
Submitted:
01 December 2023
Posted:
04 December 2023
You are already at the latest version
Abstract
Flowering in cassava is crucial for generation of botanical seed for breeding. However, most farmer preferred genotypes are poor at flowering, exhibit great disparity in time and amount of flowering or never flower. To elucidate the genetic basis of such a flowering behaviour, 293 diverse cassava accessions were evaluated for flowering traits under two locations and seasons in Uganda. Genotyping by the Diversity Array Technology Pty Ltd. (DArTseq) platform identified 24,040 single-nucleotide polymorphisms (SNPs) distributed on the 18 cassava chromosomes. Population structure analysis using principal components (PC) and kinships showed clustering, the first five PCs accounted for 49.2% of the observed genetic variation. Linkage disequilibrium (LD) estimation was averagely 0.32 at a distance of ~2850kb (kilo base pairs). Polymorphism information content (PIC) and minor allele frequency (MAF) were 0.25 and 0.23, respectively. Genome-wide association study (GWAS) analysis uncovered 53 significant marker-trait associations (MTA) with flowering traits involving 27 loci. Two loci, SNPs S5_29309724 and S15_11747301 were associated with all the flowering traits. Using five of the 27 SNPs with Phenotype_Variance_Explained (PVE) ≥ 5%, 37 candidate genes were identified in the peak SNP sites located within 50kb upstream or downstream, most were associated with branching traits. Eight of the genes, orthologous to Arabidopsis and other plant species, had known functional annotations related to flowering, e.g. eukaryotic translation initiation factor and myb family transcription factor. This study identified genomic regions associated with flowering in cassava, the identified SNPs could be useful in marker-assisted selection to overcome hybridization challenges like unsynchronized flowering, and candidate gene validation.
Keywords:
flowering traits
; first branching
; branching levels
; flowering behaviour
; functional annotations
1. Introduction
Cassava (Manihot esculenta Crantz, 2n = 36) is a valuable food crop in fighting hunger and poverty in developing countries [1,2]. Additionally, it’s a source of animal feed as well as raw materials for a diverse industries [3]. Because of this economic importance of the crop, much of the ongoing breeding efforts are directed towards its genetic improvement so as to develop cassava varieties targeting different industry and food products [4,5]. This, however, is being hampered by a number of constraints including poor flowering and limited knowledge of the genetics and inheritance of flowering traits [5,6]. To meet the growing demand for cassava as well as overcoming the challenges to its genetic improvement due to poor flowering, it is imperative to expand knowledge on the genetics of flowering so as to customize solutions to this problem.
Cassava is preferentially propagated by vegetative means using stem cuttings [7,8] due to the difficulty in obtaining botanical seed, low germination and seedling establishment and survival rates which often result in small and weak plants with comparatively low root yields [8]. Despite these seed-related challenges, seed remains an indispensable and exceedingly important resource in cassava breeding pipeline, to generate new breeding lines with genetic variabilities which may be beneficial in breeding programs [7,8,9]. In conventional cassava breeding, genetic variabilities are derived through hybridization that involve crosses under controlled or open-pollinations, resulting into full or half sib progenies [8]. Introgression of economically important traits requires selection of progenitors with desirable traits and with timely and synchronized flowering capabilities. However, many of the progenitors with desirable farmers’ preferences are often poor at flowering while others never flower at all. Besides, there is great disparity in time and amount of flowering among many cassava genotypes, with some flowering early and profusely while in others the flowering is late and scarce [5,6,10]. The early flowering or branching types tend to have shorter main stem heights while the late branching ones have taller stem heights at first branching. All this indicates that cassava flowering is complex, exhibiting varied patterns, that make synchronization of crosses difficult [11].
Flowering in cassava begins with morphological changes in plant architecture that involve branching of the main stem [10]. Flower bud formation occurs at branch points in the stem [8,10] as axillary buds below the inflorescence allow upward growth of the plant. This implies, every branching event results in flower formation [12]. The successive branching may be di-, tri-, and tetra- or penta- chotomous, resulting in several branching events [8]. The highly branching genotypes flower more prolifically than those with sparse branching, while the non-branching (erect) ones are non-flowering. Most farmer-preferred cassava genotypes are either erect or non-branching or late flowering and thus rarely produce seeds [5,12]. The correlation between branching behaviour and flower production observed in cassava suggests that these traits might share a similar genetic basis. However, knowledge of genes controlling this kind of flowering correlation and/or behaviour in the crop is scanty.
Flowering process in angiosperms is evoked by an interaction of environmental and endogenous signals [13,14,15,16]. This is known to be regulated by a sophisticated network that monitors changes in the environment to ensure that flowering occurs under suitable conditions for maximization of reproductive success and seed production [17,18]. Like in other plants, studies have indicated that flowering response in cassava is under the influence of environmental factors, such as photoperiod and temperature [19,20,21]. The understanding of the control mechanisms of flowering in many plants is mainly based on the model plant Arabidopsis thaliana, in which various genes including FLOWERING LOCUS T (FT) and proteins have been implicated [17]. Although FT orthologs such as MeGI, Cassava GI and CO-like genes (MeCOL1, MeCO, and MeCOL2) have been associated with photoperiodic induction of flowering in cassava [21,22], the precise mechanism underlying the flowering behaviour exhibited in the crop, particularly branching, branch type, branching levels, number of nodes and stem height at first branching level, is not clearly known. Thus, deciphering the genetic basis of this flowering behaviour and how it is regulated will provide important insights that could be useful in overcoming hybridization challenges, especially flowering synchronization.
Molecular marker-assisted selection (MAS) has been a primary approach in plant breeding for understanding the genetic basis as well as discovery of functional genomic regions (genes) associated with target phenotypic traits in many plant species [23]. It has been used to probe QTL (quantitative trait loci) for developing genetic linkage maps in a number of crops [24]. Several QTLs for flowering time have been detected in several crop species, for example, rapeseed (Brassica napus L.) [25]. MAS has been used to detect QTLs or genomic regions for various traits in cassava breeding [23], for example, root yield and starch content [26], plant and first branch height [27], starch biosynthesis [28], biotic stress resistance [29]. However, knowledge of genes controlling flowering and related traits in cassava is still limited.
Genome-wide association analysis (GWAS) is an alternative genomic approach that has been designed to overcome drawbacks of QTL mapping [30]. It is used to mine natural genetic variations in organisms for revelation of variabilities in genetic architectures among traits in crop plants [31,32]. It is robust and thus has been used to identify many true genotype-phenotype associations [33] in several plants, for example, Arabidopsis thaliana [32,34,35], Oryza sativa [36], Zea mays [37,38], Brassica napus [39] and Setaria italica (foxtail millet) [40]. GWAS has also been successfully utilized to unravel genetic architecture of several phenotypic traits in cassava, for example, starch content [41], dry weight content [42], beta carotene content [42,43,44], disease-resistant traits such as cassava mosaic disease (CMD) [45] and cassava brown streak disease [46,47], and drought tolerance [48].
Relatedly, GWAS has been used to elucidate the genetic basis of flowering traits in several crops. For example; flowering time variabilities in pearl millet (Pennisetum glaucum L.) [49], maize (Zea mays) [50], common bean (Phaseolus vulgaris L.) [51,52], Sesame (Sesamum indicum L.) [53], rapeseed (B. napus L.) [54] flowering and plant height under different levels of nitrogen in Indian mustard (Brassica juncea) [55] and Soybean (Glycine max) [56]. In cassava, reports on utilization of GWAS to determine the genetic basis of flowering and related traits are scanty, except first branch height [57].
Thus, in this study, we aimed at identifying genomic regions associated with flowering traits, namely; first branching; type of branching; number of branching levels; number of nodes and stem height at first branching.
2. Results
2.1. Phenotypic Variation, Distribution and Heritability Estimates of Flowering Traits
Phenotyping was done for flowers (staminate and pistillate) and five flowering traits on a total of 293 cycle two (C2) cassava accessions. This was done for two growing seasons (2019 and 2020) and two locations. Branching ranged from 0 (did not branch) to 1 (branched), branch type or habit from zero to four, branching levels were maximum at 12, both number of nodes and stem height at first branching ranged from zero to 148 and 231cm respectively (Table 1). Coefficient of variation (CV) values varied from 26.87% for branching to 54.66% for nodes.
Broad-sense (H2) and SNP (genomic) (h2) heritabilities were estimated for each trait across season and location (Table 1). Highest broad-sense heritability (H2) was exhibited by pistillates (0.83) followed by stem height at 1st branch (0.60), while staminates had the lowest heritability (0.00). SNP-based heritabilities ranged from lowest (0.00) in branch type to highest (0.35) in stem height (at 1st branch). Negatively skewed distribution trends were observed for branching and branching levels, while branch-type, nodes and stem height at first branching had positively skewed distributions (Figure 1). Results of ANOVA demonstrated that the differences among the cassava accessions (P ≤ 0.001) used in this study were highly significant for all traits, and magnitude was even higher for the seasons and locations, except branch type (Table 2).
2.2. Correlation between the Flowering Traits
Spearman’s correlation coefficient was applied to detect any correlations among the flower and flowering traits considered in this study; 1) first branching of the main stem, 2) branching type, 3) number of branching levels, 4) number of nodes, 5) stem height at the first branching, 6) flowers (pistillates and staminates). All the traits showed significant positive correlations ranging between 0.66 to 1.00 (Figure 2). Generally, relatively lower correlation values were noted between flowering traits and pistillates compared to other traits.
2.3. Genotyping and SNP Identification
Genotyping enabled the identification of 24,040 imputed SNP markers, all of which were mapped onto the 18 cassava chromosomes. Upon filtering, markers with minor allele frequency (MAF) at 1%, 22,103 SNPs were retained, distributed on all the 18 chromosomes (Figure 3). The SNP coverage per chromosome ranged from 806 SNPs on chromosome 16 to 2,065 SNPs on chromosome 1. The average values of the polymorphism information content (PIC) and MAF for the 22,103 SNP markers were 0.25 (ranging from 0.02 to 0.38) and 0.23 (ranging from 0.01 to 0.50) respectively.
2.4. Population Structure and Kinship
The population structure of the 293 accessions was estimated by using principal components analysis (PCA). The first principal component (PC) accounted for 37.3% of the observed variation (Figure 4A) while the second up to the fifth PC contributed 11.9%. Cumulatively, the first five PCs explained 49.2% of the genetic variation. Kinship among accessions estimated using a marker inferred Kinship matrix revealed familiar relationships between the accessions (Figure 4B). Both PCA and kinship analyses revealed clustering of germplasm into subgroups, three major subgroups according to the kinship heatmap (Figure 4B).
