
Submitted:
25 June 2024
Posted:
26 June 2024
You are already at the latest version
Abstract
In Southeast Asia, the incidence of Leukemia, a malignant blood cancer originating from hema-topoietic progenitor cells, is on the rise, marked by a concerning 54% mortality rate. This study focuses on enhancing early-stage prediction to improve patient recovery prospects significantly. Leveraging Machine Learning and Data Science, we employ protein sequential data from frequently mutated genes such as BCL2, HSP90, PARP, and RB to predict Chronic Myeloid Leukemia (CML). Our approach relies on robust feature extraction techniques, namely Di-peptide Composition (DPC), Amino Acid Composition (AAC), and Pseudo amino acid composition (Pse-AAC), with prior attention to addressing outliers and validating feature selection through the Pearson Corre-lation Coefficient. Data augmentation ensures a well-rounded dataset for analysis. Employing a range of Machine Learning models, including Support Vector Machine (SVM), XGBoost, Random Forest (RF), K Nearest Neighbor (KNN), Decision Tree (DT), and Logistic Regression (LR), we achieve accuracy rates spanning from 66% to 94%. These classifiers undergo comprehensive as-sessment using performance metrics such as accuracy, sensitivity, specificity, F1-score, and the confusion matrix. Our proposed solution, encompassing a user-friendly web application dashboard, presents an invaluable tool for early CML diagnosis with profound implications for practitioners, offering a deploy-able asset within healthcare institutions and hospitals.
Keywords:
Protein Sequences
; Pseudo-AAC
; AAC
; Dipeptide-C
; Machine Learning Classifiers
; Chronic Myeloid Leukemia
; Blood Cancer
1. Introduction
Leukemia is a complex medical condition influenced by genetic regulation in the production of blood cells. When hematopoietic precursor cells turn malignant [1], it gives rise to abnormal cell growth due to alterations in DNA and RNA sequences. This transformation results in the infiltration of healthy cells by malignant ones, thus causing Leukemia. The illness primarily entails the uncontrolled proliferation of specifically White Blood Cells (WBC), i.e., neutrophils, basophils, and eosinophils, while lymphocytes remain unaffected. Acute myeloid Leukemia (AML), chronic myeloid Leukemia (CML), acute lymphoblastic Leukemia (ALL), and chronic lymphocytic Leukemia (CLL) are some of the several kinds of Leukemia [2]. The only subject of our research is Chronic Myeloid Leukemia (CML).
Leukemia cancer presents a substantial health challenge due to the abnormal proliferation of White Blood Cells (WBC) [1]. While research has concentrated on detecting cancer through blood cell images, exploration of Protein Sequential data is limited. Leukemia diagnosis heavily relies on hematologists, posing limitations in regions with a scarcity of specialists. Mortality rates are on the rise, particularly in South East Asia [3], creating a demand for an early detection approach.
The motivation for driving the proposed research arises from the observation that a plethora of research has been conducted on cancer predictions—such as lung cancer, liver cancer, colon cancer, ovarian cancer, etc. utilizing MRI (magnetic resonance imaging), CT (computed tomography) scans, image processing techniques and protein sequences [4,5,6]. However, the realm of gene data in bioinformatics remains relatively uncharted, especially within the context of Chronic Myeloid Leukemia (CML). At present, no AI-based Dashboard system predicts Leukemia based on protein sequences, but developing such a system could revolutionize the diagnosis, leading to saved lives and eased healthcare burdens. Collaborative efforts between Machine Learning and Data Science can establish a robust model for accessible and timely Leukemia solutions.
As illustrated in Figure 1, the proposed research suggests the utilization of Machine Learning-based techniques to identify genes that cause Leukemia through Protein Sequences, aiming for early detection and a reduction in the mortality rate. This undertaking could emerge as a flagship initiative in health sciences, addressing the shortage of specialized hematologists. Implementation of the system would result in timely interventions and improved recovery prospects. Automating certain diagnostic processes could ease the load on specialists and enhance healthcare services. The potential impact goes beyond Leukemia diagnosis, garnering recognition, and interest from the medical community. Overall, this AI-driven research holds immense promise in reshaping healthcare and propelling the advancement of AI applications.
Because of this research, innovative insights, and progress in predicting and comprehending CML could come to fruition. This might lead to more effective diagnostic and treatment methodologies, benefiting patients and healthcare systems. Furthermore, the successful integration of bioinformatics and AI could pave the way for pioneering applications and further interdisciplinary research at the intersection of these two promising domains.
The main contribution of our proposed research is as follows:
- The current study focuses on protein sequential data rather than image data.
- The most frequently mutated genes that were responsible for chronic myeloid leukemia were discovered through a literature review.
- Datasets were formulated from the most frequently muted gene data.
- Features were extracted through physicochemical properties of Amino Acid composition, Pseudo Amino Acid Composition, and di-peptide composition.
- The study focuses on enhancing early-stage prediction to improve patient recovery prospects significantly.
- Our proposed solution encompasses a user-friendly web application dashboard that presents an invaluable tool for early CML diagnosis, offering a deploy-able asset within healthcare institutions and hospitals.
This paper follows a structured format that aims to understand the research comprehensively. Section 1, 'Introduction,' outlines the problem statement. Section 2, 'Literature Review,' discusses related research, positioning our study in the existing body of knowledge. Section 3, 'Materials and Methods,' details the dataset creation process and experimental techniques. Section 4, 'Development of Individual Classifiers,' presents our methodology and analysis. Section 5, 'Results and Discussion,' succinctly interprets the findings. Lastly, in Section 6, we offer a conclusion summarizing our contributions and outlining future research directions.
2. Literature Review
This section comprehensively discusses the recently conducted Leukemia research, focusing on Protein Sequences, RNA, and blood cell imagery. It elaborates acquiring and forming the dataset, which is pivotal in creating standardized Leukemia datasets by utilizing protein sequences. Importantly, previous researchers have not combined these three distinct feature extraction techniques while implementing a user-friendly dashboard, as done in this study.
In [7], the Random Forest model was utilized to diagnose the cancerous growth of White Blood Cells with an accuracy of 94.3%. In the research by [8], the classifier was evaluated using 60 photos, demonstrating that models like K-nearest neighbors and Naive Bayes Classifier could identify ALL with an accuracy of 92.8%. According to research [9], the Artificial Bee Colony algorithm – Back Propagation Neural Network (ABC-BPNN) scheme and Principal Component Analysis (PCA) were used to classify Leukemia cells with an average accuracy of 98.72% while also speeding up the calculation.
In reference [10] Jothi et al. investigated the identification of leukemia sub-types, particularly ALL, using BSA-based clustering and advanced classification algorithms such as decision tree (DT), K-nearest neighbor (KNN), Naive Bayes (NB), and Support Vector Machine (SVM). The SVM model exhibited an accuracy rate of 89.81%. The SVM model was used in research [11] to identify ALL, with an accuracy rate of 89.81%. The dataset was used in [12] to classify ALL using the K-nearest neighbor method, with a 96.25% accuracy rate. In study gal [13], the exploration centered around the use of ML algorithms to analyze gene expression patterns derived from RNA sequencing (RNA-seq) for accurately predicting the likelihood of CR in pediatric AML patients’ post-induction therapy.
