
Submitted:
01 December 2023
Posted:
04 December 2023
You are already at the latest version
Abstract
Wolfenden and his coworkers observed the astronomical numbers for the catalytic proficiency of some enzymes. We connected that pinnacle of biological evolution to the universal thermodynamic evolution. We added or multiplied a random noise with chosen rate constants to explore the correlation between dissipation and enzyme efficiency for ten enzymes: beta-galactosidase, glucose isomerase, β-lactamases from three bacterial strains, ketosteroid isomerase, triosephosphate isomerase, and carbonic anhydrase I, II, and T200H. The turnover number kcat and catalytic efficiency kcat/KM are proportional to the overall entropy production – the main parameter from irreversible thermodynamics. For most enzymes with the Michaelis-Menten type cycle kinetics, the best increase in the forward kcat/KM follows after increasing the equilibrium constant of substrate-enzyme association. The Discussion section emphasizes the role of biological evolution in harvesting order (high enzyme efficiency) from disorder (high noise and dissipation). It also connects the applications of the maximum partial entropy production theorem in optimizing enzyme kinetics (D. Juretić "Bioenergetics - A Bridge across Life and Universe") with the present total entropy production role analysis. De novo enzyme design and various attempts to speed up the rate-limiting catalytic steps may profit from our theoretical insights.
Keywords:
enzyme efficiency
; entropy production
; noise
; evolution
; catalytic cycle
Introduction
Why and how questions about biological evolution are often reduced to the how question, but the answer is still untrackable within realms of biology. We lack the knowledge about prebiotic chemistry and appropriate far-from-equilibrium kinetic and thermodynamic conditions to get better insights into life's origin [1,2,3]. There are more questions than quantitative answers to how biological macromolecules perform amazing catalytic feats [4,5], let alone how life emerged from inanimate matter [6,7]. This work proposes an intimate connection between why and how questions through joint thermodynamic and biological evolution. We shall show here that entropy production and kinetic parameters for enzyme performance are tightly coupled when discrete transitions are allowed between nonequilibrium quasi-steady states. Random mutations leading to amino acid substitutions are the biological mechanism for discrete changes, which can speed up evolution in combination with the selection of beneficial mutations. Does the selection process favor higher dissipation and better catalytic efficiency? The present-day enzymes are already highly evolved but may still possess hidden evolutionary potential. The simulation of noisy microscopic transitions through discrete time passage steps should help uncover it.
The passage of time is an in-build sense we all possess. Billions of our cells can cooperate in distinguishing the past from the future and in predicting the future to some extent. Unlike other senses, the passage of time mechanism remains refractory to clear insights from physical and biological research. When focused on research about elementary particles, physicists use basic equations for Newtonian or quantum mechanics, which remain unchanged when the past and future are interchanged. Irreversible time passage looks to them like an illusion. At the same time, evolution with time is an essential insight from physics, just as it is the basic tenet in biology. Still, physicists tend to relegate it to statistical mechanics and thermodynamics, the branches of physics dealing with frequently misunderstood concepts of entropy and entropy production. These concepts emerge as a holistic aid for understanding an average response when a large number of particles are exposed to internal and external perturbations. Weak or absent interactions among particles are usually assumed. Thermodynamic equilibrium or close-to-equilibrium situation is also often assumed in physics. However, nothing of interest in biology happens in the thermodynamic equilibrium when entropy production vanishes while evolution and time passage seemingly cease. Entropy production is tightly connected to the speed of evolution and irreversible time passage. There would be no evolution without dissipation.
A constant driving force is the most frequent assumption for external perturbation because it ensures that a steady, nonequilibrium state will be spontaneously established for an open system. In almost all cases, the total entropy production of the system can be calculated as the sum of force-flux products after identifying all fluxes and corresponding forces. A reader can notice that such a definition for entropy production implies a system's ability to transform the free-energy-rich input power into irreversible internal processes and the free-energy-poor output with increased entropy. Temporary free-energy storage is possible through some irreversible internal processes. Thus, we should consider entropy production as something other than a useless free-energy transformation that generates only slight heating of the system's environment. This work promotes the opposite viewpoint, namely, that high entropy production is an investment that acts as a catalyst for creating self-organized dissipative structures [8] and an enhanced turnover of organic molecules.
In biological research, one can ask about the most basic level at which we can still see a considerable increase in entropy production as the surrogate for irreversible time passage. Enzymes are housekeeping cellular macromolecules performing all biosynthetic and moving functions. Free-energy collection and transformation into the ATP currency to satisfy the cell needs would be impossible without specialized enzymes. Even the simplest bacterial cells or organelles like mitochondria in our cells can efficiently connect harvesting of free-energy sources to electron current and proton ejection, creating a strong electric field. Proton passage through a topologically closed biological membrane is then coupled to the rotatory motion of the enzyme ATP-synthase and ATP synthesis. Ultimately, all of the mentioned fluxes also produce heat, which must be exported to the environment for the cell to survive in a quasi-steady state. Naturally, a small percentage of collected free energy is temporarily stored in the cell to prepare for hard times or replication and proliferation when free-energy sources are plentiful. A whole orchestra of enzymes, memory-storage molecules (DNA and RNA), and membrane-forming molecules (lipids) must cooperate for cellular bioenergetics to continue its smooth operation [2].
During biological evolution, enzymes achieved extraordinary catalytic efficiency. There are indications that the first several hundred million years of the Archaean age were enough to develop enzymes catalyzing the same reaction with many orders of magnitude higher activity than the best inorganic catalysts [9,10,11]. That is how life accelerated spontaneous inorganic evolution billion or even billion-billion times [10,12,13,14,15,16]. The catalytic mechanism includes a substantial entropy loss on binding substrate to an enzyme, which provides free energy for the reaction rate acceleration [17]. Therefore, internal and external entropy changes must be considered in addition to free energy changes to understand the life-driven evolution of complex macromolecular structures. This work will focus on how an irreversible entropy increase can contribute to higher catalytic efficiency.
Surprisingly, inquiries about whether enzyme efficiency has anything to do with total entropy production attracted scant attention in the published literature. Offered answers are unconvincing and unclear, ranging from generalizations based on the study of two-three points and only one enzyme [18] to the lack of overall correlation between reaction thermodynamics and performance parameters for a large number of enzymes [19]. Biological and thermodynamic evolution have often been treated as separate and disconnected occurrences. To many biologists and some physicists, it looked evident that biological evolution led to a decrease in entropy, while thermodynamic evolution can lead only to an increase in entropy. Consequently, they could agree with the expectation that biological evolution should strive to produce a minimal amount of entropy. Published opposite conclusions about maximal entropy production during biological evolution [1,20] did not prevail. A reader can find rich literature sources for both viewpoints in the recent book [2].
Our previous publications (collected in [2]) examined how to model enzyme efficiency increase when partial entropy production is maximized in rate-limiting catalytic steps. Enzymes with a uni-uni catalytic mechanism convert a single substrate into a single product (Michaelis-Menten kinetics [21,22]). Their enzyme efficiency is defined as the ratio kcat/KM, where kcat is the catalytic constant, and KM is the Michaelis-Menten constant. KM is interpreted as the affinity of the enzyme to the substrate. The other name for kcat is the turnover number or cycle completion time. The other name for kcat/KM is the specificity or catalytic constant. The kcat/KM value has units M-1s-1 and can be huge for the most efficient enzymes. Thus, it should not be confused with the dimensionless efficiency from physics that is restricted to 0-1 numbers (0 to 100%).
Michaelis-Menten kinetics [21,22] survived more than 100 years of enzyme catalysis studies [23,24]. It is a good enough reason why a better connection with nonequilibrium thermodynamic parameters should be desirable. While efficiency decreases when entropy production is increased in physical systems, there is no reason to expect enzyme efficiency to decrease when catalytic activity is associated with increased partial or total dissipation. We shall explore in this paper the relationship between enzyme efficiency and its total entropy production after discrete jumps among quasi-steady states and in simulated dynamical changes in concentration of substrates, products, free enzymes, and enzyme complexes with ligands. Both quantities change after introducing variations in the microscopic rate constants. Variations we introduced are the stepway increase in chosen rate constants, uniform, or Gaussan noise. Remarkably, when equilibrium constants are not altered in catalytic steps, almost perfect proportionality is revealed between enzyme efficiency and total entropy production.
To avoid generalities, we examined the well-defined short-term evolution of chosen enzymes, their substrates, and products in the system devoid of other biological molecules. The known mechanism of action and all microscopic rate constants calculated from the experimental data were our main criteria for selecting the enzymes. We set up the initial nonequilibrium state by choosing out-of-equilibrium substrate and product concentrations. Firstly, we reproduced measured kinetic and corresponding performance parameters using different software tools. The following step was to study possible improvements in the performance parameters in our simulations. Allowing for normal noise in microscopic rate constants has several advantages. Firstly, it is a more realistic description of in vivo biochemical kinetics in a highly noisy cellular microenvironment. Secondly, it considers that experimental data are signals extracted from noise. Thirdly, we generalized the Michaelis-Menten kinetics from biochemical textbooks so that all catalytic steps are reversible. The reversibility requirement is necessary for the thermodynamic treatment [25]. Still, it does not prevent the highly irreversible nature of some catalytic steps and does not mask the enzyme dynamics when the usual quasi-steady state approximation is employed. Fourthly, it allows faster exploration of rate constant combinations associated with higher enzyme efficiency. The last but not the least important advantages of taking noise into account are the implications of coupled increases for entropy production and enzyme efficiency during biological evolution.
The proportionality between the biochemical and physical description of the enzyme's hallmarks (kcat/KM and entropy production) does not depend on noise distribution or the programming language used to incorporate noise. However, homeostatic conditions must be assumed to maintain the same proportionality line. These are physiological conditions for in vivo enzyme activity or quasi-steady state constraints for bach reactor experiments, achieved by continuously removing excess products and adding substrates (the chemiosmotic situation). In most of the presented illustrations, we regarded dissipation as the cause (x-axis values) and catalytic efficiency as the consequence (y-axis values). In the cases when some analytical function is a good fit for the efficiency-to-dissipation dependence, its shape is highly dependent on imposed constraints and on the manner of introducing noise in the system.
Methods
Selected Enzymes for the Computational Modeling
We selected the following enzymes for the simulations of their kinetics: Escherichia coli β-galactosidase (βG, 3.2.1.23) with rate constants published and estimated [26], Streptomyces murinus glucose isomerase (GI, EC 5.3.1.5) with rate constants published and calculated [27,28], β-lactamases (EC 3.5.2.6) from three bacterial strains Staphylococcus aureus, Escherichia coli, and Bacillus cereus enzymes (respectively labeled as PC1, RTEM, and Lac-1) with rate constants published and calculated [29,30], Commamonas testosteroni ketosteroid isomerase (KSI, 5.3.3.1) with estimated rate constants [31], rabbit muscle triosephosphate isomerase (TPI, EC 5.3.1.1) with rate constants from experimental data [32], and human carbonic anhydrase I and II (CAI, and CAII, EC 4.2.1.1) ) with rate constants calculated from experimental data [33].
Description of Enzyme Kinetics in Terms of Nonequilibrium Thermodynamics
To evaluate the enzyme efficiency and its total entropy production in a quasi-steady state and simulated dynamical changes in concentration of substrates, products, free enzyme, and enzyme complexes with ligands, we used the Hill’s diagram method [34,35]. Namely, each enzyme can be found in different states, either as free or in complexes, among which possible transitions are shown in Figure 1. The first-order rate constants ki characterize transitions, where i is odd in the forward direction and even in the backward direction. For the binding transitions with the substrate or product, we use ki=ki*[S] and kj=kj*[P], where ki* and kj* are the second-rate constants and [S] and [P] are concentrations of the substrate and product. The equilibrium constant Ki in the ith catalytic step is defined as the forward-to-backward rate constant ratio Ki= k2i-1/ k2i.
Entropy production for an enzyme reaction with a single cycle is given by
where J is the steady-state overall reaction flux of a reaction, X is overall steady-state thermodynamic force, and T is the absolute temperature assumed to be constant.
Reaction flux J is a function of the forward and backward reaction rate constants. For instance,
for the two-state model shown in Figure 1a),
for the three-state model shown in Figure 1b) and
for the four-state model shown in Figure 1c).
The thermodynamic force equals the sum of forces in each transition
where is the thermodynamic force of the transition i→i+1. For the two-state model
Thus,
where K= K1∙K2 is the equilibrium constant. Here R is the gas constant. For the three-state model
Then the overall thermodynamic force is
where K= K1∙K2∙K3.
In the case of four states, where
and with i given by (5),
the overall thermodynamic force becomes
where K= K1∙K2∙K3∙K4.
Hill's equations above are valid only for the steady-state kinetics. However, these steady states can be very far from equilibrium, which is the primary advantage over applications of classical irreversible thermodynamics to linear departures from thermodynamic equilibrium. All enzymes examined in this paper exhibit a nonlinear relationship between fluxes and forces. That is one reason why catalytic efficiency for most of them is very high. We also assumed that all examined systems can jump among quasi-steady states in deterministic or stochastic ways. Random state changes are expected in noisy and crowded environments typical of any living cell. Agent-based modeling better accounts for the need to consider noisy dynamics while preserving mass conservation for all forms of ligands and different enzyme conformations. Thus, we performed our simulations using the NetLogo and FORTRAN computer languages and verified for each case and each enzyme that the results mostly agreed.
Further, catalytic constants (kcat), Michaelis-Menten constants (KM), and specificity constant (kcat/KM) for all three schemes for the enzyme reactions shown in Figure 1 can be defined. Namely, for the two-state model [36]
for the three-state model [37,38]
and the four-state model [39,40]
The dissipation is defined as the product of flux and force divided by RT and expressed in the units or inverse seconds (s-1). At the constant temperature, the absolute temperature T makes the only difference between the entropy production and the dissipation function.
Software and Programs We Used in this Paper
We introduced normal noise in Hill's diagram method for two, three and four states using Box-Muller transform [41]. For FORTRAN programs, we verified that no matter how much noise was introduced, all results and all the data inserted in corresponding figures are exactly reproducible when the same program is repeatedly run for the same number of inter-state jumps. Box-Muller transform [41]
or
was used for generating normal noise in the rate constant ki= kiexpgi, where s1 and s2 are random numbers chosen from the unit interval (0,1) obtained by the standard FORTRAN generator random_number and kiexp is its experimental value. For some FORTRAN programs, shift = +1 or shift = +2 is used instead of shift = 0 to avoid negative numbers for rate constants. When random numbers s1 and s2 were called only once the corresponding Box-Muller transform was identical for rate constants to which we applied it. Noise was then canceled in ratios of selected rate constants. For instance, the expressions (kcat/KM)/Dissipation (the slope of the kcat/KM dependence on dissipation) from the Appendix contain only the ratios of rate constants. Thus, the slope never changes if random numbers s1 and s2 are called once. It resulted in perfect proportionality between catalytic efficiency and entropy production when equilibrium constants for all catalytic steps are fixed.
Noise survived only in expressions containing some of the selected constants that could not be rendered as belonging to such ratios. In other programs, random numbers s1 and s2 were called for each of the selected rate constants, and there was no noise cancelation in their ratios. In some cases, we used the Box-Muller transform to generate noise in selected equilibrium constants using the expression Ki= Kiexpgi. The legend of each figure specified how we used the Box-Muller transform to present the results. When the implicit assumption is that noise does not exist, we used the stepwise increase of the selected rate constant to cover the range, which included the observed ki value.
While FORTRAN programs do not need an introduction, agent-based modeling does [42,43,44,45]. Modeling flexibility, inherent dynamics, the ability to model individual behavior, spatial consideration, and the logical entrance of the complexity and noise in the system are some advantages of mimicking biological processes with agent-based modeling. The NetLogo (http://ccl.northwestern.edu/netlogo/) is a multi-agent simulation environment simple to use and suitable for modeling stochastic dynamics of biological processes [46,47,48,49]. We used the same parent NetLogo source code as the inspiration for all our NetLogo programs. It is the „Enzyme Kinetics“ created by Stieff and Wilensky in 2001: https://ccl.northwestern.edu/netlogo/models/EnzymeKinetics. The software simulates the traditional Michaelis-Menten model for enzyme kinetics with reversible E+S↔ES transition and irreversible complex dissociation ES→E+P. We extended it to all reversible transitions, additional conformational states EZ, EP (see Figure 1), and noisy rate constants by using a broader usage of the NetLogo tools as described and regularly updated by Prof. Wilensky's group [50].
Results
Triosephosphate Isomerase (TPI): The Favorite Enzyme for Computational Optimization of Michaelis-Menten Type Kinetics
Triosephosphate isomerase (TPI, EC 5.3.1.1) is an essential enzyme in glycolysis [51,52]. Its central housekeeping role is very fast catalytic interconversion of dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (GAP). There would be no net yield of ATP from anaerobic glucose metabolism without the TPI forward activity (DHAP → GAP). Of all enzyme-catalyzed reactions, the free-energy profile was first determined for TPI [53]. The seminal works of Jeremy Knowles [32], John Albery [54], and other authors described the TPI as a perfect enzyme in the sense that it is the perfectly evolved enzyme with catalytic efficiency close to the diffusion limit. In 1984, John Richard [55] estimated that kcat/KM for TPI increased 3∙1010 times compared to the inorganic DHAP to GAP conversion. Enzyme efficiency inside the diffusion limit was confirmed for the wild-type TPI enzymes isolated from many species [56].
As a reversible enzyme working close to thermodynamic equilibrium, the TPI can be easily induced to work in the backward direction (GAP → DHAP). Its central physiological role is maintaining the delicate balance between glycolysis and gluconeogenesis. However, since TPI belongs to the most ancient enzymes [57], the biological evolution involved it in the pentose phosphate pathway, triacylglyceride accumulation, and many other moonlighting functions [58,59]. With such a broad spectrum of activities and functions, it is not surprising that the TPI enzyme has attracted the medical community's interest. TPI inhibitors are promising as antiprotozoal drugs for the treatment of diseases caused by Trypanosoma cruzi, Trypanosoma brucei, Plasmodium falciparum, Giardia lamblia, Leishmania mexicana, Trichomonas vaginalis, and Entamoeba histolytica [60]. The upregulation of the TPI gene is common in many cancers [61]. At the same time, TPI deficiency or reduced activity causes the accumulation of DHAP connected to severe diseases, such as hemolytic anemia, recurrent infections, cardiomyopathy, and fatal neuromuscular dysfunction [62].
Stephen Blacklow asserted [63] that TPI enzyme "can improve no further as a catalyst, " assuming constraints of free diffusion and in vivo levels of its substrates. In the meantime, researchers proposed electrostatic screening [64,65], TPI oligomerization [66], elevated temperature for the TPI from thermophilic cells [67], and other mechanisms [68] how TPI catalytic efficiency can be increased above observed values. Ideally, the mutations or modifications making TPI more resistant to oxidative damage and a more efficient catalyst can help prevent and treat Alzheimer's disease [68,69].
We stressed in our previous contributions [70,71] that increasing the TPI catalytic turnover and efficiency above observed "perfect "values is theoretically possible when enzyme kinetics is connected to the maximal partial entropy production principle from irreversible thermodynamics [2]. In this chapter, we shall attempt to answer the following questions: a) Does TPI performance change after noise is taken into account? b) If it does change, is it possible to find the combination of microscopic rate constants resulting in at least ten-fold increased performance regarding the kcat/KM value calculated from the experimental data? c) How is the entropy production by TPI related to corresponding enzyme efficiency values? d) Are any published optimization methods better at finding high forward kcat/KM values than different means of noise introduction?
Let us first present observed values for TPI kinetic parameters [32,70] to easily compare all our simulations with the experimental values (Table 1). Triosephosphate isomerase can be found in four functional states [54]. According to Figure 1c) 1 is the free enzyme (E), 2 is the enzyme-substrate bound complex (ES), 3 is a transition state intermediate (EZ), and 4 is the enzyme-product bound complex (EP). The reference steady state [54] is such that the concentration of substrate is [S] = 40 μM and the concentration of product is [P] = 0.064 μM. The values of the kinetic constants k1 and k8 in Table 1 are obtained respectively from expressions k1= k1*∙[S] and k8 = k8*∙[P], where second-order rate constants k1* and k8* are measured in (Ms)-1.
The initial TPI concentration in our simulations ranged from 10 to 50 nM. Mass conservation for all enzyme conformations is always taken into account in all simulations. All NetLogo programs also required the mass conservation of ligands (substrates, products, and their intermediate TPI-bound forms). That requirement entered the FORTRAN programs as the [S]+[P] = constant condition when we allowed for changes in the concentrations of ligands. The concentration of bound ligands [ES]+[EZ]+[EP] is always much smaller than [S]initial + [P]initial concentration because bound ligands concentration cannot exceed the initial low concentration of free TPI enzymes. Thus, the mass conservation of ligands is considered a good approximation in those FORTRAN programs that examined how different parameters change after changes in the substrate and product concentrations.
Stepwise increases of rate constants from the product-release transition
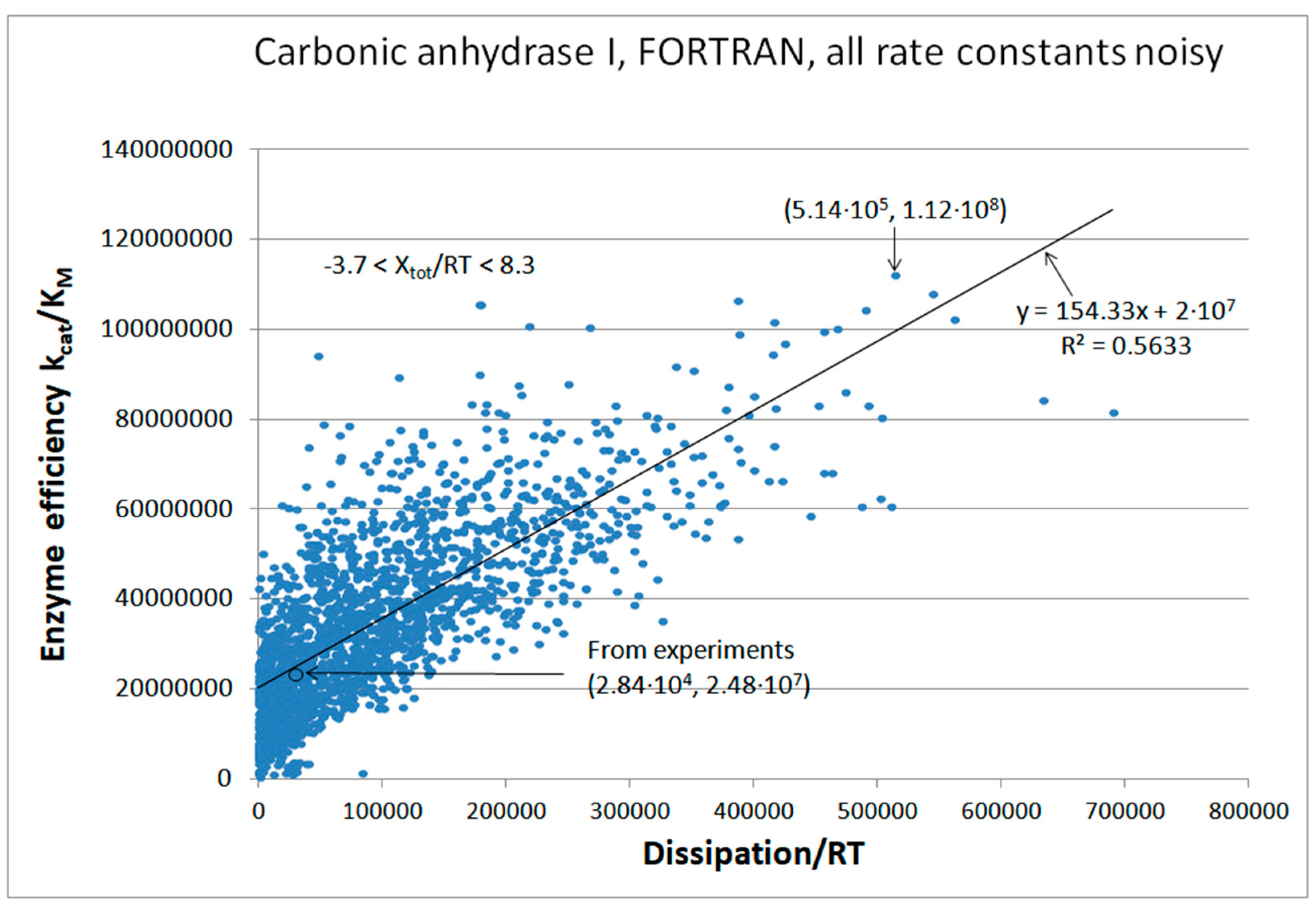
Let us first consider how catalytic efficiency depends on overall entropy production in a deterministic manner when the implicit assumption is that noise does not exist. FORTRAN program is convenient to use for such a study. For constant temperature, the dissipation function φ and total entropy production P have the absolute temperature T as the proportionality factor: φ = T∙P. Assuming that P is not the conseqeunce but the cause for the catalytic efficiency (see Introduction), we use either entropy production or dissipation term to plot the functional relationship between kcat/KM values at the y-axis and the dissipation/RT values at the x-axis. The first such plot (Figure 2) illustrates how TPI efficiency changes after the stepwise increase in the microscopic rate constant k7. All other rate constants and equilibrium constants K1, K2, and K3 are kept at their observed values (see the calculated values of rate constants and the values for the initial concentrations of substrates and products from Table 1. Since the equilibrium constant K4 = k7/k8 also goes through the stepwise increase, the expected outcome of the first simulation scenario is a regular increase in the chemical affinity or force (expressed as Xtot/RT values) from negative to positive values.
Negative force values correspond to negative backward flux (GAP → DHAP) and positive dissipation, while positive force values correspond to positive forward flux (DHAP → GAP) and positive dissipation. Both limits in the force range, negative and positive, are associated with the high dissipation. Still, only the positive limit corresponds to the maximal enzyme efficiency value of 1.25∙106 M-1s-1 (Figure 2). That result is an encouraging 1.59-fold increase over the observed value of 7.9∙105 M-1s-1 (corresponding to the Xtot/RT = 0.685), but not the significant improvement over the 1.13∙106 M-1s-1 value we obtained in the 2017 [70].
From the output of the same FORTRAN program, it is easy to select only the positive Xtot/RT values. The resulting efficiency dependence on dissipation is then well correlated (R2 = 0.944) with the straight-line proportionality (Figure 3) Thus, from zero forward catalytic efficiency and vanishing entropy production in the thermodynamic equilibrium, there must be an obligatory increase in the dissipation, which is tightly coupled to the catalytic efficiency increase. We did not ask how one can achieve the increase in only the chosen kinetic constant k7 in practice without any other change. It is unlikely that random or intentional mutations can ever do it. However, fine-tuning microwave irradiation may potentially produce the non-thermal effect of significantly accelerating the product release catalytic step. It is easier to answer why simulations presented in Figure 2 and Figure 3 dealt with the k7 stepwise increase. We assumed, as in [70], that the product release rate limits the TPI catalytic power. In our notation for rate constants (see Figure 1c), the k7 is the first-order rate constant, determining the product release rate.
We can also explore the stepwise increase of k7 and k8 when all equilibrium constants and all other rate constants maintain their observed values (see Table 1). Almost perfect proportionality is obtained between kcat/KM and corresponding entropy production values (Figure 4).
The next task is to answer how thermodynamic and kinetic parameters change for the TPI catalytic cycle when we limit deterministic changes to decreasing substrate and increasing product concentrations. The answer is provided in Figure 5, which illustrates how net flux and overall dissipation vary with the force changes.
The catalytic activity optimizations in the forward direction, when the substrate is converted into the product, are better connected with the physiological role of the TPI enzyme in glycolysis. We published one example of such optimization in 2017 [70]. It was for the fixed positive force (chemical affinity) corresponding to Xtot/RT = 0.685 (the vertical line in the insert of Figure 5). The optimization example for the reverse process (product-to-substrate conversion) leads to decreased catalytic efficiency for the forward process. The dissipation and net flux for the reverse process increase by several orders of magnitude when the applied force has a high negative value (the vertical line in the main figure). It is a pathological situation with no connection to TPI's role in cellular metabolism.
Šterk et al. result [72], after maximizing the total entropy production density, was J = - 1272 s-1 (blue point at the vertical line in the main figure). It is about 100 times higher net reaction flow in the reverse GAP→DHAP direction when compared to experimentally observed reaction rate J = 14 s-1 facilitating glycolysis [32,70]. The maximal entropy production of Šterk et al. [72] is σ/nR = 5685 s-1 (the red point at the vertical line in the main figure). Klipp and Heinrich [73] obtained even higher net reaction flow in the backward GAP→DHAP direction ranging from J = -1620 (for experimental rate constants values when [DHAP] = [GAP] = 40 μM) to J = - 4010 s-1 (for the separate limit optimization model), the result that was verified and commented by Bish and Mavrovouniotis [74]. These optimizations for highly negative flux and negative force can only ensure the non-physiological operation of the TPI enzyme and the loss of its primary function of balancing glycolysis and gluconeogenesis. For instance, optimized kcat in the forward direction of Šterk et al. [72] is kcat = 222 s-1, which is worse then kcat = 432 s-1 (experimental data [32]), while our optimized kcat = 686 s-1 [70] is an improvement over the kcat value calculated from experimental data.
It all depends on the choice of the optimization procedure. We chose to maximize the partial entropy production in the rate-limiting product-release step (the 4th catalytic step in the forward direction) [2]. We noticed in 2017 [70] how that choice led to the concomitant increase in the optimal net flux (from 14.4 to 20.77 s-1), optimal catalytic constant (from 432 to 686 s-1), optimal catalytic efficiency (from 7.86∙105 to 1.13∙106 M-1s-1), and optimal overall entropy production (from 9.9 to 14.2 s-1). Within the restriction we used (fixed equilibrium constants for each catalytic step at their values calculated from the experimental data), there was a common 30% increase in the flux, efficiency, and dissipation. Figure 4 above illustrates how a regular 30% increase between two points follows from the constant slope and perfect proportionality between enzyme kinetic parameters and its overall entropy production. We did not show the J dependence on dissipation because the dissipation function φ = J∙X, and force X was constant in the calculations we performed to construct that figure.
In the following subsection, we study the same efficiency-dissipation relationships when noise is introduced in the last catalytic step so that FORTRAN programs for calculating kinetic and thermodynamic parameters do not contain regular stepwise increases in catalytic constants k7 and k8.
Noise introduction in kinetic constants with selected restrictions
We asked what would happen after introducing random normal noise in forward (k7) and backward (k8) rate constants for the rate-limiting product-releasing step. Figure 6. illustrates the advantage of using noise when looking for the combination of rate constants corresponding to higher enzyme efficiency. The highest efficiency of 1.6∙106 M-1s-1 is associated with the highest total dissipation in the RT units (20.3 s-1) due to the perfect proportionality between the main enzyme performance parameter and the main physical parameter in irreversible thermodynamics. We used the same restrictions of constant overall force and constant equilibrium constants K1 to K4, which were required in deriving the partial entropy production theorem [2,30,70]. Random normal noise was called once in the FORTRAN program as the Box-Muller transform (see Methods, eq (25)) with the shift +2 to ensure that only positive rate constants k7 are the output. There was no need to call that function again for the multiplication with the observed k8 value because we kept the no-change requirement for all equilibrium constants Ki (i = 1, 2, 3, 4) from our 2017 paper [70].
In the next computational experiment, we introduce normal noise in the kinetic constant k7 = 4000 s-1 while taking representative initial values of rate constant k8 = 25 s−1, 32 s−1, 40 s−1, 100 s−1, and 160 s−1. Fixed equilibrium constants for each k7-k8 pair are then K4 = 160, 125, 100, 40, and 25, respectively, each calculated using experimental value k7 = 4000 s−1. Experimental data for kinetic constants of the enzyme triosephosphate isomerase shown in Table 1 are used for transitions other than transition 4. Enzyme efficiency kcat/KM as a function of dissipation/RT is shown in Figure 7 for the forces X/RT equal to 0.2389, 0.4620, 0.6852, -0.6774, and -1.1474 corresponding to the equilibrium constants K4=100, 125, 160, 40 and 25, respectively.
There is one additional condition besides those we used to create Figure 6. We assumed a constant sum of free substrates and free products. It is a good approximation for the mass conservation of ligands only if the initial free enzyme concentration (50 nM) is much smaller than the concentrations of [S]+[P] for all points and all forces. Figure 7 joins the results of five FORTRAN programs that include noise in the last forward catalytic step.
Careful examination of the case Xtot/RT = -1.1474 reveals slight curvature in the efficiency as a function of dissipation (magenta symbols). The ratios of rate constants k3/k7 and k6/k7 (from the KM expression [38]) are not constants in the output file because k3 and k6 are constants but k7 changes in a random manner. The best linear fit slope increases when negative force values approach the thermodynamic equilibrium and then decreases when positive force is increased. Thus, we examined slope changes and goodness of linear fit changes for a wider span of force values ranging from -3 to +4 (Figure 8). For that task, we constructed ten FORTRAN programs. The ratios of rate constants k3/k7 and k6/k7 (from the eq. (23) KM expression) are not constants in the output files because k3 and k6 are constants, but k7 changes randomly. The KM exhibits small changes.
Figure 8 shows the output of these programs. It illustrates how the slope and the perfection of the seemingly straight-line proportionality increase with the approach to the thermodynamic equilibrium when net force and entropy production vanish.
Computational Optimizations of the TPI Catalytic Activity when noise is included
The question we studied in this subsection is how noise introduction affects various computational optimizations for TPI catalysis. In Figure 9, variations of K1 and K4 were introduced by multiplication of K4 = k7/k8 (see Table 1) with the normal noise. The fixed force restriction X = Xtot/RT = 0.684 [70] ensured concomitant variations in K4 and K1. There was no explicit requirement for the maximal entropy production. Still, after going randomly through the 1000 quasi-steady states, our FORTRAN program finds that the maximal overall dissipation corresponds to optimal enzyme efficiency (Figure 9). The 7-fold efficiency improvement from 7.86∙105 to 5.585∙106 M-1s-1 follows after a 4-fold dissipation increase (see Table 1).
The best combination of the backward rate constants k2 and k8, which resulted in even higher kcat/KM of 8.903∙106 M-1s-1, is k2 = 74 s-1 and k8 = 2438 s-1. The enzyme working in that state has 11 times higher catalytic activity (the highest point in Figure 9) than the value of 7.86∙105 M-1s-1 calculated from the experimental data (Table 1). Required changes in rate constants are two orders of magnitude changes in k2 (decrease) and k8 (increase). These rate changes describe the inhibition of substrate release from the ES complex and stimulation of product association with the free enzyme. The corresponding overall dissipation per RT of 21.3 s-1 is approximately double the value calculated from the experimental data. Still, the dissipation needed to reach the maximal efficiency state is halved compared to maximal dissipation (Figure 9).
Interestingly, the same dissipation value of 21 to 22 s-1 is connected with two very different catalytic efficiency values of 8.9∙106 and 1.94∙106 s-1, respectively. Thus, when specific restrictions are imposed, the nonlinear system may be able to jump between two quasi-steady states characterized by high and low efficiency and a minor change in dissipation. How to force the system to live in about a 10-fold higher efficiency state with only a two-fold higher price in terms of overall dissipation is outside the scope of this paper.
Optimal efficiency can be obtained for fixed force when other pairs of equilibrium constants are varied by introducing noise. We did not show corresponding efficiency (dissipation) dependence because optimal kcat/KM values for the dissipation maximum were considerably lower from the 8.9∙106 M-1s-1 value obtained after K1-K4 variations. A reader can verify that conclusion from the Figure 10 coordinates (20.6, 1.95∙106) and (21.7, 1.9∙106) obtained after K2-K4 and K3-K4 variations. Still, the requirement that total entropy production is maximal and corresponding restrictions on equilibrium constants for chosen catalytic steps can produce higher catalytic efficiencies for fixed force than the maximal requirement for selected partial entropy productions (Figure 10).
The primary purpose of Figure 10 is to illustrate the relationships among different methods for obtaining higher than referent values for catalytic efficiency (Table 1). That task led to the map of dissipation-efficiency points when the x-axis is for dissipation and the y-axis is for enzyme efficiency. The perfect efficiency-dissipation proportionality is the straight line fit to 1000 points after each kinetic constant is multiplied with the normal noise invoked only once in the corresponding FORTRAN program. It is the consequence of assuming fixed values for all equilibrium constants Ki, meaning that the overall force is also identical for all data points (their referent values can be found in Table 1). We also used those assumptions in our previous publications [2,30,70]. We did not consider noise in these publications. The first two highlighted points (9.9, 0.79∙106) and (14.2, 1.13∙106) are centered at the linear fit. They are the dissipation and efficiency values calculated from the experimental data and the modest improvement achieved after the requirement that partial entropy production P4 in the rate-limiting product release step is maximal [2,70,71].
We discussed above the results after introducing noise in the pairs of equilibrium constants K1-K4, K2-K4, and K3-K4. These are off-line points in Figure 10, respectively: (39.4, 5.585∙106), (20.6, 1.95∙106), and (21.7, 1.9∙106). Thus, Figure 10 clearly shows the advantage of noisy substrate and product association with free enzyme (the highest point). After considering many different optimization methods for entropy production (either ours or by other authors), the K1-K4 variations with constant force restriction resulted in best theoretical increase of the TPI catalytic efficiency above its observed value [32].
Computational optimizations of TPI kinetics by some other authors [72,73] used the substrate and product concentrations similar to each other. It reversed the net flow in the direction of product → substrate due to negative flux and force and resulted in higher total entropy production values of several orders of magnitude. For instance, Šterk et al. [72] used the constraint k1*∙k3∙k5∙k7 = K+ = constant equal to the observed value. Such optimization required that the product of all kinetic constants in the forward direction and all kinetic constants in the backward direction k2∙k4∙k6∙k8* remain fixed when other parameters change. However, Šterk et al.[72] used the steady state concentrations [S] = 31.45 and [P] = 8.55 μM. The corresponding force is then highly negative Xtot/RT = - 4.47. When multiplied with the high negative flux, it produced such a high dissipation that the optimal values (5685, 1.8∙106) could not be illustrated as the (x, y) point within the confines of Figure 10. The authors found the maximum in overall entropy production, but it was about 570-fold higher than the calculated value from the experimental data. As expected, for the backward-directed enzyme turnover, the corresponding optimal efficiency for forward catalysis of 1.8∙106 M-1s-1 (the right-hand arrow pointing outside Figure 10) is substantially smaller than our best results.
Noise introduction without restrictions other than all ki>0
When normal noise without shift is introduced in all rate constants ki, some can vanish or become negative. To avoid such cases, we replaced negative with observed ki values (see Table 1). Figure 11 illustrates that reasonable proportionality exists between efficiency and entropy production when there are no other restrictions on kinetic constants and on equilibrium constants for the TPI enzyme. The advantage of calling random numbers eight times (once for each of eight kinetic constants) is an extended range of possible steady states and forces. The highest efficiency state has 30-fold better efficiency and 160-fold higher dissipation compared to values calculated from experiments. The corresponding force for that state is Xtot/RT = 6.335.
The basic assumption we used in calculating entropy production values is that each of the 10000 computational steps probes a new quasi-steady state in which all parameters of interest can be calculated by using the Terrel Hill method [34,35]. We found the maximal efficiency value in the 1078th step. It corresponds to an unusually high information entropy of 1.181 and a low Michelis-Menten constant KM = 0.000015. Interestingly, only the kinetic constants k2, k6 and k7 significantly differed from their experimental values, all being much smaller, 56, 15, and 4 times, respectively. An increase in the k1 value (from 400 to 1144 s-1) may have resulted from increased substrate concentration or increased second-order rate constant for the association between the substrate and enzyme to form the ES complex. There was no change from experimental values for the kinetic constants. k4, k5, and k8.
Enzyme turnover became slightly slower (kcat decreased from 432 to 348 s-1), but the division with considerably smaller KM (from 5.5∙10-4 to 1.474∙10-5) ensured surprisingly high efficiency. As is usually the case, the most illustrative representation is the profile of changes in the equilibrium constants or free energy changes. The equilibrium constant K1 increased about 160 times (from 0.057 to 9.07), and the K3 constant increased nearly 15 times (from 0.667 to 9.75). It led to a significant increase in the total equilibrium constant (from 1.98 to 564) despite a decrease in the K2 (from 0.333 to 0.174) and K4 (from 156.25 to 36.59).
There were 3580 points corresponding to the force X = Xtot/RT ≤ 0. Thus, for 35.8% of sets with random values for kinetic constants, the enzyme can still work in the reverse direction, converting products into substrates. Most ki octuplets simulated the major physiological role of the TPI enzyme in converting DHAP to GAP. The best case of kcat/KM = 2.36∙107 M-1s-1 is also for the forward-directed net flux. However, we used the same forward catalytic efficiency definition for X > 0 and X ≤ 0. All the experimental data in the literature were extracted for the force X > 0 and flux J > 0 under the conditions when substrate concentration greatly exceeded the product concentration. The initial concentrations were [S] = 40 μM and [P] = 0.064 μM. Variations in k1 and k8 allowed the changes either in the second-order rate constants or in concentrations. The extreme case when X = -10.19 was obtained with k1 = 3.4 s-1 and k8 = 27.0 s-1. If the change in k1 occurred only due to the change in [S], the substrate concentration would decrease almost 120 times. Therefore, although we included the points with negative force and flux in this figure and other simulations from the literature considered such cases [72,73], there is no experimental or physiological justification for retaining them.
Simulating dynamics using an agent-based modeling approach
Dynamics can be simulated using an agent-based modeling approach without solving differential equations. Agent-based model (ABM) is stochastic by nature. For instance, the stochastic noise inherent to the NetLogo computer language can be used to construct models for the stochastic interaction of an enzyme with its substrates, products, and inhibitors [75,76]. In the following example, noise is introduced in the TPI kinetics through random-float values (uniform noise) added to selected rate constants, not by Gaussian random number values (Figure 12). Additional noise in rate constants is due to random encounters among ligands and [E]free and among enzyme conformations [ES]↔[EZ] and [EZ]↔[EP], which is also specified with several different random-float values. Random changes occur in all computational steps (named „ticks“). Ticks can be in chosen time units. Agent-based programming requires dimensionless numbers as the input. However, when these numbers are specified as 40000 for substrates, 64 for products, and 50 for enzymes (for the TPI kinetics), they correspond to [S]initial = 40 μM, [P]initial =0.064 μM, and [E]initial = 0.1 μM. The mass conservation of all ligand forms ([S], [P], [ES], [EZ], and [EP]) and all enzyme forms ([E]free, [ES], [EZ], and [EP]) is an explicit requirement for each tick in all our NetLogo programs. Thus, [S]initial + [P]initial = [S] + [P] + [ES] + [EZ] + [EP] and [E]initial = [E]total = [E]free + [ES] + [EZ] + [EP] because we left the system to itself and never added ligands or enzymes.
The intermediate equilibrium constants K2 and K3 never changed from their experimental values. Due to slight changes in the substrate and product concentrations the equilibrium constants K1 and K4 also underwent small changes. However, we kept the assumption that
where is the second-order rate constant for the enzyme and substrate association, while const1 is the ratio determined from the observed value, which does not change during the simulation. Similarly,
where is the second-order rate constant for the enzyme and product association, while const2 is the ratio determined from experiments, which does not change during the simulation.
Since initial product concentration is small (64 nM), each stepwise increase in the product concentration is seen as a jump from one straight line fit to another in four steps „a“ to „d. “ It increased the product concentration to 67 nM. Thus, the proportionality between enzyme efficiency and entropy production (dissipation) remained almost perfect. Maximal efficiency values close to the 3∙106 M-1s-1 are about 4-fold higher compared to the value calculated from experiments. Similar 4-fold increase is for the corresponding dissipation.
When simulation time was extended to 2137 ticks, product concentration increased from 64 to 74 nM, while the driving force decreased from Xtot/RT = 0.68 to Xtot/RT = 0.54 with the same stepwise slope increase for the efficiency-dissipation dependence (Figure 13). The best efficiency value of 3.3∙106 M-1s-1 corresponded to the dissipation/RT = 27.2 s-1 . Free enzyme concentration dropped from 100 to 12 nM for this case of more extended simulation.