2.5. Linkage Disequilibrium Estimation
Linkage disequilibrium (LD) decay rate was measured as the chromosomal distance at which the average pairwise correlation coefficient (r2) dropped to half its maximum value. The LD and LD decay distance in all the 18 chromosomes is summarized in Figure 5. The average r2 value for 18 chromosomes was found as 0.32 at a distance of ~2850kb (kilo base pairs) which slowly dropped to its half (0.16) at distance ~4828kb, with maximum decay distance of ~32012kb on chromosome 7 and minimum decay distance of 0.009kb on chromosome 10. The lowest r2 value was 0.12 with maximum decay distance of ~39193kb on chromosome 8 and minimum decay distance of 0.006kb on chromosome 15. Meanwhile, the highest r2 value was 0.95 with maximum decay distance of ~796kb on chromosome 3 and minimum decay distance of ~33kb on chromosome 10.
2.6. Marker-Trait Association Mapping
Marker-trait association (MTA) using 293 cassava accessions and 22,103 SNP markers was undertaken using the Mixed Linear Model (MLM), which took genetic relatedness into account, greatly reduced false positives, as shown in the Manhattan and quantile–quantile (Q-Q) plots (Figure 6). The Manhattan plots of five flowering traits were based on the combination of >1000 trait de-regressed BLUPs. A total of 53 significant MTA (involving 27 SNP) was identified as being associated with flowering traits, and these were distributed mainly on chromosome 18 which had 20 of the 53 SNPs (Table 3). Branching (Branch) had 15 marker associations on chromosomes 5, 8, 11, 15, 16 and 18; branch type at first branching (Branch1_No) was associated with 08 SNPs on chromosomes 5, 8, 14, 15 and 18. Similarly, number of branching levels (Branch_Levels) had 14 marker associations on chromosomes 5, 8, 9, 11, 14, 15, and 18., while number of nodes at first branching (Branch1_Nodes) had the highest associated markers (16) on chromosomes 3, 5, 8, 9, 14, 15 and 18. Meanwhile, stem height at first branching (Branch1_Ht) had no MTA based on the Bonferroni threshold (Figure 6). The Q-Q plot showed the distribution of observed and expected p-values (association test statistics). The Q-Q-plot analysis for all traits, except stem height, showed a fairly mid-way deviation of the observed p values from those expected, towards the top left.
2.7. Identification of Candidate Genes
Gene annotation information according to Phytozome v13 (Manihot esculenta v7.1) (https://phytozome-next.jgi.doe.gov/) and National Centre for Biotechnology Information (NCBI) of cassava, Manihot esculentum genome (Manes) (https://blast.ncbi.nlm.nih.gov/) were used to determine the putative functionality of genes around the associated marker loci. Out of the 27 SNPs involved in MTA, 05 SNPs with Phenotype_Variance_Explained (PVE) ≥ 5% were selected to predict genes associated with the flowering traits. Consequently, a total of 37 candidate genes located within 50kb upstream or downstream of the five markers in the cassava genome were found to be associated with four of the five flowering traits; branching, branch type, branching levels and nodes at first branching (Table 4). SNPs S5_29309724 (on chromosome 5) and S15_11747301 (on chromosome 15) were associated with all the four traits and had 05 and 12 putative genes respectively. SNP S18_1832353 (on chromosome 18) was associated with three traits involving 10 genes, while the rest of the SNPs were associated with one trait each, but with varying numbers of putative genes. Detailed information about all significant associations is summarized in Table 5.
Marker R2 values ranged from 0.017 to 0.177, with P-values varying from 1.15×10−9 to 1.45×10−7, while PVE values considered for gene identification ranged from 4.85 to 26.85% (Table 5). Averagely, 26.86% was the highest phenotypic variance, attributed to SNP S5_29309724 (associated with four flowering traits), while 4.85% was the lowest PVE, attributed to SNP S5_22566689 (associated with Nodes at 1st branch). Whereas 10 genes (06 on SNP S3_21330906 and 04 on S5_22566689) were unique to Nodes at 1st branch, the remaining 27 genes were associated with all the four flowering traits (branching, branch type, branching levels and Nodes at 1st branch). A total of 18 gene loci were each associated with four traits, for example, Manes.15G140500.1 (eukaryotic translation initiation factor 3 subunit I) and Manes.15G140900.1 (myb family transcription factor MOF1). Similarly, 09 candidate genes such as Manes.18G016700.1 (aldehyde oxidase GLOX) and Manes.05G186700.1 (protein DETOXIFICATION 48) were associated with three traits.
On the basis of the 53 associated markers identified using GWAS, candidate genes with known functions and orthologous with Arabidopsis or other plant species were identified using five SNPs which had PVE ≥ 5% (Table 6). Among the genes found to be associated with these SNPs, we focussed only on those which were reported to be functionally controlling flowering or flowering responses in various plant species. As such, 08 genes were identified as candidates and all were associated with branching, branch type and branching level traits. For example; Manes.18G016700.1 (aldehyde oxidase GLOX) as well as Manes.18G016725.1 and Manes.18G016800.2 (lysine-specific histone demethylase 1 homolog 3), all associated with SNP S18_1832353 on chromosome 18, are involved in anther and pollen development [58] and promotion of floral transition [59] respectively in Arabidopsis thaliana. Manes.05G186700.1 (protein DETOXIFICATION 48) associated with SNP S5_29309724 on chromosome 5 is involved in specifying the lateral organ initiation rate in Arabidopsis thaliana [60]. Associated with SNP S15_11747301 on chromosome 15 are Manes.15G140500.1 (eukaryotic translation initiation factor 3 subunit I) which was reported to regulate negatively translation during flower development [61], Manes.15G140900.1 (myb family transcription factor MOF1) which plays a role in the regulation of organ identity and spikelet meristem determinacy in rice [62] and Manes.15G140300.1 (non-specific lipid-transfer protein 4.1) involved in seed and ovule maturation and development in Hordeum vulgare (Barley) and Arabidopsis thaliana [63]. Manes.15G140600.1 (short-chain dehydrogenase reductase ATA1), also located on chromosome 15 was reported to play a role in tapetum development in Arabidopsis thaliana [64]. The new genes identified in the present study are promising candidates for follow-up studies on the validation of genes controlling flowering and related traits in cassava.
3. Discussion
In this study, GWAS was used to detect genomic regions associated with flowering traits in a diverse population of 293 cassava accessions. This enabled generation of knowledge to enhance understanding of the genetics of cassava flowering traits, i.e. branching, branch type, branching levels and number of nodes of main stem at first branching.
3.1. Variation in Phenotypic Traits Related to Flowering
Ability to flower as well as early and synchronized flowering are important target traits for selection of progenitors in cassava hybridisation programmes. Branching, branch type, branching levels, number of nodes and stem height at first branch are major indicators of flowering ability in cassava. Commonly, it is through flowering and pollination that crucial genetic variation is generated, which is fundamental for success of plant breeding programs [67]. Understanding phenological variabilities exhibited in cassava flowering is essential for accurate selection of parents and planning crosses in hybridization programs. However, these different phenologies are difficult to understand due to the complex genetics of the crop.
Number of nodes followed by stem height exhibited highest variability (Table 1). There is greater potential for selection of progenitors with capacity to flower basing on these attributes compared to others. High and exploitable variability for flower and fruit numbers as well as time to flower in cassava was also reported in a previous study [11]. Conversely, the lowest variation recorded for branching indicates low exploitable variability compared to other traits.
Heritability estimates give insights into the extent of genetic control of a particular trait [67,68,69]. High heritability indicates less environmental influence [67,70]. Broad-sense heritability varied across traits, i.e. pistillates had the highest heritability (H2 = 0.83) followed by stem height at first branch (H2 = 0.60). There are not many reports on broad sense heritability estimates for flowering traits in cassava, however, heritability estimates of over 0.80 were reported for plant height at first branch, number of branches, branching levels as well as female flowers [11,71,72,73]. SNP-based heritability (h2) estimates were relatively higher for stem height at first branch (h2 = 0.35), followed by branching (h2 = 0.25) and branching levels (h2 = 0.19). These estimates could be attributed to linkage disequilibrium (LD) unevenness or heterogeneity among marker regions, due to overestimation or underestimation, as postulated by [74].
3.2. Correlation between the Flowering Traits
Correlation coefficient analysis is important for measuring the degree and direction of relationships between various traits. The positive and significant correlation among the flowering traits in this study, such as branch and branch type (r2 = 0.95) or flowers (r2 = 0.94); branching levels and pistillate flowers (r2 = 0.76), branching levels and height of first branch (r2 = 0.88), etc. was consistent with the previous findings by [75]. Thus, this reflects that selection for one trait would directly affect the expression of the other trait, hence facilitating the selection of progenitors to include in a breeding program. Relatedly, a correlation between branching levels and height of first branch was reported in a previous study on induction of cassava flowering [76]. Overall, all these traits are dependable indicators of flowering response, upon which selection of progenitors can be based.
3.3. Population Structure and Kinship
Population stratification and the degree of kinship of the samples are critical factors in genetic studies including GWAS [77,78]. Whereas population structures show presence of distinct genetic subgroups within a population [79], kinships provide valuable insights into the degree of genetic relatedness between the individuals within a population [77]. PCA and kinship analysis revealed clustering of germplasm into subgroups, three major subgroups according to the kinship heatmap (Figure 4). The clusters (within the PCA and kinship plots) show genetic relatedness within members of the subgroup, suggesting a common ancestry within the subgroup. Clusters along the diagonal line in the kinship plot show unrelatedness among groups, while pairs of individuals along the diagonal line show individuals' self-kinship. Shorter connecting lines between individuals or clusters indicate closer relationships while longer connecting lines indicate more distant relationships.
The Q-Q-plot analysis further revealed subgroups in the study population. Plots for all traits, except stem height, showed a fairly mid-way deviation of the observed p values from those expected, towards the top left (Figure 4). This indicated that, firstly, the SNPs associated with these traits were significant, secondly, the existence of genetic variations between the subpopulations. Thus, suggesting that genetic variants have different allele frequencies among subgroups of the population investigated. For stem height at first branching, the little or non-segregation between the observed and expected p values suggests that the sub populations are genetically homogeneous, indicating a similarity in the distribution of allele frequencies between the subpopulations.
3.4. Linkage Disequilibrium
Linkage disequilibrium (LD), a non-random association or correlation between alleles at different genetic loci within a population plays an integral role in genetic studies, such as genome-wide association studies (GWAS) and genomic selection [80,81]. When LD exists between a marker (e.g. a SNP) and a phenotypic trait, it implies that the marker and the causative variant of that trait are physically close to each other on the same chromosome [81].
