
Submitted:
30 November 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
The present review revisits representative studies addressed to develop efficient carbon-based heterogeneous catalysts for two important reactions, namely, the production of hydrogen from formic acid and the hydrogenation of carbon dioxide to formic acid. The HCOOH-CO2 system is considered a promising couple for a hydrogen storage system that would be involved in an ideal carbon-neutral cycle. Significant advancements have been achieved in those catalysts designed to catalyze the dehydrogenation of formic acid under mild reaction conditions, while much effort is still needed to catalyze the challenging CO2 hydrogenation reaction. The design of carbon-based heterogeneous catalysts for these reactions encompasses both the modulation of the properties of the active phase (particle size, composition, and electronic properties), as well as the modification of the supports by means of incorporation of nitrogen functional groups. These approaches are herein summarized to provide a compilation of the strategies followed in recent studies to set the basis for a hydrogen storage system attained by the HCOOH-CO2 couple.
Keywords:
Hydrogen storage
; formic acid
; CO2
; carbon materials
; heterogeneous catalysts
1. Introduction
It is well-known that fossil fuels are the main energy supply since the Industrial Revolution, and their use has led to advances in both human life and industry. However, fossil fuel depletion, their massive use due to the increasing energy demand that comes together with increasing world population and increasing prosperity, and the environmental problems associated with their use, are a global concern.
Looking back over the last two centuries, we can see how the problem has been exacerbated, mainly since the middle of the 20th century. This was the period in which globalization and consumerism began to be coined. As a result of these and due to uncontrolled population growth and the overexploitation of natural resources, an urgent change in global energy planning addressed to the decarbonization of the energy system is now mandatory [1,2]. It is generally acknowledged that the most straightforward way to achieve a decarbonized energy system lies in the utilization of renewable energy, such as solar, wind, geothermal or hydro energy. However, several technological, economic, and social issues arise when these renewable energies are thought to be used on a large scale at a reasonable cost [3].
The use of hydrogen is an auspicious option for the energy system. As for today, hydrogen is mainly produced from steam methane reforming and coal gasification, which also deliver large amounts of CO2 (for instance, steam methane reforming emits up to 8 tons of CO2 per ton of H2 produced) [4]. Also, some concerns related to hydrogen storage and transportation limit somehow the potential of hydrogen in the energy scenario. Thus, finding alternative options for the three-fold problem that limits the great potential of hydrogen (production, hydrogen, and storage) has become the focus of numerous recent investigations. Among the possible options, the use of Liquid Organic Hydrogen Carriers (LOHCs) stands out as a very interesting option that is safer, more practical and more cost-effective than the conventional technologies used for the storage and transport of hydrogen in the form of compressed hydrogen at either high pressure or low temperature [5,6,7].
Among the possible LOHCs, Formic acid (FA, HCOOH) has emerged as an ideal candidate, which is not only due to its inherent properties, such as low toxicity and high stability but also to the fact that it can be sustainably produced from biomass and biomass-derived products [8]. Another interesting pathway addressed for the production of FA is from the hydrogenation of CO2, which is particularly appealing since it would imply an ideal carbon-neutral cycle encompassing the following reaction [9].
HCOOH (l) ⇄ H2(g) + CO2 (g), ΔG298K= -32.9 kJ·mol-1
Numerous studies have already reported that the dehydrogenation of FA (1) can be achieved under moderate reaction conditions upon the selection of a proper catalyst [10,11,12,13,14,15], while the reduction of CO2 to FA would be more challenging.
Nevertheless, the decomposition of FA does not follow a unique pathway, but a second reaction (2) (dehydration reaction) may occur if no selective catalysts are used [16].
HCOOH → H2O + CO, ΔG298K= -20.7 kJ·mol-1
Due to the toxicity of carbon monoxide, and the reduction in hydrogen production caused by dehydration side reaction, the process needs to be optimized to shift the selectivity of FA towards the dehydrogenation step. The primary key to this lies in designing suitable catalysts. In literature, the highest conversions are obtained for homogeneous catalysis. However, such systems are less desirable from a practical application standpoint since the reported catalysts are large metallic complexes of noble metals such as ruthenium or iridium, with low stability and high cost [17,18]. For these reasons, much effort is being devoted to designing active and selective heterogeneous catalysts. Among those compositions explored, the use of carbon-based catalysts has aroused much interest due to their great properties as catalytic supports and the versatility of these materials [19].
However, the major challenge of this storage system is not only to obtain an active and selective catalyst for dehydrogenation but also to optimize both the catalysis of formic acid dehydrogenation and the reverse reaction attaining an ideal carbon-neutral circular system, which is schematized in Figure 1 [20].
Due to the relevance of the topic, this review presents the state-of-the-art in heterogeneous catalysis for FA decomposition and carbon dioxide reduction to FA, paying particular attention to the aspects considered towards the optimization of the catalysts.
2. Dehydrogenation of formic acid
As mentioned in the Introduction section, the appropriate pathway for the decomposition of FA towards the H2 production is the dehydrogenation reaction (1), whereas, the dehydration route (2), should be avoided due to the toxicity and poisoning effect of CO. Since the energy difference between the two decomposition pathways is very low, suitable catalysts with a high selectivity for the dehydrogenation reaction are required. Several factors influence the yield of each reaction pathway such as temperature, substrate concentration, and the presence of additives. However, it is possible to achieve interesting catalytic performances by employing a suitable catalyst [21] and adjusting the reaction conditions [22].
Traditionally, the dehydrogenation of formic acid has been studied using homogeneous catalysts. In this context, the studies performed by Beller [23,24,25] on the design of homogeneous catalysts based on transition metal complexes stand out achieving a high catalytic activity. Despite their high catalytic activity, homogeneous catalysts are problematic in terms of their recovery and applicability, and hence numerous efforts have been made to develop selective heterogeneous systems for the production of H2 from FA under mild reaction conditions.
Within the heterogeneous systems, catalysts based on supported metal nanoparticles (NPs) have garnered great and growing interest. In particular, metal NPs can significantly improve the efficiency of hydrogen production due to the increase of accessible active sites [26,27]. Among all the catalysts studied for this application, palladium-based ones have been hailed as highly promising alternatives. They have deserved significant attention, not only due to their superior CO tolerance compared to other metals but also for their ability to achieve relatively high hydrogen conversion and selectivity values at moderate temperatures [28]. Thus, numerous studies have been carried out to understand and optimize Pd-based catalytic systems considering different features, such as the size of the metal NPs, metal loading, composition, electronic properties, structure and support [29].
The Pd-support interaction has been extensively studied in these systems using porous support materials such as silica [30,31,32], metal-organic frameworks [33,34], zeolites [10,35], etc. Among the different supports, carbon materials stand out for their features such as their resistance to basic and acidic media, their modifiable porosity and the possibility of incorporating heteroatoms in their structure, which is of special interest in FA dehydrogenation. Therefore, the key strategies employed in the development of high-performance carbon-supported Pd catalysts for the dehydrogenation of FA, will be reviewed.
2.1. Carbon-supported monometallic Pd catalysts.
The study of the different properties of the active phase of the Pd-based catalysts and how they affect the catalytic activity is the starting point to achieve the development of catalysts with high performance and stability.
Among the properties of catalysts, particle size is a key parameter that can be modified by the preparation method. Li et al. conducted a study to investigate the catalytic activity and FA conversion of Pd/C catalysts with varying Pd particle sizes (ranging from 2.1 to 4.5 nm) [36]. They prepared these catalysts with controlled particle sizes using a wet impregnation method, employing NaBH4 for the reduction of the metal precursor and using sodium citrate as a stabilizing agent. The particle size variation was achieved by adjusting the reduction temperature and the sodium citrate to PdCl2 ratio. Their X-ray photoelectron spectroscopy (XPS) analysis revealed that as the particle size increased, the proportion of oxidized Pd (Pd2+ and Pd4+) species decreased, while the proportion of Pd0 increased. The catalytic activities of these Pd/C catalysts were evaluated in FA dehydrogenation in the liquid phase, and the smaller particle size catalyst (2.1 nm) exhibited the best performance with a turnover frequency (TOF) of 835 h-1. The enhanced catalytic activity of smaller Pd NPs was attributed to their higher dispersion and a larger proportion of positively charged Pd species, leading to increased coulombic interaction with formate ions.
The composition of the surface of metal nanoparticles is commonly recognized to consist of distinct site types, including high-coordination terrace atoms and low-coordination atoms found at edges and corners. In the case of Pd NPs, a cuboctahedron-shaped particle model with a cubic close-packed structure is often employed to understand the role of the different sites in the catalytic reaction [37]. The results of the values of the TOFs calculated for the different sites (low and high coordinated surface atoms) and also for the total of the surface atoms show that all the surface sites act as active sites and participate in the FA decomposition reaction. That was the case of the study reported by Navlani-García et al., who precisely controlled the size of the Pd nanoparticles by synthesizing PVP-capped colloidal NPs by the polyol method [38,39]. Pd NPs between 2.7 - 5.5 nm were obtained and the catalytic ability of the Pd/C catalysts in the FA decomposition in the liquid phase was evaluated by monitoring the H2 production in a closed liquid-phase system during 3 h at 30 ºC. The results of the catalytic activity for the samples with different average diameters of the Pd NPS are depicted in Figure 2(a), showing a volcano-like relationship between particle diameter and H2 production. To further analyze the structure-activity relationship in H2 production from formic acid, calculations were performed for the catalysts with different particle sizes assuming the particles as regular cuboctahedral in shape with a cubic close-packed structure and adopting the model of full-shell NPs. The TOF values calculated on the basis of the surface decreased in the order of Pd/C(3) (3.9 nm) > Pd/C(5) (5.5 nm) > Pd/C(2) (3.6 nm) > Pd/C(4) (4.2 nm) > Pd/C(1) (2.7 nm). This observation implies that not all Pd surface sites exhibit identical catalytic activity in FA dehydrogenation. As illustrated in Figure 2(b), the TOF on low-coordinated atoms demonstrates a significant dependence on NPs size. In contrast, when focusing solely on the highly coordinated terrace atoms as catalytically active sites, the normalized TOF was found to be independent.
It should be noted that the catalysts of these two studies [36,39] were synthesized following different protocols and also different experimental conditions were used to assess the catalytic performance. So, even though smaller nanoparticles are usually preferred in catalysis, different conclusions could be drawn under certain conditions.
The incorporation of heteroatoms in the carbon supports has been another widely investigated aspect for the design of the catalysts used in the dehydrogenation of FA.
In particular, the incorporation of nitrogen functional groups in different types of carbon materials (carbon xerogels [40], activated carbon [41,42], hierarchically porous carbon [43] mesoporous carbon [44,45]), etc.) and by following different approaches addressed to either grafting or doping the carbon material, has attracted great interest in the last years [46,47,48]. The resulting N-containing materials display improved catalytic performance in the dehydrogenation of FA compared to the N-free counterpart catalysts, which is usually attributed to electronic and acid-base properties, and also to the anchoring ability of N functional groups.
Our research group developed N-doped carbon-supported catalysts prepared from a lignocellulosic biomass residue (hemp residue) [41]. In that study, the nitrogen incorporation was carried out through an organic reaction and the surface nitrogen content was close to 2 at.%. Not only the role of the nitrogen functional groups was analyzed, but also the effect of the preparation of the catalysts was considered in that study. The results of the catalytic activity registered at 75 ºC indicated that the developed materials displayed excellent catalytic performance, with great stability at least during 6 consecutive reaction cycles and TOF of 8365 h-1, expressed per surface Pd mol.
Yao et al. prepared Pd-based catalysts supported on N-doped activated carbon (denoted as HTNC) after a high-temperature amination process [49]. The amination process was carried out by heating the activated carbon at different temperatures (from 650 to 1050 °C) in a NH3 flow. XPS spectra of N 1s show different functional groups for all the catalysts (i.e. pyridinic N, pyrrole N, and graphitic N). The highest content of total nitrogen was observed in Pd/HTNC-950. The Pd 3d XPS spectra showed the presence of Pd0 and Pd2+ and it was seen that the content in Pd2+ increased with the temperature used in the amination, which confirmed the interaction between Pd and nitrogen-doped activated carbon (AC). The catalytic performance of the as-synthesized catalysts was tested at 30 ºC in a FA/sodium formate (SF) 1:1 solution. The catalysts had remarkably different activities, and the best activity was obtained by Pd/HTNC-950 with a TOF of 1631 h-1. These results confirmed that the presence of N species in the ACs was essential to enhance the catalytic activity in FA dehydrogenation. It was claimed that the beneficial effect of N groups was due to both the electronic properties of Pd species and the high dispersion of Pd NPs achieved in the catalysts. On the one hand, it was observed that N species could modify the electron state of Pd and stabilize Pd2+ species, which interact with formate ions, key intermediates in FA dehydrogenation. Conversely, the HTNC effectively immobilized free Pd ions, which led to the creation of highly dispersed Pd nanoparticles. This process increased the prevalence of lower coordination sites in smaller particles, a factor that can be associated with improved performance in the reaction. [50]. The stability of the catalysts was measured by 5 reaction cycles. Even after the fifth run, the catalytic activity of the Pd/HTNC-950 catalyst remained consistently high, underscoring the robustness of this catalyst.
Jeon et al. developed a method to dope the AC with N groups that involved the pyrolysis of dicyandiamide using commercial Ketjen black [51]. The catalysts were prepared by a wet impregnation method with reduction assisted by NaBH4. The results obtained by TEM showed that the Pd NPs were well dispersed on the N-C support with an average size of approximately 1-2 nm. The N 1s XPS spectra of Pd/N-C displayed the presence of C-N bonds (C=N+-C and C=N), attributed to the incorporation of nitrogen atoms into the carbon structure The Pd 3d XPS demonstrated that the Pd electron density of Pd/N-C slightly increased by the electrons transferred from N to Pd species through the interaction between the metal and support. The study of the catalytic activity of these catalysts was performed at 45 ºC and Pd/N-C exhibited a better performance than Pd/C obtaining a full conversion in 100 min. Due to the excellent catalytic activity described above, it was developed a fuel cell powering system connected to the FA-based H2 generation reactor using Pd/N-C catalysts. For performing a 200 W PEMFC stack, 9.5 mL min-1 of FA was continuously supplied to the catalytic reactor. The stack received a maximum current load of 8 A, accompanied by a lower cell voltage of around 25 V. This configuration yielded a power generation of 200 W through the utilization of fuel derived from FA dehydrogenation. To establish the durability and applicability of the integrated H2 fuel cell stacks and the FA-based H2 generator system employing Pd/N-C, long-term chronopotentiometric measurements were implemented. A hydrogen production rate of approximately 3 L min-1 was achieved in galvanostatic conditions at a current of 6.45 A. The fuel cell demonstrated consistent performance for a duration of 80 minutes, with no discernible decline in stack voltage. This suggested the absence of a loss in catalytic activity for hydrogen production and indicated that there was no poisoning of the fuel cell anode catalysts, potentially caused by the formation of CO during the dehydrogenation of FA.