Research [14] Developed models for predicting and classifying different stages of colon cancer using RNA-seq data of extracellular vesicles (EV) from healthy individuals and colon cancer patients. The study employed five canonical ML and Deep Learning (DL) classifiers, achieving high accuracy rates, resulting in an accuracy of 94.6% for K-nearest neighbor, 97.33% for Random Forest, 93% for LMT, and 92% for Random Tree. In [15], the early diagnosis and distinction between types of lung cancers, i.e., Non-Small Cell Lung Cancer and Small Cell Lung Cancer, were highlighted as crucial for improving patient survival rates. The proposed diagnostic system utilized sequence-derived structural and physicochemical attributes of proteins associated with tumor types, employing feature extraction, selection, and prediction models.
The study conducted by Dhakal et al. [16] introduced a stacking classifier algorithm addressing CTS selection criteria through feature-encoding techniques, generating feature vectors that encompass k-mer nucleotide composition, dinucleotide composition, pseudo-nucleotide composition, and sequence order coupling. This innovative stacking classifier algorithm outperformed previous state-of-the-art algorithms in predicting functional miRNA targets, achieving an accuracy of 79.77%. In another study, Albitar et al. [17], Using Next Generation Sequencing (NGS) and targeted RNA sequencing along with a machine learning approach, Albitar et al. investigated the potential of discovering new biomarkers that can predict Acute graft-vs.-host disease (aGVHD). The study by Ahmad et al. [18], Predicted chronic Lymphocytic Leukemia using protein sequences with Chou’s Pseudo Amino Acid Composition (PseAAC) and statistical moments.
In the study [19], using deep learning (DL), Jian et al. constructed a prediction model for transcription factor binding sites only from original DNA base sequences. Here, a DL method based on convolutions neural network (CNN) and long short-term memory (LSTM) was proposed to investigate four Leukemia categories from the perspective of transcription factor binding sites using four large non-redundant datasets for acute, chronic, myeloid, and lymphatic Leukemia, giving an average prediction accuracy of 75%.
3. Materials and Methods
The proposed research centers on the detection of leukemia, specifically targeting Chronic Myeloid Leukemia (CML), characterized by the neoplastic proliferation of White Blood Cells (WBCs) such as neutrophils, basophils, and eosinophils, while excluding lymphocytes. As previously mentioned, CML is linked to a heightened mortality rate due to its typical diagnosis at advanced stages, posing challenges for effective recovery. In response to this concern, we aim is to create a dashboard to identify leukemia utilizing Protein Sequential data.
To achieve this goal, we collected data on the most frequently mutated genes related to leukemia cancer, leveraging the physiochemical properties of protein sequences for feature extraction. Subsequently, data augmentation techniques were applied to enhance the extracted features, while outliers were detected and removed to ensure data quality. We employed a diverse set of machine learning algorithms, including Support Vector Machine (SVM) [20,21,22,23] XG Boost, Random Forest [24,25] KNN [26,27] logistic regression, and decision tree, as comprehensively described in a study review [28,29,30,31]. The accuracy of each algorithm was evaluated, and the one exhibiting the highest accuracy was selected for integration into our system. This chosen algorithm determines the presence or absence of cancer in an individual. Finally, we serialized our model using tools such as Pickle or Joblib, facilitating the preservation of the trained model alongside its associated data. These trained models were then incorporated into a Streamlit-based dashboard, enhancing their user-friendly deployment in hospitals and other medical facilities (see Figure 2).
3.1. Block Diagram
3.2. Dataset Collection
There are many genes involved in CML. Based on the literature review, genes that are most often mutated, i.e. BCL2, HSP90, PARP and RB, were utilized for CML [20]. Moreover, the homologous samples were eliminated by maintaining 0.6 as the cutoff level [24]. HSP90functions as a chaperone protein, crucial in protein folding and degradation processes. It is up-regulation has been identified in various cancer types, including chronic myeloid leukemia (CML). Extensive research has demonstrated that inhibiting HSP90 can attenuate the growth of CML cells and enhance their susceptibility to chemotherapy and tyrosine kinase inhibitors (TKIs) [32,33]. PARP (Poly ADP-ribose polymerase) is an essential enzyme involved in DNA repair processes. Inhibiting PARP has demonstrated effectiveness in the treatment of cancers with BRCA mutations, and there is emerging evidence suggesting its potential applicability in managing chronic myeloid leukemia (CML) [34,35].
The BCL2 (B-cell lymphoma 2) protein family plays a crucial role in regulating programmed cell death, known as apoptosis. Elevated levels of BCL2 have been linked to resistance to chemotherapy in chronic myeloid leukemia (CML) cells. Studies have demonstrated that inhibiting BCL2 can reinstate apoptosis in CML cells and boost the effectiveness of tyrosine kinase inhibitors (TKIs) [36,37]. RB (Retinoblastoma) is a pivotal tumor suppressor gene involved in regulating cell cycle progression. The deactivation of RB is a prevalent characteristic in CML, and research has established that its reactivation can impede the proliferation of CML cells [38], [39]. The FASTA file format was used to extract the CML-related protein sequences from the Universal Resource of Proteins (UniProtKB) [22], [40]. A successful dataset was created as a result. The same number of negative and positive samples were gathered for CML using the opposite query phrase to create a negative dataset. Consequently, the dataset created for CML is balanced.
3.2.1. Fasta Format
In bioinformatics, the fasta format is a popular text-based format for representing proteins. It is derived from the FASTA software suite and follows a specific structure. A FASTA sequence starts with a single line that serves as a description and is followed by lines containing the sequencing data [40].
The description line is distinguished from the sequence data by the presence of a greater-than symbol (">") in the first column. The term following the ">" sign is used to identify the sequence, while the rest of the line can be used to provide an additional description, though both are optional.
3.2.2. Sample of Protein Sequence (HSP90)
Initially, protein sequences contained redundant data. We employed a benchmark method known as CD-Hit to address the issue of redundant data within the initial protein sequences (Figure 3). It is essential to utilize a benchmark algorithm for redundancy removal to ensure the validity and reliability of the data. CD-Hit, an online clustered database, was selected for this purpose, with a threshold of 0.6[41]. This threshold value helps in effectively removing redundancy while preserving the integrity of the dataset.
