Meyers, M.J., Couch, R.D. Mechanism of Action of N-Acyl and N-Alkoxy Fosmidomycin Analogs: Mono-and Bisubstrate Inhibition of IspC from Plasmodium falciparum, a Causative Agent of Malaria. ACS omega. 2021, 6(42), 27630-27639. https://doi.org/10.1021/acsomega.1c01711. [CrossRef]
- Roca, C., Avalos-Padilla, Y., Prieto-Simón, B., Iglesias, V., Ramírez, M., Imperial, S., Fernàndez-Busquets, X. Selection of an Aptamer against the Enzyme 1-deoxy-D-xylulose-5-phosphate Reductoisomerase from Plasmodium falciparum. Pharmaceutics. 2022, 14(11), 2515. https://doi.org/10.3390/pharmaceutics14112515. [CrossRef]
- Gabrielsen, M., Rohdich, F., Eisenreich, W., Gräwert, T., Hecht, S., Bacher, A., Hunter, W. N. Biosynthesis of isoprenoids: a bifunctional IspDF enzyme from Campylobacter jejuni. Eur J Biochem. 2004, 271(14), 3028-3035. https://doi.org/10.1111/j.1432-1033.2004.04234.x. [CrossRef]
- Gabrielsen, M., Bond, C.S., Hallyburton, I., Hecht, S., Bacher, A., Eisenreich, W., Hunter, W.N. Hexameric assembly of the bifunctional methylerythritol 2, 4-cyclodiphosphate synthase and protein-protein associations in the deoxy-xylulose-dependent pathway of isoprenoid precursor biosynthesis. J Biol Chem. 2004, 279(50), 52753-52761. https://doi.org/10.1074/jbc.M408895200. [CrossRef]
- Liu, Y.L., Guerra, F., Wang, K., Wang, W., Li, J., Huang, C., Zhu, W., Houlihan, K., Li, Z., Zhang, Y. Nair, S.K., Structure, function and inhibition of the two-and three-domain 4Fe-4S IspG proteins. Proc Natl Acad Sci U S A. 2012, 109(22), 8558-8563. https://doi.org/10.1073/pnas.1121107109. [CrossRef]
- Lillo, A.M., Tetzlaff, C.N., Sangari, F.J., Cane, D.E. Functional expression and characterization of EryA, the erythritol kinase of Brucella abortus, and enzymatic synthesis of L-erythritol-4-phosphate. Bioorg Med Chem Lett. 2003, 13(4), 737-739. https://doi.org/10.1016/s0960-894x(02)01032-6. [CrossRef]
- Witschel, M.C., Höffken, H.W., Seet, M., Parra, L., Mietzner, T., Thater, F., Niggeweg, R., Röhl, F., Illarionov, B., Rohdich, F. Kaiser, J. Inhibitors of the herbicidal target IspD: allosteric site binding. Angew Chem Int Ed Engl. 2011, 50(34), 7931-7935. https://doi.org/10.1002/anie.201102281. [CrossRef]
- Wu, W., Herrera, Z., Ebert, D., Baska, K., Cho, S.H., DeRisi, J.L., Yeh, E.A. Chemical rescue screen identifies a Plasmodium falciparum apicoplast inhibitor targeting MEP isoprenoid precursor biosynthesis. Antimicrob Agents Chemother. 2015, 59(1), 356-364. https://doi.org/10.1128/AAC.03342-14. [CrossRef]
- Yao, Z.K., Krai, P.M., Merino, E.F., Simpson, M.E., Slebodnick, C., Cassera, M.B., Carlier, P.R. Determination of the active stereoisomer of the MEP pathway-targeting antimalarial agent MMV008138, and initial structure–activity studies. Bioorg Med Chem Lett. 2015, 25(7), 1515-1519. https://doi.org/10.1016/j.bmcl.2015.02.020. [CrossRef]
- Imlay, L.S., Armstrong, C.M., Masters, M.C., Li, T., Price, K.E., Edwards, R.L., Mann, K.M., Li, L.X., Stallings, C.L., Berry, N.G. Plasmodium IspD (2-C-Methyl-D-erythritol 4-Phosphate Cytidyltransferase), an Essential and Drug able Antimalarial Target. ACS Infect Dis. 2015, 1(4), 157-167. https://doi.org/10.1021/id500047s. [CrossRef]
- Bowman, J.D., Merino, E.F., Brooks, C.F., Striepen, B., Carlier, P.R., Cassera, M.B. Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the malaria box. Antimicrob Agents Chemother. 2014, 58(2), 811-819. https://doi.org/10.1128/AAC.01500-13. [CrossRef]
- Ghavami, M., Merino, E.F., Yao, Z.K., Elahi, R., Simpson, M.E., Fernández-Murga, M.L., Cassera, M.B. Biological studies and target engagement of the 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase (IspD)-targeting antimalarial agent (1 R, 3 S)-MMV008138 and analogs. ACS Infect Dis. 2018, 4(4), 549-559. https://doi.org/10.1021/acsinfecdis.7b00159. [CrossRef]
- Hirsch, A.K.H., Lauw, S., Gersbach, P., Schweizer, W.B., Rohdich, F., Eisenreich, W., Bacher, A., Diederich, F. Nonphosphate inhibitors of IspE protein, a kinase in the non-mevalonate pathway for isoprenoid biosynthesis and a potential target for antimalarial therapy. Chem Med Chem. 2007, 2, 806-810. https://doi.org/10.1002/cmdc.200700014. [CrossRef]
- Ahn C.S., Pai H.S. Physiological function of IspE, a plastid MEP pathway gene for isoprenoid biosynthesis, in organelle biogenesis and cell morphogenesis in Nicotiana benthamiana. Plant Mol Biol. 2008, 66(5):503-517. https://doi.org/10.1007/s11103-007-9286-0. [CrossRef]
- Bitok, J.K., Meyers, C.F. 2 C-Methyl-D-erythritol 4-phosphate enhances and sustains cyclodiphosphate synthase IspF activity. ACS Chem Biol. 2012, 7(10), 1702-1710. https://doi.org/10.1021/cb300243w. [CrossRef]
- O’Rourke, P.E., Kalinowska-Tłuścik, J., Fyfe, P.K., Dawson, A., Hunter, W.N. Crystal structures of IspF from Plasmodium falciparum and Burkholderia cenocepacia: comparisons inform antimicrobial drug target assessment. BMC Struct Biol. 2014, 14(1), 1. https://doi.org/10.1186/1472-6807-14-1. [CrossRef]
- Crane, C.M., Kaiser, J., Ramsden, N.L., Lauw, S., Rohdich, F., Eisenreich, W., Hunter, W.N, Bacher, A., Diederich, F. Fluorescent Inhibitors for IspF, an Enzyme in the Non-Mevalonate Pathway for Isoprenoid Biosynthesis and a Potential Target for Antimalarial Therapy. Angew Chem Int Ed Engl. 2006, 45(7), 1069-1074. https://doi.org/10.1002/anie.200503003. [CrossRef]
- Geist, J.G., Lauw, S., Illarionova, V., Illarionov, B., Fischer, M., Gräwert, T., Rohdich, F., Eisenreich, W., Kaiser, J., Groll, M. Scheurer, C. Thiazolopyrimidine inhibitors of 2-methylerythritol 2, 4-cyclodiphosphate synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. Chem Med Chem. 2010, 5(7), 1092-1101. https://doi.org/10.1002/cmdc.201000083. [CrossRef]