There is limited knowledge about genetic architecture and potential markers specifically associated with flowering traits in cassava due to little research that has been conducted on the LD patterns and LD decay for these traits. In this study, the average LD among the SNP marker pairs of 293 cassava accessions was 0.32 at a distance of ~2850kb which slowly declined to 0.16 at distance ~4828kb where it became more or less stable (Figure 5). A study by [82] on evaluation of genetic diversity among 1,580 cassava accessions presented a lower LD value average of 0.014, though the initial LD decline was more rapid before it stabilized at 15 to 20 kb. Also, analysis of a panel of 876 cassava accessions belonging to the Cassava Germplasm Bank of Embrapa, a LD of r2 < 0.1 which declined to between 0.3 and 2.0 Mb was reported [83]. While a whole-genome LD decay peaked at r2 of 0.349 and dropped to an r2 of 0.212 at a distance of 10 kb in a study on genetic architecture of defensive, agro-morphological and quality-related traits involving a panel of 5130 cassava clones [84]. These observations show that the LD estimates and decay rates in the cassava genome are low and inconsistent.
The low and slow decline in LD observed in this study and the observed inconsistencies among the reported LD values of the different studies could be attributed to the genetic complexity (high heterozygosity) of the crop as well as the vegetative mode by which the crop is predominantly propagated. Additionally, the artificial selection pressures imposed by preferential selections based on specific traits, like disease resistance and high yield, tends to favour specific alleles linked to the desired traits, leading to extended LD around these beneficial variants. In potato (Solanum tuberosum L.), a clonally propagated crop, but outcrossing and highly heterozygous like cassava, LD decay was reported to be relatively fast in the short range (r2 = 0.208 at 1 kb) but slowed afterward (r2 = 0.137 at 70 kb) [85]. Perennial or clonally propagated plants including cassava, generally have long breeding cycles and, thus, show a limited number of recombination cycles. Hence, their LD decays are relatively slow [86] in spite of the outcrossing nature of these crops.
3.5. Marker–Trait Association Mapping
Association or linkage disequilibrium (LD) mapping, also known as association analysis or genome-wide association study (GWAS), is an important tool in plant breeding for understanding the genetic basis of complex traits. It involves surveying genetic variations in the whole genome to find signals of association between genetic markers, such as single-nucleotide polymorphisms (SNPs), and various phenotypic traits of interest within a diverse population [80]. It is also used in identifying candidate genes and genomic regions which are presumed to be associated with traits of interest [80,87].
Using the MLM model, we conducted a GWAS of five flowering traits for 293 cassava accessions based on 22,103 (DArTseq-derived) filtered SNP markers and detected 53 MTA on nine of the 18 chromosomes, with chromosome 18 possessing up to 20 of the 53 MTA. All traits, except stem height at first branching, had MTA on different chromosomes, suggesting pleiotropic effects. For example, branching was associated with 15 markers on chromosomes 5, 8, 11, 15, 16 and 18; while branching levels had 14 MTA on chromosomes 5, 8, 9, 11, 14, 15 and 18 (Table 3). This suggests that these traits are polygenic in nature. Meanwhile, some chromosomes had more than one MTA for some traits, for example, chromosome 18 was associated with 08, 04, 06 and 03 MTA for branching, branch type, branching levels and number of nodes at first branching respectively. This suggests that some traits are regulated by multiple genes located in close proximity to each other on the chromosome, and that, the genes may be interacting to contribute to the expression of the trait. This can be advantageous because it would enable narrowing down the search for traits of interest. A study by [83] also revealed multiple MTA on chromosome 18 for cassava starch pasting properties, indicating high LD of markers on this chromosome. Multiple MTA have also been detected for flowering traits in Indian mustard (Brassica juncea) [55] and grain yield and related traits in durum wheat (Triticum turgidum) [88]. Other traits had only one MTA on a chromosome, such as S11_32333764 on chromosome 11 (associated with branching); S15_11747301 on chromosome 15 (associated with branching, branch type, branching levels and number of nodes). This suggests that the marker may be associated with a potential gene or genomic region primarily responsible for influencing the given traits on that particular chromosome. This may be helpful particularly in genome editing.
3.6. Putative Candidate Genes Linked to Marker Loci for Flowering Traits
Using MLM in GWAS, different genomic regions (putative candidate genes) associated with flowering traits were predicted. All the predicted genes were associated with branching characteristics. These genes are novel and a new discovery, as they have not been previously reported.
Branching of shoots involves axillary bud initiation in which the shoot apical meristem is converted to axillary meristem, expansion as well as sustained branch growth, which is regulated by an interplay of genetic programs with the environmental signals, phytohormones and sugars [13,89]. Genes modulating reproductive branching have been identified in many plant species and several are broadly conserved in between species, though detailed phenotypic and developmental effects are species specific [89]. For example, LATERAL SUPPRESSOR in tomato, its Arabidopsis ortholog LAS [90], and the rice ortholog MONOCULM1 (MOC1) [91], the TEOSINTE-BRANCHED1 (TB1)/BRANCHED1(BRC1) (TB1/BRC1) gene in Arabidopsis, peas, tomato [89], BRC1 was also predicted in modulating branching in soybean [92]. In cassava, branching of the main stem is associated with transition from vegetative growth to reproductive development. This indicates on-set of flowering with flower-bud formation being preceded by a highly genetically planned apical branching [8].
In this study, three branching traits, i.e. branching, branch type and branching levels had commonality in 27 of the 37 predicted genes (Table 5). Of these, eight were reported to be functionally annotated for processes directly or indirectly leading to flowering responses in different plant species (Table 6). For example, Manes.18G016725.1 and Manes.18G016800.2 (lysine-specific histone demethylase 1 homolog 3), associated with SNP S18_1832353 on chromosome 18 was reported to influence the transition from vegetative growth to the flowering phase in Arabidopsis thaliana [59]. It reduces levels of histone methylation in chromatin of key flowering genes such as the floral repressor FLOWERING LOCUS C(FLC) which must be preferentially expressed in shoot and root apical regions during a plant’s vegetative development. This enables achievement of precise levels of FLC required for onset of a flowering response.
Manes.18G016700.1 (aldehyde oxidase GLOX, Germin-like oxalate oxidases) in close proximity with SNP S18_1832353 on chromosome 18. Though its role in plants is not yet fully understood, it is believed to be involved in anther development during which it plays a role in tapetum and pollen development in Arabidopsis thaliana [58]. It is also involved in catalyzation of the oxidation of aldehydes to the corresponding carboxylate by coupling the reaction to the reduction of dioxygen to hydrogen peroxide [65].
Manes.05G186700.1 (protein DETOXIFICATION 48) associated with SNP S5_29309724 on chromosome 5, a member of multidrug and toxic compound extrusion (MATE) gene family, is involved in specifying the lateral organ initiation rate in Arabidopsis thaliana [60]. It has been implicated in axillary bud formation in the shoot apical meristems resulting in accelerated organ formation including leaf formation and early flowering. It is also involved in detoxification or extrusion of toxins and regulation of plant development including fruit development [93].
Manes.15G140500.1 (eukaryotic translation initiation factor 3 subunit I) associated with SNP S15_11747301 on chromosome 15 is a member of eukaryotic translation initiation factor 3 (eIF3) complex [61]. It was reported to regulate negatively translation during flower development in Arabidopsis thaliana. It is responsible for initiating protein synthesis during development and germination of pollen grains and embryogenesis. Its expression was detected in plant cells and tissues differentiating into organs including inflorescences, implying its involvement in influencing transition from vegetative to reproductive growth [61]. Manes.15G140900.1 (myb family transcription factor MOF1), also associated with SNP S15_11747301, plays a role in the regulation of organ identity and spikelet meristem determinacy in rice [62]. It causes delayed transition from the spikelet to the floral meristem, which may result in defective or sterile spikelets.
Manes.15G140300.1 (non-specific lipid-transfer protein 4.1) associated with S15_11747301 on chromosome 15 plays a role in seed and ovule maturation and development, probably by regulating the fatty acids homeostasis during suberin and sporopollenin biosynthesis or deposition [94]. And Manes.15G140600.1 (short-chain dehydrogenase reductase ATA1) also near SNP S15_11747301 was reported to play a role in tapetum development in Silene latifolia and Arabidopsis thaliana [64].
4. Conclusions
The present study was undertaken to determine the genomic regions associated with flowering traits in cassava under field conditions. The interest in unravelling the genetic basis of cassava flowering is based on the premise that many breeding programmes find it still difficult to synchronize flowering time among different cassava genotypes. To date, this still remains as a significant impediment to cassava breeding. Thus, increasing our understanding of the genetics of the flowering process and related traits in cassava would greatly assist the ability to accelerate and synchronize flowering among different cassava varieties through marker-assisted breeding and/or gene editing. To this end, this study identified 53 significant MTA distributed mainly on chromosome 18, as being associated with flowering traits. Five SNPs, i.e. S3_21330906; S5_22566689; S5_29309724; S15_11747301 and S18_1832353 on chromosomes 3, 5, 15 and 18 respectively, were found to be significantly associated with number of nodes and branching traits. Eight genes, i.e. Manes.18G016700.1 (aldehyde oxidase GLOX); Manes.18G016725.1; Manes.18G016800.2 (lysine-specific histone demethylase 1 homolog 3); Manes.05G186700.1 (protein DETOXIFICATION 48); Manes.15G140500.1 (eukaryotic translation initiation factor 3 subunit I); Manes.15G140900.1 (myb family transcription factor MOF1); Manes.15G140300.1 (non-specific lipid-transfer protein 4.1) and Manes.15G140600.1 (short-chain dehydrogenase reductase ATA1) were predicted to be involved in controlling branching, branch type and branching level traits. However, further studies are required to validate the significant markers and to verify the identified candidate genes using a larger population.
5. Materials and Methods
5.1. Plant Material and Field Trials
A diverse panel, consisting of 446 cassava accessions with unknown and/or diverse flowering behaviour were used in this study. These accessions were derived from a cycle two (C2) population that was developed at NaCRRI (National Crops Resources Research Institute). Briefly, the C2 population resulted from successive cycles of selection and hybridization [95,96]. These accessions were established in trials laid out in augmented design with three checks of Mkumba, TME 204 and NAROCASS 1. The clones Mkumba and NAROCASS 1 are known for early forking and/or profuse flowering, while TME 204 is a late or non-forking. Each plot had one row of 10 plants spaced at 1m between plants and rows. The trials were established at two locations, Namulonge in Central Uganda (0.52166458oN, 32.608997564oE) at an altitude elevation of 1150m above sea level (asl) and at National Semi Arid Resources Research Institute (NaSARRI), Serere in Eastern Uganda (1.4994oN, 33.5490oE) at 1140m asl. The trial was conduvcted for two seasons, 2019/2020 and 2020/2021. All trials were managed according to the Institute’s standard agronomic practices for cassava.