3.3. Feature Extraction
This section elaborates on the feature extraction techniques using physiochemical properties of the protein sequences. These techniques enable the effective representation of protein sequences and extraction of meaningful information crucial for predicting Chronic Myeloid Leukemia. The feature extraction methods utilized in this study fall into three categories:
3.3.1. Amino Acid Composition
The presence of specific amino acids often in a protein sequence is highlighted by AAC characteristics [42,43]. The percentage frequency of an amino acid, AAC i,j, in the jth protein is calculated using the formula below:
In the above equation, n denotes the amount of amino acids type (I) found in proteins j while na, aj refers to the total amount of amino acids contained in a protein. The jth protein sequence in the AAC features dataset is represented as a 20-dimensional (20-D) feature vector as follows:
Where, demonstrates how amino acids are composed.
The technique of amino acid composition involves extracting features from our data, resulting in a 20-dimensional feature set. However, the problem with this approach lies in the limited usefulness of the features extracted. Despite employing various data science feature engineering approaches and conducting hyper-parameter tuning, accuracy remains constrained. Consequently, this approach proves less efficacious in attaining the desired outcomes.
3.3.2. Pseudo Amino Acid Composition
A 25-dimensional feature set is produced using the Pseudo Amino Acid Composition (PAAC) approach to extract features from our data[44]. The remarkable fact is that the features extracted through this method are highly valuable. By further applying data science methods and feature engineering techniques, accuracy significantly improves, reaching an impressive range of 91% to 93%. This achievement represents a remarkable success in our endeavors.
In Figure 5, we present graphs illustrating the impact of outlier removal on the dataset. Specifically, we depict the changes in data distribution before and after outlier removal. Additionally, we conducted data augmentation on the processed dataset to further enhance its accuracy.
3.3.3. Di-peptide Composition
The letters AA, AC, AD, YV, YW, and YY denote protein sequences with dipeptide characteristics. There are 400 components in these sequences. The DC feature of each component is determined as follows:
Where represents the structure of ith dipeptide for . In vector form, this feature space is represented as: . The di-peptide composition technique extracts features from our data, resulting in 400 dimensions or four hundred features. However, it became evident that not all these features were essential. By applying data science methods and feature engineering, it is concluded that only 229 features out of the initial 400 were necessary. Surprisingly, after this selection process, the accuracy of our results significantly improved, reaching an impressive 91% to 93%. This outcome marks a great success. The graphs illustrate the impact of outlier removal on the dataset, both before and after the process.
3.3.4. Data Augmentation
The Data augmentation process is initiated by segregating our dataset into positive and negative segments. The method entails isolating patients who have tested positive from those with negative results. Subsequently, a series of operations are designed to generate numerical replicas of the existing data, thereby augmenting the sample size. This augmentation enhances the machine learning algorithm's training procedure, attributed to the increased abundance of available data. However, it is important to note that the data transforms during the creation of these numerical duplicates, transitioning from its initial format into a list structure.
Consequently, the modified data is transited from this list format into a data frame. This procedural sequence ultimately leads to reintegrating the transformed data, thereby completing the data augmentation process.
4. Development of Individual Classifiers
4.1. Support Vector Machine
SVM classifier by creating a hyperplane with the greatest distance between any two points in the data [45,46,47,48,49,50]. SVM's decision surface is as follows.
We selected the parameters such as, Kernel = “rbf”, Degree =8, C =10000, gamma =100000, probability = True.
4.2. Random Forest
This method generates a substantial quantity of decision trees that are combined to arrive at a final decision. For training we selected 129,361, and for testing, 86,228 samples were selected, and we came up with the best number of estimators, i.e., n=50. In the case of dipeptide composition, we selected 2536 for training and 845 for testing, and n=150 estimators gave optimal results.
4.3. K-Nearest Neighbor (KNN)
The KNN algorithm is learned by observing samples [51,52]. Instance-based classifiers assume that the classification of unknown instances can be accomplished by comparing the unidentified instance to a known instance using a distance/similarity function [53,54,55,56].
The calculation of the Euclidean distance (below, denoted as , between two m-dimensional vectors Xi and Xj is as follows:
4.4. Naïve Bayes
Bayes rules represent this learning procedure based on the notion of independent attributes/features. The Gaussian function to train the model with equal prior probabilities in the following manner:
4.5. XGBoost
Gradient boosting is a boosting approach that significantly lowers errors by adding several classifiers to pre-existing models. The term "gradient boosting" refers to using a gradient descent strategy to minimize loss. The steps involved in gradient boosting are as follows:
4.6. Logistic Regression
In categorical binary classification, a statistical machine-learning approach called logistic regression is employed [57]. The parameters we selected were C=10, tol = 0.1, and penalty = L2.
5. Results and Discussion
5.1. Results on Pseudo Amino Acid Composition (Pse-AAC) Data
The findings of the matrices employed in the project Accuracy score, F1-score, recall [58,59], and specificity receptively on the data of Pse-AAC are displayed in Table 1 below.
Table 2 presents the results of each machine learning (ML) model concerning the data utilized, specifically the Pse-AAC data. It also includes the outcomes of additional metrics used in the research, namely Specificity and Confusion Matrix. These metrics provide insights into the True Positive, True Negative, False Positive, and False Negative values, contributing to a comprehensive evaluation of the models' performance.
5.2. Accuracy Result on Amino Acid Composition (AAC) Data
The research employs accuracy score, F1-score, recall score, and specificity as metrics on the AAC data. The outcomes of these metrics are presented in Table 3 below.
The following table (Table 4) presents the results of each machine learning (ML) model concerning the utilized data, namely AAC. Additionally, it showcases the outcomes of other metrics employed in the project, such as the Specificity and Confusion Matrix. These matrices provide essential values, including True Positive, True Negative, False Positive, and False Negative, contributing to a comprehensive assessment of the models' performance.
5.3. Accuracy Results on Di-Peptide Composition (DPC)
The table below (Table 5) displays the accuracy score, F1-score, and recall score matrices utilized in the research and their respective outcomes when applied to the DPC data.
The performance of each machine learning model is analyzed concerning the DPC data utilized. Additionally, the Specificity and Confusion Matrix results are presented (Table 6). This matrix provides essential values such as True Positive, True Negative, False Positive, and False Negative, contributing to a comprehensive evaluation of the models' performance.