Chet et al. reported an efficient procedure to obtain ultrafine Pd NPs supported on activated N-doped carbon [52]. The preparation procedure of nitrogen-doped activated carbons (NHPC-AC) was based on the mixing of activated carbon, NaHCO3 and NH4HCO3 (mass ratio 1:3.3) and then calcination at 700 ºC for 1 h in N2 flow. TEM images for the Pd/NHPC-AC catalyst indicated a good dispersion of the NPs and an average size of 1.88 nm, being smaller than for Pd/HPC-AC (2.56 nm). XPS analysis revealed the presence of pyridinic, pyrrolic, and graphitic nitrogen. The catalytic activity measured at 60 ºC with a FA:SF molar ratio of 1/1 demonstrated better behavior for Pd/HPC-NAC than for Pd/HPC-AC, reaching TOF values of 4115 h-1 at 60 ºC and 1910 h-1 at 30 ºC. The influence of SF was examined by varying the molar ratio of FA/SF. As the FA/SF molar ratio decreased, there was a significant enhancement in catalytic activity, reaching a plateau when the molar ratio fell below 1:1. Moreover, even after undergoing recycling for five cycles, Pd/NHPC-AC displayed sustained high catalytic activity, and the average size of the Pd nanoparticles NPs remained small.
Shao et al. recently investigated a method to prepare amine-functionalized hierarchically porous carbon (TPC-NH2) as a support for Pd NPs which obtained excellent results. The desired architecture of the catalysts was achieved by choosing MgO as a template and sodium hypophosphite to widen the pores achieving the hierarchically porous carbon (TPC). Then, the TPCs were functionalized with amino groups and the catalysts were prepared by a wet impregnation method with NaBH4 reduction. With this method, ultrafine Pd NPs with an average size of 2.1 nm were obtained for Pd/NH2-TPC. EDS elemental mapping analysis showed a uniform distribution of the elements C, N, O, Pd and Si. The catalytic performance of Pd/NH2-TPC at 25ºC using a 1 M FA solution without additives, revealed a high activity reaching a TOF value of 4312 h-1 and the increase of temperature to 60 ºC achieved a TOF of 12864 h-1 (see Figure 3). These results, compared with the rest of the catalysts reported for this application, using a pure FA solution without additives, show the best TOF values. The excellent catalytic activity results obtained were related to an increase of the reaction rate due to the enhancement of the mass transfer promoted by the hierarchical porous structure.
The positive effect of the presence of nitrogen functional groups has not only been observed when the reaction is performed in the liquid phase, but interesting results have also been achieved for the decomposition of FA in the gas phase. That was the case of Bulushev et al. who reported a study where the preparation of single Pd2+ cations supported on N-doped carbon (Pd/N-CM) was achieved [54]. The presence of these single sites was confirmed through aberration-corrected scanning transmission electron microscopy (ac STEM). The results of the catalytic activity measured in the gas phase for Pd/N-CM at 100 ºC showed a TOF value of 0.121 s-1. The results of X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure (NEXAFS) studies after ex-situ reduction in hydrogen at 300 ºC, showed that the Pd species exist in a Pd2+ state coordinated by nitrogen species within the support. Additionally, extended density functional theory (DFT) calculations validated the critical role of an isolated Pd atom as the active site for FA decomposition, resulting in the production of an adsorbed hydrogen atom and a carboxyl fragment. However, this activity was only viable when the Pd atoms were coordinated by a pair of pyridinic-type nitrogen atoms located at the open edge of the graphene sheet. Thus, the nitrogen doping of the carbon support plays a pivotal role in creating and stabilizing these new active Pd sites. Bulushev also reported the synthesis of catalysts based on Pd active sites on covalent triazine frameworks [55]. Pd catalysts were prepared by impregnation with the supports: 5,6,11,12,17,18-hexaazatrinaphthylene-2,8,14-tricarbonitrile (hatnCTF), 4,4′-malonyldibenzonitrile (acacCTF) and g-C3N4. The surface N/C ratio measured by XPS was much lower in Pd/acacCTF compared to Pd/hatnCTF. Similarly, the surface oxygen content is 3 times higher in Pd/acacCTF than for Pd/hatnCTF. The surface concentration ratio of pyridinic nitrogen to the overall nitrogen surface concentration was approximately 40% in both samples. However, in the Pd/g-C3N4 sample, nitrogen is predominantly found in the pyridinic configuration. By means of HRTEM and HAADF/STEM the formation of nanoparticles was observed in Pd/acacCTF and Pd/g-C3N4 with an average size of 5.2 and 3.0 nm, respectively, while for Pd/hatnCTF only single atoms were observed and for Pd/acCTF the presence of these could also be appreciated. Nevertheless, Pd/g-C3N4 only presented Pd in the form of NPs. The Pd/hatnCTF sample presented a higher proportion of Pd2+ sites than the rest of the samples, which was confirmed by both XPS and XANES. After treatment of the spectra obtained by EXAFS it was shown that the single atoms in Pd/hatnCTF were found as Pd2+-C1N3 sites and the Pd/acacCTF sample contained single-atom Pd2+-O4 sites. The presence of a significant proportion of metal NPs in Pd/acacCTF was also confirmed. The catalytic activity measured in gas phase showed the best performance for Pd/hatnCTF with a TOF of 0.28 s-1 at 180 °C. Additionally, a stability assessment was conducted for Pd/hatnCTF that showed no deactivation at 180 °C, and there was even a slight increase in FA conversion within the initial 5 hours of the test. Those results could be attributed to the presence of single-atom Pd sites (Pd-C1N3) in Pd/hatnCTF, while in Pd/acCTF the Pd single-atom Pd-O4 sites are not active for the reaction; thus, the low activity observed for this catalyst is only due to the presence of NPs.
2.2. Carbon-supported multimetallic Pd-based catalysts.
Exceptional outcomes have been attained with Pd-based bimetallic catalysts designed as alloys or core-shell structures. In these configurations, the electronic promotion of Pd species occurs through charge transfer from the other component. The favorable effects of the construction of multimetallic catalysts extend to mitigating deactivation caused by poisonous intermediates such as CO. Additionally, these catalysts weaken hydrogen adsorption on the surface of the NPs, thereby promoting the favored combination of two hydrogen atoms to form an H2 molecule [56].
Specifically, extensive research has been conducted on systems incorporating Pd in conjunction with other noble metals. Pd-Ag-based catalysts, in particular, have remarkable performance. This can be attributed to the electron-rich Pd species resulting from electron transfer from Ag to Pd by the difference in electronegativity (2.2 and 1.9 in the Pauling scale, for Pd and Ag, respectively) [57].
Navlani-García et al. studied colloidal bimetallic PdAg NPs that were prepared by using the polyol method and supported in a commercial activated carbon [58]. The study was performed with different Pd/Ag ratios, as well as different PVP/metal ratios. TEM micrographs showed how the addition of Ag in small proportions (Pd1Ag0.5, Pd1Ag1, Pd1Ag2) reduced the particle size compared to monometallic catalysts. The catalytic performance of PdAg/C catalysts in the FA dehydrogenation reaction was evaluated by monitoring the H2 produced during 3 h at 30 ºC. The high influence of the Pd/Ag ratio as well as the PVP/metal ratio could be appreciated, being the catalyst with the best activity Pd1Ag2 with PVP/M=1. EXAFS measurements for Pd1Ag2/C confirmed the heteroatomic Pd-Ag bonding in the studied catalysts. The analysis of XPS spectra indicated that Pd and Ag were predominantly present in metallic form, but a minor presence of oxidized forms (Pd2+ and Ag+) was observed for both elements. The presence of oxidized species was assigned to the electron-withdrawing property of PVP through the C=O groups, resulting in electron-deficient metal surfaces.
Recently, Kim et al. investigated different Pd:Ag compositions for elucidating the alloying effect on PdAg NPs supported on carbon nanotubes (CNT) catalysts [59]. The catalysts were prepared by a deposition-precipitation method using CNT as support. The total content of Pd and Ag was fixed at 5 wt.% while varying their molar ratios. The Pd binding energy obtained by XPS showed the highest shift to lower energy for the catalyst with an Ag/Pd ratio of 3/7, which can be attributed to increased interaction between the metals. The initial TOF values showed a volcano-type curve according to the Ag/Pd ratio, the Pd7Ag3/CNT catalyst presented the highest value. In this sense, kinetic isotopic effect (KIE) experiments were carried out to investigate the alloy effect. The KIE results showed lower values with increasing Ag content, reaching a minimum where it remains constant for Pd/Ag molar ratios of 7:3. This lower KIE (kh/kd) value can be associated with a weaker C-H bond, which facilitates the breaking of this bond of the adsorbed HCOO-, thereby easing the course overall reaction. In that study, the alloying effect of Ag on Pd was explained as an electronic modification of Pd, which excited the molecular orbital of the adsorbed HCOO− to have a weaker C–H bond. Consequently, this facilitated the cleavage of the C–H bond in HCOO−*, leading to an increased activity in the dehydrogenation of FA over the PdAg/CNT catalysts.
The positive effect of the incorporation of nitrogen functional groups has also been evidenced for bimetallic PdAg catalysts. For instance, Nabid et al. reported on the utilization of Ag-core Pd-shell NPs supported on nitrogen-doped graphene carbon nanotube aerogel (Ag@Pd/N-GCNT) [60]. In that investigation, graphene and CNTs were incorporated into a N-doped hybrid aerogel. That aerogel was then sequentially loaded with Ag and Pd in order to achieve controlled composition of the core-shell NPs. The findings revealed that, beyond the nitrogen sites, the graphene carbon nanotube aerogel provided additional stabilization points for the NPs, preventing their aggregation. Catalysts with an Ag/Pd molar ratio of 1/1 showcased the highest activity among the examined compositions, achieving a TOF value of 413 h-1 at 25 ºC.
In our research group, catalysts based on Pd and PdAg NPs were designed from activated carbons prepared from a biomass residue (almond shell) and doped with nitrogen groups [61]. Two sets of catalysts were prepared, some prepared by a wet impregnation method with reduction by NaBH4 and others in which the reduction step with NaBH4 was omitted and the metals were expected to be reduced in-situ during the reaction. The results of the catalytic activity measured at 75 °C showed significant differences between the pre-reduced catalysts and those that were not. The reduced PdAg catalysts showed better activity than the monometallic counterparts. In the non-pre-reduced bimetallic catalysts, an induction period was observed at the beginning of the gas-time curve. PdAg/N-AS showed the best performance achieving an initial TOF of 1577 h−1 and the good stability of the catalyst was demonstrated after 6 consecutive reaction cycles.
Furthermore, for Pd bimetallic catalysts, other noble metals such as Au have been used to form PdAu alloys for the dehydrogenation of FA. Jiang et al. reported a study in which AuPd NPs supported on N-doped carbon nanosheets (n-CNS) were assessed [62]. The synthesis procedure is described in Figure 4 and it consists first of a hydrothermal treatment of a solution of graphitic carbon nitride (g-C3N4) and glucose followed by calcination, using different temperatures. AuPd catalysts were prepared by wet impregnation with a reduction step. The images obtained by HAADF-STEM and STEM-EDX confirmed the homogeneous distribution of N and it was also found that the signal for Au an Pd are at the same position, which strongly confirms de existence of AuPd alloy. The highest activity was obtained for AuPd/n-CNS-Th-160 (hydrothermal treatment at 160 ºC) which achieved a TOF of 459 h-1 at 25 ºC and a record value of 1896 h-1 at 60 ºC with 100% selectivity to CO2 and H2. Furthermore, the study demonstrated that the catalytic activity did not exhibit a direct correlation with the mass of nitrogen dopant. Instead, the nitrogen bonding configurations played a crucial role, and a higher ratio of graphitic N to pyridinic N was shown to modify the electron structure distribution of the metal and enhance the interaction between AuPd metal nanoparticles and the support.
Hong et al. developed a simple method for the immobilization of PdAu NPs on commercial carbons that resulted in excellent catalytic results [63]. The method consisted of wet mixing of the metal precursors, using L-arginine (LA) as mediating reagent, with MSC-30 and Vulcan XC-72R and a reduction step. The catalyst formed by PdAu NPs supported on MSC-30 was denoted as Pd1Au1/30-LA and the one supported on Vulcan XC-72R was denoted as Pd1Au1/72-LA. Severe differences in the catalysts were observed. Firstly, the mean particle size obtained by TEM was smaller for Pd1Au1/72-LA suggesting the importance of the metal-support interaction. Pd1Au1/30-LA showed exceptional performance for the dehydrogenation of FA using a pure FA solution at 60 ºC reaching a TOF value of 8355 h-1. Moreover, the stability of that catalyst was studied after 5 consecutive reaction cycles which revealed its excellent durability. However, when the reaction was studied using a 1:3 molar FA/SF solution the catalyst with the best activity was Pd1Au1/72-LA obtaining a TOF of 11958 h-1 at 60 ºC, and also the stability test proved its high durability. In addition, LA was found to play an important role in modulating the size and dispersion of NPs.
Noble metal-based architectures are usually preferred for their reported catalytic properties. However, the scarcity and high cost of these metals have motivated the search for alternative non-noble metals like transition metals such as Co, Ni, Cu, etc.
Navlani-García et al. studied PdCo-based bimetallic systems with different Pd/Co ratios supported on (g-C3N4) [64]. The Pd-based catalysts were prepared by the standard impregnation method, mixing g-C3N4 with the appropriate amount of Na2PdCl4 and Co(NO3)2 to prepare catalysts with several Pd/Co molar ratios (Pd/Co = 1/0.1, 1/0.25, 1/0.7, 1/1 and 1.15). The catalytic activity evaluated at 75 ºC for the different compositions of the NPs showed that the gradual addition of Co resulted in outperforming catalysts until the optimal composition was reached in the sample PdCo/ g-C3N4 (1/0.7) achieving a TOF value of 1193 h-1. The additional introduction of Co was proved to be ineffective and counterproductive in terms of FA dehydrogenation capability, that could be attributed to the partial shielding of the active sites in the Pd NPs by the excess of cobalt. The XPS spectra of Pd 3d revealed a slight shift toward lower binding energies in the PdCo catalysts compared to monometallic Pd. That shift confirmed the existence of alloyed NPs and suggested the formation of electron-rich Pd species on the PdCo particles, which could attributed to the donation of electrons from Co to Pd, as indicated by their respective electronegativities (values in Pauling units of 1.88 and 2.20, respectively). The positive influence of the alloy system in catalyzing FA dehydrogenation was substantiated through potential energy profiles generated by DFT calculations, utilizing Pd43 and Pd22Co21 clusters as models for monometallic and alloy NPs, respectively. Considering the suggested reaction mechanism (Figure 5), the lower energy barriers observed for the Pd22Co21 cluster, in comparison to the monometallic Pd43, confirmed the favorable impact of bimetallic NPs in enhancing the FA dehydrogenation reaction.
