
Submitted:
30 November 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
Replication protein A (RPA) is a heterotrimeric protein complex and the main single-stranded DNA (ssDNA) binding protein in eukaryotes. RPA has key functions in most of the DNA-associated metabolic pathways and DNA damage signalling. Its high affinity for ssDNA helps to stabilise ssDNA structures and prevents the DNA sequence from nuclease attacks. RPA consists of multiple DNA-binding domains which are oligonucleotide/oligosaccharide-binding (OB)-folds, responsible for DNA binding, and interactions with proteins. These RPA-ssDNA and RPA-protein interactions are crucial for DNA replication, DNA repair, DNA damage signalling, and the conversation of the genetic information of cells. Proteins such as ATR use RPA to locate to regions of DNA damage for DNA damage signalling. Recruitment of nucleases and DNA exchange factors to sites of double strand breaks are also an important RPA function to ensure effective DNA recombination to correct these DNA lesions. Due to its high affinity to ssDNA, RPA’s removal from ssDNA is of central importance to allow these metabolic pathway to proceed and processes to exchange RPA against downstream factors are established in all eukaryotes. These facetted and multi-layered functions of RPA will be discussed in detail in the review. RPA is also a major player in a variety of human diseases, which will be discussed.
Keywords:
DNA Replication
; DNA repair
; homologous recombination
; DNA damage signalling
; replication protein A
; DNA binding
; protein interactions
Introduction
Replication protein A (RPA) is an ubiquitously expressed protein present in all eukaryotes [1]. It plays essential roles in a wide range of DNA metabolic activities, including DNA replication, DNA recombination, DNA repair, and DNA damage signalling [1,2]. RPA was initially identified in the Simian Virus 40 DNA replication system where it supported T-antigen-dependent DNA unwinding [3,4,5]. RPA efficiently and with high affinity binds to single stranded DNA (ssDNA) to avoid formations of secondary structures such as hairpins and to protect ssDNA against nuclease attacks [6,7,8,9]. Thus, as the main eukaryotic ssDNA-binding protein, RPA safeguards ssDNA but also recruits various factors to the RPA-ssDNA complex such as ATRIP for ATR-dependent DNA damage signalling [2,10]. Protein recruitment allows replication, checkpoint signalling, and repair pathways to occur [10]. Post-translational modifications may lead to conformational changes in RPA, giving rise to additional functions and characteristics [2,11].
RPA Structure, ssDNA and protein binding
RPA is a heterotrimeric protein composed of subunits RPA70, RPA32, and RPA14 with apparent molecular weights of 70kDa, 32kDa, 14kDa, respectively (Figure 1, [1,5,12]. Each subunit contains at least one of the universally conserved oligonucleotide/oligosaccharide-binding (OB)-fold characterised by coiled beta sheets and ⍺-helix capped ends. These OB-fold domains allow RPA to bind to ssDNA or target proteins involved in DNA metabolism and DNA damage signalling [10,13,14]. The three subunits bind together through a heterotrimerisation core located in DNA-binding domain-C (DBD-C) of RPA70, DBD-D of RPA32, and RPA14 (DBD-E) facilitated by hydrophobic interactions of ⍺-helices in their OB-folds in a synergistic manner, represented in Figure 1B [12,13]. RPA70 and RPA32 are the main DNA binding sites but there is some evidence that RPA14 also possesses DNA binding activity [15]. RPA14 appears to coordinate RPA70 and RPA32 association, essential for RPA function, particularly in the case of shorter DNA lengths [15].
The DNA binding domains (DBD) A and B bind ssDNA with a groove composed by the loops L12 and L45 flanking the β strands β2 and β3 of the OB-fold [13]. These two binding sites contain each two highly conserved aromatic residues, that stack with DNA bases, and two conserved hydrophobic aa. When the linker regions between the DBDs are not occupied by DNA, they become susceptible to proteolysis [13,20]. DBD-C contains crucial zinc-finger motifs for RPA structural stability, and its ssDNA binding capability [12,13]. RPA binds to ssDNA in three modes, an 8-10 nucleotide, 15-23 nucleotide, and 28-30 nucleotide mode [13,14]. Here, two to three nucleotides per DBD are enough for RPA to establish a tight association with ssDNA [7]. Binding starts with the 8-10 nt-binding mode, where DBD-A and DBD-B bind in a condensed form of RPA, followed by sequential elongation to a 12-23 nt-binding mode, which includes DBD-A, -B plus -C, and then RPA establishes a 28-30 nt-binding mode, where ssDNA sequences interact with DBD-A, -B, -C and -D [13,14]. Recent findings suggest a dynamic binding mechanism for RPA when physically interacting with ssDNA where modulation of DBD-C may cause the handoff of DBD-A and DBD-B and the affinity of RPA to ssDNA is reduced, but also additional nucleotide sequences can be bound by RPA and its mobility on ssDNA increases [7,14,21].
It has been shown that RPA preferentially binds to polypyrimidine sequences with a dissociation constant KD = ~1 nM, whereas mixed sequences of a length of 34 or 57 are bound with lower affinity, KD = 40 nM and 10 nM, respectively [1,6,8,9]. Recent studies using substrates with different oligo(dT) lengths and EMSA confirmed the modular binding approach suggesting that RPA binds dT10 and dT14 with micromolar affinities. RPA has an affinity of KD = ~10 nM for dT15 and dT20, whereas substrates with 30 or more dTs are bound with affinities KD = ~5 nM [22]. Moreover, RPA has affinity values to natural occurring telomeric oligonucleotide sequences with a length of 18 nucleotides or longer KD = ~1 nM whether they contain a G-quadruplex (G4) forming sequence or not. In contrast, the CST (CTC1-STN1-TEN1) complex, an RPA-like protein complex with ssDNA-binding and telomere synthesis function (see below), binds to linear telomeric ssDNA with similar affinities as RPA (1 to 5 nM depending on length with the longer ssDNA, ≥40 nucleotides, being preferred [7]). However, in contrast to RPA, the CST-ssDNA interactions are inhibited by G4 formation suggestion important roles for RPA telomeric DNA metabolism [7].
To fulfil its function RPA not only binds to ssDNA but also to more than 50 proteins involved in DNA replication, repair, and recombination plus DNA damage signalling [10,14,23]. Here, studies of the protein-protein binding sites of RPA have revealed that DBD-F/RPA70 N-terminal domain (NTD), aa 1-110, and adjacent N-terminal sequences, the unstructured linker region plus DBD-A, aa 111 to 290, form a hub for these interactions with more than twenty proteins have been localised to the RPA70 N-terminus (Figure 1A, and for their structural presentation see Figure 1D) bind additional proteins such as RAD51, RAD52, Polα-p180, SV40 T antigen, XPA, [10,14,16,23]). Multiple of these contacts are mediated at the side pocket and the basic and hydrophobic groove of the NTD. DBD-F/RPA70-NTD is essential for binding of proteins in DNA replication, DNA damage response (DDR), and checkpoint signalling such as helicase B, WRN (Werner helicase), BLM (Bloom helicase), SV40 T antigen (Tag), Tp53, RAD9 of RAD9–HUS1–RAD1 (9-1-1), ATRIP (ATR-interacting protein) and ETAA1, a newly identified ATR activator [10,14,23,24]. The adjacent linker region of RPA70 and DBD-A binds additional proteins such as RAD51, RAD52, Polα-p180, SV40 T antigen, XPA [10,14,23,25,26]. The WH domain of RPA32 is a second protein interaction hub of the RPA complex and binds to various proteins such as the Pol α-subunit Prim2, RAD52, Tag, UNG protein, and XPA [23,27,28,29]. Additionally, the binding of RPA to human Cdc45 and Primpol1 regulates their cellular functions [9,30,31].
In addition to the heterotrimeric ‘canonical’ RPA, researchers have found in primates a RPA32-related protein, RPA4, which can replace RPA32 in a complex with RPA70 and RPA14 forming a protein complex called alternative RPA (Alt-RPA). The biochemical activities of Alt-RPA are similar to those of canonical RPA but Alt-RPA has weaker interactions with Pol α and is unable to efficiently stimulate Pol α-dependent DNA synthesis and de novo priming by Pol α [32,33]. In contrast, Alt-RPA stimulates DNA synthesis by Pol α in the presence of RFC/PCNA, which is again contrary to canonical RPA. These findings suggested that since Alt-RPA has a widely overlapping protein interaction pattern with canonical RPA its expression in cells might be a disease-relevant phenotype [32,33]. This hypothesis was supported by recent findings that Alt-RPA expression supported CAG expansions in neurodegenerative diseases whereas canonical RPA suppressed them suggesting a Yin and Yang relationship between Alt-RPA and canonical RPA in the modulation of CAG repeat instability [34].
Roles Of RPA in DNA Replication
DNA replication is a process that requires the precise duplication of an organism’s genetic material once and once in each cell division cycle [35,36]. This process ensures that the daughter cells receive identical copies of the genetic material with each cell division, thereby maintaining genomic stability throughout numerous cell divisions [36]. Through its ssDNA binding and protection plus protein recruitment functions, RPA plays a central and essential role in eukaryotic DNA replication [35]. During the replication process, MCM2-6 (minichromosome maintenance 2 to 6) complex is loaded onto origins of DNA replication with the help of ORC1-6 (origin recognition complex 1 to 6), Cdc6 (cell division cycle 6), and Cdt1. The activation of the MCM2-6 requires the loading of Cdc45 plus the GINS complex to chromatin and the formation of CMG complex, the eukaryotic replicative helicase, which is supported by the proteins DONSON and DNA Polymerase ε (Pol ε) [37,38,39,40,41,42,43,44]. At the beginning of S phase, CMG helicase unwinds dsDNA at origins and the dsDNA is separated into two ssDNA templates to allow the DNA polymerase having access to the genetic information and to allow the duplication of DNA (summarised in Figure 2, [35]). With the help of Cdc45, RPA binds to the exposed ssDNA in a polar fashion and stabilises the ssDNA [9,30,35,45]. Due to the polar orientation of the DNA, at replication forks the unwound DNA strands give rise to template DNAs with a 5’-3’ and a 3’-5’ direction. Together with the knowledge that DNA polymerases only synthesize DNA in 5’-3’ direction with the template having a 3’-5’ directionality, the DNA is synthesised continuously forming the leading strand, whereas the second strand is synthesised discontinuously in small pieces called Okazaki fragments and forms the lagging strand as presented in Figure 2 [35].