5.2. Flowering Traits Evaluated
Plant-based counts of flowers (staminate and pistillate) were commenced at about three months after planting (MAP), and this was continued every seven days. However, this collection was done only for one season due to interruption by COVID19 lockdown in 2019 and 2020. Meanwhile, at 12 MAP data on the five traits related to flowering were collected on a per plant basis. The traits included: i) first branching of the main stem (Branch) which was scored as “1” for branched or “0” for non-branched, ii) branching type or habit (Branch1_No) as number of branches at first branching event or tier1, iii) number of branching levels of the main stem (Branch_Levels), iv) number of nodes (Branch1_Nodes), counted with tally counters up to first branching, and v) stem height to first branching (Branch1_Ht) from ground to the first branch of the main stem, measured with a meter rule or tape measure. These traits were prioritized basing on reports which inferred that stem branching was an indication and precondition for flowering in cassava [7,8,10]. Data on these traits were collected for two consecutive seasons from 2019–2021 in two locations. All data were electronically collected on tablets using a Field Book application [97].
5.3. Genomic DNA Extraction and Genotyping
Extraction of genomic DNA and genotyping was carried as described in [46]. Briefly, two young top leaves were collected from the study genotypes, folded, punched using a 5mm hand puncher and placed in 96-well plates before being shipped to Intertek, Australia for genomic DNA extraction and purification. Extraction was done using a DNA extraction protocol routinely used at Intertek-AgriTech (http://www.intertek.com/agriculture/agritech/) and the extracted DNA samples were purified and quality as well as quantity evaluated before being genotyped. Genotyping was done using the Diversity Array Technology Pty Ltd. (DArTseq) genotyping platform (https://www.diversityarrays.com/technology-and-resources/dartreseq/).
5.4. SNP Calling and Annotation
The sequences of the genomic representations were aligned to cassava reference genome v6.1 resulting in the selection of 28,434 raw SNP markers. The genotype data were then subjected to stringent filtration measures to remove genotypes with >10% and SNPs with >5% missing data or with minor allele frequency of less than 5%. Thus, a total of 24,040 high quality SNP markers were used for GWAS. The SNPs were annotated on the basis of the cassava reference genome v6.1. The markers were converted to the dosage format of 1, 0, 2, which represented alternative allele homozygotes, heterozygotes, and reference allele homozygotes, respectively.
5.5. Statistical Analyses
All phenotypic data were subjected to various statistical analyses using R Statistical Software v4.2.2 [98]. Accessions that did not have a complete set of data points across seasons and location were excluded from the statistical analyses, thus leaving 293 out of the 446 initial accessions. BLUPs for each trait were computed and used for statistical and GWAS analyses. The phenotypic datasets; number of branches, branching levels and nodes were analysed, as recommended for count data, using negative binomial distribution, [99,100] while analysis of variance (ANOVA) was performed for the parameter of stem height. Generalized Linear Mixed Models and lme4 packages for R statistical software were used. Broad-sense heritability (H2) per trait was computed using accession and location variance components extracted from the ANOVA models as:
where was the accession variance; was the variance attributed to accession by location interaction; was the model residual variance; was number of locations; and was number of seasons.
SNP-based heritability (h2) was computed as:
where was the additive genetic variance and was the residual variance.
Correlation analysis for flowering (1. first branching of the main stem; 2. branching type; 3. number of branching levels; 4. number of nodes and 5. stem height at the first branching as well as number of staminate and pistillate flowers) traits was conducted based on phenotypic BLUPs. The analyses were performed using the cor function, and visualization of the correlation matrices was done using the ‘corrplot’ package in R [98].
5.6. Population Structure and Kinship
Population structure analysis was performed to determine the genetic relatedness between individuals using 24,040 polymorphic SNP markers distributed over the whole genome. Using the Genome Association and Integrated Prediction Tool (GAPIT) 3.0 in R, we computed a genomic relationship matrix (K) (kinship) from the SNP data. Principal component analysis (PCA) on all markers was done on the genetic relationship matrix, using the prcomp function in R. The first three principal components (PC1, PC2 and PC3) values were plotted to visualize population structure.
5.7. Linkage Disequilibrium Estimation
Linkage disequilibrium (LD) (in terms of r2) analysis was performed for the whole genome as well as individually for each of the eighteen cassava chromosomes using package LDcorSV in R. To estimate LD decay, mean r2 value of each chromosome was computed and plotted against the chromosome’s physical distance to obtain a non-linear regression curve, and the LD decay distance was estimated as the physical distance between SNPs where average r2 reduced to half of the maximum LD value.
5.8. Marker-Trait Association Mapping
Search for marker-trait associations between genotypic and phenotypic data was performed for 22,103 filtered SNPs and the BLUP values obtained for the five flowering traits among the 293 filtered accessions. This was done using the Genomic Association and Prediction Integrated Tool (GAPIT) in R [98] and Mixed Linear Model (MLM) which was best at controlling type-I errors (false positives) for all of the traits evaluated. The first five PCs and marker-estimated kinship matrix in each case, were used. The genome-wide significance thresholds for each association study were assigned using Bonferroni correction set as 0.1. Significance of associations between traits and markers was assessed on the basis of p-values corrected for multiple testing. Manhattan plots as well as quantile-quantile (Q-Q) plots for association mapping were used to examine the validity of the SNPs and were visualized using the qqman package in R [98].
5.9. Identification of Putative Candidate Genes
To identify putative candidate genes residing at the close vicinity of high confidence SNPs, the associated SNPs with PVE ≥ 5% were mapped to the reference cassava genome, Manihot esculenta, v7.1 using the Phytozome tool v13 (https://phytozome-next.jgi.doe.gov/jbrowse/index.html?data=genomes/Mesculenta_v7_1). Transcripts present within 50Kb regions from both sides of associated SNPs were fetched along with their description. Identified genes were characterized for gene ontology including molecular and biological functions using UniProtKB tool (https://www.uniprot.org/). Additionally, gene and protein functions were searched using Alliance of Genomes Resources [101].
Supplementary Materials
Supporting information can be accessed at: www.cassavabase.org
Author Contributions
R.S.K. and Y.B., fund acquisition; J.K.B., methodology, investigation, data collection, curation and visualization, and writing manuscript; R.S.K., supervision of fieldwork and project administration; S.B.M., R.S.K., M.O.-S., W.E., E.N., T.A., A.O. and C.O., supervision of the general progress of the study and manuscript write-up; C.O., leader of Root Crops Program under which study was conducted. E.W. and M.K. played a role in data analysis. All authors contributed to the article and approved the submission for publication.
Funding
This research was funded by the “Next Generation Cassava Breeding Project” through a grant by the Bill & Melinda Gates Foundation (Grant INV- 007637) and the UK’s Foreign, Commonwealth & Development Office (FCDO), managed by Cornell University, through a sub-award agreement (grant number OPP1048542) between NaCRRI and Cornell University.
Data Availability Statement
Datasets generated for this study can be found in the article, supplementary material and at www.cassavabase.org
Acknowledgments
We thank the Root Crops Program and the management of NaCRRI for enabling the conduction of this research, and the field staff who assisted with the fieldwork. This research was also part of the Ph.D. thesis of the first author at Makerere University, and he thanks his supervisors and colleagues for the invaluable advice and support.
Conflicts of Interest
The authors declare no competing interests.
References
- Kaur, K.; Ahluwalia, P. Cassava as Potential Crop for the Food and Fermentation Industry: A review. Int. J. Food Ferment. Technol. 2017, 7. [Google Scholar] [CrossRef]
- Nanbol, K.K.; Namo, O.A.T. The contribution of root and tuber crops to food security: A review. J. Agric. Sci. Technol. B 2019, 9, 221–233. [Google Scholar]
- Li, S.; Cui, Y.; Zhou, Y.; Luo, Z.; Liu, J.; Zhao, M. The industrial applications of cassava: current status, opportunities and prospects. J. Sci. Food Agric. 2017, 97, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, H.; Hershey, C.; Iglesias, C.; Zhang, X. Fifty years of a public cassava breeding program: evolution of breeding objectives, methods, and decision-making processes. Theor. Appl. Genet. 2021, 134, 2335–2353. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, H.; Chareinsuk, R. Excellence in Cassava Breeding: Perspectives for the Future. Crop Breeding, Genetics and Genomics, 2020. [Google Scholar]
- Bandeira, E. , et al., Reproductive barriers in cassava: Factors and implications for genetic improvement. PLoS ONE 2021, 16, e0260576. [Google Scholar]
- Cock, J.H. and D.J. Connor, Chapter 19 - Cassava. In Crop Physiology Case Histories for Major Crops; Sadras, V.O., Calderini, D.F., Eds.; Academic Press, 2021; pp. 588–633. [Google Scholar]
- Alves, A.A.C. Cassava botany and phyiology. In Cassava: Biology, Production and Utilization; Hillocks, R.J., Thresh, J.M., Bellotti, A.C., Eds.; 2002; pp. 67–89. [Google Scholar]
- Ceballos, H.; Kulakow, P.; Hershey, C. Cassava Breeding: Current Status, Bottlenecks and the Potential of Biotechnology Tools. Trop. Plant Biol. 2012, 5, 73–87. [Google Scholar] [CrossRef]
- Perera, P.I. , et al., Comparative morphology, biology and histology of reproductive development in three lines of Manihot esculenta Crantz (Euphorbiaceae: Crotonoideae). AoB Plants 2013, 5, pls046. [Google Scholar] [CrossRef]
- Souza, L.S.; Alves, A.A.C.; de Oliveira, E.J. Phenological diversity of flowering and fruiting in cassava germplasm. Sci. Hortic. 2020, 265, 109253. [Google Scholar] [CrossRef]
- Ceballos, H.; Kawuki, R.S.; Gracen, V.E.; Yencho, G.C.; Hershey, C.H. Conventional breeding, marker-assisted selection, genomic selection and inbreeding in clonally propagated crops: a case study for cassava. Theor. Appl. Genet. 2015, 128, 1647–1667. [Google Scholar] [CrossRef] [PubMed]
- McSteen, P. and O. Leyser, Shoot Branching, in Annual Reviews. Plant Biology. 2005, Annual Reviews. 353–374.
- Gaarslev, N.; Swinnen, G.; Soyk, S. Meristem transitions and plant architecture—learning from domestication for crop breeding. Plant Physiol. 2021, 187, 1045–1056. [Google Scholar] [CrossRef]
- Sun, L.; Nie, T.; Chen, Y.; Yin, Z. From Floral Induction to Blooming: The Molecular Mysteries of Flowering in Woody Plants. Int. J. Mol. Sci. 2022, 23, 10959. [Google Scholar] [CrossRef]
- Bendahmane, M.; Dubois, A.; Raymond, O.; Le Bris, M. Genetics and genomics of flower initiation and development in roses. J. Exp. Bot. 2013, 64, 847–857. [Google Scholar] [CrossRef]
- Krishnamurthy, K.V. and B. Bahadur, Genetics of Flower Development, in Plant Biology and Biotechnology, B.B.e. al., Editor. 2015, Springer India: India. 385–407.
- Mallik, M. Flowering Control Mechanisms in Plants and Its Importance in Crop Production and Breeding. Int. J. Pure Appl. Biosci. 2018, 6, 1033–1038. [Google Scholar] [CrossRef]
- Tokunaga, H., et al., Field transcriptome analysis reveals a molecular mechanism for cassava flowering in a mountainous environment in SE Asia. Plant Molecular Biology. 2020.