Tamarany et al. studied systems based on PdNi supported in N-doped carbons [65]. The N-doped carbons (N-C) were obtained by mixing dicyanoamide with carbon black at 100 °C and then pyrolyzed at 550 °C for 4 h in N2 flow. Pd1Nix alloys supported on N–C (Pd1Nix/N–C) were prepared by dispersing N-C into aqueous solutions containing Pd(NO3)2·2H2O and Ni(NO3)2·6H2O at different Pd/Ni mole ratios (1/0.33, 1/1 and 1/3). The resulting black powders were collected and reduced under a H2/N2 flow (20% H2) at 450 ºC for 4 h, affording Pd1Nix/N–C. The catalytic activity for FA dehydrogenation was assessed at 30 °C with FA:SF solution in 1/1 molar ratio for 3 catalysts with different Pd/Ni ratios. The results showed a volcano-type behavior where the maximum catalytic activity for Pd1Ni1.3/N–C produced a TOF value of 447 h-1. In another experiment, the influence of temperature on the dehydrogenation activity with Pd1Ni1.3/N–C was studied, obtaining an initial TOF value of 4804 h-1 at 65 ºC. That value could be attributed in part to the fact that the sodium formate can potentially react with water to produce hydrogen (HCOONa + H2O → NaHCO3 + H2), particularly at temperatures higher than 65 ºC. Furthermore, the formation of PdNi alloys was confirmed by: a) elemental mapping of Pd1Ni1.3/N–C by HAADF-STEM through the overlap in the positions of Pd and Ni, and b) by the shift to lower binding energies of Pd 3d spectra obtained by XPS.
Mori et al. investigated PdCu alloy NPs supported on a macroreticular basic resin with -N(CH3)2 groups [66]. The deposition of both Pd and Cu within the basic resin was performed using a simple ion exchange method from an aqueous solution of PdCl2 and CuNO3·3H2O, and later the samples were reduced by NaBH4. EXAFS analysis revealed the presence of heteroatomic Pd-Cu bonding. The formation of PdCu nanoparticles with an average size of 1.9 nm was confirmed by TEM. The catalytic activity results measured for different Pd/Cu molar ratios show a volcano-type trend, where the Pd50Cu50 composition presents the maximum TOF value, 810 h-1 at 75 ºC using a FA/SF 9:1 aqueous solution. The improvement in catalytic activity of the bimetallic PdCu catalyst over the monometallic Pd catalyst can be explained by the energetic efficiency of transferring Cu electrons to Pd atoms due to the difference in ionization potential (Cu: 7.72 eV and Pd 8.34 eV). Additionally, that study confirms the significant impact of the basicity of the resin on the breaking of O-H bonds and the introduction of Cu is shown to influence the rate-determining step involving C-H bond dissociation from the metal-formate intermediate, attributable to the formation of electron-rich Pd species.
Pd-based trimetallic catalysts have not been as extensively studied as bimetallic catalysts, but they are an interesting alternative to achieve a Pd content reduction and modulate the electronic properties of Pd species.
Yurderi et al. designed carbon-supported trimetallic PdNiAg NPs as catalysts for FA dehydrogenation [67]. The preparation of the catalysts was achieved by a wet impregnation method with a reduction step and the precursors of the metals were mixed in different molar ratios with the commercial activated carbon. The morphology and size of the PdNiAg/C NPs were studied by TEM, obtaining a good dispersion and with an average particle size of 5.6 nm. XPS spectra showed the presence of metallic NPs and also oxidized species such as PdO, NiO, Ni(OH)2, NiOOH and Ag2O. Among the different compositions studied, the Pd0.58Ni0.18Ag0.24/C catalyst shows the best catalytic activity, reaching a TOF of 85 h-1 at 50 ºC in a 1:1 FA/SF solution. The reusability of that catalyst was studied after five consecutive cycles of reaction and it was demonstrated that 94% of the initial catalytic activity was retained.
Wang et al. prepared NiAuPd alloy supported on a commercial carbon [68]. The synthesis of the catalysts was carried out by a co-reduction method without any surfactant at 25 °C. The images obtained by HAADF-STEM showed that Ni, Au, and Pd were homogeneously distributed in each particle, pointing out that the alloy structure is indeed formed. The catalytic activity results for Ni0.40Au0.15Pd0.45/C revealed a TOF of 12.4 h-1 (calculated on the basis of the total amount of metal) at 25 °C and in a pure FA solution without additives.
Dong et al. tailored a system based on PdCoNi NPs supported on N-CN. N-CN was prepared by a combination of CN and 3-aminopropyl triethoxysilane [69]. The catalysts based on the PdCoNi NPs were synthesized by a co-reduction method. Ultrafine NPs were obtained for the Pd0.6 Co0.2Ni0.2/N-CN catalyst with an average size of 1.6 nm. XPS analysis detected the electron transfer from Co and Ni to Pd, indicating the formation of a solid solution structure of Pd0.6Co0.2Ni0.2 NPs, and the presence of pyrrolic N was also detected. The catalytic activity of Pd0.6Co0.2Ni0.2/N-CN was measured at 25°C in a 1:1 aqueous FA/SF solution obtaining an initial TOF value of 1249 h-1.
Liu et al. performed a study based on trimetallic AuPdIr nanoalloy supported on N-doped reduced graphene oxide (N-GO) [70]. AuPdIr catalysts were prepared by a co-reduction method by mixing the metal precursors with an APTS/GO solution, obtaining Au0.35Pd0.5Ir0.15/NH2-N-rGO. By XPS, the electron transfer from Pd and Ir to Au has been observed, which is consistent with their electronegativity (Au 2.4, Pd 2.2, Ir 2.2), thus confirming the nanoalloy formation. The performance in FA dehydrogenation was tested obtaining a TOF value of 12781.2 h-1 at 25 °C in a pure FA solution (Figure 6A), and the temperature increase to 60 °C gave a TOF value of 36598.4 h-1. The recycling stability of Au0.35Pd0.5Ir0.15/NH2-N-rGO was tested by adding an extra aliquot of FA dilution after completion of the previous one and showed high stability after 10 reaction cycles. To further test the long-term stability, a 150-day experiment was designed. As depicted in Figure 6D, the catalyst retained the ability to catalyze 100% FA dehydrogenation for 7.08 minutes at room temperature, even after 150 days. According to DFT calculations, that robust activity was ascribed to the incorporation of the high surface energy element iridium (Ir), which alters the initial adsorption configuration of HCOOH* and improves the overall performance of the reaction.
3. Hydrogenation of CO2 to FA
The hydrogenation of CO2 to FA is more challenging than the reverse reaction, the dehydrogenation of FA, but this reaction is essential to obtain a hydrogen charge-discharge system achieving a carbon-neutral cycle. This process is thermodynamically unfavorable, being the net reaction between gaseous H2 and CO2 to liquid HCOOH endergonic due to the strong entropic contribution (ΔG298K= 32.9 kJ·mol-1) [71,72]. A notable challenge in this process lies in activating the chemically stable CO2 molecule, with a linear molecular structure (O=C=O) and a high bond energy of 806 kJ mol-1.[73,74]. Therefore, the application of external energy is required in addition to suitable catalysts with high selectivity. Among the investigated heterogeneous catalysts, Pd-based catalysts have been extensively studied and often exhibit remarkable activity compared to other transition metals in the hydrogenation of CO2 to formic acid/formate. This superiority stems from their high stability under acidic or basic reaction conditions and a strong affinity for H2 spillover [75]. In particular, catalysts based on Pd NPs supported on carbon materials stand out for the modulation of their textural properties and ease of heteroatom incorporation.
To strategically design carbon-supported Pd-based catalysts for CO2 hydrogenation to formate/formic acid, it is crucial to identify the key factors that hinder the catalytic performance. As mentioned above, this reaction is thermodynamically unfavorable, due mainly to the transition from the gaseous phase (CO2 and H2) to liquid products, which needs a significant positive change in Gibbs free energy [76]. The most straightforward approach to overcome the thermodynamic limitation is to conduct the reaction in liquid phase (such as water, alcohol, DMSO and ionic liquids) [77]. The reaction experiences a slight exothermal shift attributed to the solvation effect (see reaction 3) [78,79]. Additionally, the hydrogenation of CO2 becomes more advantageous with the incorporation of base additives into reaction solutions (such as amines, bicarbonates, and hydroxides), which effectively inhibit the reverse reactions (the dehydration/dehydrogenation of FA).[79]. Certainly, the hydrogenation of CO2 to formic acid/formate is influenced by many factors, including the pH of the solution, reaction temperature, type of base and the pressure of CO2 and H2.
As previously mentioned, the hydrogenation of CO2 into FA/formate typically occurs in alkaline conditions, necessitating the catalyst to possess alkali-resistant properties. However, Pd NPs tend to aggregate into larger particles or leach into the solution during the catalytic reaction process. [76]. That indicates that the protection of Pd atoms is also a crucial factor in achieving high catalytic performance. To date, multiple approaches, including the anchoring of functional groups (amines or uncoordinated N as well as other basic functionalities) on supports or Pd NPs [80,81,82,83,84], doping Pd NPs with other transition metal atoms [85,86,87,88], confining the Pd NPs within pore or cavity of supports [86,89] as well as enlargement of surface area, have been employed to address challenges associated with catalytic hydrogenation of CO2 to produce FA/formate. Herein, the most significant work on the hydrogenation of CO2 to FA/formate through carbon-supported Pd-based catalysts will be revisited.
3.1. Carbon-supported monometallic Pd catalysts.
Since the hydrogenation of CO2 to FA/formate is a more challenging reaction than the reverse reaction, studies are needed to modulate the experimental conditions and catalyst properties to obtain better catalytic activity and to get insights into the reaction mechanism.
Su et al. investigated the reaction conditions for the hydrogen storage system based on ammonium bicarbonate/formate redox equilibrium over Pd catalysts [90]. First, bicarbonate and carbonate salts were compared with different cations, Na+, K+ and NH4+ for hydrogenation. It was found that the bicarbonate salts presented a better activity for hydrogenation and specifically NH4HCO3. For the study of the hydrogenation reaction with an aqueous solution of NH4HCO3 at pH 7.7 using a Pd/AC catalyst, an ammonium formate yield of 59.6% was obtained with a turnover number (TON) of 1103 after reaction for 1 h and with an initial H2 pressure of 5.5 MPa. Given that the hydrogenation of aqueous NH4HCO3 involves multiple phases (gas/liquid/solid), the rate-limiting step could be the diffusion of the reactant, H2, owing to its low solubility in water. Activated carbon is commonly a hydrophobic support and can store H2. H2 may be concentrated locally within the carbon channels or on the surface of the carbon support, facilitated by H2 spillover from the Pd metal. A C13NMR analysis confirmed that the cations of the bicarbonate salts shifted the equilibrium between carbonate or bicarbonate ions, NH4HCO3 presented the highest concentration of HCO3- in the aqueous solution, which leads to the highest formate yield of the hydrogenation reaction. In addition, the effect of reaction temperature was studied, and it was observed that the highest formate yield was achieved at room temperature, implying that the developed hydrogenation system did not require additional external energy. The ammonium formate dehydrogenation reaction was studied at 80 °C, obtaining a H2 yield of 92.1% with a TON of 1698 after 1.5 h of reaction.
Shao et al. prepared Pd-based nanocatalysts supported on mesoporous carbon nitride (mgp-C3N4) for the hydrogenation of bicarbonate to formate [80]. mgp-C3N4 was obtained using dicyandiamide and a silica template by pyrolysis treatment. The catalysts, Pd/mgp-C3N4 and a Pd/AC, were obtained by a wet chemical reduction method. The images obtained by TEM showed that the Pd NPs were well dispersed on the two supports with an average particle size of 1.9 nm for Pd/mgp-C3N4 and 2.4 nm for Pd/AC. XPS analyses showed that the N species were mainly sp2 hybridized aromatic N bonded to carbon atoms in the form of C=N-C and tertiary N bonded to carbon atoms (N-(C)3), and a small amount of NH groups was also observed. In addition, the Pd 3d XPS analysis showed the presence of Pd0 and Pd2+. The hydrogenation reaction was studied from 50 to 80 °C, p(H2) = 6.0 MPa, 20 mmol KHCO3 and 5 mL H2O. As the hydrogenation of bicarbonate ions is exothermic, the reaction rate increased with the increase in temperature, but the final formate concentration decreased. The TOF values calculated at 80 °C for Pd/mgp-C3N4 and Pd/AC were 5926 and 4672 h-1, respectively. In that study, it was suggested that the surface primary or secondary amino groups of mpg-C3N4 could aid the activation of HCO3- and promote the hydrogenation reaction of bicarbonate. The reusability of Pd/mgp-C3N4 was tested for 6 cycles showing a high stability in the reaction. In that work, the observation that only formate was formed as a product suggested that hydrogenation is going via the insertion mechanism. In this mechanism, that is shown in Figure 7, in an initial stage HCO3- can be easily adsorbed on the surface of the catalyst by the formation of O-H···N. Following this, the active H is produced over Pd NPs, which attacks the positively polarized carbon in HCO3- leading to the simultaneous formation of the formate intermediate, and the product HCO2- is released, and a new HCO3- anion is adsorbed.
Zhout et al. prepared Pd catalysts supported on g-C3N4 and modified by the addition of a Schiff base, terephthalaldehyde (TPAL), to study their effect on the catalytic activity in the hydrogenation of CO2 to formate [91]. The synthesis of the support (u-CN100) was carried out by mixing urea and TPAL and then heating it at 550 ºC in a muffle, then Pd/u-CN100 was obtained by an impregnation approach. The CO2 hydrogenation experiments were performed in an aqueous phase consisting of ethanol (15 mL) and triethylamine (Et3N) at 110 ºC with a total pressure of 7 MPa (H2/CO2 1:1). The catalytic activity results showed a great improvement in TOF value when TPAL was added, obtaining 98.9 h-1, while for Pd/u-C3N4 (without TPAL) the TOF value was 14 h-1. The stability of Pd/u-CN100, was studied after 5 consecutive cycles where the catalyst was collected and did not show a remarkable loss of activity. That marked stability may be related to the Pd species anchored to the support by the N groups. The presence of Pd nanoclusters was observed by TEM, with an average particle size for Pd/u-CN100 of 1.57 nm and for Pd/u-C3N4 of 2.22 nm. Moreover, XPS analysis showed a higher proportion of Pd2+ species in Pd/u-CN100 than in Pd/u-C3N4, confirming again the stronger interaction with N species. Thus, the addition of a Schiff base resulted in a catalytic improvement due to the modulation of the electronic properties of Pd species as well as the anchoring effect on these species.