No DNA polymerase starts DNA synthesis de novo and a special enzyme, primase often also called DNA primase, an RNA polymerase, synthesising short oligoribonucleotides with a length of ~10 nts or multimers thereof on ssDNA templates including unwound origin DNA. Primase is an essential enzyme for DNA replication and is functional as a dimer consisting of PRI1/Prim1/p49, the catalytic subunit, and PRI2/Prim2/p58, the regulatory subunit, that controls primer lengths [48,49,50]. The primase dimer forms a heterotetrameric complex with the two larger subunits PolA1/p180 carrying the catalytic DNA polymerase activity and PolA2/p68/p70/B subunit, the second regulatory subunit, which is called Polymerase ⍺ (Pol ⍺) or Polymerase ⍺-primase [51]. Pol α is the only replicative DNA polymerase complex having primase activity and thus is central for DNA replication processes. Under special circumstances such as replication fork restart Primpol may take over the restart function [52,53]. However, Pol α in association with the CST complex may also function in the replication restart [54]. Primer synthesis is initiated by the cooperation of CMG, AND-1/Ctf4/WDHD1, RPA, Pol ⍺ and primase at 5’ ssDNA (Figure 2, [38,39,55]). After the initiation, PRI2/Prim2 hands the primer to the PolA1/p180 subunit of Pol ⍺ to elongate the primer by a DNA chain of 20 nucleotides and uses RPA as a ‘fidelity clamp’ to stay on the ssDNA suggesting that RPA and the largest subunit collaborate in the polymerase transition from primase to the large subunit of Pol ⍺, PolA1/p180 [26,48,56]. After the DNA synthesis of ~20 nts, Pol ⍺ leaves the template and is replaced by replication factor C (RFC) in contact with RPA. The former then uses ATP hydrolysis to add proliferating cell nuclear antigen (PCNA) to the primed template [46,57]. In the next step, the 2nd polymerase transition, polymerase δ (Pol δ) is recruited and utilises PCNA as a loading clamp to elongate the newly synthesised DNA strand in a processive manner. For the leading strand synthesis, in a 3rd polymerase transition, the short RNA-DNA primer is then handed over to Pol ε, which is in close contact with the CMG helicase on the leading strand template and is supposed to support unwinding activity of CMG [37,58]. On the leading strand template, Pol ε synthesises the leading strand in a processive and continuous manner up to a size of a replicon [51,59]. Both Pol δ and ε are also heterotetrameric enzyme complexes but they do not have primase activity associated [51,59]. In contrast to Pol α, that does not have 3’-5’ proofreading exonuclease activity, the Pol α exonuclease domain is inactive, both Pol δ and ε contain a DNA polymerase and 3’-5’ proofreading exonuclease activity in their largest subunits, Pol D1/p125 and PolE/Pol2/p260 [51,59]. Interestingly, single molecule experiments have shown that one Pol ε, one Pol δ and one to two Pol α bound to CTF4 are functional collaborating at a replication fork [60,61].
DNA directionality of the second strand at replication fork and that all polymerase synthesise DNA 5’-3’ causes that the synthesis of new DNA strand, the lagging strand, occurs in short fragments called Okazaki fragments [51,57,59]. During lagging strand synthesis each Okazaki fragment is initiated by Pol α as described above. Taking the Okazaki fragment size and the human genome size into account, Pol α synthesise approximately 20 to 40 million primers to produce a complete round of lagging strand synthesis per cell division cycle, an enormous task. In the following, the RNA primer is handed over from the primase to PolA1/p180 by PRI2/Prim2 with the support of RPA, whereas the RNA-DNA primer is then transferred from Pol α to Pol δ [51,57,59]. The latter synthesises the Okazaki fragment until it reaches the RNA moiety of the previous Okazaki fragment. Importantly, RPA needs to protect exposed ssDNA until pol δ encounters the next primer, and a DNA ligase ligates the fragments together [47,59]. After reaching the previous Okazaki fragment, Pol δ continues synthesis of the associated Okazaki fragment into the previous fragment in a strand displacement mode. Thus, by removing the RNA primer and a part of the Pol α-synthesised DNA, the enzymes creates a 5’-flap DNA, which for efficient ligation of the two Okazaki fragments needs to be cleaved off [47,59]. The enzyme flap endonuclease 1 (FEN1) takes over this role. FEN1 binds to the 5’-flap DNA and cleaves the DNA precisely at the ssDNA-dsDNA transition leaving a nicked dsDNA remaining, an ideal substrate for LIG1 ligation [47,59]. Alternatively, RPA binds the 5’-flap ssDNA, protects the flap-DNA against FEN1 and recruits the endonuclease DNA2 to the ssDNA which cleaves the flap overhang [47,62]. Finally, the LIG1 will ligate the nicked DNA producing a new dsDNA molecule. Interestingly, FEN1, LIG1 and Pol δ can associate to the same PCNA molecule during Okazaki fragment synthesis and maturation occupying different regions of the PCNA homotrimer [47,59].
Understanding the INitiation of Okazaki fragment Synthesis at the Molecular Level
Recent cryo-EM structure studies of budding yeast and human replisomes as well as of the telomeric CST-Pol α complex have given a profound insight into the mechanism of the initiation of Okazaki fragment synthesis at replication forks and telomere sequences [38,63,64,65,66]. The replication fork structures by Jones et al show that Pol α has multiple interactions with CMG complex and the AND-1/CTF4/WDHD1 complex (summarised in Figure 2, [38]). Here, especially MCM3/P1, and the GINS subunits have direct contacts with the primase subunit PRI2/Prim2, and to the second largest Pol α subunit PolA2/Pol12 [38,67,68,69,70]. Interestingly, most of these interaction positions of the CMG complex and AND-1 are localised at the end or within intrinsic disordered regions (IDRs), which may provide flexibility in these interactions and the movement of Pol α on the lagging strand template during primer synthesis, whereas CMG advances on the leading strand template unwinding the dsDNA [38,71,72]. These protein-protein and the CMG-DNA interactions position the catalytic primase subunit of Pol α, PRI1/Prim1, close to MCM5 and the opening of the exit channel for the lagging-strand template ssDNA [38]. In the yeast CryoEM structure, the MCM5 Zn finger has physical contacts with yPRI1 but this interaction is not conserved in the human Pol α-CMG-AND-1 structure [38]. These structural biological data suggest a putative mechanism for the initiation of Okazaki fragment synthesis, but they unfortunately lack RPA, an important player of Okazaki fragment synthesis and initiation, to draw final conclusion about the mechanism.
To further understand the initiation of Okazaki fragment synthesis and to elucidate the role RPA in this process it is first important to know that primase dimers (PRI1/Prim1 and PRI2/Prim2) have a much lower affinity for ssDNA, even with a length of >50 nts (KD = ~3,000 nM for a mixed ssDNA sequence (Onwubiko & Nasheuer, unpublished results)) than that of RPA, which has ssDNA affinities in the low nanomolar range as discussed above, and it has previously been shown that RPA efficiently inhibits primase activity [8,73,74,75,76,77,78]. In the SV40 DNA replication, a mechanism for the reversion of the inhibition has been described. Similar to the interaction of RAD52 with the RPA32 WH domain, Tag interacts with the WH domain, reverses the RPA inhibition, and allows primase to synthesise primers on RPA-bound ssDNA [74,79,80]. One of the main functions in this process is the remodelling of the Pol α-primase complex from an inactive state to a priming active state [64,65,66,72,81]. This process is best understood in the initiation process of Okazaki fragment synthesis on the G strand of telomers as part of the C strand synthesis [64,65,66,72,81]. The structure of Pol α in association with CST complex, an RPA-like complex also called alpha-Accessory factor (AAF) [82], shows that the two protein complexes have numerous interactions with each other and some might be important for the remodelling of the Pol α complex to a pre-initiation complex [72,81]. Additionally, these Pol α-CST-ssDNA structures show that in the complex, the CST binds the template ssDNA and directs this ssDNA towards the catalytic centre of PRI1/Prim1 [72,81]. Considering the structural similarity of RPA and CST it is thought that RPA has similar functions as CST during the initiation of DNA synthesis on ssDNA [81].
Recently, it was shown that the STN1 subunit of CST and RPA32 alone are sufficient to stimulate Pol α initiation activity and increase its primase activity on ssDNA [73,83]. The OB-fold domain of human STN1 is sufficient to provide the stimulation of Pol α and here especially the conserved aa D157 of STN1, which is equivalent to D151 in human RPA32, is important for the stimulatory activity, whereas the ssDNA binding activity of STN1 is not important [73]. Aspartat 157 interacts with the C-terminus of PolA1 (S1365 and R1366) and might be important for the remodelling of Pol α from an inactive to an active form and thus, provide the stimulatory function for Pol α during the primase reaction [64,72,73]. The crucial aa D157 of human STN1 is conserved to human and fungal RPA32 (D151 in human RPA32 and D155 in RPA32 of Ustilago maydis (Nasheuer and Onwubiko, unpublished data)) and is part of a conserved loop between a β-strand and an α-helix structure (Figure 1B–E). Overlaying the OB-folds of RPA32 and STN1 with the published structure of the Ustilago maydis RPA complex shows that the D-containing loop (D-loop) is conserved between STN1 and these two RPA32 structures (Figure 1C, [13]). The alignment of the STN1 OB-fold with the AlphaFold-predicted human RPA structure shows this even more clearly (Figure 1E). In RPA, this D-loop lies in cleft between RPA70 and RPA14 (Figure 1, panels B to E), which may prevent access to Pol α. Thus, the interaction of the RPA32 WH domain with an mediator protein such as Tag might open the cleft and allow the binding of DBD-D in RPA32 to PolA1 C-terminus supporting the activation of Pol α. Predictions of protein-protein interactions by the Walter group (Harvard University, USA) using Alphafold also suggest an interaction of RPA32 D151 with the C-terminus of PolA1, the largest subunit of the Pol α complex [19,84]. These findings suggest that interactions of RPA32 and STN1 with PolA1 C-terminus as discussed here plus the previously suggested delivery of the ssDNA template to the catalytic domain of PRI1/Prim1 might be important for the initiation and stimulation of primase activity [72,81].
RPA and DNA rePAIR Processes
DNA replication is a complex process and errors commonly occur despite the proof-reading exonuclease activity associated with the replicative polymerases Pol δ and ε [51]. Additionally, the genetic information of cells is under constant pressure from intracellular and extracellular/environmental stresses causing DNA damage. To preserve genomic stability eukaryotic cells have developed multiple DNA repair pathways depending on the type of DNA damage to correct damaged DNA [51]. Moreover, conserved DNA damage signalling pathways are present in eukaryotic cells to maintain genome stability. All these processes require RPA to fully function [14,16,21].
- Double-Strand Break (DSB) Repair, DSBs of cellular DNA are the most deleterious and toxic lesions [85]. Eukaryotic cells have two main pathways to repair DSBs, homologous recombination (HR) and non-homologous end joining (NHEJ). HR utilizes a homologous DNA template to allow for accurate repair of DSBs whereas NHEJ only re-joins the broken ends, opening the possibility of the induction of insertions or deletions [51,85].
RPA in DNA damage Signalling
Since DNA lesions are detrimental to eukaryotic cells and the stability of their genetic information, a fast and precise response to damaged DNA is required to correct DNA lesions in chromosomal DNA of cells to avoid the appearance of genetic diseases such as cancer [2,10]. Here, under environmental or cellular stress, the protein DOCK7 promotes the accumulation of RPA at chromatin and replication forks [86]. In addition to its ssDNA binding capabilities, RPA uses its protein binding abilities to recruit certain factors to damaged sites to directly function there, or initiate downstream pathways against DNA damage [2,87]. Human Primase-polymerase 1 (hPrimpol1) is one such protein that directly interacts with RPA70’s N-terminal domain, DBD-F, at stalled replication forks, where this binding is essential for hPrimpol1 to restore DNA synthesis [10,52,53]. Replication fork stalling also causes RPA-binding to ATRIP to recruit ATR to these RPA-bound ssDNA stretches [88]. ATR in turn phosphorylates downstream targets such as CHK1 and p53 to delay the cell cycle, permitting the stabilisation of the replication fork [2].
During DNA damage the three RPA subunits are ubiquitinated. Specifically, RPA32 is modified by K63-linked ubiquitin chains which are important for ATRIP recruitment and ATR kinase activation [2]. DOCK7 is one protein that is phosphorylated by this ATR activation. In turn, Dock7 phosphorylation increases RPA association to chromatin, which again allows further ATR activation, essentially creating a positive feedback loop [86]. Cyclin dependent kinases (CDKs) and Phosphatidylinositol-3 kinase-related kinases (PIKKs) phosphorylate the RPA32 N-terminal region serine and threonine residues during the cell cycle and in response to genotoxic stress [2,89,90,91]. There are eight phosphorylation sites in this RPA32 sequence where CDK phosphorylates S23 and S29 which then stimulates ATR-dependent phosphorylation of S33. In turn, this causes the subsequent phosphorylation of the other threonine and serine residues by DNA-PK and ATM. Here, S4, S8, and T21 phosphorylation are hallmarks for replication fork breakage and this hyperphosphorylation acts as a marker for ongoing resection at double-strand break sites [2,89,90,91]. Furthermore, during the DNA damage response, Wiskott-Aldrich syndrome protein (WASp) helps to coordinate ssDNA-binding of RPA and to manage DNA stress response more optimally [92]. The association of WASp at DNA damage sites results in stable RPA-ssDNA complexes that are important for efficient DNA repair [92].