- Pala, Z.R., Saxena, V., Saggu, G.S., Garg, S. Recent advances in the [Fe–S] cluster biogenesis (SUF) pathway functional in the apicoplast of Plasmodium. Trends Parasitol. 2018, 34(9), 800-809. https://doi.org/10.1016/j.pt.2018.05.010. [CrossRef]
- Quitterer, F., Frank, A., Wang, K., Rao, G., O'Dowd, B., Li, J. Atomic-Resolution Structures of Discrete Stages on the Reaction Coordinate of the [Fe4S4] Enzyme IspG (GcpE). J Mol Biol. 2015, 427, 2220-2228. https://doi.org/10.1016/j.jmb.2015.04.002. [CrossRef]
- Rekittke, I., Olkhova, E., Wiesner, J., Demmer, U., Warkentin, E., Jomaa, H., Ermler, U. Structure of the (E)-4-hydroxy-3-methyl-but-2-enyl-diphosphate reductase from Plasmodium falciparum. FEBS Lett. 2013, 587(24), 3968-3972. https://doi.org/10.1016/j.febslet.2013.10.029. [CrossRef]
- Larkin, M.A., Blackshields, G., Brown, N., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23, 2947-2948. https://doi.org/10.1093/bioinformatics/btm404. [CrossRef]
- Foth, B.J., Ralph, S.A., Tonkin, C.J., Struck, N.S., Fraunholz, M., Roos, D.S., Cowman, A.F., McFadden, G. I. Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science. 2003, 299, 705-708. https://doi.org/10.1126/science.1078599. [CrossRef]
- Petersen, T.N., Brunak, S., von Heijne, G., and Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011, 8, 785-786. https://doi.org/10.1038/nmeth.1701. [CrossRef]
- Marchler-Bauer, A., Derbyshire, M.K., Gonzales, N.R., Lu, S., Chitsaz, F., Geer, L.Y., Geer, R.C., He, J., Gwadz, M., Hurwitz, D.I. Lanczycki, C.J. CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015, 43, 222-226. https://doi.org/10.1093/nar/gku1221. [CrossRef]
- Paysan-Lafosse, T., Blum, M., Chuguransky, S., Grego, T., Pinto, B.L., Salazar, G.A., Bateman, A. InterPro in 2022. Nucleic Acids Res. 2023, 51, 418-427. https://doi.org/10.1093/nar/gkac993. [CrossRef]
- Sigrist, C.J., Cerutti, L., Hulo, N., Gattiker, A., Falquet, L., Pagni, M., Bairoch, A., Bucher, P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002, 3, 265-274. https://doi.org/10.1093/bib/3.3.265. [CrossRef]
- Jones, D.T., Taylor, W.R., Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Computer applications in the biosciences: Comput Appl Biosci. 1992, 8(3), 275-282. https://doi.org/10.1093/bioinformatics/8.3.275. [CrossRef]
- Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018 1;35(6):1547-1549. https://doi.org/10.1093/molbev/msy096. [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985, 783-791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x. [CrossRef]
- Söding, J., Biegert, A., Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, 244-248. https://doi.org/10.1093/nar/gki408. [CrossRef]
- Sali, A. Blundell, T.L. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993, 234(3), 779-815. https://doi.org/10.1006/jmbi.1993.1626. [CrossRef]
- Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A.E., Berendsen, H.J. GROMACS: fast, flexible, and free. J Comput Chem. 2005, 26, 1701-1718. https://doi.org/10.1002/jcc.20291. [CrossRef]
- Yang, J., Roy, A., Zhang, Y. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2013, 41, 1096-1103. https://doi.org/10.1093/nar/gks966. [CrossRef]
- Thomsen, R., Christensen, M.H. Mol Dock: a new technique for high-accuracy molecular docking. J Med Chem. 2006, 49, 3315-3321. https://doi.org/10.1021/jm051197e. [CrossRef]
- Antonova-Koch, Y., Meister, S., Abraham, M., Luth, M.R., Ottilie, S., Lukens, A.K., Winzeler, E.A. Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science. 2018, 362(6419), eaat9446. https://doi.org/10.1126/science.aat9446. [CrossRef]
Figure 1.
Isoprenoid biosynthesis by mevalonate dependent (MVA) and non-mevalonate pathway (MEP) in a typical cell. Isoprenoids are derived from the basic 5-carbon isoprenoid building blocks IPP and its isomer, DMAPP. In the MVA pathway IPP and DMAPP are synthesized by condensation of acetyl CoA. Enzymes participating in this pathway are ACT, HMGS, HMGR, MVK, PMK, and MDS. Statins inhibit HMGR, the rate-limiting enzyme of the mevalonate pathway, and 6FMVA-PP and BPNs inhibit the other steps of the pathway. In the MEP pathway, IPP and DMAPP are generated from pyruvate and GA3P. Enzymes of this pathway are named here according to their E. coli homologs (DXS, IspC, IspD, IspE, IspF, IspG, and IspH). Ketoclomazone and Fosmidomycin inhibit the rate-limiting steps of this pathway and block isoprenoid biosynthesis. (Abbreviation details are provided in the supplementary data).
Figure 1.
Isoprenoid biosynthesis by mevalonate dependent (MVA) and non-mevalonate pathway (MEP) in a typical cell. Isoprenoids are derived from the basic 5-carbon isoprenoid building blocks IPP and its isomer, DMAPP. In the MVA pathway IPP and DMAPP are synthesized by condensation of acetyl CoA. Enzymes participating in this pathway are ACT, HMGS, HMGR, MVK, PMK, and MDS. Statins inhibit HMGR, the rate-limiting enzyme of the mevalonate pathway, and 6FMVA-PP and BPNs inhibit the other steps of the pathway. In the MEP pathway, IPP and DMAPP are generated from pyruvate and GA3P. Enzymes of this pathway are named here according to their E. coli homologs (DXS, IspC, IspD, IspE, IspF, IspG, and IspH). Ketoclomazone and Fosmidomycin inhibit the rate-limiting steps of this pathway and block isoprenoid biosynthesis. (Abbreviation details are provided in the supplementary data).
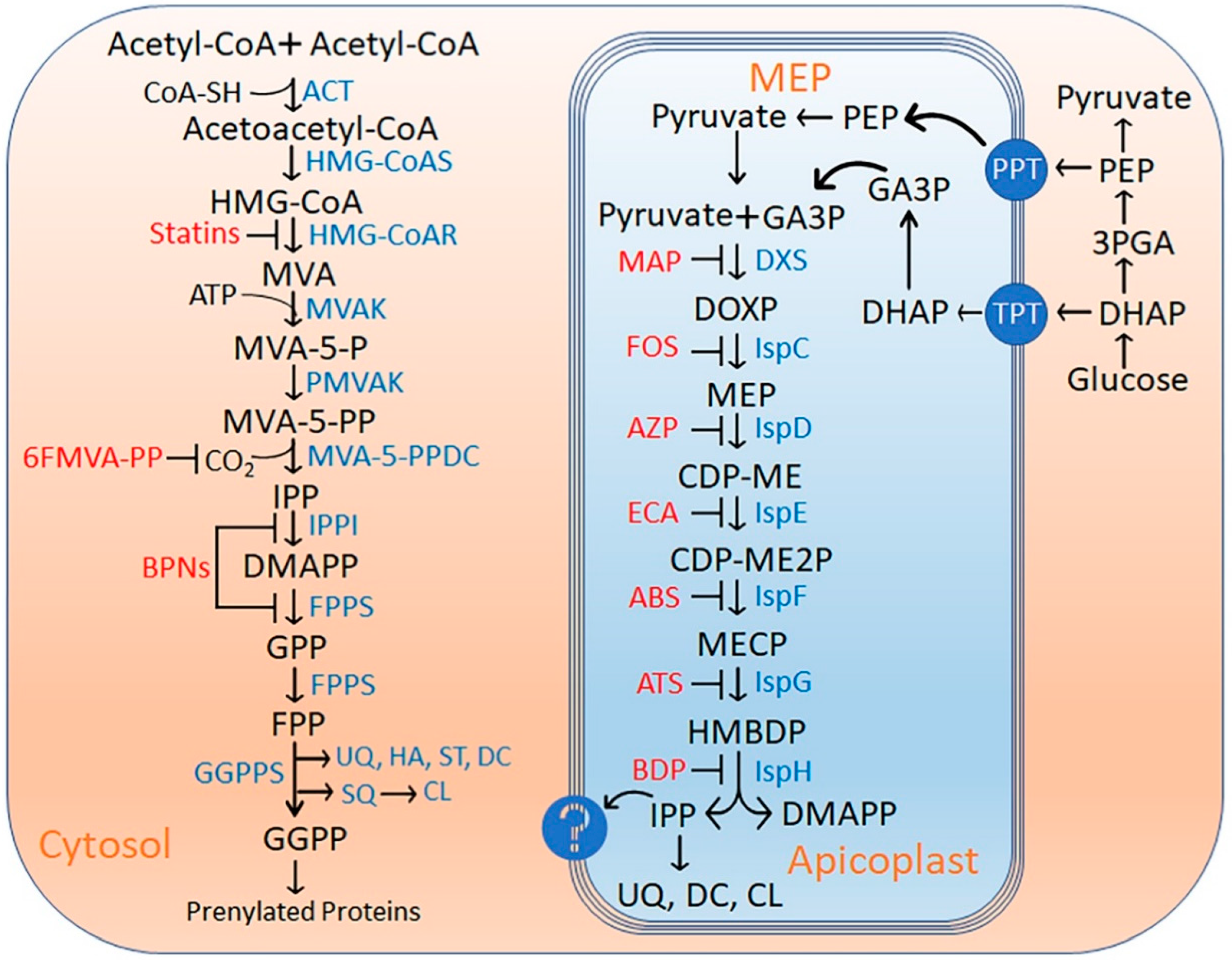
Figure 2.