Wang et al. designed a palladium-based system supported on N-doped mesoporous carbon (NMC) as a bifunctional catalyst for formate-mediated hydrogen storage [82]. NMC was prepared by a hard templating method using a mesoporous silica as a template and carbonized under ammonia flow for nitridation. Palladium-supported catalysts were prepared by a wet chemical reduction method. The catalytic performance for bicarbonate hydrogenation for Pd/NMC-8 (nitridation treatment at 873 K) obtained a turn-over number (TON) of 1598 at 80 °C in 2 h and a conversion of 69.7 % in aqueous KHCO3 solution and with a H2 pressure of 6 MPa. The reusability of the catalyst was measured after 4 runs, in the fourth run the formate yield decreased by 13.3%. The bicarbonate hydrogenation was studied for Pd/NMC-8 at 80 ºC obtaining a TOF in the first 10 min of 2416 h-1 in a HCOOK aqueous solution. The average particle size obtained for the Pd NPS in Pd/NMC-8 by STEM was 2.4 nm, which was smaller than for the catalyst without N groups. XPS analysis detected pyridine, nitrile, pyrrole and quaternary N species. In addition, a higher contribution of Pd2+ was detected in the N-doped support demonstrating the interaction between the nitrogen species with the Pd NPs. That work demonstrates how the nitrogen groups in the support play a crucial role in promoting the bicarbonate/formate redox equilibrium.
Song et al. prepared catalysts based on Pd NPs supported on chitin by a wet impregnation method followed by reduction [83]. The morphology of Pd/chitin was determined by SEM yielding a fibers form and some of them are disorderly gathered to form flowerlike architecture. SEM analysis revealed that the Pd NPs were well distributed in the chitin fibers and the average particle size was 1.84 nm. Analysis of the Pd 3d XPS spectra showed the presence of Pd0 and Pdδ+; the oxidized species were attributed to the electronic donation of Pd to the acetoamide nitrogen groups of chitin. Thus, the interaction of the acetoamide groups of chitin with Pd was confirmed. The CO2 hydrogenation reaction was carried out in an aqueous solution using Na2CO3 as an additive under a total pressure of 4 MPa and at 60 °C. Under these conditions, Pd/chitin with 0.25% Pd content obtained the highest TOF value, 257 h-1. The recyclability study showed good stability after 5 reaction cycles.
Some other studies report the ability of the developed materials to catalyze both the dehydrogenation of FA and the hydrogenation of CO2 or bi/carbonates. For example, Lee et al. designed a system based on Pd NPS supported on mpg-C3N4 for H2 generation and storage via FA dehydrogenation and CO2 hydrogenation [81]. The Pd NPSs were immobilized onto mpg-C3N4 by a wet impregnation method with H2 reduction. The Pd NPs were found to be well distributed and with an average size of 1.7 nm, as indicated by HRTEM. Pd/mpg-C3N4 achieved a TOF for the FA dehydrogenation reaction of 324 h-1 at 55 °C, in an aqueous phase without any additives. To study the CO2 hydrogenation reaction, as FA can react with H2 resulting in products such as methanol and methane, Et3N was used to trap the FA formed. The total pressure of the gases was regulated at 40 bar, although this value was increased by heating the mixture up to 100 °C and 150 °C. The amount of FA formed depended on the reaction conditions. It was found that a decrease in CO2 pressure did not noticeably affect FA production, while the activity decreased when the H2 pressure was low. It was also observed that the activity improved when the temperature increased. The results obtained by 13 C NMR after an experiment where CO2 was pressurized in a reactor containing Et3N and deuterium verified the formation of Et3N-CO2. The best activity was observed upon utilization of the CO2 and H2 pressures of 13 bar and 27 bar at 150 ºC, respectively, to give 4.74 mmol of FA.
Bi et al. designed a system for the development of a formate-based hydrogen battery mediated by Pd catalysts supported on reduced graphene oxide (r-GO) [92]. Pd/r-GO was synthesized by a one-step co-reducing method. The TEM images showed spherical and uniform Pd particles with a mean size of 2.4 nm dispersed on the r-GO sheets. The predominant presence of Pd0 on r-GO was confirmed by XPS analysis. The underlying support surface may cause a structural effect on the electronic structure of Pd, creating a lattice microstrain, which could be related to a higher activity for dehydrogenation of formate. This fact was confirmed by XRD in Pd/r-GO. The dehydrogenation reaction was studied at 80 ºC in an aqueous solution of 5 mL HCOOK 4.8 M, in these conditions, Pd/r-GO with 1 wt% Pd, achieved a TOF value of 11299 h-1. The hydrogenation reaction to potassium formate was performed in the same aqueous solution, and with a total pressure (H2/CO2 1:1) of 80 bar at 100 ºC, the Pd/r-GO catalyst reached a TON value of 2420 and the almost complete conversion was achieved in 10 h. In view of the good catalytic results, cyclic operation tests were carried out by coupling the two reactions. Therefore, alternating pressurized charging (hydrogenation reaction) and pressure-free discharging (dehydrogenation reaction) procedures were employed to evaluate the feasibility in a commercially available standard autoclave. It was noted that up to six consecutive identical cycles of H2 release and the hydrogenation of the resulting KHCO3 were achieved.
Koh et al. synthesized small Pd NPs supported on mesoporous N-doped carbon (PDMC) for hydrogen storage and release based on bicarbonate/formate [84]. The N-doped support was prepared by a PANI-assisted method using silica as a template. The resulting material was pyrolyzed in Ar atmosphere and then the product was treated with NaOH solution in an autoclave at 100 °C for 18 h to remove the silica template. Catalysts with formula Pd/PDMC-T-x (T represents the pyrolysis temperature and x represents the amount of colloidal silica template), were prepared by a wet impregnation method followed by a reduction in N2/H2 atmosphere. The average particle size measured by TEM for Pd/PDMC-800-16 was 1.6 nm, proving that the use of colloidal silica templates indirectly helped to obtain small NPs. The dehydrogenation reaction of formate was carried out in a 5 mL 1M aqueous solution of HCOONa at 800 ºC. The highest catalytic activity was obtained for the catalyst Pd/PDMC-800-16, obtaining a TOF value of 2562 h-1. Additionally, it was observed that the catalytic activity of the catalysts followed a volcano-like trend with the pyrolysis temperature, reaching a maximum when the temperature was 800 ºC. Furthermore, the reusability study measured for 3 cycles demonstrated the stability of the catalyst. The hydrogenation reaction of sodium bicarbonate was performed in a 1M aqueous solution of NaHCO3, with a H2 pressure of 40 bar and at 80 ºC. Under these conditions, Pd/PDMC-800-16 presented the best activity with a TON value of 1625. The surface chemistry of the catalysts was studied by XPS, and the presence of pyridinic-N, pyrrolic-N, quaternary-N and pyrdinic-N+-O- groups was detected. For Pd/PDMC-800-16 a higher proportion of pyrrolic-N and pyridinic-N was detected. The EXAFS study indicated that the coordination numbers (CNs) of Pd-Pd bonding in all the Pd/PDMC-T-x materials (CN Pd-Pd ≈ 2 to 4) are notably lower than that of bulk Pd foil (CN Pd-Pd ≈ 12). That confirmed the existence of well-dispersed Pd nanoclusters, each containing only a few atoms, on the N-doped carbon support materials.
3.2. Carbon-supported multimetallic Pd-based catalysts
For CO2 hydrogenation, there are not many studies where multimetallic catalysts are used. However, alloying Pd with other metals can be an interesting option since pure Pd can be easily poisoned by some reaction intermediates such as CO [93,94].
Yang et al. investigated PdAg alloy NPs encapsulated in N-doped microporous hollow carbon spheres (NMHCS) for the hydrogenation of CO2 to formate [86]. The NMHCS were prepared using ethylenediamine as the nitrogen source and SiO2 as the template. The catalysts were prepared by impregnation and then reduced with NaBH4 to form PdAg@NMHCS spheres. FE-SEM images show uniform spheres without impurities. It was observed by TEM that PdAg@NMHCS-0.6-500 (carbonized at 500 ºC) presents hollow cavities with an approximate diameter of 340 nm and a N-doped carbon shell of 30 nm of thickness. The PdAg NPS have an average size of 7.5 nm and are uniformly encapsulated in the hollow cavities. The presence of PdAg alloy was confirmed by STEM elemental maps. The N 1s orbital spectra determined by XPS showed that with increasing carbonization temperature from 400 to 500 °C the N-pyridinic fractions decreased while the N-pyrrole fraction increased. That means that part of the N-pyridinic was transformed into N-pyrrole at 500 ºC. The hydrogenation reaction of CO2 to formate was carried out using a 15 mL aqueous solution of 1.0 M NaHCO3 at 100 °C and a total pressure of 2.0 MPa (H2/CO2 1:1). For PdAg@NMHCS-0.6-500 a TON of 640 and 2750 was obtained after 2 and 24 h, respectively, presenting a better catalytic activity than the catalyst without hollow structure, indicating that this structure plays an important role to obtain a high catalytic activity. In addition, the reaction was studied using other bases such as NaOH, NH4HCO3, Na2CO3 and only H2O. The best result was obtained for NaHCO3 and the use of only pure water resulted in a sharp decline in activity. This fact showed that alkaline solutions facilitate the absorption of CO2 in the solution for the formation of HCO3-.
Masuda et al. developed a catalyst based on PdAg NPs supported on phenylamine-functionalized mesoporous carbons (MSCs) for FA and CO2 interconversion [87]. MSC was treated with an aqueous nitric acid solution and then functionalized with p-phenylenediamine to produce amine-MSC (see Figure 8). PdAg-based catalysts were prepared by an impregnation method with the supports and subsequent reduction with NaBH4. Good dispersion of the Pd and Ag NPs was observed with an average size of 1.2 nm measured by HAADF-STEM. The slight shift of XPS spectra towards lower binding energies for the 3d Pd peaks in PdAg/amine-MSC compared to PdAg/MSC demonstrated that the amine groups affect the electronic states of Pd, confirming their interaction. In addition, PdAg alloying could also be verified by XPS and EXAFS. From the results obtained by EXAFS, it could be elucidated that in PdAg/amine-MSC the Ag atoms are preferentially located in the core region with the Pd atoms located in the shell. The FA dehydrogenation reaction was measured in an aqueous FA/SF solution in a 9:1 molar ratio at 75 °C, for the PdAg/amine-MSC catalyst a TOF value of 5638 h-1 was obtained, on the basis of the total amount of Pd. The hydrogenation of CO2 to form FA was assessed with an aqueous NaHCO3 solution under a total pressure of 2 MPa (H2:CO2 = 1:1, volume ratio) at 100 ºC, and PdAg/amine-MSC showed a selectivity >99% and a TOF and reaction rate of 839 h-1 and 328 mmolh-1gPd-1, respectively. Potential energy profiles calculated by DFT verified that the amine-functionalized catalyst favor both reactions. The graphitized phenylamine group positively influences the O-H bond dissociation in the dehydrogenation of FA, whereas the interaction between HCO3- and phenylamine molecules plays a crucial role in stabilizing the reaction intermediate in the hydrogenation of CO2. A catalyst sample was initially employed for the dehydrogenation of FA over 15 minutes and subsequently collected through centrifugation after the reaction. Reversibility during FA and CO2 interconversion was examined for PdAg/amine-MSC. The recovered catalyst was subsequently redispersed in an aqueous NaHCO3 solution (1.0 M) and employed for the hydrogenation reaction to produce FA. After 24 hours, the catalyst was once more collected by centrifugation, dispersed in distilled water, and employed again for the dehydrogenation of FA. The reversibility of FA and CO2 interconversion was assessed for PdAg/amine-MSC. The catalyst reversibility was confirmed through three consecutive cycles, demonstrating no inherent loss of catalytic activity.
Yang et al. prepared PdCu alloy NPs confined in mesoporous hollow carbon spheres (MHCS) for the hydrogenation of CO2 to formate [85]. The method of preparation of these catalysts was similar to that presented in the previous work of these authors [86]. TEM images show hollow spheres of approximately 220 nm diameter and 40 nm shell thickness. HAADF-STEM observations for Pd2Cu14-N@MHCS showed the NPs encapsulated within hollow cavities with an average particle size of 5 nm. The Pd/Cu NPs exhibit lattice fringes, which are identified for the Cu(111) plane (lattice spacing 0.21nm), indicating the dispersion of Pd atoms in Cu NPs. The surface species were determined by XPS, confirming the presence of reduced Cu and Pd, and the electron transfer from Cu to Pd was verified. In addition, it was possible to verify by EXAFS that Pd is dispersed in the Cu particles as an alloy. The hydrogenation reaction of CO2 to formate was carried out under a total pressure of 2 MPa in an aqueous solution of 1 M NaHCO3 at 100 ºC. For the Pd2Cu14-N@MHCS catalyst a TON value of 1432 was obtained after 24 h. Furthermore, kinetic investigations were carried out by varying the reaction conditions, which revealed that bicarbonate adsorbs readily on Cu atoms. These studies suggested the following reaction mechanism for this catalyst: 1) adsorption and activation of H2 on Pd on PdCu, 2) gaseous CO2 dissolves in the aqueous solution to form HCO3-, 3) HCO3- adsorbs on the Cu atoms of the PdAu alloy, 4) nucleophilic hydrogen atom formed on Pd reacts with HCO3- adsorbed on Cu, forming formate ion, 5) OH- adsorbs on MHCS, 6) H remaining on Pd reacts with OH- to form H2O, which regenerates the catalyst. The computational calculations obtained by DFT demonstrated that alloying Cu with Pd facilitates the dissociation of H2, and also influences the activation energy for the reduction of HCO3-.
Nguyen et al. developed a catalyst based on PdNi alloy supported on carbon nanotube-graphene (CNT-GR) for the hydrogenation of CO2 to FA [84]. PdNi supported on CNT-GR composite were obtained by a wet impregnation method followed by reduction. Previously, the optimum composition for these catalysts was found to be Pd3Ni7. The PdNi NPs were found by TEM to be uniform and well dispersed with an average particle size of 4 nm. After HRTEM analysis, the presence of the alloy was confirmed by PdNi (111) at a lattice spacing of 0.222 nm. The presence of mixed valences for Pd (Pd0, Pd2+ and Pd4+) could be observed, but the most oxidized species are found in a lower proportion. The CO2 hydrogenation reaction was carried out for 15 h in an aqueous solution of distilled water without any base additive. When the reaction was studied with Pd3Ni7/CNT-GR at 40 °C and 50 bar total pressure (H2/CO2 1:1) the highest FA yield, 1.93 mmol, was achieved. Furthermore, higher Pd contents, including the monometallic composition, did not result in a better yield, highlighting the synergistic effect of the optimal composition obtained with Pd3Ni7. On the other hand, the study of different reaction temperatures revealed that increasing the temperature above 40 °C was counterproductive. This could be explained considering that the reaction between CO2 and H2 at high temperatures normally results in the formation of CO and H2O or the back decomposition of FA. Furthermore, at temperatures below 40 °C, the reaction kinetics were not favorable for overcoming the activation barrier of the reaction. The stability study revealed that after the first cycle, the activity decreases significantly, but the yield stabilized from the second to the seventh cycle with negligible deactivation.