RPA in DNA Repair Pathways
To avoid genome instability by bulky DNA adducts or major DNA structure distortions Nucleotide excision repair (NER) is an important DNA repair pathway (summarised in Figure 3, [93,94,95]). The importance of the NER pathway is underlined by the existence of a rare genetic disorder called Xeroderma pigmentosum, in which genes coding for proteins involved in the NER pathway are mutated [94]. These proteins are called XPA to XPG and XPV, which is a DNA polymerase called Pol η [94,95]. In global genome NER, XPC and RAD23B first recognise DNA lesions, then the ten subunit complex TFIIH is recruited, where XPB translocase and XPD helicase unwind the dsDNA [95]. XPA and RPA join the complex stabilising the ssDNA. These two proteins are also responsible for correctly positioning the ERCC1-XPF and XPG endonucleases. When ERCC1-XPF incises DNA at the 5’-end, RPA helps to recruit RFC for PCNA loading and DNA synthesis by Pol δ and/or ε (Figure 3). XPG allows 3’-incision to occur so a ligase can bind the DNA once the DNA synthesis is completed and the damaged DNA strand is replaced [95].
In contrast, small DNA lesions such as oxidative, deamination and alkylation lesions, that cause distortion in the DNA helix, are repaired by another pathway, BER [93]. BER starts with enzymes, called DNA glycosylases, which cleave the bond between deoxyribose and the modified DNA base removing the damaged base and leaving an abasic site (loss of nucleobase by hydrolysis) behind. Then AP endonuclease recognises these abasic sites and cleave the 5’-phosphodiester bond on the abasic site, which leaves a deoxyribose at the 5’-end [93]. In following process, called short patch BER, Pol β is the major enzyme involved and fills in the abasic site with the correct nucleobase. Finally, DNA ligase 1 or 3 ligates the nicked DNA [93]. As an alternative to the described short-patch repair, long-patch BER takes place. Pol β, or Pol δ/ε depending on the proliferation state of the cell fill in the abasic site with the correct nucleobase, but these DNA polymerases extend the DNA synthesis step beyond the abasic site and synthesise DNA in a strand-displacement mode. Fen1 then removes the produced 5’- flap and Lig1 ligates the nicked DNA [93]. In BER, uracil-DNA glycosylase (UNG), an important BER protein, interacts with RPA via the WH domain of RPA32. RPA’s exact role in BER is unclear but it is thought that RPA is involved in the gap filling stage, similar to NER [2].
The MMR pathway repairs base-base mismatches and deletion or insertions that occur during the replication process and increases the fidelity of the replication process by a factor of at least 100 [93,96]. Mutations in MMR proteins lead to a hypermutation phenotype of organisms and may cause early onset of cancer [93,96]. In MMR, MutSα and MutSβ heterodimers recognise one nucleotide and large mismatches, respectively, with the help of MutLα and RPA [93,97]. EXO1 exonuclease then excises the damaged DNA and RPA stabilises the resulting ssDNA gaps and becomes hyperphosphorylated [93,97]. In the following, at the 3’-end of the DNA at dsDNA-ssDNA transition RFC-PCNA complexes support the association of Pol δ, which then fills the gap with the correct nucleobases. After the DNA synthesis, the resulting nicked dsDNA is ligated [93,96,97].
RPA and DNA Recombination
DSBs are one of the most deleterious and toxic DNA lesions in cells. Eukaryotic cells have developed multiple pathways to repair these lesions in error-free and error-prone manner [93,98]. Here, we focus on homologous recombination (HR), a conserved and error-free pathway to repair DSBs. To initiate HR at DSBs the MRN (MRE11-RAD50-NBS1) complex recognises and binds the DSB followed by the recruitment of ATM and TIP60 to DNA [93,98]. The activated ATM phosphorylates H2AX, which in turn recruits MDC1 that is then phosphorylated by ATM. Next, the phosphorylated MDC1 recruits the ubiquitin E3 ligases RNF8 and RNF168, which together ubiquitinate H2AX [98]. Ubiquitinated chromatin sequesters 53BP1 and BRCA1. In the S/G2 phase, BRCA1 successfully counteracts 53BP1 initiating the ubiquitination of the downstream components such as CtIP and RPA (see Figure 4 [93,98]). The ubiquitin ligase, RING finger and WD repeat domain 3 (RFWD3), ubiquitinates RPA70 and RPA32 at sites of DNA damage, which promotes HR [99]. In the following helicases and nucleases unwind the damaged dsDNA and resect the 5’-ends of the DNA strands at a DSB site to create an 3’- overhang which forms RAD51-ssDNA filaments for homology-dependent binding a complementary DNA followed by DNA synthesis and resolution of the resulting DNA structure [93,99,100].
In the beginning of the DSB repair, MRN with its endonuclease activity and the help of CtIP starts the initial resection, whereas long-range resection occurs via BLM helicase/DNA2 or EXO1. In one resection pathway, BLM helicase unwinds dsDNA at DSBs in an ATP-dependent manner and DNA2 resects the DNA in an RPA-controlled 5’-3’ directionality, as shown in Figure 4. MRN has a stimulatory effect by recruiting the BLM helicase to DNA ends and initiating the resection process [93,99]. The resection is terminated by the phosphorylation of RPA at the N-terminus of RPA70 by interrupting its interaction with BLM causing a reduction of BLM helicase activity [10,99]. Alternatively, in the EXO1-dependent HR pathway, MRN recruits EXO1 to DNA ends and stimulates its 5’-3’ digestion process. This MRN stimulation is increased in the presence of RPA. The addition of BLM helicase further stimulates EXO1 activity. This EXO-catalysed resection is also used in mismatch repair of DNA [99]. Additionally, to stimulating and directing the resection reaction, RPA coats the 3′ ssDNA overhang producing an RPA-ssDNA filament. Next, these RPA-bound ssDNA overhangs are exchanged to RAD51-ssDNA filaments to form the active RAD51 recombinase and to search for the complementary DNA [100,101,102,103]. RPA’s higher ssDNA affinity creates a kinetic block between RPA and RAD51. N.B. RPA has ssDNA affinities of KD = 1 – 20 nM depending on the ssDNA sequence composition (see above), whereas RAD51•ATP binds to ssDNA with an affinity KD = ~200 nM [1,6,8,9,22,104]. So, replacement of RPA by RAD51 needs mediator factors such as BRCA2 in mammals and RAD52 in yeast [100,101,103,105,106].
In mammals, BRCA2 mediates the transfer of RPA to RAD51-coated ssDNA where BRCA2 supports the nucleation of RAD51-ssDNA filaments causing an outstretched structure able to search for homologous DNA [100,101,103,105,106]. Although BRCA2 does not directly physically interact with RPA, its partner DSS1, which contains a solvent-exposed acidic domain, binds to RPA using this acidic domain to mimic DNA [105]. DSS1 performs multiple physical interactions with the DBD-A, B, C and F and thus, interferes with the ssDNA capability of RPA70 reducing the affinity of the RPA to ssDNA and allowing BRCA2 to load RAD51 onto the 3’-overhang ssDNA replacing RPA [105]. Recently, Bell et al determined that the BRC repeats of BRCA2 pre-assembled RAD51 nuclei, accelerating RAD51 nucleation [103]. Post-translational modifications have multiple functions in the transfer from RPA-ssDNA to RAD51-ssDNA filaments. It has been suggested that extensive poly(ADP-ribosyl)ation of RPA reduces its high ssDNA affinity and that RAD51 preferentially binds to phosphorylated RPA [2]. Interestingly, poly(ADP-ribosyl)ation of RPA enhances the recruitment of repair proteins to DNA damage sites [11,87]. Additionally, post DNA damage SUMOylation of RPA70 causes a favourable binding of RAD51 to RPA [2]. In the presence of incorrectly functioning BRCA2, RAD51 is unable to replace RPA on ssDNA. The accumulation of RPA on ssDNA will result in a continuous activation of ATR signalling and downstream phosphorylation [2,93]. In contrast, de-phosphorylation of RPA2 by PP2A and PP4 phosphatases is a sign of completion of HR-dependent DSB repair and suggests an interplay between resection termination and RPA phosphorylation [23]. Correct HR completion is essential since dysregulation of HR by WASp deficiency can cause genome instability [92]. Importantly the availability of RPA in the cells ensures that DNA undergoes HR instead of the error-prone microhomology-mediated end joining (MMEJ) where translocation and deletions may occur [2].
In yeast, RAD52 mediates the exchange of RPA-ssDNA to RAD51-ssDNA filaments. When RPA binds to ssDNA the individual DBDs are in a variety of dynamic conformational ssDNA-bound states [2,14,106]. In the RPA-ssDNA complex, this flexibility allows access to the 5’- and the 3’-segment of the RPA-ssDNA complex. During the HR process, the recombination mediator RAD52 interacts with RPA bound to 3’-overhang ssDNA of a DSB via the RPA32 WH domain (see Figure 1, [106]). This RAD52-RPA32 interaction specifically changes the ssDNA-binding dynamics of DBD-D (see Figure 1) and reduces the affinity of RPA to ssDNA, which in turn allows the nucleation of RAD52-bound RAD51 on the RPA-bound ssDNA. This further decreases the binding capacity of RPA to ssDNA and finally results in a RAD52-mediated removal of RPA and the exchange of RPA-ssDNA filaments into RAD51-ssDNA filaments, the HR-active components. These RAD51-filaments now will start to search for homologous DNA sequences on dsDNA located in close vicinity [106]. The RAD51 recombinase with the associated ssDNA having a free 3’-OH invades a nearby dsDNA and forms a D-loop [93,107]. In the following RAD51 is removed and the 3′-OH end bound to a template DNA serves as a primer for synthesis by Pol δ, κ and ν. The resulting DNA structure is resolved in a well-defined process as reviewed by Chatterjee and Walker [93].
RPA in Health Prospects
Neurodegenerative diseases are caused by G-rich tandem repeat sequences. Repeated expansions such as CAGCTG can be seen in diseases like Huntington’s Disease (HD) [108]. The progression of diseases such as HD occurs from incorrect or lack of slip-out excision of these expansion mutations [34]. RPA enhances correct slipped-DNA repair and FAN1-controlled slip-out excision in HD mouse brains, protecting cells from CAG-expanded repeats and in turn from certain neurodegenerative diseases. In contrast, expression of Alt-RPA inhibits correct repair of slipped-CAG intermediates. Interestingly, both types of RPA are overexpressed in HD and spinocerebellar ataxia type 1 (SCA1) patient brains. However, Alt-RPA is strongly whereas canonical RPA is mildly overexpressed in these brains. In this contest, canonical RPA prevents whereas Alt-RPA promotes somatic repeats expansions [34]. The latter worsens the disease outcome. Interestingly, SCA1 mice showed reduced disease phenotype after RPA overexpression supporting the link between RPA modulating neurodegenerative disease pathogenesis [34].
To establish new drugs against cancers that have become resistant to existing therapies including chemotherapy Van der Vere-Carozza et al have created RPA inhibitors that target its OB-folds [109]. RPA is a central player in eukaryotic DDR and high levels of RPA expression act as a negative biomarker for patient survival in smoking-related lung cancer [109]. Additionally, increased levels of RPA in cancer chemotherapy may serve as an adaptive process to protect cells against genotoxic stress suggesting that having active RPA and a fully functional DDR, cells have a chance to survive DNA damage e.g., in cancer chemotherapy, whereas, when inhibiting both RPA functions and DDR, cancer-treated cells become sentenced to apoptosis or other death pathways. Thus, using a combination of RPA and DDR inhibition ensures that a high degree of cancer cells with damaged DNA resulted from established DNA-damaging chemotherapies are eliminated [109].