A comparative analysis of the DXS enzyme across diverse organisms has been undertaken to investigate its conserved catalytic activity and elucidate its significance as a potential therapeutic target. PfDXS enzymes conserved domain analysis: TPP (thiamine pyrophosphate binding domain; 412-551), PYR (pyrimidine binding domain; 828-994), and transketolase C domain (1064-1191) (a), multiple sequence alignment of PfDXS sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfDXS enzyme structure obtained from Alpha fold server, repeated asparagine residues forms a loop in the central region (d), Interaction of PfDXS conserved residues with substrate TPP (e). In-silico inhibition studies revealed the binding of ß-fluropyruvate (f) and Methylacetylphosphonate (g) with conserved residues of PfDXS. *Conserved amino acids residues across various organisms.
Figure 2.
A comparative analysis of the DXS enzyme across diverse organisms has been undertaken to investigate its conserved catalytic activity and elucidate its significance as a potential therapeutic target. PfDXS enzymes conserved domain analysis: TPP (thiamine pyrophosphate binding domain; 412-551), PYR (pyrimidine binding domain; 828-994), and transketolase C domain (1064-1191) (a), multiple sequence alignment of PfDXS sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfDXS enzyme structure obtained from Alpha fold server, repeated asparagine residues forms a loop in the central region (d), Interaction of PfDXS conserved residues with substrate TPP (e). In-silico inhibition studies revealed the binding of ß-fluropyruvate (f) and Methylacetylphosphonate (g) with conserved residues of PfDXS. *Conserved amino acids residues across various organisms.
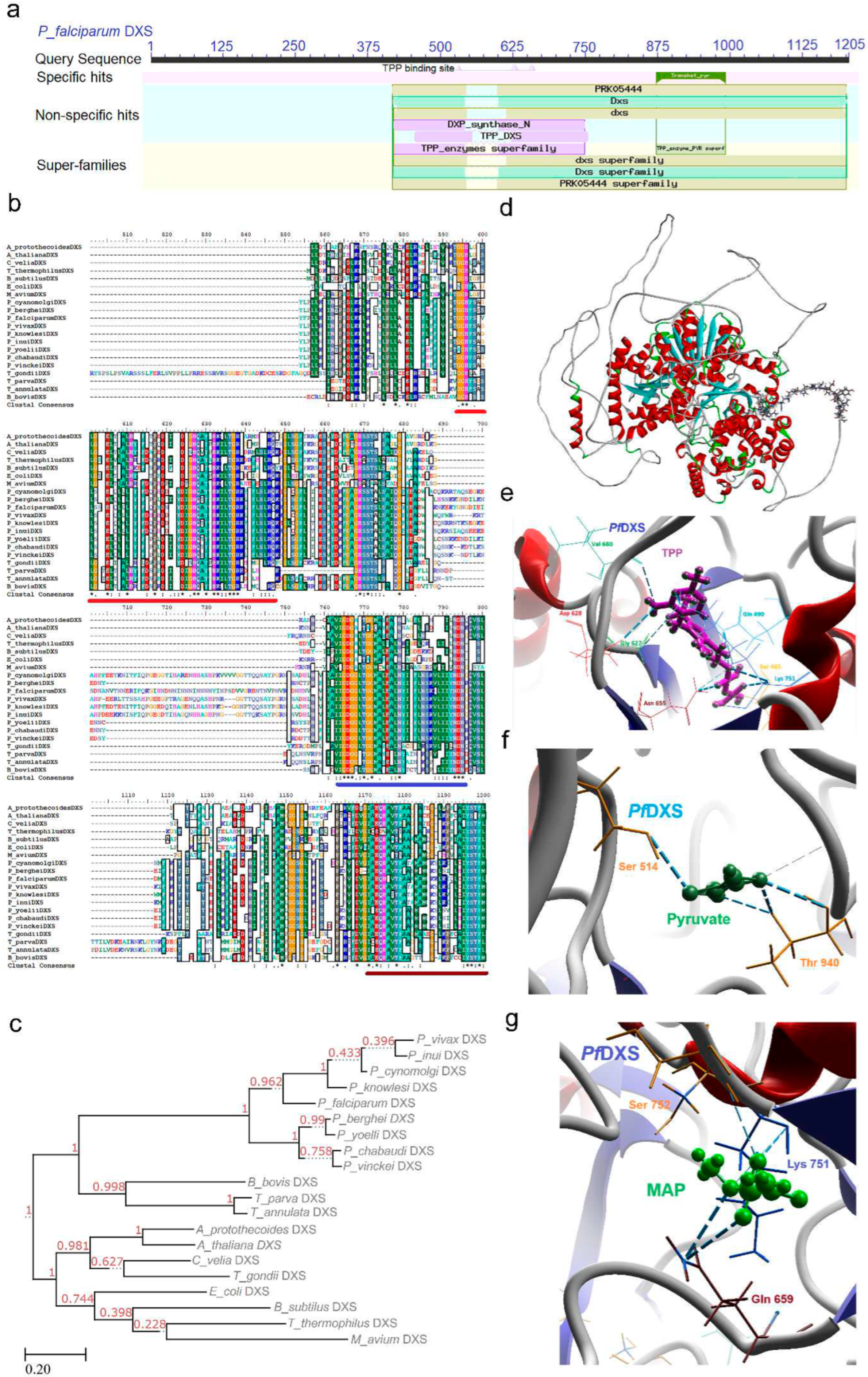
Figure 3.
A comparative analysis of the IspC enzyme across diverse organisms has been conducted to examine its conserved catalytic activity and ascertain its significance as a prospective therapeutic target. PfIspC enzyme conserved domain analysis: DXP reductoisomerase (80-212), DXP reductoisomerase catalytic domain (227-323), and DXP reductoisomerase C-terminal (357-475) (a), multiple sequence alignment of PfIspC sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspC enzyme structure retrieved from protein data bank server (Id no. 4KP7) (d). Interaction of PfIspC conserved residues with substrate DOXP (e), Interaction of PfIspC with Fos (f). *Conserved amino acids residues across various organisms.
Figure 3.
A comparative analysis of the IspC enzyme across diverse organisms has been conducted to examine its conserved catalytic activity and ascertain its significance as a prospective therapeutic target. PfIspC enzyme conserved domain analysis: DXP reductoisomerase (80-212), DXP reductoisomerase catalytic domain (227-323), and DXP reductoisomerase C-terminal (357-475) (a), multiple sequence alignment of PfIspC sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspC enzyme structure retrieved from protein data bank server (Id no. 4KP7) (d). Interaction of PfIspC conserved residues with substrate DOXP (e), Interaction of PfIspC with Fos (f). *Conserved amino acids residues across various organisms.