4. Conclusions
The present review summarizes recent work on heterogeneous Pd-based catalytic systems for hydrogen storage mediated by the HCOOH-CO2 couple. Given the great relevance that H2 has acquired as an energy vector in recent years, this topic is of great importance to alleviate the problems related to H2 production and storage. The dehydrogenation of FA has been exhaustively studied, delving into various factors such as the importance of the properties of the metal active phase and the support, as well as the experimental conditions. Moreover, theoretical investigations have been performed to disclose important features for the design of catalysts with high catalytic activity and to obtain information about the reaction mechanism. Among the different heterogeneous catalysts studied, those based on Pd NPs stand out for their high selectivity and conversion towards H2 at moderate temperatures. As for the support, carbon materials are an outstanding option for their different tailored characteristics and the possibility of including heteroatoms in their structure. Considering the varied properties of the active phase, the pursuit of cost-effective catalysts has conducted the exploration of nanoarchitectures to attain a substantial proportion of surface Pd active sites. In addition, it has been evidenced that the incorporation of basic nitrogen functionalities in the support can greatly improve the catalytic activity and stability by different effects: i) anchoring the Pd NPs in the support; ii) modifying the electronic properties of Pd species that are involved in the reaction mechanism improving the rate of the catalytic steps; iii) interacting with the molecules of FA. The formation of Pd-based multimetallic catalysts through the formation of alloys by the incorporation of one or more metals has also been studied. It has been confirmed that after the formation of alloys with Pd, the electronic properties can be modulated and the resistance to CO poisoning can be improved. Despite all the improvements that have been implemented to obtain high performance for this reaction, there are still shortcomings that limit the practical application of large-scale systems. Among these limitations are the use of additives, usually formates (HCOONa or HCOOK), as well as the loss of stability after several reaction cycles. These limitations should be the focus of research for future studies. On the other hand, the hydrogenation of CO2 to FA/formate on carbon-supported Pd-based catalysts has not been as extensively studied as the reverse reaction due to the unfavorable thermodynamics of the reaction. However, the studies revisited in this review have focused on adjusting the experimental conditions to promote the exothermicity of the reaction and to understand the mechanism of the reaction in order to modulate the properties of the catalysts. The study of CO2 hydrogenation in aqueous media with additives such as NaHCO3 and KHCO3, has shown an improvement in the course of the reaction due to the decrease in the Gibbs energy in the aqueous phase. Concerning the properties of the active metal phase and the support, the same aspects as those considered for the optimization of the performance of the catalysts studied for the dehydrogenation of FA (particle size reduction, incorporation of another metal, doping with nitrogen groups) have also resulted in an improvement in the catalytic activity of the catalysts towards the hydrogenation of CO2. Despite the knowledge gained about this reaction, efforts are still needed for the design of catalysts with high activity and stability to allow the reduction of high temperatures and pressures, as well as the use of base additives.
The objective of future research would be to develop a catalyst with high activity and stability for both reactions and operating under mild conditions. In an ideal scenario, a single system would perform the dehydrogenation and hydrogenation reactions, to be applied to deliver or store H2, thus functioning as a charge-discharge system to obtain H2 on demand, and generating an ideal carbon-neutral cycle. Hence, it is expected that in the coming years, more and more research groups will be interested in this promising research topic, which will be crucial to developing an additional alternative for the energy future based on an environmentally sustainable approach.
Author Contributions
P. R.-G. wrote the manuscript, M.N.-G. designed the structure of the manuscript and reviewed the paper, and D.C.-A. reviewed the paper. All authors approved the manuscript for publication.
Funding
This work is part of the R+D+I project PID2021-123079OB-I00 funded by MCIN/AEI/ 10.13039/501100011033 and by “ERDF A way of making Europe” and also the project TED2021-131324B-C22 funded by MCIN/AEI/ 10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR”. MNG would like to thank the grant RYC2021-034199-I funded by MCIN/AEI/ 10.13039/501100011033 and by “ESF Investing in your future”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Armaroli, N. and Balzani , V., The future of energy supply: Challenges and opportunities, Angew Chem Int Ed Engl, 2007, 46, 52–66. [CrossRef]
- Chow, J., Kopp, R. J., Portney, P. R., Energy Resources and Global Development, Science , 2023, 302, 1528–1531. [CrossRef]
- Papadis E., Tsatsaronis G., Challenges in the decarbonization of the energy sector, Energy, 2020, 205, 118025. [CrossRef]
- Proost, J., Critical assessment of the production scale required for fossil parity of green electrolytic hydrogen, Int J Hydrogen Energy, 2020, 45, 17067–17075. [CrossRef]
- Muthukumar, P., Kumar, A., Afzal, M., Bhogilla, S., Sharma, P., Parida, A., Jana, S., Kumar, E., Pai, R.K., Jain, I.P, Review on large-scale hydrogen storage systems for better sustainability, Int J Hydrogen Energy, 2023, 48, 33223–33259. [CrossRef]
- Aakko-Saksa, P. T., Cook, C., Kiviaho, J., Repo, T., Liquid organic hydrogen carriers for transportation and storing of renewable energy – Review and discussion, J Power Sources, 2018, 396, 803–823. [CrossRef]
- Rao, P. C. Yoon, M., Potential Liquid-Organic Hydrogen Carrier (LOHC) Systems: A Review on Recent Progress, Energies, 2020, 13, 6040. [CrossRef]
- Bulushev, D. A., Ross, J. R. H., Towards Sustainable Production of Formic Acid, ChemSusChem, 2018, 11, 821–836. [CrossRef]
- Enthaler, S., Von Langermann, J., Schmidt, T., Carbon dioxide and formic acid —the couple for environmental-friendly hydrogen storage ?, Energy Environ Sci, 2020, 3, 1207–1217.
- Navlani-García, M., Martis, M., Lozano-Castelló, D., Cazorla-Amorós, D., Mori, K., and Yamashita, H., Investigation of Pd nanoparticles supported on zeolites for hydrogen production from formic acid dehydrogenation, Catal Sci Technol, 2014, 5, 364–371. [CrossRef]
- Wu, Y., Wen, M., Navlani-García, M., Kuwahara, Y., Mori, K. and Yamashita, H., Palladium Nanoparticles Supported on Titanium-Doped Graphitic Carbon Nitride for Formic Acid Dehydrogenation, Chem Asian J, 2017, 12, 860–867. [CrossRef]
- Navlani-García, M., Salinas-Torres, D., Vázquez-Álvarez, F. D., Cazorla-Amorós, D., Formic acid dehydrogenation attained by Pd nanoparticles-based catalysts supported on MWCNT-C3N4 composites, Catal Today, 2022, 397–399, 428–435. [CrossRef]
- Ortega-Murcia, A., Navlani-García, M., Morallón, E., Cazorla-Amorós, D., MWCNT-Supported PVP-Capped Pd Nanoparticles as Efficient Catalysts for the Dehydrogenation of Formic Acid, Front Chem, 2020, 8, 534710. [CrossRef]
- Sun, Q., Chen, B., Wang, N, He, Q., Chang, A., Yang, C.M,. Asakura, H., Tanaka, T., Hülsey, M.J, Yu, J, Yan, N., Zeolite-Encaged Pd–Mn Nanocatalysts for CO2 Hydrogenation and Formic Acid Dehydrogenation, Angewandte Chemie International Edition, 2020, 59, 20183–20191.
- Jin-Jeon H., Chung, Y. M., Hydrogen production from formic acid dehydrogenation over Pd/C catalysts: Effect of metal and support properties on the catalytic performance, Appl Catal B, 2017, 210, 212–222.
- Navlani-García, M., Salinas-Torres, D., Cazorla-Amorós, D., Hydrogen Production from Formic Acid Attained by Bimetallic Heterogeneous PdAg Catalytic Systems, Energies 2019, 12, 4027. [CrossRef]
- Yadav, M., Xu Q., Liquid-phase chemical hydrogen storage materials, Energy Environ Sci, 2012, 5, 9698–9725.
- Jiang, H. L., Singh, S. K., Yan, J. M., Zhang, X. B., Xu, Q., Liquid-Phase Chemical Hydrogen Storage: Catalytic Hydrogen Generation under Ambient Conditions, ChemSusChem, 2010, 3, 541–549. [CrossRef]
- Navlani-García, M., Mori, K., Kuwahara, Y., Yamashita, H., Recent strategies targeting efficient hydrogen production from chemical hydrogen storage materials over carbon-supported catalysts, NPG Asia Materials, 2018, 10, 277–292. [CrossRef]
- Enthaler, S., Von Langermann, J., Schmidt, T., Carbon dioxide and formic acid —the couple for environmental-friendly hydrogen storage, Energy Environ Sci, 2010, 3, 1207–1217. [CrossRef]
- Li, S.-J., Zho, Y-T., Kang, X.,Liu, D-X., Gu, L., Zhang, Q-H., Yan, J-M., Jiang, Q., A Simple and Effective Principle for a Rational Design of Heterogeneous Catalysts for Dehydrogenation of Formic Acid, Advanced Materials, 2019, 31, 1806781. [CrossRef]
- Wakai, C., Yoshida, K., Tsujino, Y., Matubayasi, N., Nakahara, M., Effect of concentration, acid, temperature, and metal on competitive reaction pathways for decarbonylation and decarboxylation of formic acid in hot water, Chem Lett, 2004, 33, 572–573. [CrossRef]
- Mellmann, D., Sponholz, P., Junge, H., Beller, M., Formic acid as a hydrogen storage material – development of homogeneous catalysts for selective hydrogen release, Chem Soc Rev, 2016, 45, 3954–3988. [CrossRef]
- Sordakis, K., Tang, C., Vogt, L.K., Junge, H., Dyson, P.j., Beller, M., Laurenczy, G., Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols, Chem Rev, 2018, 118, 372–433. [CrossRef]
- Onishi, N., Laurenczy, G., Beller, M., Himeda, Y., Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol, Coord Chem Rev, 2018, 373, 317–332. [CrossRef]
- Sun, Q., Wang, N., Xu, Q., Yu, J., Nanopore-Supported Metal Nanocatalysts for Efficient Hydrogen Generation from Liquid-Phase Chemical Hydrogen Storage Materials, Adv Mater, 2020, 32. [CrossRef]
- Xie, W., Schlücker, S., Surface-enhanced Raman spectroscopic detection of molecular chemo- and plasmo-catalysis on noble metal nanoparticles, Chemical Communications, 2018, 54, 2326–2336. [CrossRef]
- He, N., Li, Z. H., Palladium-atom catalyzed formic acid decomposition and the switch of reaction mechanism with temperature, Physical Chemistry Chemical Physics, 2016, 18, 10005–10017. [CrossRef]
- Navlani-García, M., Mori, K., Salinas-Torres, D., Kuwahara, Y., Yamashita, H., New approaches toward the hydrogen production from formic acid dehydrogenation over pd-based heterogeneous catalysts, Front Mater, 2019, 6, 439363. [CrossRef]
- Mori, K., Masuda, S., Tanaka, H., Yoshizawa, K., Che, M., Yamashita, H., Phenylamine-functionalized mesoporous silica supported PdAg nanoparticles: a dual heterogeneous catalyst for formic acid/CO2 -mediated chemical hydrogen delivery/storage, Chemical Communications, 2017, 53, 4677–4680. [CrossRef]
- Jin, M. H., Park, J. H., Oh, D., Park, J. S., Lee, K. Y, Lee, D. W., Effect of the amine group content on catalytic activity and stability of mesoporous silica supported Pd catalysts for additive-free formic acid dehydrogenation at room temperature, Int J Hydrogen Energy, 2019, 44, 4737–4744. [CrossRef]
- Bulut, A., Yurderi, M., Karatas, Y., Zahmakiran, M., Kivrak, H., Gulcan, M., Kaya, M., Pd-MnOx nanoparticles dispersed on amine-grafted silica: Highly efficient nanocatalyst for hydrogen production from additive-free dehydrogenation of formic acid under mild conditions, Appl Catal B, 2015, 164, 324–333.
- Wen, M., Mori, K., Kuwahara, Y., Yamashita, H., Plasmonic Au@Pd nanoparticles supported on a basic metal-organic framework: Synergic boosting of H2 production from formic acid, ACS Energy Lett, 2017, 2, 1–7. [CrossRef]
- Zhang, A., Xia, J., Yao, Q., Lu, Z. H., Pd–WOx heterostructures immobilized by MOFs-derived carbon cage for formic acid dehydrogenation, Appl Catal B, 2022, 309.
- Sun, Q., Chen, B., Wang, N, He, Q., Chang, A., Yang, C.M,. Asakura, H., Tanaka, T., Hülsey, M.J, Yu, J, Yan, N., Zeolite-Encaged Pd–Mn Nanocatalysts for CO2 Hydrogenation and Formic Acid Dehydrogenation, Angewandte Chemie International Edition, 2020, 59, 20183–20191.