Telomeres at the end of chromosomes shorten with each cell cycle due to the primer removal on the lagging stand during chromosomal DNA replication [110,111]. To overcome this limitation, cancer cells and stem cells recruit the enzyme telomerase to telomeres for their elongation and supporting cell immortality [110,111]. As telomeres naturally contain an ssDNA sequence, cells need to prevent RPA binding to telomeres and DDR induction to maintain normal cell growth and to shelter cells from negative reactions [112]. The protein protection of telomeres 1 (POT1) is a part of the SHELTERIN complex and involved in the telomere length control in of cells [112]. In POT1 mutant cells, RPA-dependent activation of ATR DDR causes telomerase-mediated hyper-elongation of telomeres supporting immortalisation of cancer cells [112].
Conclusion s
The central roles of RPA in a variety of metabolic processes show the importance of future research using different angles regarding RPA functions and cooperations with replication, repair and recombination proteins. The main future prospects will be the examination of RPA levels and/or post-translational modification as biomarkers, and testing its inhibition to combat cancers and other diseases. Additionally, the structural biological insights into the various metabolic processes including RPA at replication forks, RPA in complex with DNA recombination mediators on ssDNA to name a few will significantly enhance field and give insights into the mechanisms of these central reactions.
Acknowledgements
This work was supported by a grant from the Else-Kröner-Fresenius Foundation.
Abbreviations
| 9-1-1 complex | Rad 9-Hus 1-Rad 1 complex (fission yeast and human) equivalent to the Rad 17-Mec3-Ddc1 complex in budding yeast | ||
| aa | aminoacid | ||
| AND-1/CTF4/ | acidic and nucleoplasmic DNA- binding protein/Chromosome Trans- | ||
| WDHD1 | mission Fidelity 4/WD repeat and HMG-box DNA-binding protein 1 | ||
| A-T | Ataxia telangiectasia | ||
| ATM | Ataxia telangiectasia-mutated | ||
| ATR | ATM-Rad 3-related protein | ||
| ATRIP | ATR interacting protein | ||
| BER | base excision repair | ||
| BLM | Bloom’s helicase | ||
| CDC | cell division cycle | ||
| CDK | cyclin-dependent kinase | ||
| CMG | CDC45-MCM2-6-GINS | ||
| CST | CTC1-STN1-TEN1 | ||
| DBD | DNA-binding domain | ||
| DNA-PK | DNA-dependent protein kinase | ||
| DDR | DNA damage response | ||
| DSB | double-strand break | ||
| dsDNA | double-stranded DNA | ||
| G4 | G-quadruplexes | ||
| HR | homologous recombination | ||
| mt | mitochondrial | ||
| MCM | minichromosome maintenance | ||
| MMEJ | microhomology-mediated end joining | ||
| hPolprim1 | human primase-polymerase 1 | ||
| MMR | mismatch repair | ||
| NER | nucleotide excision repair | ||
| Nt | nucleotide | ||
| NTD | N-terminal domain | ||
| OB-fold | oligosaccharide/oligonucleotide-binding fold | ||
| PCNA | proliferating cell nuclear antigen | ||
| PIKK | phosphotnositol-3 kinase-like protein kinase | ||
| Pol α | DNA polymerase α–primase | ||
| Pol δ | DNA polymerase δ | ||
| Pol ε | DNA polymerase ε | ||
| POT1 | protector of telomeres 1 | ||
| RAD | radiation-induced mutation | ||
| RFC | replication factor C | ||
| RFWD3 | RING finger and WD repeat | ||
| RPA | replication protein A | ||
| ROS | reactive oxygen species | ||
| SSB | single-stranded DNA-binding protein | ||
| ssDNA | single-stranded DNA | ||
| S. cerivisiae | Saccheromyces cerevisiae | ||
| SV40 | simian virus 40 | ||
| TFIIH | transcription factor II H | ||
| TopBP1 | topoisomerase II-binding protein 1 | ||
References
- Wold, M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res 2015, 25, 9–23. [Google Scholar] [CrossRef]
- Kenny, M.K.; Lee, S.H.; Hurwitz, J. Multiple functions of human single-stranded-DNA binding protein in simian virus 40 DNA replication: single-strand stabilization and stimulation of DNA polymerases alpha and delta. Proc Natl Acad Sci USA 1989, 86, 9757–9761. [Google Scholar] [CrossRef] [PubMed]
- Fairman, M.P.; Stillman, B. Cellular factors required for multiple stages of SV40 replication in vitro. EMBO J. 1988, 7, 1211–1218. [Google Scholar] [CrossRef]
- Wold, M.S.; Kelly, T.J. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc.Natl.Acad.Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef]
- Kim, C.; Snyder, R.O.; Wold, M.S. Binding properties of replication protein A from human and yeast cells. Mol. Cell. Biol. 1992, 12, 3050–3059. [Google Scholar] [PubMed]
- Olson, C.L.; Barbour, A.T.; Wuttke, D.S. Filling in the blanks: how the C-strand catches up to the G-strand at replicating telomeres using CST. Nature structural & molecular biology 2022, 29, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Onwubiko, N.O.; Borst, A.; Diaz, S.A.; Passkowski, K.; Scheffel, F.; Tessmer, I.; Nasheuer, H.P. SV40 T antigen interactions with ssDNA and replication protein A: a regulatory role of T antigen monomers in lagging strand DNA replication. Nucleic Acids Res 2020, 48, 3657–3677. [Google Scholar] [CrossRef] [PubMed]
- Szambowska, A.; Tessmer, I.; Prus, P.; Schlott, B.; Pospiech, H.; Grosse, F. Cdc45-induced loading of human RPA onto single-stranded DNA. Nucleic Acids Res, 1093. [Google Scholar] [CrossRef]
- Wu, Y.; Fu, W.; Zang, N.; Zhou, C. Structural characterization of human RPA70N association with DNA damage response proteins. eLife 2023, 12. [Google Scholar] [CrossRef]
- Zhao, J.; Tian, S.; Guo, Q.; Bao, K.; Yu, G.; Wang, X.; Shen, X.; Zhang, J.; Chen, J.; Yang, Y.; et al. A PARylation-phosphorylation cascade promotes TOPBP1 loading and RPA-RAD51 exchange in homologous recombination. Mol Cell 2022, 82, 2571–2587. [Google Scholar] [CrossRef]
- Bochkareva, E.; Korolev, S.; Lees-Miller, S.P.; Bochkarev, A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. The EMBO journal 2002, 21, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Pavletich, N.P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev 2012, 26, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Salas, T.R.; Petruseva, I.; Lavrik, O.; Saintome, C. Evidence for direct contact between the RPA3 subunit of the human replication protein A and single-stranded DNA. Nucleic Acids Res 2009, 37, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Broderick, S.; Rehmet, K.; Concannon, C.; Nasheuer, H.P. Eukaryotic single-stranded DNA binding proteins: central factors in genome stability. Subcell Biochem 2010, 50, 143–163. [Google Scholar]
- Stephan, H.; Concannon, C.; Kremmer, E.; Carty, M.P.; Nasheuer, H.P. Ionizing radiation-dependent and independent phosphorylation of the 32-kDa subunit of replication protein A during mitosis. Nucleic Acids Res 2009, 37, 6028–6041. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. The EMBO journal 1993, 12, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: making protein folding accessible to all. Nat Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Pestryakov, P.E.; Weisshart, K.; Schlott, B.; Khodyreva, S.N.; Kremmer, E.; Grosse, F.; Lavrik, O.I.; Nasheuer, H.P. Human replication protein A: The C-terminal RPA70 and the central RPA32 domains are involved in the interactions with the 3'-end of a primer-template DNA. J Biol Chem 2003, 278, 17515–17524. [Google Scholar]
- Chen, R.; Wold, M.S. Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Shen, J.; Zhao, Y.; Zhong, S.; Lai, L.; Niu, H.; Qi, Z. ssDNA accessibility of Rad51 is regulated by orchestrating multiple RPA dynamics. Nat Commun 2023, 14, 3864. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Replication protein A: a multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Taneja, P.; Boche, I.; Hartmann, H.; Nasheuer, H.P.; Grosse, F.; Fanning, E.; Weisshart, K. Different activities of the largest subunit of replication protein A cooperate during SV40 DNA replication. FEBS Lett 2007, 581, 3973–3978. [Google Scholar] [CrossRef] [PubMed]
- Dornreiter, I.; Erdile, L.F.; Gilbert, I.U.; von Winkler, D.; Kelly, T.J.; Fanning, E. Interaction of DNA polymerase alpha-primase with cellular replication protein A and SV40 T antigen. The EMBO journal 1992, 11, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.A.; Lao, Y.; He, Z.; Ingles, C.J.; Wold, M.S. Role of protein-protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase α by multiple mechanisms. Biochemistry 1997, 36, 8443–8454. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhou, J.; Fan, J.; Shen, Z.; Chen, X. Streamline proteomic approach for characterizing protein-protein interaction network in a RAD52 protein complex. J Proteome Res 2009, 8, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Weisshart, K.; Pestryakov, P.; Smith, R.W.; Hartmann, H.; Kremmer, E.; Lavrik, O.; Nasheuer, H.P. Coordinated regulation of replication protein A activities by its subunits p14 and p32. J Biol Chem 2004, 279, 35368–35376. [Google Scholar] [CrossRef] [PubMed]
- Vaithiyalingam, S.; Warren, E.M.; Eichman, B.F.; Chazin, W.J. Insights into eukaryotic DNA priming from the structure and functional interactions of the 4Fe-4S cluster domain of human DNA primase. Proc Natl Acad Sci U S A 2010, 107, 13684–13689. [Google Scholar] [CrossRef]
- Broderick, R.; Rainey, M.D.; Santocanale, C.; Nasheuer, H.P. Cell cycle-dependent formation of Cdc45-Claspin complexes in human cells are compromized by UV-mediated DNA damage. FEBS J 2013, 280, 4888–4902. [Google Scholar] [CrossRef]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO reports 2013, 14, 1104–1112. [Google Scholar] [CrossRef]
- Kemp, M.G.; Mason, A.C.; Carreira, A.; Reardon, J.T.; Haring, S.J.; Borgstahl, G.E.; Kowalczykowski, S.C.; Sancar, A.; Wold, M.S. An alternative form of replication protein A expressed in normal human tissues supports DNA repair. J Biol Chem 2010, 285, 4788–4797. [Google Scholar] [CrossRef]
- Mason, A.C.; Roy, R.; Simmons, D.T.; Wold, M.S. Functions of alternative replication protein A in initiation and elongation. Biochemistry 2010, 49, 5919–5928. [Google Scholar] [CrossRef] [PubMed]
- Gall-Duncan, T.; Luo, J.; Jurkovic, C.M.; Fischer, L.A.; Fujita, K.; Deshmukh, A.L.; Harding, R.J.; Tran, S.; Mehkary, M.; Li, V.; et al. Antagonistic roles of canonical and Alternative-RPA in disease-associated tandem CAG repeat instability. Cell 2023, 186, 4898–4919. [Google Scholar] [CrossRef]
- Bleichert, F.; Botchan, M.R.; Berger, J.M. Mechanisms for initiating cellular DNA replication. Science 2017, 355, eaah6317. [Google Scholar] [CrossRef] [PubMed]