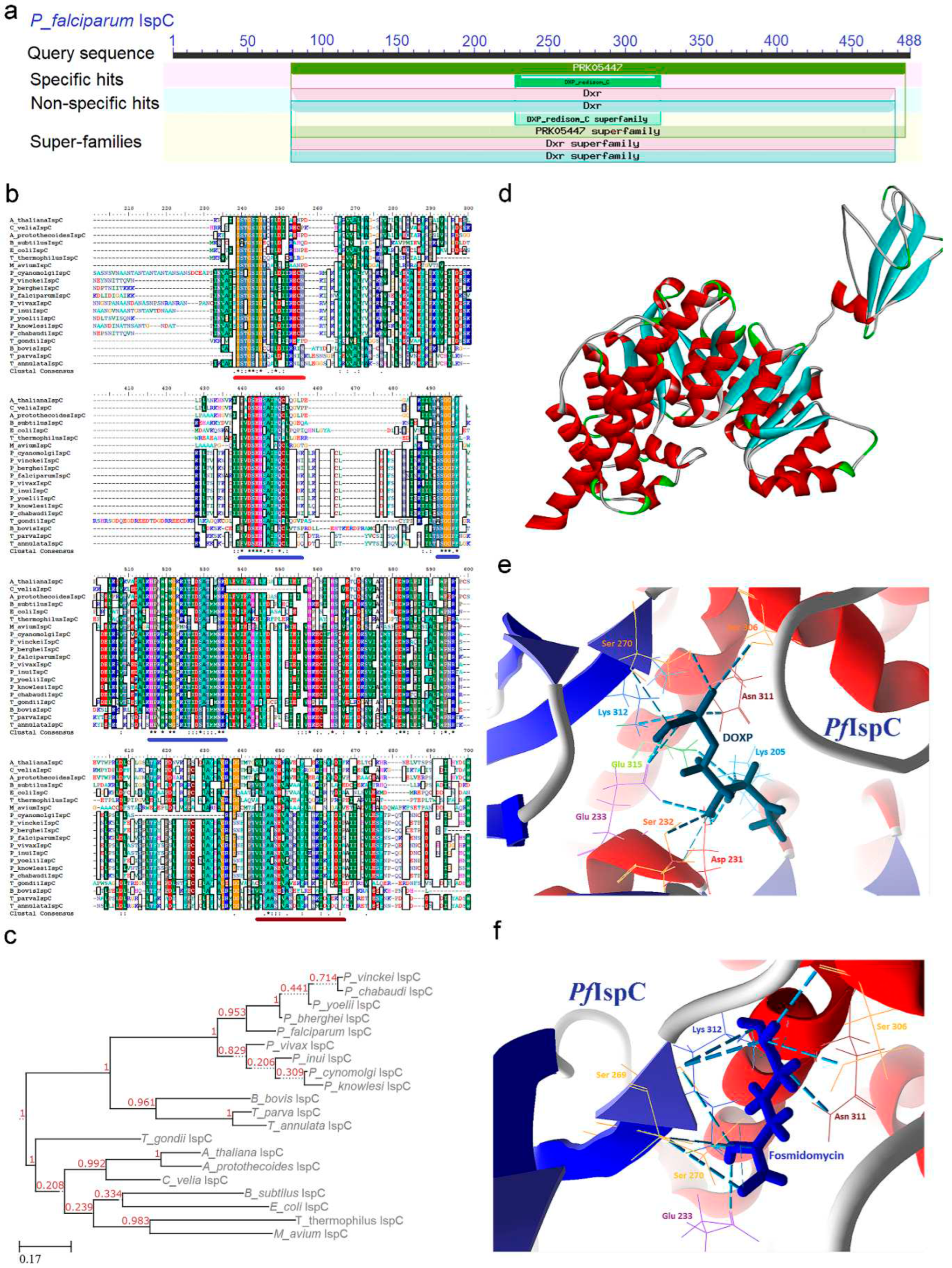
Figure 4.
A comparative analysis of the IspD enzyme across diverse organisms has been employed to investigate its conserved catalytic activity and assess its significance as a potential therapeutic target. PfIspD enzyme conserved CDP-ME synthase domain (512-677) (a), multiple sequence alignment of PfIspD sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspD enzyme structure predicted with computational methods, beta sheets S1-S7, S4 contains the conserved signature motifs (d), Interaction of PfIspD conserved residues with substrate MEP (e) and CTP (f). In-silico inhibition studies revealed the binding of Azolopyrimidines (g) and Fos (h) with conserved residues of PfIspD. *Conserved amino acids residues across various organisms.
Figure 4.
A comparative analysis of the IspD enzyme across diverse organisms has been employed to investigate its conserved catalytic activity and assess its significance as a potential therapeutic target. PfIspD enzyme conserved CDP-ME synthase domain (512-677) (a), multiple sequence alignment of PfIspD sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspD enzyme structure predicted with computational methods, beta sheets S1-S7, S4 contains the conserved signature motifs (d), Interaction of PfIspD conserved residues with substrate MEP (e) and CTP (f). In-silico inhibition studies revealed the binding of Azolopyrimidines (g) and Fos (h) with conserved residues of PfIspD. *Conserved amino acids residues across various organisms.
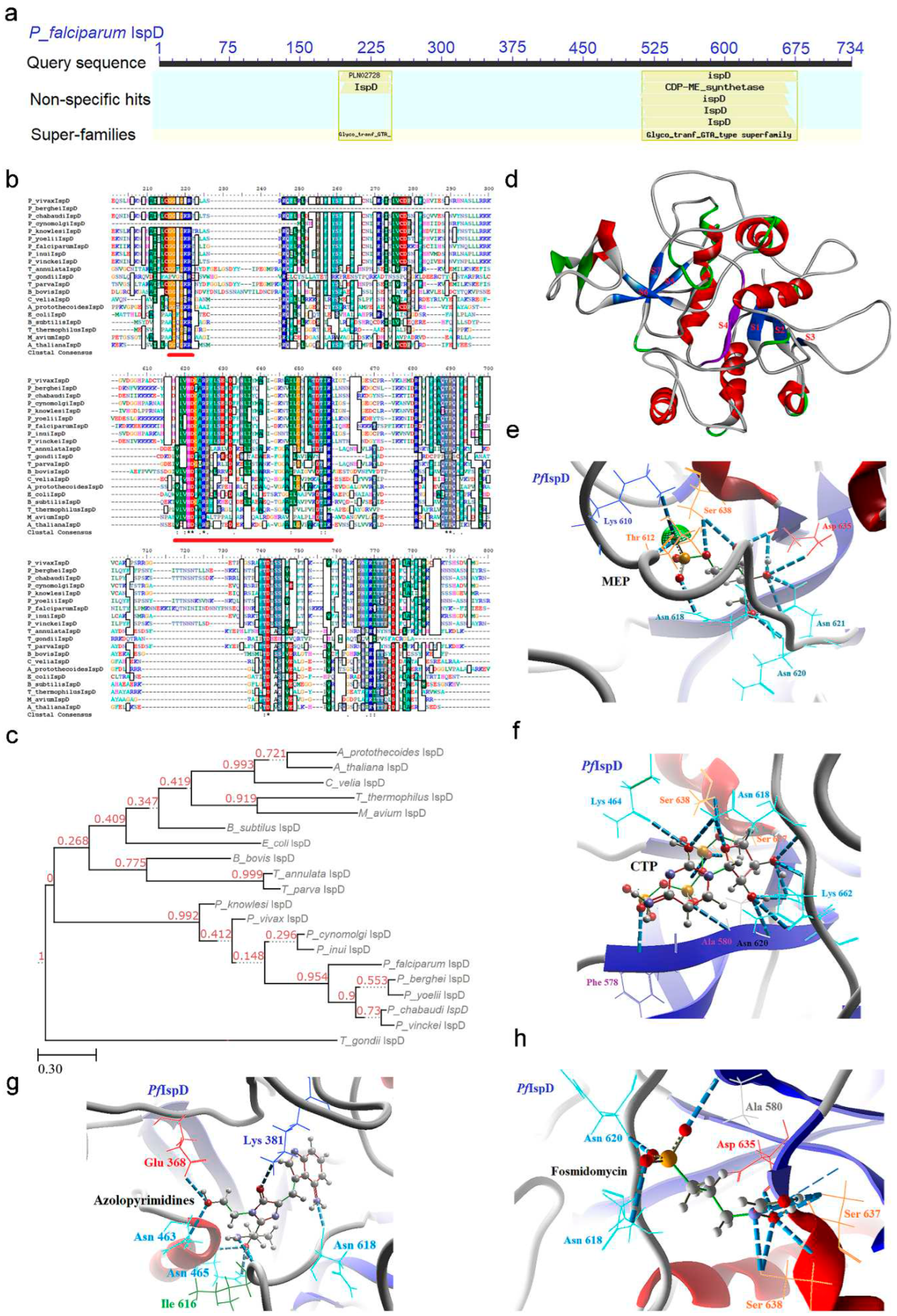
Figure 5.