The insight from these figures would be that maximal catalytic efficiency remains approximately the same during the system relaxation toward the thermodynamic equilibrium (when chemical affinity, net flux, and total entropy production all reach their zero values). The slope of the efficiency-dissipation line keeps increasing toward an infinitely high value at the thermodynamic equilibrium when dissipation vanishes. Also, the perfection of the straight line approximation for the fit connecting all (x, y) values keeps increasing in discrete jumps (for each unit change in the product concentration) while chemical affinity decreases. The same time-development rule holds when equilibrium is spontaneously approached from the high positive or negative initial forces (see Figure 8). Better efficiency to dissipation proportionality for positive forces stems from the definition of catalytic efficiency, where both the catalytic constant and the Michaelis-Menten constant are defined for the forward direction . It is easy to see that nonlinear is considerably better than linear fit for efficiency-dissipation proportionality in the case of higher negative X (see Figure 8).
For longer simulations, the concentrations of enzyme conformations ES, EZ, and EP after each step (tick) go through the typical Michaelis-Menten kinetics: slow initial increase, faster, nearly constant increase, a broad maximum with minor changes, and prolonged decrease. That pattern repeats itself with the ES complex, after some delay with the EZ complex, and finally with the EP complex.
We next examined if a broader scope search for better enzyme performance is possible when Gaussian noise gi (see Methods) is multiplied with each microscopic rate constant ki (Figure 14). The best catalytic efficiency of kcat/KM = 2.22∙107 M-1s-1 is indeed better than previous NetLogo simulations and similar to the best result we obtained after FORTRAN simulation for the TPI kinetics (Figure 11).
Ketosteroid Isomerase (KSI) Case: What is Different when the Operating Range is Farther from Equilibrium?
Paul Talalay discovered in 1951 [77] the Pseudomonas testosteroni bacterium (presently named Commamonas testosteroni [78]) from soil beneath a rosebush on the Berkeley campus. The bacterium could grow in a medium containing testosterone as its only carbon and energy source. That was a clever and brave approach because, at that time, many steroid metabolites were known, but enzymic transformations of steroid hormones and metabolites were yet undiscovered. Paul Talalay and his collaborators purified highly active small bacterial enzyme ketosteroid isomerase from that bacterium and reported their findings from 1955 onward [77]. The alternative name for the KSI enzyme is 3-oxo-Δ5-steroid isomerase (EC:5.3.3.1).
Anna Radzicka and Richard Wolfenden reported typical high values for the
catalytic constant, catalytic efficiency, and catalytic proficiency of KSI as respectively 6.6∙104 s-1, 3.0∙108 M-1s-1, and 1.8∙1015 M-1 [12]. The catalytic proficiency is the catalytic efficiency rate enhancement (kcat/KM)/kuncat when a nonenzymatic reaction rate constant kuncat can be found for a corresponding spontaneous chemical reaction without the enzyme (1.7∙10-7 s-1 in our case). Thus, KSI is one of the fastest enzymes with extraordinary catalytic power. The formation of essential steroid hormones would take months to millions of years without enzymes such as KSI [79]. The equilibrium constant Keq = 2400 [80] corresponds to the far from equilibrium conditions, high positive force, and the preference for the forward isomerization rate of 5-androstene-3,17-dione (a substrate for KSI) to its conjugate isomer 4-androstene-3,17-dione. Elucidating how the KSI reaction mechanism is connected to structure, kinetics, electrostatics, and thermodynamics was a highly challenging but worthy task through the last 50 years [81,82,83,84]. Hopefully, the rational design of KSI enzymes with augmented catalytic efficiency would benefit green chemistry goals for the pharmaceutical industry in manufacturing specialized steroid chemicals [85].
Mammalian steroid isomerases have multifunctional activity and a more complex structure than bacterial KSI enzymes [86]. Although crucial in all mammals, their structure-function connection has not been as extensively examined as in the case of the model enzyme KSI from bacteria. Thus, we shall use the best predicted KSI rate constants for bacterial KSI [31] that agree well with those reported earlier by [80,87].
Our first task was a wide exploration of possible system states when noise is introduced into each of the eight rate constants for the 4-state kinetic scheme (Figure 15). Our FORTRAN simulation kept the concentrations of substrates and products fixed at their initial values (Table 2, second column: [S] = 10-4 M, [P] = 5∙10-5 M). Nevertheless, due to random changes in all rate constants, the force changed in the range 0.72 < Xtot/RT < 17.17. Repeated runs produced an identical output. The third best efficiency value from the (1.71∙104, 1.66∙109) point reveals that 5-fold higher efficiency can be achieved when corresponding entropy production is 10-fold smaller than their experimental values. That is a rare case when the choice of rate constants results in high catalytic activity despite low dissipation for the KSI enzyme.
As for the case of triosephosphate isomerase, the perfect efficiency-dissipation proportionality followed after the no-change requirement in the equilibrium constants for all catalytic steps (not shown). When noise is called only once, a nearly perfect linear fit survives for the efficiency-dissipation dependence, no matter how many rate constants ki are multiplied with the normal noise function (not shown). The consequence of fixed equilibrium constants Ki is constant overall force, too.
Regular dependence of enzyme efficiency on overall dissipation follows when noise is introduced only into one or two kinetic constants without fixed Ki requirements (Figure 16 and Figure 17). However, that dependence is very different if the overall force Xtot/RT is allowed to vary too (Figure 16) and when overall force is kept at the constant initial value Xtot/RT = 8.426 (Figure 17, see Table 2). Figure 17 confirms the observation from Figure 9 that the maximum in overall entropy production exists when variations in K1 and K4 equilibrium constants are introduced and fixed overall force is maintained in all steps of FORTRAN simulation. Total entropy production is maximal in the point (1.3∙105, 4.7∙108)(Figure 17). The corresponding optimal efficiency is about 50% higher than the observed value of. 3.02∙108 M-1s-1. Still, the point with the highest efficiency (1.96∙104, 8.15∙108) corresponds to 5.9 times smaller dissipation than the value 1.16∙105 s-1 calculated from the experimental data. That is another rare case when randomly chosen equilibrium constants within imposed restriction (constant overall force) resulted in a high catalytic efficiency despite low dissipation for the KSI enzyme.
Agent-based modeling extended and confirmed the simulation results for the KSI kinetics using the FORTRAN software. The advantage of NetLogo simulation (Figure 18) is that random changes in the concentrations of substrates, products, free enzymes, and enzyme complexes are allowed. Typical Michaelis-Menten kinetics for concentration changes, which we described for the NetLogo simulation of the TPI kinetics, is also seen for the KSI kinetics (not shown). Initial concentrations were [E]free = 5 μM, [S]free = 100 μM, [P]free = 50 μM. Final foncentrations at the 6977th tick were [E]free = 4 μM, [S]free = 95 μM, [P]free = 54 μM, [ES] = 0.3 μM, [EX] = 0.4 μM, [EP] = 0.3 μM. The mass conservation conditions [Etot] = [Efree] + [ES] + [EX] + [EP] and [ligands] = [S]free + [P]free + [ES] + [EX] + [EP] were satisfied through all time jumps (ticks). Freedom to change equilibrium constants in each tick enabled the exploration of a wide range for overall force (1.1 < Xtot/RT <16.8), catalytic efficiency, and overall dissipation. The best pair of dissipation-efficiency values (4.6∙105, 2.6∙109) corresponded to approximately 4-fold higher dissipation and almost 10-fold higher efficiency in the comparison with values calculated from observed data (1.2∙105, 3.0∙108).
CA I, CAII, CAII-T200H chapter (also 4-state enzymes)
Carbon dioxide conversion into biomass is essential for the survival and spreading of life in all terrestrial environments. Carbon sequestration is also crucial for the survival of our carbon dioxide-producing civilization, which is unfortunately addicted to fossil fuels burning and breaking all life-supporting balances the biosphere has developed through eons. Nature developed multiple means and different organic structures for the fast conversion of carbon dioxide to bicarbonate – the first step toward carbon fixation. Carbonic anhydrases (CAs) are universal enzymes responsible for that process in all three life domains: Bacteria, Archaea, and Eukarya [88]. With rare exceptions [89], CAs are metalloenzymes containing the metal ion (most often zinc) in their central active-site cavity. From their discovery in red blood cells in 1932, the scientific interest in CAs continued to grow, as seen from the abundance of more than 900 solved CA structures deposited in the Protein Data Bank [90].
The spontaneous reaction of CO2 with water can produce bicarbonate , but that reaction is too slow to support respiration [91,92] and other biological processes catalyzed by different CAs. Eight unrelated families of carbonic anhydrase (CA) enzymes represent different ways nature performed the feat of fast catalytic inter-conversion between carbon dioxide and carbonic oxide [93], reaching the catalytic turnover of 1 μs-1 or even higher [94]. There is little or no sequence homology among CA families α, β, γ, δ, ζ, η, θ, and ι [89,95]. Molecular biologists concluded that convergent biological evolution performed the spectacular function-enhancing feat at least seven times because different CAs evolved to perform an identical function [93,96,97].
Mammals possess 16 different CA isoenzymes from the alpha class family [98]. All are metalloenzymes, with the Zn II hydride located at the enzyme center anchored by three histidines. CA isoforms are involved in a variety of physiological functions. Human CA isoforms are well-recognized drug targets for designing isoenzyme-specific inhibitors [99,100] to help fight glaucoma, epilepsy, obesity, cancer, and other diseases. Also, human CA II is one of the most efficient known enzymes. Its calculated catalytic efficiency from experimental data is 1.5∙108 M-1s-1 [100]. Earlier efficiency calculations also positioned CA among “perfect” enzymes working close to the diffusion limit [14,56].
Genetic defects of specific CA isoforms can cause osteopetrosis, cerebral calcifications, retinal problems, hyperammonemia, hyperchlorhidrosis, neurodegenerative and other metabolic diseases [101], which is a good enough reason to look for CA activators [98] or other means for increasing the activity of these isoforms. Memory enhancement can be achieved through CA activation [102]. It opens the possibility for targeted improvement of brain CA performance to enhance cognition and slow the aging process [98,103]. Some CA mutants can accelerate the proton transfer, the rate-limiting step for CA turnover [94]. Another reason for increasing CA activity is the urgent need for green ways of industrial CO2 sequestration [104] we mentioned above.
This subsection deals with the theoretical possibilities for catalytic efficiency improvements of human CAs I, II, and the T200H variant of CAII with His200 replacing Thr200 [105]. There may be better models than the four-state kinetic model for reversible Michaelis-Menten-type kinetics (Figure 19). Still, it is based on the publication [105] that contains all microscopic rate constants needed to calculate and compare the enzyme’s performance with associated dissipation. Referent (initial) state values can be found in Table 3.
The FORTRAN simulation of noisy CA I kinetics did not change any of the initial concentrations (Table 3), and it still found in the 246th step the dissipation-efficiency point (5.14∙105, 1.12∙108) with the 4.5-times higher catalytic efficiency from the calculated value based on the observed kinetic data. The corresponding overall force was positive (Xtot/RT = 5.0) and closer to the upper end of the force range (Xtot/RT = 8.3). However, the substantial efficiency increase (4.5-fold) was „paid for “ with the 18 times higher overall dissipation. Closer inspection of the performance parameters from the 246th computational step (concerning observed initial values) revealed the 6.3-fold increase in the turnover number and 2.8-fold increase in the overall force as the main reason for the improved efficiency.
Figure 20.
The FORTRAN simulation of the relationship between catalytic efficiency and overall dissipation for the carbonic anhydrase I, when each of eight rate constants ki is multiplied with independently called normal noise function gi without shift (see Methods, eq (25)). Only positive gi values were allowed while the program ran through two thousand steps. There was no change in the substrate, product, and buffer concentration from their initial values (Table 3).
Figure 20.
The FORTRAN simulation of the relationship between catalytic efficiency and overall dissipation for the carbonic anhydrase I, when each of eight rate constants ki is multiplied with independently called normal noise function gi without shift (see Methods, eq (25)). Only positive gi values were allowed while the program ran through two thousand steps. There was no change in the substrate, product, and buffer concentration from their initial values (Table 3).
The NetLogo simulation of noisy CA I kinetics (Figure 21) slightly changed initial substrate and product concentrations (Table 3). The 6-fold efficiency increase point, which we found halfway through the simulation, was „paid-for“ with the 22-fold dissipation increase. That quasi-steady state corresponded to about a 3-fold increase in the kcat and overall force.
Human red cell isoenzyme CAII is superior to CAI when their catalytic efficiencies are compared [105] (see Table 3). Thus, simulations for the CAII will have the advantage of starting from a better initial state. Here, we show only the NetLogo simulation (Figure 22). The CAII mutant T200H, constructed by Behravan et al. [105], was an attempt to find the single amino acid substitution that would lead toward catalytic parameters of CAI. The NetLogo simulation (Figure 23) indicates the evolutionary potential for improving the performance of CA-T200H as being indeed between CAI and CAII but closer to CAII (Figure 24).
We compared enzyme performance and associated dissipation for three CAs isoenzymes: CAI, CAII, and the T200H mutant of CAII (Figure 24). Values calculated from observed rate constants [105] are confined near the origin of that figure, while the best-simulated values are expanded in the order CAII > CAII T200H > CAI. Improved catalytic efficiency is associated with increased dissipation in the same order.
Evolutionary Related β-Lactamases
This subsection is the extension of our earlier studies [30,106] when we examined the evolutionary relationship among bacterial β-lactamases, their kinetic performance parameters, and entropy production. The evolution of β-lactamases, as an example of adaptation in bacteria, is not just of academic interest. Diverse classes of β-lactamases inactivate the antibiotics (for instance, ampicillins and cephalosporins) by performing the hydrolysis of their beta-lactam bridge [107]. The rapid global spread of beta-lactamase-mediated bacterial resistance in hospitals has become a severe challenge in treating bacterial infections [108].
We used here the same set of microscopic rate constants for S. aureus, E-coli, and B. cereus enzymes (respectively labeled as PC1, RTEM, and Lac-1) determined during 1980s [29] together with our estimate for missing backward rate constants [30] that were needed for the calculation of nonequilibrium steady state quantities in the reversible three-state Michaelis-Menten kinetic scheme (Figure 1b). The natural evolution of β-lactamases took place millions of years before the widespread use of penicillin-based antibiotics (β-lactam antibiotics) had a chance to accelerate it in the wild-type bacterial species studied during the 1980s [109]. It probably developed as a defense from naturally occurring beta-lactam antibiotics produced by some fungi and bacteria [110]. Thus, evolutionary distances based on β-lactamases sequences determined in the 1970s [2,106,111] should be suitable to study possible connection to the total entropy production as the most important quantity in the nonequilibrium thermodynamics.
How appropriate is the „perfect“ enzyme name for the 3-state scheme with some rate constants observed or calculated as representing fast transitions in the case of β-lactamases [29,112]? That general claim about β-lactamases as almost perfect enzymes has been supported for Lac-1 but not for the RTEM and PC1 enzymes [113]. Perfect enzymes supplied with their best substrate should be able to operate close to or inside the range 108 – 1010 M-1s-1 predicted for diffusion-limited enzyme reactions [63]. Collected kcat/KM values for the hydrolysis of some characteristic β-lactams by various class A β-lactamases [114] are considerably smaller from the lower end of the diffusion limit despite „close to the diffusion-limit, i.e. 108 M-1s-1“ assertion by these authors. However, the latent potential for these lactamases to evolve further toward higher turnover number and catalytic efficiency exists when thermodynamic principles are considered together with kinetic restrictions [30,106].
We used the Gaussian noise to explore the combinations of microscopic rate constants and associated dissipation, leading to substantially improved catalytic activity for the PC1, RTEM, and Lac-1 β-lactamases. We also wanted to answer the question of whether efficiency-dissipation proportionality exists for the three-state kinetic scheme named the Haldane reversible three-step model (Figure 1b) [115,116]. The serendipitous discovery from this subsection is that a linear-like relationship survives between total entropy production increase and evolutionary distance increase (from a putative common ancestor) even after dissipation is calculated for the maximal catalytic efficiency points reached after noise introduction.
PC1-β-Lactamase
When we maintain the same restrictions of unchanged initial values for the equilibrium constants (except for the changes in the substrate and product concentrations), identical normal noise introduction in all forward kinetic constants leads to only slight changes in the nearly perfect proportionality between catalytic efficiency and dissipation (Figure 25). Besides noise, an additional reason for changes in k1 and k6 is the decrease in the free substrate concentration and an increase in the free product concentration during enzyme cycling scheme E+S ↔ ES ↔ EP ↔ E+P. It produces a slight decrease in the first equilibrium constant K1 = k1*∙[S]/k2 and an increase in the third equilibrium constant K3 = k5/(k6*∙[P]. Increased product concentration is the main reason for the gradual force decrease, from initial Xtot/RT = 11.4 to final Xtot/RT = 10.8 after 5168 ticks of the NetLogo simulation. At the 381st NetLogo simulation tick, we found the best efficiency value of kcat/KM = 4.18∙107 M−1s−1, which corresponds to forward rate constants k1 = 1.35∙105 s-1, k3 = 717 s-1, k5 = 398 s-1, catalytic constant kcat = 252 s-1, and dissipation/RT = 2823.6 s-1. That is the same 4.1-fold increase for all of these parameters with respect to their observed values (see Table 4).
The next goal is to look for limits to the evolvability of PC1 β-lactamase subject to variable rate constants k1 and k2 in the first catalytic step (association-dissociation between the free substrate and free enzyme: E+S ↔ ES). Figure 26 illustrates how the multiplication of k1 and k2 with the, respectively, Box-Muller normal noise functions named g1 and g2 can find a quasi-steady state with 6.5 times higher catalytic efficiency and merely 1.2 times higher dissipation in comparison with those values calculated from experiments (Table 4). That is a significantly better result than all previous optimizations [30] based on the requirement of maximal partial entropy production in the proton transfer catalytic steps 2 (ES ↔ EP) and 3 (EP ↔ E+P). For instance, joint optimizations of both catalytic steps for maximal transitional entropy production in these steps find about 2-fold higher efficiency, which is "paid for" by the 183 times higher dissipation. The maximum total entropy production requirement combined with the obligatory K+ = k1∙k3∙k5 = const constraint leads to 333 times lower catalytic efficiency (Figure 26, and [30]).
No further gain in enzyme efficiency follows after normal noise is independently introduced in four or all six rate constants. The maximal kcat/KM was in a narrow range from 5.9∙107 to 6.2∙107 M-1s-1. Thus, we shall keep the best result from Figure 26 (6.5∙107 M-1s-1) to compare evolutionary potential with other enzymes.
RTEM-β-Lactamase
Figure 27 and Figure 28 for the NetLogo simulations of the RTEM β-lactamase kinetics are analogues to Figure 25 and Figure 26 for the PC1 β-lactamase kinetics. Since RTEM β-lactamase is evolutionary more advanced enzyme [30,106], the simulations had a head start and ended with higher values for the best catalytic efficiency. The kcat/KM = 9.02∙107 M-1s-1 point from Figure 27 also corresponds to the highest dissipation, to the highest values for forward rate constants, and to only slightly lower Xtot/RT = 7.71 compared to the initial value Xtot/RT = 7.74. That followed from an early 572nd tick when all performance parameters increased about 3.8 times from their initial values (see Table 4).
As for the NetLogo simulation of the PC1 β-lactamase kinetics, variations in the kinetic constants k1 and k2 resulted in the exponential dependence of the catalytic efficiency on overall dissipation (Figure 28). It is essential to introduce twice the normal noise, once in the forward direction and once in the backward direction (see Methods). The beneficial consequence is the possibility of separating the enzyme efficiency increase from the dissipation increase in favor of a former quantity. The best catalytic efficiency is already well inside the diffusion-limited range.
Lac1-β-Lactamase
For the case of the Lac1-β-lactamase kinetics, we explored several options for the independent noise introduction in each kinetic constant from chosen pairs (Figure 29, Figure 30 and Figure 31). It turned out that the k1, k2 pair is the best choice because it led to the catalytic efficiency value of 1.25∙108 M-1s-1, which is also inside the range 108–1010 M-1s-1 for diffusion-limited enzyme reactions [63].
When normal noise is introduced only once in the forward rate constants k1, k3, and k5 with the proviso that the equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* do not change from their observed values, the perfect proportionality follows for all efficiency-dissipation pairs (Figure 32). Due to higher initial product concentration (see Table 4), the constraints K1* = const1, K2 = const2, K3* = const3 are almost equivalent to the requirement that initial equilibrium constants K1, K2, K3 never change during the NetLogo simulation for the Lac-1 β-lactamase kinetics. Since (kcat/KM)/dissipation expression depends only on equilibrium constants and the ratios of rate constants (see Appendix), there is no reason for the slope change in the efficiency-dissipation dependence (Figure 32). The best efficiency value of 9.68∙107 M-1s-1 is close to the diffusion-limit range's lower end (108 M-1s-1). It was reached at the 2182nd tick for the Xtot/RT = 8.23 and 3.7-fold higher turnover kcat = 7086 s-1.
To summarize, we have seen nearly perfect kinetic-thermodynamic proportionality for the PC1 (Figure 25), RTEM (Figure 27), and Lac-1 (Figure 32). Corresponding FORTRAN programs confirmed it for all three β-lactamases (Figure 42). We also confirmed the efficiency-dissipation proportionality for the triosephosphate isomerase kinetics (Figure 4, Figure 6, Figure 7, Figure 10, Figure 12 and Figure 13). It will likely hold whenever the no-change condition is imposed for the equilibrium constants in all catalytic steps (see Appendix for more details). The capture-release initial step leads to different relationships when the no-change condition is imposed on all first and second-order rate constants except k1 and k2 (Figure 26, Figure 28 and Figure 31 for β-lactamases). A fast enzyme efficiency increase can then occur for limited dissipation. The potential for the exponential-like efficiency increase is likely to be a general phenomenon for all Michaelis-Menten enzymes after lowering the activation barrier for the E+S→ES transition and increasing the activation barrier for the reverse ES→E+S transition.
Figure 33 illustrates the relationship between evolutionary distance and overall entropy production for PC1, RTEM, and Lac-1 lactamase. We found blue points and corresponding fit lines (black) from the simulation of experimental data [30,106]. The dissipations associated with the red points (and red fit line) are from the best catalytic efficiency points in Figure 26, Figure 28 and Figure 31. The dissipation increased in an almost linear manner for more evolved β-lactamases. Noise introduction and searching for the highest enzyme efficiency confirmed the proportionality between the time passage (evolutionary distance) and overall entropy production. It is to be expected if we can consider the cumulative entropy production as a surrogate for time passage.
Dissipation from observed data and from simulated maximal catalytic efficiency are both proportional to the evolutionary distance of β-lactamases
β-galactosidase
β-galactosidase (βG, 3.2.1.23) also belongs to universal enzymes used by microbes and mammals. Microbial βG has a unique role in molecular biology, firstly due to Jacob and Monod's model for the regulation of gene expression [117], secondly because of numerous molecular biology procedures using its bright blue reaction product, and thirdly for the confirmation of Michaelis-Menten mechanism at the single molecule level [118,119,120]. No less important is the βG role in the food industry [121]. The conventional βG use for the preparation of dairy products with reduced lactose content has been recently extended as a catalyst for lactose upgrading into valuable sweet glycosides, which support the growth of beneficial gut microbes [121,122]. In this subsection, we used published microscopic rate constants [26,119,123] to study how βG catalytic efficiency depends on its entropy production (Table 5). Our contribution to Table 5 was calculating all relevant kinetic and thermodynamic parameters using initial published values. For kcat/KM and P, we verified that other authors obtained identical results using different methods (Case A from [18]).
In the following NetLogo simulation (Figure 34), noise is introduced only in the encounters among substrates and enzymes to form or dissociate the ES complex. It amounts to independent variations in k1 and k2. As expected, there was a steep increase in catalytic efficiency for moderate dissipation increase, as we already observed for substrate capture-release in the case of β-lactamases. The maximal efficiency point has the coordinates 104 s-1 and 5.2∙107 M-1s-1 in the efficiency-dissipation plot (Figure 34). It is close to the point associated with the highest dissipation.
The next task was to examine a vast efficiency-dissipation space by introducing changes in all four rate constants ki (Figure 35). The best value we found of kcat/KM = 8.59∙107 M-1s-1 is close to the diffusion limit. The NetLogo runs are not completely reproducible. For instance, the second run with the identical agent-based software finds better enzyme efficiency of 1.09∙108 M-1s-1 during a smaller number of re-setting steps (Table 6).
We can notice from Table 6 first-run ki quadruplets and Table 5 initial values that the 40-fold decrease in the rate constant k2 for the ES complex dissociation back to the free enzyme and free substrate is the best strategy for getting about 40-times efficiency increase. Similarly, from Table 6 second-run ki quadruplets, the 70-times k2 decrease brings about a 60-times efficiency increase. The k1 increase up to three times also contributes to stronger substrate-enzyme binding and the simulation result of higher catalytic activity. The second conclusion from these NetLogo simulations is that significantly increased catalytic efficiency does not need maximal nor close to maximal dissipation. The third conclusion is that any means for increasing the irreversibility of the first catalytic step in the forward direction would increase enzyme efficiency since the enzyme already acts according to the standard Michaelis-Menten kinetics by having the highly irreversible product release step. We also presented the turnover numbers (kcat) in Table 6. In the two-state model for generalized (reversible) Michaelis-Menten kinetics, the turnover number kcat equals the forward rate constant k3 for dissociating the ES complex into enzyme E and product P. Our NetLogo simulation did not change much the initial (observed) k3 = 730 s-1. Thus, the best enzyme efficiency increase in both Table 6 runs is mainly due to considerably smaller Michaelis-Menten constant KM than observed KM (see Table 5 and Table 6).
The following Figure 36 illustrates how the concentrations of [S], [E], [ES], and [P] change in the second NetLogo simulation (Table 6), together with noisy changes in the overall affinity (force) during 8021 ticks. The last state, „L, “ is our subjective choice for ending the program run. It does not have any special meaning. Still, there is a slow relaxation of overall force and dissipation during the program run (only the force relaxation is shown in Figure 36). Initially, faster and clearly nonlinear relaxation occurs when transformations among different conformations are speeded up (not shown).
We also performed FORTRAN simulation in the presence of noise with the same initial values (Figure 37). We called random numbers s1 and s2 only once (see Methods). We multiplied identical Box-Muller transform gi containing shift +1 with each of four kinetic constants ki to eliminate the cases of negative ki. Mass conservation for ligands was approximately satisfied with the condition [S]+[P]=const for all ten thousand computational steps. We obtained the best result for the highest efficiency at the 9532nd step. It was less impressive (9.48∙106 M-1s-1) when compared to the best result from the NetLogo simulation. The force decrease during 10000 steps exhibited a similar gradual decrease from higher initial values as in Figure 36 (not shown).
A considerably more comprehensive search for the best dissipation-efficiency coordinate pairs occurred when we called the Box-Muller transform separately four times, once for each of the four rate constants ki. The kigi products gained the freedom to vary independently of each other. The output of such a FORTRAN program (Figure 38) contains two catalytic efficiency values close to the lower range of the diffusion limit (108 M-1s-1).
Glucose isomerase
Glucose isomerase (GI abbreviation) fulfills nutritional requirements mainly in bacteria [124]. It is also known as xylose isomerase because GI reversibly isomerizes D-glucose and D-xylose to D-fructose and D-xylulose, respectively. Glucose to-fructose conversion is rather inefficient but critical for the commercial production of high-fructose corn syrup (HFCS) due to the specificity (the absence of non-metabolizable or toxic side products) and mild ambient conditions [125]. Together with other industrial applications through decades, such as bioethanol production [126], the GI maintained a high market share of the food industry among other industrial enzymes despite its inherently low activity [124,127]. That is why there are so many research attempts to use molecular engineering to improve GI performance for different applications [125,126,128,129,130]. Besides academic interest, that is also the reason for theoretical research devoted to improving GI performance when kinetic and thermodynamic limits are taken into account.
Considering previous examples for other enzymes, the best option is to initiate the research with all the microscopic rate constants inferred from the observed data. Unfortunately, the best such example [27] is for the GI preparation from Streptomyces murinus with very low measured activity. Nevertheless, the principles we employed in this section to significantly improve the GI performance may be applicable for predicting activity gains of the most promising GI variants for green industry applications. We also wanted to test our hypothesis about catalytic efficiency proportionality to dissipation by using the example of an inefficient enzyme working close to the thermodynamic equilibrium. The drawback is the restriction to the two-state model (Figure 1a), that is, the reversible Briggs-Haldane mechanism used in early and recent proposals for the kinetic mechanism of immobilized GI [27,131,132,133,134]. In the pseudo-steady state, the solution is the Michaelis-Menten equation and corresponding performance parameters kcat, KM, and kcat/KM. In our notation (Figure 1a) k3 = kcat and KM = (k2 + k3)/k1s , where k1s is the second order rate constant. Unusual experimental conditions used to determine these kinetic parameters include an elevated temperature (65 ◦C) besides the immobilization of enzymes. These conditions are not responsible for observed low kcat and kcat/KM values. If anything, they slightly increase the performance parameters.

Table 7.
Initial values of microscopic rate constants from experimental and estimated data [27,28] for GI to which we added our calculations of other initial kinetic and thermodynamic parameters in the case of Streptomyces murinus catalyzed conversion of glucose (substrate) to fructose (product) at 65 ◦C.
Table 7.
Initial values of microscopic rate constants from experimental and estimated data [27,28] for GI to which we added our calculations of other initial kinetic and thermodynamic parameters in the case of Streptomyces murinus catalyzed conversion of glucose (substrate) to fructose (product) at 65 ◦C.
| Rate constants |
Observed values [27] | Calculated values [28] |
| k1* | 3.8 M−1min−1 | 0.063 M−1s−1 |
| k2 | 1.23 min−1 | 0.021 s−1 |
| k3 | 1.75 min−1 | 0.029 s−1 |
| k4* | 4.9 M−1min−1 | 0.082 M−1s−1 |
| Other relevant parameters |
Initial values (this paper) |
|
| [S] | 2.0 M | |
| [P] | 0.2 M | |
| [E] | 0.01 M | |
| k1 | 0.126 s−1 | |
| k4 | 0.0164 s−1 | |
| kcat | 0.029 s-1 | |
| KM | 0.794 M | |
| kcat/KM | 0.0365 M−1s−1 | |
| Keqtot | 10.61 | |
| Xtot/RT | 2.31 | |
| Initial value (this paper) |
||
| P | 0.0392 s-1 |
Figure 39 NetLogo simulation with noise independently introduced in all rate constants illustrates the absence of a strong proportionality relationship between catalytic efficiency and entropy production for an inefficient enzyme, such as glucose isomerase. The best point with the coordinates (0.21, 0.21) was found at the 1715th tick. It is associated with the Xtot/RT = 4.7, about 2.5-fold higher k1, 9-fold smaller k2, 24-fold higher equilibrium constant K1, and approximately 11-fold higher partial entropy production P1. Thus, the association and dissociation of substrate with enzyme should be shifted toward complex ES formation for the significant 5.8-fold increase in enzyme efficiency and 2.3-fold increase in the turnover number.
Imposing some constraints on the system can recover the efficiency-dissipation relationship. For instance, rate constants k1 and k2 can be independently multiplied with the normal noise without changes in the rate constants k3 and k4 other than those caused by increased product concentration. The correlation R2 then jumps to 0.881 for kcat/KM dependence on dissipation. However, NetLogo simulation goes through restricted search space and finds somewhat lower values for the best efficiency (not shown).
We also performed the FORTRAN simulations to verify that different software and ways for noise introduction can still produce an approximately linear response in the catalytic efficiency to the dissipation expressed as forcing XJ combination of external force and internal current (Figure 40). Normal noise was called only once and used to multiply all four rate constants. The best catalytic efficiency of 0.18 M-1s-1 was comparable to the best result for the NetLogo simulations with noise. It was also considerably better than the 0.0215 M-1s-1 catalytic efficiency, easily calculated from the Dobovišek et al. optimization [28]. Incidentally, Dobovišek's result was obtained for the positive force Xtot/RT = 0.51 when the net flow is in the forward direction, and our two-state expressions for kcat and KM (see Methods) are appropriate to use for the calculation of kcat/KM. It emerged due to the special nature of the quasi-steady state these authors obtained after imposing the no-change constraint for the product of forward rate constants: = const.
Normal noise with shift +1 (see Methods) was called four times in the next FORTRAN simulation so that each rate constant was multiplied with its own Box-Muller transform (Figure 41). The best catalytic efficiency of 0.226 M-1s-1 result was found for lower overall dissipation in the RT units (0.12 s-1) compared to the previous NetLogo simulation (Figure 39).
The best fold improvements for the catalytic efficiency after noise introduction and the analysis of corresponding changes in rate constants
When variations are allowed in concentrations and microscopic rate constants, an artificial evolution of enzymes is, in theory, possible. We can then visually pick up the conditions with high catalytic efficiency or high turnover numbers. That can be automated in the future using an iterative procedure in which the best performance parameters are chosen as initial until no further improvements occur. Starting from experimentally observed parameters, Table 8 illustrates that enzymes differ in their evolutionary potential but have similar associations among their kinetic and thermodynamic parameters. Namely, a joint increase of kcat/KM and total entropy production argues for the fundamental connection between more efficient free energy transduction into essential biochemical reactions and the level of thermodynamic irreversibility.
Dissipation, Evolution, and Catalytic Power of Enzymes
The evolution of the Universe can be described as the universal evolution. It created time, space, and myriad beautiful objects such as galaxies, stars, planets, and living beings [135]. The invisible but not less important product of universal evolution is increased entropy and entropy production. Through time passage, the product of absolute temperature and entropy production (the dissipation) kept rising from the mysterious initial singularity, which must have had very low entropy, extremely high temperature, and zero entropy production. A new phase of universal evolution started with the appearance of objects that can be associated with the huge jump in entropy production. The first class of such objects are black holes, astronomical objects originating after the explosive death of some massive stars. The second class of objects originated in an aqueous environment endowed with rich chemistry as the first living cells. The same volume of some bacterial cells, mitochondria, or chloroplast produces many orders of magnitude higher dissipation than an equivalent average volume of a sun-like star despite the star's much higher temperature [136,137]. The specific variety of complex life and mineralogy we enjoy here on Earth is not likely to exist anywhere else in the Universe [138]. Thus, we should protect it, study it, and, if possible, understand it as a natural consequence of universal evolution.
It is hard to imagine life without housekeeping enzymes. The simplest and the most successful description of how many such enzymes work is generalized Michaelis-Menten kinetics [36,139,140,141]. The increased complexity of life through eons required means for increasing the catalytic efficiency of such enzymes. According to our knowledge, the scientific literature has never seriously considered how catalytic efficiency is related to an enzyme's entropy production. Banerjee and Bhattacharyya's finding [18] that the more efficient enzyme involves higher total dissipation is in accord with the results presented in this paper. The finding is based only on three pairs of dissipation-efficiency values for a single enzyme (β-galactosidase). Still, it is gratifying that their different method for calculating overall entropy production produced the same result (2553 s-1) as Terrel Hill's approach [34] we used throughout this paper (see the last row from Table 5). How changes in dissipation can lead to an increased catalytic efficiency was not the main interest of these authors.
Martyushev and Seleznev [142] anticipated a fruitful connection between optimal kinetics parameters and entropy production for strongly nonequilibrium processes. However, it is surprising that the relationship between overall dissipation and frequently measured specificity constant kcat/KM was never thoroughly examined. These two parameters connect laboratory biochemistry with the fundamental thermodynamics of nonequilibrium processes. Wofenden and coworkers found with other authors that nothing makes a sharper distinction between life and non-life from the massive jump in the catalytic power, which enzymes show when the speed and specificity of the reaction they catalyze is compared to an equivalent reaction in the presence of inorganic catalysts [10,12,13,14,15,16,143,144,145].
Great strides have been made in uncovering the 3D structure of proteins with enzymatic activity [146]. Still, dynamic changes essential for understanding their catalytic activity are difficult to trace structurally [147]. Structural studies did not help as much as we hoped in answering how enzymes work [4,148]. Initial expectations that entropy changes are the most important contribution to the stabilization of the transition state [149,150] were not confirmed [151], nor the expectations that determining an enzyme's mechanism of action is enough to understand its catalytic power [152].
Despite all the complexity of the enzymes we mentioned here, they are simpler than organisms for investigating evolution [153]. That "cleaner" opportunity brings us back to our approach to the mystery of how enzymes work. Evolution in physics is firmly connected to entropy production. At the same time, the marvelous biochemistry of enzymes is tied to the evolutionary enhancements of enzymes' catalytic rate (up to 1026-fold, according to Edwards et al. [16]). Corresponding catalytic proficiency for alkylsulfatase is an astronomical number: (kcat/KM)/kuncat = 1029 M-1. The mystery lies in the vast increase of enzyme efficiency kcat/KM during pre-biological and biological evolution. Since an increase in entropy production speeds up the physical evolution of any nonequilibrium system (it undergoes faster relaxation from the initial far-from-equilibrium state), we can assume the connection with the evolution of catalytic efficiency.
The initial impetus for the evolution must be the relaxation tendency, that is, the appearance of fluxes in the system, which spontaneously dissipate external force gradients. All products of forces and corresponding fluxes together are nothing else but overall dissipation. Thus, the dissipation is a dynamic phenomenon including external driving forces, fluxes through the system, and output fluxes toward the system's exterior. A system in a quasi-steady nonequilibrium state is an almost perfect converter. It converts free energy influx into internal dissipative fluxes, and external entropy increase, which is usually detected as the heat flux. A small percentage of free energy influx can be converted into transient free energy storage within the system. For biological systems in nearly stationary, far-from-equilibrium states, a hierarchy of internal free-energy conversions can lead to dissipative adaptation [154]. A slight change in the system's properties increases its ability to absorb free energy from the environment. At the same time, the system must produce a slight increase in its dissipative output to remain in a stable quasi-steady state. After long enough free-energy accumulation, some systems acquire the essential biological property of self-replication.
A large outflow of entropy from the growing cells is the major thermodynamic process [155]. Only a small portion of available free energy is used by cells for synthetic and mechanistic goals. For instance, the free energy converted into chemical bonds is a minor contribution compared to the free energy change from catabolism. Still, an almost perfect correlation exists between the total heat released and the amount of dry mass grown or the total amount of oxygen consumed during the aerobic growth of a yeast culture [156]. Theoretical studies also concluded that a higher maximal growth rate would be achieved by replicating a system capable of producing more heat [154,157]. Thus, higher entropy production can be an advantage during the evolution of organisms. As a rule, total entropy production reaches its maximum value before a slow decrease when microorganisms are fully supplied with free-energy sources and engaged in vigorous growth during their short-term evolution in batch experiments. This pattern is recapitulated in the life of every individual organism. Metabolic heat production per surface area reaches the maximal value early, with a subsequent decline over the lifetime [158].
The metabolic heat production is due to enzymes. Using the microcalorimetry method, Sica et al. found in 1987 [159] the proportionality between enzyme activity kcat and observed heat flow. Thermal power was directly proportional to the reaction rate for dihydrofolate reductase (EC 1.5.1.3). Todd and Gomez [160] extended that observation to representative enzymes from each EC classification (a total of 11 different enzymes), assuming the validity of the Michaelis-Menten equation [21]. Isothermal titration microcalorimetry can be used for direct, nondestructive, and precise reaction rate measurement. Todd and Gomez [160] found a reasonably good agreement between kinetic parameters kcat, KM, and kcat/KM assayed colorimetrically and with other methods. Riedel et al. [161] confirmed the agreement of calorimetric and kinetic parameters kcat and KM for catalase, urease, alkaline phosphatase, and triosephosphate isomerase. These authors also studied the feedback effect of released heat in enzyme-catalyzed reactions. Surprisingly, some enzymes asymmetrically release enough heat to increase their diffusion in the presence of substrates.
It is the chemotactic behavior. The enzyme preferentially diffuses towards higher substrate gradients, thus increasing its activity due to increased dissipation. Therefore, nature found a way to channel dissipated and "useless "energy released by enzyme catalysis towards helpful purposes, such as directed motion. For instance, directional water ejection of hexokinase classifies it as a biological pump despite being a cytoplasmic enzyme [162,163]. Hexokinase-2 (HK-II) in humans has been described as a gatekeeper between life and death [2] because it integrates glycolysis with oxidative phosphorylation in healthy cells, contributing to stronger respiratory control. HK-II chemotaxis, due to the entropically favored expulsion of water outside the enzyme pocket, is probably instrumental in its preferential access to ATP generated by mitochondrion.
Assuming that KM does not change for a chosen enzyme, the observed proportionality between the enzyme's entropy production and the turnover number kcat implies a linear increase in catalytic efficiency kcat/KM with dissipation. When noise is present in rate constants, approximate KM constancy will still hold for no changes in k3/k1 ratio (2-state equation (17)), or k5/k1 and k5/k4 ratio (3-state equation (20)), or k3/k1, k3/k5, and k5/k7 ratio (4-state equation (23)) when equilibrium constants do not change.
Through this paper, we could have presented both kcat and kcat/KM proportionality with overall entropy production for each enzyme. One example of the proportionality between kcat and dissipation is for three β-lactamases (Figure 42). Besides kcat to dissipation/RT proportionality (in the units of inverse seconds), that figure also illustrates nearly linear connection between the evolutionary distances of PC1 (1.19), RTEM (1.44), and Lac1 (1.60) lactamase and either kcat or overall dissipation (see Figure 33, [30], and [106] for the evolutionary distances we put in parenthesis). We obtained the same result after comparing experimental results for kinetic and thermodynamic parameters of the A-class β-lactamases and after looking for the maximal partial dissipation in the rate-limiting steps [2,106] In these and other publications [30], we stressed that the optimization for the turnover numbers should be based on the physical principle of maximum transitional entropy production, not on the uncritical acceptance of the maximal catalytic efficiency or maximal catalytic constant as the selection or optimization criterion.
There was no need in the present study to make a priory assumption of either physical or biological principle reigning supreme. We only required some mechanism for reasonable variations in the microscopic rate constants. A crowded cellular milieu and unavoidable errors in translation and transcription offer several such means for noise introduction in kinetic parameters. Stochastic fluctuations are always present and are relevant for applying the Michaelis-Menten type kinetics inside cells [164]. Our simulations are, admittedly, a crude and artificial way of considering the noise. Better methods for dealing with physical and biological noise sources are undoubtedly possible. However, we were primarily interested in whether different means of noise introduction can uncover regular relationships between the most important thermodynamic and kinetic parameters for highly active enzymes that work arbitrarily far from equilibrium. Making use of thermal and non-thermal noise through stochastic fluctuations and dynamic disorder [165,166] may well have been beneficial during biological evolution [167,168,169,170].
Standard evolutionary theory [171] has a simple answer to the question of how new variations can arise: random mutations and natural selection, ensuring the adaptation of the organisms to their environment. Thus, a particular noise class (chance genetic changes) is adapted to provide a better fit among organisms and environments. That view has been extended recently by taking into account the physicochemical evolutionary driving forces [172], including the maximization of dissipation [1].
Figure 42.
FORTRAN simulations for the kcat dependence on overall dissipation in the case of three β-lactamases (PC1, RTEM, and Lac1). Each forward rate constant was multiplied with the identical normal noise function while corresponding backward rate constants were determined from the no-change requirement to equilibrium constants. Table 4 parameters were used for each enzyme. Concentrations were not allowed to change from their initial (observed) values. As for other figures, the Ptot label at the x-axis is the dissipation/RT in inverse seconds. The figure illustrates the proportional increase or decrease of the turnover number with dissipation from observed (calculated) points for PC1 (689, 61), RTEM (6757, 975) and Lac-1 (14526, 1905) (see Table 4 and Juretić et al., 2019 [106]). The highest points have coordinates (3035, 268) for PC1, (3∙104, 4303) for RTEM, and (6.4∙104, 8394) for Lac-1. As for catalytic efficiencies (Figure 33), both observed, and the highest points (dissipation, kcat) are nearly proportional to the evolutionary distance from the putative common ancestor in the order PC1 (1.19) < RTEM (1.44) < Lac1 (1.60).
Figure 42.