5.5. Machine Learning Based Dashboard
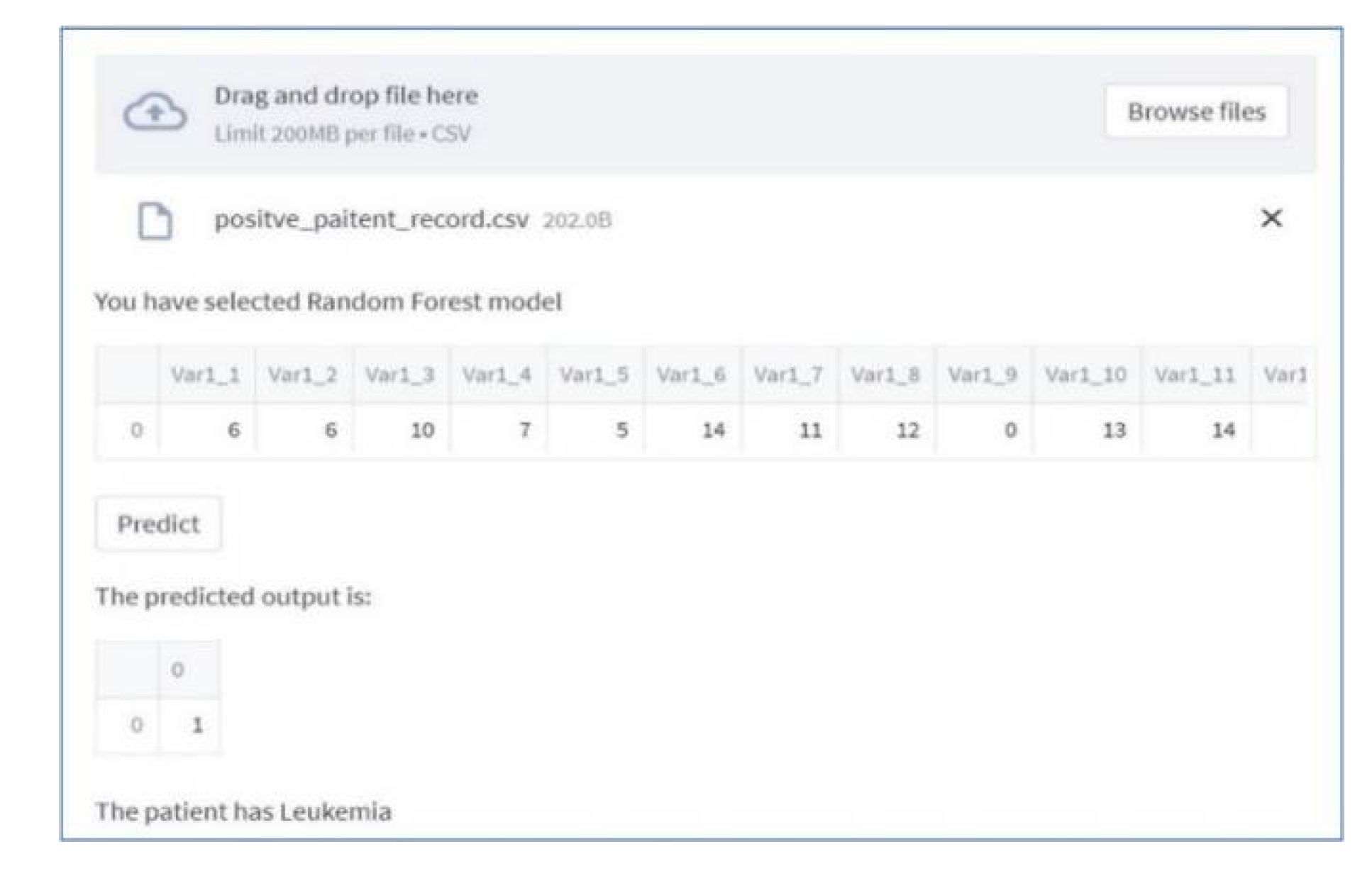
In Figure 4, we provide an overview of the dashboard developed using Streamlit, which is accessible through Streamlit Cloud. This interactive dashboard enables users to select their preferred model for analysis. Within this user-friendly interface, individuals are prompted to upload patient records directly through the web application and select a specific prediction model. Subsequently, users can review the results to ascertain whether an individual is affected by leukemia. Users can effortlessly select and upload patient records from their computer by simply clicking the browse button. Once the data is uploaded, users gain access to both the raw data and predictive outcomes, as illustrated in Figure 5.
Figure 4.
Screenshot of dashboard.
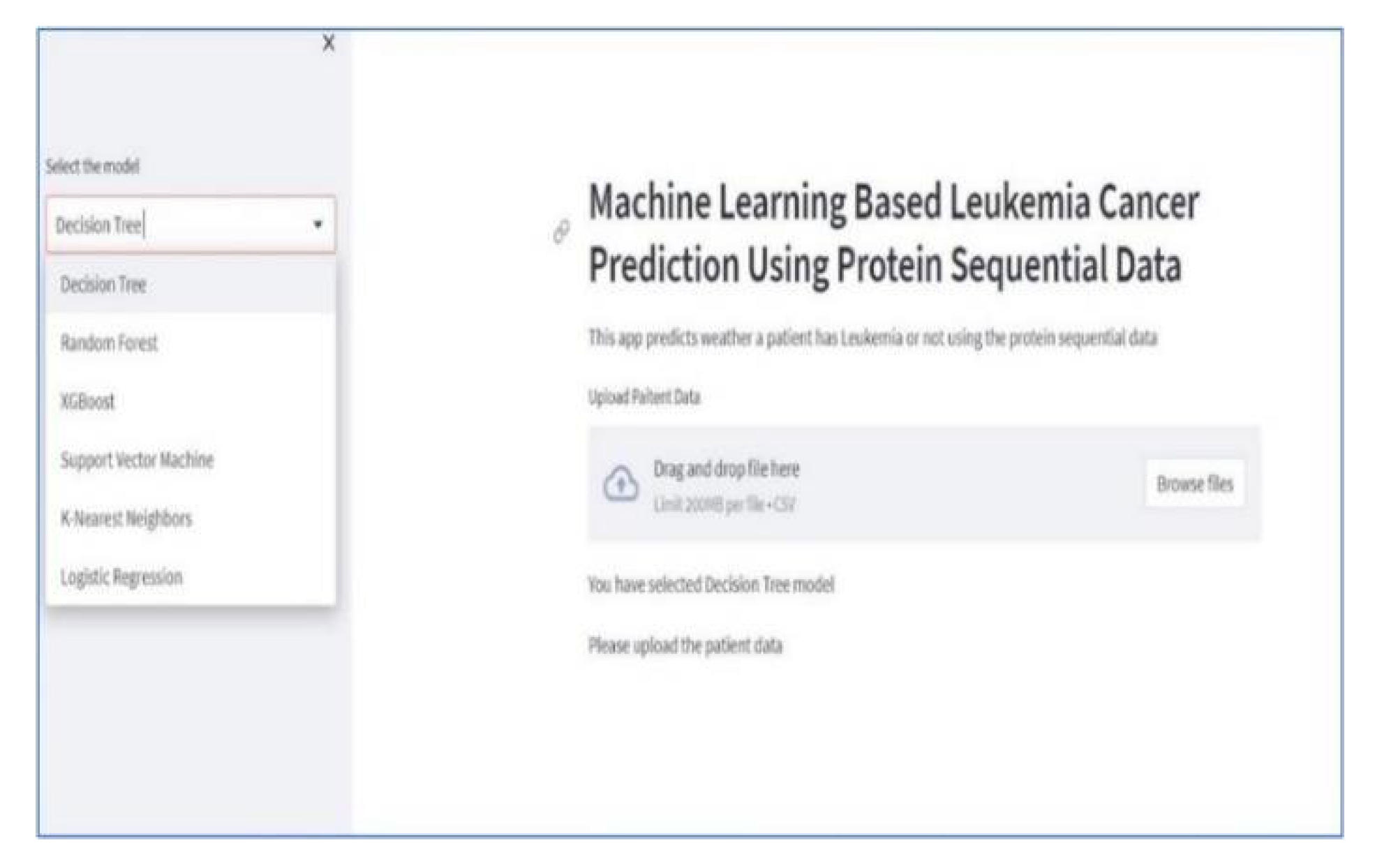
Figure 5.