A comparative analysis of the IspE enzyme across diverse organisms has been undertaken to scrutinize its conserved catalytic activity and elucidate its significance as a potential therapeutic target. PfIspE enzyme conserved GHMP domain (92-516): N-terminal ribosomal protein S5 domain (133-352) and the C-terminal region (359-503) (a), multiple sequence alignment of PfIspE sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspE enzyme structure predicted with computational methods (d), Interaction of PfIspE conserved residues with substrate CDP-ME (e), ATP (f) and inhibitor ethyl {3-[4-amino-5-{3-[(cyclopropyl sulfonyl)amino] prop-1-yn-1-yl}-2-oxopyrimidin-1(2H)-yl] oxetan-3-yl}acetate (g). *Conserved amino acids residues across various organisms.
Figure 5.
A comparative analysis of the IspE enzyme across diverse organisms has been undertaken to scrutinize its conserved catalytic activity and elucidate its significance as a potential therapeutic target. PfIspE enzyme conserved GHMP domain (92-516): N-terminal ribosomal protein S5 domain (133-352) and the C-terminal region (359-503) (a), multiple sequence alignment of PfIspE sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspE enzyme structure predicted with computational methods (d), Interaction of PfIspE conserved residues with substrate CDP-ME (e), ATP (f) and inhibitor ethyl {3-[4-amino-5-{3-[(cyclopropyl sulfonyl)amino] prop-1-yn-1-yl}-2-oxopyrimidin-1(2H)-yl] oxetan-3-yl}acetate (g). *Conserved amino acids residues across various organisms.
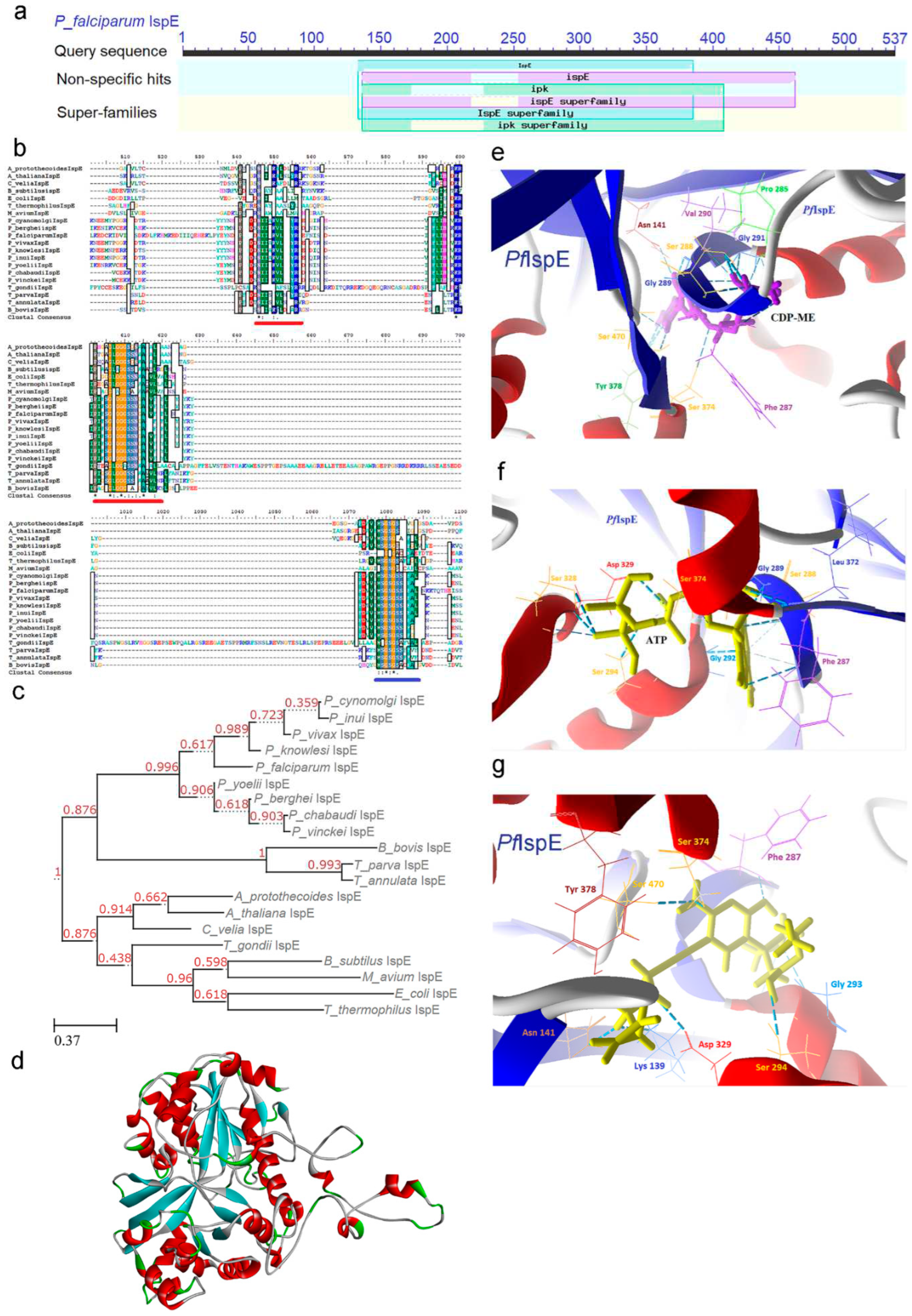
Figure 6.
A comparative analysis of the IspF enzyme across diverse organisms has been undertaken to scrutinize its conserved catalytic activity and elucidate its significance as a potential therapeutic target. PfIspF enzyme conserved MECDP synthase domain (64-236) (a), multiple sequence alignment of PfIspF sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspF enzyme structure predicted with computational methods (d), Interaction of PfIspF conserved residues with substrate CDP-ME-2P (e). In-silico inhibition studies revealed the binding of bis sulfonamide (f) and hydroxy benzylidene (g) with conserved residues of PfIspF. *Conserved amino acids residues across various organisms.
Figure 6.
A comparative analysis of the IspF enzyme across diverse organisms has been undertaken to scrutinize its conserved catalytic activity and elucidate its significance as a potential therapeutic target. PfIspF enzyme conserved MECDP synthase domain (64-236) (a), multiple sequence alignment of PfIspF sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), phylogenetic tree (c), PfIspF enzyme structure predicted with computational methods (d), Interaction of PfIspF conserved residues with substrate CDP-ME-2P (e). In-silico inhibition studies revealed the binding of bis sulfonamide (f) and hydroxy benzylidene (g) with conserved residues of PfIspF. *Conserved amino acids residues across various organisms.