- Nasheuer, H.P.; Smith, R.; Bauerschmidt, C.; Grosse, F.; Weisshart, K. Initiation of eukaryotic DNA replication: regulation and mechanisms. Prog Nucleic Acid Res Mol Biol 2002, 72, 41–94. [Google Scholar] [PubMed]
- Vipat, S.; Gupta, D.; Jonchhe, S.; Anderspuk, H.; Rothenberg, E.; Moiseeva, T.N. The non-catalytic role of DNA polymerase epsilon in replication initiation in human cells. Nat Commun 2022, 13, 7099. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Aria, V.; Baris, Y.; Yeeles, J.T.P. How Pol α-primase is targeted to replisomes to prime eukaryotic DNA replication. Mol Cell 2023. [Google Scholar] [CrossRef]
- Jones, M.L.; Baris, Y.; Taylor, M.R.G.; Yeeles, J.T.P. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. The EMBO journal 2021, 40, e108819. [Google Scholar] [CrossRef]
- Evrin, C.; Alvarez, V.; Ainsworth, J.; Fujisawa, R.; Alabert, C.; Labib, K.P. DONSON is required for CMG helicase assembly in the mammalian cell cycle. EMBO Rep 2023, 24, e57677. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Sadano, K.; Miyata, N.; Ito, H.; Tanaka, H. Novel role of DONSON in CMG helicase assembly during vertebrate DNA replication initiation. The EMBO journal 2023, 42, e114131. [Google Scholar] [CrossRef]
- Kingsley, G.; Skagia, A.; Passaretti, P.; Fernandez-Cuesta, C.; Reynolds-Winczura, A.; Koscielniak, K.; Gambus, A. DONSON facilitates Cdc45 and GINS chromatin association and is essential for DNA replication initiation. Nucleic Acids Res 2023, 51, 9748–9763. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Tamayo-Orrego, L.; Schmid, E.; Tarnauskaite, Z.; Kochenova, O.V.; Gruar, R.; Muramatsu, S.; Lynch, L.; Schlie, A.V.; Carroll, P.L.; et al. In silico protein interaction screening uncovers DONSON's role in replication initiation. Science 2023, 381, eadi3448. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Sonneville, R.; Jenkyn-Bedford, M.; Ji, L.; Alabert, C.; Hong, Y.; Yeeles, J.T.P.; Labib, K.P.M. DNSN-1 recruits GINS for CMG helicase assembly during DNA replication initiation in Caenorhabditis elegans. Science 2023, 381, eadi4932. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, O.I.; Kolpashchikov, D.M.; Nasheuer, H.P.; Weisshart, K.; Favre, A. Alternative conformations of human replication protein A are detected by crosslinks with primers carrying a photoreactive group at the 3'-end. FEBS Lett. 1998, 441, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Yuzhakov, A.; Kelman, Z.; Hurwitz, J.; O'Donnell, M. Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. The EMBO journal 1999, 18, 6189–6199. [Google Scholar] [CrossRef] [PubMed]
- Prindle, M.J.; Loeb, L.A. DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen 2012, 53, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lisova, A.E.; Morstadt, L.M.; Babayeva, N.D.; Tahirov, T.H. Insight into RNA-DNA primer length counting by human primosome. Nucleic Acids Res 2022, 50, 6264–6270. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Babayeva, N.D.; Zhang, Y.; Gu, J.; Suwa, Y.; Pavlov, Y.I.; Tahirov, T.H. Mechanism of Concerted RNA-DNA Primer Synthesis by the Human Primosome. J Biol Chem 2016, 291, 10006–10020. [Google Scholar] [CrossRef]
- Zerbe, L.K.; Kuchta, R.D. The p58 subunit of human DNA primase is important for primer initiation, elongation, and counting. Biochemistry 2002, 41, 4891–4900. [Google Scholar] [CrossRef]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr Opin Struct Biol 2018, 53, 77–87. [Google Scholar] [CrossRef]
- Bainbridge, L.J.; Teague, R.; Doherty, A.J. Repriming DNA synthesis: an intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Res 2021, 49, 4831–4847. [Google Scholar] [CrossRef]
- Zabrady, K.; Li, A.W.H.; Doherty, A.J. Mechanism of primer synthesis by Primase-Polymerases. Curr Opin Struct Biol 2023, 82, 102652. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep 2012, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Baris, Y.; Taylor, M.R.; Yeeles, J.T.P. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. The EMBO journal 2023, 42, e115685. [Google Scholar] [CrossRef]
- Maga, G.; Frouin, I.; Spadari, S.; Hübscher, U. Replication protein A as a "fidelity clamp" for DNA polymerase alpha. J Biol Chem 2001, 276, 18235–18242. [Google Scholar] [CrossRef]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol 2016, 26, 640–654. [Google Scholar] [CrossRef]
- Jenkyn-Bedford, M.; Jones, M.L.; Baris, Y.; Labib, K.P.M.; Cannone, G.; Yeeles, J.T.P.; Deegan, T.D. A conserved mechanism for regulating replisome disassembly in eukaryotes. Nature 2021, 600, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu Rev Biochem 2017, 86, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, N.; El-Hajj, Z.W.; Zheng, H.; Beattie, T.R.; Yu, A.; Reyes-Lamothe, R. Processive Activity of Replicative DNA Polymerases in the Replisome of Live Eukaryotic Cells. Mol Cell 2020, 80, 114–126. [Google Scholar] [CrossRef]
- Lewis, J.S.; Spenkelink, L.M.; Schauer, G.D.; Yurieva, O.; Mueller, S.H.; Natarajan, V.; Kaur, G.; Maher, C.; Kay, C.; O'Donnell, M.E.; et al. Tunability of DNA Polymerase Stability during Eukaryotic DNA Replication. Mol Cell 2020, 77, 17–25. [Google Scholar] [CrossRef]
- Sun, H.; Ma, L.; Tsai, Y.F.; Abeywardana, T.; Shen, B.; Zheng, L. Okazaki fragment maturation: DNA flap dynamics for cell proliferation and survival. Trends Cell Biol 2023, 33, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, R.E.; Schauer, G.D.; Yao, N.Y.; Langston, L.D.; Yurieva, O.; Zhang, D.; Finkelstein, J.; O'Donnell, M.E. Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. eLife 2015, 4, e04988. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Lin, X.; Chavez, B.L.; Agrawal, S.; Lusk, B.L.; Lim, C.J. Structures of the human CST-Polα–primase complex bound to telomere templates. Nature 2022, 608, 826–832. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Song, H.; Chan, H.; Liu, B.; Wang, Y.; Su<monospace>š</monospace>ac, L.; Zhou, Z.H.; Feigon, J. Structure of Tetrahymena telomerase-bound CST with polymerase α-primase. Nature 2022, 608, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.W.; Zinder, J.C.; Svetlov, V.; Bush, M.W.; Nudler, E.; Walz, T.; de Lange, T. Cryo-EM structure of the human CST–Polα/primase complex in a recruitment state. Nature structural & molecular biology 2022, 29, 813–819. [Google Scholar] [CrossRef]
- Thömmes, P.; Fett, R.; Schray, B.; Burkhart, R.; Barnes, M.; Kennedy, C.; Brown, N.C.; Knippers, R. Properties of the nuclear P1 protein, a mammalian homologue of the yeast Mcm3 replication protein. Nucleic Acids Res. 1992, 20, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Maine, G.T.; Sinha, P.; Tye, B.K. Mutants of S. cerevisiae defective in the maintenance of minichromosomes. Genetics 1984, 106, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Tye, B.K. The MCM2-3-5 proteins: are they replication licensing factors? Trends Cell Biol 1994, 4, 160–166. [Google Scholar] [CrossRef]
- Takayama, Y.; Kamimura, Y.; Okawa, M.; Muramatsu, S.; Sugino, A.; Araki, H. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev 2003, 17, 1153–1165. [Google Scholar] [CrossRef]
- Kilkenny, M.L.; Simon, A.C.; Mainwaring, J.; Wirthensohn, D.; Holzer, S.; Pellegrini, L. The human CTF4-orthologue AND-1 interacts with DNA polymerase α/primase via its unique C-terminal HMG box. Open biology 2017, 7. [Google Scholar] [CrossRef]
- Nasheuer, H.P.; Onwubiko, N.O. Lagging Strand Initiation Processes in DNA Replication of Eukaryotes-Strings of Highly Coordinated Reactions Governed by Multiprotein Complexes. Genes (Basel) 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Ganduri, S.; Lue, N.F. STN1-POLA2 interaction provides a basis for primase-pol α stimulation by human STN1. Nucleic Acids Research 2017, 45, 9455–9466. [Google Scholar] [CrossRef]
- Arunkumar, A.I.; Klimovich, V.; Jiang, X.; Ott, R.D.; Mizoue, L.; Fanning, E.; Chazin, W.J. Insights into hRPA32 C-terminal domain--mediated assembly of the simian virus 40 replisome. Nature structural & molecular biology 2005, 12, 332–339. [Google Scholar]
- Melendy, T.; Stillman, B. An interaction between replication protein A and SV40 T antigen appears essential for primosome assembly during SV40 DNA replication. J. Biol. Chem. 1993, 268, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Onwubiko, N.O.; Scheffel, F.; Tessmer, I.; Nasheuer, H.P. SV40 T antigen helicase domain regions responsible for oligomerisation regulate Okazaki fragment synthesis initiation. FEBS Open Bio 2022, 12, 649–663. [Google Scholar] [CrossRef]
- Smith, R.W.; Steffen, C.; Grosse, F.; Nasheuer, H.P. Species specificity of simian virus 40 DNA replication in vitro requires multiple functions of human DNA polymerase alpha. J Biol Chem 2002, 277, 20541–20548. [Google Scholar]
- Weisshart, K.; Förster, H.; Kremmer, E.; Schlott, B.; Grosse, F.; Nasheuer, H.P. Protein-protein interactions of the primase subunits p58 and p48 with simian virus 40 T antigen are required for efficient primer synthesis in a cell-free system. J Biol Chem 2000, 275, 17328–17337. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weiner, B.E.; Zhang, H.; Fuller, B.E.; Gao, Y.; Wile, B.M.; Zhao, K.; Arnett, D.R.; Chazin, W.J.; Fanning, E. Structure of a DNA polymerase alpha-primase domain that docks on the SV40 helicase and activates the viral primosome. J Biol Chem 2010, 285, 17112–17122. [Google Scholar] [CrossRef]
- Vaithiyalingam, S.; Arnett, D.R.; Aggarwal, A.; Eichman, B.F.; Fanning, E.; Chazin, W.J. Insights into eukaryotic primer synthesis from structures of the p48 subunit of human DNA primase. Journal of molecular biology 2014, 426, 558–569. [Google Scholar] [CrossRef]
- Barbour, A.T.; Wuttke, D.S. RPA-like single-stranded DNA-binding protein complexes including CST serve as specialized processivity factors for polymerases. Curr Opin Struct Biol 2023, 81, 102611. [Google Scholar] [CrossRef]
- Casteel, D.E.; Zhuang, S.; Zeng, Y.; Perrino, F.W.; Boss, G.R.; Goulian, M.; Pilz, R.B. A DNA Polymerase-{alpha}{middle dot}Primase Cofactor with Homology to Replication Protein A-32 Regulates DNA Replication in Mammalian Cells. J Biol Chem 2009, 284, 5807–5818. [Google Scholar] [CrossRef] [PubMed]
- Lue, N.F.; Chan, J.; Wright, W.E.; Hurwitz, J. The CDC13-STN1-TEN1 complex stimulates Pol alpha activity by promoting RNA priming and primase-to-polymerase switch. Nat Commun 2014, 5, 5762. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.; Walter, J.C. Predictomics webpage. Availabe online: (accessed on 28.11.).