Prediction on Data.
5. Conclusion
This research is focused on Chronic Myeloid Leukemia (CML), a condition characterized by genetic mutations leading to abnormal proliferation of white blood cells, red blood cells, and platelets. While MRI and CT scans have been extensively used in cancer detection, research on protein sequence data in this domain is limited. By leveraging information from mutated genes like BCL2, HSP90, PARP, and RB, the research aims to revolutionize early CML prediction. Through rigorous data preprocessing and feature extraction techniques, we achieved an impressive accuracy rate of 92–94%. The proposed approach integrates diverse machine learning algorithms such as SVM, Decision Trees, XGBoost, Random Forest, and KNN, each offering unique strengths in pattern recognition and prediction. The resulting dashboard facilitates easy prediction of CML in patients, enhancing clinical workflows and potentially saving lives. This study sheds light on critical scientific challenges in CML research, offering insights into disease mechanisms and biomarker identification. we envision expanding this research to encompass multi-cancer detection, integrating AI and bioinformatics with healthcare systems for enhanced cancer diagnosis and improved patient outcomes.
Authors' Contributions: All the authors contributed equally and substantially to this manuscript.
Funding
The authors extend their appreciation to the Research and Innovations of Datamatics Technologies, Dubai, UAE, for funding this work.
Acknowledgments
The authors would like to thank anonymous referees for giving very helpful comments and suggestions that have greatly improved this paper. Additionally, the authors would like to thank the King Salman Center for Disability Research for their valuable input.
Conflicts of Interest
The authors have no conflicts of interest to report regarding the present study.
References
- R. L. Siegel, K. D. Miller, H. E. Fuchs, and A. Jemal, "Cancer statistics, 2021," Ca Cancer J Clin, vol. 71, no. 1, pp. 7-33 %@ 1542-4863, 2021.
- N. Bibi, M. Sikandar, I. Ud Din, A. Almogren, and S. Ali, "IoMT-based automated detection and classification of leukemia using deep learning," Journal of healthcare engineering, vol. 2020, pp. 1-12 %@ 2040-2309, 2020.
- I. IafRoC, "Leukaemia Source: Globocan 2020 2020 [Available from: https://gco. iarc. fr/today/data/factsheets/cancers/36-Leukaemia-fact-sheet. pdf," ed: Accessed, 2022.
- C. R. Munteanu, A. L. Magalhães, E. Uriarte, and H. González-Díaz, "Multi-target QPDR classification model for human breast and colon cancer-related proteins using star graph topological indices," Journal of theoretical biology, vol. 257, no. 2, pp. 303-311 %@ 0022-5193, 2009. [CrossRef]
- R. G. Ramani and S. G. Jacob, "Improved classification of lung cancer tumors based on structural and physicochemical properties of proteins using data mining models," PloS one, vol. 8, no. 3, pp. e58772 %@ 1932-6203, 2013. [CrossRef]
- J.-Y. Yang et al., "Predicting time to ovarian carcinoma recurrence using protein markers," The Journal of clinical investigation, vol. 123, no. 9, pp. 3740-3750 %@ 0021-9738, 2013.
- H. Mohamed et al., "Automated detection of white blood cells cancer diseases," 2018: IEEE, pp. 48-54 %@ 1538650835.
- S. Kumar, S. Mishra, P. Asthana, and Pragya, "Automated detection of acute leukemia using k-mean clustering algorithm," 2018: Springer, pp. 655-670 %@ 9811037728.
- R. Sharma and R. Kumar, "A novel approach for the classification of leukemia using artificial bee colony optimization technique and back-propagation neural networks," 2019: Springer, pp. 685-694 %@ 9811312168.
- G. Jothi, H. H. Inbarani, A. T. Azar, and K. R. Devi, "Rough set theory with Jaya optimization for acute lymphoblastic leukemia classification," Neural Computing and Applications, vol. 31, pp. 5175-5194 %@ 0941-0643, 2019. [CrossRef]
- Z. Moshavash, H. Z. Moshavash, H. Danyali, and M. S. Helfroush, "An automatic and robust decision support system for accurate acute leukemia diagnosis from blood microscopic images," Journal of digital imaging, vol. 31, pp. 702-717 %@ 0897-1889, 2018. [CrossRef]
- D. Umamaheswari and S. Geetha, "A framework for efficient recognition and classification of acute lymphoblastic leukemia with a novel customized-KNN classifier," Journal of computing and information technology, vol. 26, no. 2, pp. 131-140 %@ 1330-1136, 2018. [CrossRef]
- O. Gal, N. Auslander, Y. Fan, and D. Meerzaman, "Predicting complete remission of acute myeloid leukemia: machine learning applied to gene expression," Cancer informatics, vol. 18, pp. 1176935119835544 %@ 1176-9351, 2019. [CrossRef]
- E. Bostanci, E. Kocak, M. Unal, M. S. Guzel, K. Acici, and T. Asuroglu, "Machine learning analysis of RNA-seq data for diagnostic and prognostic prediction of colon cancer," Sensors, vol. 23, no. 6, pp. 3080 %@ 1424-8220, 2023. [CrossRef]
- F. Hosseinzadeh, A. H. KayvanJoo, M. Ebrahimi, and B. Goliaei, "Prediction of lung tumor types based on protein attributes by machine learning algorithms," SpringerPlus, vol. 2, pp. 1-14, 2013. [CrossRef]
- P. Dhakal, H. Tayara, and K. T. Chong, "An ensemble of stacking classifiers for improved prediction of miRNA–mRNA interactions," Computers in Biology and Medicine, vol. 164, pp. 107242 %@ 0010-4825, 2023. [CrossRef]
- M. Albitar et al., "Bone Marrow-Based Biomarkers for Predicting aGVHD Using Targeted RNA Next Generation Sequencing and Machine Learning," Blood, vol. 138, pp. 2892 %@ 0006-4971, 2021. [CrossRef]
- W. Ahmad, M. Hameed, M. Bilal, and A. Majid, "ML-Pred-CLL: Machine Learning based prediction of Chronic Lymphocytic Leukemia using protein sequential data," 2022: IEEE, pp. 1-7 %@ 1665491035.