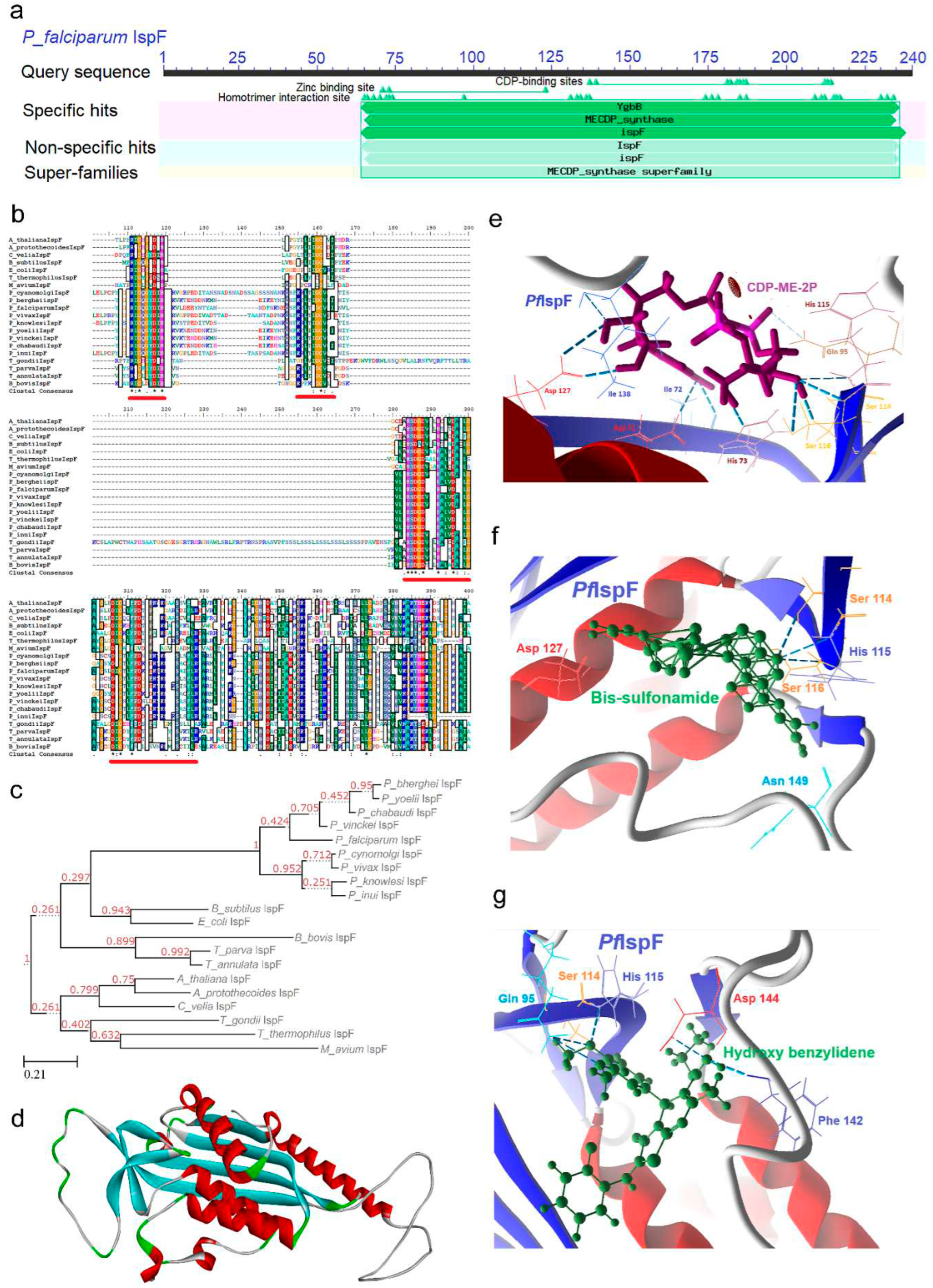
Figure 7.
A comparative analysis of the IspG enzyme across diverse organisms has been initiated to investigate its conserved catalytic activity and discern its significance as a potential therapeutic target. PfIspG is a GcpE superfamily enzyme with TIM barrel domain (MECP substrate binding; 112-403) and C-terminal domain for (iron-sulfur clusters;716-817) (a). multiple sequence alignment of PfIspG sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), PfIspG enzyme structure predicted with computational methods (c), Interaction of PfIspG conserved residues with substrate MECP(d) and 4Fe-4S cluster (e). In-silico inhibition studies revealed the binding of Fos (f), Atorvastatin (g), and Alkyne diphosphate (h) with conserved residues of PfIspG. Phylogenetic tree (i). *Conserved amino acids residues across various organisms.
Figure 7.
A comparative analysis of the IspG enzyme across diverse organisms has been initiated to investigate its conserved catalytic activity and discern its significance as a potential therapeutic target. PfIspG is a GcpE superfamily enzyme with TIM barrel domain (MECP substrate binding; 112-403) and C-terminal domain for (iron-sulfur clusters;716-817) (a). multiple sequence alignment of PfIspG sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), PfIspG enzyme structure predicted with computational methods (c), Interaction of PfIspG conserved residues with substrate MECP(d) and 4Fe-4S cluster (e). In-silico inhibition studies revealed the binding of Fos (f), Atorvastatin (g), and Alkyne diphosphate (h) with conserved residues of PfIspG. Phylogenetic tree (i). *Conserved amino acids residues across various organisms.
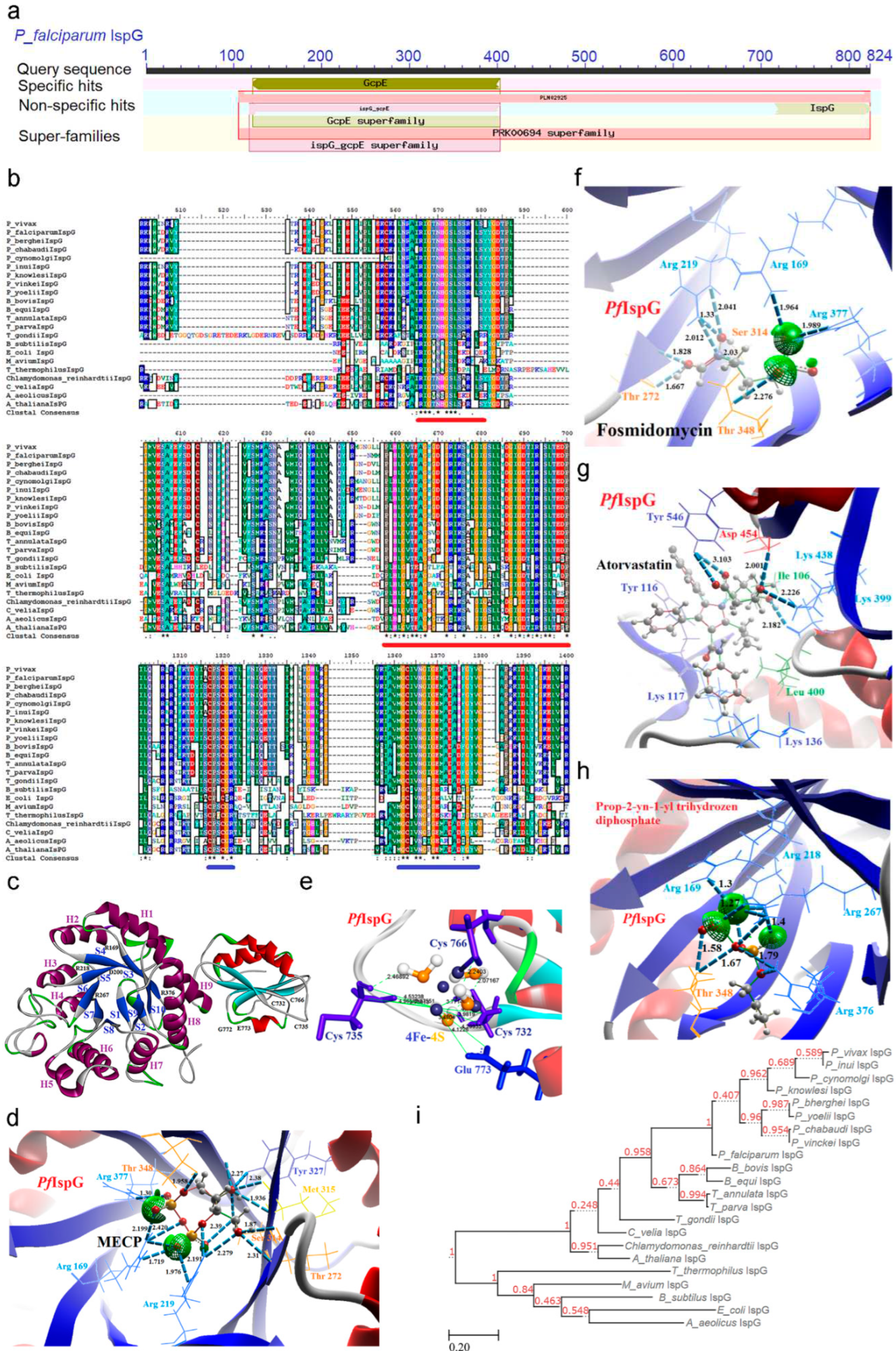
Figure 8.