- J. Li, Chen, W., Zhao, H., Zheng, X., Wu, L., Pan, H., Zhu, J., Chen, Y., Lu, j., Size-dependent catalytic activity over carbon-supported palladium nanoparticles in dehydrogenation of formic acid, J Catal, 2017, 352, 371–381. [CrossRef]
- Mori, K., Hara T., Mizugaki, T., Ebitani, K., Kaneda, K., Hydroxyapatite-supported palladium nanoclusters: A highly active heterogeneous catalyst for selective oxidation of alcohols by use of molecular oxygen, J Am Chem Soc, 2004, 126, 10657–10666. [CrossRef]
- Navlani-García, M., Mori, K., Wen, M., Kuwahara, Y., Yamashita,H., Size Effect of Carbon-Supported Pd Nanoparticles in the Hydrogen Production from Formic Acid, BCSJ, 2015, 88, 1500–1502. [CrossRef]
- Navlani-García, M., Mori, K., Nozaki, A., Kuwahara, Y., Yamashita, H., Investigation of Size Sensitivity in the Hydrogen Production from Formic Acid over Carbon-Supported Pd Nanoparticles, ChemistrySelect, 2016, 1, 1879–1886. [CrossRef]
- Navlani-García, M., Salinas-Torres,D., Mori, K., Léonard, A., Kuwahara, Y., Job, N., Yamashita, H., Insights on palladium decorated nitrogen-doped carbon xerogels for the hydrogen production from formic acid, Catal Today, 2019, 324, 90–96. [CrossRef]
- Chaparro-Garnica, J.A., Navlani-García, M., Salinas-Torres, D., Morallón, E., Cazorla-Amorós, D., Highly Stable N-Doped Carbon-Supported Pd-Based Catalysts Prepared from Biomass Waste for H2 Production from Formic Acid, ACS Sustain Chem Eng, 2020, 8, 15030–15043. [CrossRef]
- Chaparro-Garnica, J.A., Navlani-García, M., Salinas-Torres, D., Morallón, E., Cazorla-Amorós, D., H2 production from formic acid using highly stable carbon-supported Pd-based catalysts derived from soft-biomass residues: Effect of heat treatment and functionalization of the carbon support, Materials, 2021, 14, 6506. [CrossRef]
- Shao, X., Miao, X., Tian, F., Bai, M., Guo, X., Wang, W., Zhao, Z., Ji, X., Li, M., Deng, F., Amine-functionalized hierarchically porous carbon supported Pd nanocatalysts for highly efficient H2 generation from formic acid with fast-diffusion channels, Journal of Energy Chemistry, 2023, 76, 249–258. [CrossRef]
- Masuda, S., Mori, K., Futamura, Y., Yamashita, H., PdAg Nanoparticles Supported on Functionalized Mesoporous Carbon: Promotional Effect of Surface Amine Groups in Reversible Hydrogen Delivery/Storage Mediated by Formic Acid/CO2, ACS Catal, 2018, 8, 2277–2285. [CrossRef]
- Kim, Y., Kim, D. H., Hydrogen production from formic acid dehydrogenation over a Pd supported on N-doped mesoporous carbon catalyst: A role of nitrogen dopant, Appl Catal A Gen, 2020, 608, 117887. [CrossRef]
- Navlani-García M., Mori, K., Kuwahara, Y., . Yamashita, H., Recent strategies targeting efficient hydrogen production from chemical hydrogen storage materials over carbon-supported catalysts, NPG Asia Materials, 2018, 10, 277–292.
- Salinas-Torres, D., Navlani-García, M., Mori, K., Kuwahara, Y., Yamashita, H., Nitrogen-doped carbon materials as a promising platform toward the efficient catalysis for hydrogen generation, Appl Catal A Gen, 2019, 571, 25–41. [CrossRef]
- Nishchakova, A. D., Bulushev, D.A., Stonkus, O.A., Asanov, I.P., Ishchenko, A.V., Okotrub, A.V., Bulusheva, L.G., Effects of the Carbon Support Doping with Nitrogen for the Hydrogen Production from Formic Acid over Ni Catalysts, Energies, 2019, 12, 4111. [CrossRef]
- Yao, M., Liang, W., Chen, H., Zhang X., Efficient Hydrogen Production from Formic Acid Using Nitrogen-Doped Activated Carbon Supported Pd, Catal Letters, 2020, 150, 2377–2384. [CrossRef]
- Wang, H., Gu, X.K., Zheng, X., Pan, H., Zhu, J., Chen, S., Cao, L., Li, W-X., Lu, J., “Disentangling the size-dependent geometric and electronic effects of palladium nanocatalysts beyond selectivity,” Sci Adv, 2019, 5. [CrossRef]
- Jeon, M., Han, D.J., Lee, K-S., Choi, S.H, Han, J., Nam, S.W., Jang, S.C., Park, H.S., Yoon, C.W., Electronically modified Pd catalysts supported on N-doped carbon for the dehydrogenation of formic acid, Int J Hydrogen Energy, 2016, 15453–15461. [CrossRef]
- Chen, Y., Li, X., Wei, Z., Mao, S., Deng, J., Cao, Y., Wang, Y., Efficient synthesis of ultrafine Pd nanoparticles on an activated N-doping carbon for the decomposition of formic acid, Catal Commun, 2018, 108, 55–58. [CrossRef]
- Shao, X., Miao, X., Tian, F., Bai, M., Guo, X., Wang, W., Zhao, Z., Ji, X., Li, M., Deng, F., Amine-functionalized hierarchically porous carbon supported Pd nanocatalysts for highly efficient H2 generation from formic acid with fast-diffusion channels, Journal of Energy Chemistry, 2023, 76, 249–258. [CrossRef]
- Bulushev, D.A., Zacharska, M.,Shlyakhova, E.V., Chuvilin, A.L., Guo Y., Beloshapkin, S., Okotrub, A.V., Bulesheva, L.G., “Single Isolated Pd2+ Cations Supported on N-Doped Carbon as Active Sites for Hydrogen Production from Formic Acid Decomposition,” ACS Catal, 2016, 6, 681–691. [CrossRef]
- Bulushev, D. A., Golub, F.S., Trubina, S.V, Zvereva, V.V, Gerasimov, E.Y., Prosvirin, I.P, Navlani-García, M., Jena, H.S., Pd Active Sites on Covalent Triazine Frameworks for Catalytic Hydrogen Production from Formic Acid, ACS Appl Nano Mater, 2023, 6, 13551–13560. [CrossRef]
- Liu, D., Gao, Z. Y., Wang, X. C., Zeng, J., Li, Y. M., DFT study of hydrogen production from formic acid decomposition on Pd-Au alloy nanoclusters, Appl Surf Sci, 2017, 426, 194–205. [CrossRef]
- Navlani-García, M., Salinas-Torres, D., Cazorla-Amorós, D., Hydrogen Production from Formic Acid Attained by Bimetallic Heterogeneous PdAg Catalytic Systems, Energies, 2019,12, 4027. [CrossRef]
- Navlani-García, M., Mori, K., Nozaki, A., Kuwahara, Y., Yamashita, H., Screening of Carbon-Supported PdAg Nanoparticles in the Hydrogen Production from Formic Acid, Ind Eng Chem Res, 2016, 55, 7612–7620. [CrossRef]
- Kim Y., Kim D. H., Elucidating the alloying effect of PdAg/CNT catalysts on formic acid dehydrogenation with kinetic isotope effect, Molecular Catalysis, 2023, 547, 113343.
- Nabid, M. R., Bide, Y., Etemadi, B., Ag@Pd nanoparticles immobilized on a nitrogen-doped graphene carbon nanotube aerogel as a superb catalyst for the dehydrogenation of formic acid, New Journal of Chemistry, 2017, 41, 10773–10779. [CrossRef]
- Chaparro-Garnica, J., Navlani-García, M., Salinas-Torres, D., Berenguer-Murcia, Á., Morallón, E., Cazorla-Amorós, D., Efficient production of hydrogen from a valuable CO2-derived molecule: Formic acid dehydrogenation boosted by biomass waste-derived catalysts, Fuel, 2022, 320, 123900. [CrossRef]
- Jiang, Y., Fan, X., Chen, M., Xiao, X., Zhang, Y., Wang, C., Chen, L., AuPd Nanoparticles Anchored on Nitrogen-Decorated Carbon Nanosheets with Highly Efficient and Selective Catalysis for the Dehydrogenation of Formic Acid, Journal of Physical Chemistry C, 2018, 122, 4792–4801. [CrossRef]
- Hong, W., Kitta, M., Tsumori, N., Himeda Y., Autrey T., Xu Q., Immobilization of highly active bimetallic PdAu nanoparticles onto nanocarbons for dehydrogenation of formic acid, J Mater Chem A Mater, 2019, 7, 18835–18839. [CrossRef]
- Navlani-García, M., Salinas-Torres, D., Mori, K., Kuwahara, Y., Yamashita, H., Enhanced formic acid dehydrogenation by the synergistic alloying effect of PdCo catalysts supported on graphitic carbon nitride, Int J Hydrogen Energy, 2019, 44, 28483–28493. [CrossRef]
- Tamarany, R., Shin, D.Y., Kang, S., Jeong, H., Kim, J., Kim, J., Yoon, C.W., Lim, D-H., Formic acid dehydrogenation over PdNi alloys supported on N-doped carbon: synergistic effect of Pd–Ni alloying on hydrogen release, Phys. Chem. Chem. Phys., 2021, 23, 11515–11527. [CrossRef]
- 66. Mori, K., Tanaka, H., Dojo, M., Yoshizawa, K., Yamashita, H., “Synergic Catalysis of PdCu Alloy Nanoparticles within a Macroreticular Basic Resin for Hydrogen Production from Formic Acid, Chemistry – A European Journal, 2015, 21, 12085–12092. [CrossRef]
- Yurderi, M., Bulut, A., Zahmakiran, M., Kaya, M., Carbon supported trimetallic PdNiAg nanoparticles as highly active, selective and reusable catalyst in the formic acid decomposition, Appl Catal B, 2014, 160–161, 514–524.
- Wang, Z. L., Ping, Y., Yan, J. M., Wang, H. L., Jiang, Q., Hydrogen generation from formic acid decomposition at room temperature using a NiAuPd alloy nanocatalyst, Int J Hydrogen Energy, 2014, 39, 4850–4856. [CrossRef]
- Dong, Z., Li, F., He, Q., Xiao, X., Chen, M., Wang, C., Fan, X., Chen, L., PdCoNi nanoparticles supported on nitrogen-doped porous carbon nanosheets for room temperature dehydrogenation of formic acid, Int J Hydrogen Energy, 2019, 44, 11675–11683. [CrossRef]
- Liu, D. X., Zhou, Y. T., Zhu, Y. F., Chen, Z. Y., Yan, J. M., Jiang, Q., Tri-metallic AuPdIr nanoalloy towards efficient hydrogen generation from formic acid, Appl Catal B, 2022, 309, 121228. [CrossRef]
- Yan, N., Philippot, K., Transformation of CO2 by using nanoscale metal catalysts: cases studies on the formation of formic acid and dimethylether, Curr Opin Chem Eng, 2018, 20, 86–92. [CrossRef]
- Verma, P., Zhang, S., Song, S., Mori, K., Kuwahara, Y., Wen, M., Yamashita, H., An, T.,, Recent strategies for enhancing the catalytic activity of CO2 hydrogenation to formate/formic acid over Pd-based catalyst, Journal of CO2 Utilization, 2021, 54, 101765. [CrossRef]
- Kumarave, V. l., Bartlett, J., Pillai, S. C., Photoelectrochemical Conversion of Carbon Dioxide (CO2) into Fuels and Value-Added Products, ACS Energy Lett, 2020, 486–519. [CrossRef]
- Sun, Z., Ma T., Tao, H., Fan, Q., Han, B., Fundamentals and Challenges of Electrochemical CO2 Reduction Using Two-Dimensional Materials, Chem, 2017, 3, 560–587. [CrossRef]
- Bulushev, D. A., H. Ross, J. R., Heterogeneous catalysts for hydrogenation of CO2 and bicarbonates to formic acid and formates, Catalysis Reviews, 2018, 60, 566–593.
- Jessop, P. G., Ikariya, T., Noyori, R., Homogeneous Hydrogenation of Carbon Dioxide, Chem Rev, 1995, 95, 259–272. [CrossRef]
- Sun, R., Liao, Y., Bai, S-T., Zhen, M., Zhou, C., Zhang, T., Sels, B.F., Heterogeneous catalysts for CO2 hydrogenation to formic acid/formate: from nanoscale to single atom, Energy Environ Sci, 2021, 14, 1247–1285. [CrossRef]
- Jessop, P. G., Joó F., Tai, C. C., Recent advances in the homogeneous hydrogenation of carbon dioxide, Coord Chem Rev, 2004, 248,. 2425–2442. [CrossRef]
- Álvarez, A., Bansode, A., Urakawa, A., Bavykina, A.V, Wezendonk, T.A, Makkee, M., Gascon, J., Kapteijn, F., Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem Rev, 2017, 117, 9804–9838.
- Shao, X., Xu, J., Huang, Y., Su, X., Duan, H., Wang, X., Zhang, T., Pd@C3N4 nanocatalyst for highly efficient hydrogen storage system based on potassium bicarbonate/formate, AIChE Journal, 2016, 62, 2410–2418.
- Lee, J.H., Ryu, J., Kim, J.Y., Nam, S-W., Han, J.H., Lim, T-H., Gautam, S., Chae, K.H., Yoon, C.W., Carbon dioxide mediated, reversible chemical hydrogen storage using a Pd nanocatalyst supported on mesoporous graphitic carbon nitride, J Mater Chem A Mater, 2014, 2, 9490–9495. [CrossRef]
- Wang, F., Xu, J., Shao, X., Su, X., Huang, Y., Zhang, T., Palladium on Nitrogen-Doped Mesoporous Carbon: A Bifunctional Catalyst for Formate-Based, Carbon-Neutral Hydrogen Storage, ChemSusChem, 2016, 9, 246–251.
- Song, H., Zhang, N., Zhong, C., Liu, Z., Xiao, M., Gai, H., Hydrogenation of CO2 into formic acid using a palladium catalyst on chitin, New Journal of Chemistry, 2017, 41, 9170–9177. [CrossRef]
- Koh, K., Jeon, M., Chevrier, D. M., Zhang, P., Yoon, C. W., Asefa, T. Novel nanoporous N-doped carbon-supported ultrasmall Pd nanoparticles: Efficient catalysts for hydrogen storage and release, Appl Catal B, 2017, 203, 820–828. [CrossRef]
- Yang, G., Kuwahara, Y., Mori, K., Louis, C., Yamashita, H., Pd-cu alloy nanoparticles confined within mesoporous hollow carbon spheres for the hydrogenation of CO2 to formate, Journal of Physical Chemistry C, 2021, 125, 3961–3971. [CrossRef]
- Yang, G., Kuwahara, Y., Mori, K., Louis, C., Yamashita, H., PdAg alloy nanoparticles encapsulated in N-doped microporous hollow carbon spheres for hydrogenation of CO2 to formate, Appl Catal B, 2021, 283, 119628. [CrossRef]
- S. Masuda, K. Mori, Y. Futamura, and H. Yamashita, “PdAg Nanoparticles Supported on Functionalized Mesoporous Carbon: Promotional Effect of Surface Amine Groups in Reversible Hydrogen Delivery/Storage Mediated by Formic Acid/CO2,” ACS Catal, 2018, 8, 2277–2285. [CrossRef]
- Nguyen, L.T.M., Park, H., Banu, M., Kim, J.Y., Youn, D. H., Magesh, G., Kim, W. Y., Lee, J.S., Catalytic CO2 hydrogenation to formic acid over carbon nanotube-graphene supported PdNi alloy catalysts, RSC Adv, 2015, 5, 105560–105566. [CrossRef]
- Yang, G., Kuwahara, Y., Mori, K., Louis, C., Yamashita, H., PdAg alloy nanoparticles encapsulated in N-doped microporous hollow carbon spheres for hydrogenation of CO2 to formate, Appl Catal B, 2021, 283, 119628. [CrossRef]
- Su, J., Yang, L., Lu, M., Lin., H., Highly Efficient Hydrogen Storage System Based on Ammonium Bicarbonate/Formate Redox Equilibrium over Palladium Nanocatalysts, ChemSusChem, 2015, 8, 813–816. [CrossRef]
- Zhou, Y., Huang, Y., Jin, B., Luo, X., Liang, Z., Pd Nanoclusters-Based Catalysts with Schiff Base Modifying Carrier for CO2 Hydrogenation to Formic Acid, Ind Eng Chem Res, 2019, 58, 44–52. [CrossRef]
- Bi, Q-Y., Lin, J-D., Liu, Y-M., Du, X-L., Wang, J-Q., He, H-Y., Cao, Y., An Aqueous Rechargeable Formate-Based Hydrogen Battery Driven by Heterogeneous Pd Catalysis, Angewandte Chemie International Edition, 2014, 53, 13583–13587. [CrossRef]
- Soma-Noto, Sachtler, Y. W. M. H., Infrared spectra of carbon monoxide adsorbed on supported palladium and palladium-silver alloys, J Catal, 1974, 32, 315–324.