- J. He, X. Pu, M. Li, C. Li, and Y. Guo, "Deep convolutional neural networks for predicting leukemia-related transcription factor binding sites from DNA sequence data," Chemometrics and Intelligent Laboratory Systems, vol. 199, pp. 103976 %@ 0169-7439, 2020. [CrossRef]
- D. Rodríguez et al., "Mutations in CHD2 cause defective association with active chromatin in chronic lymphocytic leukemia," Blood, The Journal of the American Society of Hematology, vol. 126, no. 2, pp. 195-202 %@ 0006-4971, 2015. [CrossRef]
- A. Ashraf, Q. Zhao, W. H. Bangyal, and M. Iqbal, "Analysis of Brain Imaging Data for the Detection of Early Age Autism Spectrum Disorder Using Transfer Learning Approaches for Internet of Things," IEEE Transactions on Consumer Electronics, 2023. [CrossRef]
- R. Apweiler et al., "UniProt: the universal protein knowledgebase," Nucleic acids research, vol. 32, no. suppl_1, pp. D115-D119 %@ 0305-1048, 2004. [CrossRef]
- W. Bangyal, J. Ahmad, and Q. Abbas, "Recognition of off-line isolated handwritten character using counter propagation network," International Journal of Engineering and Technology, vol. 5, no. 2, p. 227, 2013. [CrossRef]
- L. Fu, B. Niu, Z. Zhu, S. Wu, and W. Li, "CD-HIT: accelerated for clustering the next-generation sequencing data," Bioinformatics, vol. 28, no. 23, pp. 3150-3152 %@ 1367-4803, 2012. [CrossRef]
- P.-M. Feng, H. Lin, and W. Chen, "Identification of antioxidants from sequence information using naive Bayes," Computational and mathematical methods in medicine, vol. 2013 %@ 1748-670X, 2013. [CrossRef]
- P.-M. Feng, H. Ding, W. Chen, and H. Lin, "Naive Bayes classifier with feature selection to identify phage virion proteins," Computational and mathematical methods in medicine, vol. 2013 %@ 1748-670X, 2013. [CrossRef]
- J. Jia, Z. Liu, X. Xiao, B. Liu, and K.-C. Chou, "pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach," Journal of theoretical biology, vol. 394, pp. 223-230 %@ 0022-5193, 2016.
- W.-Z. Lin, J.-A. Fang, X. Xiao, and K.-C. Chou, "iDNA-Prot: identification of DNA binding proteins using random forest with grey model," PloS one, vol. 6, no. 9, pp. e24756 %@ 1932-6203, 2011.
- A.M. Ali and M. A. Mohammed, "A Comprehensive Review of Artificial Intelligence Approaches in Omics Data Processing: Evaluating Progress and Challenges," International Journal of Mathematics, Statistics, and Computer Science, vol. 2, pp. 114-167, 2024. [CrossRef]
- Z. H. Arif and K. Cengiz, "Severity Classification for COVID-19 Infections based on Lasso-Logistic Regression Model," International Journal of Mathematics, Statistics, and Computer Science, vol. 1, pp. 25-32, 2022. [CrossRef]
- K. Qu, K. Han, S. Wu, G. Wang, and L. Wei, "Identification of DNA-binding proteins using mixed feature representation methods," Molecules, vol. 22, no. 10, pp. 1602 %@ 1420-3049, 2017. [CrossRef]
- K. V. Khajapeer and R. Baskaran, "Hsp90 inhibitors for the treatment of chronic myeloid leukemia," Leukemia research and treatment, vol. 2015 %@ 2090-3219, 2015.
- R. Alves et al., "Alvespimycin Inhibits Heat Shock Protein 90 and Overcomes Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines," Molecules, vol. 28, no. 3, pp. 1210 %@ 1420-3049, 2023. [CrossRef]
- L. W. Ellisen, "PARP inhibitors in cancer therapy: promise, progress, and puzzles," Cancer cell, vol. 19, no. 2, pp. 165-167 %@ 1535-6108, 2011.
- Y. Liu, H. Song, H. Song, X. Feng, C. Zhou, and Z. Huo, "Targeting autophagy potentiates the anti-tumor effect of PARP inhibitor in pediatric chronic myeloid leukemia," AMB Express, vol. 9, pp. 1-9, 2019. [CrossRef]
- D. Kaloni, S. T. Diepstraten, A. Strasser, and G. L. Kelly, "BCL-2 protein family: Attractive targets for cancer therapy," Apoptosis, vol. 28, no. 1-2, pp. 20-38 %@ 1360-8185, 2023. [CrossRef]
- T. K. Ko, C. T. H. Chuah, J. W. J. Huang, K.-P. Ng, and S. T. Ong, "The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors," Oncotarget, vol. 5, no. 19, p. 9033, 2014. [CrossRef]
- L. Zhou et al., "Post-translational modifications on the retinoblastoma protein," Journal of Biomedical Science, vol. 29, no. 1, pp. 1-16 %@ 1423-0127, 2022. [CrossRef]
- D.-D. Yin et al., "Notch signaling inhibits the growth of the human chronic myeloid leukemia cell line K562," Leukemia research, vol. 33, no. 1, pp. 109-114 %@ 0145-2126, 2009. [CrossRef]
- Y.-D. Cai and K.-C. Chou, "Predicting subcellular localization of proteins in a hybridization space," Bioinformatics, vol. 20, no. 7, pp. 1151-1156 %@ 1367-4811, 2004. [CrossRef]
- K.-C. Chou, "Impacts of bioinformatics to medicinal chemistry," Medicinal chemistry, vol. 11, no. 3, pp. 218-234 %@ 1573-4064, 2015. [CrossRef]
- K. C. Chou, "Prediction of protein cellular attributes using pseudo-amino acid composition," Proteins: Structure, Function, and Bioinformatics, vol. 43, no. 3, pp. 246-255 %@ 0887-3585, 2001. [CrossRef]
- Y. D. Khan, F. Ahmad, and M. W. Anwar, "A neuro-cognitive approach for iris recognition using back propagation," World Applied Sciences Journal, vol. 16, no. 5, pp. 678-685 %@ 1818-4952, 2012.
- A.S. o. C. O. (ASCO). "Genes and Cancer." Cancer.net. https://www.cancer.net/navigating-cancer-care/cancer-basics/genetics/genes-and-cancer (accessed 11, 2023).