A comparative analysis of the IspH enzyme across diverse organisms has been undertaken to probe its conserved catalytic activity and ascertain its significance as a potential therapeutic target. PfIspH is a LytB superfamily enzyme with HMBDP substrate binding; 220-535 and iron-sulfur cluster binding domain (a). multiple sequence alignment of PfIspH sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), Phylogenetic tree (c), PfIspH enzyme structure predicted with computational methods (d), Interaction of PfIspH conserved residues with substrates [3Fe-4S] clusters (e) and substrate HMBDP (f). In-silico inhibition studies revealed the binding of (E)-4-mercapto-3-methyl but-2-enyl diphosphate (g) and Phenolphthalein diphosphate pyridine (h) with conserved residues of PfIspH. *Conserved amino acids residues across various organisms.
Figure 8.
A comparative analysis of the IspH enzyme across diverse organisms has been undertaken to probe its conserved catalytic activity and ascertain its significance as a potential therapeutic target. PfIspH is a LytB superfamily enzyme with HMBDP substrate binding; 220-535 and iron-sulfur cluster binding domain (a). multiple sequence alignment of PfIspH sequence with other apicomplexans, bacterial and plant homologs (b; all conserved domains are underlined with colored bar.), Phylogenetic tree (c), PfIspH enzyme structure predicted with computational methods (d), Interaction of PfIspH conserved residues with substrates [3Fe-4S] clusters (e) and substrate HMBDP (f). In-silico inhibition studies revealed the binding of (E)-4-mercapto-3-methyl but-2-enyl diphosphate (g) and Phenolphthalein diphosphate pyridine (h) with conserved residues of PfIspH. *Conserved amino acids residues across various organisms.
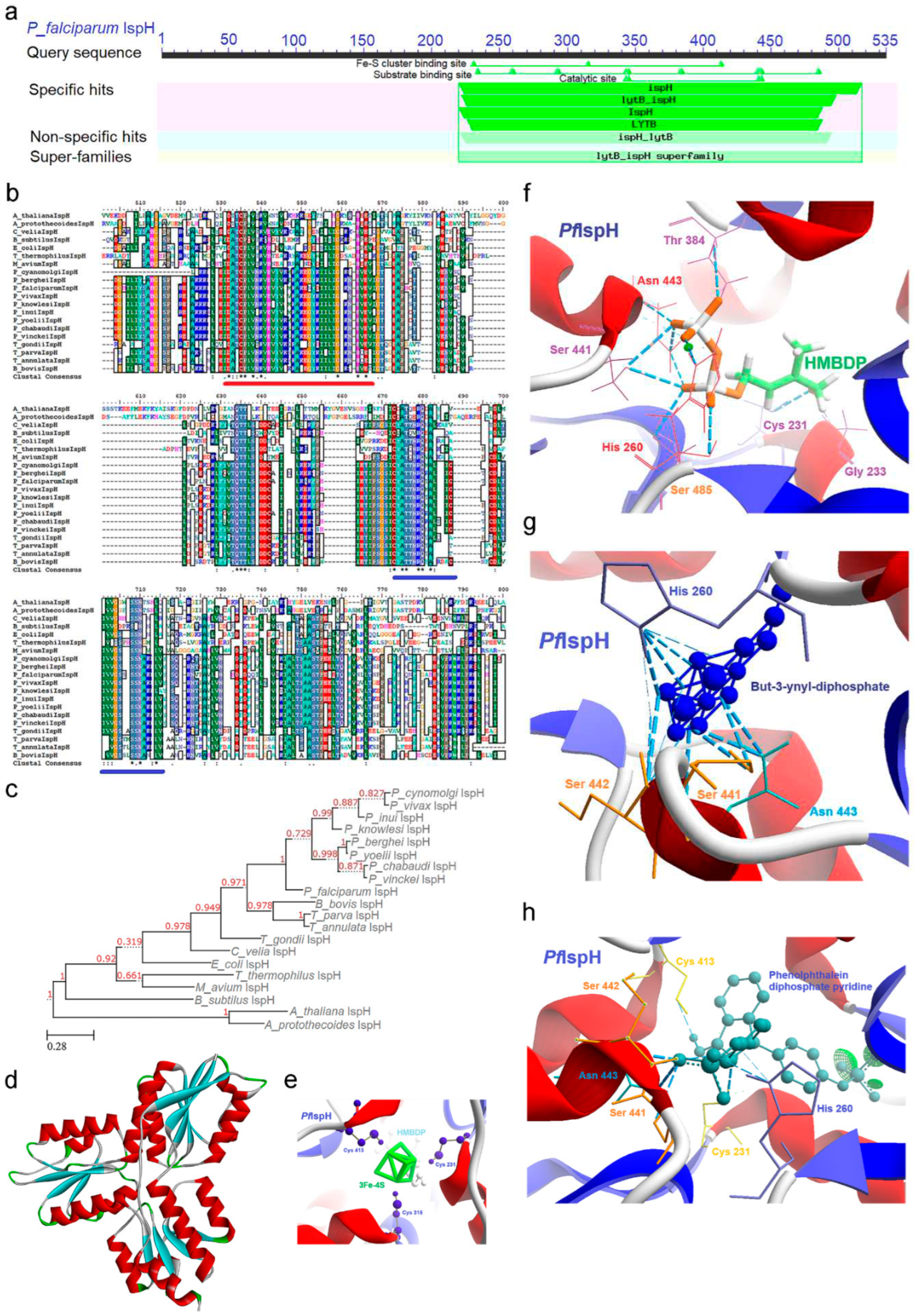

Table 1.
Comparative analysis of various MEP pathway enzymes in Prokaryotes (E. coli; B. subtilus), plants (A. thaliana), and various Plasmodium species to understand the presence of conserved domain, signature motifs, gene annotation, and encoded region. PlasmoDB accession numbers correspond to the Plasmodium parasite, ToxoDB IDs are utilized for T. gondii, and NCBI accession numbers are assigned to prokaryotes.
Table 1.