FORTRAN simulations for the kcat dependence on overall dissipation in the case of three β-lactamases (PC1, RTEM, and Lac1). Each forward rate constant was multiplied with the identical normal noise function while corresponding backward rate constants were determined from the no-change requirement to equilibrium constants. Table 4 parameters were used for each enzyme. Concentrations were not allowed to change from their initial (observed) values. As for other figures, the Ptot label at the x-axis is the dissipation/RT in inverse seconds. The figure illustrates the proportional increase or decrease of the turnover number with dissipation from observed (calculated) points for PC1 (689, 61), RTEM (6757, 975) and Lac-1 (14526, 1905) (see Table 4 and Juretić et al., 2019 [106]). The highest points have coordinates (3035, 268) for PC1, (3∙104, 4303) for RTEM, and (6.4∙104, 8394) for Lac-1. As for catalytic efficiencies (Figure 33), both observed, and the highest points (dissipation, kcat) are nearly proportional to the evolutionary distance from the putative common ancestor in the order PC1 (1.19) < RTEM (1.44) < Lac1 (1.60).
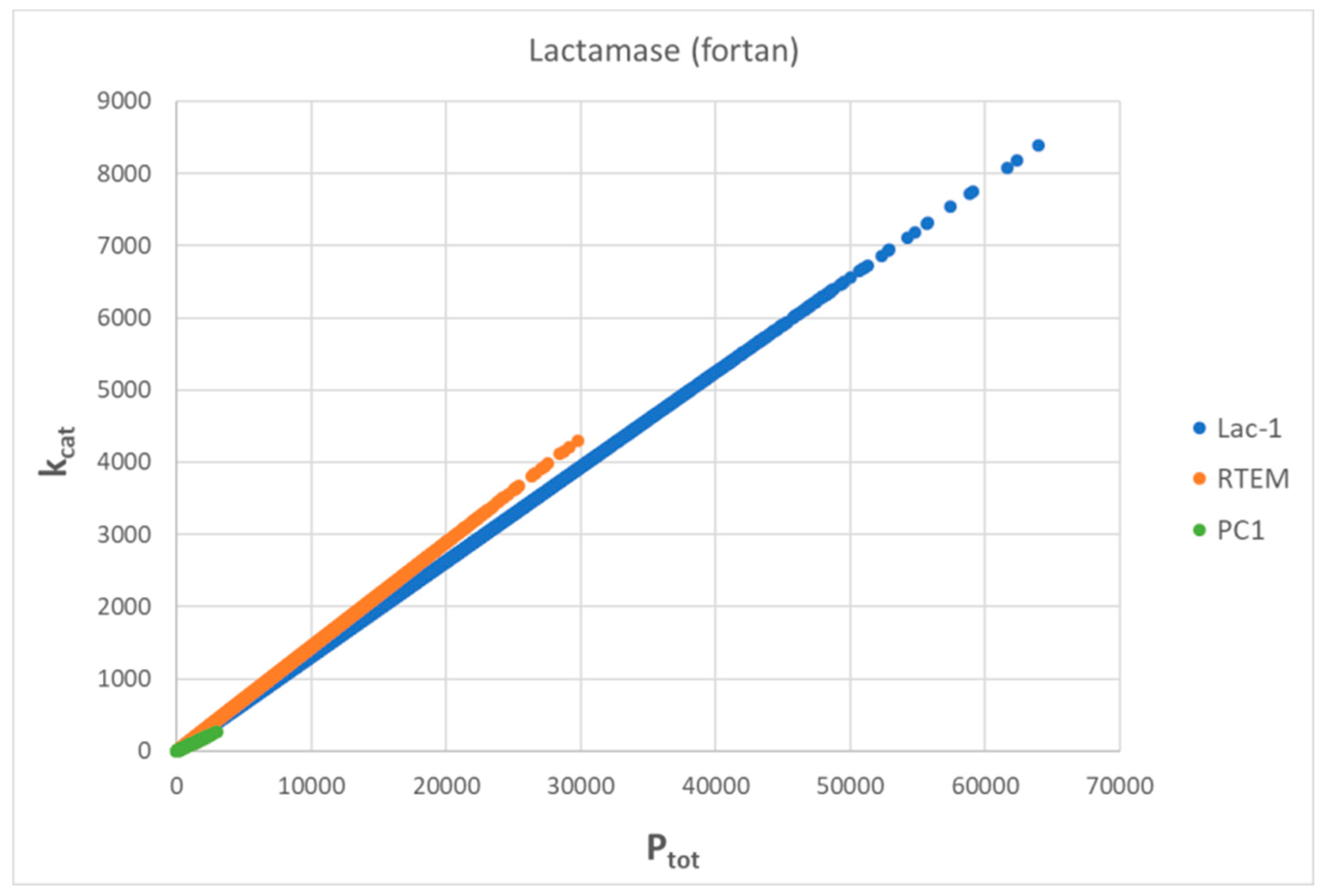
Computational Improvements of the Catalytic Power for Specific Enzymes
The catalytic power of enzymes is measured as kcat or kcat/KM. Some authors did not recommend using kcat/KM as an index for comparing the catalytic effectiveness of enzymes [173]. The majority consensus is that kcat/KM is the appropriate measure for the specificity of noncooperative Michaelis-Menten enzymes [24,174]. In rare cases when all microscopic rate constants have been determined [175], kcat and kcat/KM can be connected to partial and total entropy production when an enzyme reversibly cycles through all of its functionally important conformations (this work and [2]). Moreover, after variations of rate constants around their observed values, we can analyze optimal rate constants ki and dissipations associated with the highest performance parameters kcat and kcat/KM. What are, if any, common features of the states with the highest enzyme efficiency, and how do the thermodynamic and kinetic parameters of these states differ from the same values calculated or inferred from the experimental data? Table 9 helps deal with that question. Our choice in this paper was to examine the best kcat/KM values for corresponding ki, partial, and total dissipation. Table 9 gives the partial entropy production in the first forward catalytic step because it exhibited the highest increase regarding the observed value. That is the consequence of increased forward rate constant k1 and decreased backward constant k2 for the substrate-to-enzyme association and dissociation.
In the case of triosephosphate isomerase (TPI), there was a 1454-fold increase of the partial entropy production P1 for the E+S↔ES transition, which became the 42% instead of 6% contribution to the total dissipation (NetLogo result from Figure 14). For other enzymes and software simulations in the presence of noise, P1 increased one to two orders of magnitude, and its percentage also increased for the best enzyme efficiency results.
The single exception is the carbonic anhydrase. For CAI, CAII, and the T200H CAII mutant, an absolute increase in P1 was not accompanied by an increase in its percentage. A possible reason is a different kinetic scheme for the CA enzyme (Figure 19) and an inadequacy of the standard kcat and kcat/KM expression (equations (22)-(24) ) for that scheme. Krishnamurthy et al. [92] (Table 1 from [92]) compared all known CA enzymes for their and values for the catalytic hydration of CO2 and the dehydration of bicarbonate:
That is the first half-reaction. The buffer is considered the second substrate in the overall two-substrate ping-pong reaction, which recovers free enzymes.
The best efficiency-fold improvement is seen for β-galactosidase, which also reaches the highest efficiency-to-dissipation fold ratio (Table 8). However, ketosteroid isomerase has the best evolutionary potential in our simulations. That can be connected to the two proton transfer reactions catalyzed by KSI [176,177,178] and a powerful electric field [152]. Electric field catalysis needs a strong and correctly oriented field. The measured field of 1.44∙1010 V/m is enough to account for 72% of the total acceleration rate [152]. The transient appearance of billion volts per meter electric field strength in the interior of active proton-shuffling enzymes frequently speeds up catalysis [2]. The isomerization of 5-androstene-3,17-dione in solution through the same mechanism utilized by KSI is slow. That is why KSI catalytic proficiency is so high. As mentioned in the main text, Radzicka and Wolfenden [12] estimated it as 1.8∙1015 M-1 based on kuncat = 6∙10-7 s-1.
Interestingly, the efficiency-fold improvement (Table 8) is similar for the best (KSI) and worst enzyme (glucose isomerase). The kcat = 0.029 s-1 (experimental) and kcat = 0.031 s-1 or 0.068 s-1 (optimal) values for the k3 in Table 9 GI results are two orders of magnitude smaller than the turnover numbers 2 s-1 and 11 s-1 reported in the literature [128,179]. The Converti et al. [27] data we used to initiate simulations pertain to weakly active GI working close to thermodynamic equilibrium. Nevertheless, our method for the theoretical increase in the catalytic activity is robust enough to ensure its close to 10-fold increase (from 0.0365 to 0.2262 M-1s-1, Figure 41).
Possible Benefits of Considering Unanswered Questions
Most enzymes did not use their potential to evolve higher catalytic efficiencies due to the absence of selection pressure to maximize it for individual enzymes [19]. When metabolic demand existed, the superstars of enzyme evolution developed, often named perfect enzymes [175]. Our simulations suggested the theoretical possibility of increasing the kcat/KM of either moderately efficient or perfect enzymes. In practice, more than one amino acid substitution is needed to improve the performance parameters. Several orders of magnitude improvement typically requires at least 5 to 10 beneficial mutations [180].
Living far from equilibrium is an essential asymmetry of present-day life [181,182]. Total entropy production is a convenient measure of how far the system is removed from thermodynamic equilibrium. A certain distance from equilibrium must be maintained to increase the beneficial catalytic efficiency after increased dissipation by whatever means for enzymes we studied in this paper. The plausible inference is that some abiotic driving forces, such as proton gradients in alkaline hydrothermal vents, must have operated to maintain far-from-equilibrium situations and high entropy production during life emergence on Earth. According to that assumption, bioenergetics and vectorial biochemistry are older than the genetic code and the first universal common ancestor [2]. It enabled enzyme-less and cell-less synthesis of amino acids, sugars, nucleotides, and lipids. Nonlinearity and far-from-equilibrium conditions are two requirements for driving proto-metabolism toward autocatalysis and self-organization. Accelerated accumulation of organic molecules followed in the presence of long-lived abiotic protonmotive force to jump-start the development of life [183]. The efficiency of organic synthesis with proto-enzymes was surely low compared to present-day enzymatic catalysis. However, such self-reinforcing reactions increased the efficiency of dissipating available free-energy gradients. The present-day connection between dissipation and catalytic efficiency we studied in this paper is thus likely to reflect the linkage between the higher dissipation potential and accelerated synthesis of ever more complex organic compounds, which was already present at the origin of life. Entropy production increases faster due to the enzyme's activity, albeit in the microscopic world.
Within biology, we cannot find the answer to why dissipation was crucial for the emergence of life, as it is essential for the present-day catalytic efficiency of uni-uni enzymes. Can entropy production have an autocatalytic role too? Namely, did increased entropy production promote the selection of the organic structures capable of increasing entropy production? That question has yet to be answered in biology or physics of nonequilibrium processes. The evolution of all systems in the universe may be coupled to decreasing their free energy in the least possible time [184]. Thus, the living systems and biological macromolecules can be regarded as manifestations of physical principles about dissipation intensity rather than ends in themselves [185].
We mainly dealt with the academic interest in answering the why and how of life emergence during universal thermodynamic evolution. However, there is also a practical goal of enhancing the desired activity of natural enzymes or competing with nature in a rational design of artificial enzymes with better catalytic performance. These research fields are still in their infancy. Microwave irradiation can enhance enzyme activity and entropy production under chemiosmotic conditions [186]. Also, the enhancement in these biochemical and physical parameters can result from distal mutations that do not change individual equilibrium constants for each catalytic step or the overall equilibrium constant of the reaction [187]. De novo enzyme design for green chemistry and medical goals has a huge potential [188,189,190,191,192,193]. It has been recently explored by combining computational methods and directed evolution experiments [194]. Still, something needs to be added to our insights about enzymatic catalysis. Artificial enzymes are generally inferior in catalytic efficiency compared to their natural counterparts [190]. While role or reorganization energy is recognized in rational protein design [190], that is not the case with the catalytic efficiency to dissipation proportionality for uni-uni enzymes we described in this paper. After all other means are employed to identify possible beneficial mutations for increasing the catalytic efficiency with a given substrate, the computer-aided enzyme design can be extended with an additional selection for higher overall entropy production. In principle, mutations can be predicted based on their contribution to total entropy production, not only their contribution to the transition state stabilization and reorganization energy.
Conclusions
Our results stress the hallmark of uni-cycle enzymes as the dissipation gates. Enzymes are not Maxwell's demons that fight the mechanical tendency toward disorder, as Jacob argued in his book "Logic of Life " [195]. Just the opposite, the enzymes open gates for the incomparably faster equilibration of concentrations than in their absence. When such gates opened during biological evolution, they sped up the spontaneous free-energy transduction into dissipative catalytic cycling for many orders of magnitude.
Selecting enzyme structures exhibiting high catalytic efficiency kcat/KM is the hallmark of biological evolution through natural selection. Together with the production of small molecules essential for life, it is indeed an order-creating function of enzymes. Still, it arises through opening the dissipation gates for a vast increase in disorder. A search to open dissipation avalanches implies random structural changes (mutations) and a way to simultaneously fix the advantageous changes causing higher enzyme efficiency and dissipation. Thus, random noise and the increase in overall entropy production are prerequisites rather than hindrances to the evolution of complex life.
There are no known rules for repeating the miracle of biological evolution in increasing or improving enzyme efficiency [180,196]. However, a better connection of observed performance parameters with overall or partial dissipation and introducing dynamic disorder can help find such rules. We performed simulations with five well-known "perfect "enzymes cycling through generalized Michaelis-Menten-type kinetics near the diffusion limit. The take-home message is that increased catalytic efficiency results from higher entropy production.
The changes in enzyme activity and specificity depend on noise channeling constraints. Enzyme efficiency is more or less proportional to overall entropy production when we allow less or more freedom in the choice of restrictions. The efficiency-dissipation proportionality is perfect when we do not permit the change in the driving force and equilibrium constants in each catalytic step. When translated into biological terms, it is the requirement that identical enzymes work in steady or quasi-steady state homeostatic conditions.
Dissecting entropy production contributions suggested the formation of the Michaelian complex ES as the critical catalytic step. Increased equilibrium constant for the substrate-enzyme association can increase the catalytic efficiency in the forward direction (S→P), partial entropy production of that step, and overall dissipation better than other means for increasing activity for most enzymes.
Thus, within physics, we can find the answer to why dissipation was crucial for the emergence of life, as it is essential for the present-day catalytic efficiency of uni-uni enzymes. An increased catalytic efficiency is the outcome of higher entropy production. It is impossible to separate enzyme catalytic rate, efficiency, or power from its overall dissipation. Biological evolution is not different from thermodynamic evolution despite its ability to accelerate the latter. The origin of enzymes' prodigious catalytic power is the synergy between thermodynamic and biological evolution. If increasing enzyme efficiency is the natural evolutionary target for some enzymes and the target for the directed evolution of designed enzymes, researchers can explore beneficial mutations based on their contribution to partial and total entropy production.
Author Contributions
Conceptualization, D.J.; methodology, software, validation, investigation, and writing, D.J. and Ž.B.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
FORTRAN and NetLogo source codes as well as the output excel files are available on request.
Acknowledgments
We acknowledge the validation of initial results by Andrea Gelemanović.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix
We examined the conditions for nearly perfect efficiency to dissipation proportionality. Examples are kinetics simulations for some enzymes, which illustrate the remarkably linear dependency of catalytic efficiency on overall entropy production (Figure 4, Figure 6, Figure 7, Figure 10, Figure 12, Figure 13, Figure 25, Figure 27, Figure 32, Figure 37 and Figure 40). For other enzymes we studied in this paper, such linear dependence was not explicitly shown, although we obtained it when identical assumptions were used. Since such proportionality can be the previously unmentioned hallmark of all enzymes exhibiting generalized Michaelis-Menten kinetics, it is worthwhile to analyze the assumptions and their mathematical consequences.
In this appendix, we first give the expressions for the dependence of efficiency on dissipation for two states
three states
and four states
The next task is to examine the conditions when these expressions for the efficiency to dissipation slope are less likely to change due to variations in the microscopic rate constants. When equilibrium constants for each catalytic step are kept fixed to their observed values, the change in slope can happen only in the case of variable temperature, variable concentrations, or variable ratios of rate constants belonging to different catalytic steps. For selected quasi-steady states, there should be no change in temperature, but the substrate and product concentration do change for an active enzyme if not fixed to their initial values. It all depends on how jumps between steady states are defined. When random numbers s1 and s2 are called only once in the Box-Muller transform (see Methods), the slope never changes for the constant concentration of ligands because the noise cancells in all ratios of rate constants ki/kj, where i and j do not have to be from the same catalytic step. There are slight changes in the slope after allowing for minor changes in the concentration of substrates, products, and enzyme-ligand complexes within the same assumption that identical noise is introduced into one or more catalytic steps. Still, the slope only undergoes a limited increase due to the increase in product concentration and slight force decrease. An entirely different situation occurs when noise is introduced independently two or more times in one or more pairs of rate constants. The constraint of fixed or slightly variable equilibrium constants in catalytic steps is then abandoned, and the goodness of the straight line fit for the proportional dependence of enzyme efficiency on dissipation varies among enzymes but is still strongly affected if any other constraint is imposed.
References
- Gusev, O.A.; Martyushev, L.M. An Evolution Based on Various Energy Strategies. Entropy 2021, 23, 317. [Google Scholar] [CrossRef]
- Juretić, D. Bioenergetics: A Bridge Across Life and Universe; CRC Press Taylor & Francis Group: Boca Raton, Florida, USA, 2021. [Google Scholar]
- Pross, A.; Pascal, R. On the Emergence of Autonomous Chemical Systems through Dissipation Kinetics. Life 2023, 13, 2171. [Google Scholar] [CrossRef]
- Blow, D. So do we understand how enzymes work? Structure 2000, 8, R77–R81. [Google Scholar] [CrossRef]
- Herschlag, D.; Natarajan, A. Fundamental challenges in mechanistic enzymology: progress toward understanding the rate enhancements of enzymes. Biochemistry 2013, 52, 2050–67. [Google Scholar] [CrossRef]
- Schrödinger, E. What is Life. The Physical Aspect of the Living Cell; Cambridge Univ. Press: Cambridge, UK, 1944. [Google Scholar]
- Straškraba, M.; Jørgensen, S.E.; Patten, B.C. Ecosystems emerging: 2. Dissipation. Ecol. Model. 1999, 117, 3–39. [Google Scholar] [CrossRef]
- Nicolis, G.; Prigogine, I. Self-Organization in Non-Equilibrium Systems: From Dissipative Structures to Order Through Fluctuations; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Abramov, O.; Mojzsis, S.J. Microbial habitability of the Hadean Earth during the late heavy bombardment. Nature 2009, 459, 419–422. [Google Scholar] [CrossRef]
- Wolfenden, R. Benchmark reaction rates, the stability of biological molecules in water, and the evolution of catalytic power in enzymes. Annu. Rev. Biochem. 2011, 80, 645–667. [Google Scholar] [CrossRef]
- Goldman, A.D.; Kacar, B. Cofactors are Remnants of Life’s Origin and Early Evolution. J. Mol. Evol. 2021, 89, 127–133. [Google Scholar] [CrossRef]
- Radzicka, A.; Wolfenden, R. A proficient enzyme. Science 1995, 267, 90–93. [Google Scholar] [CrossRef]
- Wolfenden, R. Thermodynamic and extrathermodynamic requirements of enzyme catalysis. Biophys. Chem. 2003, 105, 559–572. [Google Scholar] [CrossRef]
- Snider, M.G.; Temple, B.S.; Wolfenden, R. The path to the transition state in enzyme reactions: a survey of catalytic efficiencies. J. Phys. Org. Chem. 2004, 17, 586–591. [Google Scholar] [CrossRef]
- Stockbridge, R.B.; Lewis Jr., C.A.; Yuan, Y.; Wolfenden, R. Impact of temperature on the time required for the establishment of primordial biochemistry, and for the evolution of enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 22102–22105. [CrossRef]
- Edwards, D.R.; Lohman, D.C.; Wolfenden, R. Catalytic proficiency: the extreme case of S-O cleaving sulfatases. J. Am. Chem. Soc. 2012, 134, 525–531. [Google Scholar] [CrossRef]
- Jencks, W.P. From chemistry to biochemistry to catalysis to movement. Annu. Rev. Biochem. 1997, 66, 1–18. [Google Scholar] [CrossRef]
- Banerjee, K.; Bhattacharyya, K. States with identical steady dissipation rate in reaction networks: A non-equilibrium thermodynamic insight in enzyme efficiency. Chem. Phys. 2014, 438, 1–6. [Google Scholar] [CrossRef]
- Davidi, D.; Longo, L.M.; Jabłońska, J.; Milo, R.; Tawfik, D.S. A Bird's-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations. Chem. Rev. 2018, 118, 8786–8797. [Google Scholar] [CrossRef]
- Martyushev, L.M.; Seleznev, V.D. Maximum entropy production principle in physics, chemistry and biology. Phys. Rep. 2006, 426, 1–45. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M. L. Kinetics of invertase action. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Michaelis, L.; Menten M., L.; Johnson K., A.; Goody R., S. The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry. 2011, 50, 8264–9. [Google Scholar] [CrossRef]
- Johnson, K.A. A century of enzyme kinetic analysis, 1913 to 2013. FEBS Letters 2013, 587, 2753–2766. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. One hundred years of Michaelis–Menten kinetics. Perspectives in Science 2015, 4, 3–9. [Google Scholar] [CrossRef]
- Voorsluijs, V.; Avanzini, F.; Esposito, M. Thermodynamic validity criterion for the irreversible Michaelis-Menten equation. arXiv arXiv:2006.06476v1. [CrossRef]
- Das, B.; Banerjee, K.; Gangopadhyay, G. Propensity approach to nonequilibrium thermodynamics of a chemical reaction network: Controlling single E-coli β-galactosidase enzyme catalysis through the elementary reaction steps. J. Chem. Phys. 2013, 139, 244104. [Google Scholar] [CrossRef]
- Converti, A.; Borghi, M.D. Kinetics of glucose isomerization to fructose by immobilized glucose isomerase in the presence of substrate protection. Bioprocess Engineering 1998, 18, 27–33. [Google Scholar] [CrossRef]
- Dobovišek, A.; Vitas, M.; Brumen, M.; Fajmut, A. Energy conservation and maximal entropy production in enzyme reactions. Biosystems. 2017, 158, 47–56. [Google Scholar] [CrossRef]
- Christensen, H.; Martin, M.T.; Waley, G. β-lactamases as fully efficient enzymes. Determination of all the rate constants in the acyl-enzyme mechanism. Biochem. J. 1990, 266, 853–861. [Google Scholar]
- Juretić, D.; Bonačić Lošić, Ž.; Domagoj Kuić, D.; Juraj Simunić, J.; Dobovišek, A. The maximum entropy production requirement for proton transfers enhances catalytic efficiency for β-lactamases. Biophys. Chem. 2019, 244, 11–21. [Google Scholar] [CrossRef]
- Toney, M.D. Carbon Acidity in Enzyme Active Sites. Front. Bioeng. Biotechnol. 2019, 7, 25. [Google Scholar] [CrossRef]
- Knowles, J.R.; Albery, W.J. Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc. Chem. Res. 1977, 10, 105–111. [Google Scholar] [CrossRef]
- Behravan, G.; Jonsson, B.H.; Lindskog, S. Fine tuning of the catalytic properties of carbonic anhydrase. Studies of a Thr200 → His variant of human isoenzyme II. Eur. J. Biochem. 1990, 190, 351–357. [Google Scholar] [CrossRef]
- Hill, T.L. Free Energy Transduction in Biology. The Steady State Kinetic and Thermodynamic Formalism; Academic Press: New York, NY, USA, 1977. [Google Scholar]
- Hill, T.L. Free Energy Transduction and Biochemical Cycle Kinetics; Dover Publications, Inc.: Mineola, New York, USA, 2005. [Google Scholar]
- Wilhelm, T.; Hoffmann-Klipp, E.; Heinrich, R. An evolutionary approach to enzyme kinetics: optimization of ordered mechanisms. Bull. Math. Biol. 1994, 56, 65–106. [Google Scholar] [CrossRef]
- Ge, H.; Qien, M. Steady-state cycle kinetics of single enzymes: Competing substrates and multi-conformations. J. Theor. Comput. Chem. 2008, 7, 1001–1027. [Google Scholar] [CrossRef]
- Toney, M. D. Common Enzymological Experiments Allow Free Energy Profile Determination. Biochemistry. 2013, 52, 5952–5965. [Google Scholar] [CrossRef]
- Heinrich, R.; Schuster, S.; Holzhütter, H.-G. Mathematical analysis of enzymic reaction systems using optimmization principles. Eur. J. Biochem. 1991, 201, 1–21. [Google Scholar] [CrossRef]
- Johnson, K.A. 1 Transient-State Kinetic Analysis of Enzyme Reaction Pathways. In The Enzymes; Sigman, D.S., Ed.; Academic Press, 1992; Volume 20, pp. 1–61. [Google Scholar]
- Box, G.E.P.; Muller, M.E. A note on the generation of random normal derivatives. Ann. Math. Stat. 1958, 29, 610–611. [Google Scholar] [CrossRef]
- Wilensky, U.; Reisman, K. Thinking Like a Wolf, a Sheep, or a Firefly: Learning Biology Through Constructing and Testing Computational Theories—An Embodied Modeling Approach. Cognition and Instruction 2006, 24, 171–2009. [Google Scholar] [CrossRef]
- Apte, A. Computational modeling of biochemical systems using cellular automata. Ph.D. thesis, Virginia Commonwealth University, 2009. [Google Scholar] [CrossRef]
- Ishida, T. Possibility of Controlling Self-Organized Patterns with Totalistic Cellular Automata Consisting of Both Rules like Game of Life and Rules Producing Turing Patterns. Micromachines (Basel) 2018, 9, 339. [Google Scholar] [CrossRef]
- Koopmans, L.; Youk, H. Predictive landscapes hidden beneath biological cellular automata. J. Biol. Phys. 2021, 47, 355–369. [Google Scholar] [CrossRef]
- Sklar, E. NetLogo, a multi-agent simulation environment. Artif. Life 2007, 13, 303–311. [Google Scholar] [CrossRef]
- Dong, X.; Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Agent-Based Modeling of Endotoxin-Induced Acute Inflammatory Response in Human Blood Leukocytes. PLoS ONE 2010, 5, e9249. [Google Scholar] [CrossRef]
- Bravo, R.; Axelrod, D.E. A calibrated agent-based computer model of stochastic cell dynamics in normal human colon crypts useful for in silico experiments. Theor. Biol. Med. Model 2013, 10, 66. [Google Scholar] [CrossRef]
- Dutot, A.; Olivier, D. Swarm Problem-Solving. In Agent-Based Spatial Simulation with NetLogo; Banos, A., Lang, C., Marilleau, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2, pp. 117–172. [Google Scholar] [CrossRef]
- Wilensky, U. NetLogo; Center for Connected Learning and Computer-Based Modeling, Northwestern University: Evanston, IL, USA, 1999; Available online: http://ccl.northwestern.edu/netlogo/.
- Wierenga, R.K.; Kapetaniou, E.G.; Venkatesan, R. Triosephosphate isomerase: a highly evolved biocatalyst. Cell. Mol. Life Sci. 2010, 67, 3961–3982. [Google Scholar] [CrossRef]
- Olivares-Illana, V.; Riveros-Rosas, H.; Nallely Cabrera, N.; de Gómez-Puyou, M.T.; Ruy Pérez-Montfort, R.; Costas, M.; Gómez-Puyou, A. A guide to the effects of a large portion of the residues of triosephosphate isomerase on catalysis, stability, druggability, and human disease. Proteins. 2017, 85, 1190–1211. [Google Scholar] [CrossRef]
- Gerlt, J. A. Evolution of Enzyme Function and the Development of Catalytic Efficiency: Triosephosphate Isomerase, Jeremy R. Knowles, and W. John Albery. Biochemistry 2021, 60, 3529–3538. [Google Scholar] [CrossRef]
- Albery, W.J.; Knowles, J.R. Evolution of enzyme function and the development of catalytic efficiency. Biochemistry 1976, 15, 5631–5640. [Google Scholar] [CrossRef]
- Richard, J.P. Acid-Base Catalysis of the Elimination and Isomerization Reactions of Triose Phosphates. J. Am. Chem. Soc. 1984, 106, 4926–4936. [Google Scholar] [CrossRef]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 2011, 50, 4402–4410. [Google Scholar] [CrossRef]
- Webster, K.A. Evolution of the coordinate regulation of glycolytic enzyme genes by hypoxia. J. Exp. Biol. 2003, 206, 2911–2922. [Google Scholar] [CrossRef]
- Orozco, J.M.; Krawczyk, P.A.; Scaria, S.M.; Cangelosi, A.L.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. Dihydroxyacetone phosphate signals glucose availability to mTORC1. Nat. Metab. 2020, 2, 893–901. [Google Scholar] [CrossRef]
- Myers, T.D.; Palladino, M.J. Newly discovered roles of triosephosphate isomerase including functions within the nucleus. Mol. Med. 2023, 29, 18. [Google Scholar] [CrossRef]
- Vázquez-Jiménez, L.K.; Moreno-Herrera, A.; Juárez-Saldivar, A.; González-González, A.; Ortiz-Pérez, E.; Paz-González, A.D.; Palos, I.; Ramírez-Moreno, E.; Rivera, G. Recent Advances in the Development of Triose Phosphate Isomerase Inhibitors as Antiprotozoal Agents. Curr. Med. Chem. 2022, 29, 2504–2529. [Google Scholar] [CrossRef]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2014, 34, 3751–3759. [Google Scholar] [CrossRef]
- Ationu, A.; Humphries, A. The feasibility of replacement therapy for inherited disorder of glycolysis: triosephosphate isomerase deficiency (review). Int. J. Mol. Med. 1998, 6, 701–704. [Google Scholar] [CrossRef]
- Blacklow, S.C.; Raines, R.T.; Lim, W.A.; Zamore, P.D.; Knowles, J.R. Triosephosphate isomerase catalysis is diffusion controlled. Biochemistry 1988, 27, 1158–1165. [Google Scholar] [CrossRef]
- Wade, R.C.; Davis, M.E.; Luty, B.A.; Madura, J.D.; McCammon, J.A. Gating of the active site of triose phosphate isomerase: Brownian dynamics simulations of flexible peptide loops in the enzyme. Biophys J. 1993, 64, 9–15. [Google Scholar] [CrossRef]
- Wade, R.C.; Gabdoulline, R.R.; Lüdemann, S.K.; Lounnas, V. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proc. Natl. Acad. Sci. USA. 1998, 95, 5942–5949. [Google Scholar] [CrossRef]
- Katebi, A.R.; Jernigan, R.L. The critical role of the loops of triosephosphate isomerase for its oligomerization, dynamics, and functionality. Protein Sci. 2014, 23, 213–228. [Google Scholar] [CrossRef]
- Sharma, P.; Guptasarma, P. ‘Super-perfect’ enzymes: structural stabilities and activities of recombinant triose phosphate isomerases from Pyrococcus furiosus and Thermococcus onnurineus produced in Escherichia coli. Biochem. Biophys. Res. Commun. 2015, 460, 753–758. [Google Scholar] [CrossRef]
- Schachner, L.F.; Soye, B.D.; Ro, S.; Kenney, G.E.; Ives, A.N.; Su, T.; Goo, Y.A.; Jewett, M.C.; Rosenzweig, A.C.; Kelleher, N.L. Revving an Engine of Human Metabolism: Activity Enhancement of Triosephosphate Isomerase via Hemi-Phosphorylation. ACS Chem. Biol. 2022, 17, 2769–2780. [Google Scholar] [CrossRef]
- Tajes, M.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Palomer, E.; Guix, F.X.; Muñoz, F.J. The pathophysiology of triose phosphate isomerase dysfunction in Alzheimer's disease. Histol. Histopathol. 2013, 28, 43–51. [Google Scholar] [CrossRef]
- Bonačić Lošić, Ž.; Donđivić, T.; Juretić, D. Is the catalytic activity of triosephosphate isomerase fully optimized? An investigation based on maximization of entropy production. J. Biol. Phys. 2017, 43, 69–86. [Google Scholar] [CrossRef]
- Juretić, D.; Bonačić Lošić, Ž. Comments on ’Flexibility of enzymatic transitions as a hallmark of optimized enzyme steady-state kinetics and thermodynamics’. Comput. Biol. Chem. 2021, 95, 107571. [Google Scholar] [CrossRef]
- Šterk, M.; Markovič, R.; Marhl, M.; Fajmut, A.; Dobovišek, A. Flexibility of enzymatic transitions as a hallmark of optimized enzyme steady-state kinetics and thermodynamics. Comput. Biol. Chem. 2021, 91, 107449. [Google Scholar] [CrossRef]
- Klipp, E.; Heinrich, R. Evolutionary optimization of enzyme kinetic parameters; effect of constraints. J. Theor. Biol. 1994, 171, 309–323. [Google Scholar] [CrossRef]
- Bish, D.R.; Mavrovouniotis, M.L. Enzymatic reaction rate limits with constraints on equilibrium constants and experimental parameters. Biosystems 1998, 47, 37–60. [Google Scholar] [CrossRef]
- Stieff, M.; Wilensky, U. NetLogo Enzyme Kinetics model; Center for Connected Learning and Computer-Based Modeling, Northwestern University: Evanston, IL, USA, 2001; Available online: http://ccl.northwestern.edu/netlogo/models/EnzymeKinetics.
- Chapleau, R.R.; Robinson, P.J.; Schlager, J.J.; Gearhart, J.M. Potential new therapeutic modality revealed through agent-based modeling of the neuromuscular junction and acetylcholinesterase inhibition. Theor. Biol. Med. Model. 2014, 11, 42. [Google Scholar] [CrossRef]
- Talalay, P. A fascination with enzymes: the journey not the arrival matters. J. Biol. Chem. 2005, 280, 28829–28847. [Google Scholar] [CrossRef]
- Pollack, R.M. Enzymatic mechanisms for catalysis of enolization: ketosteroid isomerase. Bioorg. Chem. 2004, 32, 341–353. [Google Scholar] [CrossRef]
- Fried, S.D.; Boxer, S.G.; Fayer, M.D.; Solomon, E.I. On the origins of catalysis by ketosteroid isomerase. PhD by Fried, S.D. 2014. Available online: http://purl.stanford.edu/bf687qj8918.