- A.H. Butt, S. A. Khan, H. Jamil, N. Rasool, and Y. D. Khan, "A prediction model for membrane proteins using moments based features," BioMed research international, vol. 2016 %@ 2314-6133, 2016. [CrossRef]
- Y. D. Khan, F. Ahmed, and S. A. Khan, "Situation recognition using image moments and recurrent neural networks," Neural Computing and Applications, vol. 24, pp. 1519-1529 %@ 0941-0643, 2014. [CrossRef]
- G. Hu, Y. Zheng, L. Abualigah, and A. G. Hussien, "DETDO: An adaptive hybrid dandelion optimizer for engineering optimization," Advanced Engineering Informatics, vol. 57, p. 102004, 2023. [CrossRef]
- L. Abualigah, S. Ekinci, D. Izci, and R. A. Zitar, "Modified elite opposition-based artificial hummingbird algorithm for designing FOPID controlled cruise control system," Intelligent Automation & Soft Computing, 2023. [CrossRef]
- A.H. Butt, N. Rasool, and Y. D. Khan, "A treatise to computational approaches towards prediction of membrane protein and its subtypes," The Journal of membrane biology, vol. 250, pp. 55-76 %@ 0022-2631, 2017. [CrossRef]
- W. Bangyal, J. Ahmad, and Q. Abbas, "Analysis of learning rate using CPN algorithm for hand written character recognition application," International Journal of Engineering and Technology, vol. 5, no. 2, p. 187, 2013. [CrossRef]
- Y. D. Khan, S. A. Khan, F. Ahmad, and S. Islam, "Iris recognition using image moments and k-means algorithm," The Scientific World Journal, vol. 2014 %@ 2356-6140, 2014. [CrossRef]
- M. Sugiyama, Introduction to statistical machine learning. Morgan Kaufmann, 2015.
- S. Theodoridis, Machine learning: a Bayesian and optimization perspective. Academic press, 2015.
- V. Vapnik, The nature of statistical learning theory. Springer science & business media, 1999.
- P. E. Hart, D. G. Stork, and R. O. Duda, Pattern classification. Wiley Hoboken, 2000.
- W. H. Bangyal et al., "Detection of fake news text classification on COVID-19 using deep learning approaches," Computational and mathematical methods in medicine, vol. 2021, pp. 1-14, 2021. [CrossRef]
- O.A. Montesinos López, A. Montesinos López, and J. Crossa, Multivariate statistical machine learning methods for genomic prediction. Springer Nature, 2022.
- Y. Jiao and P. Du, "Performance measures in evaluating machine learning based bioinformatics predictors for classifications," Quantitative Biology, vol. 4, pp. 320-330 %@ 2095-4689, 2016. [CrossRef]
- T. Fawcett, "ROC graphs: Notes and practical considerations for researchers," Machine learning, vol. 31, no. 1, pp. 1-38, 2004.
Figure 1.
Various stages of chronic Myeloid leukemia classification.
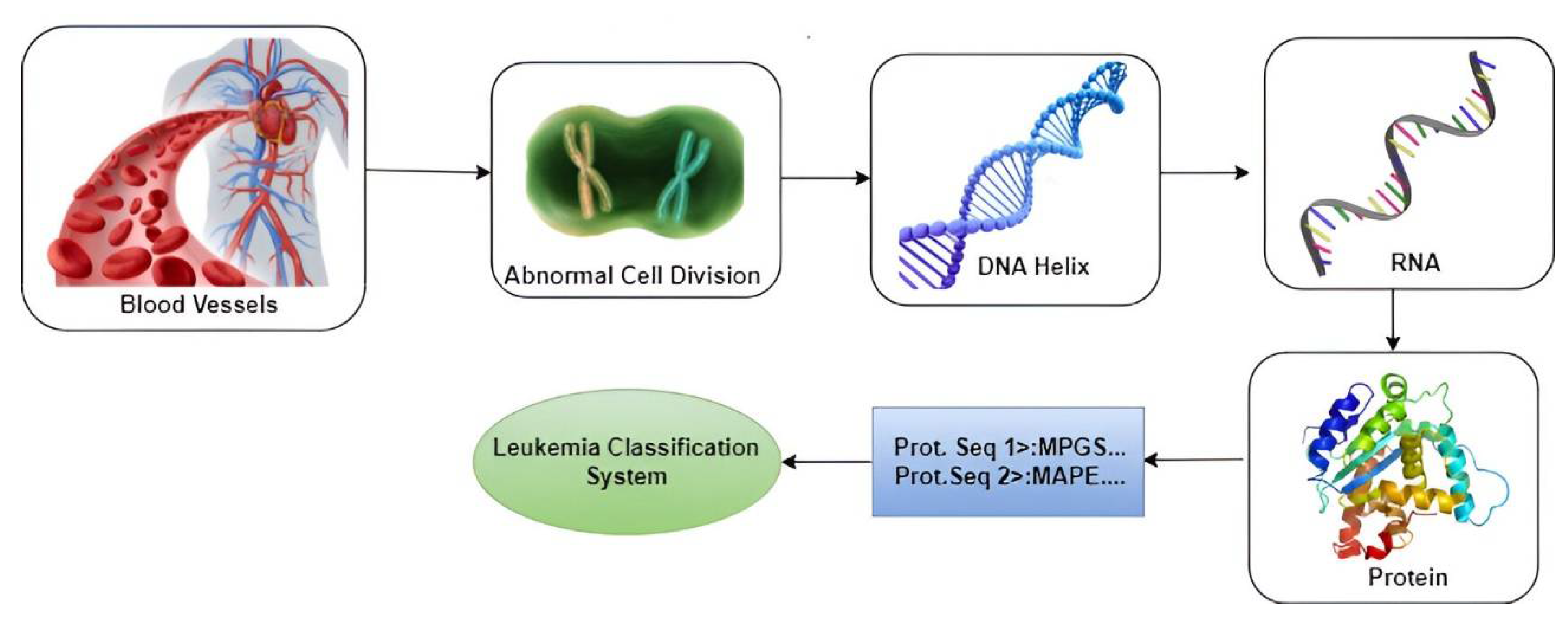
Figure 2.
Block Diagram of Designed System.
