Submitted:
01 December 2023
Posted:
04 December 2023
You are already at the latest version
Abstract
The pro-protein convertase FURIN (PCSK3) is implicated in a wide range of normal and pathological biological processes such as infectious disease, cancer and cardiovascular disease. Previously, we performed a systemic inhibition of FURIN in a mouse model of atherosclerosis and demonstrated significant plaque reduction and alterations in macrophage function. To understand the cellular mechanisms affected by FURIN inhibition in myeloid cells, we optimized a CRISPR-mediated gene deletion protocol for successfully deriving hemizygous (HZ) and nullizygous (NZ) FURIN knockout clones in U937 monocytic cells using lipotransfection based procedures and a dual guide RNA delivery strategy. We observed differences in monocyte and macrophage functions involving phagocytosis, lipid accumulation, cell migration, inflammatory gene expression, cytokine release patterns, secreted proteomics (cytokines) and whole-genome transcriptomics between wild-type, HZ and NZ FURIN clones. These studies provide a mechanistic basis on the possible roles of myeloid cell FURIN in cardiovascular disorders.
Keywords:
myeloid cells
; FURIN protease
; gene editing
; functional assays
; genomics
; cytokine secretion
1. Introduction
The pro-protein convertase, FURIN (PCSK3) belongs to a family of proteins that affect a wide variety of biological functions through proteolytic cleavage and activation of target proteins [1,2]. Through genetic association analyses, we previously identified FURIN as a gene candidate associated with coronary artery disease in humans [3]. This genetic finding was subsequently validated in two independent mouse models of atherosclerosis, where systemic inhibition of FURIN was found to provide atheroprotection as well as reduce restenosis [4]. An associated finding was that FURIN inhibition reduced the number of macrophages in atherosclerotic plaques of the coronary artery, suggesting that FURIN inhibition may play a specific role in myeloid cell function. Notably, FURIN expression is increased in several cell types in human atherosclerotic lesions, including macrophages [4,5]. Based on these observations, we wanted to examine the effects of FURIN gene editing on key functions in cultured monocytes/macrophages.
Based on the innate immune response mechanisms in bacteria, clustered regularly interspersed short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) gene-editing technologies have evolved as a powerful and popular methodology for genome editing both in vivo and in vitro. When two or more sgRNAs are co-expressed with a single Cas9 protein, different genomic loci can be targeted simultaneously, thereby greatly improving the efficiency of gene editing, including gene deletions and the generation of hemizygous (HZ, containing a wildtype and a mutant allele) and nullizygous (NZ, both copies of genes deleted) clones [6,7,8]. These advantages have greatly popularized the CRISPR/Cas9 systems as indispensable tools in the field of genome editing and have shown great promise even for gene-targeted therapy [9].
THP-1 and U937 are two of the most commonly studied pro-monocytic human leukemia cell lines that are capable of differentiating into macrophages or dendritic cells in vitro [10]. However, the phagocytic properties of monocytes often lead to the degradation of exogenously introduced DNA, accompanied by cell death [11]. The three inter-related problems of low transfection efficiency, low cell viability and low success of stable transfections make monocytes/macrophages refractory to DNA transfections in general, including CRISPR-mediated genome editing [11,12]. Some advanced methods exist, but they generally require special equipment that is often expensive and proprietary, and therefore not available in all research settings. Additionally, they are also often associated with significant cell toxicity. There remains a need for optimizing simpler techniques to enable successful genome editing in normally refractory cell types such as monocytes.
In this manuscript, we report on an optimized protocol for CRISPR-editing in U937 monocytes using standard lipid-based transfection reagents, resulting in hemizygous and nullizygous clones for FURIN and observed important differences in several targeted cellular phenotypes as a function of FURIN gene dosage. Additionally, we carried out whole-genome transcriptomic and targeted proteomic characterization of HZ and NZ FURIN clones compared to wild-type cells (WT), to characterize the effects of FURIN on myeloid cell function on a broader scale.
2. Materials and Methods
Cells and reagents
U937 cells (catalog no. CRL-1593.2,) and THP-1 monocytic cells (catalog no. TIB-202) were purchased from ATCC (Manassas, USA) . Plasmids for CRISPR-related cell transfection were a gift from Dr. Shang Li, Duke-NUS. gRNAs and PCR primers were purchased from IDT(Singapore). RPMI media, Opti-mem, L-glutamine and Lipofectamine were purchased from ThermoFisher (Singapore). Fetal bovine serum was purchased from Research Instrument (Singapore). 96-well plates for cell culture were obtained from Practical Mediscience (Singapore), and 24- and 6-well plates were from Scimed (Asia) (Singapore). DAPI solution was purchased from BD (Franklin Lakes, USA). PCR reactions were performed on a CFX96 (Bio-rad, Hercules, USA) using PCR mastermix from Promega (Madison, USA). Western blotting was performed via the Semi-Dry Rapid Blot Transfer system from Bio-rad.
Plasmid preparation
Two single guide RNAs (sgRNA) targeting the 5’ and 3’ ends of the FURIN genomic region (chr15:91411885-91426687, hg19) were designed using an online software (https://zlab.bio/guide-design-resources from crispr.mit.edu, currently unavailable). The sgRNA sequences were as follows (gRNA1-Forward: 5’ - CCAAGGAGACGGGCGCTCCA - 3’; gRNA1-Reverse: 5’ - TGGAGCGCCCGTCTCCTTGG - 3’; gRNA2-Forward: 5’ - GCCGGGGCAAGCTGCCCTAA - 3’; gRNA2-Reverse: 5’ - TTAGGGCAGCTTGCCCCGGC - 3’). The plasmid construct is shown in Supplementary Figure S1a and the expected cleavage sites in the FURIN gene are shown in Supplementary Figure S1b. The engineering of a single vector co-expressing spCas9-T2A-GFP and the two sgRNAs was performed as described previously [13,14]. Briefly, the plasmids pX330A-1x2 Cas9-2A-GFP and pX330S-2 were digested with BbsI restriction enzyme. The sgRNA1 was inserted into the pX330A-1x2 Cas9-2A-GFP plasmid, while the sgRNA2 was inserted into the pX330S-2 plasmid. The U6 cassette (U6-sgRNA2-gRNA scaffold) from pX330A-1x2 was then inserted down stream of U6 cassette (U6-sgRNA1-gRNA scaffold) pX330A-1x2 Cas9-2A-GFP vector using BsaI restriction enzyme by golden gate assembly procedure [13]. This results in the GFP being cloned to the C-terminal of CRISPR plasmid (PX330A Cas9-2A), such that GFP is only expressed when the full length plasmid had been successfully transcribed from its N- to C-terminal. Cells that express GFP are therefore expected to also contain the CRISPR Cas 9 gene product, as well as the two gRNAs transcribed (Supplementary Figure S2).
Cell culture, transfection and cell proliferation assays
THP1 and U937 monocytes were cultured in RPMI 1640 media containing L-glutamine with 10% FBS and 1% pen-strep. Cells were passaged every 3 days on average prior to transfection. Both U937 and THP1 cells were transfected for 24 hours without changing the media followed by screening for GFP expression via florescence microscopy (Supplementary Figure S3). A Bio-rad cell counter TC20 was used to perform all cell counting. Nuclear counterstaining was performed via DAPI. GFP positive cells were sorted into single cells into a 96 well plate based on DAPI negative (preferentially stained dead cells) and GFP positive staining, as determined by FACS. In all transfection assays, Hek293 cells were used as a positive control. Cell proliferation was monitored via Cell Titre Glo reagent (Promega), as per the manufacturer’s protocol, on 1000 cells/100 ul in 96-well plates and measurements taken at 24 hours intervals after seeding.
PCR screening for CRISPR
PCR screening of the transfected clones was performed via a set of internal and external primers designed around the FURIN gene. Primer sequences were as follows: (Internal Forward: 5’ 5’ - GACGGCTACACCAACAGTATC - 3’; Internal Reverse: 5’ - CACAGAGTGCCTTCTACCTAAC - 3’; External Forward: 5’ – AGTCTTCATCCTGCTTCTTCT - 3’; External Reverse: 5’ - GCCTGGATGGGACCATTATT - 3’). For internal primers (expected amplicon size 993 bp), the standard PCR protocol was used. For PCR involving external primers (expected amplicon size ~7.3kb) the GoTag Long PCR kit from Promega (cat: M4021) was used, and PCR was performed according to the manufacturer’s instructions.
Western blot
Proteins were separated in 10.5% polyacrylamide gels containing Sodium Dodecyl Sulfate (SDS) and transferred to PVDF membranes (Bio-Rad) using the semi-dry blotting system (Bio-Rad) and the Transblot Turbo 5X Transfer buffer (all from Bio-Rad), following the manufacturer's protocol. After transfer, the membrane was blocked with 5% milk (Bio-Rad) for 1 hour at room temperature. The membranes were incubated with a rabbit monoclonal anti-FURIN antibody (1:5000 dilution, Cell Signaling, Danvers, USA) overnight. The blots were washed three times with Tris-buffered saline with 0.1% Tween-20 (TBST) and then incubated with a goat anti-mouse polyclonal antibody (1:10,000 dilution) conjugated with horseradish peroxidase (HRP) for 1 hour at room temperature (BD). Subsequently, they were visualized using enhanced chemiluminescence (Pierce).
Lipid uptake assay
Lipid uptake by the macrophages was performed using Dil-Ox LDL (L34358) obtained from Life Technology, following the manufacturer's protocol. U937 cells were differentiated into macrophages using 100nM phorbol 12-myristate 13-acetate (PMA) for 72 hours. Subsequently, they were exposed to fluorescently-labeled oxidized lipids (Dil-Ox LDL) for 4-8 hours. The extent of cellular lipid uptake per cell (intensity density) was captured through fluorescent imaging using a Leica microscope (Wetzlar, Germany) and then quantified using ImageJ software [15] by comparing the lipid-associated red fluorescence to the fluorescence from nuclear staining with Hoechst 33342 stain.
Phagocytosis assay
Phagocytosis assays were performed using Phrodo green E.coli conjugate obtained from Life Technologies. Prior to the assay, U937 cells were differentiated into macrophages using 100nM PMA for 72 hours. Phagocytosis assays were conducted as per the manufacturer’s instructions. The extent of phagocytosis was quantified through the ImageJ software by comparing green fluorescence from engulfed E.coli to fluorescence from nuclear staining with Hoechst 33342 stain.