- Pollack, R.M.; Zeng, B.; Mack, J.P.G.; Eldin, S. Determination of the microscopic rate constants for the base catalyzed conjugation of 5-androstene-3,17-dione. J. Am. Chem. Soc. 1989, 111, 6419–6423. [Google Scholar] [CrossRef]
- Pollack, R.M.; Thornburg, L.D.; Wu, Z.R.; Summers, M.F. Mechanistic insights from the three-dimensional structure of 3-oxo-Delta(5)-steroid isomerase. Arch. Biochem. Biophys. 1999, 370, 9–15. [Google Scholar] [CrossRef]
- Fried, S.D.; Bagchi, S.; Boxer, S.G. Extreme electric fields power catalysis in the active site of ketosteroid isomerase. Science 2014, 346, 1510–1514. [Google Scholar] [CrossRef]
- Wu, Y.; Boxer, S.G. A Critical Test of the Electrostatic Contribution to Catalysis with Noncanonical Amino Acids in Ketosteroid Isomerase. J. Am. Chem. Soc. 2016, 138, 11890–11895. [Google Scholar] [CrossRef]
- Wu, Y.; Fried, S.D. Boxer, S.G. A Preorganized Electric Field Leads to Minimal Geometrical Reorientation in the Catalytic Reaction of Ketosteroid Isomerase. J. Am. Chem. Soc. 2020, 142, 9993–9998. [Google Scholar] [CrossRef]
- Ito, M.; Brinck, T. Novel Approach for Identifying Key Residues in Enzymatic Reactions: Proton Abstraction in Ketosteroid Isomerase. J. Phys. Chem. B 2014, 118, 13050–13058. [Google Scholar] [CrossRef]
- Thomas, J.L.; Evans, B.W.; Blanco, G.; Mason, J.I.; Strickler, R.C. Creation of a fully active, cytosolic form of human type I 3β-hydroxysteroid dehydrogenase/isomerase by the deletion of a membrane-spanning domain. J. Mol. Endocrinol 1999, 23, 231–239. [Google Scholar] [CrossRef]
- Hawkinson, D.C.; Eames, T.C.; Pollack, R.M. Energetics of 3-oxo-Δ5-steroid isomerase: source of the catalytic power of the enzyme. Biochemistry 1991, 30, 10849–10858. [Google Scholar] [CrossRef]
- Kupriyanova, E.; Pronina, N.; Los, D. Carbonic anhydrase — a universal enzyme of the carbon-based life. Photosynthetica 2017, 55, 3–19. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Senda, M. Fukuda, K.; Yu, H.Y.; Ishida, M.; Taira, M.; Kinbara, K.; Senda, T. Characterization of a novel type of carbonic anhydrase that acts without metal cofactors. BMC Biol. 2021, 19, 105. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Andring, J.T.; McKenna, R. Crystallography and Its Impact on Carbonic Anhydrase Research. Int. J. Med. Chem. 2018, 2018, 9419521. [Google Scholar] [CrossRef]
- Sanyal, G.; Maren, T.H. Thermodynamics of carbonic anhydrase catalysis. A comparison between human isoenzymes B and C. J. Biol. Chem. 1981, 256, 608–612. [Google Scholar] [CrossRef]
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein–Ligand Binding. Chem. Rev. 2008, 108, 946–1051. [Google Scholar] [CrossRef]
- McDevitt, M.E.; Lambert, L.A. Molecular evolution and selection pressure in alpha-class carbonic anhydrase family members. Biochim. Biophys. Acta 2011, 1814, 1854–18561. [Google Scholar] [CrossRef]
- Fisher, S.Z.; Tu, C.; Bhatt, D.; Govindasamy, L. Agbandje-McKenna, M.; McKenna, R.; Silverman, D.N. Speeding up proton transfer in a fast enzyme: kinetic and crystallographic studies on the effect of hydrophobic amino acid substitutions in the active site of human carbonic anhydrase II. Biochemistry 2007, 46, 3803–3813. [Google Scholar] [CrossRef]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Hewett-Emmett, D.; Hopkins, P.J.; Tashian, R.E.; Czelusniak, J. Origins and molecular evolution of the carbonic anhydrase isozymes. Ann. N. Y. Acad. Sci. 1984, 429, 338–358. [Google Scholar] [CrossRef]
- Liljas, A.; Laurberg, M. A wheel invented three times. The molecular structures of the three carbonic anhydrases. EMBO Repb. 2000, 1, 16–17. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef]
- Cabaleiro-Lago, C.; Lundqvist, M. The Effect of Nanoparticles on the Structure and Enzymatic Activity of Human Carbonic Anhydrase I and II. Molecules 2020, 25, 4405. [Google Scholar] [CrossRef] [PubMed]
- Diez-Fernandez, C.; Rüfenacht, V.; Santra, S.; Lund, A.M.; Santer, R.; Lindner, M.; Tangeraas, T.; Unsinn, C.; de Lonlay, P.; Burlina, A.; van Karnebeek, C.D.; Häberle, J. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early-onset life-threatening metabolic crisis. Genet. Med. 2016, 18, 991–1000. [Google Scholar] [CrossRef]
- de Souza, L.C.; Provensi, G.; Vullo, D.; Carta, F.; Scozzafava, A.; Costa, A.; Schmidt, S.D.; Passani, M.B.; Supuran, C.T.; Blandina, P. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017, 118, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.D.; Nachtigall, E.G.; Marcondes, L.A.; Zanluchi, A.; Furini, C.R.G.; Passani, M.B.; Supuran, C.T.; Blandina, P.; Izquierdo, I.; Provensi, G.; de Carvalho Myskiw, J. Modulation of Carbonic Anhydrases Activity in the Hippocampus or Prefrontal Cortex Differentially Affects Social Recognition Memory in Rats. Neuroscience 2022, 497, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, S.; Fang, X.; Salmon, S. Carbonic Anhydrase Enhanced UV-Crosslinked PEG-DA/PEO Extruded Hydrogel Flexible Filaments and Durable Grids for CO2 Capture. Gels 2023, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Behravan, G.; Jonsson, B.H.; Lindskog, S. Fine tuning of the catalytic properties of carbonic anhydrase. Studies of a Thr200 → His variant of human isoenzyme II. Eur. J. Biochem. 1990, 190, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Juretić, D.; Simunić, J.; Bonačić Lošić, Ž. Maximum entropy production theorem for transitions between enzyme functional states and its application. Entropy 2019, 21, 743. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Endimiani, A.; Hujer, K.M.; Bonomo, R.A. The continuing challenge of ESBLs, Curr. Opin. Pharmacol. 2007, 7, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Medicin. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076–18. [Google Scholar] [CrossRef]
- Liras, P.; Martin, J.F. Gene clusters for beta-lactam antibiotics and control of their expression: why have clusters evolved, and from where did they originate? Int. Microbiol. 2006, 9, 9–19. [Google Scholar]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R Soc. Lond. B Biol. Sci. 1980, 289, 321–331. [Google Scholar]
- Burbaum, J.J.; Raines, R.T.; Albery, W.J.; Knowles, J.R. Evolutionary optimization of the catalytic effectiveness of an enzyme. Biochemistry 1989, 28, 9293–9305. [Google Scholar] [CrossRef]
- Brocklehurst, K.; Topham, C.M. Kinetic parameters of the acyl-enzyme mechanism and conditions for quasi-equilibrium and for optimal catalytic characteristics. Biochem. J. 1990, 270, 561–3. [Google Scholar] [CrossRef]
- Matagne, A.; Dubus, A.; Galleni, M. Frère, J.M. The beta-lactamase cycle: a tale of selective pressure and bacterial ingenuity. Nat. Prod. Rep. 1999, 16, 1–19. [Google Scholar] [CrossRef]
- Haldane, J.B.S. Enzymes. Longmans: London, UK, 1930. [Google Scholar]
- Brocklehurst, K. Evolution of enzyme catalytic power. Characteristics of optimal catalysis evaluated for the simplest plausible kinetic model. Biochem J. 1977, 163, 111–116. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Walter, N. Michaelis-Menten is dead, long live Michaelis-Menten! Nat. Chem. Biol. 2006, 2, 66–67. [Google Scholar] [CrossRef]
- English, B.P.; Min, W.; van Oijen, A.M.; Lee, K.T.; Luo, G.; Sun, H.; Cherayil, B.J.; Kou, S.C.; Xie, X.S. Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited. Nat. Chem. Biol. 2006, 2, 87–94. [Google Scholar] [CrossRef]
- Juers, D.H.; Matthews, B.W.; Huber, R.E. LacZ β-galactosidase: structure and function of an enzyme of historical and molecular biological importance. Protein Sci. 2012, 21, 1792–1807. [Google Scholar] [CrossRef]
- Vera, C.; Guerrero, C.; Aburto, C.; Cordova, A.; Illanes, A. Conventional and non-conventional applications of β-galactosidases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140271. [Google Scholar] [CrossRef]
- Muñoz-Labrador, A.; Lebron-Aguilar, R.; Quintanilla-Lopez, J.E.; Galindo-Iranzo, P. M.; Azcarate, S.A.; Kolida, S.; Kachrimanidou, V.; Garcia-Cañas, V.; Methven, L.; Rastall, R.A.; Moreno, F.J.; Hernandez-Hernandez, O. Prebiotic Potential of a New Sweetener Based on Galactooligosaccharides and Modified Mogrosides. J. Agric. Food Chem. 2022, 70, 9048–9056. [Google Scholar] [CrossRef]
- Das, B.; Gangopadhyay, G. Large deviation theory for the kinetics and energetics of turnover of enzyme catalysis in a chemiostatic flow. J. Chem. Phys. 2018, 148, 174104. [Google Scholar] [CrossRef]
- Bhosale, S.H.; Rao, M.B.; Deshpande, V.V. Molecular and Industrial Aspects of Glucose Isomerase. Microbiol. Rev. 1996, 60, 280–300. [Google Scholar] [CrossRef]
- Nam, K.H. Glucose Isomerase: Functions, Structures, and Applications. Appl. Sci. 2022, 12, 428. [Google Scholar] [CrossRef]
- Seike, T.; Kobayashi, Y.; Sahara, T.; Ohgiya, S.; Kamagata, Y.; Fujimori, K.E. Molecular evolutionary engineering of xylose isomerase to improve its catalytic activity and performance of micro-aerobic glucose/xylose co-fermentation in Saccharomyces cerevisiae. Biotechnol. Biofuels 2019, 12, 139. [Google Scholar] [CrossRef]
- DiCosimo, R.; McAuliffe, J.; Ayrookaran, J.P.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–74. [Google Scholar] [CrossRef]
- Meng, M.; Bagdasarian, M. Zeikus, J.G. The role of active-site aromatic and polar residues in catalysis and substrate discrimination by xylose isomerase. Proc. Natl. Acad. Sci. USA. 1993, 90, 8459–8463. [Google Scholar] [CrossRef]
- Hartley, B.S.; Hanlon, N.; Jackson, R.J.; Rangarajan, M. Glucose isomerase: insights into protein engineering for increased thermostability. Biochim. Biophys. Acta 2000, 1543, 294–335. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M. Epting, K.L., Kelly, R.M. N-terminal fusion of a hyperthermophilic chitin-binding domain to xylose isomerase from Thermotoga neapolitana enhances kinetics and thermostability of both free and immobilized enzymes. Biotechnol. Prog. 2010, 26, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Boersma, J.G.; Vellenga, K.; De Wilt, H.G.J.; Joosten, G.E.H. Mass transfer effects on the rate of isomerization of D-glucose into D-fructose, catalyzed by whole-cell immobilized glucose isomerase. Biotechnol. and Bioeng. 1979, 21, 1711–1724. [Google Scholar] [CrossRef]
- Kikkert, A.; Vellenga, K.; De Wilt, H.G.J.; Joosten, G.E.H. The isomerization of D-glucose into D-fructose catalyzed by whole-cell immobilized glucose isomerase. The dependence of the intrinsic rate of reaction on substrate concentration, pH, and temperature. Biotechnol. Bioeng. 1981, 23, 1087–1101. [Google Scholar] [CrossRef]
- Chen, K.-C.; Wu, J.-Y. Substrate protection of immobilized glucose isomerase. Biotechnol. Bioeng. 1987, 30, 817–824. [Google Scholar] [CrossRef]
- Carrazco-Escalante, M.; Caro-Corrales, J.; Iribe-Salazar, R.; Ríos-Iribe, E.; Vázquez-López, Y.; Gutiérrez-Dorado, R.; Hernández-Calderón, O. A new approach for describing and solving the reversible Briggs-Haldane mechanism using immobilized enzyme. Can. J. Chem. Eng. 2020, 98, 316–329. [Google Scholar] [CrossRef]
- Smolin, L. The Life of the Cosmos. Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Metzner, H. Bioelectrochemistry of photosynthesis: a theoretical approach. Bioelectrochem. Bioenerg. 1984, 13, 183–190. [Google Scholar] [CrossRef]
- Martyushev, L.M. Life Defined in Terms of Entropy Production: 20th Century Physics Meets 21st Century Biology. Bioessays 2020, 42, e2000101. [Google Scholar] [CrossRef]
- Hystad, G.; Downs, R.T.; Grew, E.S.; Hazen, R.M. Statistical analysis of mineral diversity and distribution: Earth’s mineralogy is unique. Earth Planet. Sci. Lett. 2015, 426, 154–157 doiorg/101016/jepsl201506028. [Google Scholar] [CrossRef]
- Keleti, T. Two rules of enzyme kinetics for reversible Michaelis-Menten mechanisms. FEBS Lett. 1986, 208, 109–112. [Google Scholar] [CrossRef]
- Brooks, S.P.; Storey, K.B. A kinetic description of sequential, reversible, Michaelis-Menten reactions: practical application of theory to metabolic pathways. Mol. Cell. Biochem. 1992, 115, 43–48. [Google Scholar] [CrossRef]
- Pettersson, G. Evolutionary optimization of the catalytic efficiency of enzymes. Eur. J. Biochem. 1992, 206, 289–295. [Google Scholar] [CrossRef]
- Martyushev, L.M.; Seleznev, V.D. Maximum entropy production: application to crystal growth and chemical kinetics. Curr. Opin. Chem. Eng. 2015, 7, 23–31. [Google Scholar] [CrossRef]
- Lee, J.K.; Houk, K.N. A proficient enzyme revisited: the predicted mechanism for orotidine monophosphate decarboxylase. Science 1997, 276, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S. Catalytic Efficiency of Enzymes: A Theoretical Analysis. Biochemistry. 2013, 52, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Amyes, T.L.; Goryanova, B.; Zhai, X. Enzyme Architecture: On the Importance of Being in a Protein Cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vrielink, A.; Sampson, N. Sub-Angstrom resolution enzyme X-ray structures: is seeing believing? Curr. Opin. Struct. Biol. 2003, 13, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Skopintsev, P.; James, D.; Dworkowski, F.; Panepucci, E.; Kekilli, D.; Furrer, A.; Brünle, S.; Mous, S.; Ozerov, D.; Nogly, P.; Wang, M.; Standfuss, J. Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 2019, 365, 61–65. [Google Scholar] [CrossRef]
- Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D.G. How enzymes work: analysis by modern rate theory and computer simulations. Science 2004, 303, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Page, M.I.; Jencks, W.P. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. USA 1971, 68, 1678–1683. [Google Scholar] [CrossRef]
- Jencks, W.P. From chemistry to biochemistry to catalysis to movement. Annu. Rev. Biochem. 1997, 66, 1–18. [Google Scholar] [CrossRef]
- Villa, J.; Strajbl, M.; Glennon, T.M.; Sham, Y.Y.; Chu, Z.T.; Warshel, A. How important are entropic contributions to enzyme catalysis? Proc. Natl. Acad. Sci. USA. 2000, 97, 11899–904. [Google Scholar] [CrossRef]
- Fried, S.D.; Boxer, S.G. Electric Fields and Enzyme Catalysis. Annu. Rev. Biochem. 2017, 86, 387–415. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.J.; Thornton, J.W. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat. Rev. Genet. 2013, 14, 559–571. [Google Scholar] [CrossRef] [PubMed]
- England, J.L. Statistical physics of self-replication. J. Chem. Phys. 2013, 139, 121923. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, J.; Wagensberg, J. On the entropy production in microbiological stationary states. J. Theor. Biol. 1986, 122, 347–358. [Google Scholar] [CrossRef]
- von Stockar, U. Biothermodynamics of live cells: a tool for biotechnology and biochemical engineering. J. Non-Equilib. Thermodyn. 2010, 35, 415–475. [Google Scholar] [CrossRef]
- Saadat, N. P.; Nies, T.; Rousset, Y.; Ebenhöh, O. Thermodynamic Limits and Optimality of Microbial Growth. Entropy 2020, 22, 277. [Google Scholar] [CrossRef]
- Aoki, I. Entropy principle for human development, growth and aging. J. Theor. Biol. 1991, 150, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Sica, L.; Gilli, R.; Briand, C.; Sari, J.C. A flow microcalorimetric method for enzyme activity measurements: application to dihydrofolate reductase. Anal. Biochem. 1987, 165, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Todd, M.J.; Gomez, J. Enzyme kinetics determined using calorimetry: a general assay for enzyme activity? Anal. Biochem. 2001, 296, 179–187. [Google Scholar] [CrossRef]
- Riedel, C.; Gabizon, R.; Wilson, C.A.M.; Hamadani, K.; Tsekouras, K.; Marqusee, S.; Pressé, S.; Bustamante, C. The heat released during catalytic turnover enhances the diffusion of an enzyme. Nature 2015, 517, 227–230. [Google Scholar] [CrossRef]
- Sengupta, S.; Dey, K.K.; Muddana, H.S.; Tabouillot, T.; Ibele, M.E.; Butler, P.J.; Sen, A. Enzyme molecules as nanomotors. J. Am. Chem. Soc. 2013, 135, 1406–1414. [Google Scholar] [CrossRef]
- Mohajerani, F.; Zhao, X.; Somasundar, A.; Velegol, D.; Sen, A. A theory of enzyme chemotaxis: From experiments to modeling. Biochemistry 2018, 57, 6256–6263. [Google Scholar] [CrossRef]
- Grima, R.; Walter, N.G. Schnell, S. Single-molecule enzymology à la Michaelis-Menten FEBS J. 2014, 281, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Zwanzig, R. Rate processes with dynamical disorder. Acc. Chem. Res. 1990, 23, 148–152. [Google Scholar] [CrossRef]
- Kou, S.C.; Cherayil, B.J.; Min, K.; English, B.P.; Xie, X.S. Single-Molecule Michaelis−Menten Equations. J. Phys. Chem. B 2005, 109, 19068–19081. [Google Scholar] [CrossRef]
- Astumian, R.D.; Chock, P.B.; Tsong, T.Y.; Chen, Y.D.; Westerhoff, H.V. Can free energy be transduced from electric noise? Proc. Natl. Acad. Sci. USA. 1987, 84, 434–438. [Google Scholar] [CrossRef]
- Qian, H. Cooperativity in Cellular Biochemical Processes: Noise-Enhanced Sensitivity, Fluctuating Enzyme, Bistability with Nonlinear Feedback, and Other Mechanisms for Sigmoidal Responses. Annu. Rev. Biophys. 2012, 41, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Milias-Argeitis, A.; Khammash, M. Dynamic disorder in simple enzymatic reactions induces stochastic amplification of substrate. J. R. Soc. Interface 2017, 14, 20170311. [Google Scholar] [CrossRef]
- Ariga, T.; Tateishi, K.; Tomishige, M.; Mizuno, D. Noise-Induced Acceleration of Single Molecule Kinesin-1. Phys. Rev. Lett. 2021, 127, 178101. [Google Scholar] [CrossRef]
- Laland, K.; Uller, T.; Feldman, M.; Sterelny, K.; Müller, G.B.; Moczek, A.; Jablonka, E.; Odling-Smee, J.; Wray, G.A.; Hoekstra, H.E.; Futuyma, D.J.; Lenski, R.E.; Mackay, T.F.C.; Schluter, D.; Strassmann, J.E. Does evolutionary theory need a rethink? Nature 2014, 514, 161–164. [Google Scholar] [CrossRef]
- Auboeuf, D. Physicochemical Foundations of Life that Direct Evolution: Chance and Natural Selection are not Evolutionary Driving Forces. Life 2020, 10, 7; [Google Scholar] [CrossRef]
- Eisenthal, R.; Danson, M.J.; Hough, D.W. Catalytic efficiency and kcat/KM: a useful comparator? Trends Biotechnol. 2007, 25, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A.; Cárdenas, M.L. Specificity of Non-Michaelis-Menten Enzymes: Necessary Information for Analyzing Metabolic Pathways. J. Phys. Chem. B 2010, 114, 16209–16213. [Google Scholar] [CrossRef]
- Bar-Even, A.; Milo, R.; Noor, E.; Tawfik, D.S. The Moderately Efficient Enzyme: Futile Encounters and Enzyme Floppiness. Biochemistry 2015, 54, 4969–4977. [Google Scholar] [CrossRef] [PubMed]
- Kraut, D.A.; Sigala, P.A.; Pybus, B.; Liu, C.W.; Ringe, D.; Petsko, G.A.; Herschlag, D. Testing electrostatic complementarity in enzyme catalysis: hydrogen bonding in the ketosteroid isomerase oxyanion hole. PLoS Biol. 2006, 4, e99. [Google Scholar] [CrossRef]
- Chakravorty, D.K.; Hammes-Schiffer, S. Impact of Mutation on Proton Transfer Reactions in Ketosteroid Isomerase: Insights from Molecular Dynamics Simulations. J. Am. Chem. Soc. 2010, 132, 7549–7555. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.T.; Lewandowski, E.M.; Chen, Y. Low barrier hydrogen bonds in protein structure and function. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140557. [Google Scholar] [CrossRef]
- Asbóth, B.; Náray-Szabó, G. Mechanism of action of D-xylose isomerase. Curr. Protein Pept. Sci. 2000, 1, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.; Tawfik, D.S. Enzyme engineering: reaching the maximal catalytic efficiency peak. Curr. Opin. Struct. Biol. 2017, 47, 140–150. [Google Scholar] [CrossRef]
- Branscomb, E.; Russell, M.J. Frankenstein or a Submarine Alkaline Vent: Who Is Responsible for Abiogenesis?: Part 1: What is life-that it might create itself? Bioessays 2018, 40, 1700179. [Google Scholar] [CrossRef]
- Branscomb, E.; Russell, M.J. Frankenstein or a submarine alkaline vent: who is responsible for abiogenesis? Part 2: As life is now, so it must have been in the beginning. BioEssays 2018, 40, 1700182. [Google Scholar] [CrossRef]
- Peretó, J. Prebiotic chemistry that led to life. In Handbook of Astrobiology; Kolb, V.M., Ed.; CRC Press: Boca Raton, USA, 2019; pp. 219–233. [Google Scholar]
- Wang, Q.A. Maximum entropy change and least action principle for nonequilibrium systems. Astrophys. Space Sci. 2006, 305, 273–281. [Google Scholar] [CrossRef]
- Annila, A.; Baverstock, K. Genes without prominence: a reappraisal of the foundations of biology. J. R. Soc. Interface 2014, 11, 20131017. [Google Scholar] [CrossRef]
- Yu, D.; Wu, H.; Zhang, A.; Tian, L.; Liu, L.; Wang, C.; Fang, X. Microwave irradiation-assisted isomerization of glucose to fructose by immobilized glucose isomerase. Process Biochemistry 2011, 46, 599–603. [Google Scholar] [CrossRef]
- Wang, L.; Tharp, S.; Selzer, T.; Benkovic, S.J.; Kohen, A. Effects of a distal mutation on active site chemistry. Biochemistry 2006, 45, 1383–92. [Google Scholar] [CrossRef]
- Khersonsky, O.; Röthlisberger, D.; Wollacott, A.M.; Murphy, P.; Dym, O.; Albeck, S.; Kiss, G.; Houk, K.N.; Baker, D.; Tawfik, D.S. Optimization of the in silico designed Kemp eliminase KE70 by computational design and directed evolution. J. Mol. Biol. 2011, 407, 391–412. [Google Scholar] [CrossRef]
- Labas, A.; Szabo, E.; Mones, L.; Fuxreiter, M. Optimization of reorganization energy drives evolution of the designed Kemp eliminase KE07. Biochim. Biophys. Acta 2013, 1834, 908–917. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Mones, L. The role of reorganization energy in rational enzyme design. Curr. Opin. Chem. Biol. 2014, 21, 34–41. [Google Scholar] [CrossRef]
- Frushicheva, M.P.; Mills, M.J.L.; Schopf, P.; Singh, M.K.; Warshel, A. Computer Aided Enzyme Design and Catalytic Concepts. Curr. Opin. Chem. Biol. 2014, 21, 56–62. [Google Scholar] [CrossRef]
- Crean, R.M.; Gardner, J.M.; Kamerlin, S.C.L. Harnessing Conformational Plasticity to Generate Designer Enzymes. J. Am. Chem. Soc. 2020, 142, 11324–11342. [Google Scholar] [CrossRef]
- Mondal, D.; Kolev, V.; Warshel, A. Combinatorial Approach for Exploring Conformational Space and Activation Barriers in Computer-Aided Enzyme Design. ACS Catal. 2020, 10, 6002–6012. [Google Scholar] [CrossRef]
- Yang, K.K. Wu, Z.; Arnold, F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Jacob, F. The logic of life. Pantheon Books: New York, NY, USA, 1973. [Google Scholar]
- Agarwal, P.K. A Biophysical Perspective on Enzyme Catalysis. Biochemistry 2019, 58, 438–449. [Google Scholar] [CrossRef]
Figure 1.
Schemes for the enzyme reactions of Michaelis-Menten type with a) two, b) three, and c) four states.
Figure 1.
Schemes for the enzyme reactions of Michaelis-Menten type with a) two, b) three, and c) four states.
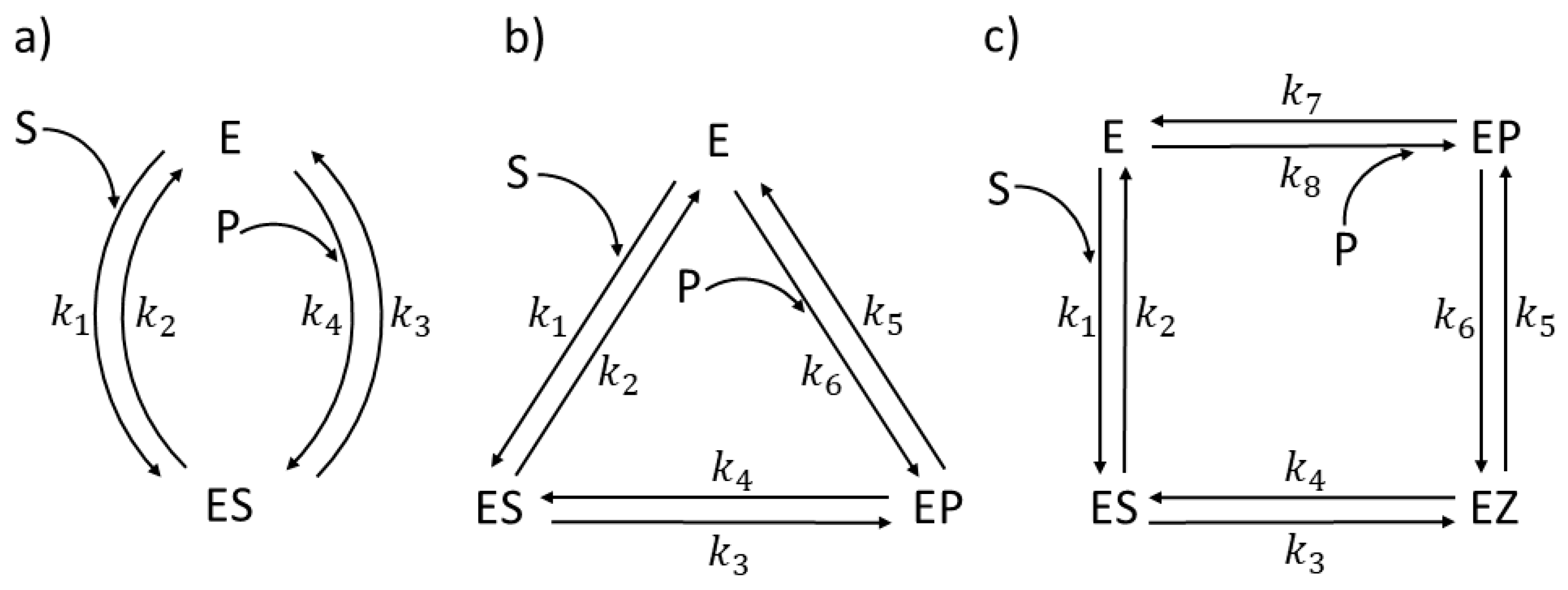
Figure 2.
The catalytic efficiency dependence on dissipation after stepwise increase of the last forward rate constant k7. The k7 jumped for 10.0 units in each of the 1000 deterministic steps in the FORTRAN program, starting with the k7 = 10 s-1. The K4 = k7/k8 is then calculated from fixed k8 and variable k7. There are no changes in other equilibrium constants. Their values follow from Table 1 as K1 = k1/k2, K2 = k3/k4, and K3 = k5/k6. The X/RT values also go through the stepwise increase from negative -5.3 to positive 1.6, thus passing through the vanishing value at the thermodynamic equilibrium and near equilibrium value 0.685, which we kept constant in our previous simulation of the TPI kinetics [70]. There was no change in initial concentrations of substrates (40 μM) and products (0.064 μM).
Figure 2.
The catalytic efficiency dependence on dissipation after stepwise increase of the last forward rate constant k7. The k7 jumped for 10.0 units in each of the 1000 deterministic steps in the FORTRAN program, starting with the k7 = 10 s-1. The K4 = k7/k8 is then calculated from fixed k8 and variable k7. There are no changes in other equilibrium constants. Their values follow from Table 1 as K1 = k1/k2, K2 = k3/k4, and K3 = k5/k6. The X/RT values also go through the stepwise increase from negative -5.3 to positive 1.6, thus passing through the vanishing value at the thermodynamic equilibrium and near equilibrium value 0.685, which we kept constant in our previous simulation of the TPI kinetics [70]. There was no change in initial concentrations of substrates (40 μM) and products (0.064 μM).
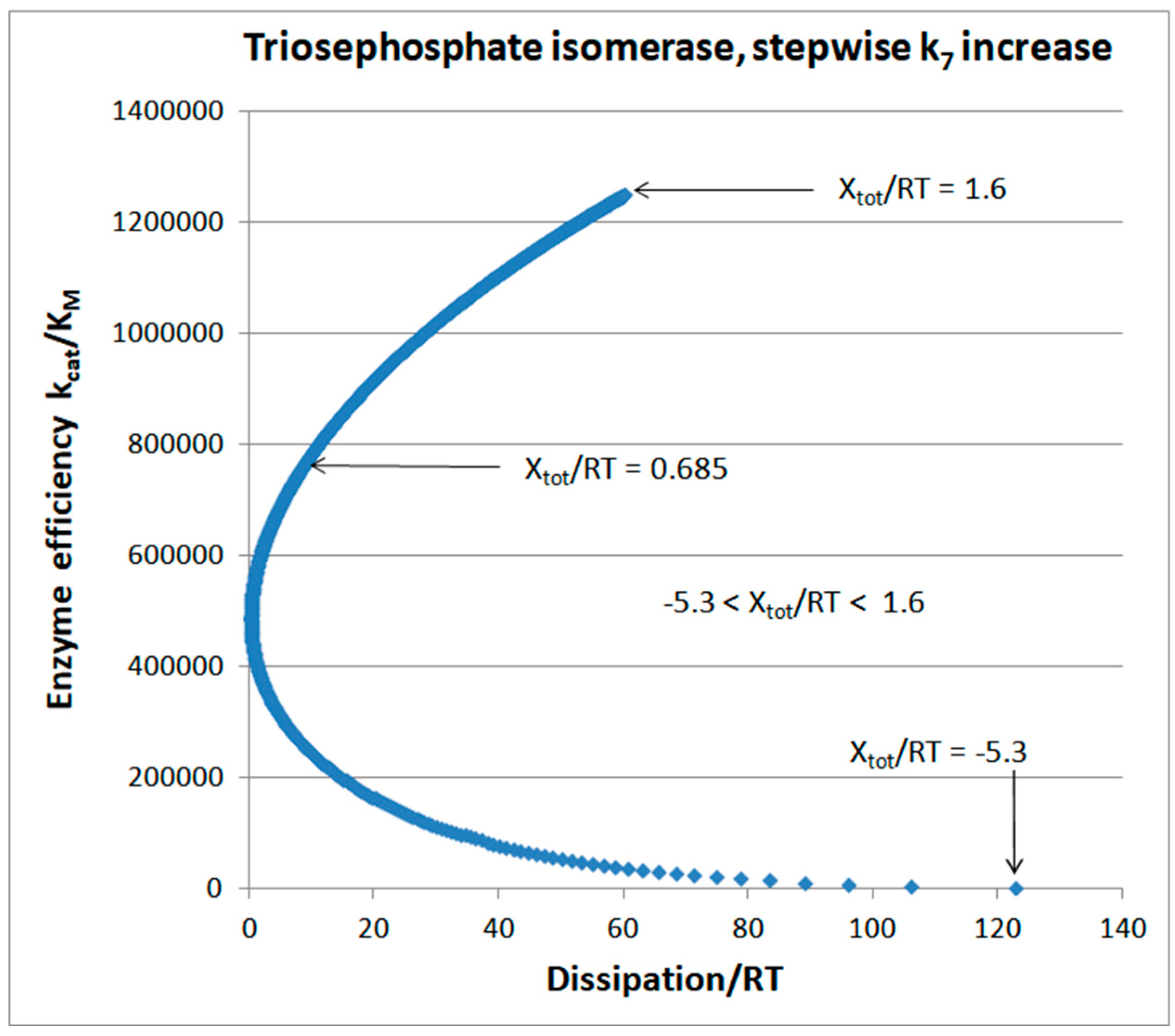
Figure 3.
The catalytic efficiency dependence on dissipation after stepwise increase of the last forward rate constant k7, for positive force values. The same FORTRAN program and identical conditions conditions are used as described in the legend of Figure 2. We deleted all negative Xtot values and corresponding efficiency and dissipation values to get this figure. The figure illustrates the nonlinearity of the efficiency(dissipation) function despite a surprisingly good linear fit with R2 = 0.9442.
Figure 3.
The catalytic efficiency dependence on dissipation after stepwise increase of the last forward rate constant k7, for positive force values. The same FORTRAN program and identical conditions conditions are used as described in the legend of Figure 2. We deleted all negative Xtot values and corresponding efficiency and dissipation values to get this figure. The figure illustrates the nonlinearity of the efficiency(dissipation) function despite a surprisingly good linear fit with R2 = 0.9442.
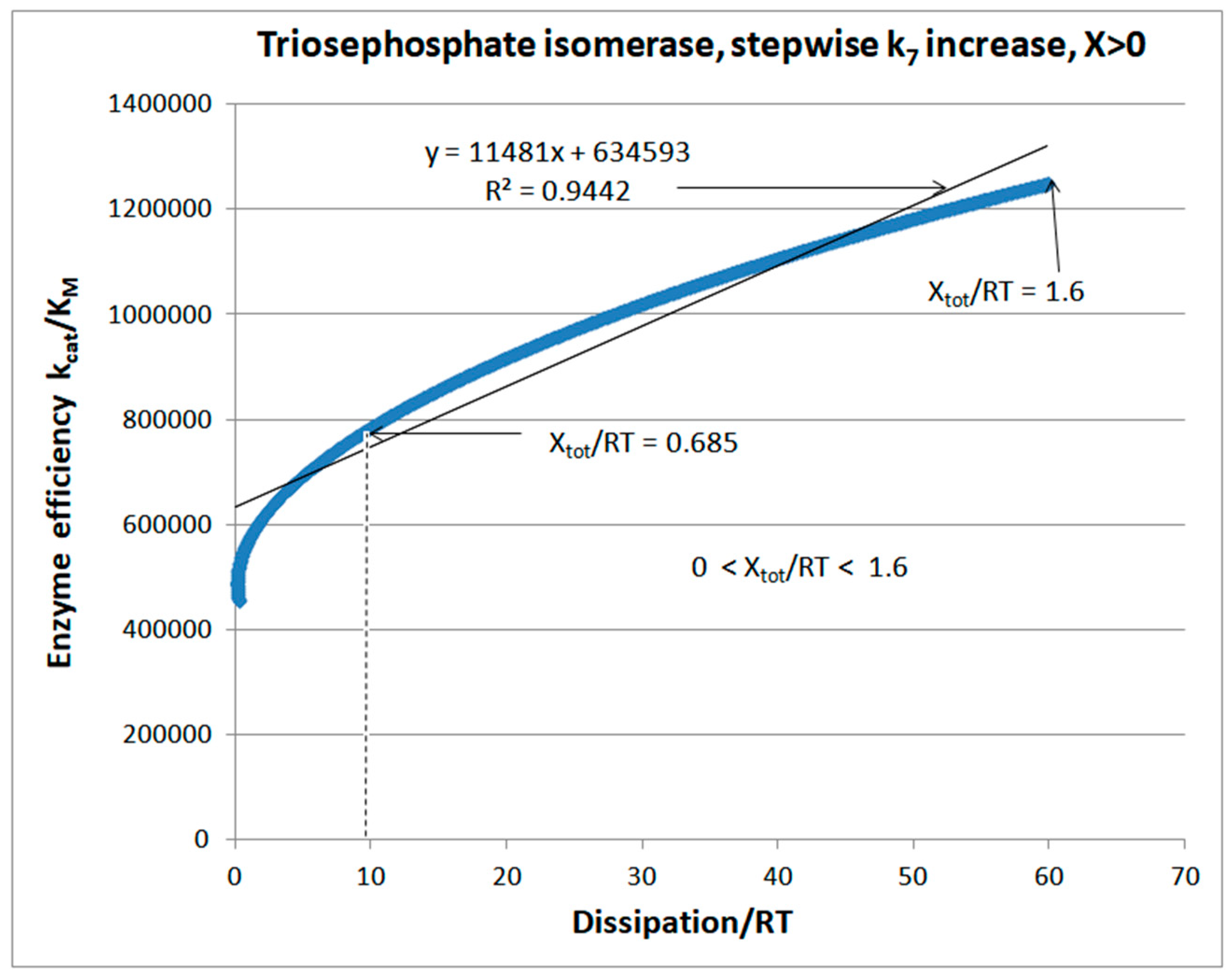
Figure 4.
The catalytic efficiency dependence on dissipation for deterministic jumps between steady states such that increases in the forward rate constant k7 (for whatever reason) are constrained by the requirement that equilibrium constant K4 does not change from the observed value K4 = 156 [32]. Thus, the backward rate constant k8 in the last catalytic step must also go through stepwise increases calculated from the K4 = const requirement. Other rate and equilibrium constants remain equal to their initial values (see Table 1). The total force Xtot/RT remains equal to its initial value of 0.685 through all jumps between 1000 steady states. The figure illustrates the perfect proportionality between enzyme efficiency and entropy production when change is not allowed in equilibrium constants for cyclic catalytic steps.
Figure 4.
The catalytic efficiency dependence on dissipation for deterministic jumps between steady states such that increases in the forward rate constant k7 (for whatever reason) are constrained by the requirement that equilibrium constant K4 does not change from the observed value K4 = 156 [32]. Thus, the backward rate constant k8 in the last catalytic step must also go through stepwise increases calculated from the K4 = const requirement. Other rate and equilibrium constants remain equal to their initial values (see Table 1). The total force Xtot/RT remains equal to its initial value of 0.685 through all jumps between 1000 steady states. The figure illustrates the perfect proportionality between enzyme efficiency and entropy production when change is not allowed in equilibrium constants for cyclic catalytic steps.
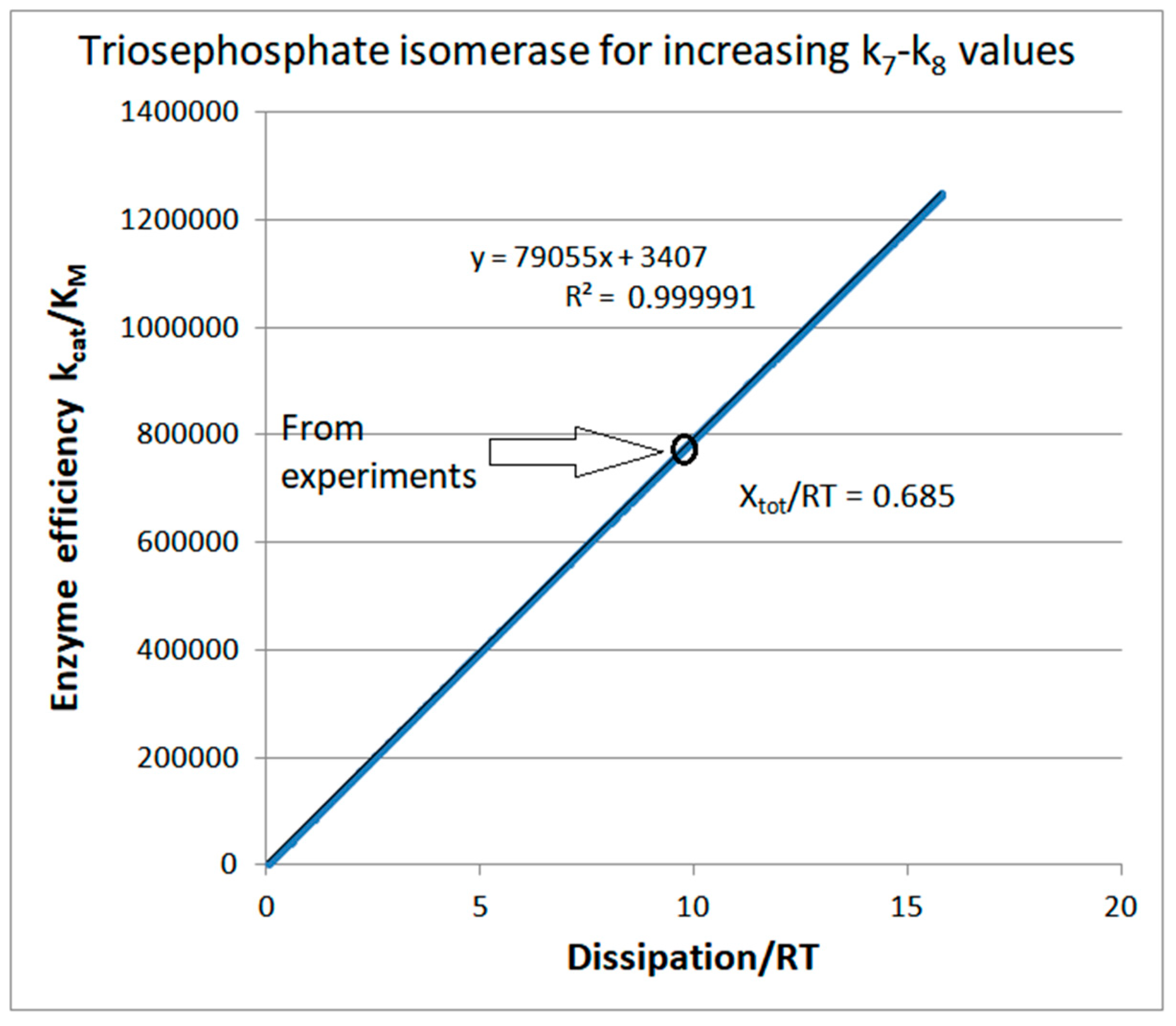
Figure 5.
Triosephosphate isomerase thermodynamics and kinetics. Stepwise changes in the substrate and product concentrations are the only cause for the change in the chemical affinity (force) at the x-axis and of entropy production and net flux (both in units of inverse seconds) at the y-axis. We assumed that the initial sum of substrate and product concentrations does not change. Therefore, the decrease in the substrate concentration from its initial concentration of 40 μM is always accompanied by the increase in the product concentration from its initial concentration of 0.064 μM. Other parameters in the FORTRAN program are the same as in our 2017 paper [70], and we used the symbols from that paper to facilitate the comparison with that and other older simulations. In the figure, we compared our results (the vertical line for the positive force with arrows in the insert) with the simulation results of Šterk et al. [72] (the vertical line for the negative force with arrows in the main figure).
Figure 5.
Triosephosphate isomerase thermodynamics and kinetics. Stepwise changes in the substrate and product concentrations are the only cause for the change in the chemical affinity (force) at the x-axis and of entropy production and net flux (both in units of inverse seconds) at the y-axis. We assumed that the initial sum of substrate and product concentrations does not change. Therefore, the decrease in the substrate concentration from its initial concentration of 40 μM is always accompanied by the increase in the product concentration from its initial concentration of 0.064 μM. Other parameters in the FORTRAN program are the same as in our 2017 paper [70], and we used the symbols from that paper to facilitate the comparison with that and other older simulations. In the figure, we compared our results (the vertical line for the positive force with arrows in the insert) with the simulation results of Šterk et al. [72] (the vertical line for the negative force with arrows in the main figure).
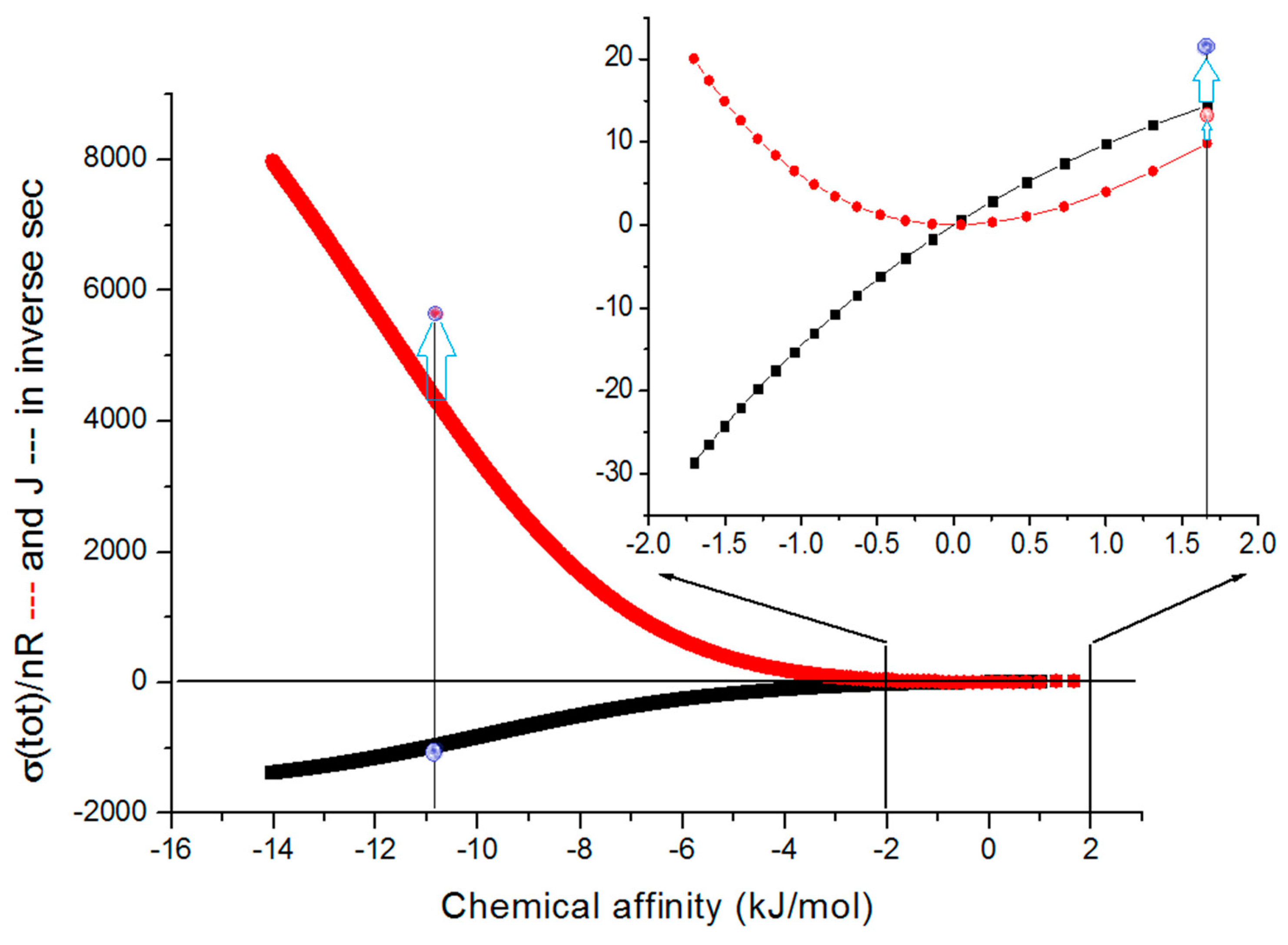
Figure 6.
The catalytic efficiency dependence on dissipation for nondeterministic jumps between steady states when forward rate constant k7 = 4000 s-1 is multiplied with the Box-Muller transform for the random normal noise with the shift +2 (see Methods). The k8 variation follows from the requirement k8=k7/156.25 (K4=156.25), that is, from the no-change restriction for the calculated K4 value from the observed data [32]. The FORTRAN program used to prepare this figure also required fixed K1, K2, K3, and all other parameters at their initial values (see Table 1) for all of the 10000 computational steps.
Figure 6.
The catalytic efficiency dependence on dissipation for nondeterministic jumps between steady states when forward rate constant k7 = 4000 s-1 is multiplied with the Box-Muller transform for the random normal noise with the shift +2 (see Methods). The k8 variation follows from the requirement k8=k7/156.25 (K4=156.25), that is, from the no-change restriction for the calculated K4 value from the observed data [32]. The FORTRAN program used to prepare this figure also required fixed K1, K2, K3, and all other parameters at their initial values (see Table 1) for all of the 10000 computational steps.
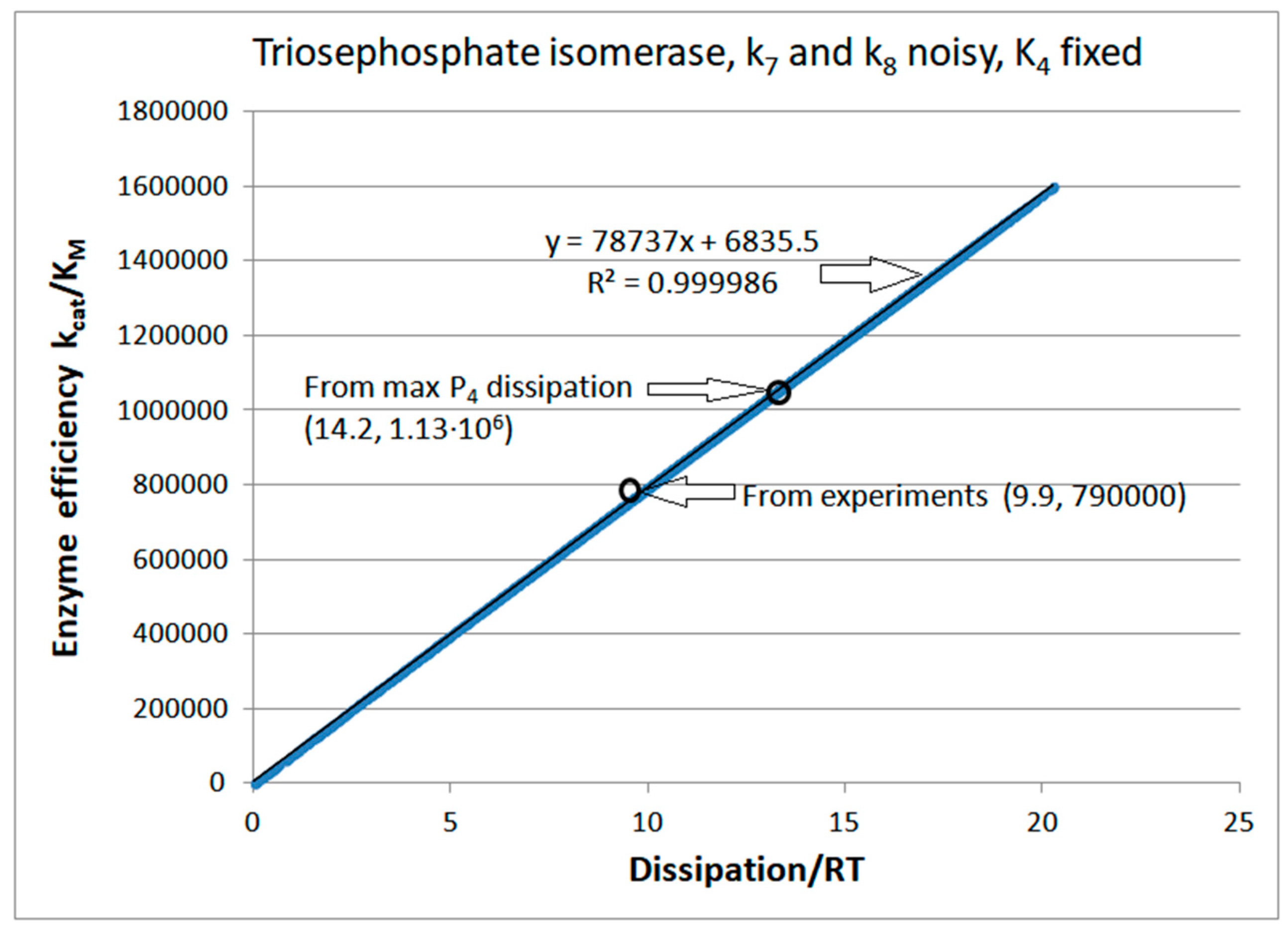
Figure 7.
Enzyme efficiency kcat/KM as a function of dissipation/RT for the triosephosphate isomerase when kinetic constants k7 and k8 in the last transition vary due to the introduction of Gaussian noise. Representative initial values of rate constant k8 are 25 s−1, 32 s−1, 40 s−1, 100 s−1, and 160 s−1. As before, we used the K4 = const restriction and the experimental data for kinetic constants shown in Table 1 for transitions between other catalytic steps. Figure 7 differs from Figure 6 because we allowed five different equilibrium constants K4 = 160, 125, 100, 40, and 25 to span five force values Xtot/RT ≡ X/RT from negative to positive (see inserted X/RT values and corresponding symbols).
Figure 7.