- Tedsree, K., Li, T., Jones, S., Chan, C. W. A., Yu, K. M. K., Bagot, P. A. J., Marquis, E.A., Smith, G.D.W., Tsang, S. C. E., Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst, Nat Nanotechnol, 2011, 6, 302–307. [CrossRef]
Figure 1.
Chemical H2 storage cycle using formic acid mediated by a catalyst. In green, the hydrogenation CO2 reaction, and in blue, is the formic acid dehydrogenation.
Figure 1.
Chemical H2 storage cycle using formic acid mediated by a catalyst. In green, the hydrogenation CO2 reaction, and in blue, is the formic acid dehydrogenation.
Figure 2.
(a) Relationship between H2 production after 3 h of reaction at 30 ºC and the average NP size. (b) Normalized TOF values calculated on the basis of high-coordinated (terrace) and low-coordinated (edge and corner) as a function of the average Pd NP size. Adapted from [39].
Figure 2.
(a) Relationship between H2 production after 3 h of reaction at 30 ºC and the average NP size. (b) Normalized TOF values calculated on the basis of high-coordinated (terrace) and low-coordinated (edge and corner) as a function of the average Pd NP size. Adapted from [39].
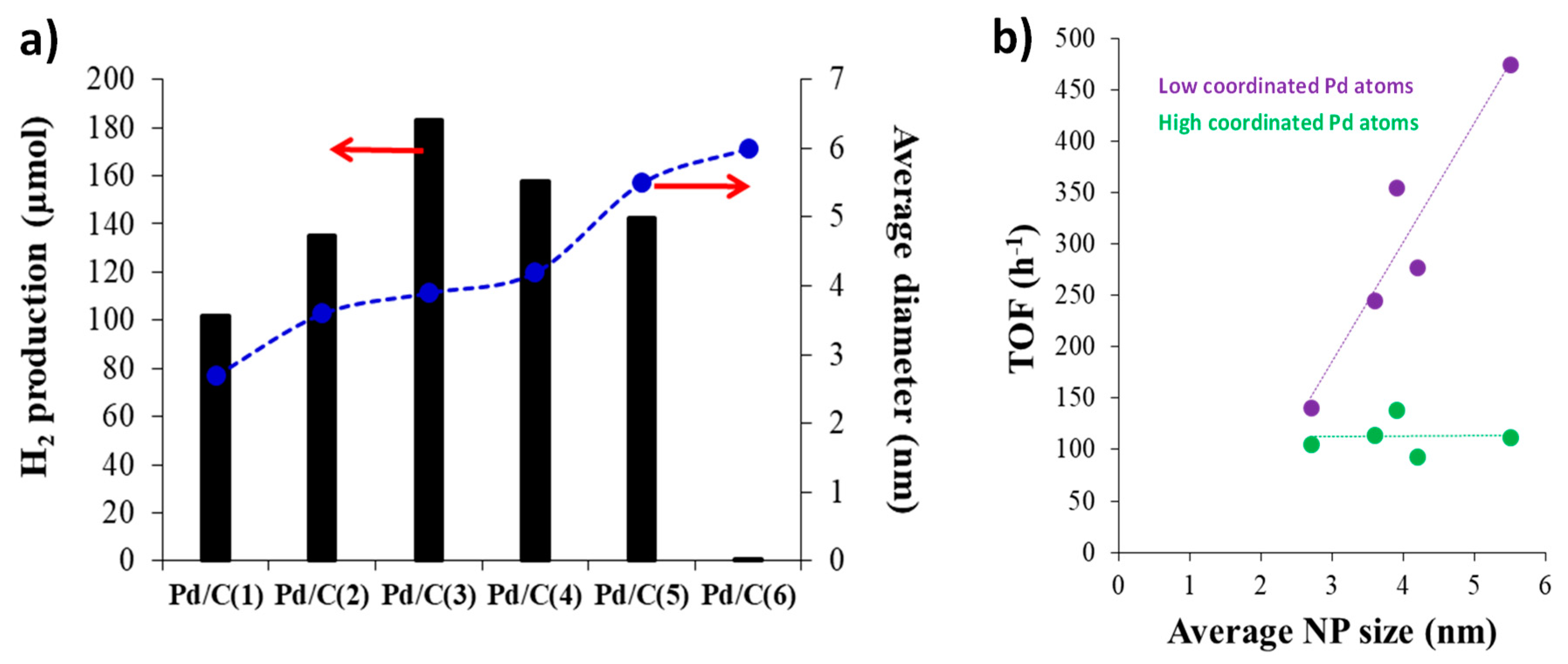
Figure 3.
(a) Time-dependent gas production for various Pd-based catalysts during the decomposition of FA and (b) the related TOF values. (c) Plot of gas generation from FA as a function of time at different temperatures. (d) Arrhenius plot of FA dehydrogenation. (Rection conditions: 5 mL, 1.0 M FA, nPd/nFA = 0.0037). (e) A comparison of the catalysts for additive-free FA dehydrogenation described in the literature. Reprtinted with permisision from [53].
Figure 3.
(a) Time-dependent gas production for various Pd-based catalysts during the decomposition of FA and (b) the related TOF values. (c) Plot of gas generation from FA as a function of time at different temperatures. (d) Arrhenius plot of FA dehydrogenation. (Rection conditions: 5 mL, 1.0 M FA, nPd/nFA = 0.0037). (e) A comparison of the catalysts for additive-free FA dehydrogenation described in the literature. Reprtinted with permisision from [53].
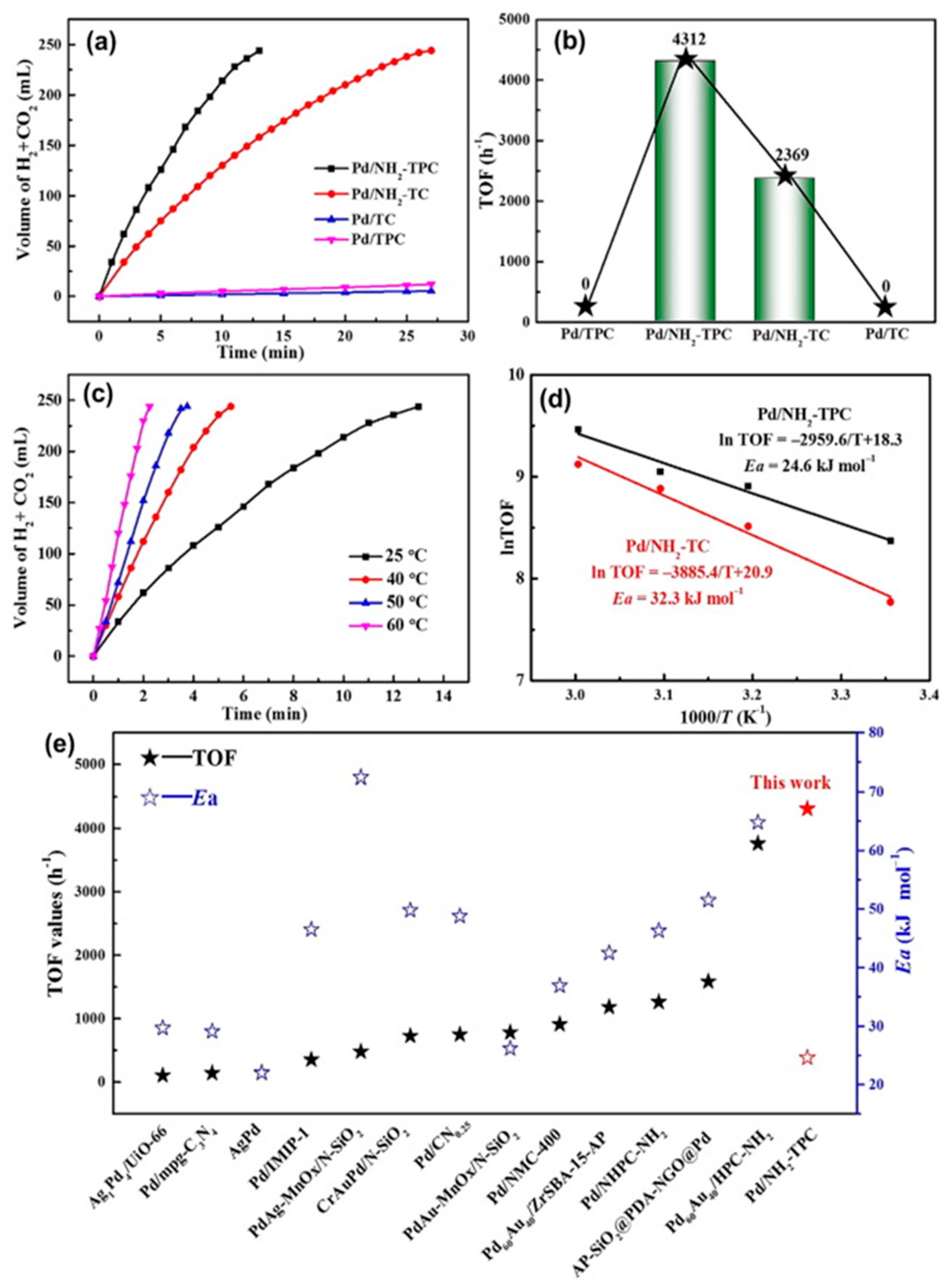
Figure 4.
Schematic illustration for the preparation procedure of AuPd/n-CNS nanocatalyst. Reprinted with permission from [62].
Figure 4.
Schematic illustration for the preparation procedure of AuPd/n-CNS nanocatalyst. Reprinted with permission from [62].
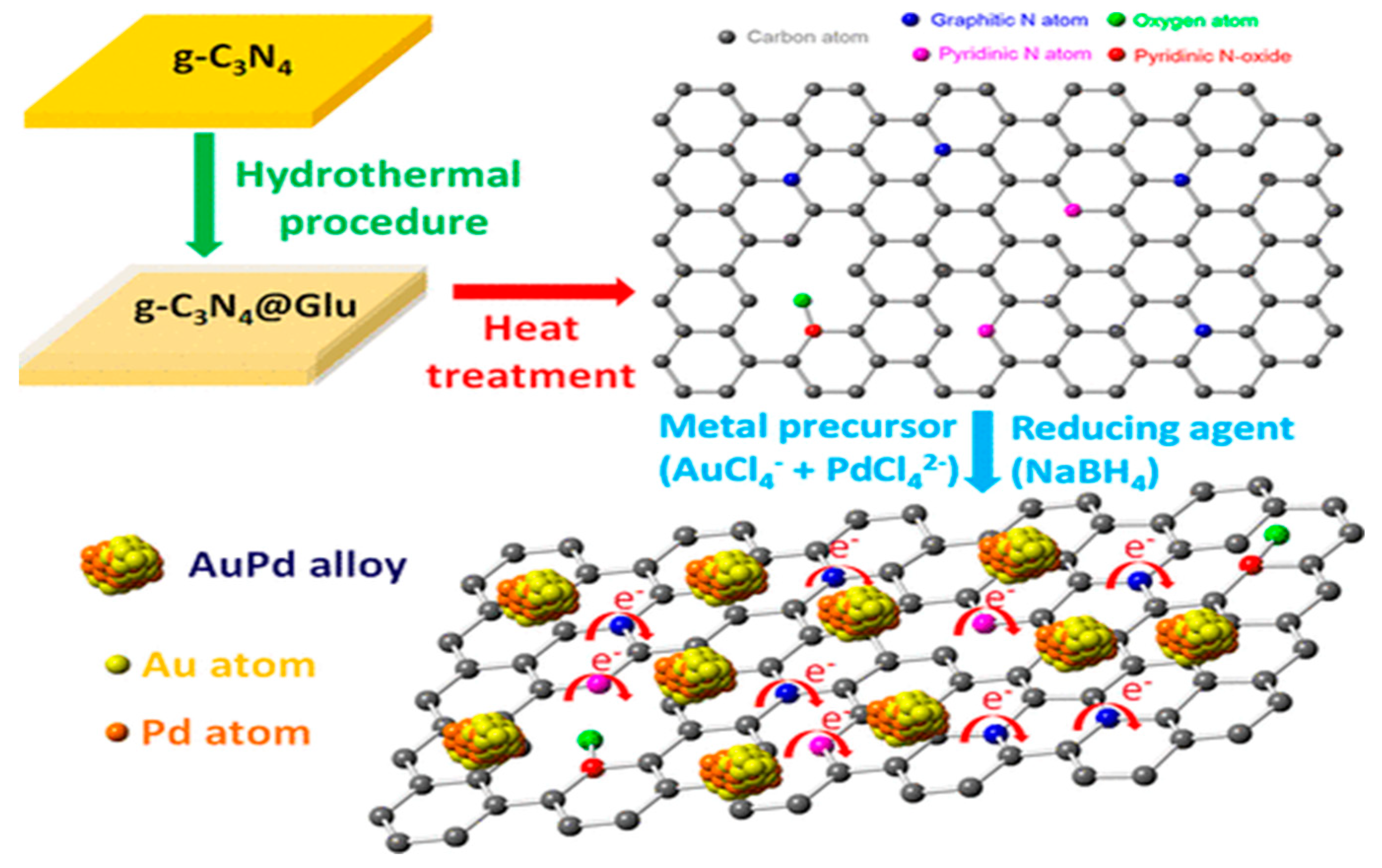
Figure 5.
(a) Possible reaction pathway; (b) Potential energy profiles for dehydrogenation for FA dehydrogenation over Pd43 and Pd22Co21 cluster models. Reprinted with permission from [64].
Figure 5.
(a) Possible reaction pathway; (b) Potential energy profiles for dehydrogenation for FA dehydrogenation over Pd43 and Pd22Co21 cluster models. Reprinted with permission from [64].