Migration assay
In the trans-well cell migration assay, we employed Transwell 6.5mm inserts with a 24-well plate configuration, containing 8.0μm polycarbonate membranes (Sigma, St. Louis, USA). The lower chamber was loaded with 600μL of media containing 10% FBS and 10nM CXCL12 (SDF-1α) as a chemoattractant sourced from Sigma, with an incubation duration of 8 hours. The upper chamber was seeded with 150,000 U937 monocytes suspended in 100μL of media lacking FBS. The number of migrated cells was quantified via the Cell Counting Kit-8 from Dojindo (Kumamoto, Japan).
Inflammatory gene expression via quantitative PCR assays
Total RNAs were extracted from WT, HZ and NZ FURIN clones of U937 cells via TRIzol (Life Technologies). Single strand cDNA was prepared by reverse-transcription using random hexamers via the IScript cDNA synthesis kit (Bio-rad). Gene expression was measured by quantitative RT-PCR (CFX96) using the ssoAdvUniversal Sybrgreen master mix (Bio-rad). Both the signals and relative gene expression were normalized to corresponding cyclophilin gene expression controls. Fold-change of gene expression was analysed via the 2(Delta Delta -C(T)) method [15]. Primer sequences for qPCR for all genes are listed in Supplementary Table ST1.
RNA sequencing analysis
Total RNA was prepared from triplicate cell cultures of WT, HZ and NZ FURIN clones of U937 cells as described above. RNA sequencing was performed at Novogene (Singapore) on the Illumina Novaseq 6000 platform with 150-base, paired end sequencing strategy.
The quality of RNA-seq reads was ascertained via FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The median sequencing depth was 52 million reads per sample, with a median per-base quality > 30 for all samples. Adapter trimming was performed via BBDuk (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbduk-guide/). Sequencing reads were mapped to the human reference genome (GRCh38) via STAR (2.7.7.a) (Dobin et al., 2013), with an average mapping rate of 74.5%. Raw count matrices of RNA sequencing data were obtained via the featureCounts [16] package in R and further processed for gene quantification and identification of differentially expressed genes using the limma package [17]. Gene counts were log2 transformed and normalized for sequencing depth via the trimmed means of M values (TMM) method [18]. The TMM-normalized data was used to identify potential sample outliers via Principal Components Analysis through the princomp package in R. Genes with at least one countper million (CPM) reads in three or more samples were retained for further analysis, resulting in 13,582 genes. The mean-variance relationship of gene-wise standard deviation to average log CPM gene signal was assessed via ‘voom’ [19] to generate precision weights for each observation, which were then used to generate empirical Bayes moderated t-statistics estimates for the identification of differentially expressed genes. To adjust for multiple testing, adjusted p-values were calculated via the false discovery rate (FDR) [20]. Genes with absolute fold-change≥ 2-fold, and adjusted p-value <0.01 were considered as differentially expressed. The full RNA sequencing data have been deposited to Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/info/) with series accession number GSEXXXX.
Pathway enrichment analysis
Pathway enrichment analysis on differentially expressed genes was conducted via gene-set enrichment analysis (GSEA) [21], using pathway databases from KEGG and Gene Ontology Biological Process (GOBP), obtained from MSigDB [22]. GSEA was run in the pre-ranked mode with genes ranked by their log fold-change between comparisons. Analysis was restricted to pathways containing between 15 and 250 genes, and the enrichment statistic was computed by the ‘classic’ method. Pathways with an adjusted p-value ≤ 0.25 were considered to be significantly regulated.
Self-organizing maps (SOM) analysis
SOM analysis was performed on a subset of differentially expressed genes, including genes with a minimum adjusted p-value <0.01, a maximum fold-change >2-fold and maximum average group expression (logCPM) >2 across HZ vs. WT and NZ vs. WT comparisons. A total of 1515 genes met these conditions. SOM analysis was carried out via the som package in R (https://cran.r-project.org/web/packages/som/index.html) by setting the neighbourhood parameter to ‘Gaussian’, along with a rectangular topology and linear initiation. A subset of the SOM gene clusters were further analyzed for biological pathway enrichment through the Bioplanet database [23] in the Enrichr enrichment analysis tool [24]. We also queried two additional databases in Enrichr (‘ENCODE_and_ChEA_Consensus_TFs_from_ChIP-X’, and ‘TF_perturbations_followed_by_expression’) to identify putative transcription factors whose regulation by cellular FURIN status could give rise to the gene expression patterns observed across the different SOM clusters.
Targeted proteomic analysis
Quantitative sandwich-based antibody arrays (RayBio® Human Cytokine Array C3) were used to quantify the levels of 42 cytokines secreted from WT, HZ and NZ FURIN clones of U937 cells, before and after stimulation with LPS (5 ug/ml, overnight stimulation). Briefly, 1 ml of media from each experiment were added into each array blot and incubated with capture antibodies overnight at 4oC. After washing, the arrays were incubated with a biotin-conjugated anti-cytokine antibody mix overnight at 4oC. HRP-conjugated streptavidin was added to bind with biotin from the detection antibodies for 2hrs at room temperature, and the chemiluminescent signal was detected using a Universal Hood III (Bio-rad). After 40 seconds of exposure, signal values for each spot were captured and quantified via ImageJ. For each blot, background counts were subtracted from the raw counts of each spot. Background-corrected counts were further normalized by a scaling factor derived by equalizing the average intensity of the positive control spots from each blot. The background-subtracted, scaled spot intensities were used for linear regression modeling to identify significant changes in protein levels as a function of LPS treatment and FURIN genotype, along with any interaction effects between them.
3. Results
3.1. Transfection efficiency and post-transfection viability of THP-1 and U937 monocytes3.1.1.
To compare transfection efficiencies, both THP-1 and U937 cells were transfected with 1.5ul of lipofectamine and 1ug of plasmid for 24 hours (Figure 1a). Although the overall transfection efficiency was very low, U937 cells demonstrated a better transfection efficiency (0.71%) compared to THP1 cells (0.54%), as determined by the number of GFP-positive cells post transfection (Figure 1b-c). The FACS-estimated mean GFP expression signal was also higher in U937 cells (7289.44 units) compared to THP1 cells (1237.56 units), suggesting greater expression of GFP (and therefore, the co-transcribed gRNAs) in U937 cells (Figure 1d). At 24 hours post-transfection, cell debris was observed in the transfected THP1, but not U937 cells. Cell viability assessment via Trypan blue staining showed a significant reduction in THP1 cell growth (Figure 1e) at 24- and 48-hrs post-transfection, compared to U937 cells.
3.2. Transfection optimization
To optimize the transfection efficiency for U937, several transfection conditions were tested. Optimization experiments were conducted with 120,000 cells seeded into each well of 24-well plates in a final volume of 500ul. The amount of lipofectamine 3000 was varied from 0.75ul to 1.5ul, whereas the transfectant plasmid was used at 1ug, 3ug or 5ug per transfection. The amount of ‘lipo-complex’ that was added into each well was also tested at 50ul and 75ul volumes. All experiments were conducted in triplicate and experiments were further duplicated on two different days. Of all the conditions tested, the largest transfection efficiency (0.71%) was observed with 1.5ul lipofectamine 3000 with 1ug plasmid and 50ul of lipo-complex (Figure 2). Increasing the plasmid amount to 3ug or 5ug always led to lower transfection, at all levels of lipofectamine or lipo-complex tested. We used a non-myeloid, human embryonic kidney cell-line (HEK-293T) as a positive control for transfection and observed it to have a transfection efficiency of 54% and cell viability of 86% at 24 hours post transfection. Although the transfection efficiency in U937 cells was much lower than that for the easily transfectable HEK-293T cells, the observations were reproducible and led to successful identification of HZ and NZ knockouts of FURIN gene in U937 cells.
3.3. PCR screening of CRISPR candidate clones
GFP-positive clones were seeded into 96-well plates at 1 cell/well and allowed to grow for 2 weeks before screening for gene deletion via PCR. RNA was prepared from candidate clones and subjected to PCR analysis by external and internal primer pairs as described earlier. A schematic of the primer locations with respect to the FURIN gene is shown in Figure 3a, and PCR results from 18 candidate clones are shown in Figure 3b. Of these, one clone was identified as a NZ knockout of FURIN (lane 10 from the left, lane 1 being molecular weight markers), and 5 clones were putative HZ clones (lanes 11,12,15,16, and 18), with one copy of the gene deleted. Further confirmation of the HZ and NZ clones was obtained via direct DNA sequencing of the PCR fragments (Supplementary Figure S4a-b).
3.4. Validation of NZ and HZ clones via qPCR and protein blotting
We quantified FURIN mRNA expression in WT, HZ and NZ U937 clones via quantitative PCR. Interestingly, the level of FURIN message was similar between WT and HZ clone, whereas no message could be detected in the NZ clone (Figure 3c). FURIN protein levels were assessed via Western blotting on cell lysates from WT, HZ and NZ clones using a goat anti-human anti-FURIN antibody (Figure 3d). FURIN protein expression was observed in the WT and HZ clones (lanes 1,2) but no signal was detected in the NZ clone (lane 3). We compared growth rates of WT, HZ and NZ FURIN clones and found that while the proliferation of WT and HZ clones were comparable, the total absence of FURIN in the NZ clones was associated with a reduced rate of growth. Thus, over 4 days, the growth rate of the NZ clone was less than 50% of the rates observed for WT and HZ clones, suggesting that a complete lack of FURIN is detrimental to cell growth (Figure 3e).
3.5. Loss of FURIN does not modulate macrophage phagocytosis
A comparative analysis of phagocytic capacity was performed on the WT, HZ or NZ FURIN clones (differentiated into macrophages). The results are shown visually in Figure 4a (green fluorescence from phagocytosed E.coli, blue fluorescence from Hoechst 33342 nuclear staining) and further quantified in Figure 4b. No significant differences were observed in the abilities of these clones to phagocytose fluorescently labeled E.coli, suggesting FURIN activity to be dispensable for this function (p>0.05 for all comparisons to WT).
3.6. Loss of FURIN reduces macrophage LDL accumulation
A comparison of the abilities of the WT, HZ and NZ FURIN clones to accumulate lipid was performed by first differentiating the clones into macrophages and then exposing them to fluorescently-labeled oxidized LDL particles for 4-8 hrs. The results suggest a reduced, but not statistically significant, lipid accumulation capacity in the HZ clone compared to WT (p>0.05). However, a significantly reduced lipid accumulation was observed in the NZ clone (p<0.05), suggesting a potential role of FURIN in regulating lipid accumulation in macrophages (Figure 5a-b).