- Hyde, P.T. and T.L. Setter, Long-day photoperiod and cool temperature induce flowering in cassava: Expression of signaling genes. Front. Plant Sci. 2022, 13, 973206. [Google Scholar] [CrossRef] [PubMed]
- Adeyemo, O.S.; Hyde, P.T.; Setter, T.L. Identification of FT family genes that respond to photoperiod, temperature and genotype in relation to flowering in cassava (Manihot esculenta, Crantz). Plant Reprod. 2018, 32, 181–191. [Google Scholar] [CrossRef]
- Bull, S.E.; Alder, A.; Barsan, C.; Kohler, M.; Hennig, L.; Gruissem, W.; Vanderschuren, H. FLOWERING LOCUS T Triggers Early and Fertile Flowering in Glasshouse Cassava (Manihot esculenta Crantz). Plants 2017, 6, 22. [Google Scholar] [CrossRef]
- Hasan, N.; Choudhary, S.; Naaz, N.; Sharma, N.; Laskar, R.A. Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. J. Genet. Eng. Biotechnol. 2021, 19, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.E., S. Song, and A.H. Schnell, Model-based linkage analysis of a quantitative trait. Statistical Human Genetics: Methods and Protocols. 2017; 283–310.
- Xu, Y. , et al., Quantitative Trait Locus Mapping and Identification of Candidate Genes Controlling Flowering Time in Brassica napus L. Front. Plant Sci. 2020, 11, 626205. [Google Scholar] [CrossRef]
- Sraphet, S. , et al., Quantitative trait loci underlying root yield and starch content in an F1 derived cassava population (Manihot esculenta Crantz). The Journal of Agricultural Science 2016, 155, 569–581. [Google Scholar] [CrossRef]
- Hmwe, N.H.; Sraphet, S.; Srisawad, N.; Smith, D.; Triwitayakorn, K. Identification of QTL Underlying Plant Height and First Branch Height of Cassava. Philipp. J. Sci. 2022, 151. [Google Scholar] [CrossRef]
- Tappiban, P.; Smith, D.R.; Triwitayakorn, K.; Bao, J. Recent understanding of starch biosynthesis in cassava for quality improvement: A review. Trends in Food Science & Technology 2019, 83, 167–180. [Google Scholar]
- Garcia-Oliveira, A.L.; Kimata, B.; Kasele, S.; Kapinga, F.; Masumba, E.; Mkamilo, G.; Sichalwe, C.; Bredeson, J.V.; Lyons, J.B.; Shah, T.; et al. Genetic analysis and QTL mapping for multiple biotic stress resistance in cassava. PLoS ONE 2020, 15, e0236674. [Google Scholar] [CrossRef]
- Challa, S. and N.R.R. Neelapu, Genome-Wide Association Studies (GWAS) for Abiotic Stress Tolerance in Plants, in Biochemical, Physiological and Molecular Avenues for Combating Abiotic Stress Tolerance in Plants. 2018; 135–150.
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Kostova, D.; Bulut, M.; Fernie, A.R. Genome-wide association studies: assessing trait characteristics in model and crop plants. Cell. Mol. Life Sci. 2021, 78, 5743–5754. [Google Scholar] [CrossRef]
- Uffelmann, E. , et al., Genome-wide association studies. Nature Reviews Methods Primers 2021, 1. [Google Scholar] [CrossRef]
- Togninalli, M.; Seren, .; Meng, D.; Fitz, J.; Nordborg, M.; Weigel, D.; Borgwardt, K.; Korte, A.; Grimm, D.G. The AraGWAS Catalog: a curated and standardized Arabidopsis thaliana GWAS catalog. Nucleic Acids Res. 2018, 46, D1150–D1156. [CrossRef]
- Sasaki, E.; Köcher, T.; Filiault, D.L.; Nordborg, M. Revisiting a GWAS peak in Arabidopsis thaliana reveals possible confounding by genetic heterogeneity. Heredity 2021, 127, 245–252. [Google Scholar] [CrossRef]
- Yano, K.; Morinaka, Y.; Wang, F.; Huang, P.; Takehara, S.; Hirai, T.; Ito, A.; Koketsu, E.; Kawamura, M.; Kotake, K.; et al. GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proc. Natl. Acad. Sci. 2019, 116, 21262–21267. [Google Scholar] [CrossRef]
- Ruanjaichon, V.; Khammona, K.; Thunnom, B.; Suriharn, K.; Kerdsri, C.; Aesomnuk, W.; Yongsuwan, A.; Chaomueang, N.; Thammapichai, P.; Arikit, S.; et al. Identification of Gene Associated with Sweetness in Corn (Zea mays L.) by Genome-Wide Association Study (GWAS) and Development of a Functional SNP Marker for Predicting Sweet Corn. Plants 2021, 10, 1239. [Google Scholar] [CrossRef] [PubMed]
- Tomkowiak, A.; Bocianowski, J.; Wolko, .; Adamczyk, J.; Mikołajczyk, S.; Kowalczewski, P.. Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.). Plants 2019, 8, 330. [CrossRef] [PubMed]
- Zhou, H.; Xiao, X.; Asjad, A.; Han, D.; Zheng, W.; Xiao, G.; Huang, Y.; Zhou, Q. Integration of GWAS and transcriptome analyses to identify SNPs and candidate genes for aluminum tolerance in rapeseed (Brassica napus L.). BMC Plant Biol. 2022, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Gupta, S.; Gahlaut, V.; Muthamilarasan, M.; Bandyopadhyay, T.; Ramchiary, N.; Prasad, M. Genome-Wide Association Study of Major Agronomic Traits in Foxtail Millet (Setaria italica L.) Using ddRAD Sequencing. Sci. Rep. 2019, 9, 5020. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.D.D.; e Sousa, M.B.; Brito, A.C.; de Oliveira, E.J. Genome-wide association studies for waxy starch in cassava. Euphytica 2020, 216, 1–17. [Google Scholar] [CrossRef]
- Rabbi, I.Y.; Udoh, L.I.; Wolfe, M.; Parkes, E.Y.; Gedil, M.A.; Dixon, A.; Ramu, P.; Jannink, J.; Kulakow, P. Genome-Wide Association Mapping of Correlated Traits in Cassava: Dry Matter and Total Carotenoid Content. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Ikeogu, U.N. , et al. Genetic Correlation, Genome-Wide Association and Genomic Prediction of Portable NIRS Predicted Carotenoids in Cassava Roots. Front. Plant Sci. 2019, 10, 1570. [Google Scholar] [CrossRef] [PubMed]
- Esuma, W.; Herselman, L.; Labuschagne, M.T.; Ramu, P.; Lu, F.; Baguma, Y.; Buckler, E.S.; Kawuki, R.S. Genome-wide association mapping of provitamin A carotenoid content in cassava. Euphytica 2016, 212, 97–110. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Rabbi, I.Y.; Egesi, C.; Hamblin, M.; Kawuki, R.; Kulakow, P.; Lozano, R.; Del Carpio, D.P.; Ramu, P.; Jannink, J.-L. Genome-Wide Association and Prediction Reveals Genetic Architecture of Cassava Mosaic Disease Resistance and Prospects for Rapid Genetic Improvement. Plant Genome 2016, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nandudu, L.; Kawuki, R.; Ogbonna, A.; Kanaabi, M.; Jannink, J.-L. Genetic dissection of cassava brown streak disease in a genomic selection population. Front. Plant Sci. 2023, 13, 5627. [Google Scholar] [CrossRef]
- Kayondo, S.I.; Del Carpio, D.P.; Lozano, R.; Ozimati, A.; Wolfe, M.; Baguma, Y.; Gracen, V.; Offei, S.; Ferguson, M.; Kawuki, R.; et al. Genome-wide association mapping and genomic prediction for CBSD resistance in Manihot esculenta. Sci. Rep. 2018, 8, 1549. [Google Scholar] [CrossRef]
- Silva, P.P.d.S.; e Sousa, M.B.; de Oliveira, E.J.; Morgante, C.V.; de Oliveira, C.R.S.; Vieira, S.L.; Borel, J.C. Genome-wide association study of drought tolerance in cassava. Euphytica 2021, 217, 1–26. [Google Scholar] [CrossRef]
- Diack, O.; Kanfany, G.; Gueye, M.C.; Sy, O.; Fofana, A.; Tall, H.; Serba, D.D.; Zekraoui, L.; Berthouly-Salazar, C.; Vigouroux, Y.; et al. GWAS unveils features between early- and late-flowering pearl millets. BMC Genom. 2020, 21, 777. [Google Scholar] [CrossRef]
- Yuan, Y.; Cairns, J.E.; Babu, R.; Gowda, M.; Makumbi, D.; Magorokosho, C.; Zhang, A.; Liu, Y.; Wang, N.; Hao, Z.; et al. Genome-Wide Association Mapping and Genomic Prediction Analyses Reveal the Genetic Architecture of Grain Yield and Flowering Time Under Drought and Heat Stress Conditions in Maize. Front. Plant Sci. 2019, 9, 1919. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Habyarimana, E.; Karaköy, T.; Baloch, F.S. Genetic dissection of days to flowering via genome-wide association studies in Turkish common bean germplasm. Physiol. Mol. Biol. Plants 2021, 27, 1609–1622. [Google Scholar] [CrossRef] [PubMed]
- Raggi, L.; Caproni, L.; Carboni, A.; Negri, V. Genome-Wide Association Study Reveals Candidate Genes for Flowering Time Variation in Common Bean (Phaseolus vulgaris L.). Front. Plant Sci. 2019, 10, 962. [Google Scholar] [CrossRef]
- Sabag, I.; Morota, G.; Peleg, Z. Genome-wide association analysis uncovers the genetic architecture of tradeoff between flowering date and yield components in sesame. BMC Plant Biol. 2021, 21, 549. [Google Scholar] [CrossRef] [PubMed]
- Xu, L. , et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res. 2016, 23, 43–52. [Google Scholar] [PubMed]
- Akhatar, J.; Goyal, A.; Kaur, N.; Atri, C.; Mittal, M.; Singh, M.P.; Kaur, R.; Rialch, I.; Banga, S.S. Genome wide association analyses to understand genetic basis of flowering and plant height under three levels of nitrogen application in Brassica juncea (L.) Czern & Coss. Sci. Rep. 2021, 11, 4278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, Q.; Cregan, P.B.; Nelson, R.L.; Wang, X.; Wu, J.; Jiang, G.-L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 2015, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, X.; Lu, C.; Ye, J.; Zou, M.; Lu, K.; Feng, S.; Pei, J.; Liu, C.; Zhou, X.; et al. Genome-Wide Association Studies of 11 Agronomic Traits in Cassava (Manihot esculenta Crantz). Front. Plant Sci. 2018, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The MYB80 Transcription Factor Is Required for Pollen Development and the Regulation of Tapetal Programmed Cell Death inArabidopsis thaliana. Plant Cell 2011, 23, 2209–2224. [Google Scholar] [CrossRef]
- Jiang, D. , et al. Arabidopsis relatives of the human lysine-specific Demethylase1 repress the expression of FWA and FLOWERING LOCUS C and thus promote the floral transition. Plant Cell 2007, 19, 2975–2987. [Google Scholar] [CrossRef]