Enzyme efficiency kcat/KM as a function of dissipation/RT for the triosephosphate isomerase when kinetic constants k7 and k8 in the last transition vary due to the introduction of Gaussian noise. Representative initial values of rate constant k8 are 25 s−1, 32 s−1, 40 s−1, 100 s−1, and 160 s−1. As before, we used the K4 = const restriction and the experimental data for kinetic constants shown in Table 1 for transitions between other catalytic steps. Figure 7 differs from Figure 6 because we allowed five different equilibrium constants K4 = 160, 125, 100, 40, and 25 to span five force values Xtot/RT ≡ X/RT from negative to positive (see inserted X/RT values and corresponding symbols).
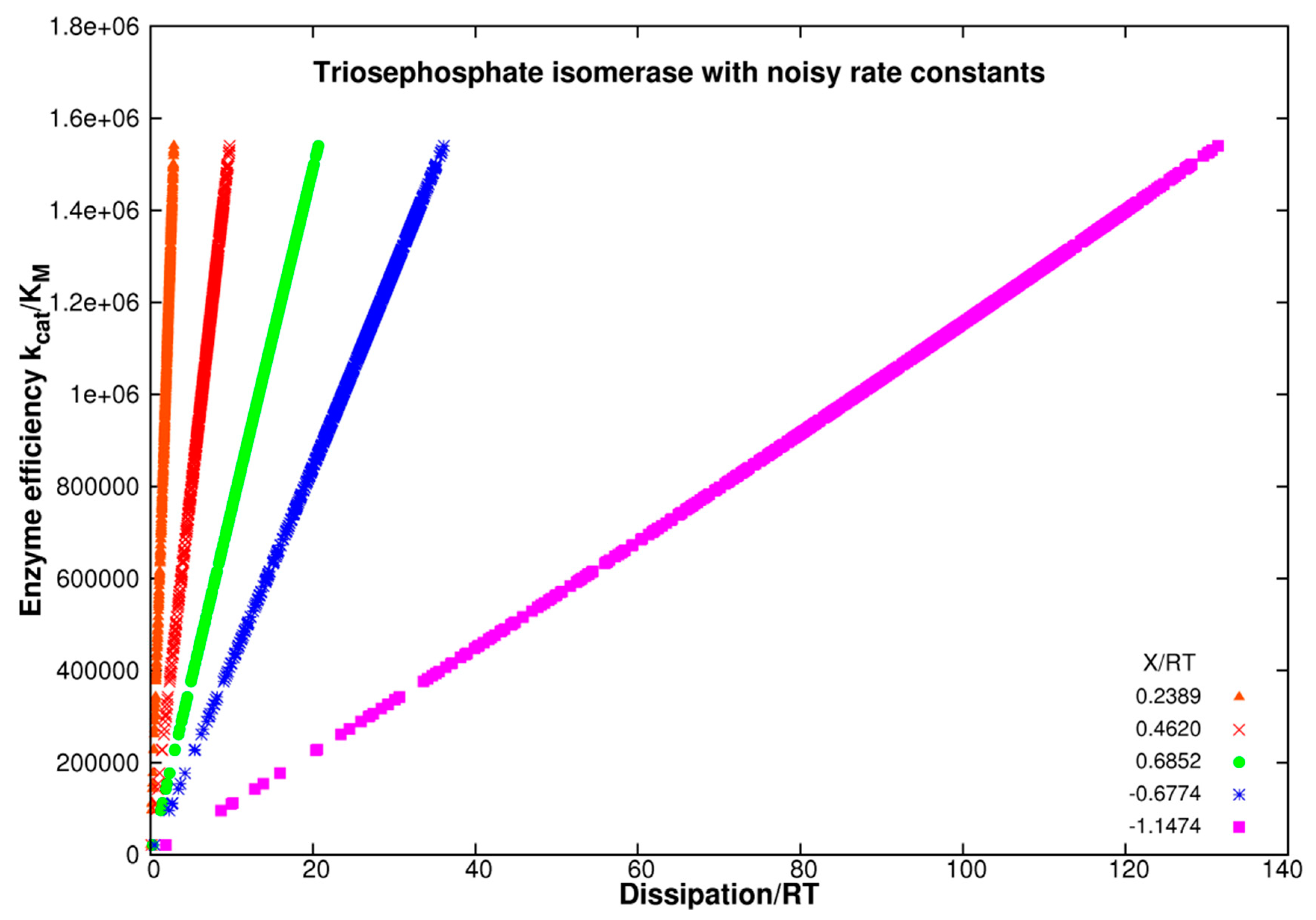
Figure 8.
The figure is constructed after calculating linear fit and efficiency to dissipation slope for 10 different X/RT values from -2.980 to 3.928 (corresponding to the total Keq range from 4 to 4000). The physiological range for Xtot/RT values when TPI enzyme works in the cellular environment, is likely to be even more restricted to low and mainly positive X values. See legends of Figure 6 and Figure 7 for details about the construction of 10 FORTRAN programs used to get Figure 8.
Figure 8.
The figure is constructed after calculating linear fit and efficiency to dissipation slope for 10 different X/RT values from -2.980 to 3.928 (corresponding to the total Keq range from 4 to 4000). The physiological range for Xtot/RT values when TPI enzyme works in the cellular environment, is likely to be even more restricted to low and mainly positive X values. See legends of Figure 6 and Figure 7 for details about the construction of 10 FORTRAN programs used to get Figure 8.
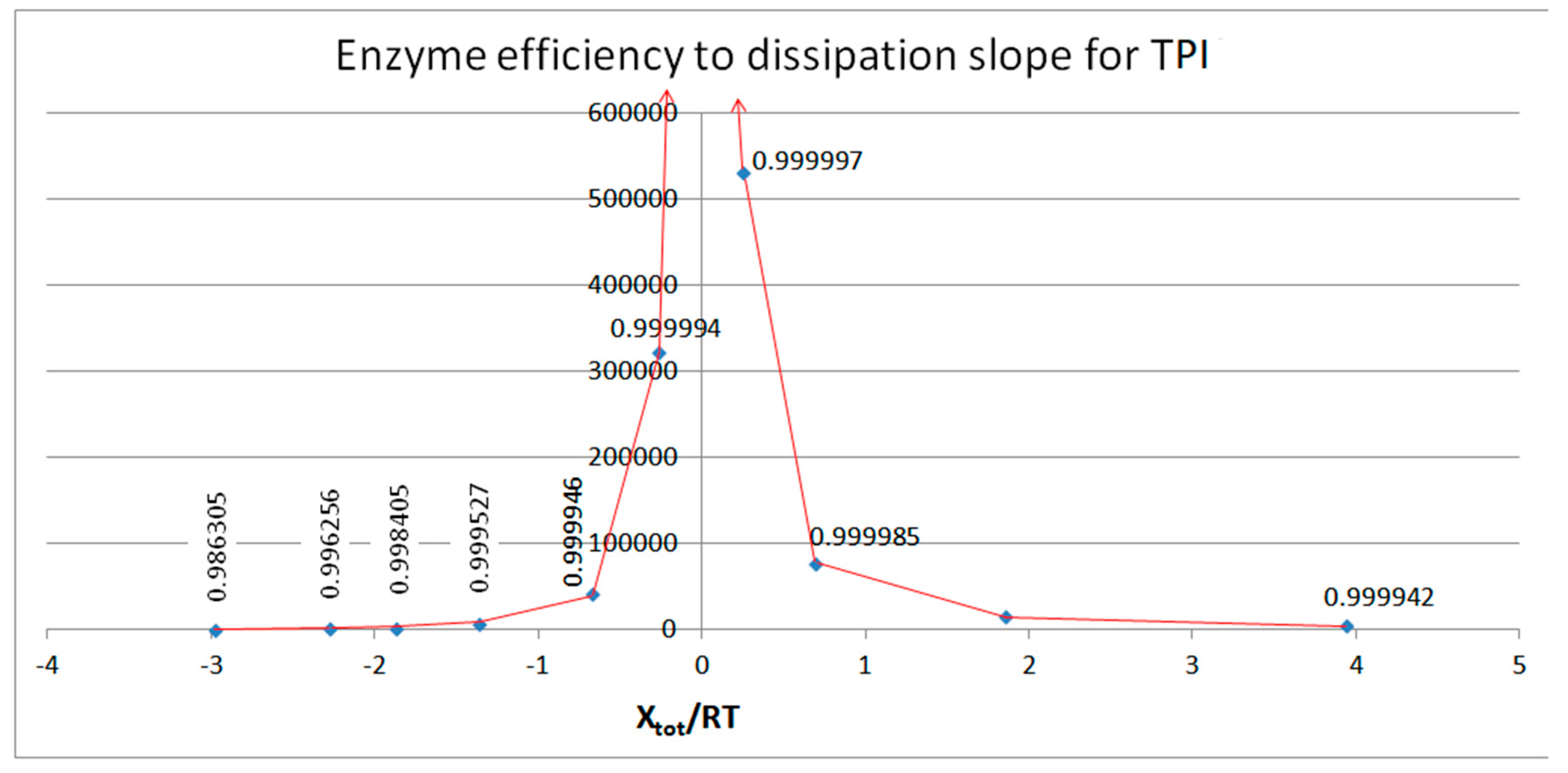
Figure 9.
Enzyme efficiency kcat/KM as a function of dissipation/RT for noisy TPI kinetics with variations in K1 and K4 equilibrium constants and fixed overall force Xtot/RT. Each of the 1000 points in this figure corresponds to a randomly established steady state when K4 is multiplied with the normal noise function with a +2 shift. The FORTRAN program found a unique steady state with optimal enzyme efficiency and maximal total entropy production, such that the optimal efficiency (higher arrow y coordinate) is considerably higher than calculated catalytic efficiency using experimental data (lower arrow y coordinate from this Figure and Figure 6).
Figure 9.
Enzyme efficiency kcat/KM as a function of dissipation/RT for noisy TPI kinetics with variations in K1 and K4 equilibrium constants and fixed overall force Xtot/RT. Each of the 1000 points in this figure corresponds to a randomly established steady state when K4 is multiplied with the normal noise function with a +2 shift. The FORTRAN program found a unique steady state with optimal enzyme efficiency and maximal total entropy production, such that the optimal efficiency (higher arrow y coordinate) is considerably higher than calculated catalytic efficiency using experimental data (lower arrow y coordinate from this Figure and Figure 6).
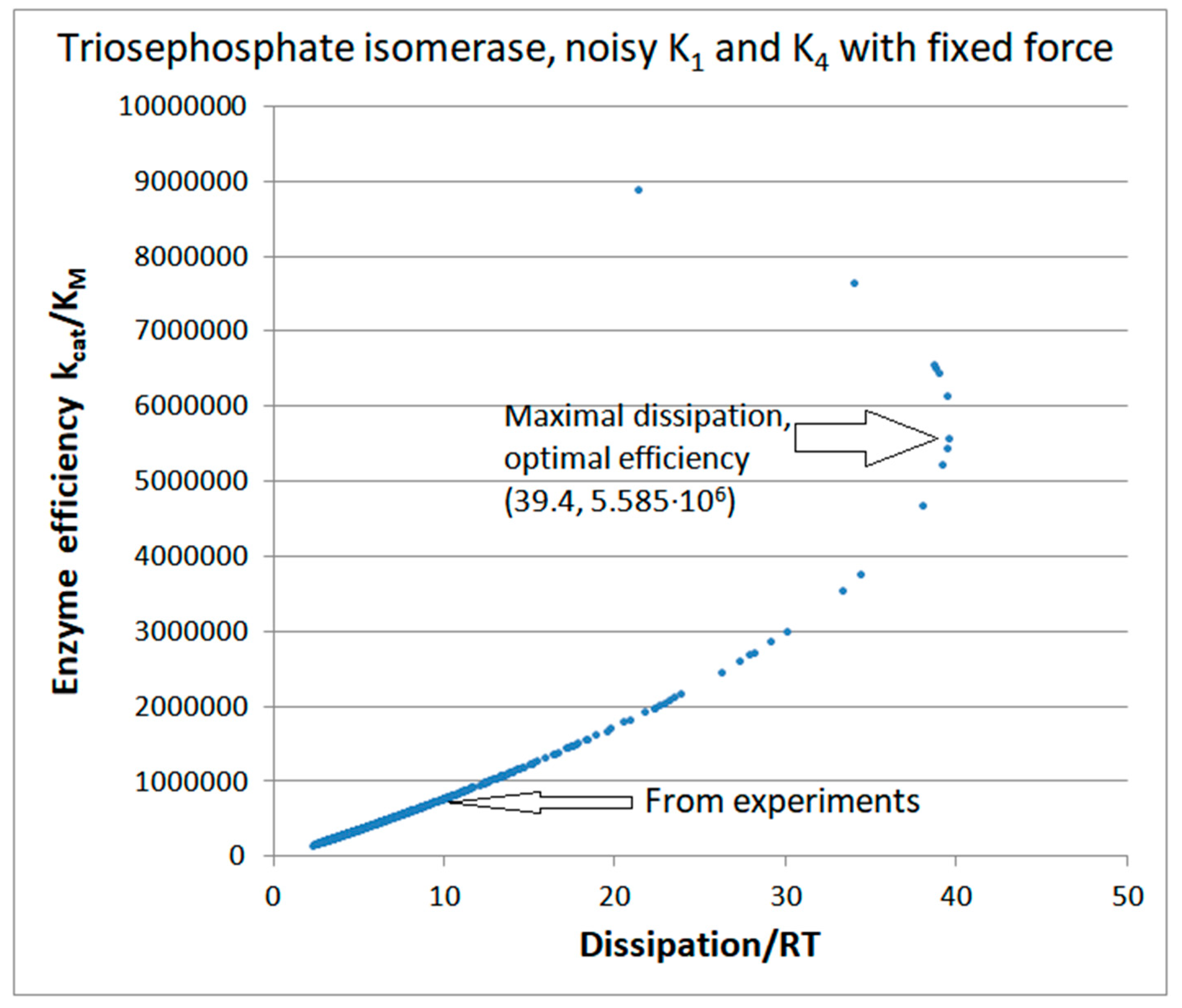
Figure 10.
The map of dissipation-efficiency values for different constraints in the case of TPI kinetics. The straight-line efficiency dependence on dissipation follows after the same restrictions we described in the legend of Figure 6. Pairs of (x, y) values with higher (dissipation/RT, kcat/KM) values from those in Figure 6 resulted because we introduced the same normal noise in all kinetic constants, not just in the k7, k8 pair. Specifically, we multiplied each of the four forward kinetic constants with the same Box-Muller transform containing two random numbers and a positive shift of +2 (see Methods), which was called only once in the program. The +2 shift ensured the absence of negative values for some kinetic constants. The corresponding FORTRAN program calculated backward rate constants from the constant K1 to K4 requirement (their referent values can be calculated from corresponding rate constant values in Table 1). That requirement ensured, combined with the normal noise introduction in each forward ki, that a) noise is canceled in the ratio of kinetic constants in each catalytic step and b) all catalytic constants are different in each of 1000 changes among steady states. See the main text for the meaning of points (highlighted circles) obtained using different restrictions or optimizations with or without introduced noise.
Figure 10.
The map of dissipation-efficiency values for different constraints in the case of TPI kinetics. The straight-line efficiency dependence on dissipation follows after the same restrictions we described in the legend of Figure 6. Pairs of (x, y) values with higher (dissipation/RT, kcat/KM) values from those in Figure 6 resulted because we introduced the same normal noise in all kinetic constants, not just in the k7, k8 pair. Specifically, we multiplied each of the four forward kinetic constants with the same Box-Muller transform containing two random numbers and a positive shift of +2 (see Methods), which was called only once in the program. The +2 shift ensured the absence of negative values for some kinetic constants. The corresponding FORTRAN program calculated backward rate constants from the constant K1 to K4 requirement (their referent values can be calculated from corresponding rate constant values in Table 1). That requirement ensured, combined with the normal noise introduction in each forward ki, that a) noise is canceled in the ratio of kinetic constants in each catalytic step and b) all catalytic constants are different in each of 1000 changes among steady states. See the main text for the meaning of points (highlighted circles) obtained using different restrictions or optimizations with or without introduced noise.
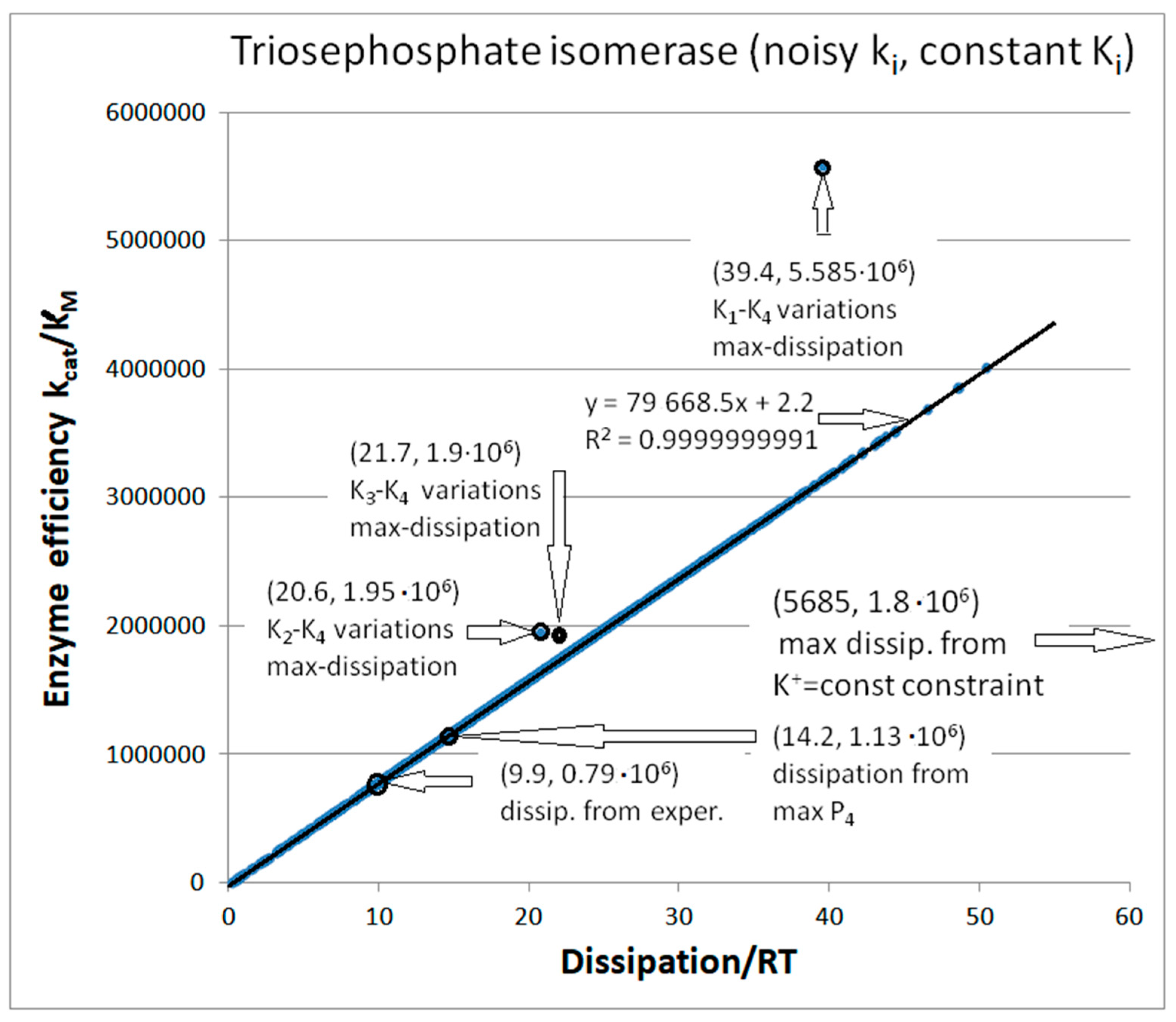
Figure 11.
The dependence of catalytic efficiency on dissipation when all rate constants are noisy. Box-Muller transforms gi without shift (see Methods) were called and multiplied with each of eight rate constants ki (i = 1,2,..8). The multiplication with gi > 0 introduced normal noise in these constants. For the gi values that did not satisfy the gi > 0 condition, we kept observed ki values (see Table 1). The main loop from the FORTRAN program contained 10000 steps. After all steps, we examined the kinetic and thermodynamic parameters for maximal values in catalytic efficiency, overall entropy production, and possible correlation between enzyme efficiency and total dissipation.
Figure 11.
The dependence of catalytic efficiency on dissipation when all rate constants are noisy. Box-Muller transforms gi without shift (see Methods) were called and multiplied with each of eight rate constants ki (i = 1,2,..8). The multiplication with gi > 0 introduced normal noise in these constants. For the gi values that did not satisfy the gi > 0 condition, we kept observed ki values (see Table 1). The main loop from the FORTRAN program contained 10000 steps. After all steps, we examined the kinetic and thermodynamic parameters for maximal values in catalytic efficiency, overall entropy production, and possible correlation between enzyme efficiency and total dissipation.
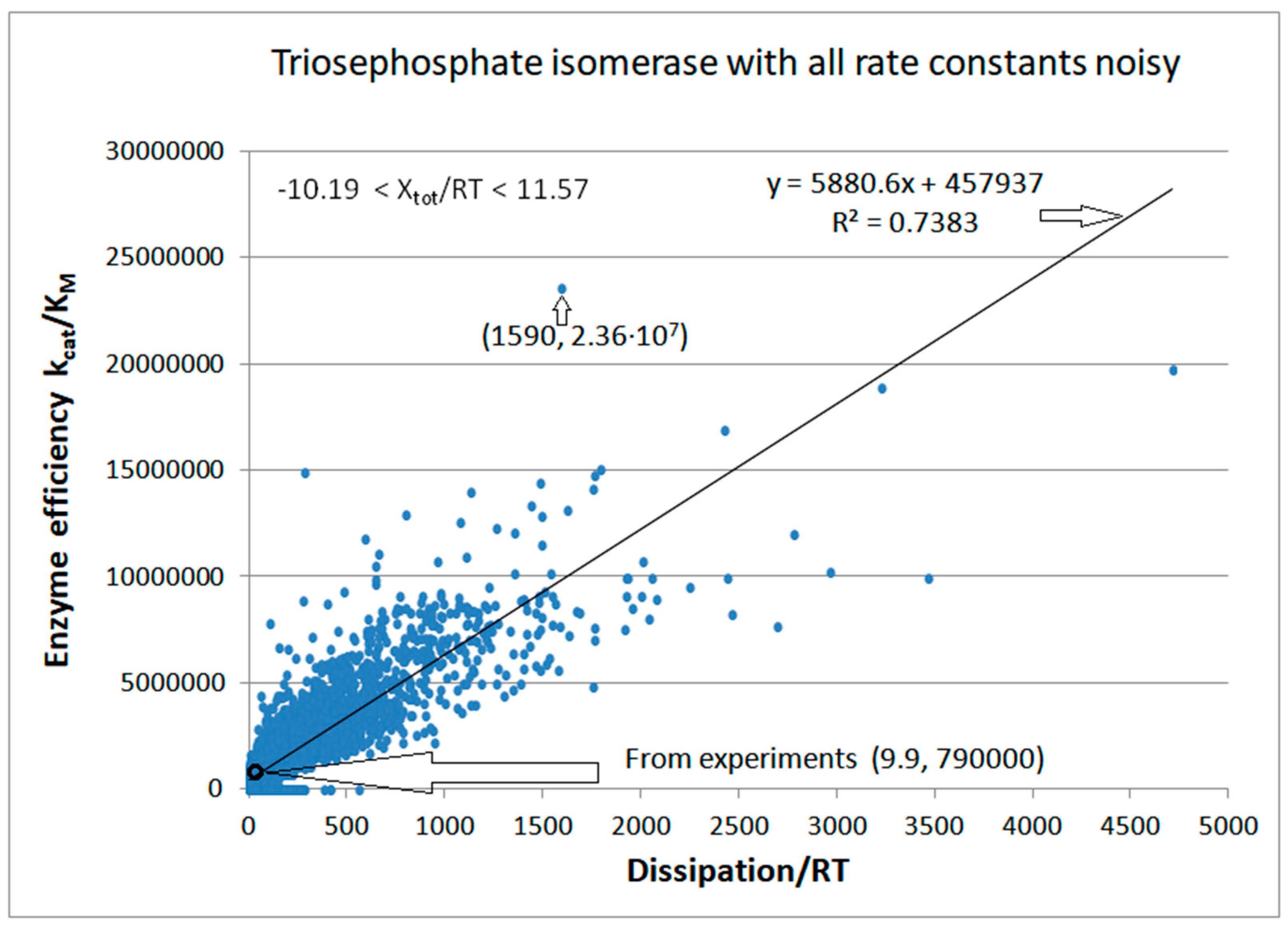
Figure 12.
NetLogo simulation of the catalytic efficiency dependence on dissipation for the TPI kinetics. The initial enzyme concentration was 100 nM. All other initial values and assumptions were identical to those we used previously [70]. Due to the dynamics inherent to the NetLogo agent-based language, the assumption about unchanged equilibrium constants from that paper could be only partially retained. Noise is introduced through different random-float values, not by Gaussian random number values. Additional noise is due to random encounters among ligands and [enzyme]free and among enzyme conformations [ES]-[EX] and [EX]-[EP] also specified with several different random-float values.
Figure 12.
NetLogo simulation of the catalytic efficiency dependence on dissipation for the TPI kinetics. The initial enzyme concentration was 100 nM. All other initial values and assumptions were identical to those we used previously [70]. Due to the dynamics inherent to the NetLogo agent-based language, the assumption about unchanged equilibrium constants from that paper could be only partially retained. Noise is introduced through different random-float values, not by Gaussian random number values. Additional noise is due to random encounters among ligands and [enzyme]free and among enzyme conformations [ES]-[EX] and [EX]-[EP] also specified with several different random-float values.
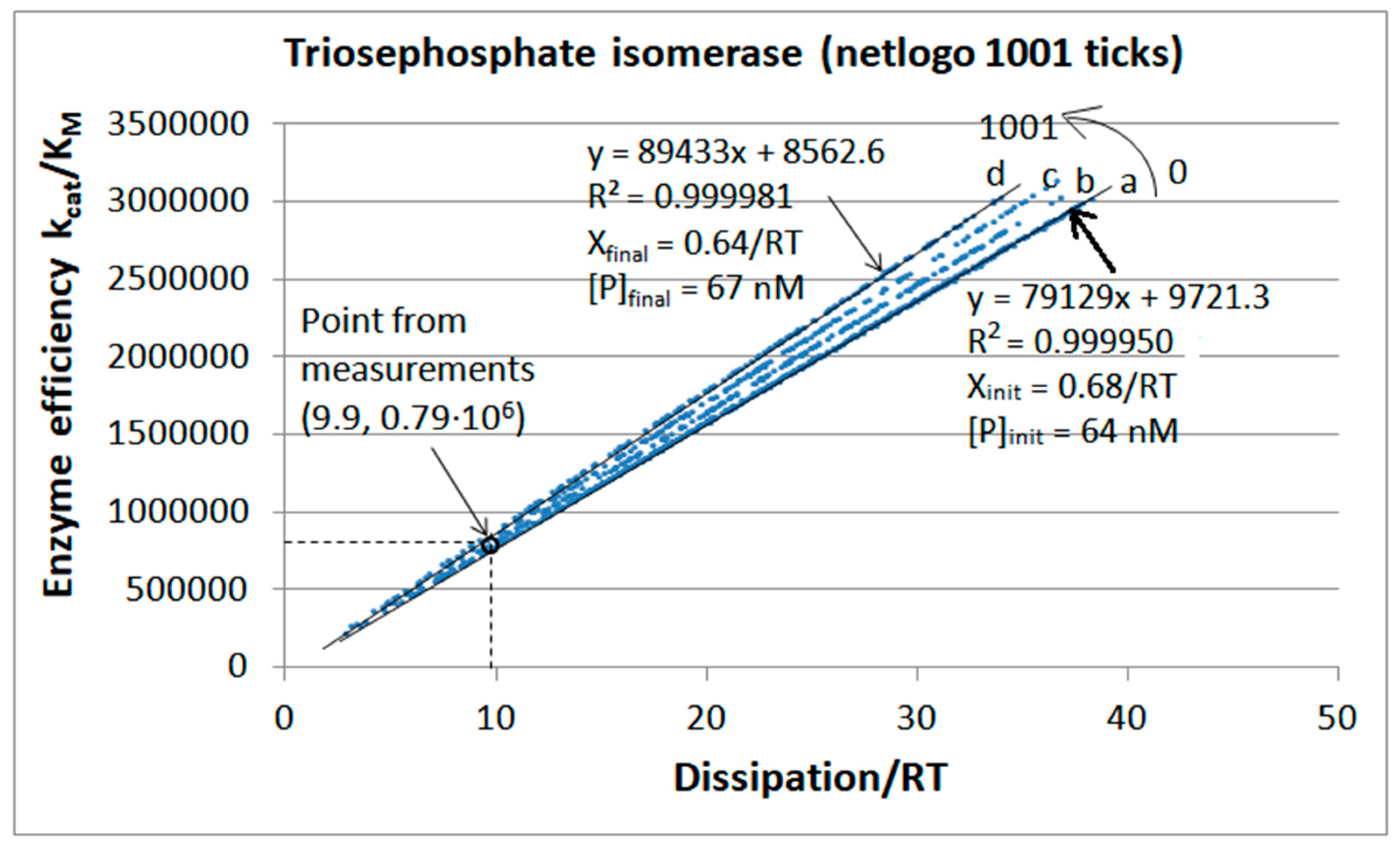
Figure 13.
The more extended simulation run using the same software and identical conditions we described in Figure 12. The program was stopped at 2137 ticks when initial concentrations [S]init = 40 μM, [P]init = 0.064 μM, and [enzyme]free-init = 100 nM changed to the [S]final = 39.902 μM, [P]final = 0.074 μM, [enzyme]free-final = 12 nM. Final concentrations of enzyme-ligand complexes were [ES] = 19 nM, [EZ] = 35 nM, and [EP] = 34 nM. Overall force in the RT units (X/RT) dropped from the initial 0.685 to the final 0.538.
Figure 13.
The more extended simulation run using the same software and identical conditions we described in Figure 12. The program was stopped at 2137 ticks when initial concentrations [S]init = 40 μM, [P]init = 0.064 μM, and [enzyme]free-init = 100 nM changed to the [S]final = 39.902 μM, [P]final = 0.074 μM, [enzyme]free-final = 12 nM. Final concentrations of enzyme-ligand complexes were [ES] = 19 nM, [EZ] = 35 nM, and [EP] = 34 nM. Overall force in the RT units (X/RT) dropped from the initial 0.685 to the final 0.538.
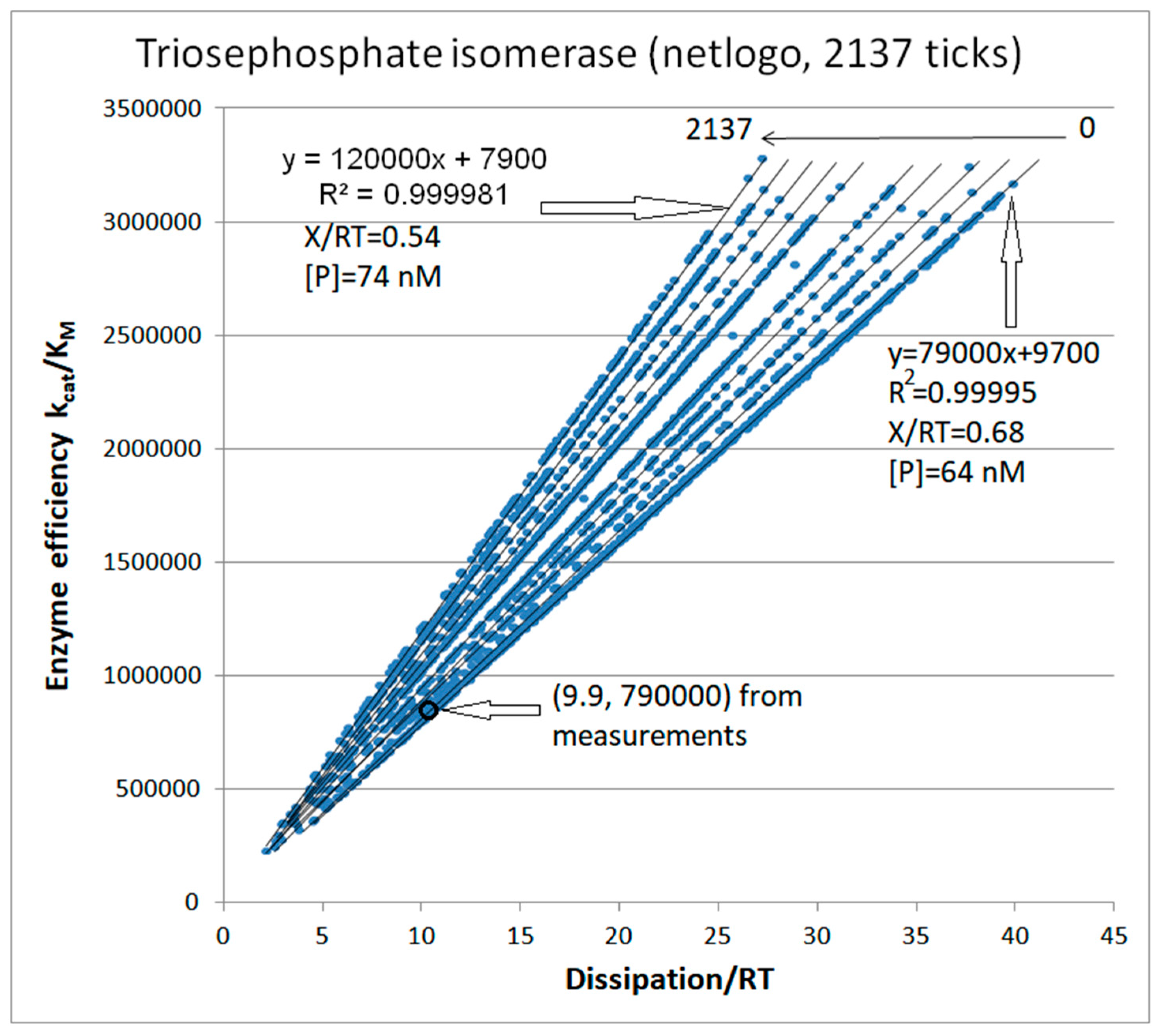
Figure 14.
The catalytic efficiency dependence on dissipation when each kinetic constant ki is independently multiplied with the Gaussian noise function gi (see Methods) for the reversible 4-state triosephosphate kinetic scheme. Initial conditions were the same as in Table 1 ([E]free = 50 nM). The program was stopped at the 1800th tick. The final concentrations of enzyme conformations were [Efree] = 30 nM, [ES] = 6 nM, [EX] = 8 nM, and [EP] = 6 nM.
Figure 14.
The catalytic efficiency dependence on dissipation when each kinetic constant ki is independently multiplied with the Gaussian noise function gi (see Methods) for the reversible 4-state triosephosphate kinetic scheme. Initial conditions were the same as in Table 1 ([E]free = 50 nM). The program was stopped at the 1800th tick. The final concentrations of enzyme conformations were [Efree] = 30 nM, [ES] = 6 nM, [EX] = 8 nM, and [EP] = 6 nM.
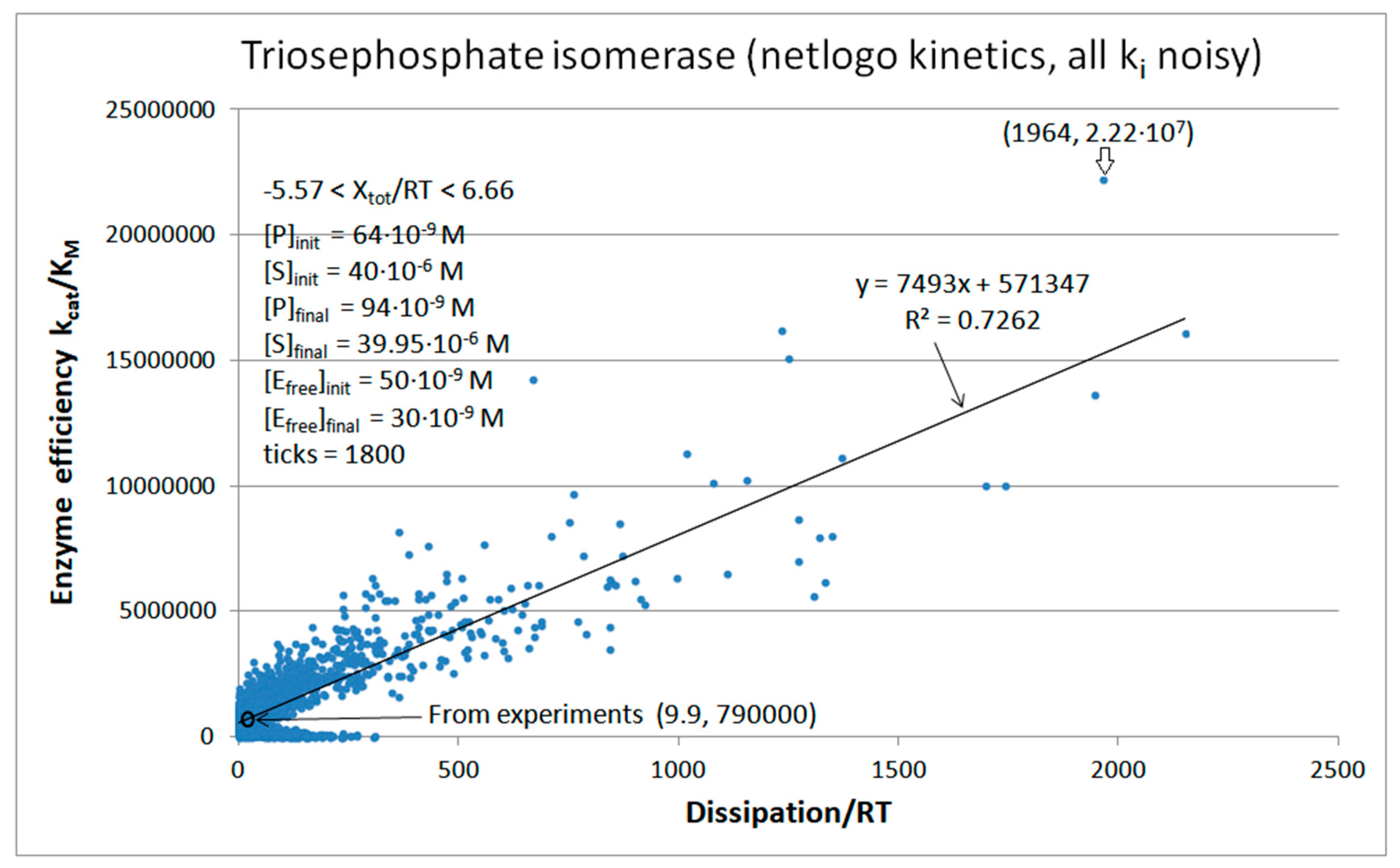
Figure 15.
The catalytic efficiency dependence on dissipation when all rate constants are noisy for the ketosteroid isomerase kinetics. A spectrum of quasi-steady states with different ki octuplets resulted after the multiplication of each observed ki (see Table 2) with a separately called normal noise gi. There was no shift in eight Box-Muller transforms with the cosine function (see Methods q. 25). The if-else condition in the FORTRAN simulation ensured that negative or zero ki values were replaced with their experimental values. The program went through the 1000 steps, requiring overall force X to be positive in each step.
Figure 15.
The catalytic efficiency dependence on dissipation when all rate constants are noisy for the ketosteroid isomerase kinetics. A spectrum of quasi-steady states with different ki octuplets resulted after the multiplication of each observed ki (see Table 2) with a separately called normal noise gi. There was no shift in eight Box-Muller transforms with the cosine function (see Methods q. 25). The if-else condition in the FORTRAN simulation ensured that negative or zero ki values were replaced with their experimental values. The program went through the 1000 steps, requiring overall force X to be positive in each step.
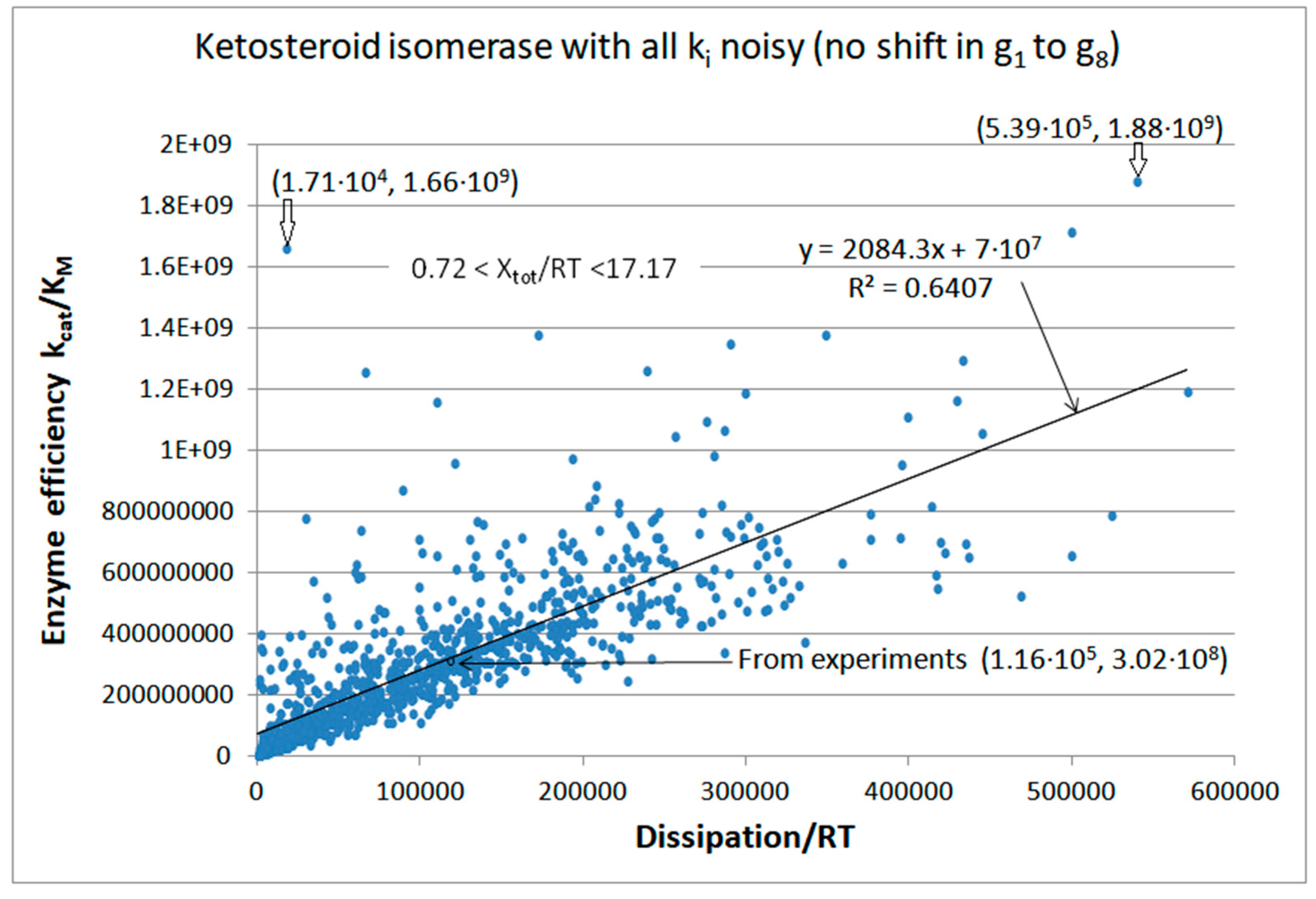
Figure 16.
When dissipation increases, the enzyme efficiency increases even faster in the presence of noisy associations between substrates and enzymes. We multiplied ki with the normal noise function, which included the shift = +2 (see Methods). The FORTRAN simulation runs through a thousand steps without any other changes from the initial calculated values (Table, second column). Thus, noisy Xtot/RT values in the range from 3.87 to 10.06 resulted from noisy K1 values in the range from 0.01 to 4.93.
Figure 16.