3.7. Loss of FURIN reduces monocyte trans-migration
We quantified the number of U937 monocytes (WT, HZ or NZ for FURIN) undergoing transmembrane migration to a chemoattractant (CXCL12) through the transwell migration assay. Beginning with the same number of cells (150,000), the HZ FURIN clone showed a statistically non-significant reduction compared to the WT clone at all timepoints tested. However, there was a significant impairment of cell migration in the NZ FURIN clone when compared to WT U937 cells, beginning from 4 hours of migration (Figure 6). This suggests a clear role of FURIN in monocyte migration capacity that is mostly preserved even with one functional copy of the FURIN gene.
3.8. Loss of FURIN shows variable effects on inflammatory gene expression
WT, HZ and NZ U937 monocyte clones were stimulated with LPS for up to 6 hours, followed by quantitative PCR analysis of selected inflammatory gene expression in cell lysates. The changes in gene expression fold-changes varied widely, ranging from a less than 4-fold change for CD68 to over a 100-fold change observed for IL-6 (6 hour values), although for several genes noticeable changes in gene expression were only observed at the 6 hour timepoint (Figure 7). The pattern of gene expression also varied between genes and could be classified into three main classes. Genes such as CD68, COX1 and AMAC1 showed a similar time-dependent increase in gene expression in all samples (compared to expression levels in WT samples at baseline), independent of the gene-editing status of FURIN. Genes such as IL-6 and IL-8 displayed significantly higher changes in expression in WT samples compared to both types of FURIN-edited clones. Interestingly, for TGFB and LPL, the HZ clone showed maximal changes at 6 hour followed by the NZ and WT clones.
3.9. RNA sequencing analysis
3.9.1. Loss of FURIN impacts transcriptomic landscape
RNA sequencing analysis identified significant differences in transcriptome expression across the three clone types. Principal components analysis showed a clear separation between the WT, HZ and NZ FURIN monocyte clones along the first principal component, with the HZ clones clustering between the WT and NZ clones (Supplementary Figure S5). Nearly 49% of the variation in gene expression was captured in the first principal component, suggesting FURIN copy number to be a major driver in the transcriptomic landscape. A further separation of the HZ clones with respect to the WT and NZ clones was observed in the second principal component, accounting for nearly 25% of the residual gene expression variance. A total of 968 and 2019 genes were differentially expressed when comparing WT samples to HZ and NZ FURIN clones, respectively, based on a cutoff of at least 2-fold up- or down-regulation and an adjusted p-value of 0.01, suggesting a progressively increasing effect on gene transcription due to loss of 1 or both copies of FURIN. An overlap comparison of the differentially expressed genes showed 674 genes to be regulated in common in both HZ and NZ clones compared to WT, whereas 294 and 1345 genes were uniquely regulated in HZ and NZ compared to WT, respectively (Figure 8a). The extent of differential gene expression is depicted via volcano plots for WT vs HZ FURIN clones compared to WT cells (Figure 8b-c), with the top 20 most differentially expressed genes annotated for each comparison (sorted by adjusted p-values for differential expression). The per-sample expression of the top 50 most differentially expressed genes (selection based on adjusted p-value) are further depicted via heatmaps (Figure 8d,e), and show that whereas about an equal number of genes were up- and down-regulated between WT and HZ clones, a very large majority (47 out of 50) of the top differentially expressed genes were downregulated in NZ clones compared to WT cells (additional details of transcriptome analysis provided in Supplementary Table ST2).
3.9.2. Loss of FURIN affects biological pathways
In order to identify relevant biological mechanisms enriched among the differentially expressed genes, we performed gene-set enrichment analysis (GSEA) on KEGG and GOBP pathways (Supplementary Table ST3). For HZ vs WT comparison, 5 KEGG pathways and 1 GOBP pathway was significant at an adjusted p-value <0.2, whereas for NZ vs WT, 4 KEGG and 2 GOBP pathways were significant at the same statistical threshold (Figure 8f). Of these, the KEGG pathway termed ‘complement and coagulation cascades’ was significantly downregulated both in HZ and NZ clones compared to WT cells, as shown by their respective enrichment plots (Figure 8g,h). We further considered the genes contributing to the enrichment of this pathway and plotted their log fold-change values in HZ vs. WT and NZ vs. WT on a KEGG map of the pathway (Figure 8i,j). From the figure it is clear that genes in both the “intrinsic pathway” arm of the coagulation cascade, and “alternative pathway” arm of the complement cascade were differentially regulated in both comparisons.
3.9.3. Loss of FURIN leads to different gene expression patterns
In order to identify patterns of gene expression as a function of the copy number of FURIN, we performed self-organizing maps (SOM) analysis on a subset of genes (minimum adjusted p-value <=0.01, maximum absolute fold-change >=2.0 and maximum average logCPM >=2 across the 3 groups) and identified 8 distinct patterns of gene expression clusters (Figure 9a). Individual gene-cluster associations are provided in Supplementary Table ST4. We performed pathway enrichment analysis via Enrichr on the SOM clusters and identified enrichment of several pathways (adjusted p-value <=0.1) among the different patterns (Figure 9 b-f). Pathways such as ‘complement and coagulation cascade’ and ‘extracellular matrix organization’ were enriched in clusters 2-3 where gene expression was progressively decreased with reduction or loss of FURIN, whereas pathways related to TGF-1 beta signaling, IL-1 signaling and chromosome maintenance were enriched for genes that had increased expression upon reduction of FURIN (clusters 6-8). The full list of pathways impacted by loss of FURIN is available in Supplementary Table S5.
3.10. Loss of FURIN variably modulates cytokine secretion
We investigated changes in selected cytokine secretion from U937 cells as a function of FURIN copy-number, either in basal or LPS-stimulated conditions. Figure 10 shows the changes in secretion levels for each cytokine under basal and LPS-stimuation in each of the WT, HZ and NZ clones. For each cytokine, the statistical significance of secretion differences was estimated via linear regression models. Compared to NZ, the top 5 cytokines showing a statistically significant difference of secretion in HZ under baseline and LPS stimulated conditions included ANG (reduced in both baseline and LPS), TNF alpha, MDC and EGF (increased in both baseline and LPS), and GMCSF (increased in baseline and reduced under LPS). Similarly, the top 5 cytokines showing significant differences in secretion between WT and NZ cells included angiogenin, SDF1, IL6 (reduced in both baseline and LPS), TARC and SCF (reduced in baseline and increased in LPS). The top 5 cytokines affected by LPS stimulation independent of FURIN genotype included TARC, TGFB, Leptin (reduced in LPS), GMCSF (reduced in WT and HZ, increased in NZ), and SCF (increased in WT, reduced in HZ and NZ). In addition to these main effects, interaction effects were also observed for some cytokines. For example, the top 5 statistically significant HZ:LPS interactions were observed for GMCSF, GCSF, PDGFBB, ENA78, and TGFB, whereas the top 5 interactions for WT:LPS were observed for GMCSF, TARC, SCF, IL2 and ENA78. Overall, we observed heterogeneity in cytokine secretion in response to FURIN copy number. The full results from linear modeling are provided in Supplementary Table ST6. We further compared the cytokines on the array to a predicted list of FURIN substrates (PMID 23335997) and identified overlap for only 3 cytokines (PDGFB, TGFB and IGF1). For both TGFB and IGF1, loss of FURIN (HZ or NZ) resulted in increased cytokine secretion under basal conditions that was suppressed by LPS stimulation. For the other cytokines, the observed effects are presumed to be secondary, not related to their proteolytic cleavage by FURIN (although the possibility of them being as yet unknown FURIN substrates cannot be ruled out).
4. Discussion
Through proteolytic cleavage and activation of diverse protein substrates in the secretory pathway, intracellular FURIN acts as a central regulator of several biological functions. Cell-surface expressed FURIN also cleaves a diverse array of pathogenic substrates including bacterial toxins and viral fusion peptides [25,26] thereby providing protection from infections. Conversely, FURIN activity can also aid in viral infection, the most notable recent example being that of coronavirus SARS-Cov2 [27]. Together, these make FURIN an attractive target for therapeutic intervention in multiple disorders [1,28,29]. Previously, we identified FURIN as a coronary artery disease-associated gene candidate based on genetic association analysis of the CardiOgram cohort [3,30]. We further demonstrated that systemic inhibition of FURIN in mouse models of atherogenesis resulted in lower atherosclerotic lesion area and reduction in severe lesions, whereas exogenous introduction of FURIN protein led to increased intimal plaque thickness [4]. Additionally, we observed reduced monocyte migration and reduced inflammatory and cytokine gene expression in macrophages upon FURIN inhibition. These results prompted us to further investigate the effects of FURIN inhibition in the U937 human monocytic cell line, through a combination of cell biology, proteomic and genomic approaches. Through an optimised lipotransfection-based protocol, we were able to successfully perform CRISPR-driven gene editing in U937cells and obtained cells containing either heterozygous or homozygous knockouts of the FURIN gene.
An examination of several cellular functions in U937 cells with wild-type (WT), hemizygous (HZ) or nullizygous (NZ) FURIN status revealed several interesting observations. Compared to WT cells, cell-growth rate was similar in HZ but reduced in NZ cells. Cell viability was also similar between WT and HZ cells but reduced in NZ cells, suggesting a role of FURIN in macrophage cell proliferation and survival. However, the reduction or loss of FURIN did not affect the phagocytic abilities of the cells, suggesting that inhibition of FURIN spares this innate immunity function of macrophages related to the ingestion and degradation of bacteria, dead cells, debris, tumor cells, and foreign materials [31].
One key function of monocytes/macrophages related to atherosclerosis involves the scavenging of oxidized lipids and transformation into foam cells, followed by the production of proinflammatory mediators [32]. We therefore tested the effects of FURIN gene dosage on oxidized lipid accumulation by U937 cells and observed that the accumulation of scavenged lipids was reduced in HZ and NZ cells (although reduction in HZ was not statistically significant). Another important early event in atherosclerosis is the increased recruitment of monocyte-derived cells into the subendothelial space where they subsequently differentiate into macrophages and contribute to the dynamic progression of atherosclerotic plaques [33]. We tested the ability of WT, HZ and NZ U937 clones for trans-well migration and observed consistently reduced migration for both HZ and NZ clones at all timepoints tested (although the reduction in HZ was not statistically significant). Selected inflammatory gene expression analysis via qPCR of U937 clones identified a heterogeneity of gene regulation in response to LPS stimulation, with genes such as IL6 and IL8 showing marked reduction in transcription in response to loss of one of both copies of FURIN, whereas IL23, LPL and TGFB displayed increased transcription in both HZ and NZ clones compared to WT.