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Guo, G.; Huang, J.; Hou, X.; Ham, H.; Kim, W.; Zhao, F.; Tu, X.; Zhou, Q.; Zhang, C.; et al. DOCK7 protects against replication stress by promoting RPA stability on chromatin. Nucleic Acids Res 2021, 49, 3322–3337. [Google Scholar] [CrossRef] [PubMed]
- Illuzzi, G.; Fouquerel, E.; Ame, J.C.; Noll, A.; Rehmet, K.; Nasheuer, H.P.; Dantzer, F.; Schreiber, V. PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic Acids Res 2014, 42, 7776–7792. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.L.; Myers, J.S.; Cortez, D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell 2005, 16, 2372–2381. [Google Scholar] [CrossRef] [PubMed]
- Borgstahl, G.E.; Brader, K.; Mosel, A.; Liu, S.; Kremmer, E.; Goettsch, K.A.; Kolar, C.; Nasheuer, H.P.; Oakley, G.G. Interplay of DNA damage and cell cycle signaling at the level of human replication protein A. DNA repair 2014, 21, 12–23. [Google Scholar] [CrossRef]
- Binz, S.K.; Wold, M.S. Regulatory Functions of the N-terminal Domain of the 70-kDa Subunit of Replication Protein A (RPA). J Biol Chem 2008, 283, 21559–21570. [Google Scholar] [CrossRef] [PubMed]
- Nuss, J.E.; Patrick, S.M.; Oakley, G.G.; Alter, G.M.; Robison, J.G.; Dixon, K.; Turchi, J.J. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry 2005, 44, 8428–8437. [Google Scholar] [CrossRef]
- Han, S.S.; Wen, K.K.; García-Rubio, M.L.; Wold, M.S.; Aguilera, A.; Niedzwiedz, W.; Vyas, Y.M. WASp modulates RPA function on single-stranded DNA in response to replication stress and DNA damage. Nat Commun 2022, 13, 3743. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Aguilera, A.; Gellert, M.; Hanawalt, P.C.; Hays, J.B.; Lehmann, A.R.; Lindahl, T.; Lowndes, N.; Sarasin, A.; Wood, R.D. DNA repair: from molecular mechanism to human disease. DNA repair 2006, 5, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 2013, 5, a012609. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu Rev Biochem 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, Y.; Yuan, F.; Gao, Y.; Gu, L.; Wong, I.; Li, G.M. Regulation of replication protein a functions in DNA mismatch repair by phosphorylation. J Biol Chem 2006, 281, 21607–21616. [Google Scholar] [CrossRef]
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front Genet 2022, 13, 793884. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Kwon, Y.; Rösner, H.; Zhao, W.; Selemenakis, P.; He, Z.; Kawale, A.S.; Katz, J.N.; Rogers, C.M.; Neal, F.E.; Badamchi Shabestari, A.; et al. DNA binding and RAD51 engagement by the BRCA2 C-terminus orchestrate DNA repair and replication fork preservation. Nat Commun 2023, 14, 432. [Google Scholar] [CrossRef]
- Le, H.P.; Heyer, W.D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Foertsch, F.; Kache, T.; Drube, S.; Biskup, C.; Nasheuer, H.P.; Melle, C. Determination of the number of RAD51 molecules in different human cell lines. Cell Cycle 2019, 18, 3581–3588. [Google Scholar] [CrossRef]
- Bell, J.C.; Dombrowski, C.C.; Plank, J.L.; Jensen, R.B.; Kowalczykowski, S.C. BRCA2 chaperones RAD51 to single molecules of RPA-coated ssDNA. Proc Natl Acad Sci U S A 2023, 120, e2221971120. [Google Scholar] [CrossRef] [PubMed]
- Špírek, M.; Mlcoušková, J.; Belán, O.; Gyimesi, M.; Harami, G.M.; Molnár, E.; Novacek, J.; Kovács, M.; Krejci, L. Human RAD51 rapidly forms intrinsically dynamic nucleoprotein filaments modulated by nucleotide binding state. Nucleic Acids Res 2018, 46, 3967–3980. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Vaithiyalingam, S.; San Filippo, J.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol Cell 2015, 59, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, N.; Caldwell, C.C.; Corless, E.I.; Tillison, E.A.; Tibbs, J.; Jocic, N.; Tabei, S.M.A.; Wold, M.S.; Spies, M.; Antony, E. Dynamics and selective remodeling of the DNA-binding domains of RPA. Nature structural & molecular biology 2019, 26, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Donnianni, R.A.; Zhou, Z.X.; Lujan, S.A.; Al-Zain, A.; Garcia, V.; Glancy, E.; Burkholder, A.B.; Kunkel, T.A.; Symington, L.S. DNA Polymerase Delta Synthesizes Both Strands during Break-Induced Replication. Mol Cell 2019, 76, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H. Repeat expansion diseases. Handb Clin Neurol 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- VanderVere-Carozza, P.S.; Gavande, N.S.; Jalal, S.I.; Pollok, K.E.; Ekinci, E.; Heyza, J.; Patrick, S.M.; Masters, A.; Turchi, J.J.; Pawelczak, K.S. In Vivo Targeting Replication Protein A for Cancer Therapy. Front Oncol 2022, 12, 826655. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat Rev Mol Cell Biol 2021, 22, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Zaug, A.J.; Goodrich, K.J.; Song, J.J.; Sullivan, A.E.; Cech, T.R. Reconstitution of a telomeric replicon organized by CST. Nature 2022, 608, 819–825. [Google Scholar] [CrossRef]
- Takasugi, T.; Gu, P.; Liang, F.; Staco, I.; Chang, S. Pot1b -/- tumors activate G-quadruplex-induced DNA damage to promote telomere hyper-elongation. Nucleic Acids Res 2023, 51, 9227–9247. [Google Scholar] [CrossRef]
Figure 1.
Structure of RPA A. The bars represent the three RPA subunits with DNA-binding domains (DBDs) DBD-A to DBD-F highlighted and their borders indicated by amino acid numbering of the human protein sequences [14,16]. Each DBD contains a highly conserved OB-fold. The structure of one of the DBDs is highlighted in the bubble within the panel. The domains that are coloured in orange and darker orange are able to bind to ssDNA. DBD-F, the N-terminal domain of RPA70 and the pink winged helix domain (WH) of RPA32 are mainly protein-protein interaction sites. DBD-C contains the Zinc domains represented by the protruding Zn. The N-terminus of RPA32, shown in light pink and with the letter P, has recognition sites for multiple protein kinases including CDKs and PIKKs [17]. The lines between the three subunits represent the points of interaction between the subunits through the heterotrimerisation core (Adapted from [12,18]). The human RPA aa sequence is used for the numbering. Panel B presents an image of the RPA trimer including RPA70 in green, RPA32 in light cyan, RPA14 in brown, and oligo(dT) ssDNA in orange (structure derived from PDB 4gop [13] and presented using Pymol). Human STN1 and RPA32 OB-fold structures were predicted by AlphaFold [19] and are shown in light magenta and yellow, respectively. C. Human STN1 and RPA32 were aligned to Ustilago maydis RPA (structure derived from 4gop) using Pymol (for a better overview aa N93 to T124 of STN1, which did not align to any structure, were omitted in the presentation). The loops containing D157 of STN1, D151 of human RPA32 and D155 of Ustilago maydis RPA32 are shown in blue. Panels D and E show an AlphaFold prediction of the human RPA complex without DNA and in a similar position as Ustilago maydis RPA. RPA70 is in green with DNA-F domain being in the back coloured in light green. The unstructured linker between DBD-F and DBD-A is located at the top ends of the panels. RPA32 is shown in light blue with the phosphorylation sequence in dark blue. The WD domain, which is lacking in Ustilago maydis RPA, is shown in the back of the figure. RPA14 is in orange (lower left of the panels). The RPA32 loop containing D151, called D loop, is shown in red. In panel E, the OB-fold of STN1 shown in light pink is aligned to human RPA using PyMol (for a better overview aa N93 to T124 of STN1, which did not align to any structure, were omitted in the presentation). The loop containing the conserved D157 (D loop) is presented in red.
Figure 1.
Structure of RPA A. The bars represent the three RPA subunits with DNA-binding domains (DBDs) DBD-A to DBD-F highlighted and their borders indicated by amino acid numbering of the human protein sequences [14,16]. Each DBD contains a highly conserved OB-fold. The structure of one of the DBDs is highlighted in the bubble within the panel. The domains that are coloured in orange and darker orange are able to bind to ssDNA. DBD-F, the N-terminal domain of RPA70 and the pink winged helix domain (WH) of RPA32 are mainly protein-protein interaction sites. DBD-C contains the Zinc domains represented by the protruding Zn. The N-terminus of RPA32, shown in light pink and with the letter P, has recognition sites for multiple protein kinases including CDKs and PIKKs [17]. The lines between the three subunits represent the points of interaction between the subunits through the heterotrimerisation core (Adapted from [12,18]). The human RPA aa sequence is used for the numbering. Panel B presents an image of the RPA trimer including RPA70 in green, RPA32 in light cyan, RPA14 in brown, and oligo(dT) ssDNA in orange (structure derived from PDB 4gop [13] and presented using Pymol). Human STN1 and RPA32 OB-fold structures were predicted by AlphaFold [19] and are shown in light magenta and yellow, respectively. C. Human STN1 and RPA32 were aligned to Ustilago maydis RPA (structure derived from 4gop) using Pymol (for a better overview aa N93 to T124 of STN1, which did not align to any structure, were omitted in the presentation). The loops containing D157 of STN1, D151 of human RPA32 and D155 of Ustilago maydis RPA32 are shown in blue. Panels D and E show an AlphaFold prediction of the human RPA complex without DNA and in a similar position as Ustilago maydis RPA. RPA70 is in green with DNA-F domain being in the back coloured in light green. The unstructured linker between DBD-F and DBD-A is located at the top ends of the panels. RPA32 is shown in light blue with the phosphorylation sequence in dark blue. The WD domain, which is lacking in Ustilago maydis RPA, is shown in the back of the figure. RPA14 is in orange (lower left of the panels). The RPA32 loop containing D151, called D loop, is shown in red. In panel E, the OB-fold of STN1 shown in light pink is aligned to human RPA using PyMol (for a better overview aa N93 to T124 of STN1, which did not align to any structure, were omitted in the presentation). The loop containing the conserved D157 (D loop) is presented in red.