- Burko, Y.; Geva, Y.; Refael-Cohen, A.; Shleizer-Burko, S.; Shani, E.; Berger, Y.; Halon, E.; Chuck, G.; Moshelion, M.; Ori, N. From Organelle to Organ: ZRIZI MATE-Type Transporter is an Organelle Transporter that Enhances Organ Initiation. Plant Cell Physiol. 2011, 52, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Wang, Y.; Li, W.; Chen, Y.; Deng, Y.; Zhang, X.; Chen, L.; Ye, D. The Arabidopsis eukaryotic translation initiation factor 3, subunit F (AteIF3f), is required for pollen germination and embryogenesis. Plant J. 2010, 63, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Rao, Y.; Yu, H.; Xu, Q.; Cui, Y.; Xia, S.; Yu, X.; Liu, H.; Hu, H.; Xue, D.; et al. MORE FLORET1 Encodes a MYB Transcription Factor That Regulates Spikelet Development in Rice. Plant Physiol. 2020, 184, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Edstam, M.M.; Edqvist, J. Involvement of GPI-anchored lipid transfer proteins in the development of seed coats and pollen inArabidopsis thaliana. Physiol. Plant. 2014, 152, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lebel-Hardenack, S. , et al. Conserved expression of a TASSELSEED2 homolog in the tapetum of the dioecious Silene latifolia and Arabidopsis thaliana. Plant J. 1997, 12, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Guan, X.; Xu, Y.; Wang, Y. Characterization of novel gene expression related to glyoxal oxidase by agro-infiltration of the leaves of accession Baihe-35-1 of Vitis pseudoreticulata involved in production of H2O2 for resistance to Erysiphe necator. Protoplasma 2013, 250, 765–777. [Google Scholar] [CrossRef]
- Wang, R. , et al. A subgroup of MATE transporter genes regulates hypocotyl cell elongation in Arabidopsis. J Exp Bot. 2015, 66, 6327–6343. [Google Scholar] [CrossRef]
- Taneva, K., V. Bozhanova, and I. Petrova. Variability, heritability and genetic advance of some grain quality traits and grain yield in durum wheat genotypes. Bulgarian Journal of Agricultural Science 2019, 25, 288–295. [Google Scholar]
- Young, A.I. Solving the missing heritability problem. PLOS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef]
- Ndukauba, J. , et al. Variability in Egusi-Melon Genotypes (Citrullus lanatus (Thumb) Matsum and Nakai) in Derived Savannah. International Journal of Plant Research 2015, 5, 19–26. [Google Scholar]
- Schmidt, P.; Hartung, J.; Bennewitz, J.; Piepho, H.-P. Heritability in Plant Breeding on a Genotype-Difference Basis. Genetics 2019, 212, 991–1008. [Google Scholar] [CrossRef]
- Favour, E.; Emeka, N.; Chiedozie, E.; Bunmi, O.; Emmanuel, O. Genetic variability, heritability and variance components of some yield and yield related traits in second backcross population (BC2) of cassava. Afr. J. Plant Sci. 2017, 11, 185–189. [Google Scholar] [CrossRef]
- Rodrmguez, E.P.B.; Morante, N.; Salazar, S.; Hyde, P.T.; Setter, T.L.; Kulakow, P.; Aparicio, J.S.; Zhang, X. Flower-inducing technology facilitates speed breeding in cassava. Front. Plant Sci. 2023, 14, 1172056. [Google Scholar] [CrossRef] [PubMed]
- Aquilino, L.; Pariyo, A.; Baguma, Y.; Edema, R.; Gibson, P.; Bisikwa, J. Genotypes by environment effect on flowering and seed set in cassava, Manihot esculenta Crantz in Uganda. J. Food, Nutr. Agric. 2021, 26–37. [Google Scholar] [CrossRef]
- Ren, D.; Cai, X.; Lin, Q.; Ye, H.; Teng, J.; Li, J.; Ding, X.; Zhang, Z. Impact of linkage disequilibrium heterogeneity along the genome on genomic prediction and heritability estimation. Genet. Sel. Evol. 2022, 54, 47. [Google Scholar] [CrossRef] [PubMed]
- Leelawijitkul, S. , et al. Correlation Between Relative Gene Expression Patterns of Two Flowering locus T (MeFT1 and MeFT2) in Cassava Leaf and Flowering Traits Under Different Flowering Induction Conditions. Pak J Biol Sci. 2022, 25, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Morante, N.; Salazar, S.; Cuásquer, J.; Hyde, P.T.; Setter, T.L.; Ceballos, H. Induction of Earlier Flowering in Cassava through Extended Photoperiod. Agronomy 2020, 10, 1273. [Google Scholar] [CrossRef]
- Ochoa, A.; Storey, J.D. Estimating FST and kinship for arbitrary population structures. PLOS Genet. 2021, 17, e1009241. [Google Scholar] [CrossRef]
- Meisner, J.; Albrechtsen, A. Inferring Population Structure and Admixture Proportions in Low-Depth NGS Data. Genetics 2018, 210, 719–731. [Google Scholar] [CrossRef]
- Li, H.; Ralph, P.L. Local PCA Shows How the Effect of Population Structure Differs Along the Genome. Genetics 2018, 211, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Bangarwa, S.K. , et al. Association Mapping in Crops Plants. International Journal of Current Microbiology and Applied Sciences 2020, 9, 1313–1325. [Google Scholar] [CrossRef]
- Vos, P.G.; Paulo, M.J.; Voorrips, R.E.; Visser, R.G.F.; van Eck, H.J.; van Eeuwijk, F.A. Evaluation of LD decay and various LD-decay estimators in simulated and SNP-array data of tetraploid potato. Theor. Appl. Genet. 2017, 130, 123–135. [Google Scholar] [CrossRef]
- de Albuquerque, H.Y.G. and C.D. do Carmo, Genetic diversity of Manihot esculenta Crantz germplasm based on SNP markers. Annals of Applied Biology 2018, 1–14. [Google Scholar]
- Santos, C.S.D. , et al. Genome-wide association study of cassava starch paste properties. PLoS ONE 2022, 17, e0262888. [Google Scholar] [CrossRef]
- Rabbi, I.Y.; Kayondo, S.I.; Bauchet, G.; Yusuf, M.; Aghogho, C.I.; Ogunpaimo, K.; Uwugiaren, R.; Smith, I.A.; Peteti, P.; Agbona, A.; et al. Genome-wide association analysis reveals new insights into the genetic architecture of defensive, agro-morphological and quality-related traits in cassava. Plant Mol. Biol. 2020, 109, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Simko, I.; Haynes, K.G.; Jones, R.W. Assessment of Linkage Disequilibrium in Potato Genome With Single Nucleotide Polymorphism Markers. Genetics 2006, 173, 2237–2245. [Google Scholar] [CrossRef]
- D’hoop, B.B.; Paulo, M.J.; Kowitwanich, K.; Sengers, M.; Visser, R.G.F.; van Eck, H.J.; van Eeuwijk, F.A. Population structure and linkage disequilibrium unravelled in tetraploid potato. Theor. Appl. Genet. 2010, 121, 1151–1170. [Google Scholar] [CrossRef]
- Braulio, J. and S. Cloutier. Association Mapping in Plant Genomes. In Genetic Diversity in Plants; p. 2012.
- Mulugeta, B.; Tesfaye, K.; Ortiz, R.; Johansson, E.; Hailesilassie, T.; Hammenhag, C.; Hailu, F.; Geleta, M. Marker-trait association analyses revealed major novel QTLs for grain yield and related traits in durum wheat. Front. Plant Sci. 2023, 13, 1009244. [Google Scholar] [CrossRef]
- Wang, B.; Smith, S.M.; Li, J. Genetic Regulation of Shoot Architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef]
- Greb, T. , et al. Molecular analysis of the LATERAL SUPPRESSOR gene in Arabidopsis reveals a conserved control mechanism for axillary meristem formation. Genes Dev. 2003, 17, 1175–1187. [Google Scholar] [CrossRef]
- Shao, G.; Lu, Z.; Xiong, J.; Wang, B.; Jing, Y.; Meng, X.; Liu, G.; Ma, H.; Liang, Y.; Chen, F.; et al. Tiller Bud Formation Regulators MOC1 and MOC3 Cooperatively Promote Tiller Bud Outgrowth by Activating FON1 Expression in Rice. Mol. Plant 2019, 12, 1090–1102. [Google Scholar] [CrossRef]
- Shim, S. , et al. GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill). Int J Mol Sci. 2019, 20. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Liu, D.; Ma, S.; Dai, Y.; Zhang, X.; Wang, Y.; Hu, T.; Xiao, M.; Zhou, Y.; et al. Identification and Expression of the Multidrug and Toxic Compound Extrusion (MATE) Gene Family in Capsicum annuum and Solanum tuberosum. Plants 2020, 9, 1448. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, X.; Lu, C.; Zeng, X.; Li, Y.; Fu, D.; Wu, G. Non-specific lipid transfer proteins in plants: presenting new advances and an integrated functional analysis. J. Exp. Bot. 2015, 66, 5663–5681. [Google Scholar] [CrossRef]
- Ozimati, A.; Kawuki, R.; Esuma, W.; Kayondo, I.S.; Wolfe, M.; Lozano, R.; Rabbi, I.; Kulakow, P.; Jannink, J.-L. Training Population Optimization for Prediction of Cassava Brown Streak Disease Resistance in West African Clones. G3 Genes|Genomes|Genetics 2018, 8, 3903–3913. [Google Scholar] [CrossRef] [PubMed]
- Ozimati, A.; Kawuki, R.; Esuma, W.; Kayondo, S.I.; Pariyo, A.; Wolfe, M.; Jannink, J.-L. Genetic Variation and Trait Correlations in an East African Cassava Breeding Population for Genomic Selection. Crop. Sci. 2019, 59, 460–473. [Google Scholar] [CrossRef]
- Rife, T.W.; Poland, J.A. Field Book: An Open-Source Application for Field Data Collection on Android. Crop. Sci. 2014, 54, 1624–1627. [Google Scholar] [CrossRef]
- R-Core-Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Lloyd-Smith, J.O. Maximum Likelihood Estimation of the Negative Binomial Dispersion Parameter for Highly Overdispersed Data, with Applications to Infectious Diseases. PLOS ONE 2007, 2, e180. [Google Scholar] [CrossRef] [PubMed]
- Towers, S. Negative Binomial Likelihood Fits for Overdispersed Count Data. 2018. [Google Scholar]
- Alliance of Genome Resources, C. Alliance of Genome Resources Portal: unified model organism research platform. Nucleic Acids Res. 2020, 48, D650–D658. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Density distribution of variation for flower and flowering traits in cassava.