A broader global transcriptome profiling of WT, HZ and NZ clones identified clear differences in gene transcription as a function of FURIN copy number, with progressively increasing effects on gene transcription due to loss of 1 or both copies of FURIN gene. Pathway enrichment analysis identified biological mechanisms related to complement and coagulation being significantly downregulated in both HZ and NZ clones compared to WT, suggestive of alterations in key macrophage functions involving innate immunity and hemostasis [34]. Additionally, an analysis of gene expression patterns based on self-organizing maps identified pathways related to TGF-1 beta signaling, IL-1 signaling and chromosome maintenance to be enriched among genes with higher expression in HZ and NZ compared to WT clones. Finally, a focused analysis of the secreted proteome revealed differing patterns of cytokine release from the WT, HZ and NZ U937 clones under basal and LPS-stimulated conditions. Notably, FURIN depletion resulted in an increase in the secretion of IGF-1, a key factor associated with inflammation resolution in macrophages [35] and directly implicated in reducing atherosclerotic burden in mice [36,37]. Another finding of potential importance is the upregulation of ANG (Angiogenin) in FURIN depleted cells, given that endothelial ANG has been recently identified as an anti-atherogenic factor [38].
In summary, we had previously showed that FURIN inhibition decreased atherosclerotic lesions in two independent mouse models. We show here that some pathways by which FURIN inhibition may confer atheroprotection is via reducing monocyte migration, macrophage proliferation and foam cell formation. and by reducing specific atherogenic inflammatory cytokine production. We further show global scale transcriptomic and transcriptionally-dictated pathway alterations consequent to FURIN inactivation. Finally, proteomic analysis revealed alterations in the secretion of cytokines, some of which are directly implicated in atherogenic processes. These results provide further insights into function of FURIN in myeloid cells and suggest that monocyte/macrophage-specific targeted FURIN inhibition may be a useful therapeutic strategy for atherosclerosis and related disorders.
Supplementary Materials
The following supporting information can be downloaded at: https://github.com/sg3451/FURIN_U937_study/tree/main: Figure S1a: Design of plasmid vector used for CRISPR studies; Figure S1b: Design of guide RNA sequences for targeting FURIN via CRISPR; Figure S2: Schematic of plasmid construction expressing GFP and two guide RNAs; Figure S3: FACS analysis of U937 cell transfections; Figure S4a: Design of sequencing primers to verify FURIN gene status in U937 cells; Figure S4b: Results from DNA sequencing of U937 clones subjected to CRISPR-mediated FURIN gene modification; Figure S5: Principal components analysis (PCA) of gene expression in WT, HZ and NZ U937 clones; ST1 – Sequences of PCR primers used for quantifying candidate gene expression; ST2 - Differentialy gene expression analysis using limma; ST3 – Gene Set Enrichment Analysis (GSEA) results; ST4 – Results from Self Organizing Maps (SOM) analysis; ST5 – Pathway enrichment analysis of SOM clusters; ST6 – Cytokine secretion data from proteomics study.
Author Contributions
Conceptualization, R.C and S.G.; methodology, R.C.; formal analysis, R.C., L.W. and S.G..; investigation, R.C.; resources, S.G.; data curation, L.W. and S.G.; writing—original draft preparation, R.C. and S.G.; writing—review and editing, R.S., R.C. and S.G.; supervision, S.G.; project administration, S.G.; funding acquisition, S.G. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in Github (https://github.com/sg3451/FURIN_U937_study/tree/main). Gene expression data has been submitted to GEO ().
Acknowledgments
We thank Dr. Tay Hwee Goon for help with FACS analysis, and Dr. Chutima Rattanasopa for some cell-based studies. We also thank Professor Shang Li and Dr Amrita Singha for assistance with plasmid construction and guide RNA design.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Couture F, Kwiatkowska A, Dory YL, Day R. Therapeutic uses of furin and its inhibitors: A patent review. Expert opinion on therapeutic patents. 2015;25(4):379-96. Epub 20150107. [CrossRef] [PubMed]
- Ren K, Jiang T, Zheng XL, Zhao GJ. Proprotein convertase furin/PCSK3 and atherosclerosis: New insights and potential therapeutic targets. Atherosclerosis. 2017;262:163-70. Epub 20170407. [CrossRef] [PubMed]
- Ghosh S, Vivar J, Nelson CP, Willenborg C, Segre AV, Makinen VP, et al. Systems Genetics Analysis of Genome-Wide Association Study Reveals Novel Associations Between Key Biological Processes and Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2015;35(7):1712-22. Epub 20150514. [CrossRef] [PubMed] [PubMed Central]
- Yakala GK, Cabrera-Fuentes HA, Crespo-Avilan GE, Rattanasopa C, Burlacu A, George BL, et al. FURIN Inhibition Reduces Vascular Remodeling and Atherosclerotic Lesion Progression in Mice. Arterioscler Thromb Vasc Biol. 2019;39(3):387-401. [CrossRef] [PubMed] [PubMed Central]
- Stawowy P, Kallisch H, Borges Pereira Stawowy N, Stibenz D, Veinot JP, Grafe M, et al. Immunohistochemical localization of subtilisin/kexin-like proprotein convertases in human atherosclerosis. Virchows Arch. 2005;446(4):351-9. Epub 20050309. [CrossRef] [PubMed]
- Do PT, Nguyen CX, Bui HT, Tran LTN, Stacey G, Gillman JD, et al. Demonstration of highly efficient dual gRNA CRISPR/Cas9 editing of the homeologous GmFAD2-1A and GmFAD2-1B genes to yield a high oleic, low linoleic and alpha-linolenic acid phenotype in soybean. BMC Plant Biol. 2019;19(1):311. Epub 20190715. [CrossRef] [PubMed] [PubMed Central]
- Pathak B, Zhao S, Manoharan M, Srivastava V. Dual-targeting by CRISPR/Cas9 leads to efficient point mutagenesis but only rare targeted deletions in the rice genome. 3 Biotech. 2019;9(4):158. Epub 20190328. [CrossRef] [PubMed] [PubMed Central]
- 8. Adikusuma F, Pfitzner C, Thomas PQ. Versatile single-step-assembly CRISPR/Cas9 vectors for dual gRNA expression. PLoS ONE. 2017;12(12):e0187236. Epub 20171206. [CrossRef] [PubMed] [PubMed Central]
- Tao Y, Hou X, Zuo F, Li X, Pang Y, Jiang G. Application of nanoparticle-based siRNA and CRISPR/Cas9 delivery systems in gene-targeted therapy. Nanomedicine (Lond). 2019;14(5):511-4. Epub 2019/02/27. [CrossRef] [PubMed]
- Chanput W, Peters V, Wichers H. THP-1 and U937 Cells. In: Verhoeckx K, Cotter P, López-Expósito I, Kleiveland C, Lea T, Mackie A, et al., editors. The Impact of Food Bioactives on Health: In vitro and ex vivo models. Cham: Springer International Publishing; 2015. p. 147-59.
- Zhang X, Edwards JP, Mosser DM. The expression of exogenous genes in macrophages: Obstacles and opportunities. Methods Mol Biol. 2009;531:123-43. [CrossRef] [PubMed] [PubMed Central]
- Burke B, Sumner S, Maitland N, Lewis CE. Macrophages in gene therapy: Cellular delivery vehicles and in vivo targets. J Leukoc Biol. 2002;72(3):417-28. [CrossRef] [PubMed]
- Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Scientific reports. 2014;4:5400. Epub 20140623. [CrossRef] [PubMed] [PubMed Central]
- Yan T, Ooi WF, Qamra A, Cheung A, Ma D, Sundaram GM, et al. HoxC5 and miR-615-3p target newly evolved genomic regions to repress hTERT and inhibit tumorigenesis. Nature communications. 2018;9(1):100. Epub 2018/01/10. [CrossRef] [PubMed] [PubMed Central]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402-8. [CrossRef] [PubMed]
- Liao Y, Smyth GK, Shi W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923-30. Epub 20131113. [CrossRef] [PubMed]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. Epub 20150120. [CrossRef] [PubMed] [PubMed Central]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. Epub 20100302. [CrossRef] [PubMed] [PubMed Central]
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. Epub 20140203. [CrossRef] [PubMed] [PubMed Central]
- Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19(3):368-75. [CrossRef] [PubMed]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-50. Epub 20050930. [CrossRef] [PubMed] [PubMed Central]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739-40. Epub 20110505. [CrossRef] [PubMed] [PubMed Central]
- Huang R, Grishagin I, Wang Y, Zhao T, Greene J, Obenauer JC, et al. The NCATS BioPlanet - An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front Pharmacol. 2019;10:445. Epub 20190426. [CrossRef] [PubMed] [PubMed Central]
- Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc. 2021;1(3):e90. [CrossRef] [PubMed] [PubMed Central]
- Molloy SS, Anderson ED, Jean F, Thomas G. Bi-cycling the furin pathway: From TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 1999;9(1):28-35. [CrossRef] [PubMed]
- Tian S, Huang Q, Fang Y, Wu J. FurinDB: A database of 20-residue furin cleavage site motifs, substrates and their associated drugs. International journal of molecular sciences. 2011;12(2):1060-5. Epub 20110208. [CrossRef] [PubMed] [PubMed Central]
- Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021;591(7849):293-9. Epub 20210125. [CrossRef] [PubMed] [PubMed Central]
- Wu C, Zheng M, Yang Y, Gu X, Yang K, Li M, et al. Furin: A Potential Therapeutic Target for COVID-19. iScience. 2020;23(10):101642. Epub 20201005. [CrossRef] [PubMed] [PubMed Central]
- Douglas LEJ, Reihill JA, Montgomery BM, Martin SL. Furin as a therapeutic target in cystic fibrosis airways disease. Eur Respir Rev. 2023;32(168). Epub 20230503. [CrossRef] [PubMed] [PubMed Central]
- Preuss M, Konig IR, Thompson JR, Erdmann J, Absher D, Assimes TL, et al. Design of the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet. 2010;3(5):475-83. Epub 20101005. [CrossRef] [PubMed] [PubMed Central]
- Hirayama D, Iida T, Nakase H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. International journal of molecular sciences. 2017;19(1). Epub 20171229. [CrossRef] [PubMed] [PubMed Central]
- 32. Gruber EJ, Aygun AY, Leifer CA. Macrophage uptake of oxidized and acetylated low-density lipoproteins and generation of reactive oxygen species are regulated by linear stiffness of the growth surface. PLoS ONE, 0260. [CrossRef] [PubMed] [PubMed Central]
- Lin P, Ji HH, Li YJ, Guo SD. Macrophage Plasticity and Atherosclerosis Therapy. Front Mol Biosci. 2021;8:679797. Epub 20210507. [CrossRef] [PubMed] [PubMed Central]
- Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71-9. [CrossRef] [PubMed] [PubMed Central]
- Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, et al. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol Ther. 2015;23(7):1189-200. Epub 20150421. [CrossRef] [PubMed] [PubMed Central]
- Snarski P, Sukhanov S, Yoshida T, Higashi Y, Danchuk S, Chandrasekar B, et al. Macrophage-Specific IGF-1 Overexpression Reduces CXCL12 Chemokine Levels and Suppresses Atherosclerotic Burden in Apoe-Deficient Mice. Arterioscler Thromb Vasc Biol. 2022;42(2):113-26. Epub 20211202. [CrossRef] [PubMed] [PubMed Central]
- Higashi Y, Sukhanov S, Shai SY, Danchuk S, Tang R, Snarski P, et al. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E-Deficient Mice. Circulation. 2016;133(23):2263-78. Epub 20160506. [CrossRef] [PubMed] [PubMed Central]
- Su E, Yu P, Zhang B, Zhang A, Xie S, Zhang C, et al. Endothelial Intracellular ANG (Angiogenin) Protects Against Atherosclerosis by Decreasing Endoplasmic Reticulum Stress. Arterioscler Thromb Vasc Biol. 2022;42(3):305-25. Epub 20220120. [CrossRef] [PubMed]
Figure 1.