Comparative analysis of various MEP pathway enzymes in Prokaryotes (E. coli; B. subtilus), plants (A. thaliana), and various Plasmodium species to understand the presence of conserved domain, signature motifs, gene annotation, and encoded region. PlasmoDB accession numbers correspond to the Plasmodium parasite, ToxoDB IDs are utilized for T. gondii, and NCBI accession numbers are assigned to prokaryotes.
| Gene name | Features | P. falciparum | P. cynomolgi | P. berghei | T. gondii | P. vivax | E. coli | B. subtilus | A. thaliana |
|---|---|---|---|---|---|---|---|---|---|
| DXS | Accession No. | PF3D7_1337200 | PCYB_122560 | PBANKA_1351000 | TGRH88_022190 | PVX_082790 | EDV65838 | AGG61826 | AEE83625 |
| Gene size (bp) / Encoded protein size (KDa) |
3618/132 | 2964/108.5 | 2532/92 | 4239/155.3 | 3336/122.2 | 1863/68.2 | 1302/47.6 | 2154/78.8 | |
| CDD region | TPP DXS | ||||||||
| (425-750) | (367-695) | (200-425) | (600-925) | (360-679) | (45-285) | (40-280) | (115-365) | ||
| TPP PYR DXS | |||||||||
| (825-990) | (775-925) | (500-650) | (1020-1175) | (757-913) | (320-480) | (325-475) | (400-560) | ||
| Transketolase C | |||||||||
| (1065-1175) | (765-920) | (700-825) | (1300-1380) | (955-1097) | (495-605) | (493-615) | (578-700) | ||
| Signature motif R-x(3)-[LIVMTA]- [DENQSTHKF]-x(5,6)-[GSN]-G-H-[PLIVMF]-[GSTA]-x(2)-[LIMC]-[GS] |
935-951 (GEDGATHQGIYDLSYLG) |
874-890 (GEDGATHQGIYDLTFLG) |
597-613 (GEDGATHQGIYDLSYLG) |
1111- 1132 (GPDGSTHQ GSFDLAYLG) |
860-876 (GEDGATHQGIYDLTFLG) |
28-47 (RQFLITSL SASGGHIG PNLG) |
36-55 (RRYLLD SVSRSSG HFASGL G) |
102-121 (RSDVIFNV SKTGGHLG SSLG) |
|
| IspC | Accession No. | PF3D7_1467300 | PCYB_124770 | PBANKA_1330600 | TGME49_214850 | PVX_117100 | EDV66507 | AGG61028 | Q9XFS9 |
| Gene size (bp) / Encoded protein size (KDa) |
1467/53.6 | 1557/56.9 | 1245/45.6 | 1899/69.5 | 1587/58 | 1197/43.7 | 1152/42.1 | 1434/52.4 | |
| CDD region | DXP reductoisom | ||||||||
| (80-210) | (135-262) | (30-165) | (190-320) | (118-250) | (4-132) | (5-130) | (85-210) | ||
| DXP redisom C | |||||||||
| (225-322) | (280-375) | (175-270) | (382-470) | (265-361) | (146-238) | (145-225) | (225-307) | ||
| DXPR C | |||||||||
| (360-480) | (410-480) | (305-410) | (500-620) | (396-516) | (270-390) | (260-380) | (340-465) | ||
| Signature motif | Not identified | ||||||||
| IspD | Accession No. | PF3D7_0106900 | PCYB_021710 | PBANKA_0206400 | TGME49_306260 | PVX_081425 | EDV68085 | AGG59433 | P69834 |
| Gene size (bp) / Encoded protein size (KDa) |
2205/80.7 | 1248/45.6 | 867/31.7 | 1137/41.5 | 1857/67.9 | 711/25.9 | 699/25.5 | 909/33.2 | |
| CDD region | CDP-ME Synthetase (GTA type super family) | ||||||||
| (510-675) | (140-350) | (95-235) | (255-365) | (191-558) | (7-222) | (5-220) | (80-292) | ||
| Signature motif [IVT]-[LIVMC]-[IVT]- [HS]-D-[SGAV]-[AV]-R |
512-519 (ILIHDGAR) |
221-228 (ILVHDGAR) |
94-101 (ILVHDAAR) |
265-272 (VMIHDAAR) |
424-431 (ILVHDGAR) |
102-109 (VLVHDAAR) | 100-107 (VLVHDGAR) |
175-182 (VCIHDSAR) |
|
| IspE | Accession No. | PF3D7_0503100 | PCYB_103980 | PBANKA_1102800 | TGME49_306550 | PVX_097660 | YP_489475 | AGG59389 | AAG01340 |
| Gene size (bp) / Encoded protein size (KDa) |
1614/59 | 1581/57.8 | 303/12.1 | 3648/133.6 | 1524/55.7 | 852/31.1 | 870/31.7 | 1152/42.1 | |
| CDD region | IspE (135-380) | PRK00650 (135-495) |
PRK02534 (6-70) |
PHA03247 (135-350) |
PRK00650 (131-497) |
GHMP kinases N | |||
| (92-148) | (85-142) | (160-215) | |||||||
| GHMP kinases C | |||||||||
| (188-262) | (195-275) | ||||||||
| Signature motif | Not identified | ||||||||
| IspF | Accession No. | PF3D7_0209300 | PCYB_042060 | PBANKA_0306400 | TGME49_255690 | PVX_003920 | YP_490955 | AGG59434 | NP_850971 |
| Gene size (bp) / Encoded Protein size (KDa) |
723/26.4 | 840/30.6 | 540/19.6 | 1017/37.1 | 864/31.5 | 480/17.4 | 477/17.3 | 696/25.4 | |
| CDD region | MECDP Synthase | ||||||||
| (65-235) | (90-270) | (5-175) | (175-305) | (106-282) | (2-154) | (3-155) | (75-227) | ||
| Signature motif S-[DN]-[GA]-D-LIVAP]-[LIVAG]-x-H-[STAC]-x(2)-[DNT]-[SAG]-x(2)-[SGA] |
116-131 (SDGDIIYHSIVDSILG) |
152-167 (SDGDVVFHALVDALLG) |
56-71 (SDGDIIYHALVDSILG) |
186-201 (SDGDVLLHAVCDAVFG) |
163-178 (SDGDVIFHALVDALLG) |
35-50 (SDGDVALHALTDALLG) |
36-51 (SDADVLLHTVADACLG) |
109-124 (SDGDVLLHCVVDAILG) |
|
| IspG | Accession No. | PF3D7_1022800 | PCYB_061710 | PBANKA_0507000 | TGME49_262430 | PVX_111575 | EDV66700 | AGG61906 | AAL91150 |
| Gene size (bp) / Encoded protein size (KDa) |
2475/90.6 | 1797/65.7 | 2460/90 | 3852/141.3 | 2463/90.2 | 1119/40.9 | 1134/41.4 | 2226/81.5 | |
| CDD region | GcpE super family | ||||||||
| (125-400 & 720-820) |
(30-180 & 490-590) |
(125-400 & 660-810) |
(385-680 & 1150-1280) | (110-385 & 712-813) | (1-360) | (1-360) | (87-270 & 600-700) |
||
| Signature motif | Not identified | ||||||||
| IspH | Accession No. | PF3D7_0104400 | PCYB_021940 | PBANKA_0208700 | TGME49_227420 | PVX_081535 | EDV65216 | WP_003237046 | Q94B35 |
| Gene size (bp) / Encoded protein size (KDa) |
1608/58.8 | 1311/47.9 | 1050/38.3 | 2157/78.9 | 1281/46.8 | 951/34.7 | 945/34.5 | 1401/51.2 | |
| CDD region | LytB IspH | ||||||||
| (225-495) | (135-390) | (30-310) | (390-660) | (112-388) | (2-282) | (2-282) | (115-450) | ||
| Signature motif [LIVMAC]-[LIVFYWA]- {DYP}-[DN]-P-P-[FYW] |
497-503 (LLTNPPF) |
110-118 (CGSGACGGC) |
304-310 (VLTNPPY) |
601-609 (VVGSEASSN) |
387-393 (VLTGEPF) |
219-227) (VVGSKNSSN) |
220-228 (VVGDPKSNN) |
373-381 (VVGGWNSSN) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