When dissipation increases, the enzyme efficiency increases even faster in the presence of noisy associations between substrates and enzymes. We multiplied ki with the normal noise function, which included the shift = +2 (see Methods). The FORTRAN simulation runs through a thousand steps without any other changes from the initial calculated values (Table, second column). Thus, noisy Xtot/RT values in the range from 3.87 to 10.06 resulted from noisy K1 values in the range from 0.01 to 4.93.
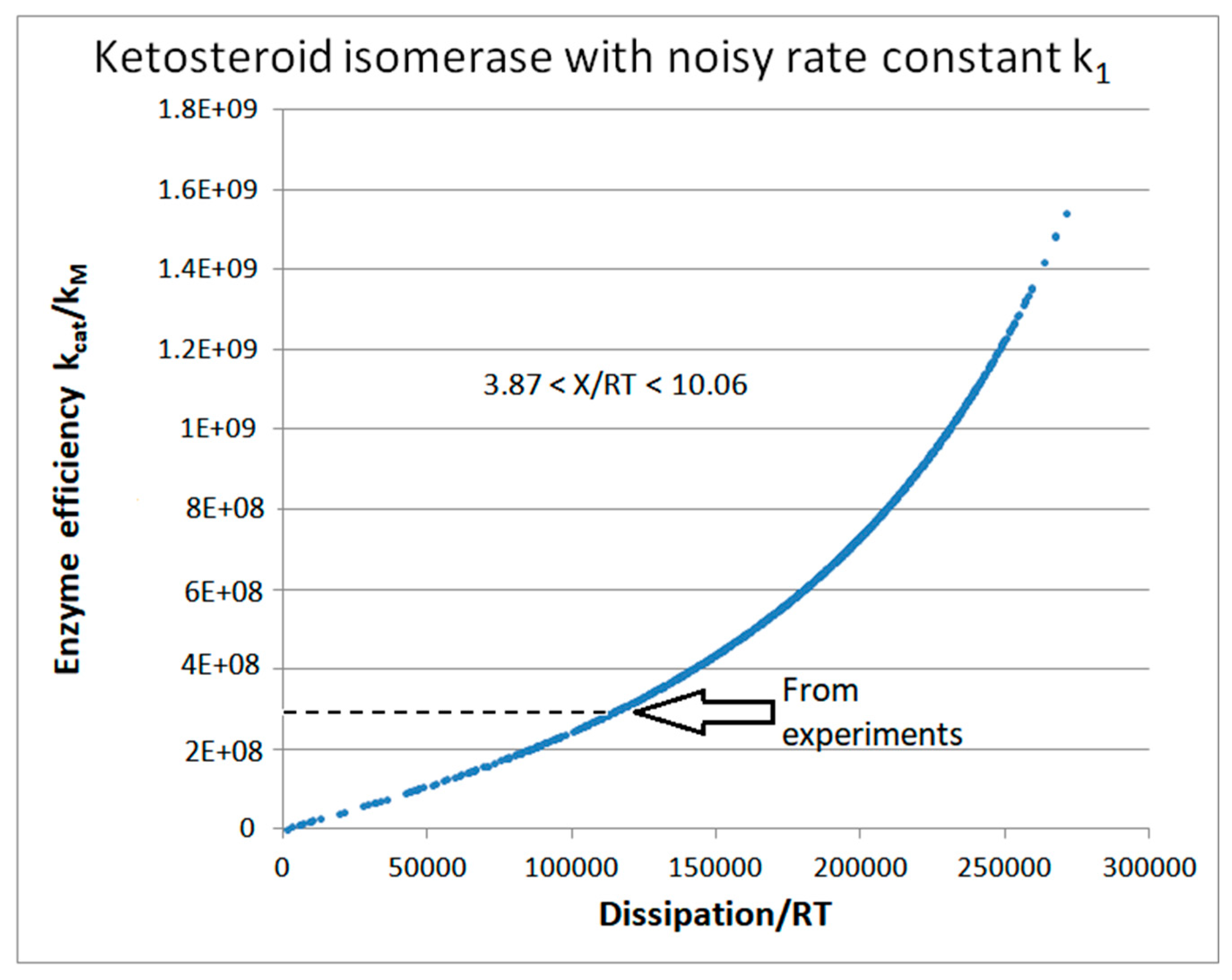
Figure 17.
Maximal overall entropy production and associated optimal enzyme efficiency follow from the Xtot/RT = 8.426 requirement (see Table 2) in the presence of noisy association-dissociation of the enzyme with the substrate or product. FORTRAN simulation went through 1000 steps.
Figure 17.
Maximal overall entropy production and associated optimal enzyme efficiency follow from the Xtot/RT = 8.426 requirement (see Table 2) in the presence of noisy association-dissociation of the enzyme with the substrate or product. FORTRAN simulation went through 1000 steps.
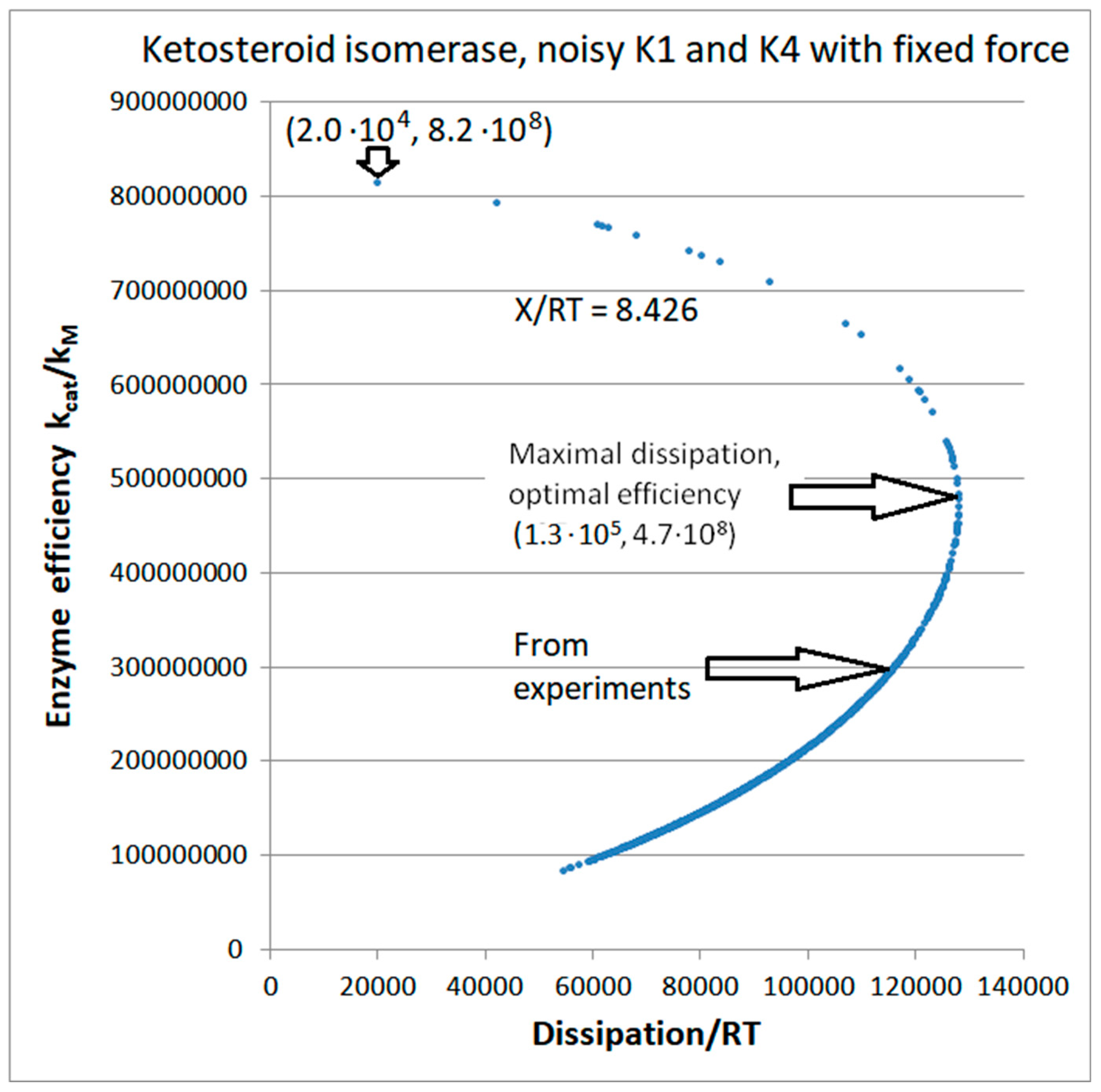
Figure 18.
NetLogo simulation of KSI kinetics, when the Box-Muller transform without shift is invoked eight times to multiply each of eight rate constants ki. Initial concentrations were [S]init = 10-4 M, [P]init = 5∙10-5 M, and [E]free = 5∙10-6 M. Final concentrations at the 6977th tick were [S]init = 9.5∙10-5 M, [P]init = 5.4∙10-5 M, [E]free = 4∙10-6 M, [ES] = 3∙10-7 M, [EZ] = 4∙10-7 M, and [EP] = 3∙10-7 M. We found the highest catalytic efficiency value from this extensive search at the 3911th tick. It was kcat/KM = 2.59∙109 M-1s-1; thus, it was well inside the diffusion limit range from 108 to 1010 M-1s-1. The corresponding overall force was Xtot/RT = 12.6.
Figure 18.
NetLogo simulation of KSI kinetics, when the Box-Muller transform without shift is invoked eight times to multiply each of eight rate constants ki. Initial concentrations were [S]init = 10-4 M, [P]init = 5∙10-5 M, and [E]free = 5∙10-6 M. Final concentrations at the 6977th tick were [S]init = 9.5∙10-5 M, [P]init = 5.4∙10-5 M, [E]free = 4∙10-6 M, [ES] = 3∙10-7 M, [EZ] = 4∙10-7 M, and [EP] = 3∙10-7 M. We found the highest catalytic efficiency value from this extensive search at the 3911th tick. It was kcat/KM = 2.59∙109 M-1s-1; thus, it was well inside the diffusion limit range from 108 to 1010 M-1s-1. The corresponding overall force was Xtot/RT = 12.6.
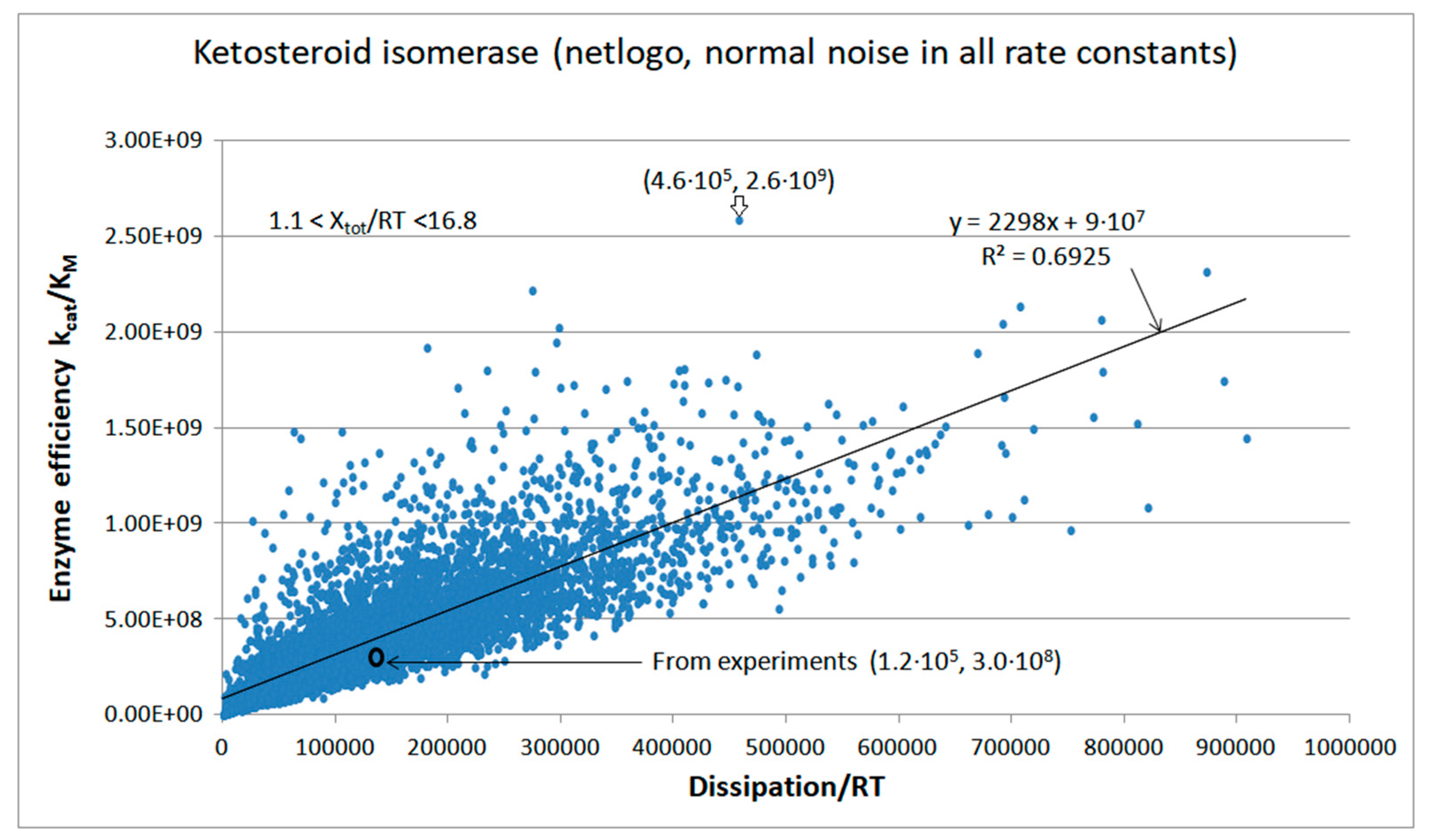
Figure 19.
The four-state reversible kinetic scheme for three CAs isoenzymes. The inclusion of buffer B in both transitions of the last forward step reflects the substantial difference in the performance of all CAs for different buffers [105].
Figure 19.
The four-state reversible kinetic scheme for three CAs isoenzymes. The inclusion of buffer B in both transitions of the last forward step reflects the substantial difference in the performance of all CAs for different buffers [105].
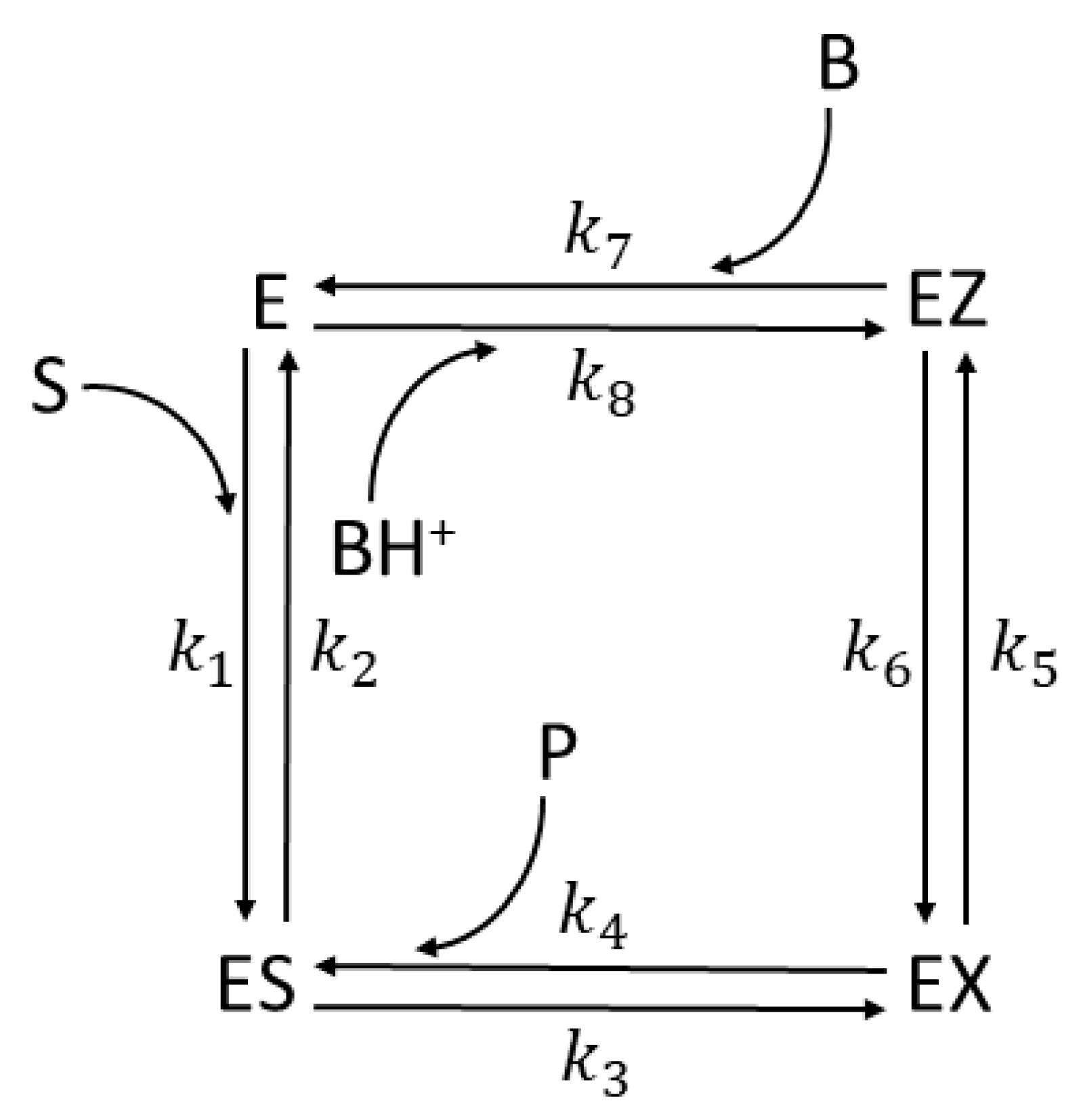
Figure 21.
The NetLogo simulation of the relationship between catalytic efficiency and overall dissipation for the carbonic anhydrase I, when each of eight rate constants ki is multiplied with independently introduced normal noise function gi without shift (see Methods, eq (25)). The best efficiency value of 1.47∙108 M-1s-1 resulted in the 2774th tick for Xtot/RT = 6.5. The overall force in the RT units ranged from -5.0 to 8.5, but there was no significant force decrease with the time passage (ticks). From the initial 100 μM free enzyme concentration, the conversion during 5521 ticks ended up with less than 1 μM free enzyme concentration, [ES] = 1 μM, [EX] = 45 μM, and [EZ] = 54 μM. The Michaelis-Menten time dependence pattern is seen for the ES complex concentration (fast rise followed by plateauing and slow decrease). During the simulation, there was little change in the substrate (CO2) and product (HCO3-) concentration.
Figure 21.
The NetLogo simulation of the relationship between catalytic efficiency and overall dissipation for the carbonic anhydrase I, when each of eight rate constants ki is multiplied with independently introduced normal noise function gi without shift (see Methods, eq (25)). The best efficiency value of 1.47∙108 M-1s-1 resulted in the 2774th tick for Xtot/RT = 6.5. The overall force in the RT units ranged from -5.0 to 8.5, but there was no significant force decrease with the time passage (ticks). From the initial 100 μM free enzyme concentration, the conversion during 5521 ticks ended up with less than 1 μM free enzyme concentration, [ES] = 1 μM, [EX] = 45 μM, and [EZ] = 54 μM. The Michaelis-Menten time dependence pattern is seen for the ES complex concentration (fast rise followed by plateauing and slow decrease). During the simulation, there was little change in the substrate (CO2) and product (HCO3-) concentration.
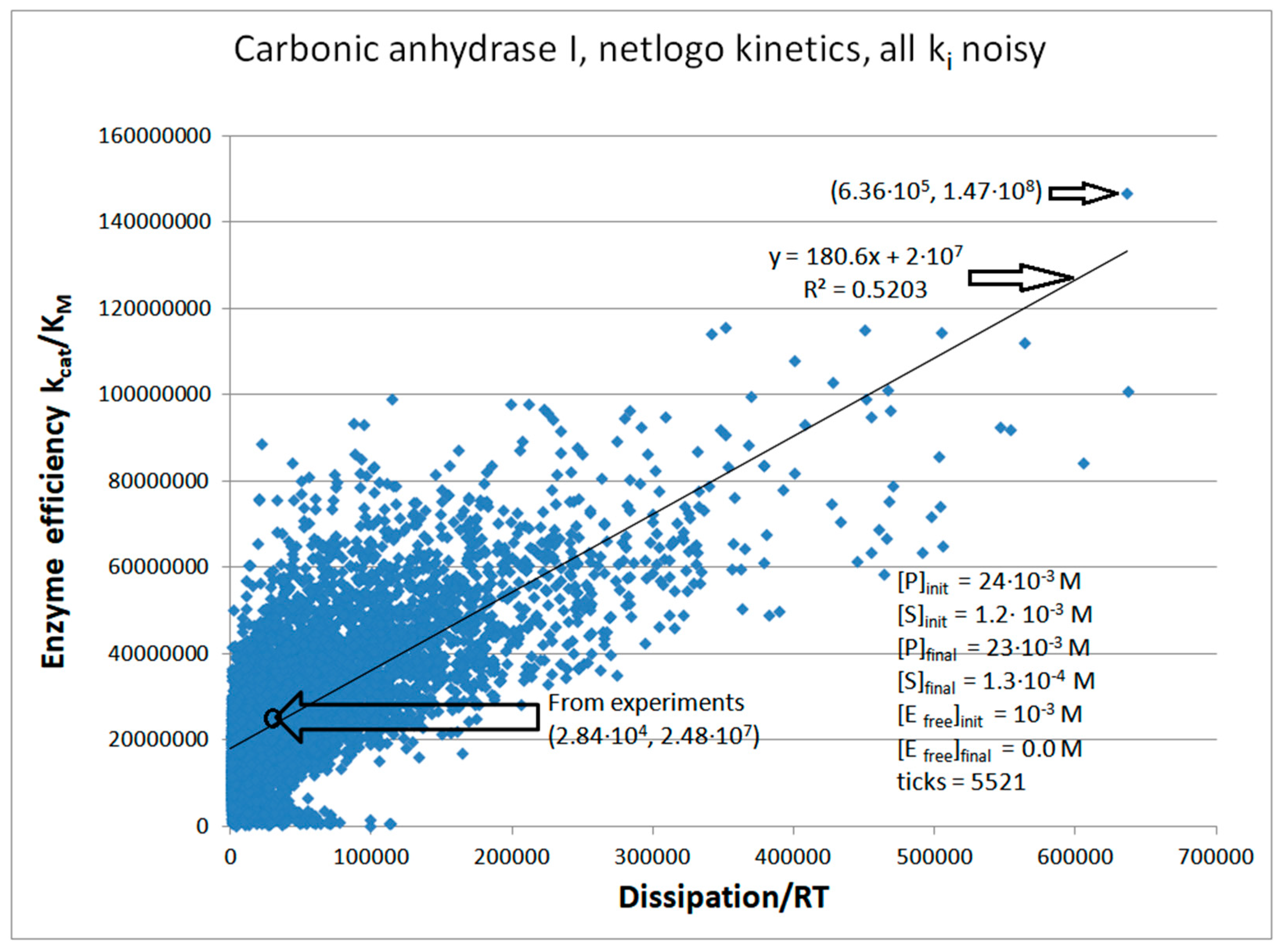
Figure 22.
The NetLogo simulation of the relationship between catalytic efficiency and overall dissipation for the carbonic anhydrase II, when each of eight rate constants ki is multiplied with independently introduced normal noise function gi without shift (see Methods, eq (25)). The best efficiency value of 4.25∙108 M-1s-1 from the 288th tick is for Xtot/RT = 4.76. There was no clear force decrease with the time passage (ticks). From the initial 100 μM free enzyme concentration, the conversion during 8007 ticks ended up with less than 1 μM free enzyme concentration, [ES] = 1 μM, [EX] = 50 μM, and [EZ] = 49 μM. The Michaels-Menten time dependence pattern was the same as seen for CAI.
Figure 22.
The NetLogo simulation of the relationship between catalytic efficiency and overall dissipation for the carbonic anhydrase II, when each of eight rate constants ki is multiplied with independently introduced normal noise function gi without shift (see Methods, eq (25)). The best efficiency value of 4.25∙108 M-1s-1 from the 288th tick is for Xtot/RT = 4.76. There was no clear force decrease with the time passage (ticks). From the initial 100 μM free enzyme concentration, the conversion during 8007 ticks ended up with less than 1 μM free enzyme concentration, [ES] = 1 μM, [EX] = 50 μM, and [EZ] = 49 μM. The Michaels-Menten time dependence pattern was the same as seen for CAI.
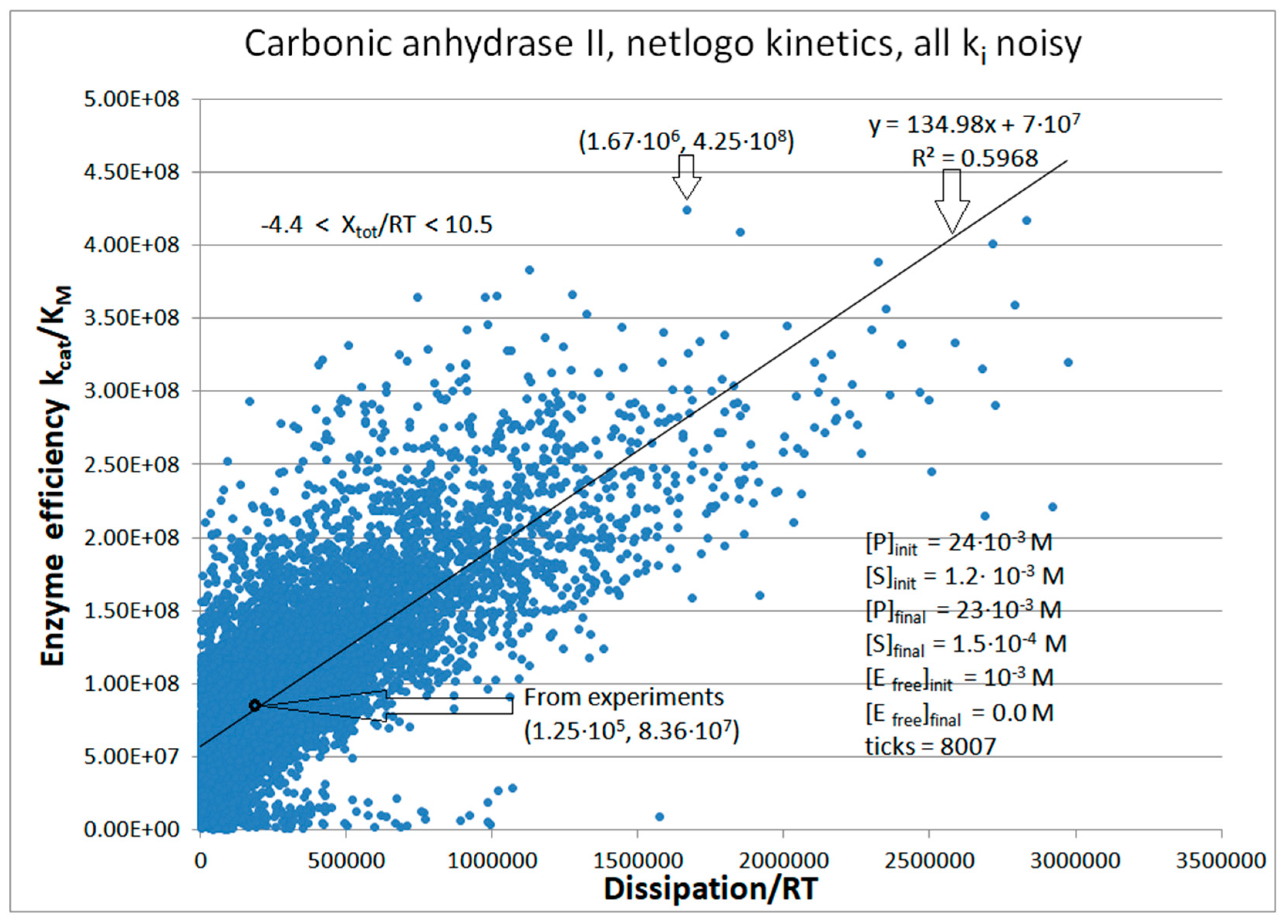
Figure 23.
The catalytic efficiency dependence on dissipation for the T200H mutant of carbonic anhydrase II.
Figure 23.
The catalytic efficiency dependence on dissipation for the T200H mutant of carbonic anhydrase II.
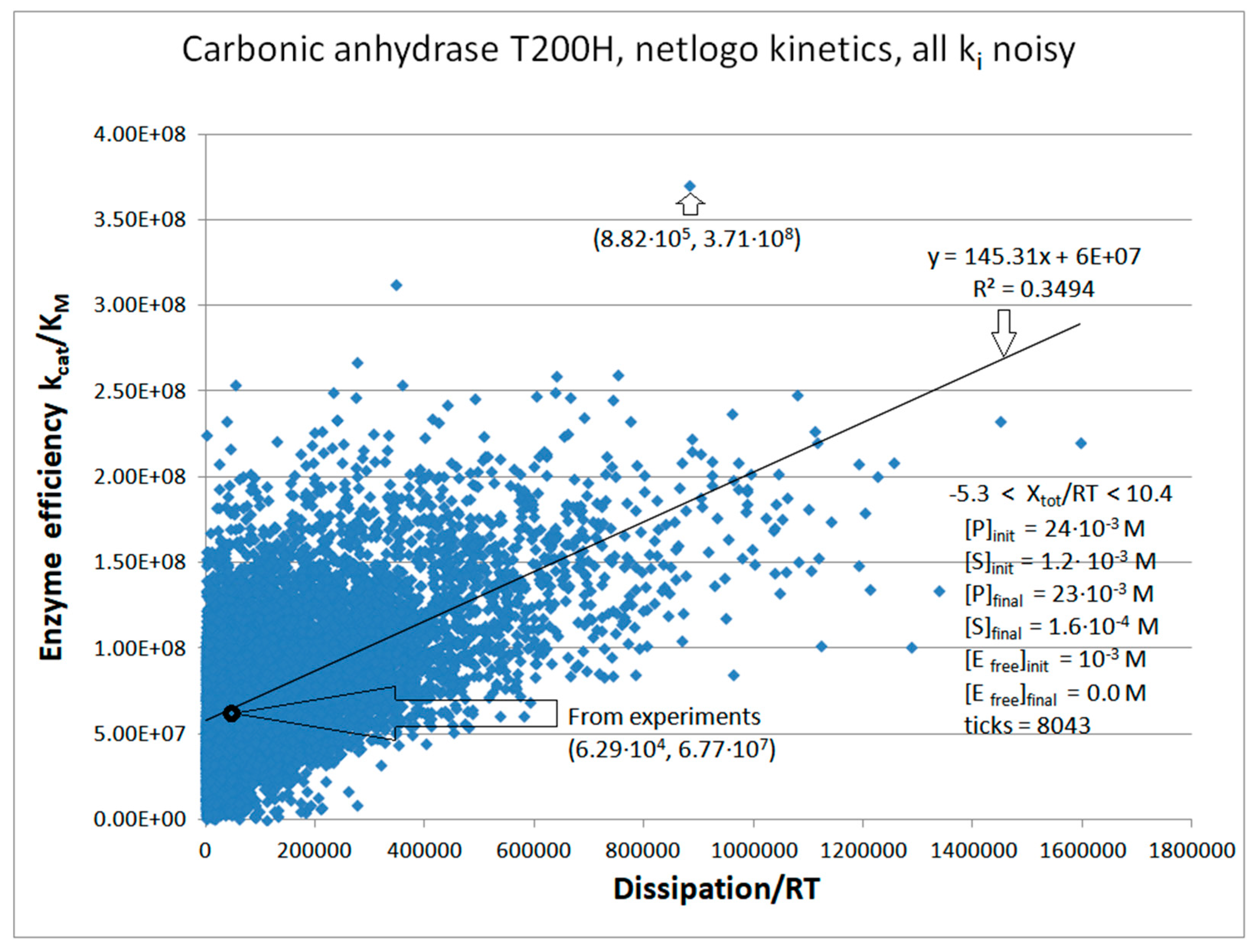
Figure 24.
The observed [105] and the best-predicted values for the performance of carbonic anhydrases CAI, CAII, and the T200H mutant of CAII. We collected the NetLogo simulation results from Figure 21, Figure 22, and Figure 23 to easily compare enzyme efficiency and overall dissipation calculated from observed rate constants (dashed circle) and the best-simulated values when noise was introduced in all ki (rounded rectangle). The arrows connect such points for different isoenzymes (blue for CAI, orange for CAII, and green for the T200H mutant). The small red arrow shows the performance and dissipation decrease for the Thr200 → His substitution mutant of carbonic anhydrase II. In contrast, the green arrow indicates the possibility of improving its performance above the observed value for CAII. By the way, the potential to enhance the CAII performance (yellow arrow) appears much higher than for CAI (blue arrow).
Figure 24.
The observed [105] and the best-predicted values for the performance of carbonic anhydrases CAI, CAII, and the T200H mutant of CAII. We collected the NetLogo simulation results from Figure 21, Figure 22, and Figure 23 to easily compare enzyme efficiency and overall dissipation calculated from observed rate constants (dashed circle) and the best-simulated values when noise was introduced in all ki (rounded rectangle). The arrows connect such points for different isoenzymes (blue for CAI, orange for CAII, and green for the T200H mutant). The small red arrow shows the performance and dissipation decrease for the Thr200 → His substitution mutant of carbonic anhydrase II. In contrast, the green arrow indicates the possibility of improving its performance above the observed value for CAII. By the way, the potential to enhance the CAII performance (yellow arrow) appears much higher than for CAI (blue arrow).
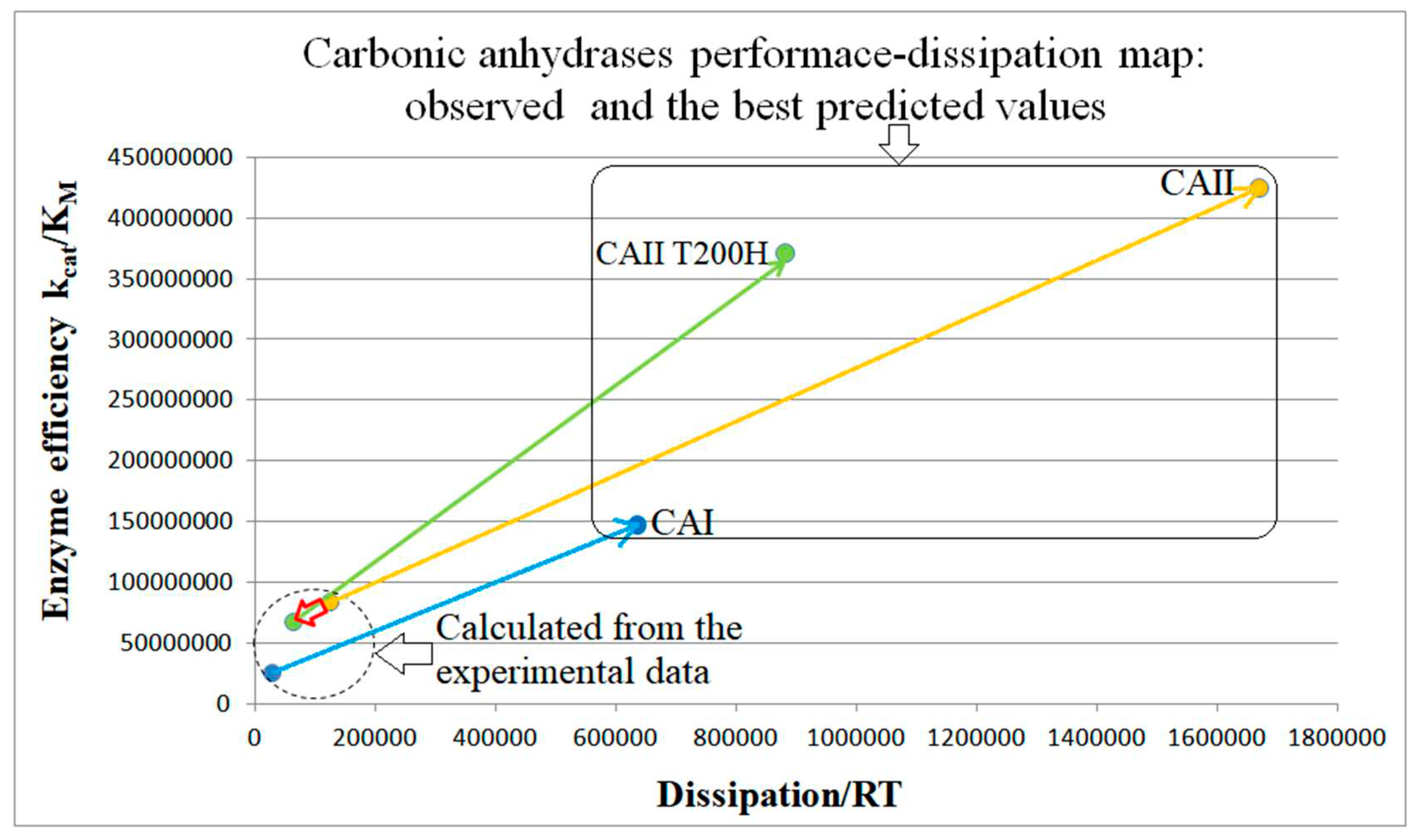
Figure 25.
The catalytic efficiency dependence on dissipation when all forward rate constants are noisy but all equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* maintain their observed values [30] for the PC1 β-lactamase. As expected for strong and constant positive force, there is close to perfect efficiency-dissipation proportionality despite introduced and inherent noise in encounters among the enzyme and ligand conformers in the NetLogo simulation when identical normal noise is introduced in k1, k2, and k3. The force decreased from 11.4 to 10.8, as the NetLogo simulation proceeded through 5168 ticks. The best point with the highest dissipation (2.82∙103 s-1) and the highest enzyme efficiency (4.18∙107 M−1s−1) occurred when an overall force of 11.3 was very similar to the initial force (11.4).
Figure 25.
The catalytic efficiency dependence on dissipation when all forward rate constants are noisy but all equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* maintain their observed values [30] for the PC1 β-lactamase. As expected for strong and constant positive force, there is close to perfect efficiency-dissipation proportionality despite introduced and inherent noise in encounters among the enzyme and ligand conformers in the NetLogo simulation when identical normal noise is introduced in k1, k2, and k3. The force decreased from 11.4 to 10.8, as the NetLogo simulation proceeded through 5168 ticks. The best point with the highest dissipation (2.82∙103 s-1) and the highest enzyme efficiency (4.18∙107 M−1s−1) occurred when an overall force of 11.3 was very similar to the initial force (11.4).
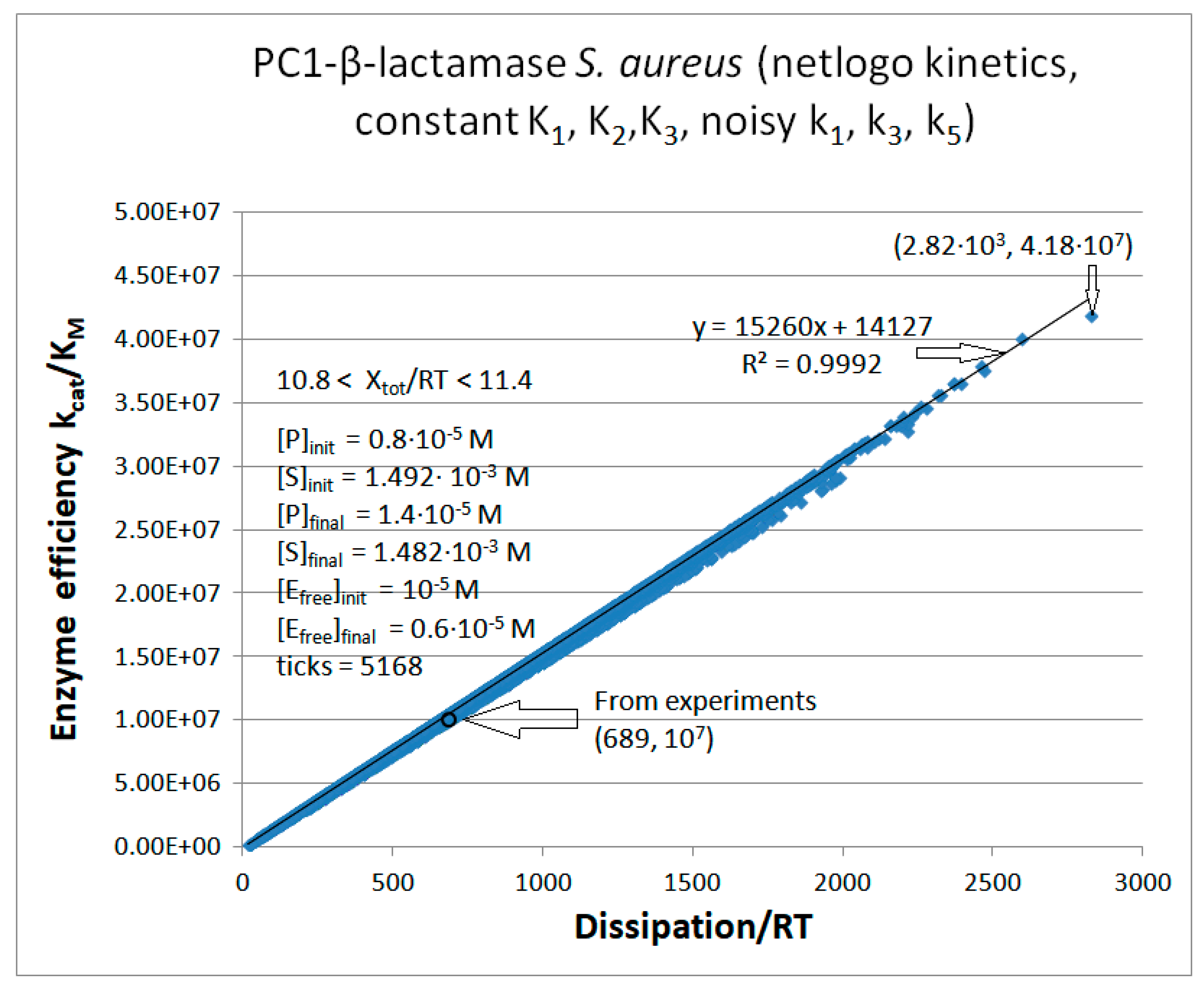
Figure 26.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation - in the forward rate constant k1 and the backward rate constant k2. Variations only in the substrate-enzyme association-dissociation produced close to an exponential increase in the enzyme efficiency after dissipation increased. The best efficiency value of 6.5∙107 M-1s-1 is now close to the lower-end diffusion-limit range (108 M-1s-1). There was no change in the catalytic constant from observed kcat = 60.8 1s-1 during the 3016 simulation ticks because forward kcat (S→P) = k3∙k5/(k3+k4+k5) does not depend on the rate constants k1 and k2 for the first catalytic step. Descriptions and arrows in red color depict the optimization results from Juretić et al. [106]. The best efficiency value of 6.5∙107 M-1s-1 corresponds to 6.1-fold higher k1 and 3.5-fold smaller k2, which increased 21-fold the first equilibrium constant K1 = k1/k2 from observed 167.5 to the optimal value of 3582.3, with the concomitant increase in the overall force Xtot/RT from the referent value of 11.4 to the optimal value of 14.1.
Figure 26.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation - in the forward rate constant k1 and the backward rate constant k2. Variations only in the substrate-enzyme association-dissociation produced close to an exponential increase in the enzyme efficiency after dissipation increased. The best efficiency value of 6.5∙107 M-1s-1 is now close to the lower-end diffusion-limit range (108 M-1s-1). There was no change in the catalytic constant from observed kcat = 60.8 1s-1 during the 3016 simulation ticks because forward kcat (S→P) = k3∙k5/(k3+k4+k5) does not depend on the rate constants k1 and k2 for the first catalytic step. Descriptions and arrows in red color depict the optimization results from Juretić et al. [106]. The best efficiency value of 6.5∙107 M-1s-1 corresponds to 6.1-fold higher k1 and 3.5-fold smaller k2, which increased 21-fold the first equilibrium constant K1 = k1/k2 from observed 167.5 to the optimal value of 3582.3, with the concomitant increase in the overall force Xtot/RT from the referent value of 11.4 to the optimal value of 14.1.