Comparison of transfection efficiency in monocytic and non-monocytic cell-lines. (a) GFP fluourescence indicating extent of transfection of GFP-containing plasmid vector in Hek293T, THP1 and U937 cells. The Hek293T cells were used as a positive transfection control and display robust plasmid transfection, whereas transfection is detectable but much lower in the monocytic cell-lines. (b) Quantification of transfection efficiencies (percent of cells transfected) in Hek293T, THP1 and U937 cells. (c) Comparison of fluorescence signals (copies of GFP per cell) between Hed293T, THP1 and U937 cells, showing greater GFP expression in U937 compared to THP1 cells. (d) Effect of transfection on cell proliferation. Untransfected (U937-C, THP1-C) and transfected (U937-T, THP-T) cells were monitored for cell proliferation over a period of 48 hrs post-transfection. (e) Effect of transfection on viability of U937 and THP1 cells. Untransfected (U937-C, THP1-C) and transfected (U937-T, THP-T) cells were followed for 48hrs post-transfection and the number of viable cells were quantified via Trypan blue staining.
Figure 1.
Comparison of transfection efficiency in monocytic and non-monocytic cell-lines. (a) GFP fluourescence indicating extent of transfection of GFP-containing plasmid vector in Hek293T, THP1 and U937 cells. The Hek293T cells were used as a positive transfection control and display robust plasmid transfection, whereas transfection is detectable but much lower in the monocytic cell-lines. (b) Quantification of transfection efficiencies (percent of cells transfected) in Hek293T, THP1 and U937 cells. (c) Comparison of fluorescence signals (copies of GFP per cell) between Hed293T, THP1 and U937 cells, showing greater GFP expression in U937 compared to THP1 cells. (d) Effect of transfection on cell proliferation. Untransfected (U937-C, THP1-C) and transfected (U937-T, THP-T) cells were monitored for cell proliferation over a period of 48 hrs post-transfection. (e) Effect of transfection on viability of U937 and THP1 cells. Untransfected (U937-C, THP1-C) and transfected (U937-T, THP-T) cells were followed for 48hrs post-transfection and the number of viable cells were quantified via Trypan blue staining.
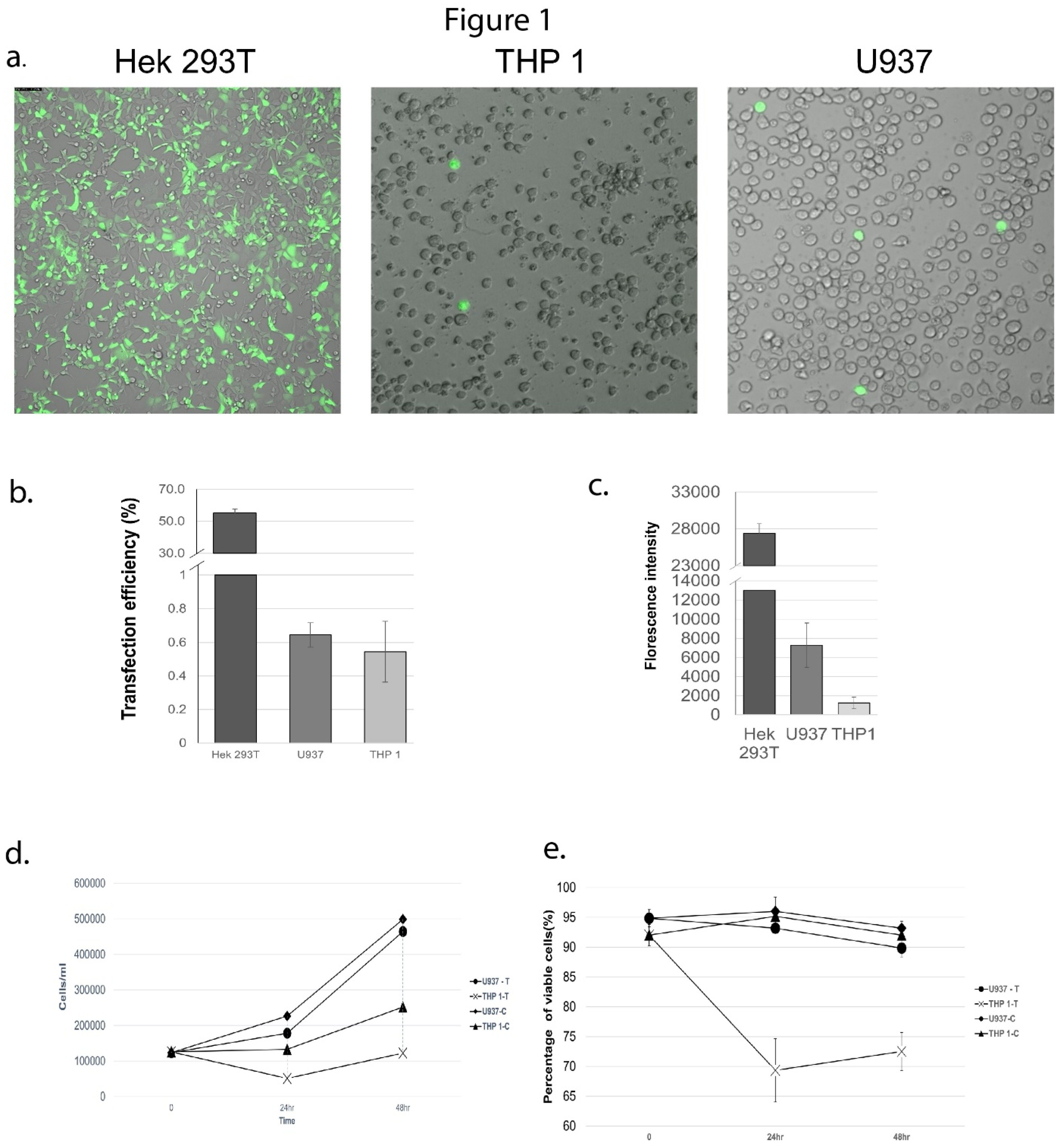
Figure 2.
Optimization of transfection in U937 cells. Different combinations of the transfection agent (Lipofectamine), transfection plasmid and the lipo-complex were tested and the efficiency of transfection monitored via GFP expression in U937 cells. The x-axis describes each of the 12 conditons tested as noted in the table below the plot. Transfection efficiency (% cells transfected) is quantified on the y-axis. All experiments were conducted in triplicate and further duplicated on two different days.
Figure 2.
Optimization of transfection in U937 cells. Different combinations of the transfection agent (Lipofectamine), transfection plasmid and the lipo-complex were tested and the efficiency of transfection monitored via GFP expression in U937 cells. The x-axis describes each of the 12 conditons tested as noted in the table below the plot. Transfection efficiency (% cells transfected) is quantified on the y-axis. All experiments were conducted in triplicate and further duplicated on two different days.
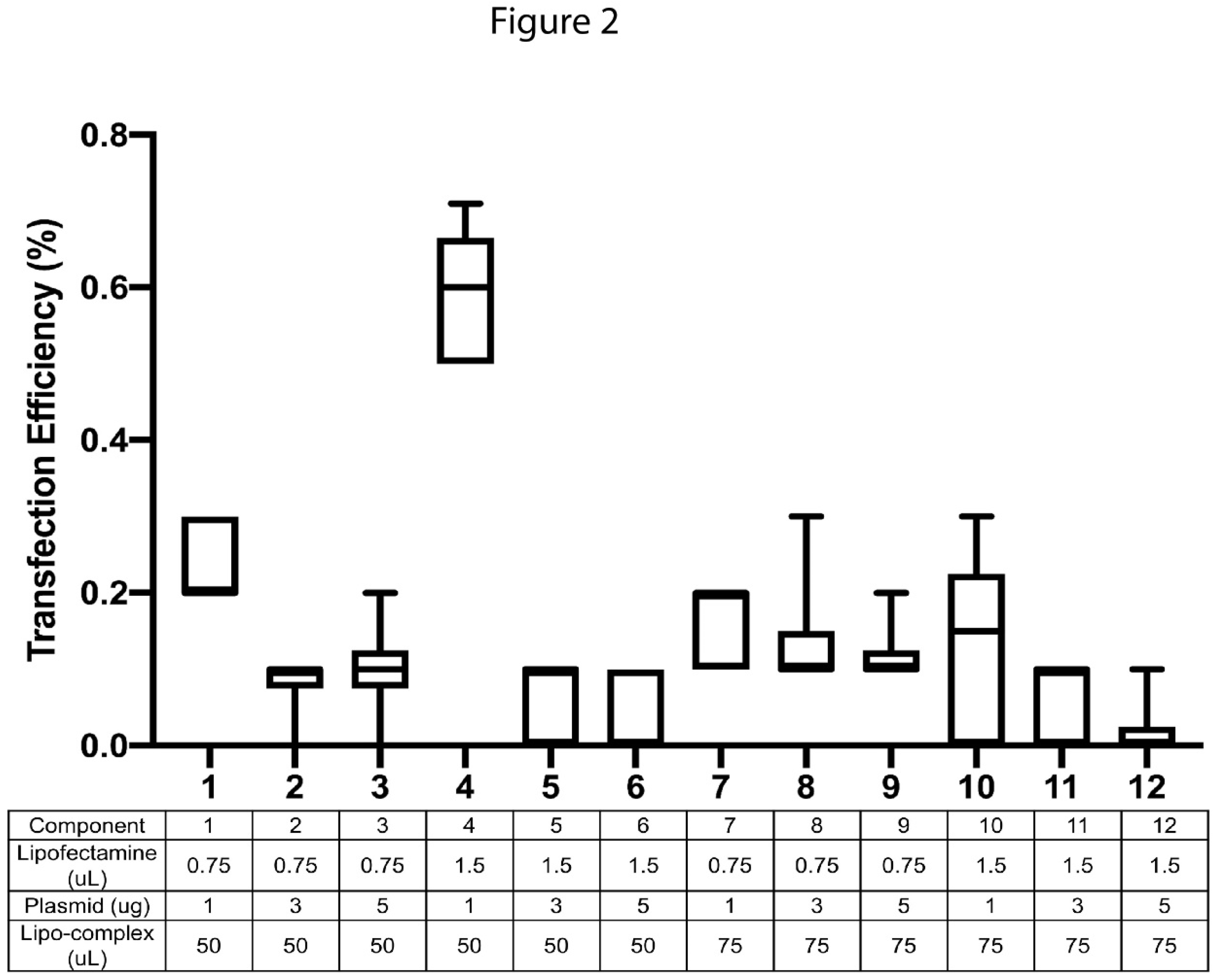
Figure 3.