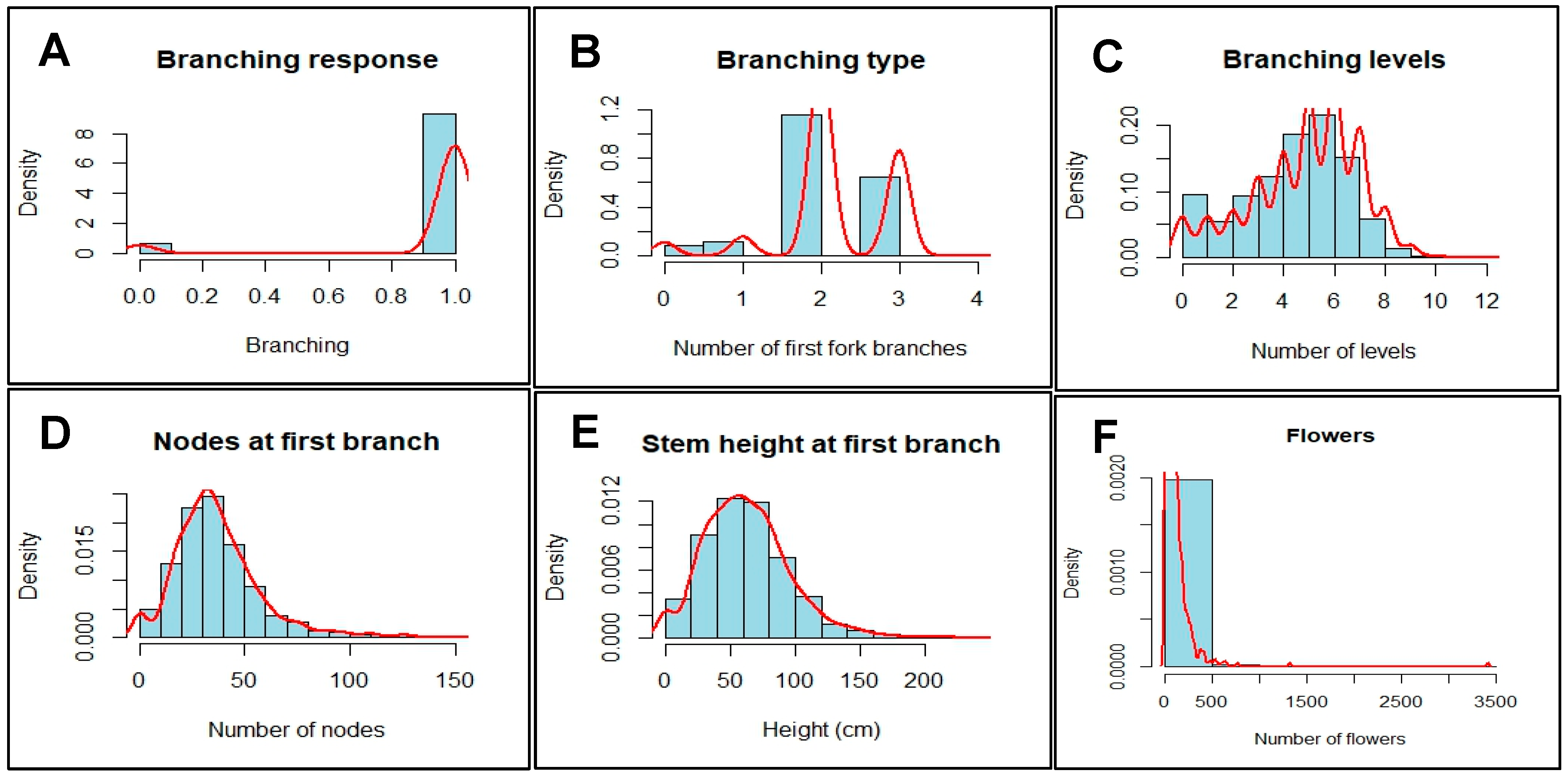
Figure 2.
Spearman correlation coefficient (r) for flowering traits and flowers evaluated across two seasons and locations. Branch1_No = branch type at first branching; Branch1_Nodes = number of nodes at first branching; Branch1_Ht = stem height at first branching.
Figure 2.
Spearman correlation coefficient (r) for flowering traits and flowers evaluated across two seasons and locations. Branch1_No = branch type at first branching; Branch1_Nodes = number of nodes at first branching; Branch1_Ht = stem height at first branching.
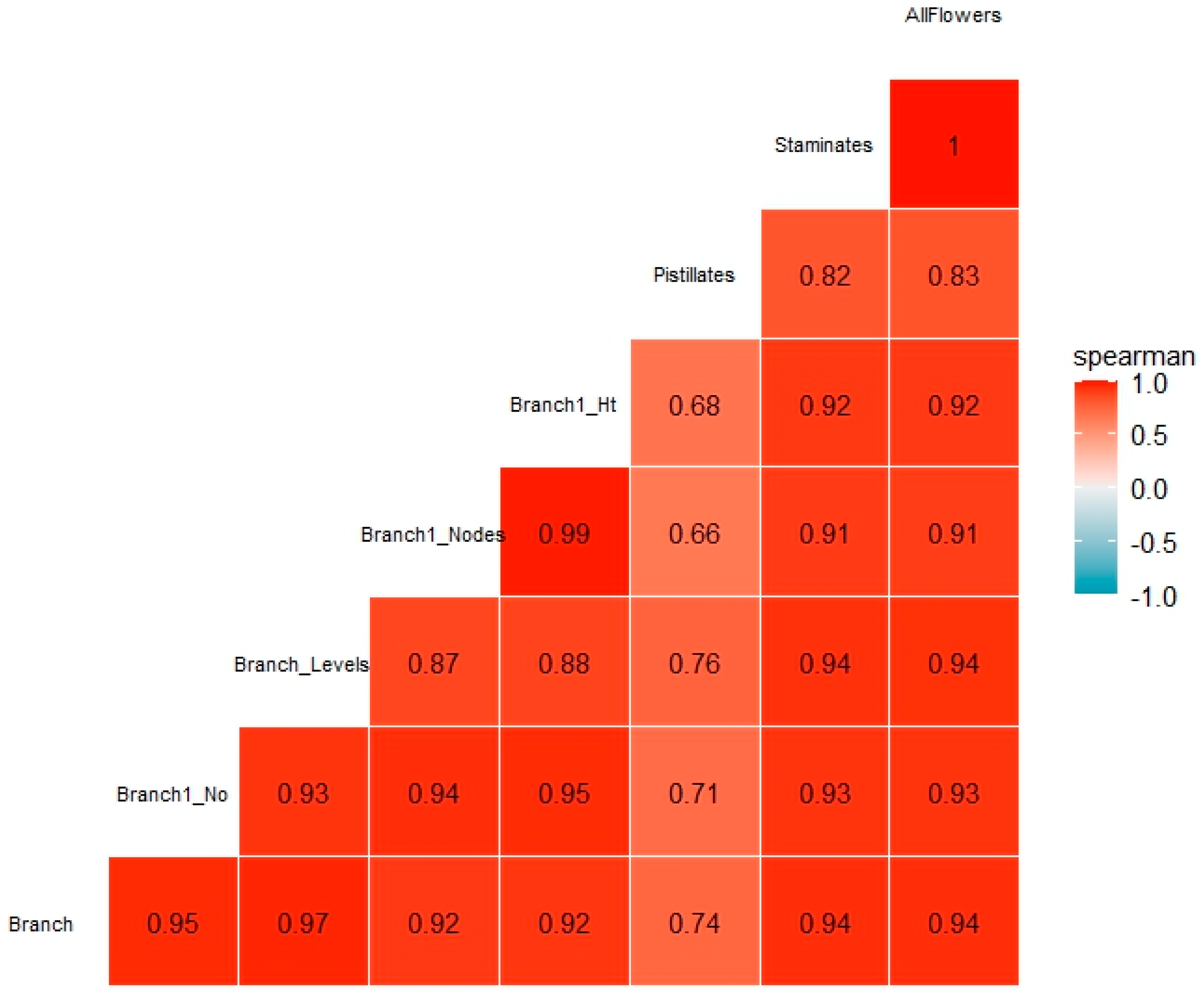
Figure 3.
Distribution of SNP markers across the 18 chromosomes. The graph represents the number of imputed SNPs (24040) within a 1 mega base window, horizontal axis displays the chromosome length.
Figure 3.
Distribution of SNP markers across the 18 chromosomes. The graph represents the number of imputed SNPs (24040) within a 1 mega base window, horizontal axis displays the chromosome length.
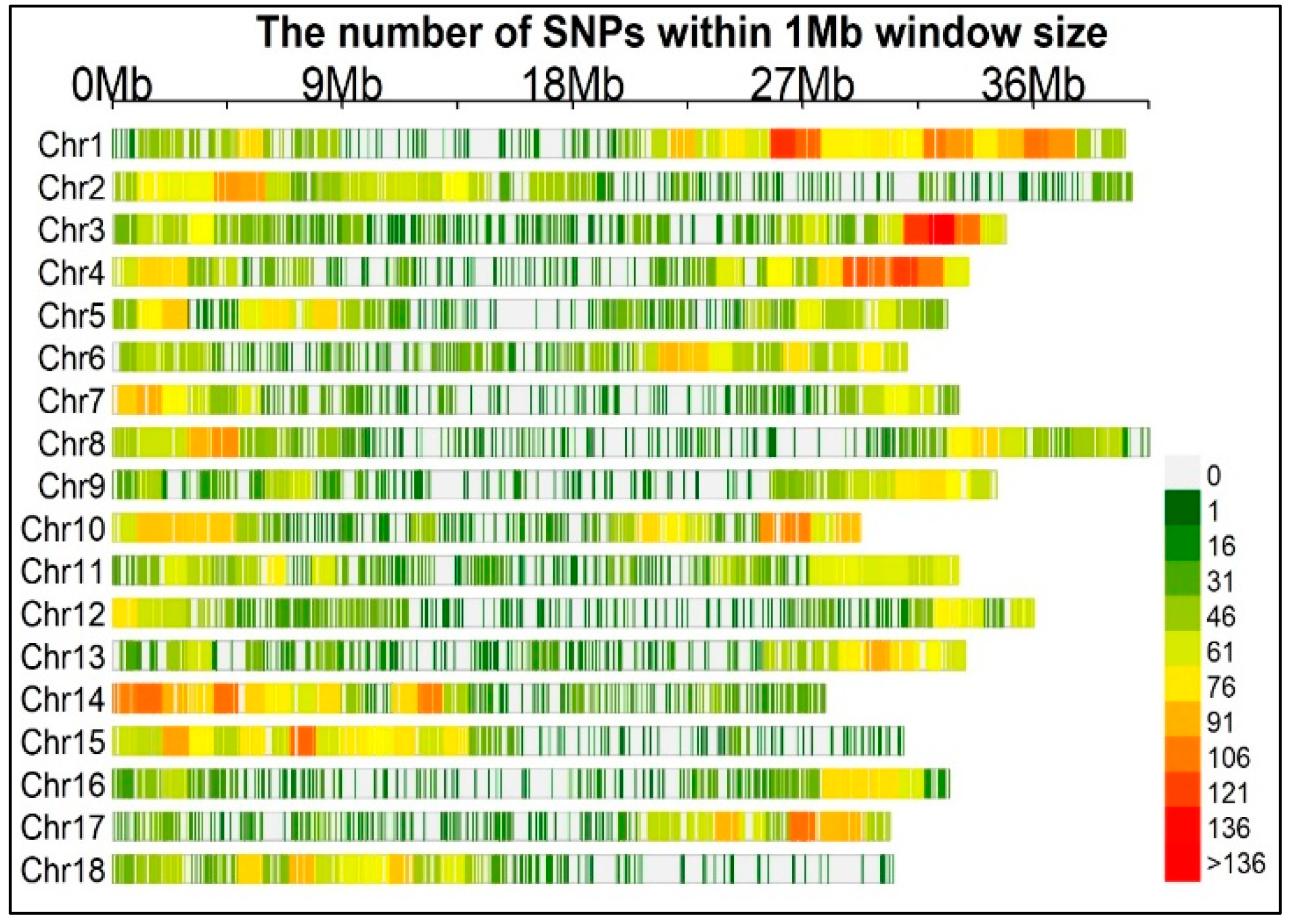
Figure 4.