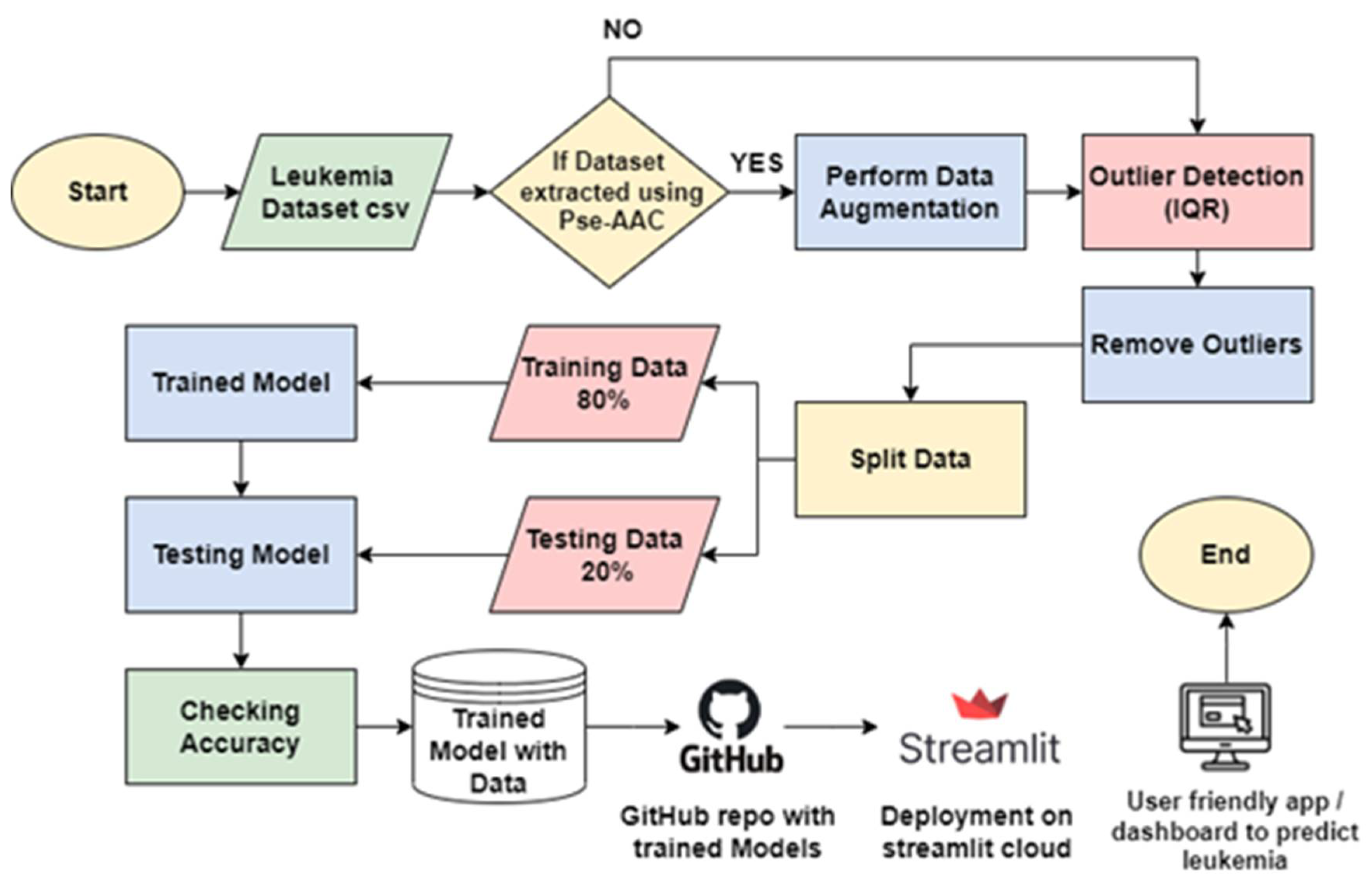
Figure 3.
Gene Sample.


Table 1.
Results on Pseudo Amino Acid Composition (Pse-AAC) Data.
| Name of Algorithm | Accuracy | F1-Score | Recall | Specificity |
|---|---|---|---|---|
| Support Vector Classifier | 92~94% | 91~92% | 91~93% | 92~94% |
| Extreme Gradient Boost | 79~85% | 63~70% | 51~55% | 92~94% |
| Logistic Regression | 66~69% | 10~20% | 6~10% | 97~98% |
| Decision Tree | 81~84% | 73~76% | 74~76% | 84~86% |
| Random Forest | 87~91% | 85~87% | 80~83% | 96~97% |
| K Nearest Neighbor | 82~86% | 72~74% | 61~64% | 93~95% |
Table 2.
Confusion Matrix.
| Name of Algorithm | Confusion Matrix | |
|---|---|---|
| Support Vector Classifier | True Negative = 424 False Negative = 14 |
False Positive = 28 True Positive = 211 |
| Extreme Gradient Boost | True Negative = 26159 False Negative = 3435 |
False Positive = 2271 True Positive = 10890 |
| Logistic Regression | True Negative = 25817 False Negative = 11010 |
False Positive = 2849 True Positive = 3445 |
| Decision Tree | True Negative = 24388 False Negative = 3803 |
False Positive = 4278 True Positive = 10652 |
| Random Forest | True Negative = 28014 False Negative = 2753 |
False Positive = 808 True Positive = 11546 |
| K Nearest Neighbor | True Negative = 419 False Negative = 95 |
False Positive = 23 True Positive = 140 |
Table 3.
Result on Amino Acid Composition (AAC) Data.
| Name of Algorithm | Accuracy | F1-Score | Recall | Specificity |
|---|---|---|---|---|
| Support Vector Classifier | 54.95% | 14.3% | 0.7% | 100% |
| Extreme Gradient Boost | 56.8% | 52.9% | 45.9% | 69% |
| Logistic Regression | 51.1% | 27.6% | 19.1% | 81.7% |
| Decision Tree | 54.4% | 52.25% | 52.9% | 55.8% |
| Random Forest | 50.6% | 41.1% | 35.4% | 64.9% |
| K Nearest Neighbor | 54.2% | 54.8% | 57% | 51% |
Table 4.
Confusion Matrix.
| Name of Algorithm | Confusion Matrix | |
|---|---|---|
| Support Vector Classifier | True Negative = 271 False Negative = 121 |
False Positive = 0 True Positive = 62 |
| Extreme Gradient Boost | True Negative = 409 False Negative = 119 |
False Positive = 23 True Positive = 103 |
| Logistic Regression | True Negative = 9028 False Negative = 8519 |
False Positive = 2022 True Positive = 2025 |
| Decision Tree | True Negative = 124 False Negative = 95 |
False Positive = 98 True Positive = 107 |
| Random Forest | True Negative = 12612 False Negative = 11832 |
False Positive = 6817 True Positive = 6510 |
| K Nearest Neighbor | True Negative = 112 False Negative = 89 |
False Positive = 105 True Positive = 118 |
Table 5.
Results on Pseudo Amino Acid Composition (Pse-AAC) Data.
| Name of Algorithm | Accuracy | F1-Score | Recall | Specificity |
|---|---|---|---|---|
| Support Vector Classifier | 92~94% | 87~88% | 91~93% | 90~93% |
| Extreme Gradient Boost | 79~84% | 66~68% | 55~57% | 92~94% |
| Logistic Regression | 66~69% | 0~0% | 6~10% | 100% |
| Decision Tree | 81~84% | 70~73% | 56~59% | 96~97% |
| Random Forest | 82~84% | 67~68% | 57~58% | 94~95% |
| K Nearest Neighbor | 72~73% | 31~32% | 20~21% | 95~97% |
Table 6.
Confusion Matrix.
| Name of Algorithm | Confusion Matrix | |
|---|---|---|
| Support Vector Classifier | True Negative = 416 False Negative = 17 |
False Positive = 37 True Positive = 207 |
| Extreme Gradient Boost | True Negative = 413 False Negative = 105 |
False Positive = 25 True Positive = 134 |
| Logistic Regression | True Negative = 453 False Negative = 224 |
False Positive = 0 True Positive = 0 |
| Decision Tree | True Negative = 433 False Negative = 54 |
False Positive = 16 True Positive = 134 |
| Random Forest | True Negative = 437 False Negative = 93 |
False Positive = 23 True Positive = 124 |
| K Nearest Neighbor | True Negative = 438 False Negative = 179 |
False Positive = 15 True Positive = 45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