Identification of CRISPR-mediated U937 clones hemizygous (HZ) and nullizygous (NZ) for FURIN, and detection of FURIN transcript and protein. (a) Schematic of the internal primer (internal to FURIN coding region) and external primers (external to and flanking the FURIN gene) used to test for the presence of CRISPR-mediated FURIN gene deletion via PCR. The expected amplicon lengths in the presence or absence of FURIN gene cleavage are indicated. (b) Results from PCR analysis of a selection of 18 CRISPR-edited U937 clones. The top panel depicts PCR amplicons generated by the internal primer and the bottom panel depicts amplicons generated by the external primer. Molecular weight markers are shown in lane 1 of each electropherogram. Lane 9 in both images correspond to a homozygous FURIN deletion (NZ), whereas lanes 10,11,14,15 and 17 refer to potential heterozygotes with one copy of FURIN gene intact (HZ). Other lanes refer to clones with wild-type FURIN status. The expected amplicon sizes for wild-type (WT), hemizygous (HZ) and nullizygous (NZ) FURIN statuses are shown in the table below. (c) Quantification of FURIN messenger RNA in WT, HZ and NZ cell clones via qPCR. Results are depicted as fold-change with respect to the clone containing wild-type FURIN, and are calculated by the delta-delta Ct method. (d) Western blot analysis of FURIN protein expression in WT, HZ and NZ clones. For each blot, molecular weight markers are indicated on the leftmost lane. The left panel shows FURIN expression in WT and HZ clones, along with that of beta-ACTIN as loading control. The right panel quantifies FURIN expression in WT and NZ clones along with beta-ACTIN controls (no FURIN expression observed in NZ sample). (e) Quantification of cell proliferation in WT (gray circle), HZ (gray diamond) and NZ (gray triangle) U937 clones over 96 hours post seeding.
Figure 3.
Identification of CRISPR-mediated U937 clones hemizygous (HZ) and nullizygous (NZ) for FURIN, and detection of FURIN transcript and protein. (a) Schematic of the internal primer (internal to FURIN coding region) and external primers (external to and flanking the FURIN gene) used to test for the presence of CRISPR-mediated FURIN gene deletion via PCR. The expected amplicon lengths in the presence or absence of FURIN gene cleavage are indicated. (b) Results from PCR analysis of a selection of 18 CRISPR-edited U937 clones. The top panel depicts PCR amplicons generated by the internal primer and the bottom panel depicts amplicons generated by the external primer. Molecular weight markers are shown in lane 1 of each electropherogram. Lane 9 in both images correspond to a homozygous FURIN deletion (NZ), whereas lanes 10,11,14,15 and 17 refer to potential heterozygotes with one copy of FURIN gene intact (HZ). Other lanes refer to clones with wild-type FURIN status. The expected amplicon sizes for wild-type (WT), hemizygous (HZ) and nullizygous (NZ) FURIN statuses are shown in the table below. (c) Quantification of FURIN messenger RNA in WT, HZ and NZ cell clones via qPCR. Results are depicted as fold-change with respect to the clone containing wild-type FURIN, and are calculated by the delta-delta Ct method. (d) Western blot analysis of FURIN protein expression in WT, HZ and NZ clones. For each blot, molecular weight markers are indicated on the leftmost lane. The left panel shows FURIN expression in WT and HZ clones, along with that of beta-ACTIN as loading control. The right panel quantifies FURIN expression in WT and NZ clones along with beta-ACTIN controls (no FURIN expression observed in NZ sample). (e) Quantification of cell proliferation in WT (gray circle), HZ (gray diamond) and NZ (gray triangle) U937 clones over 96 hours post seeding.
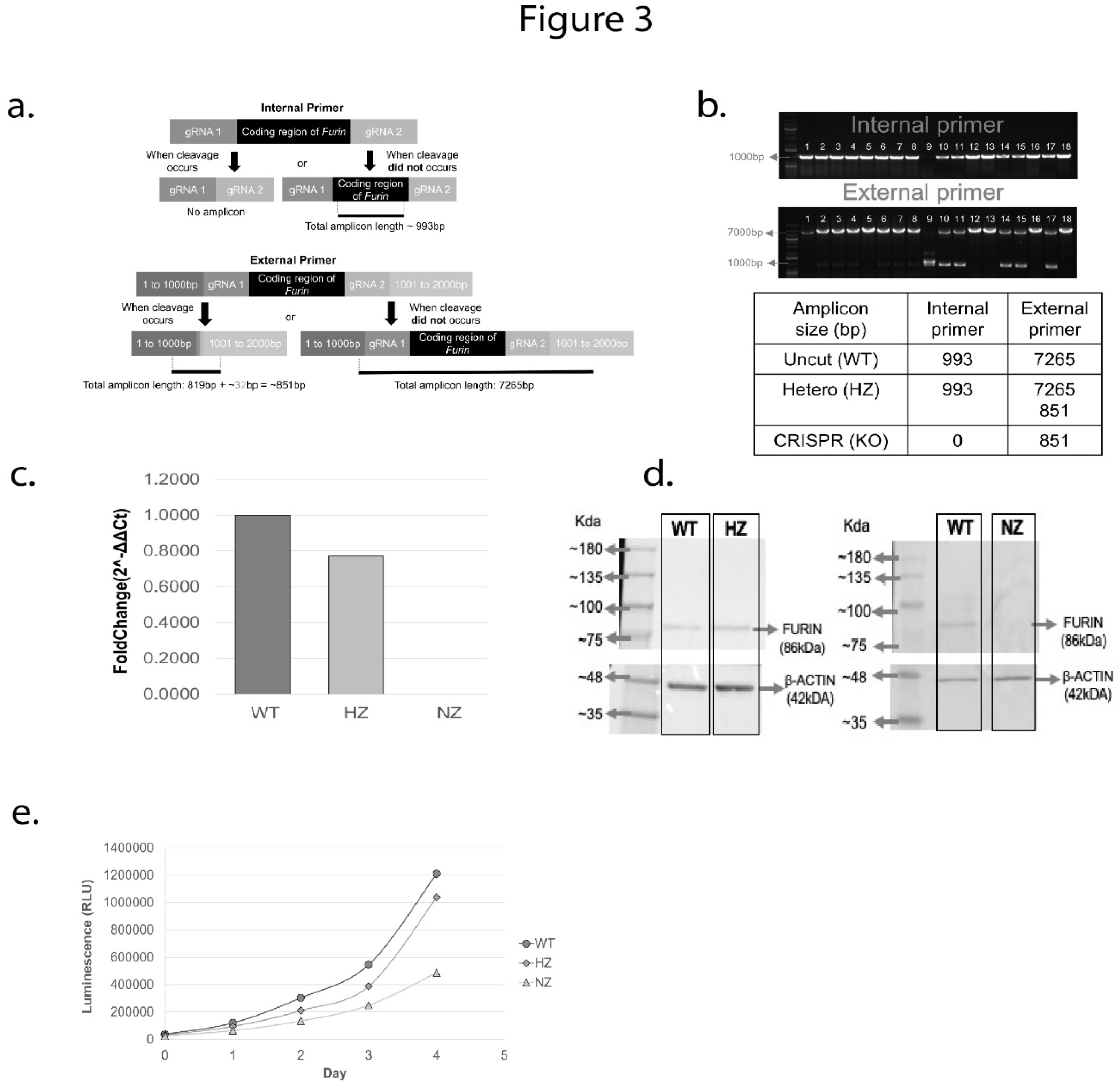
Figure 4.
Effectf of FURIN gene status on phagocytic activity of U937 cells. (a) WT, HZ and NZ U937 clones were differentiated into macrophages and tested for engulfment of pHrodo green E.coli bioparticles conjugate. Nuclear staining was performed via Hoechst 33342. (b) Quantification of phagocytosis via intensity density ratio plot (pHrodo fluorescence / nuclear fluorescence). Experiments were conducted in triplicate.
Figure 4.
Effectf of FURIN gene status on phagocytic activity of U937 cells. (a) WT, HZ and NZ U937 clones were differentiated into macrophages and tested for engulfment of pHrodo green E.coli bioparticles conjugate. Nuclear staining was performed via Hoechst 33342. (b) Quantification of phagocytosis via intensity density ratio plot (pHrodo fluorescence / nuclear fluorescence). Experiments were conducted in triplicate.