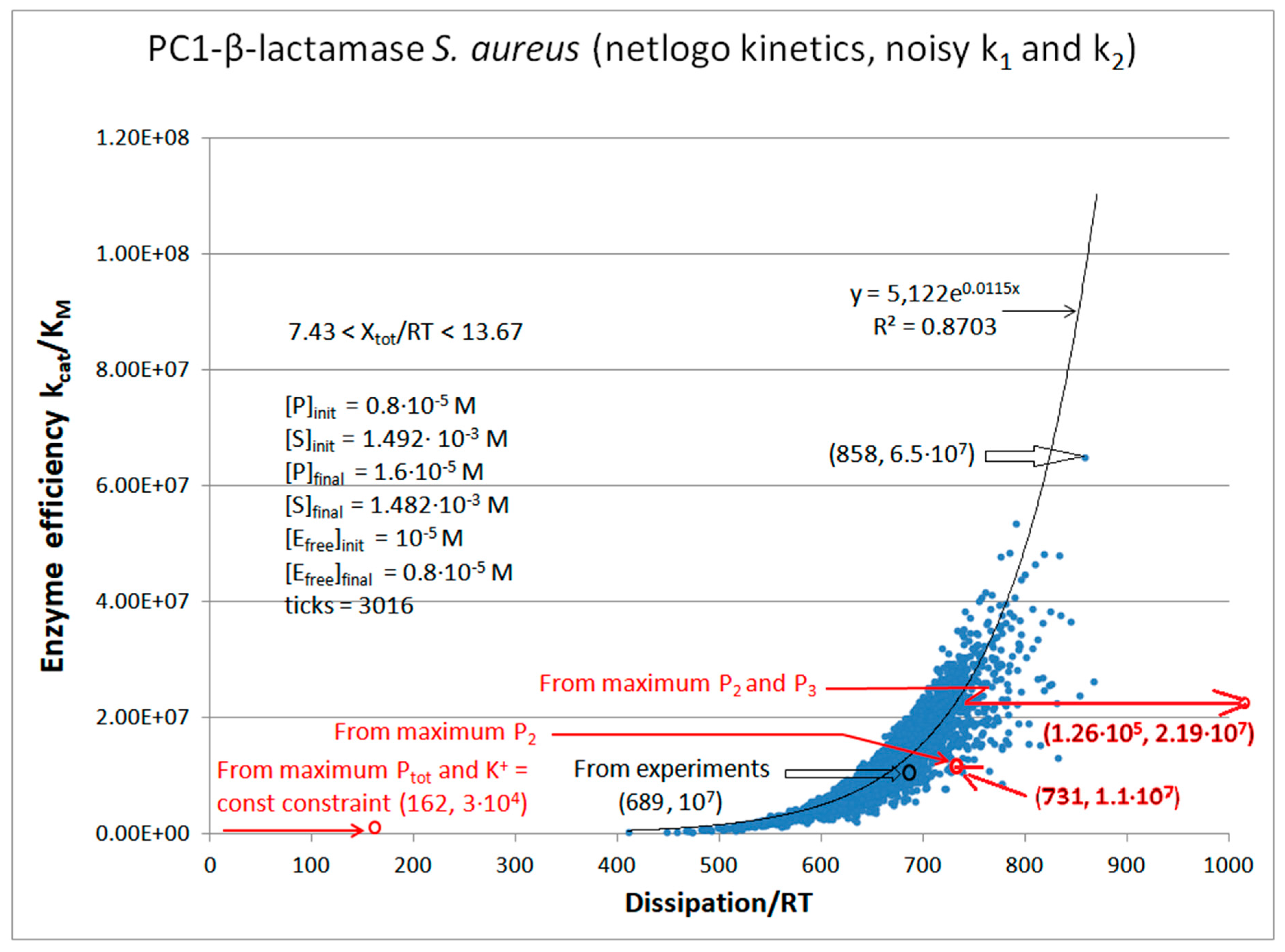
Figure 27.
The catalytic efficiency dependence on dissipation when all forward rate constants are noisy but all equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* maintain their observed values [30] for the RTEM β-lactamase (see Table 4). At the 572nd tick, the NetLogo simulation found the quasi-steady state with the highest catalytic efficiency and dissipation. Due to an excellent joint linear increase in the catalytic efficiency and dissipation, all other kinetic parameters also increased for the similar 3.8-fold factor from their initial Table 4 values.
Figure 27.
The catalytic efficiency dependence on dissipation when all forward rate constants are noisy but all equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* maintain their observed values [30] for the RTEM β-lactamase (see Table 4). At the 572nd tick, the NetLogo simulation found the quasi-steady state with the highest catalytic efficiency and dissipation. Due to an excellent joint linear increase in the catalytic efficiency and dissipation, all other kinetic parameters also increased for the similar 3.8-fold factor from their initial Table 4 values.
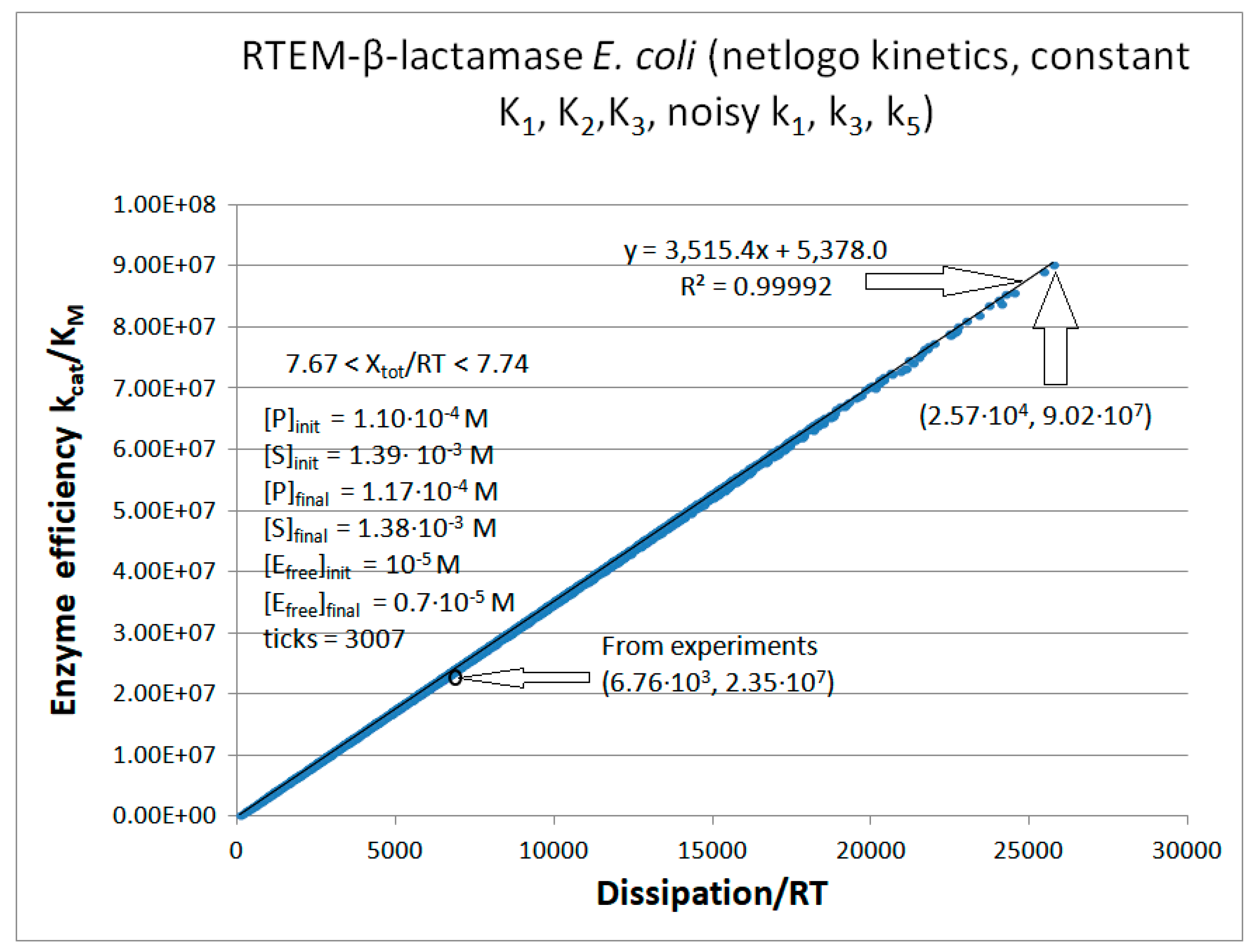
Figure 28.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the RTEM β-lactamase kinetics - in the forward rate constant k1 and in the backward rate constant k2. The best catalytic efficiency from this NetLogo simulation (2.26∙108 M-1s-1) corresponds to the highest dissipation/RT: 1.08∙104 s-1. Notice that the highest dissipation is not so high as for the best quasi-steady state from the previous simulation (Figure 27). Optimal k1 increased 2.4-fold, while optimal k2 decreased 14-fold, producing a significant increase in the irreversibility of the substrate interaction with the enzyme. The best-case efficiency is also associated with the highest Xtot/RT = 11.18, found only for the 3163rd tick. .
Figure 28.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the RTEM β-lactamase kinetics - in the forward rate constant k1 and in the backward rate constant k2. The best catalytic efficiency from this NetLogo simulation (2.26∙108 M-1s-1) corresponds to the highest dissipation/RT: 1.08∙104 s-1. Notice that the highest dissipation is not so high as for the best quasi-steady state from the previous simulation (Figure 27). Optimal k1 increased 2.4-fold, while optimal k2 decreased 14-fold, producing a significant increase in the irreversibility of the substrate interaction with the enzyme. The best-case efficiency is also associated with the highest Xtot/RT = 11.18, found only for the 3163rd tick. .
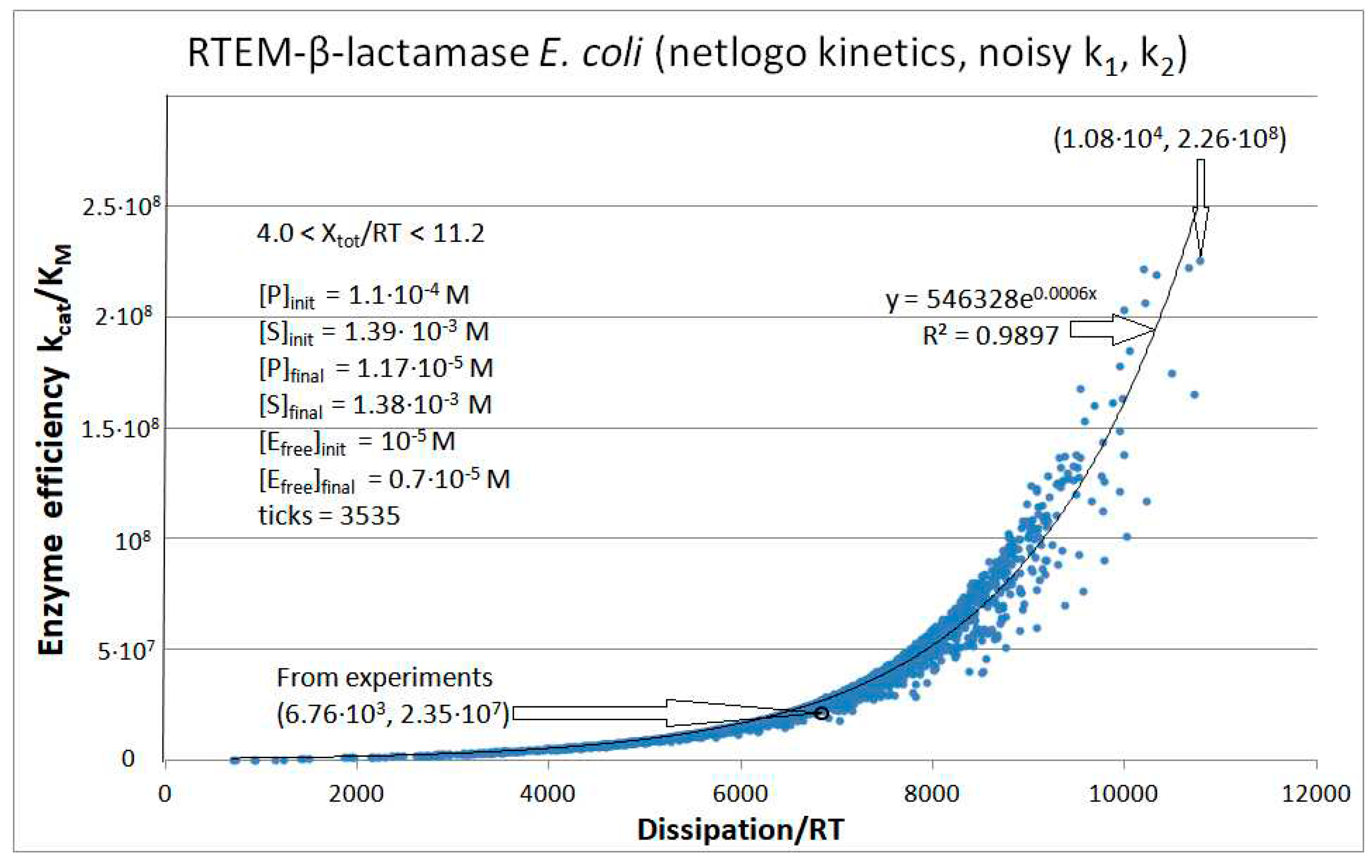
Figure 29.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the Lac-1 β-lactamase kinetics - in the forward rate constant k3 and in the backward rate constant k4.
Figure 29.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the Lac-1 β-lactamase kinetics - in the forward rate constant k3 and in the backward rate constant k4.
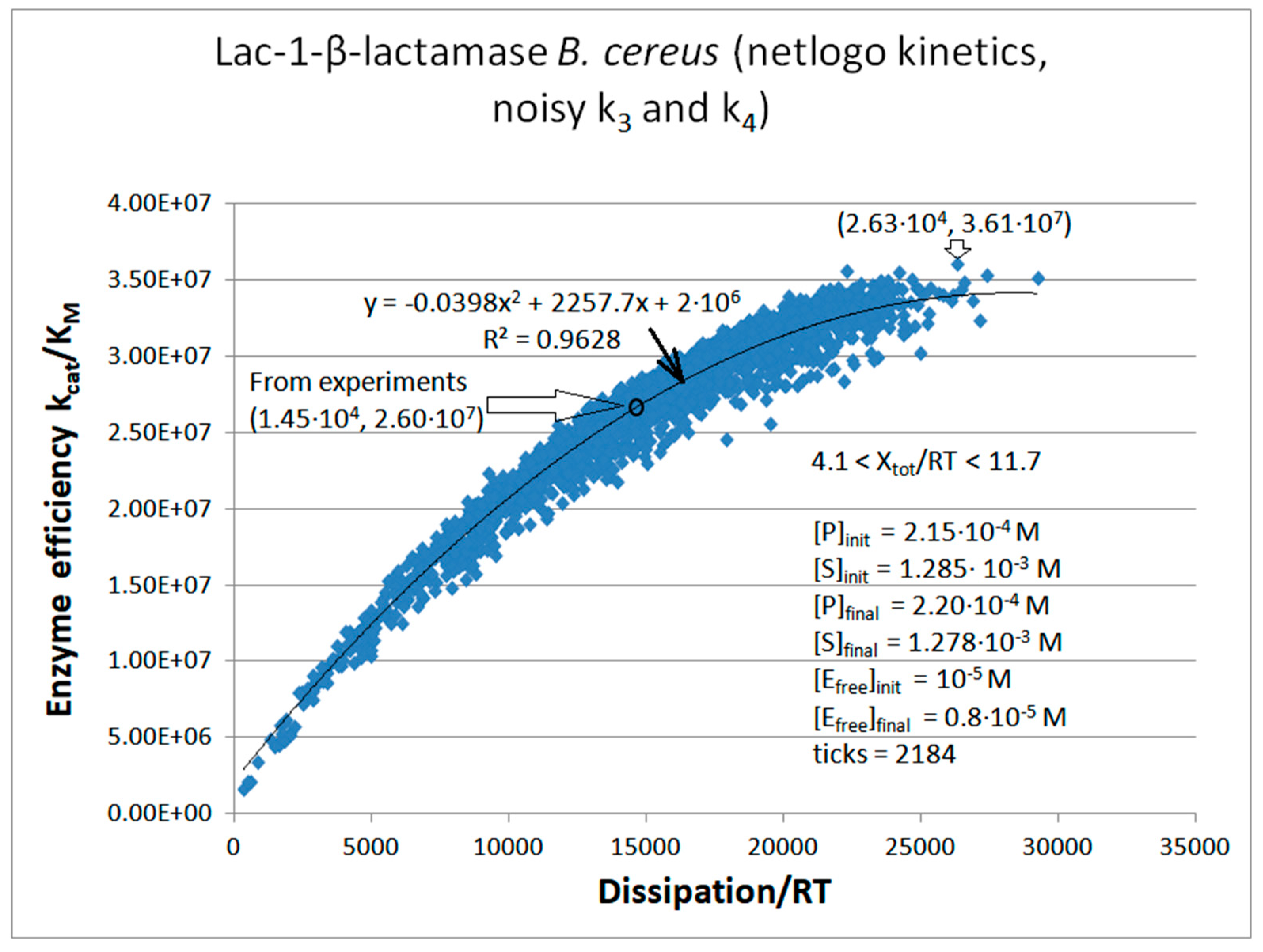
Figure 30.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the Lac-1 β-lactamase kinetics - in the forward rate constant k1 and in the backward rate constant k6.
Figure 30.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the Lac-1 β-lactamase kinetics - in the forward rate constant k1 and in the backward rate constant k6.
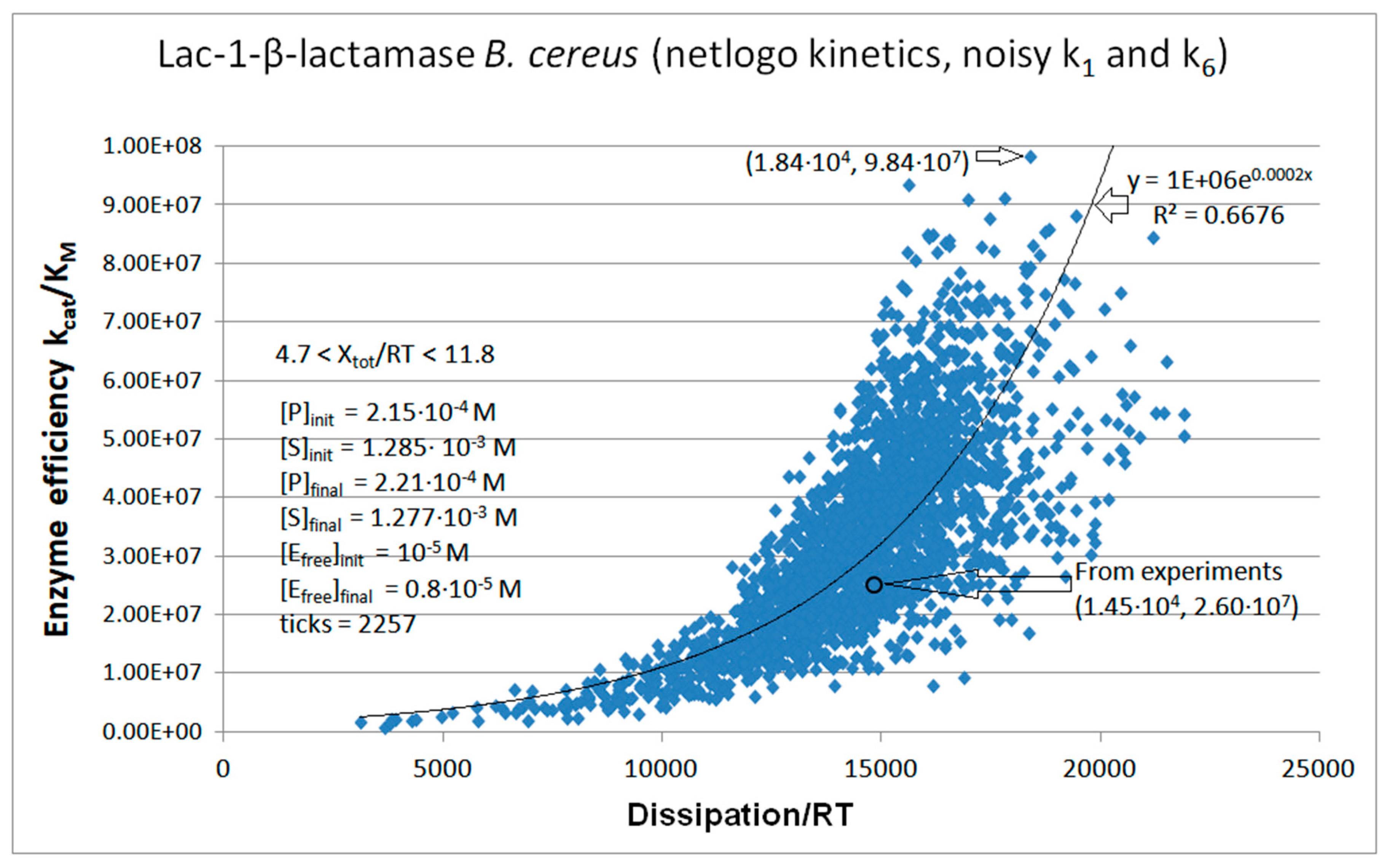
Figure 31.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the Lac-1 β-lactamase kinetics - in the forward rate constant k1 and in the backward rate constant k2.
Figure 31.
The catalytic efficiency dependence on dissipation when noise is introduced twice in the NetLogo simulation for the Lac-1 β-lactamase kinetics - in the forward rate constant k1 and in the backward rate constant k2.
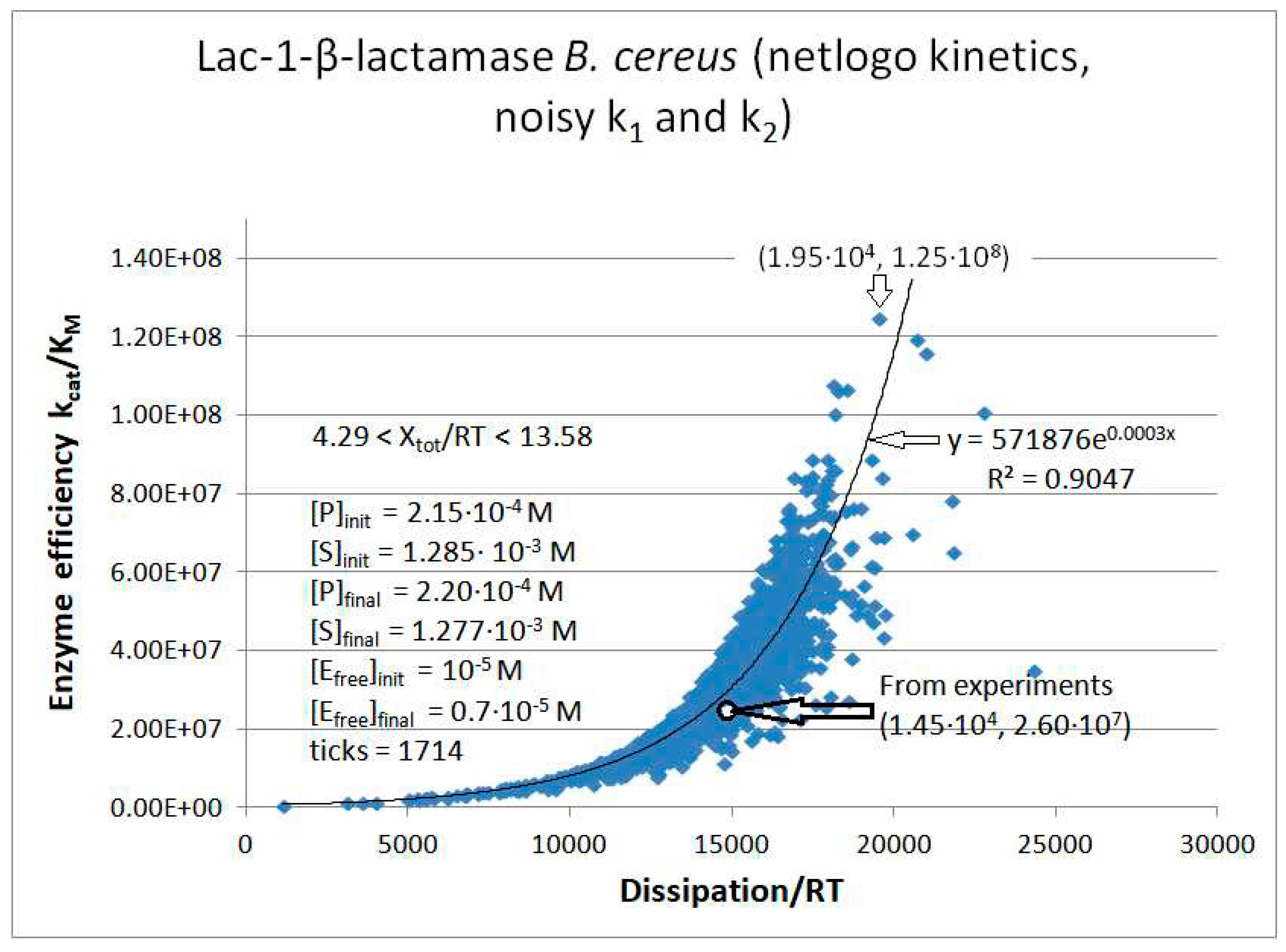
Figure 32.
The catalytic efficiency dependence on dissipation when all forward rate constants are noisy but all equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* maintain their observed values [30] for the Lac-1 β-lactamase. The best coordinates were found at the 2182nd tick. The corresponding optimal values for the rate constants were 3.7 times higher than the initial values (Table 4).
Figure 32.
The catalytic efficiency dependence on dissipation when all forward rate constants are noisy but all equilibrium constants K1* = k1*/k2, K2 = k3/k4, and K3* = k5/k6* maintain their observed values [30] for the Lac-1 β-lactamase. The best coordinates were found at the 2182nd tick. The corresponding optimal values for the rate constants were 3.7 times higher than the initial values (Table 4).
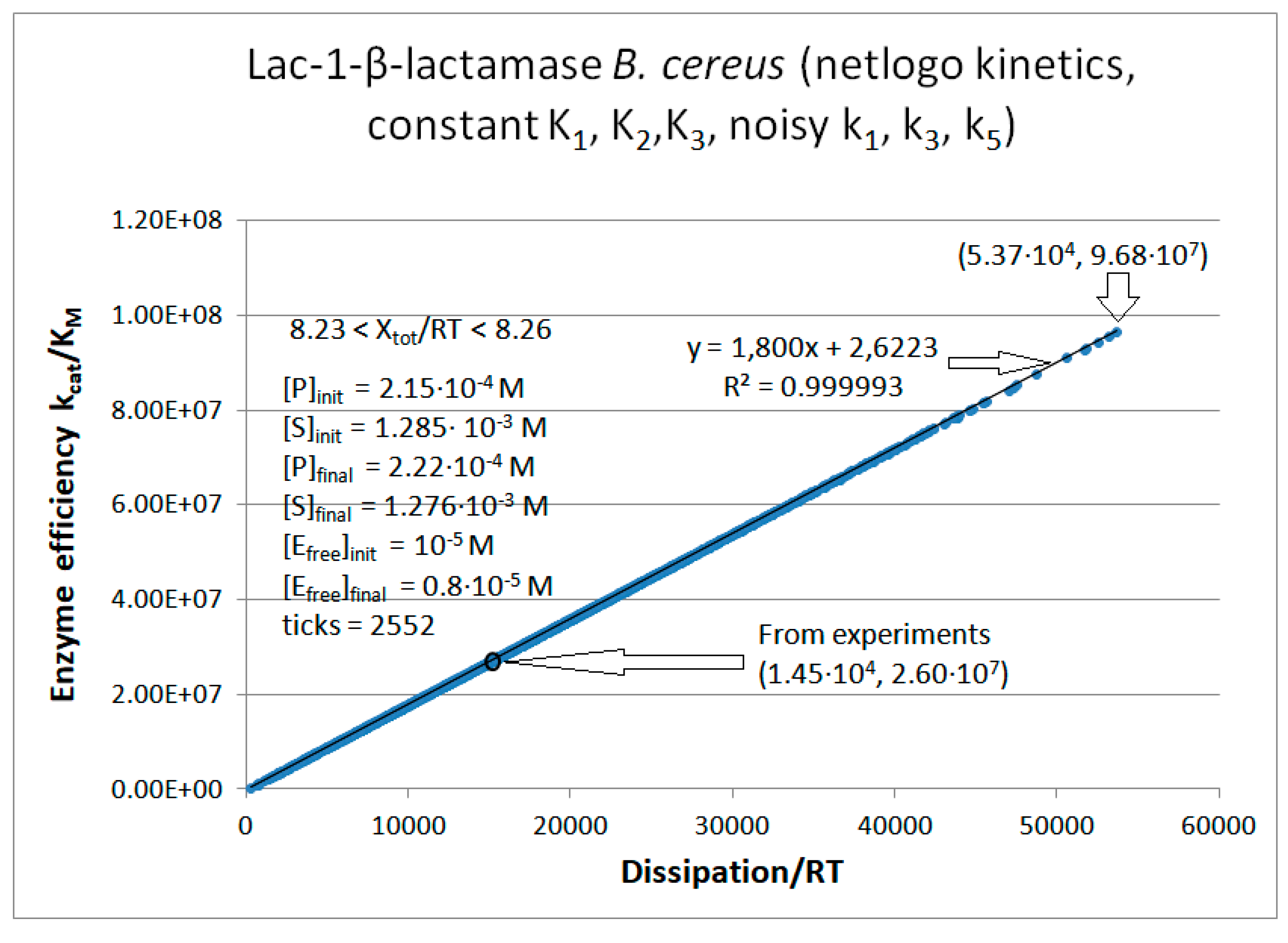
Figure 33.
We compared evolutionary distances 1.19, 1.44, and 1.60 for, respectively, β-lactamases PC1, RTEM, and Lac-1 [2,30,106] with numerical values for the total entropy production either calculated from experimental data (blue points) ([30] and Table 4) or for cases of maximal catalytic efficiency when normal noise is present in the E+S ↔ ES step (red points) (see Figure 26, Figure 28 and Figure 31). The figure illustrates the proportionality between overall entropy production and evolutionary distance when natural or artificial evolution produces the maximal possible catalytic efficiency.
Figure 33.
We compared evolutionary distances 1.19, 1.44, and 1.60 for, respectively, β-lactamases PC1, RTEM, and Lac-1 [2,30,106] with numerical values for the total entropy production either calculated from experimental data (blue points) ([30] and Table 4) or for cases of maximal catalytic efficiency when normal noise is present in the E+S ↔ ES step (red points) (see Figure 26, Figure 28 and Figure 31). The figure illustrates the proportionality between overall entropy production and evolutionary distance when natural or artificial evolution produces the maximal possible catalytic efficiency.
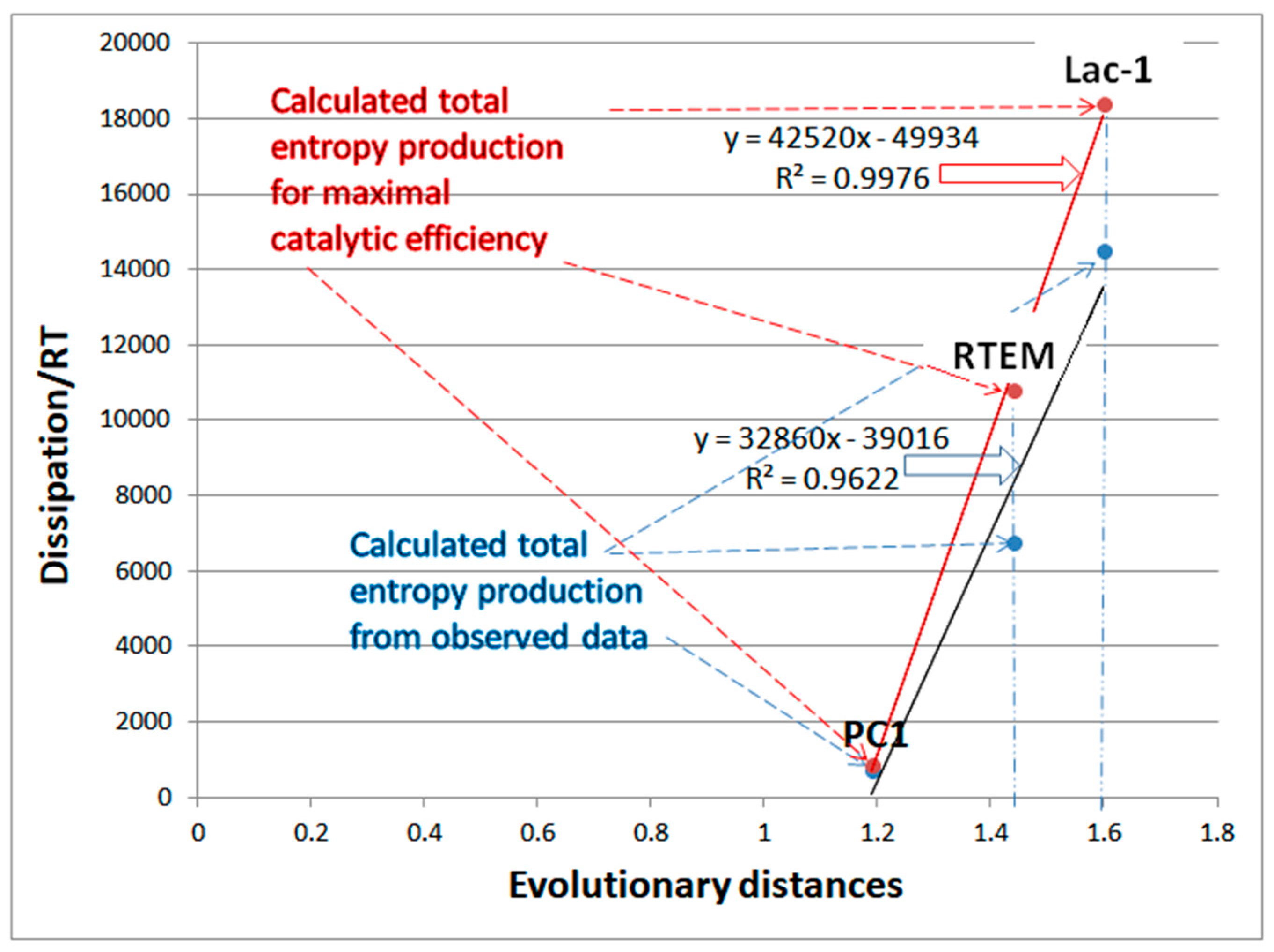
Figure 34.
The catalytic efficiency dependence on dissipation when normal noise is introduced twice, in the forward rate constant k1 as k1∙g1, and, independently in the corresponding backward rate constant k2 as k2∙g2 for the first catalytic step (E+S↔ES) in the case of β-galactosidase kinetics (see Methods). The initial concentration of enzymes was higher than the initial concentration of products, but that quickly changed after less than 100 ticks when product concentration increased about 24 times. The subscript „free“ means the enzyme is free from substrate or product.
Figure 34.
The catalytic efficiency dependence on dissipation when normal noise is introduced twice, in the forward rate constant k1 as k1∙g1, and, independently in the corresponding backward rate constant k2 as k2∙g2 for the first catalytic step (E+S↔ES) in the case of β-galactosidase kinetics (see Methods). The initial concentration of enzymes was higher than the initial concentration of products, but that quickly changed after less than 100 ticks when product concentration increased about 24 times. The subscript „free“ means the enzyme is free from substrate or product.
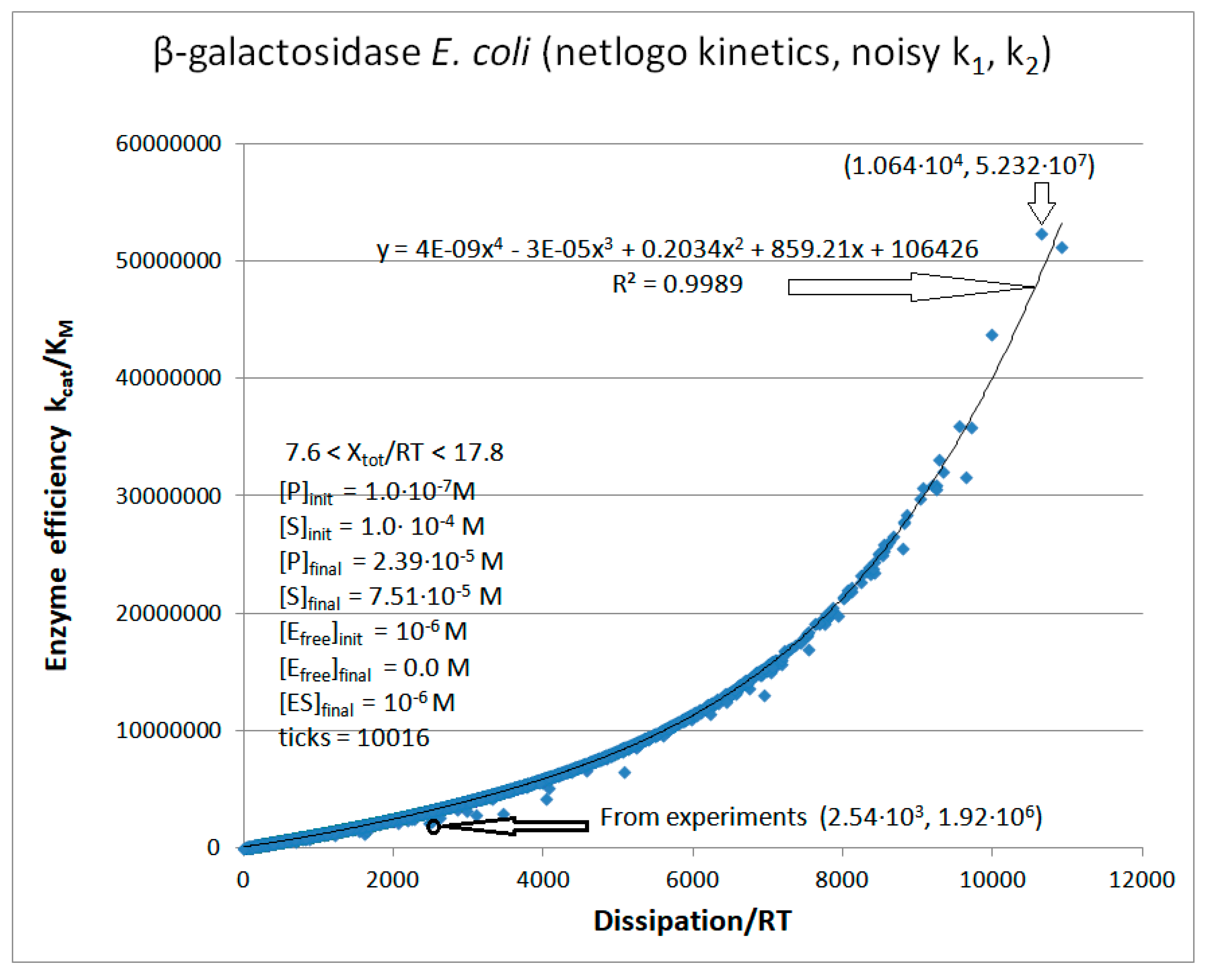
Figure 35.
The catalytic efficiency dependence on dissipation when normal noise is introduced independently in all kinetic constants ki for the β-galactosidase kinetics (see Methods and Table 5). We decreased the probability of stochastic jumps between enzyme conformational states and enzyme to ligand association-dissociation to examine the initial system states with small changes in the substrate concentration. The highest catalytic efficiency was found at the 390th tick when the overall force was at the upper end of its range Xtot/RT = 25.3.
Figure 35.
The catalytic efficiency dependence on dissipation when normal noise is introduced independently in all kinetic constants ki for the β-galactosidase kinetics (see Methods and Table 5). We decreased the probability of stochastic jumps between enzyme conformational states and enzyme to ligand association-dissociation to examine the initial system states with small changes in the substrate concentration. The highest catalytic efficiency was found at the 390th tick when the overall force was at the upper end of its range Xtot/RT = 25.3.
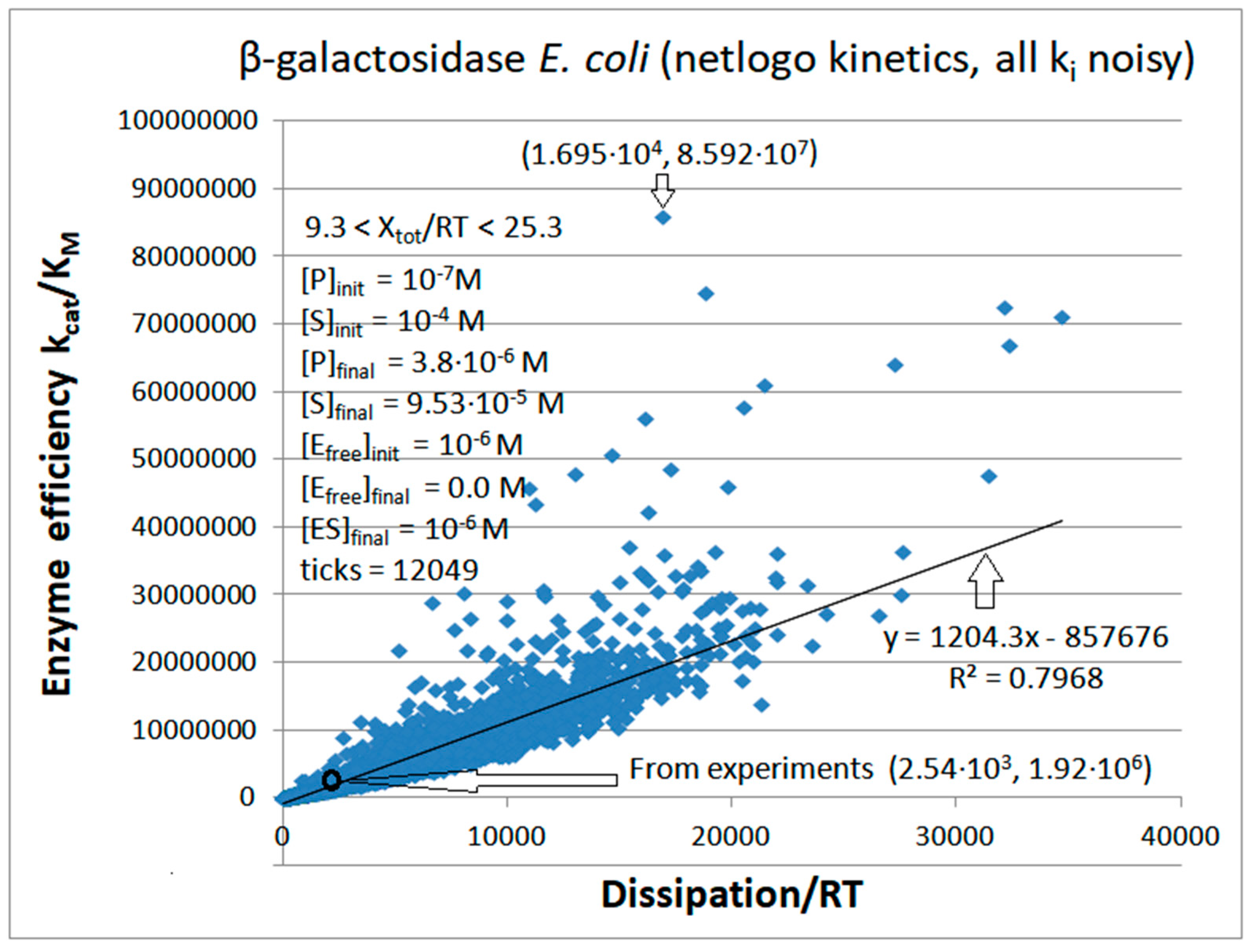
Figure 36.