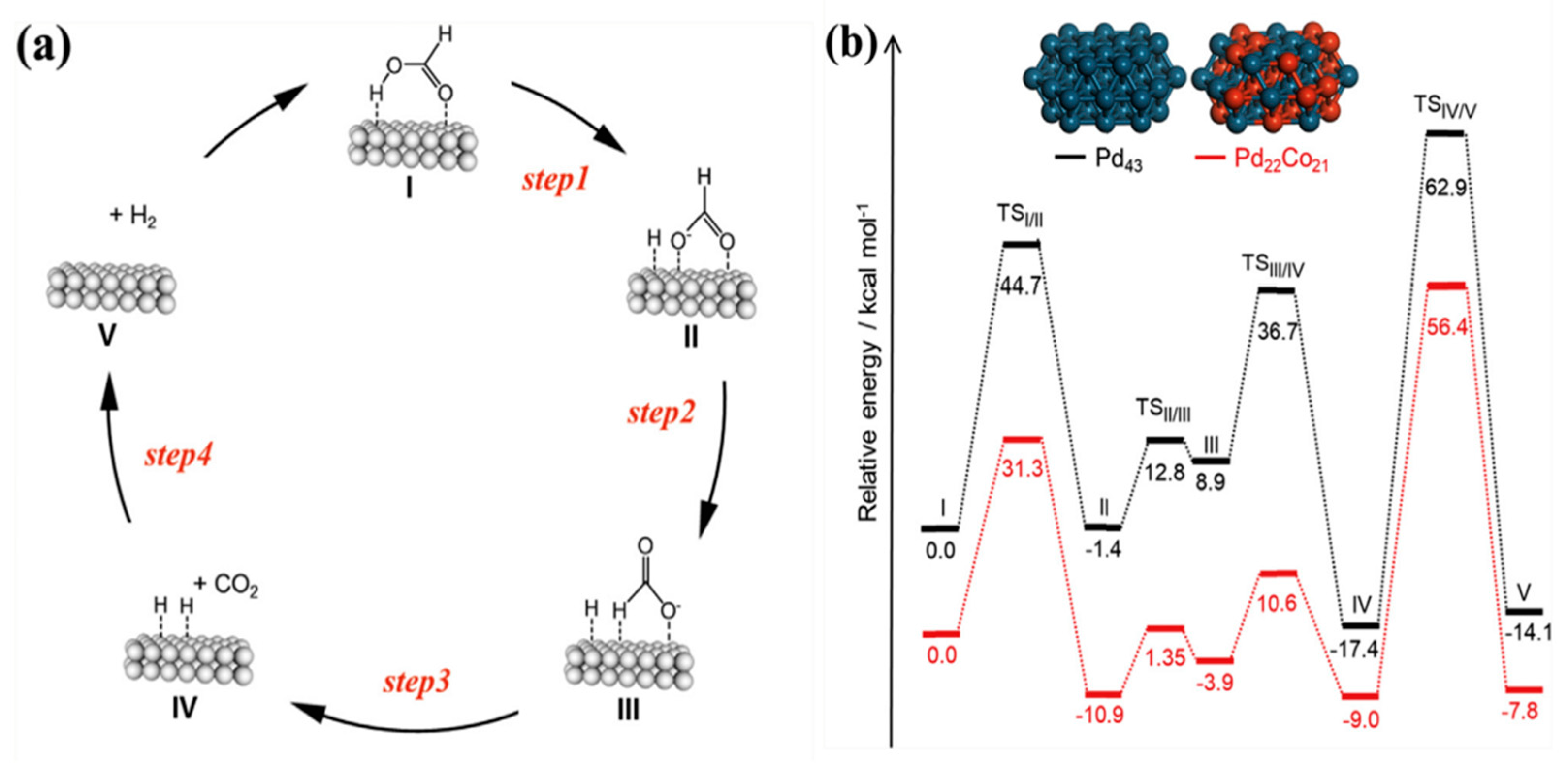
Figure 6.
(a) Gas generation from FA dehydrogenation at 298 K, with the corresponding TOF values inset. (b) Mass spectrometry of released gas from the dehydrogenation of FA over Au0.35Pd0.5Ir0.15/NH2-N-rGO. (c) Continuous recycling tests and (d) 150-day long-term recycling test of Au0.35Pd0.5Ir0.15/NH2-N-rGO toward H2 generation from FA solution, and inset is the photo of catalysts dispersed in the reaction solution on the 1st and 150th day. Reprinted with permission from [70].
Figure 6.
(a) Gas generation from FA dehydrogenation at 298 K, with the corresponding TOF values inset. (b) Mass spectrometry of released gas from the dehydrogenation of FA over Au0.35Pd0.5Ir0.15/NH2-N-rGO. (c) Continuous recycling tests and (d) 150-day long-term recycling test of Au0.35Pd0.5Ir0.15/NH2-N-rGO toward H2 generation from FA solution, and inset is the photo of catalysts dispersed in the reaction solution on the 1st and 150th day. Reprinted with permission from [70].
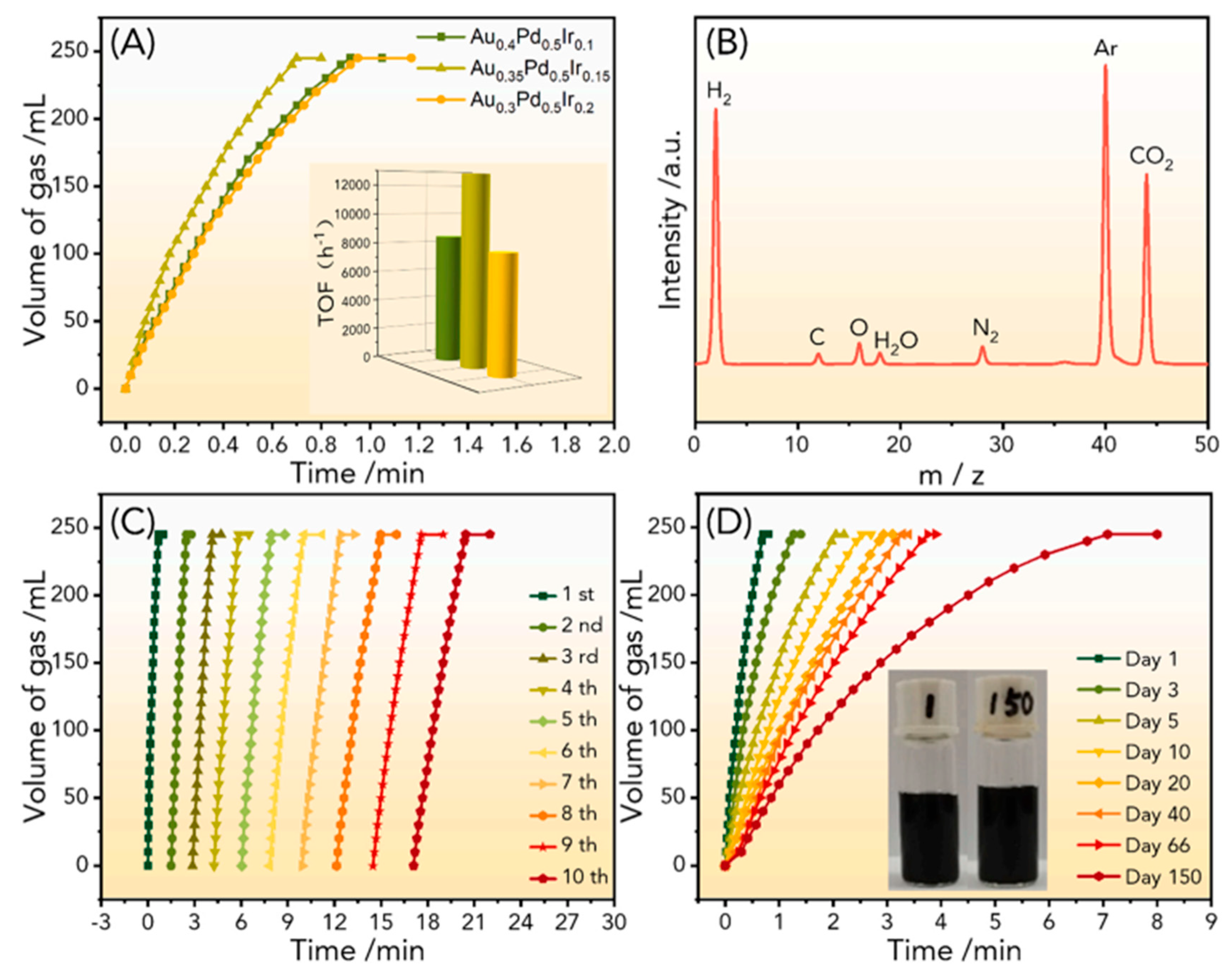
Figure 7.
Possible reaction mechanism of bicarbonate hydrogenation over Pd/mpg-C3N4. Reprinted with permission of [80].
Figure 7.
Possible reaction mechanism of bicarbonate hydrogenation over Pd/mpg-C3N4. Reprinted with permission of [80].
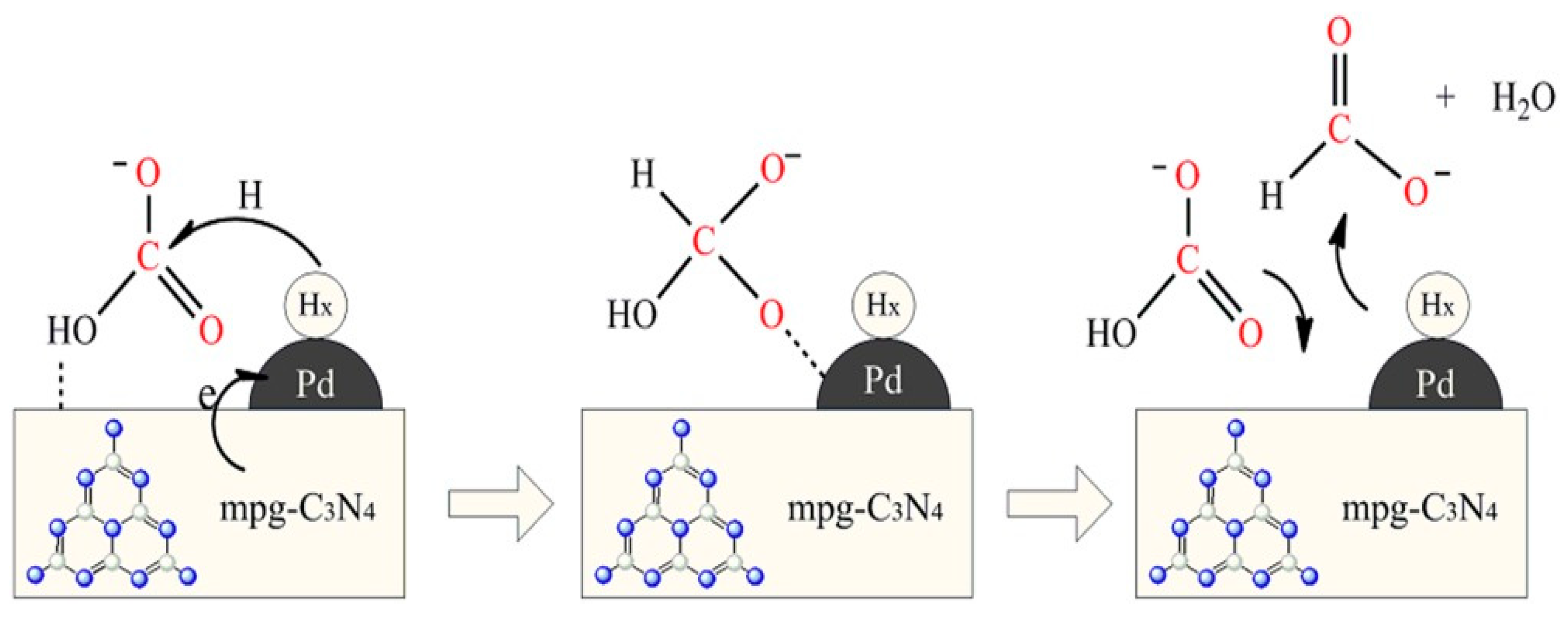
Figure 8.
Scheme of PdAg/amine-MSC synthesis. Reprinted with permission from [87].
Figure 8.
Scheme of PdAg/amine-MSC synthesis. Reprinted with permission from [87].
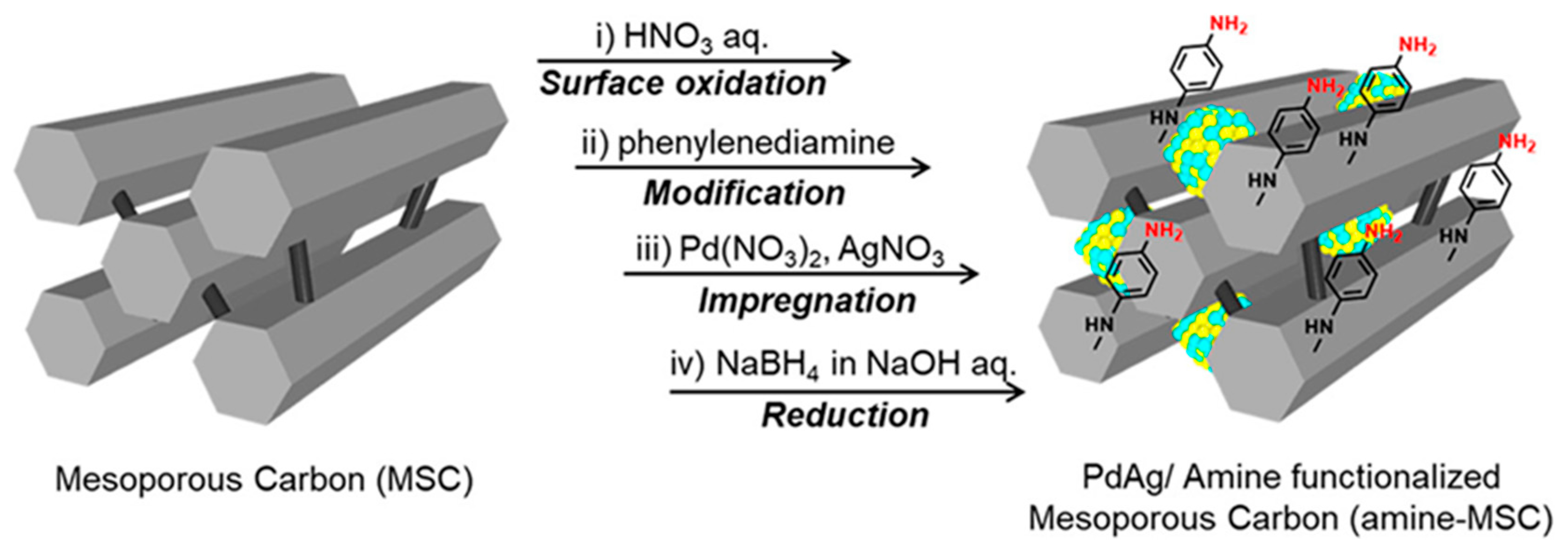
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