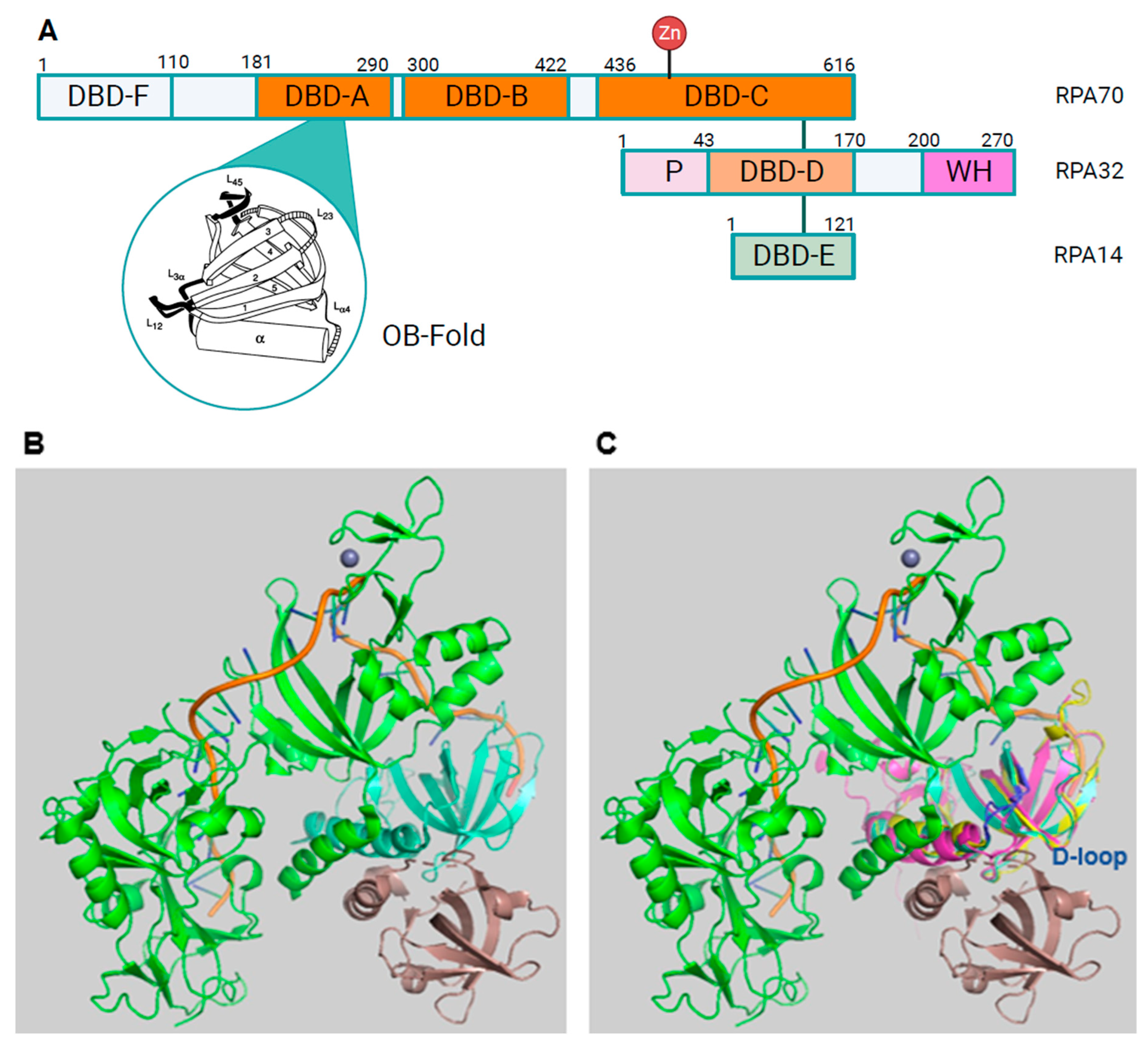
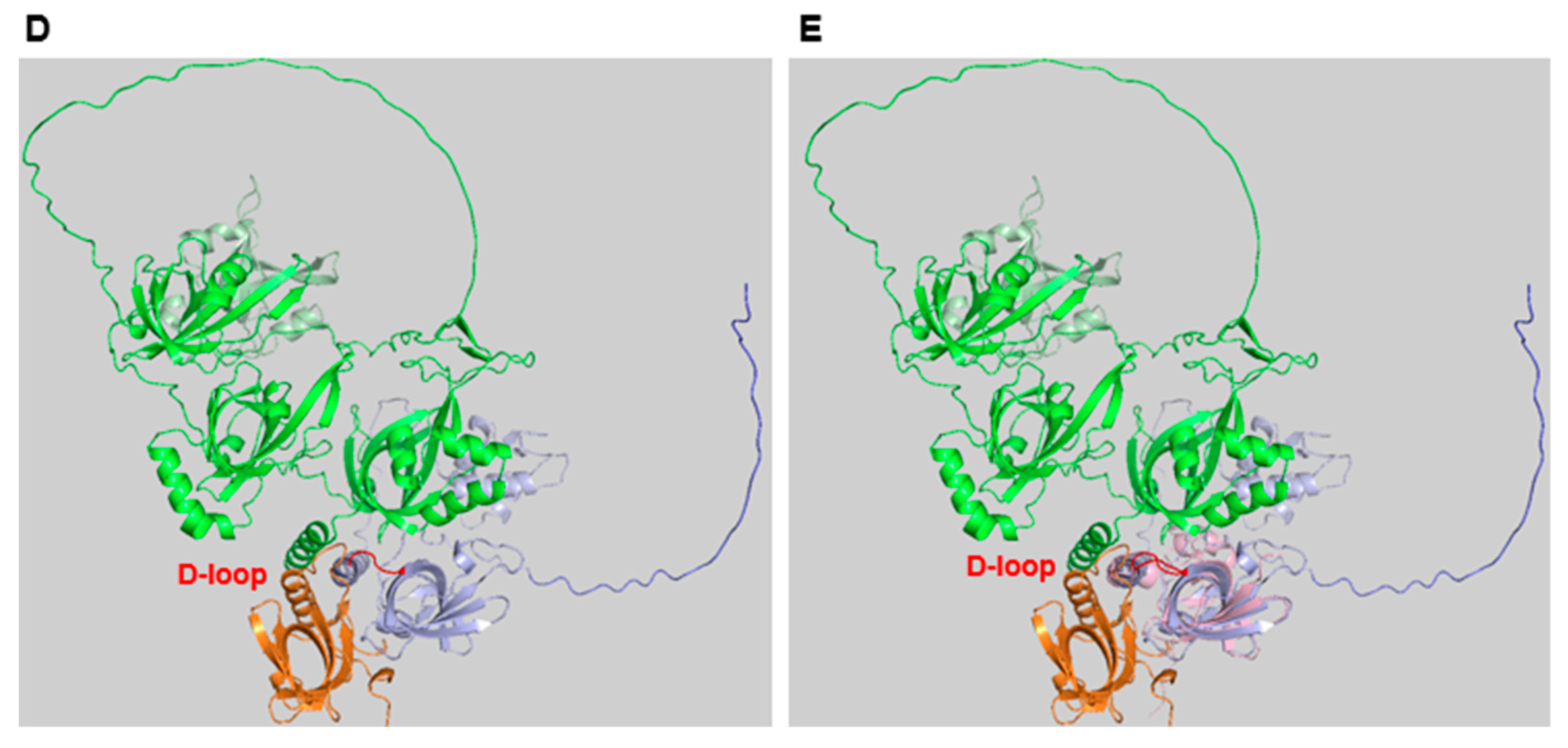
Figure 2.
DNA synthesis at replication forks of eukaryotic chromosomes. This schematic drawing of a eukaryotic replication fork shows CMG helicase (orange) unwinding dsDNA into the leading plus lagging strand templates and the replication proteins involved in DNA synthesis at the fork. Heterotrimeric RPA binds the resulting ssDNAs. The navy blue DNA strands are the parental strands. Pol ε (grey) newly synthesises the leading strand (royal blue DNA) in 5’ to 3’ direction as indicated by the arrow, and is associated with the CMG. Through the associated AND-1/CTF4/WDHD1 protein (dark grey, named AND-1) links CMG to the Pol α complex. The primer (red) synthesised by primase function of Pol α allows for replication synthesis initiation by the DNA polymerase activity of Pol ⍺ (pink) in a 5’-3’ direction to start Okazaki fragment synthesis for the lagging strand synthesis. PCNA (purple) and RFC (turquoise) replace Pol ⍺, making a landing site for Pol δ (maroon) to bind to the RNA-DNA and PCNA and elongate it until this complex reaches the next Okazaki on the parent strand allowing the maturation of the Okazaki fragments to occur (Adapted from [35,38,46,47]).
Figure 2.
DNA synthesis at replication forks of eukaryotic chromosomes. This schematic drawing of a eukaryotic replication fork shows CMG helicase (orange) unwinding dsDNA into the leading plus lagging strand templates and the replication proteins involved in DNA synthesis at the fork. Heterotrimeric RPA binds the resulting ssDNAs. The navy blue DNA strands are the parental strands. Pol ε (grey) newly synthesises the leading strand (royal blue DNA) in 5’ to 3’ direction as indicated by the arrow, and is associated with the CMG. Through the associated AND-1/CTF4/WDHD1 protein (dark grey, named AND-1) links CMG to the Pol α complex. The primer (red) synthesised by primase function of Pol α allows for replication synthesis initiation by the DNA polymerase activity of Pol ⍺ (pink) in a 5’-3’ direction to start Okazaki fragment synthesis for the lagging strand synthesis. PCNA (purple) and RFC (turquoise) replace Pol ⍺, making a landing site for Pol δ (maroon) to bind to the RNA-DNA and PCNA and elongate it until this complex reaches the next Okazaki on the parent strand allowing the maturation of the Okazaki fragments to occur (Adapted from [35,38,46,47]).
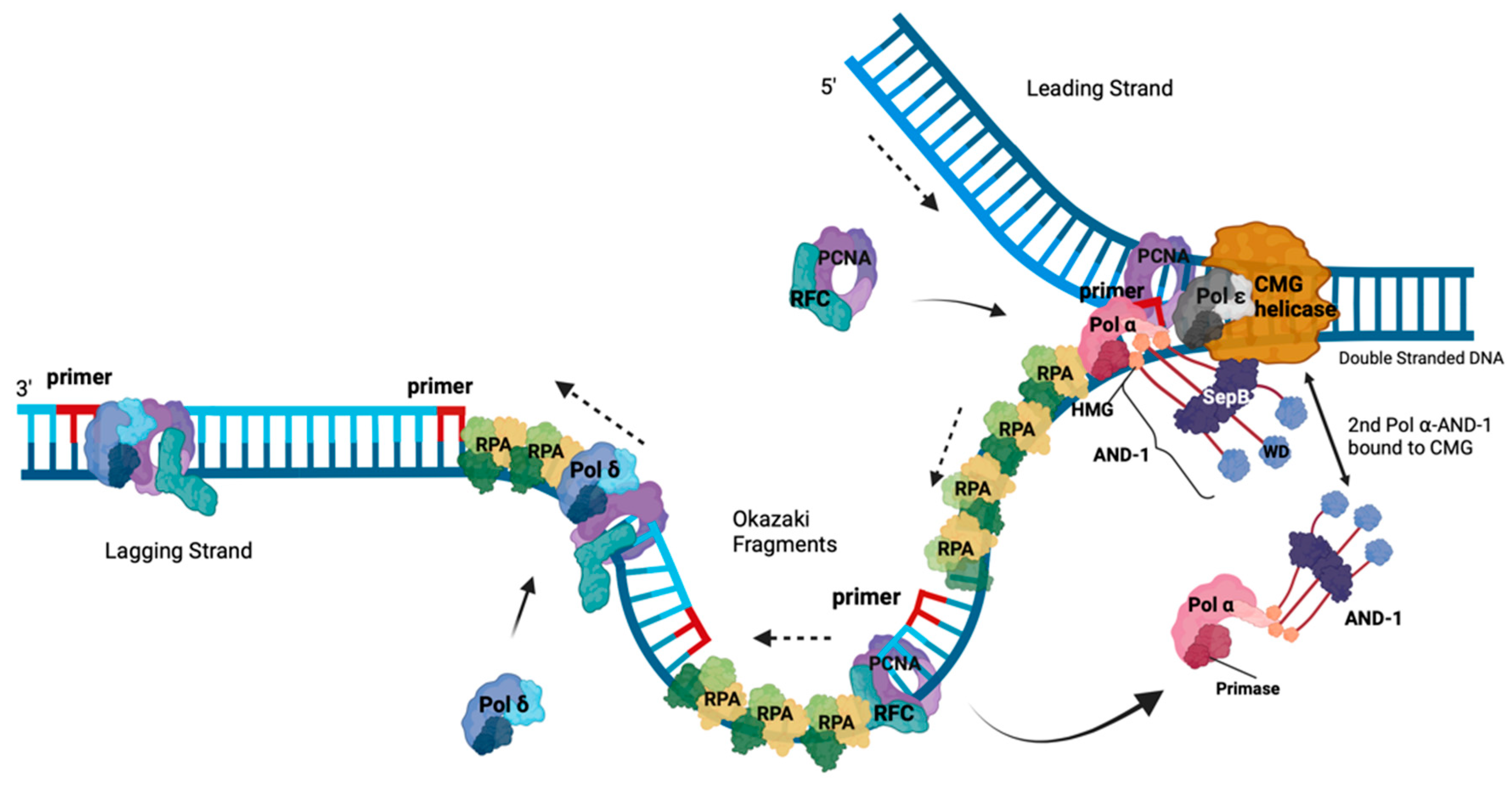
Figure 3.