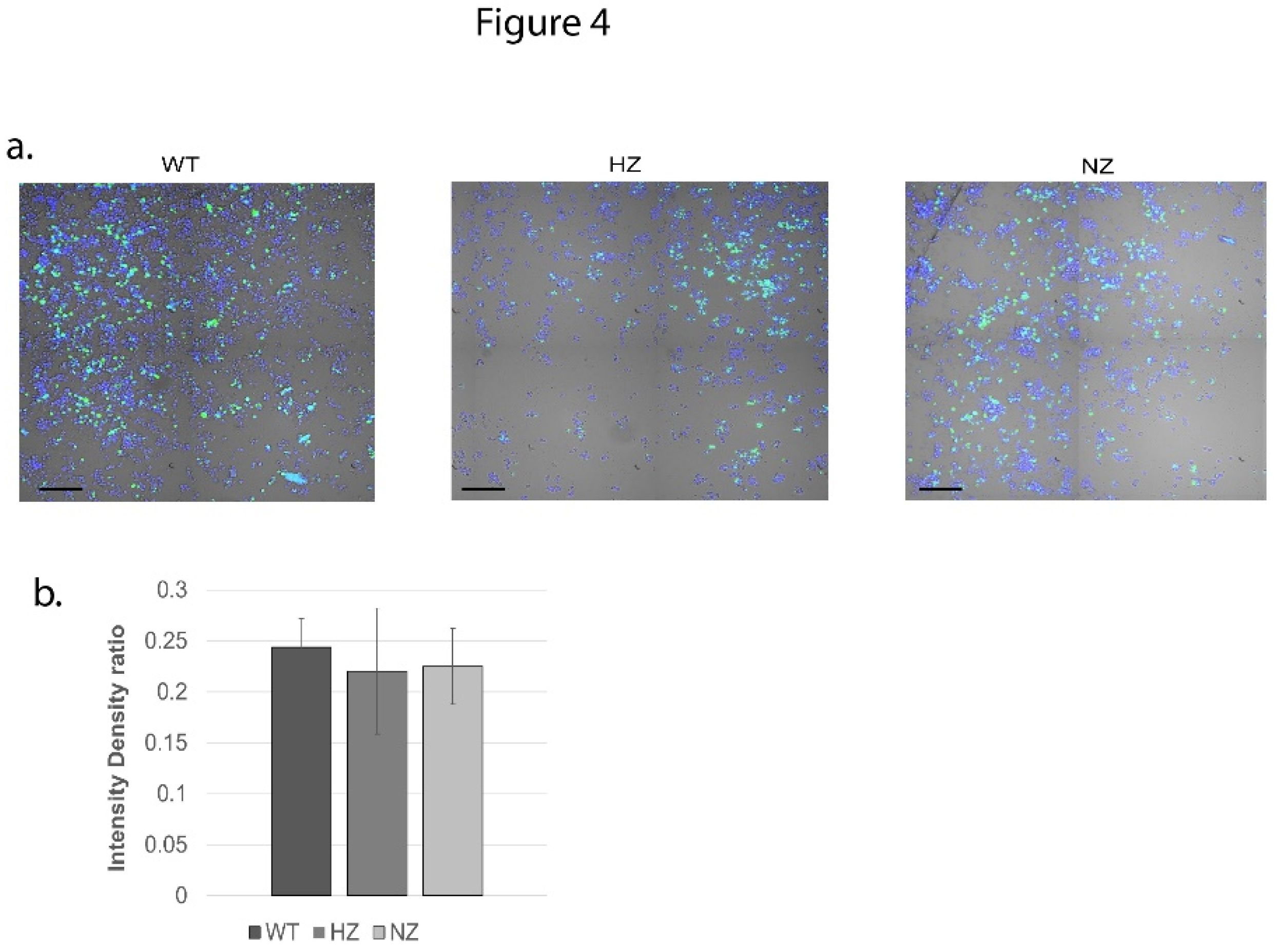
Figure 5.
Uptake of oxidized lipid as a function of FURIN gene status. (a) WT, HZ and NZ U937 clones were differentiated into macrophages and exposed to fluorescently labeled oxidized LDL particles for 4-8 hrs. Nuclear counterstaining was performed via Hoeschst 33342. (b) Quantification of oxidized lipid uptake via intensity density ratio plot (pHrodo fluorescence / nuclear fluorescence). Experiments were conducted in triplicate. .
Figure 5.
Uptake of oxidized lipid as a function of FURIN gene status. (a) WT, HZ and NZ U937 clones were differentiated into macrophages and exposed to fluorescently labeled oxidized LDL particles for 4-8 hrs. Nuclear counterstaining was performed via Hoeschst 33342. (b) Quantification of oxidized lipid uptake via intensity density ratio plot (pHrodo fluorescence / nuclear fluorescence). Experiments were conducted in triplicate. .

Figure 6.
Effect of FURIN gene status on chemoattractant induced migration. WT (black square), HZ (gray circle) and NZ (gray triangle) U937 monocyte clones were seeded in a transwell chamber and subjected to CXCL12 induced migration over 8 hrs. The number of cells migrating to the lower chamber of the transwell was quantified via Cell Counting Kit 8. Experiments were conducted in triplicate.
Figure 6.
Effect of FURIN gene status on chemoattractant induced migration. WT (black square), HZ (gray circle) and NZ (gray triangle) U937 monocyte clones were seeded in a transwell chamber and subjected to CXCL12 induced migration over 8 hrs. The number of cells migrating to the lower chamber of the transwell was quantified via Cell Counting Kit 8. Experiments were conducted in triplicate.

Figure 7.
Effect of FURIN gene status on inflammation-related gene expression. WT (gray circle), HZ (gray diamond) and NZ (gray triangle) U937 monocyte clones were assayed for the expression of selected inflammation-related genes following LPS-stimulation, via qPCR. All results are reported as the fold-change in gene expression with respect to the basal 0hr expression. Experiments were conducted in triplicate and fold-change in gene expression was calculated via the delta-delta Ct method. HZ, heterozygous ; NZ, homozygous knockout (nullizygous). .
Figure 7.
Effect of FURIN gene status on inflammation-related gene expression. WT (gray circle), HZ (gray diamond) and NZ (gray triangle) U937 monocyte clones were assayed for the expression of selected inflammation-related genes following LPS-stimulation, via qPCR. All results are reported as the fold-change in gene expression with respect to the basal 0hr expression. Experiments were conducted in triplicate and fold-change in gene expression was calculated via the delta-delta Ct method. HZ, heterozygous ; NZ, homozygous knockout (nullizygous). .
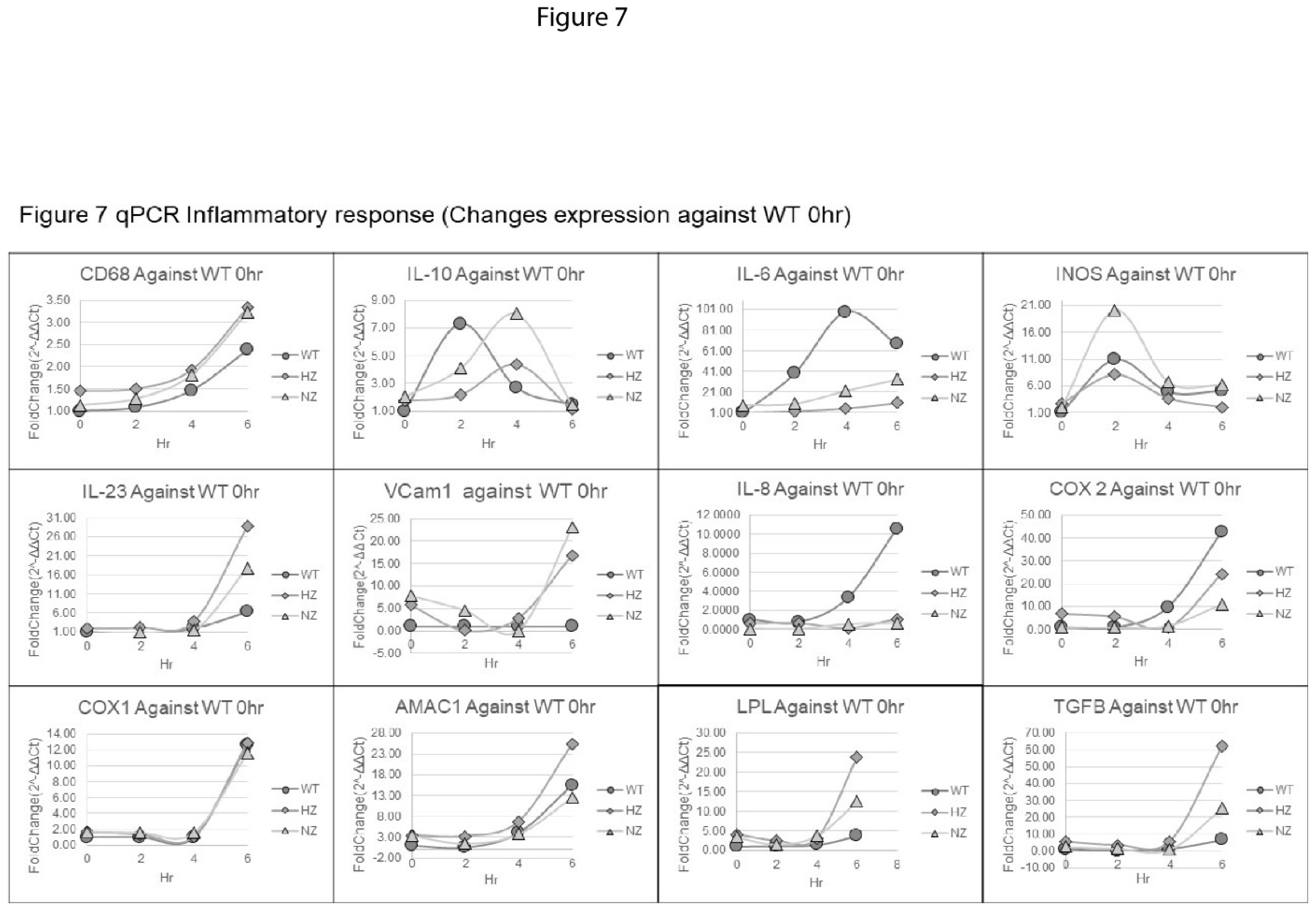
Figure 8.
Effect of FURIN gene status on whole-genome transcriptome expression in U937 cells. (a) Unique and overlapping differentially expressed genes (absolute fold-change >2, adj.p-value < 0.01) in HZ and NZ clones compared to WT. (b) Volcano plots of log2 fold-change (x-axis) vs. log10 p-value in HZ vs. WT and NZ vs. WT comparisons, respectively. (c) Heatmaps of the top 40 most differentially expressed genes (by adjusted p-value) in HZ vs. WT and NZ vs. WT, respetively. (d) Table of the top up- and downregulated KEGG and Gene Ontology Biological Process pathways identified from Gene Set Enrichment Analysis (adjusted p-value < 0.2) of HZ vs. WT and NZ vs. WT. (e,f) Enrichment plots for the ‘Complement and Coagulation Cascades’ pathway (KEGG) in HZ vs. WT and NZ vs. WT analyses, respectively, depicting the significant downregulation of this pathway in HZ and NZ samples. For each comparison, the x-axis depicts the ranks of all genes based on log fold-change (larger positive fold-changes shown in red), with the rank positions of pathway genes indicated by black lines. The y-axis represents a running metric for pathway enrichment (enrichment score) across the full gene list. (g,h) Corresponding KEGG pathway maps for the ‘Complement and Coagulation Cascades’ pathway, with the genes contributing to pathway enrichment highlighted in blue. .
Figure 8.