Force and concentration dependence on time steps (ticks) through 8021 ticks from the second Netlogo simulation (Table 6) as the system relaxes from the initial state values. From the 6323rd tick onward, all free enzymes have been converted into the ES complex. Still, the substrate-to-product conversion reached a stable state with the 28-times substrate excess because we intentionally slowed down the conversion to increase the chance of finding the catalytic efficiency value inside the diffusion limit.
Figure 36.
Force and concentration dependence on time steps (ticks) through 8021 ticks from the second Netlogo simulation (Table 6) as the system relaxes from the initial state values. From the 6323rd tick onward, all free enzymes have been converted into the ES complex. Still, the substrate-to-product conversion reached a stable state with the 28-times substrate excess because we intentionally slowed down the conversion to increase the chance of finding the catalytic efficiency value inside the diffusion limit.
Figure 37.
FORTRAN simulation of enzyme efficiency dependence on dissipation in the presence of noisy rate constants and small changes in the substrate and product concentrations.
Figure 37.
FORTRAN simulation of enzyme efficiency dependence on dissipation in the presence of noisy rate constants and small changes in the substrate and product concentrations.
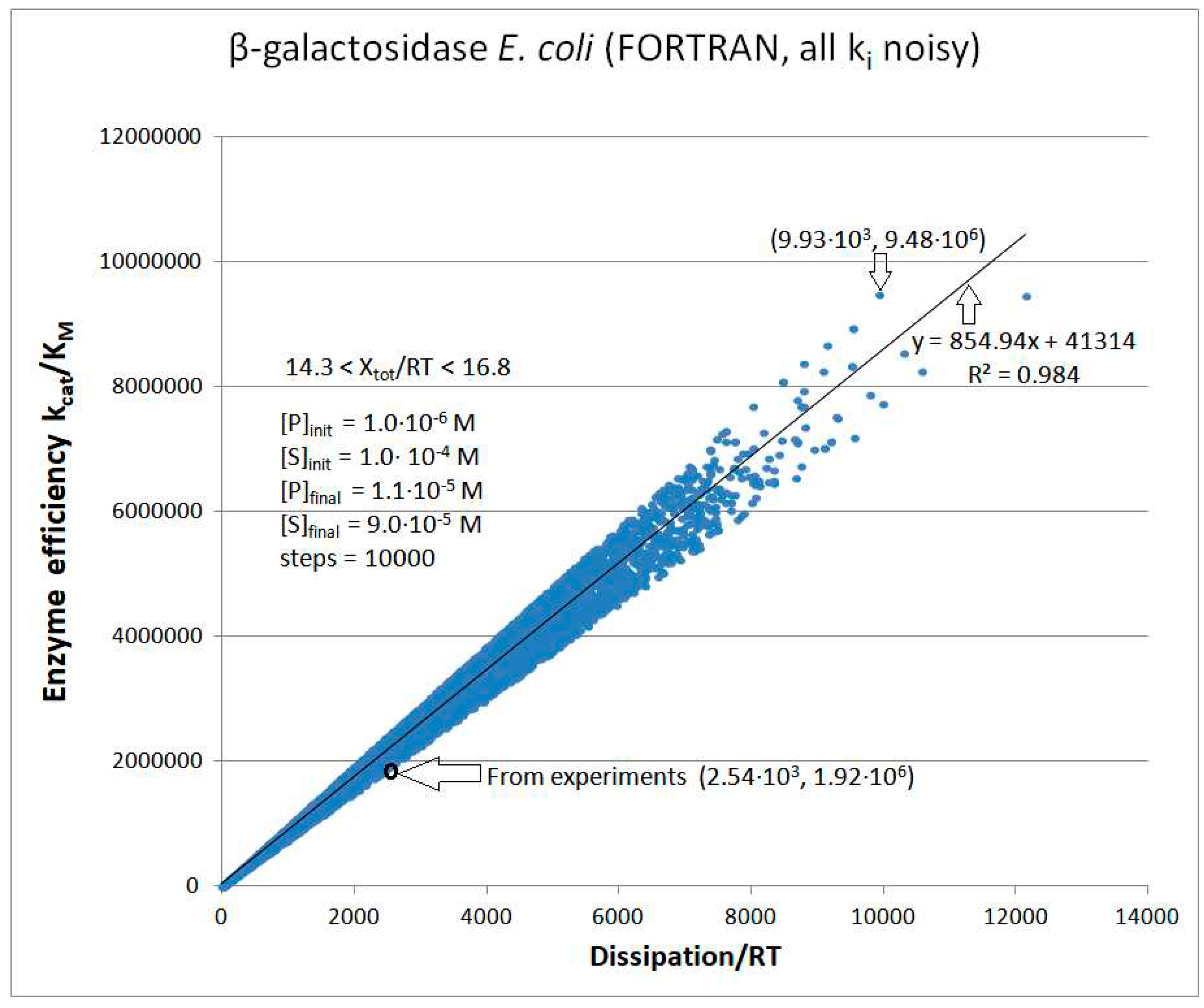
Figure 38.
The catalytic efficiency dependence on dissipation when normal noise function gi with shift +1 is called separately four times to multiply each rate constant ki (i = 1, 2, 3, 4) in the FORTRAN simulation for the β-galactosidase kinetics. The highest catalytic efficiency was found at the 6540th computational step when the total force was Xtot/RT = 20.8.
Figure 38.
The catalytic efficiency dependence on dissipation when normal noise function gi with shift +1 is called separately four times to multiply each rate constant ki (i = 1, 2, 3, 4) in the FORTRAN simulation for the β-galactosidase kinetics. The highest catalytic efficiency was found at the 6540th computational step when the total force was Xtot/RT = 20.8.
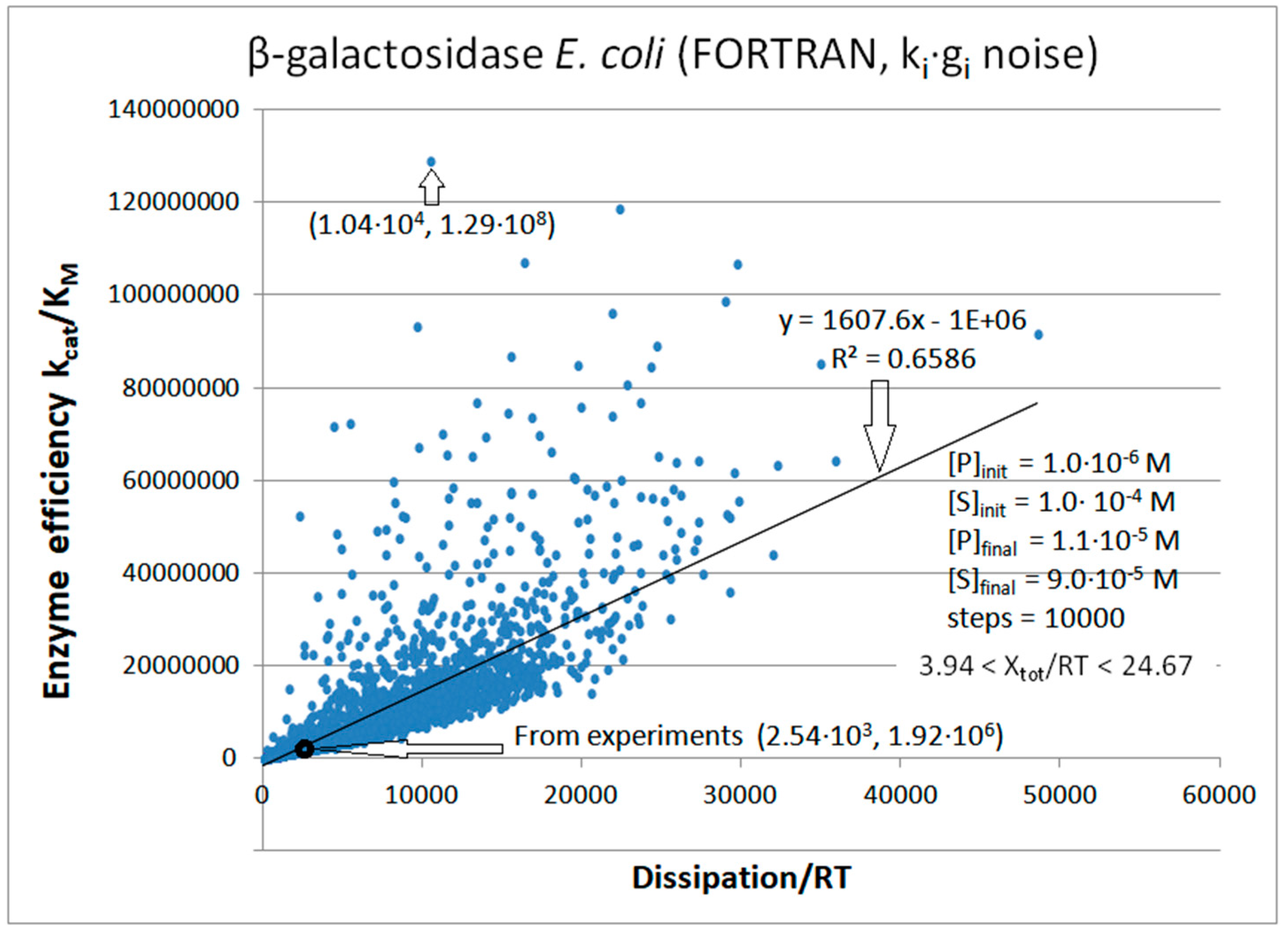
Figure 39.
The catalytic efficiency dependence on dissipation in the case of glucose isomerase kinetics when normal noise is introduced independently in all rate constants ki. The assumed temperature was 65 ◦C. Data for kinetic constants (in moles) are from Converti and Del Borghi [27] and Dobovišek et al. [28], while initial substrate [S]initial and product [P]initial concentrations of, respectively, 2 molar and 0.2 molar, are chosen so that the enzyme works mainly in the forward direction. The simulation with 100 enzymes corresponds to a 100 mM initial concentration of free enzymes. That number quickly drops to zero [E]free concentration because all free enzymes are converted into the 100 mM [ES] complex concentration after only 29 ticks. Due to discrete NetLogo simulation steps, [E]free = 0 can be any concentration smaller than 0.5 mM. The maximal value of catalytic efficiency of 0.213 M-1s-1 is reached in the 1715th step (tick). That efficiency is 5.8 times higher than the value calculated from initial parameters in the absence of noise.
Figure 39.
The catalytic efficiency dependence on dissipation in the case of glucose isomerase kinetics when normal noise is introduced independently in all rate constants ki. The assumed temperature was 65 ◦C. Data for kinetic constants (in moles) are from Converti and Del Borghi [27] and Dobovišek et al. [28], while initial substrate [S]initial and product [P]initial concentrations of, respectively, 2 molar and 0.2 molar, are chosen so that the enzyme works mainly in the forward direction. The simulation with 100 enzymes corresponds to a 100 mM initial concentration of free enzymes. That number quickly drops to zero [E]free concentration because all free enzymes are converted into the 100 mM [ES] complex concentration after only 29 ticks. Due to discrete NetLogo simulation steps, [E]free = 0 can be any concentration smaller than 0.5 mM. The maximal value of catalytic efficiency of 0.213 M-1s-1 is reached in the 1715th step (tick). That efficiency is 5.8 times higher than the value calculated from initial parameters in the absence of noise.
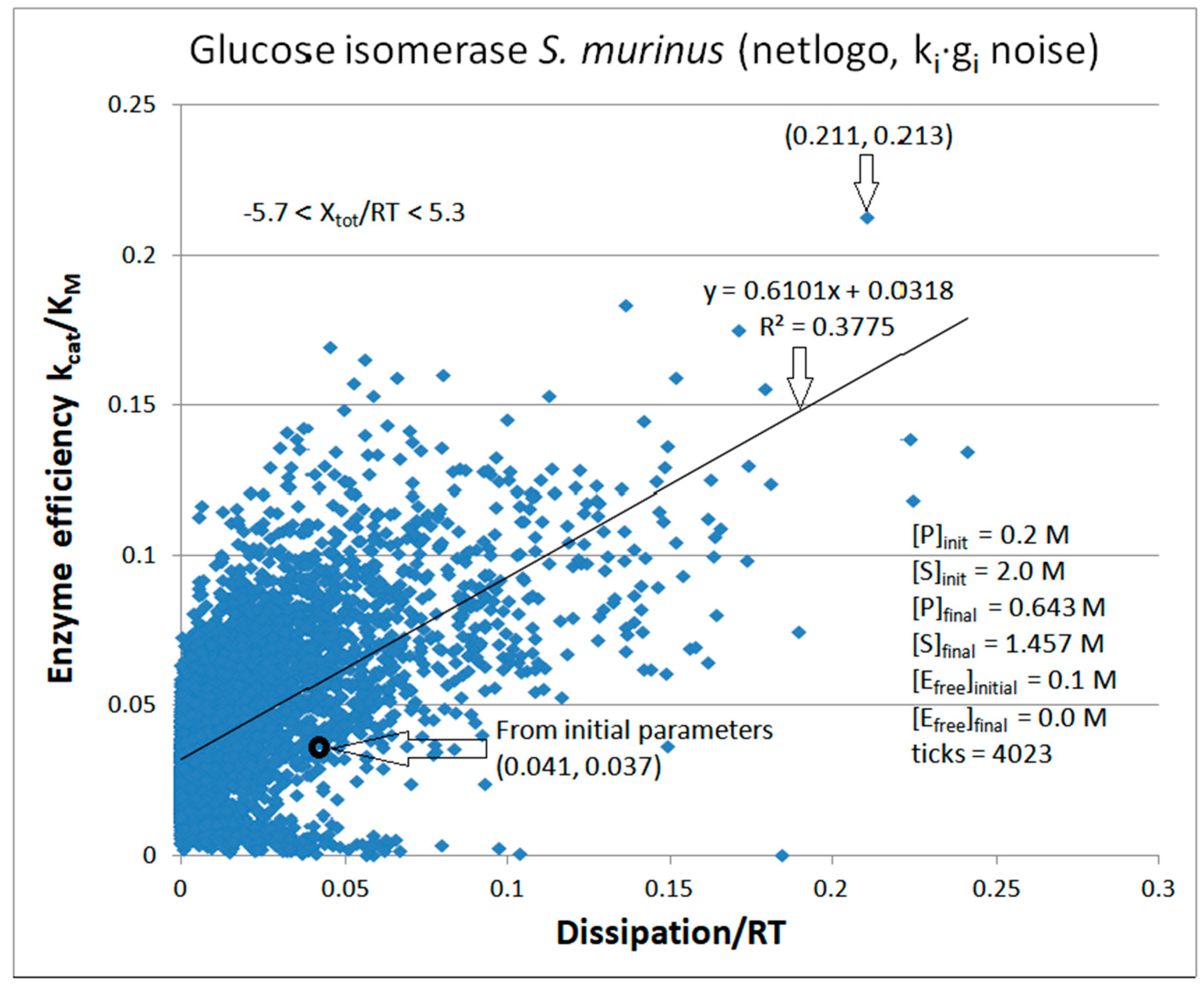
Figure 40.
The catalytic efficiency dependence on dissipation when rate constants ki (i = 1, 2, 3, 4) are noisy in the FORTRAN simulation. Box-Muller transform g1 with shift +1 (see Methods) was called only once and multiplied with each ki. The main loop from the FORTRAN program contained the 10000 steps.
Figure 40.
The catalytic efficiency dependence on dissipation when rate constants ki (i = 1, 2, 3, 4) are noisy in the FORTRAN simulation. Box-Muller transform g1 with shift +1 (see Methods) was called only once and multiplied with each ki. The main loop from the FORTRAN program contained the 10000 steps.
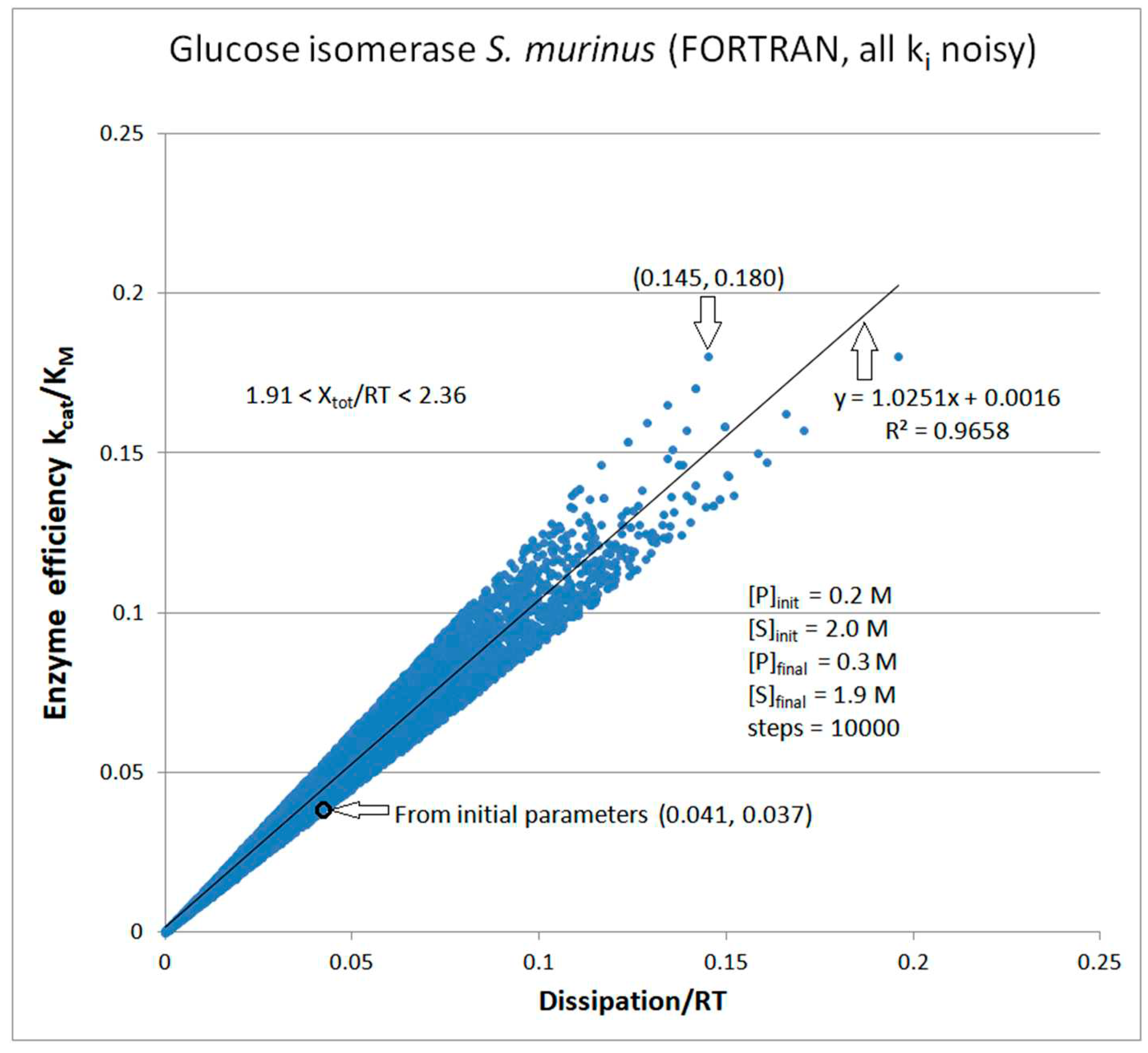
Figure 41.
The catalytic efficiency dependence on dissipation when normal noise function gi with shift +1 is called separately four times to multiply each rate constant ki (i = 1, 2, 3, 4) in the FORTRAN simulation for the glucose isomerase kinetics.
Figure 41.
The catalytic efficiency dependence on dissipation when normal noise function gi with shift +1 is called separately four times to multiply each rate constant ki (i = 1, 2, 3, 4) in the FORTRAN simulation for the glucose isomerase kinetics.
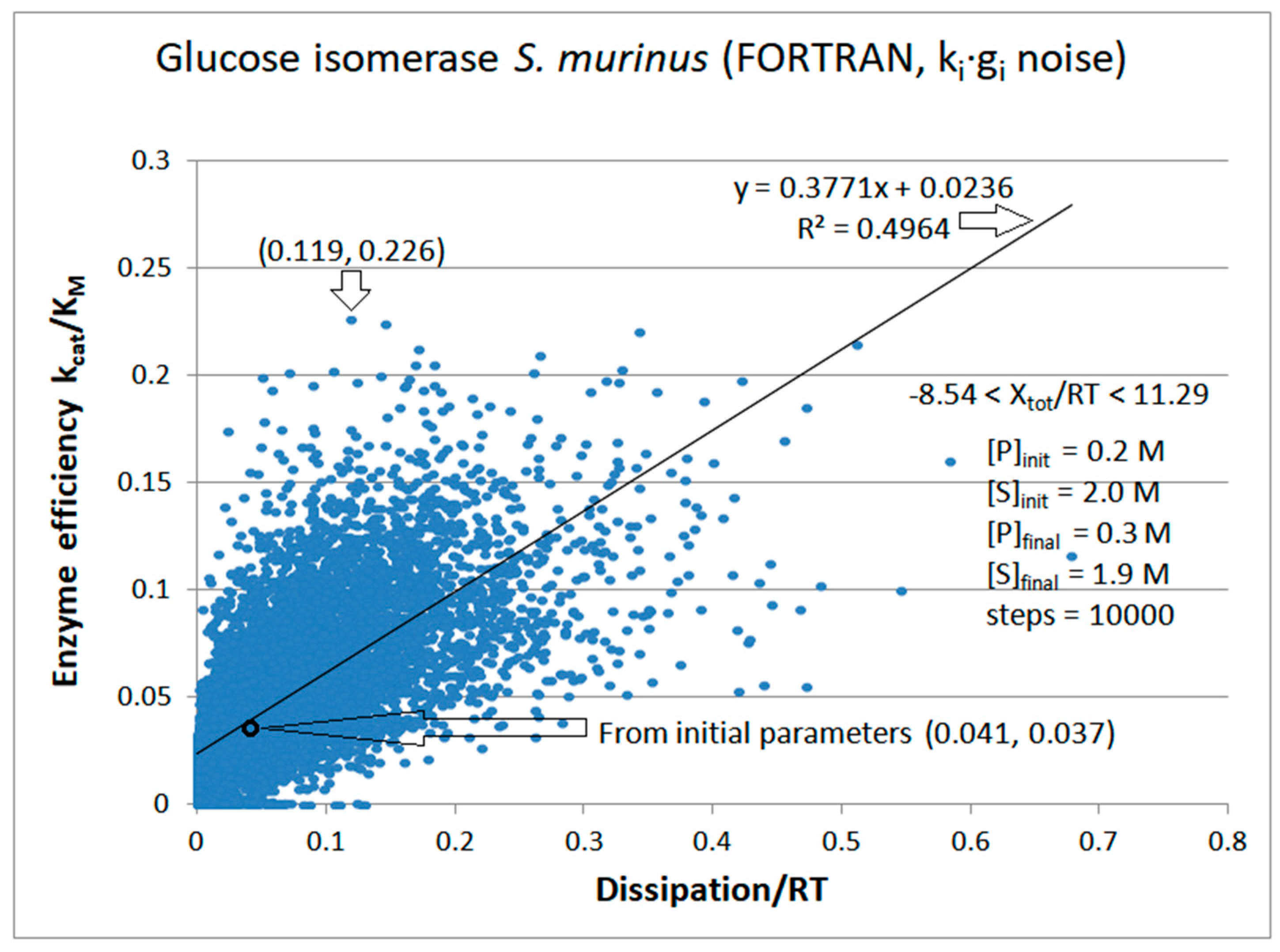
Table 1.
Calculated microscopic rate constants and kinetic parameters from the experimental data [32] in the case of TPI isomerase catalyzed conversion of DHAP (substrate) to GAP (product) at 25 ◦C. The substrate and product concentration were respectively [S] = 40 μM and [P] = 0.064 μM.
Table 1.
Calculated microscopic rate constants and kinetic parameters from the experimental data [32] in the case of TPI isomerase catalyzed conversion of DHAP (substrate) to GAP (product) at 25 ◦C. The substrate and product concentration were respectively [S] = 40 μM and [P] = 0.064 μM.
| Rate constants |
Calculated Values [32,70] |
Kinetic parameters | Calculated initial values [70] |
| k1* | 107 M-1s-1 | k1 | 400 s-1 |
| k2 | 7000 s-1 | k8 | 25.60 s-1 |
| k3 | 2000 s-1 | [S] | 4·10-5 M |
| k4 | 6000 s-1 | [P] | 6.4·10-8 M |
| k5 | 60000 s-1 | [E] | 5·10-8 M |
| k6 | 90000 s-1 | kcat | 432 s-1 |
| k7 | 4000 s-1 | KM | 5.5·10-4 M |
| k8* | 4·108 M-1s-1 | kcat/ KM | 7.86·105 M-1s-1 |
| Keqtot | 3.2·10-3 | ||
| Xtot/RT | 0.685 | ||
| P | 9.9 s-1 |
Table 2.
Calculated microscopic rate constants and kinetic parameters from experimental data [87] and global optimization of experimental data [31] in the case of 3-oxo-Δ5-steroid isomerase catalyzed conversion of 5-androstene-3,17-dione (substrate) to 4-androstene-3,17-dione (product) at 25 ◦C.
| Rate constants |
Calculated values [87] | Calculated values [31] |
| k1* | 8.6·108 M−1s−1 | 8.3·108 M−1s−1 |
| k2 | 8.6·104 s−1 | 8.6·104 s−1 |
| k3 | 1.7·105 s−1 | 1.8·105 s−1 |
| k4 | > 3·105 s−1 | 1.7·106 s−1 |
| k5 | > 1·105 s−1 | 6.4·105 s−1 |
| k6 | 40 s−1 | 43 s−1 |
| k7 | 1.3·105 s−1 | 1.5·105 s−1 |
| k8* | 8.6·108 M−1s−1 | 1·109 M−1s−1 |
| Kinetic parameters | Initial values (this paper) |
|
| [S] | 10-4 M | 10-4 M |
| [P] | 5·10-5 M | 5·10-5 M |
| [E] | 5·10-6 M | 5·10-6 M |
| k1 | 8.3·104 s−1 | |
| k8 | 5·104 s−1 | |
| kcat | 3.5·104 s-1 | |
| KM | 1.16·10-4 M | |
| kcat/KM | 3·108 M−1s−1 | |
| Keqtot | 2281 | |
| Xtot/RT | 8.426 | |
| Initial value (this paper) |
||
| P | 1.16∙105 s−1 |
Table 3.
Calculated microscopic rate constants and kinetic parameters from experimental data [105] (Behravan-1990) in the case of substrate (CO2) to product interconversion and proton-transfer buffer (B)-dependent step catalyzed by carbonic anhydrase isoenzymesat 25 ◦C.
Table 3.
Calculated microscopic rate constants and kinetic parameters from experimental data [105] (Behravan-1990) in the case of substrate (CO2) to product interconversion and proton-transfer buffer (B)-dependent step catalyzed by carbonic anhydrase isoenzymesat 25 ◦C.
| Rate constants [105] |
Calculated values CA I |
Calculated values CA II |
Calculated values CA II T200H |
| k1* | 3.4·107 M−1s−1 | 1.3·108 M−1s−1 | 8.2·107 M−1s−1 |
| k2 | 3.8·104 s−1 | 1.8·106 s−1 | 5.4·104 s−1 |
| k3 | 2.9·105 s−1 | 1.7·107 s−1 | 3.0·105 s−1 |
| k4* | 2.6·107 M−1s−1 | 2.0·108 M−1s−1 | 9.0·106 M−1s−1 |
| k5 | 9.0·105 s−1 | 1.2·106 s−1 | 2.7·106 s−1 |
| k6 | 9.0·106 s−1 | 1.2·106 s−1 | 2.1·107 s−1 |
| k7 | 1.1·108 M−1s−1 | 4.0·108 M−1s−1 | 3.6·108 M−1s−1 |
| k8* | 9.0·105 M−1s−1 | 2.0·107 M−1s−1 | 1.8·107 M−1s−1 |
| Kinetic parameters | Initial values CA I (this paper) |
Initial values CA II (this paper) |
Initial values CA II T200H (this paper) |
| [S] | 1.2∙10-3 M | 1.2∙10-3 M | 1.2∙10-3 M |
| [P] | 2.4·10-2 M | 2.4·10-2 M | 2.4·10-2 M |
| [B] | 5.0·10-2 M | 5.0·10-2 M | 5.0·10-2 M |
| [E] | 1.0·10-4 M | 1.0·10-4 M | 1.0·10-4 M |
| k1 | 4.08·104 s-1 | 1.56·105 s−1 | 9.84·104 s−1 |
| k4 | 6.24·105 s-1 | 4.80·106 s−1 | 2.16·105 s−1 |
| k7 | 5.50·106 s-1 | 2.00·107 s−1 | 1.80·107 s−1 |
| k8 | 4.50·104 s-1 | 1.00·106 s−1 | 9.00·105 s−1 |
| kcat | 7.77·104 s-1 | 8.05·105 s−1 | 2.10·105 s-1 |
| KM | 3.13·10-3 M | 9.63·10-3 M | 3.10·10-3 M |
| kcat/KM | 2.48·107 M−1s−1 | 8.36·107 M−1s−1 | 6.77·107 M−1s−1 |
| Keqtot | 6.10 | 6.14 | 6.51 |
| Xtot/RT | 1.81 | 1.81 | 1.87 |
| CAI (this paper) |
CAII (this paper) |
CA T200H (this paper) |
|
| Pinitial | 2.84∙104 s−1 | 1.25∙105 s−1 | 6.29∙104 s−1 |
Table 4.
Calculated microscopic rate constants, performance parameters, and dissipation from experimental data [2,29,30] in the case of benzylpenicillin substrate hydrolysis catalyzed at 20 ◦C by the A class β-lactamases.
| Rate constants [106] |
Calculated values PC1 |
Calculated values RTEM |
Calculated values Lac-1 |
| k1* | 2.2·107 M−1s−1 | 1.23·108 M−1s−1 | 4.1·107 M−1s−1 |
| k2 | 196 s−1 | 1.18·104 s−1 | 2.32·103 s−1 |
| k3 | 173 s−1 | 2.8·103 s−1 | 4.09·103 s−1 |
| k4 | 4.0 s−1 | 6.0 s−1 | 50 s−1 |
| k5 | 96 s−1 | 1.5·103 s−1 | 3.61·103 s−1 |
| k6* | 1.0·106 M−1s−1 | 4.0·107 M−1s−1 | 8.0·106 M−1s−1 |
| Kinetic parameters | Initial values PC1 (this paper) |
Initial values RTEM (this paper) |
Initial values Lac-1 (this paper) |
| [S] | 1.492∙10-3 M | 1.390∙10-3 M | 1.285∙10-3 M |
| [P] | 8.0·10-6 M | 1.1·10-4 M | 2.15·10-4 M |
| [E] | 10-5 M | 10-5 M | 10-5 M |
| k1 | 3.28·104 s-1 | 1.71·105 s-1 | 5.27·104 s-1 |
| k6 | 8.0 s-1 | 4.4·103 s−1 | 1.72·103 s−1 |
| kcat | 61 s-1 | 9.75·102 s-1 | 1.91·103 s-1 |
| KM | 6.0·10-6 M | 4.15·10-5 M | 7.32·10-5 M |
| kcat/KM | 1.01·107 M−1s−1 | 2.35·107 M−1s−1 | 2.60·107 M−1s−1 |
| Keqtot | 8.69·104 | 2.3·103 | 3.9·103 |
| Xtot/RT | 11.4 | 7.74 | 8.3 |
| Initial value PC1 [106] |
Initial value RTEM [106] | Initial value Lac-1 [106] |
|
| P | 689 s-1 | 6757 s-1 | 14526 s-1 |
Table 5.
Initial values of microscopic rate constants from experimental data [26] and our calculations of other initial kinetic and thermodynamic parameters in the case of E. coli β-galactosidase catalyzed conversion of resorufin-b-D-galactopyranoside (substrate) to a fluorescent resorufin (product) at 25 ◦C.
Table 5.
Initial values of microscopic rate constants from experimental data [26] and our calculations of other initial kinetic and thermodynamic parameters in the case of E. coli β-galactosidase catalyzed conversion of resorufin-b-D-galactopyranoside (substrate) to a fluorescent resorufin (product) at 25 ◦C.
| Rate constants |
Calculated values [26] | Calculated values (this work and [26] |
| k1* | 5.0·107 M−1s−1 | 5.0·107 M−1s−1 |
| k2 | 1.83·104 s−1 | 1.83·104 s−1 |
| k3 | 7.3·102 s−1 | 7.3·102 s−1 |
| k4* | 10 M−1s−1 | 10 M−1s−1 |
| Other relevant parameters |
Initial values (this paper) |
|
| [S] | 10-4 M | 10-4 M |
| [P] | 10-7 M | |
| [E] | 10-6 M | |
| k1 | 5.0·103 s−1 | |
| k4 | 10-5 s−1 | |
| kcat | 730 s−1 | |
| KM | 3.81·10-4 M | |
| kcat/KM | 1.92·106 M−1s−1 | |
| Keqtot | 2.0·107 | |
| Xtot/RT | 16.81 | |
| Initial value (this paper) |
||
| P | 2.55·103 s−1 |
Table 6.
Kinetic and thermodynamic parameters for the NetLogo runs for noisy k1, k2, k3, and k4. The first run corresponds to Figure 35. The second run corresponds to Figure 36. The „Best B“ notation is for the parameters in the quasi-steady state with the highest catalytic efficiency. The „Last L“ notation is for the steady state when we stopped the program after concentrations did not change for many ticks.
Table 6.
Kinetic and thermodynamic parameters for the NetLogo runs for noisy k1, k2, k3, and k4. The first run corresponds to Figure 35. The second run corresponds to Figure 36. The „Best B“ notation is for the parameters in the quasi-steady state with the highest catalytic efficiency. The „Last L“ notation is for the steady state when we stopped the program after concentrations did not change for many ticks.
| Run # Best |
Total ticks | B or L tick | [S]free (M) | [P]free (M) | k1 (s-1) |
k2 (s-1) |
k3 (s-1) |
k4 (s-1) |
Xtot/RT | KM (M) | Dissip/RT (s-1)* | kcat/KM (M-1s-1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 12049 | 390 | 9.996·10-5 | 1.3·10-7 | 14109 | 467 | 726 | 2.25·10-7 | 25.3 | 8.45·10-6 | 16948 | 8.59·107 |
| 2 | 8021 | 373 | 9.996 ·10-5 | 1.3·10-7 | 14325 | 251 | 784 | 1.26 ·10-6 | 24.3 | 7.22·10-6 | 17759 | 1.09·108 |
| Run # Last |
||||||||||||
| 1 | 12049 | 12049 | 9.530·10-5 | 3.80·10-6 | 2843 | 3.428·104 | 224 | 1.04·10-4 | 12.1 | 1.16·10-3 | 207 Av: 1310 |
1.94 ·105 |
| 2 | 8021 | 8021 | 9.564·10-5 | 3.46·10-6 | 5084 | 4390 | 780 | 8.32·10-5 | 16.2 | 9.73·10-5 | 6268 Av: 2852 |
8.02·106 |
* An average (Av) dissipation/RT value is calculated only for the last 10 ticks.
Table 8.
Fold-improvement for enzyme efficiency and corresponding fold-increase of overall dissipation in the best cases concerning values found from experiments.
Table 8.
Fold-improvement for enzyme efficiency and corresponding fold-increase of overall dissipation in the best cases concerning values found from experiments.
| Enzyme (functional states, Fig. #) | Simulation software abbreviation (noisy ki) |
Efficiency fold- improvement |
Dissipation fold increase |
Eff/Disssip. (fold factor)* | Best eff. (M-1s-1) |
|---|---|---|---|---|---|
| Glucose isomerase (2, 39) | GI-NetLogo-kin-simul (all ki noisy) |
5.8 | 5.2 | 1.0 (1.1) | 0.213 |
| Glucose isomerase (2, 41) | GI-FORTRAN-kin-simul (all ki noisy) | 6.1 | 2.9 | 1.9 (2.1) | 0.226 |
| β-galactosidase (2, 35) | GAL-NetLogo-kin-simul (all ki noisy) | 44.8 | 6.7 | 5.1∙103 (6.7) | 8.59∙107 |
| β-galactosidase (2, 38) | GAL-FORTRAN-kin-simul (all ki noisy) | 67.2 | 4.1 | 1.2∙104 (16.4) | 1.29∙108 |
| Lac1-β-lactamase (3, 31) | Lac-1-NetLogo-kin-simul (noisy k1,k2) | 4.8 | 1.3 | 6.4∙103 (3.6) | 1.3∙108 |
| RTEM-β-lactamase (3, 28) | RTEM-lac-NetLogo-simul (noisy k1,k2) | 9.6 | 1.6 | 2.1∙104 (6.0) | 2.3∙108 |
| PC1-β-lactamase (3, 26) | PC1-lac-NetLogo-simul (noisy k1,k2) | 6.5 | 1.2 | 7.6∙104 (5.2) | 6.5∙107 |
| Carbonic anhydrase I (4, 20) | CA-I-FORTRAN-kin-simul (all ki noisy) | 4.5 | 18.1 | 218 (0.25) | 1.1∙108 |
| Carbonic anhydrase I (4, 21) | CA-I-NetLogo-kin-simul (all ki noisy) | 5.9 | 22.4 | 231 (0.26) | 1.47∙108 |
| Carbonic anhydrase II (4, 22) | CA-II-NetLogo-kin-simul (all ki noisy) | 5.1 | 13.4 | 254 (0.38) | 4.25∙108 |
| Carbonic anhydrase T200H (4, 23) | CA-T200H-NetLogo-kin-simul (all ki noisy) | 5.5 | 14.0 | 421 (0.39) | 3.71∙108 |
| Ketosteroid isomerase (4, 15) |
KSI-FORTRAN-kin-simul (all ki noisy) | 6.2 | 4.6 | 3.5∙103 (1.34) | 1.88∙109 |
| Ketosteroid isomerase (4, 18) |
KSI-NetLogo-kin-simul (all ki noisy) |
8.6 | 3.8 | 5.7∙103 (2.26) | 2.59∙109 |
| Triophosphate isomerase (4, 11) |
TPI-FORTRAN-kin-simul (all ki noisy) | 29.9 | 160.6 | 1.5∙104 (0.19) | 2.4∙107 |
| Triophosphate isomerase (4, 14) |
TPI-NetLogo-kin-simul (all ki noisy) |
28.1 | 198.4 | 1.1∙104 (0.14) | 2.2∙107 |
* The fold factor is the ratio of the best efficiency/dissipation and observed efficiency/dissipation.Discussion.
Table 9.
Kinetic and thermodynamic parameters for the best NetLogo and FORTRAN results concerning values found from experiments. The green highlight denotes increased, yellow decreased, and orange equals the value of the experimental one.
Table 9.
Kinetic and thermodynamic parameters for the best NetLogo and FORTRAN results concerning values found from experiments. The green highlight denotes increased, yellow decreased, and orange equals the value of the experimental one.
| Figure | Enzyme | Software | k1 (s-1) | k2 (s-1) | P1 (s-1) (%P) | P (s-1) | k3 (s-1) | k4 (s-1) | k5 (s-1) | k6 (s-1) | k7 (s-1) | k8 (s-1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TPI | Exper&calc. | 400 | 7.0·103 | 0.573 (6) | 9.883 | 2.0·103 | 6.0·103 | 6.0·104 | 9.0·104 | 4·103 | 25.60 | |
| 14 | NetLogo | 1.05·103 | 303 | 833 (42) | 1.96·103 | 5.4·103 | 12.6·103 | 9.4·104 | 9.97·104 | 6.5·103 | 128 | |
| 11 | FORTRAN | 1.14·103 | 126 | 435 (27) | 1.59·103 | 1.05·103 | 6.0·103 | 6.0·104 | 6.15·103 | 937 | 25.60 | |
| KSI | Exper&calc. | 8.3·104 | 8.6·104 | 6.22·103 (5) (5) | 1.16·105 | 1.8·105 | 1.7·106 | 6.4·105 | 43 | 1.5·105 | 5.0·104 | |
| 18 | NetLogo | 2.77·105 | 3.7·104 | 7.9·104 (18) | 4.50·105 | 4.95·105 | 7.1·105 | 1.02·106 | 16 | 6.9·104 | 7.7·104 | |
| 15 | FORTRAN | 2.30·105 | 2.5·104 | 7.17·104 (13) | 5.39·105 | 1.8·105 | 3.97·105 | 6.4·105 | 43 | 1.5·105 | 2.9·104 | |
| CA I | Exper&calc. | 4.08·104 | 3.8·104 | 1.48·104 (52) | 2.84·104 | 2.9·105 | 6.24·105 | 9.0·105 | 9.0·106 | 5.5·106 | 4.5·104 | |
| 21 | NetLogo | 2.0·105 | 2.3·104 | 2.51·105(40) | 6.36·105 | 4.7·105 | 6.55·105 | 2.0·106 | 9.3·106 | 8.8·106 | 1.8·104 | |
| 20 | FORTRAN | 1.53·105 | 1.5·104 | 2.13·105 (41) | 5.14·105 | 1.95·106 | 7.71·105 | 2.3·106 | 8.5·106 | 6.6·106 | 3.1·104 | |
| CA II | Exper&calc. | 1.56·105 | 1.8·106 | 5.33·104 (43) | 1.25·105 | 1.7·107 | 4.80·106 | 1.2·106 | 1.2·106 | 2.0·107 | 1.0·106 | |
| 22 | NetLogo | 6.38·105 | 2.5·106 | 5.61·105 (34) | 1.67·106 | 3.7·107 | 3.93·106 | 1.5·106 | 1.5·106 | 3.0·107 | 6.6·105 | |
| CA T200H | Exper&calc. | 9.84·104 | 5.4·104 | 4.03·104 (64) | 6.3·104 | 3.0·105 | 2.16·105 | 2.7·106 | 2.1·107 | 1.8·107 | 9.0·105 | |
| 23 | NetLogo | 6.49·105 | 6.7·104 | 4.05·105(46) | 8.82·105 | 7.98·105 | 7.4·104 | 3.2·106 | 2.9·107 | 7.4·106 | 4.6·105 | |
| PC1 | Exper&calc. | 3.28·104 | 196 | 37 (5) | 689 | 173 | 4.0 | 96 | 8.0 | |||
| 26 | NetLogo | 1.15·105 | 32 | 111 (13) | 858 | 173 | 4 | 96 | 11 | |||
| RTEM | Exper&calc. | 1.71·105 | 1.18·104 | 185 (3) | 6.76·103 | 2.8·103 | 6.0 | 1.5·103 | 4.4·103 | |||
| 28 | NetLogo | 4.07·105 | 851 | 1.4·103 (13) | 1.08·104 | 2.8·103 | 6 | 1.5·103 | 4.7·103 | |||
| Lac-1 | Exper&calc. | 5.27·104 | 2.32·103 | 1.8·103 (12) | 1.45·104 | 4.09·103 | 50 | 3.61·103 | 1.72·103 | |||
| 31 | NetLogo | 1.98·105 | 976 | 3.1·103 (16) | 1.95·104 | 4.09·103 | 50 | 3.61·103 | 1.76·103 | |||
|
β-galacto- sidase |
Exper&calc. | 5.0·103 | 1.83·104 | 5.84 (0.2) | 2.55·103 | 730 | 1.0·10-5 | |||||
| 35 | NetLogo | 1.4·104 | 467 | 628 (4) | 1.70∙104 | 726 | 2.25∙10-7 E07 | |||||
| 38 | FORTRAN | 1.3·104 | 61 | 1.12·103 (11) | 1.04∙104 | 520 | 0.0001 | |||||
| Glucose isomerase | Exper&calc. | 0.126 | 0.021 | 0.0126 (31) | 0.0392 | 0.029 | 0.016 | |||||
| 39 | NetLogo | 0.320 | 0.002 | 0.143 (68) | 0.211749 | 0.068 | 0.088 | |||||
| 41 | FORTRAN | 0.499 | 0.004 | 0.057 (48) | 0.119 | 0.031 | 0.045 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