Nuclear excision repair. The schematic drawing depicts the global genomic NER (ggNER) pathway the most prominent NER pathway in cells [93,94,95]. Eukaryotic cells also repair bulky lesions via the transcription-coupled NER (tcNER) pathway which only occurs in a transcription-depen-dent manner on the transcribed strand [93,94,95]. The tcNER pathway feeds into the pre-sented pathway at diagram but was omitted for simplification reasons. A. DNA damage can result in bulky DNA lesions. B. RAD23B and XPC (pink) recognise these damaged sites in dsDNA. C. They recruit the multi-subunit protein complex TFIIH (transcription facto II H, light green) that contains XPB in purple and XPD in dark green to verify and extend the opening of the dsDNA. D. XPA, in orange, and the multi-coloured RPA are recruited to the DNA damage site to bind and protect newly unwound ssDNA. The positioning of RPA and XPA ensure the correct localisation of XPF and XPG, in darker pink colours, to undertake their incision functions as shown in panel E, F & G. DNA elongation occurs after binding of RFC (grey) and loading of PCNA (purple), and DNA polymerase (dark grey) recruitment (panel G). H. Ligase (purple) binds the newly synthesised DNA at the incision site and ligates the nick in the dsDNA to yield repaired dsDNA. (Adapted from Schärer, 2013 [95]).
Figure 3.
Nuclear excision repair. The schematic drawing depicts the global genomic NER (ggNER) pathway the most prominent NER pathway in cells [93,94,95]. Eukaryotic cells also repair bulky lesions via the transcription-coupled NER (tcNER) pathway which only occurs in a transcription-depen-dent manner on the transcribed strand [93,94,95]. The tcNER pathway feeds into the pre-sented pathway at diagram but was omitted for simplification reasons. A. DNA damage can result in bulky DNA lesions. B. RAD23B and XPC (pink) recognise these damaged sites in dsDNA. C. They recruit the multi-subunit protein complex TFIIH (transcription facto II H, light green) that contains XPB in purple and XPD in dark green to verify and extend the opening of the dsDNA. D. XPA, in orange, and the multi-coloured RPA are recruited to the DNA damage site to bind and protect newly unwound ssDNA. The positioning of RPA and XPA ensure the correct localisation of XPF and XPG, in darker pink colours, to undertake their incision functions as shown in panel E, F & G. DNA elongation occurs after binding of RFC (grey) and loading of PCNA (purple), and DNA polymerase (dark grey) recruitment (panel G). H. Ligase (purple) binds the newly synthesised DNA at the incision site and ligates the nick in the dsDNA to yield repaired dsDNA. (Adapted from Schärer, 2013 [95]).
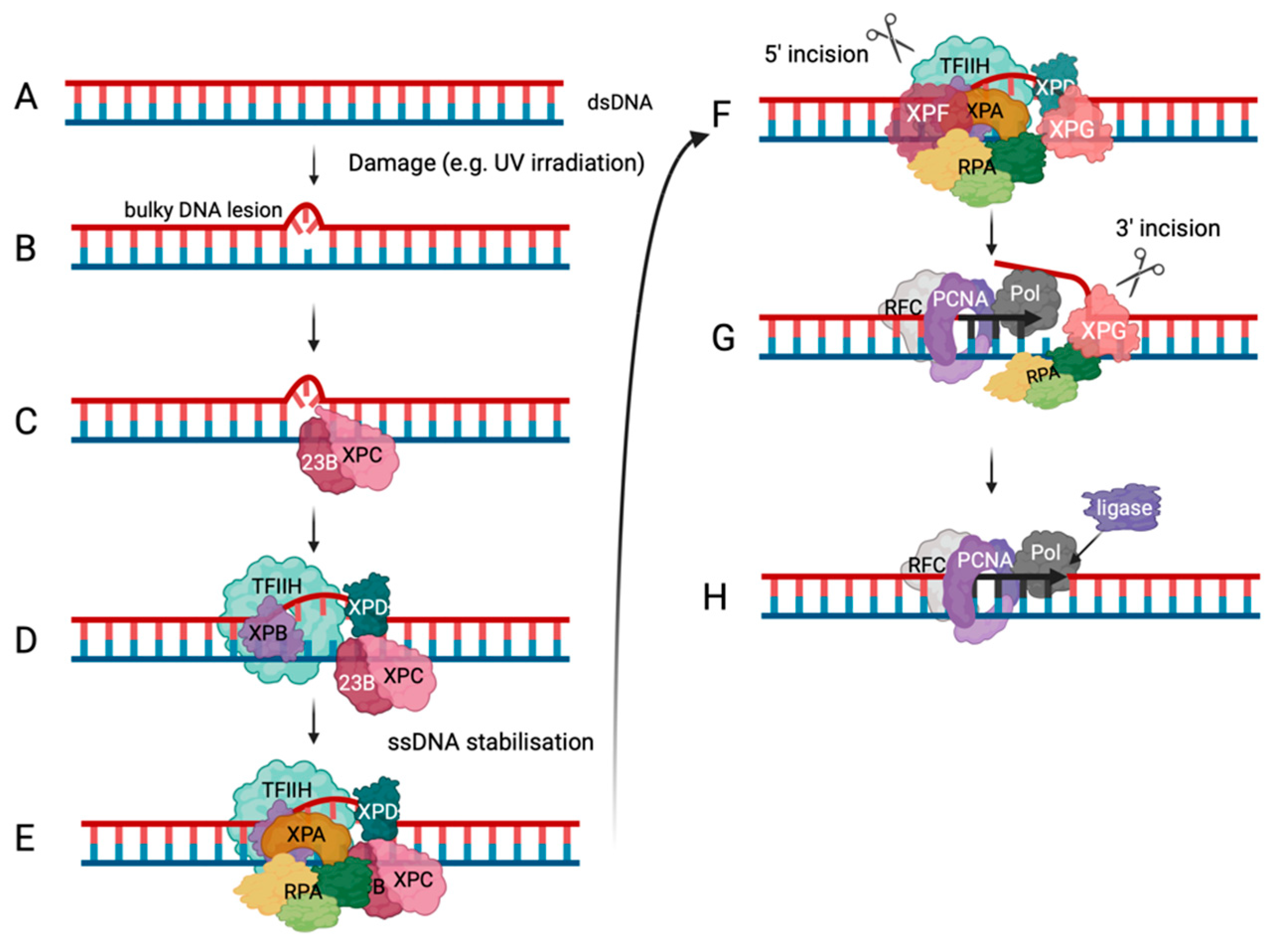
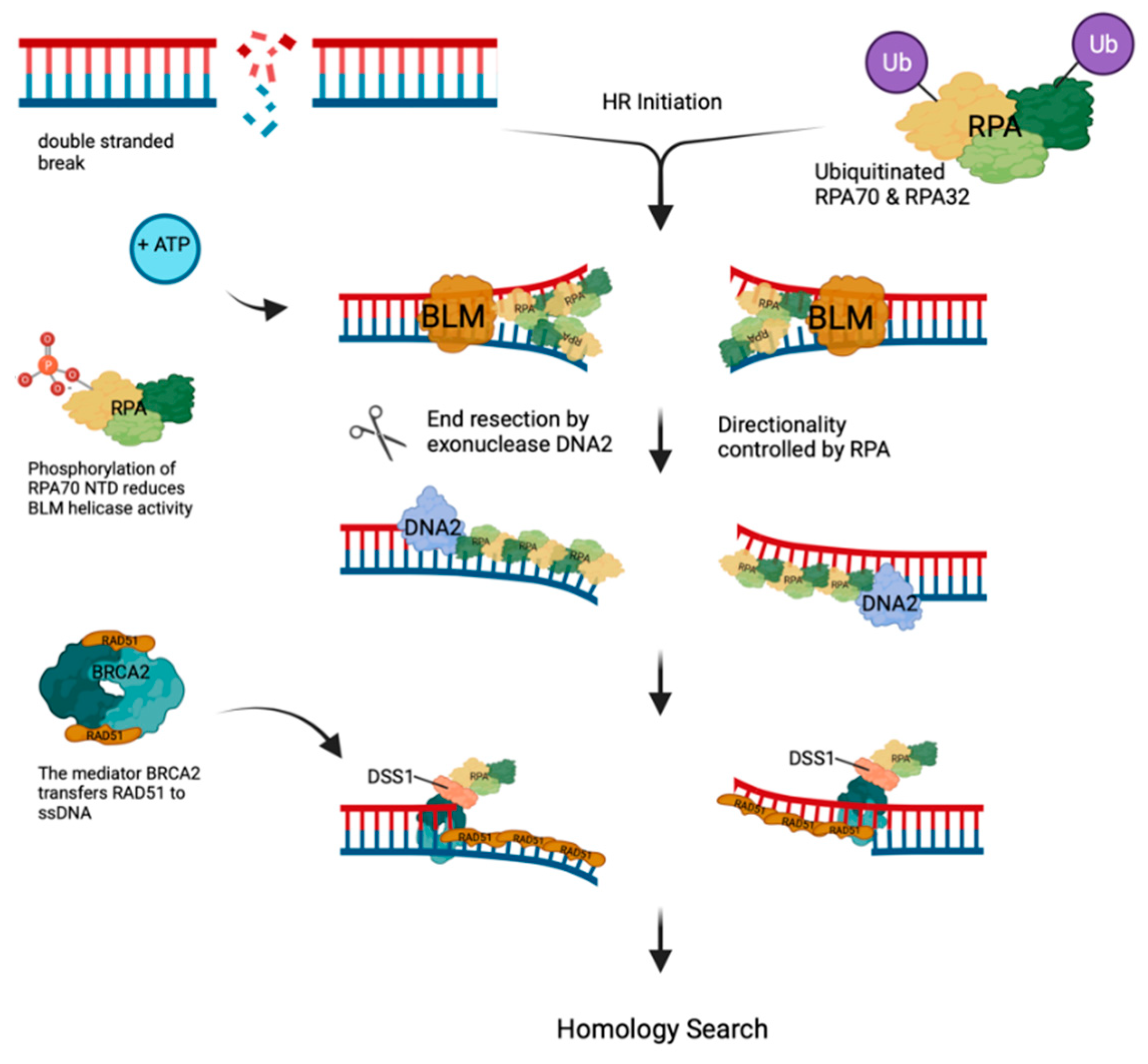
Figure 4.
Homologous recombina-tion. Double strand breaks (DSBs) are highly toxic for cells and occur in DNA after high energy radiation or cellular processes such as antibody develop-ment. Following DSB, binding of MRN and chromatin modifications including ubiquitination of cellular proteins such as RPA70 and RPA32 allows HR to occur. Then BLM helicase (orange) un-winds the dsDNA in an ATP-dependent fashion and RPA binds to ssDNA. DNA2 (purple) interacts with RPA and degrades one DNA strand in 5’-3’ direction controlled by RPA. Phosphorylation of RPA70 NTD alters the RPA-BLM interactions causing a reduction of BLM activity. The mediator BRCA2 via its partner DSS1 interacts with resulting RPA-ssDNA filaments and exchanges RPA towards RAD51. The RAD51-ssDNA filament then start homology search of these overhangs (Adapted from [99]).
Figure 4.
Homologous recombina-tion. Double strand breaks (DSBs) are highly toxic for cells and occur in DNA after high energy radiation or cellular processes such as antibody develop-ment. Following DSB, binding of MRN and chromatin modifications including ubiquitination of cellular proteins such as RPA70 and RPA32 allows HR to occur. Then BLM helicase (orange) un-winds the dsDNA in an ATP-dependent fashion and RPA binds to ssDNA. DNA2 (purple) interacts with RPA and degrades one DNA strand in 5’-3’ direction controlled by RPA. Phosphorylation of RPA70 NTD alters the RPA-BLM interactions causing a reduction of BLM activity. The mediator BRCA2 via its partner DSS1 interacts with resulting RPA-ssDNA filaments and exchanges RPA towards RAD51. The RAD51-ssDNA filament then start homology search of these overhangs (Adapted from [99]).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