Effect of FURIN gene status on whole-genome transcriptome expression in U937 cells. (a) Unique and overlapping differentially expressed genes (absolute fold-change >2, adj.p-value < 0.01) in HZ and NZ clones compared to WT. (b) Volcano plots of log2 fold-change (x-axis) vs. log10 p-value in HZ vs. WT and NZ vs. WT comparisons, respectively. (c) Heatmaps of the top 40 most differentially expressed genes (by adjusted p-value) in HZ vs. WT and NZ vs. WT, respetively. (d) Table of the top up- and downregulated KEGG and Gene Ontology Biological Process pathways identified from Gene Set Enrichment Analysis (adjusted p-value < 0.2) of HZ vs. WT and NZ vs. WT. (e,f) Enrichment plots for the ‘Complement and Coagulation Cascades’ pathway (KEGG) in HZ vs. WT and NZ vs. WT analyses, respectively, depicting the significant downregulation of this pathway in HZ and NZ samples. For each comparison, the x-axis depicts the ranks of all genes based on log fold-change (larger positive fold-changes shown in red), with the rank positions of pathway genes indicated by black lines. The y-axis represents a running metric for pathway enrichment (enrichment score) across the full gene list. (g,h) Corresponding KEGG pathway maps for the ‘Complement and Coagulation Cascades’ pathway, with the genes contributing to pathway enrichment highlighted in blue. .
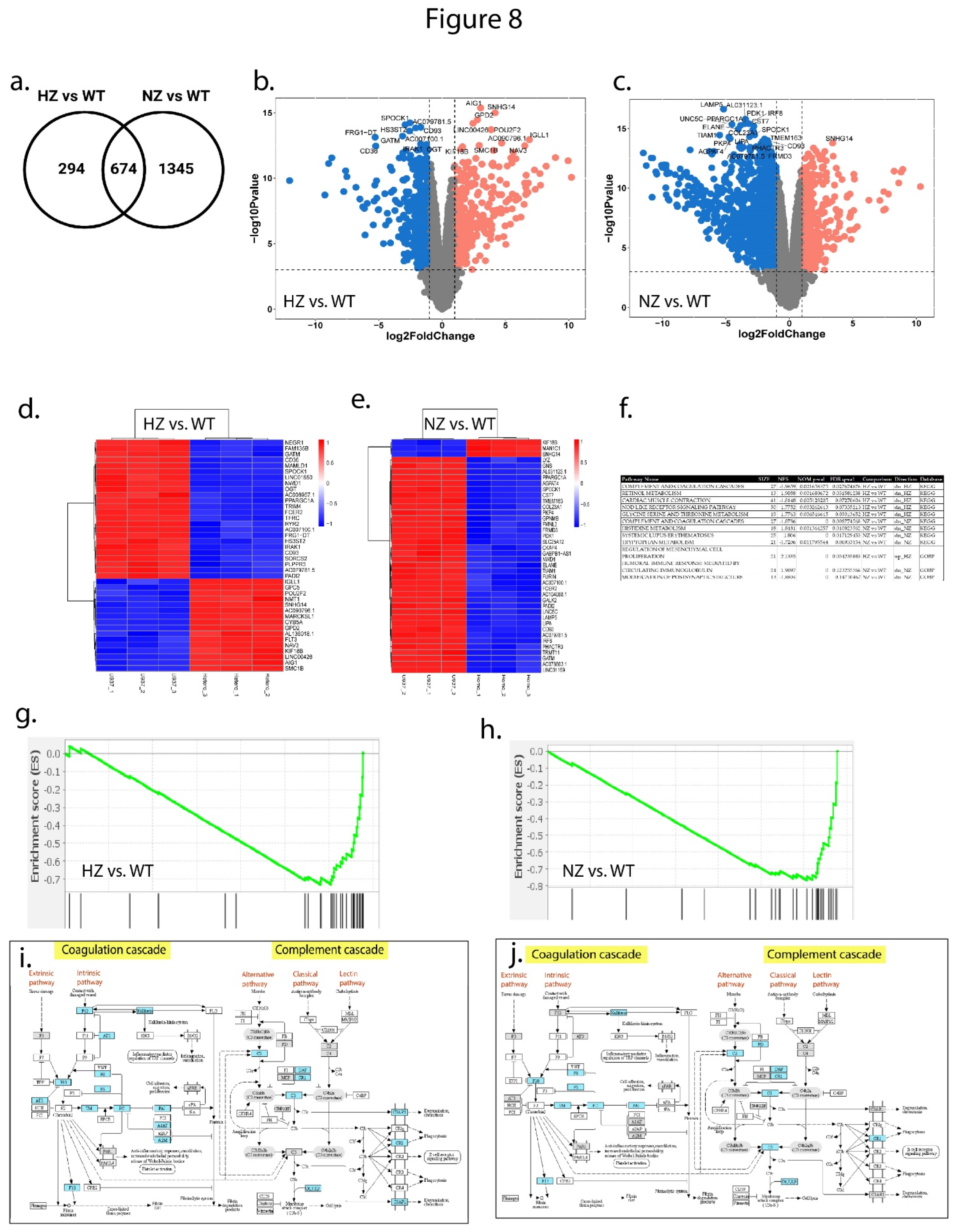
Figure 9.
Patterns of gene expression as a function of FURIN gene status. (a) Gene expression profiles across WT, HZ and NZ U937 cells were clustered via self-organizing maps (SOMs), depicting 8 distinct patterns. (b-f) Pathway enrichment analysis of gene-sets in selected clusters. Each heatmap plots the expression of cluster-specific gene names (rows) and average gene expression across the U937 clones (columns). Expression values are row-normalized and color-coded from low (blue) to high (red) expression. The pathway name is indicated at the top of each heatmap and the SOM cluster is indicated in parentheses. .
Figure 9.
Patterns of gene expression as a function of FURIN gene status. (a) Gene expression profiles across WT, HZ and NZ U937 cells were clustered via self-organizing maps (SOMs), depicting 8 distinct patterns. (b-f) Pathway enrichment analysis of gene-sets in selected clusters. Each heatmap plots the expression of cluster-specific gene names (rows) and average gene expression across the U937 clones (columns). Expression values are row-normalized and color-coded from low (blue) to high (red) expression. The pathway name is indicated at the top of each heatmap and the SOM cluster is indicated in parentheses. .
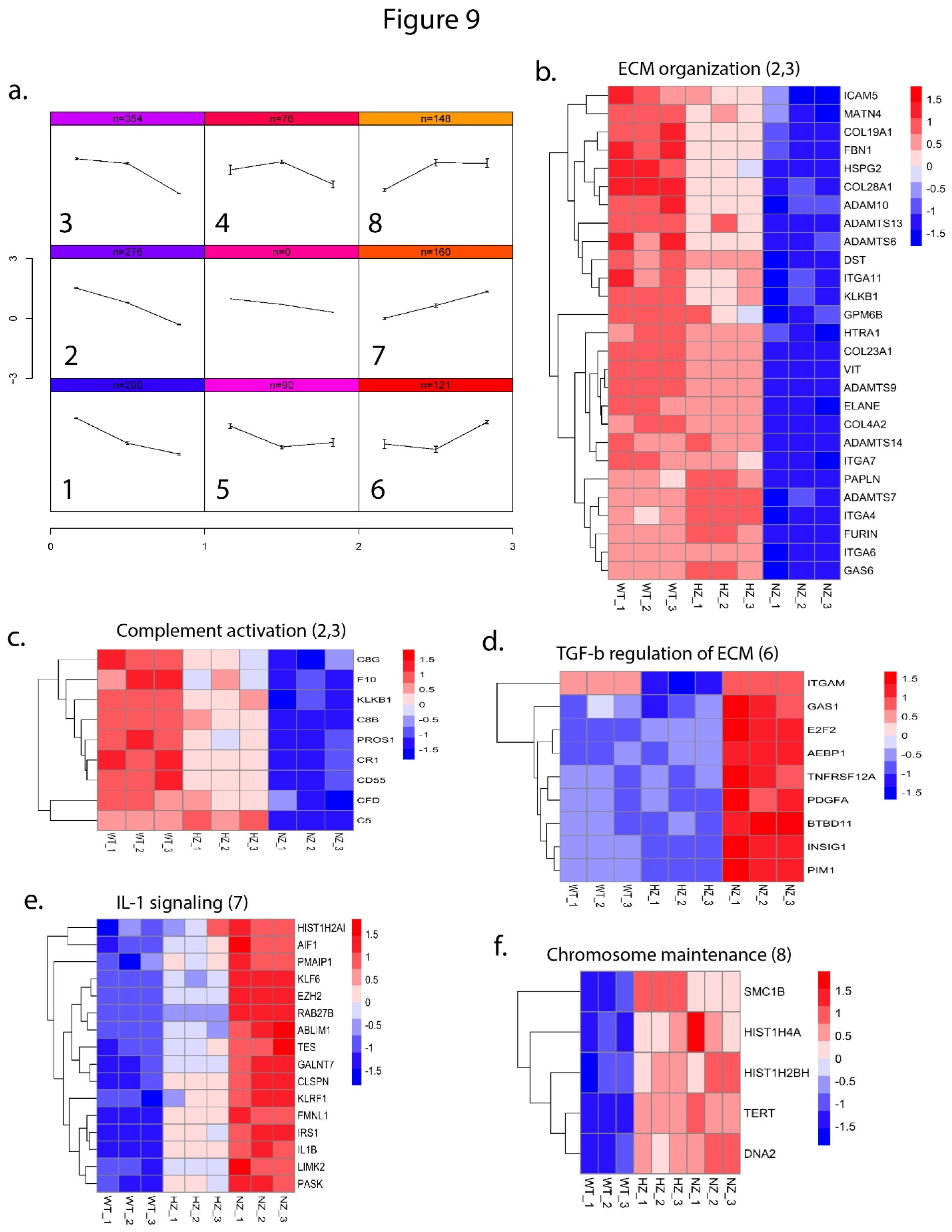
Figure 10.
Proteomics analysis of the effects of FURIN gene status on cytokine secretion under basal and LPS-stimulated conditions. Cytokine secretion was assessed on antibody arrays and quantified after normalization through background subtraction and scaling. Cytokine names are indicated at the top of each subplot. Bars are color coded by a combined identifier containing the FURIN gene status and LPS-stimulation status. The x-axis lists the combined identifiers and the y-axis represents the normalized cytokine secretion levels (results are averaged over duplicates).
Figure 10.
Proteomics analysis of the effects of FURIN gene status on cytokine secretion under basal and LPS-stimulated conditions. Cytokine secretion was assessed on antibody arrays and quantified after normalization through background subtraction and scaling. Cytokine names are indicated at the top of each subplot. Bars are color coded by a combined identifier containing the FURIN gene status and LPS-stimulation status. The x-axis lists the combined identifiers and the y-axis represents the normalized cytokine secretion levels (results are averaged over duplicates).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