Plots of principle components and kinship: A) displays the distribution of clones in PC1-PC2; B) Heatmap showing pairwise genomic relationship matrix of 293 accessions, from no relationship (yellow) to high relationship (red).
Figure 4.
Plots of principle components and kinship: A) displays the distribution of clones in PC1-PC2; B) Heatmap showing pairwise genomic relationship matrix of 293 accessions, from no relationship (yellow) to high relationship (red).
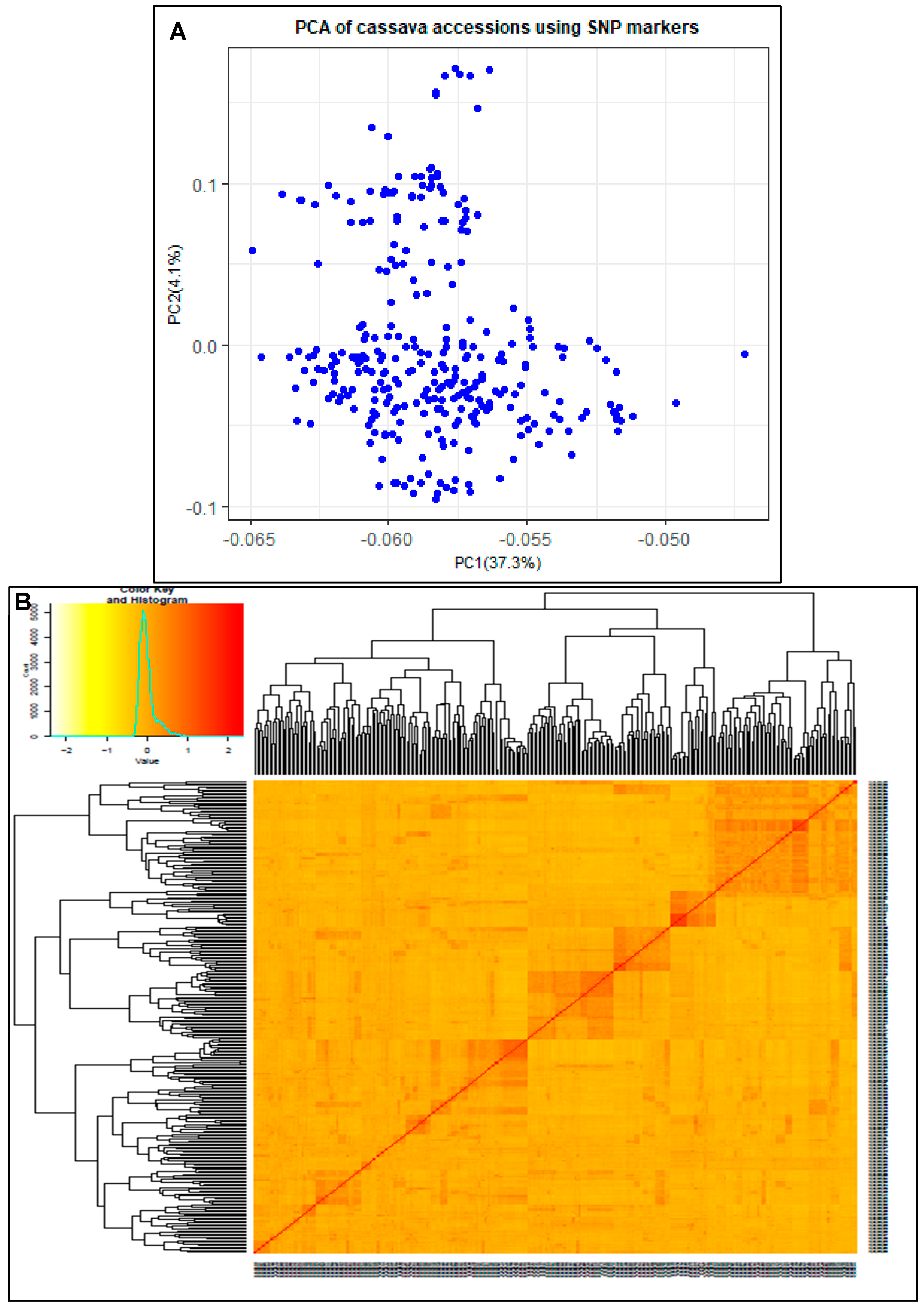
Figure 5.
Plot of LD decay of 22,103 SNPs on 18 cassava chromosomes.
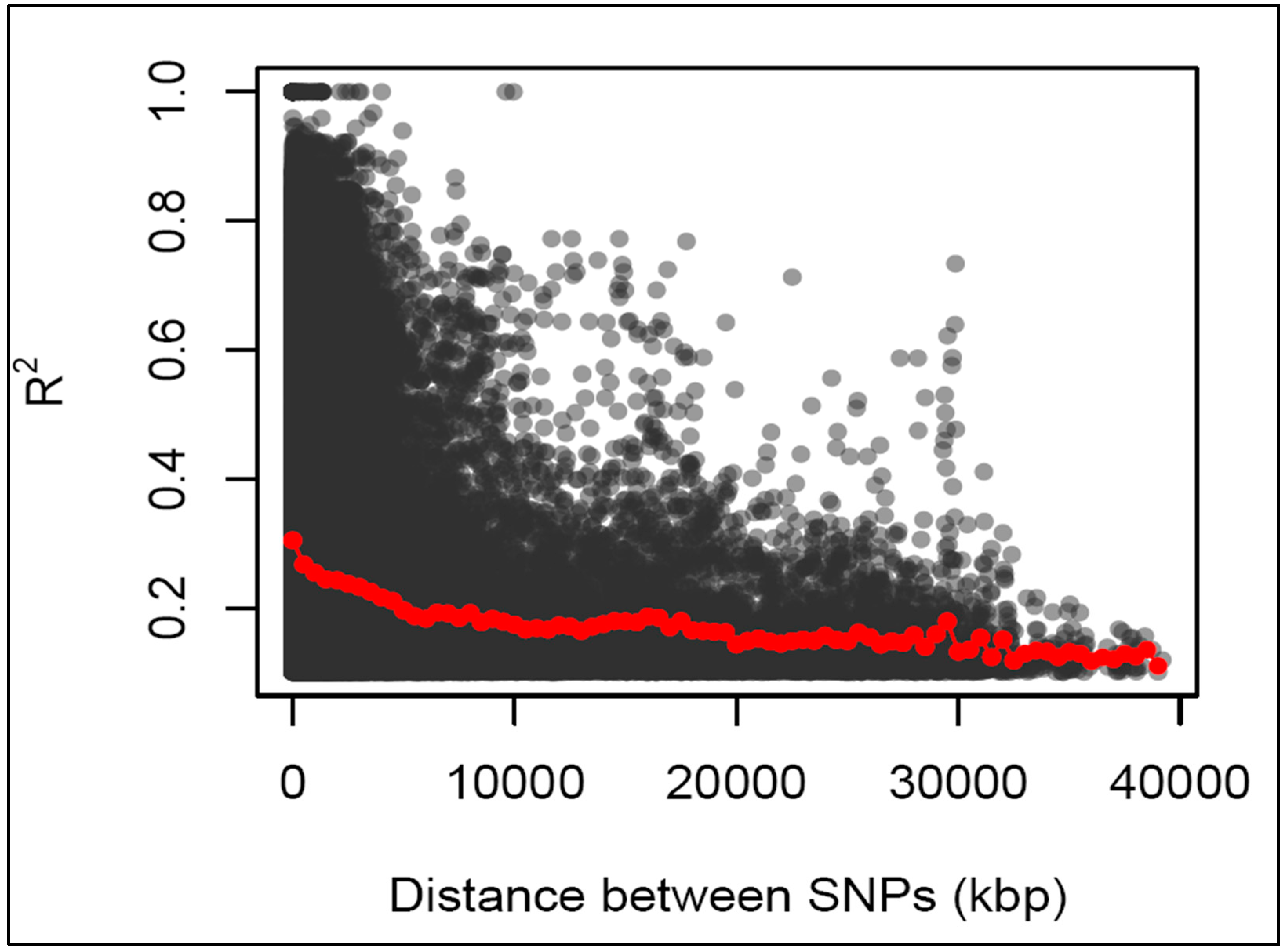
Figure 6.
GWAS of five flowering traits across 18 chromosomes in 293 cassava accessions: (A – E) Manhattan plots of chromosomal positions; (F – J) quantile-quantile (Q-Q) plots. In all Manihatttan plots, the green solid horizontal line indicates a significant line denoting the (-log10(P) value significance threshold and the green dashed line indicates a suggestive Bonferroni threshold (-log10(P) = 0.1). Plots in red outline compare effects of different models on the same trait.
Figure 6.
GWAS of five flowering traits across 18 chromosomes in 293 cassava accessions: (A – E) Manhattan plots of chromosomal positions; (F – J) quantile-quantile (Q-Q) plots. In all Manihatttan plots, the green solid horizontal line indicates a significant line denoting the (-log10(P) value significance threshold and the green dashed line indicates a suggestive Bonferroni threshold (-log10(P) = 0.1). Plots in red outline compare effects of different models on the same trait.
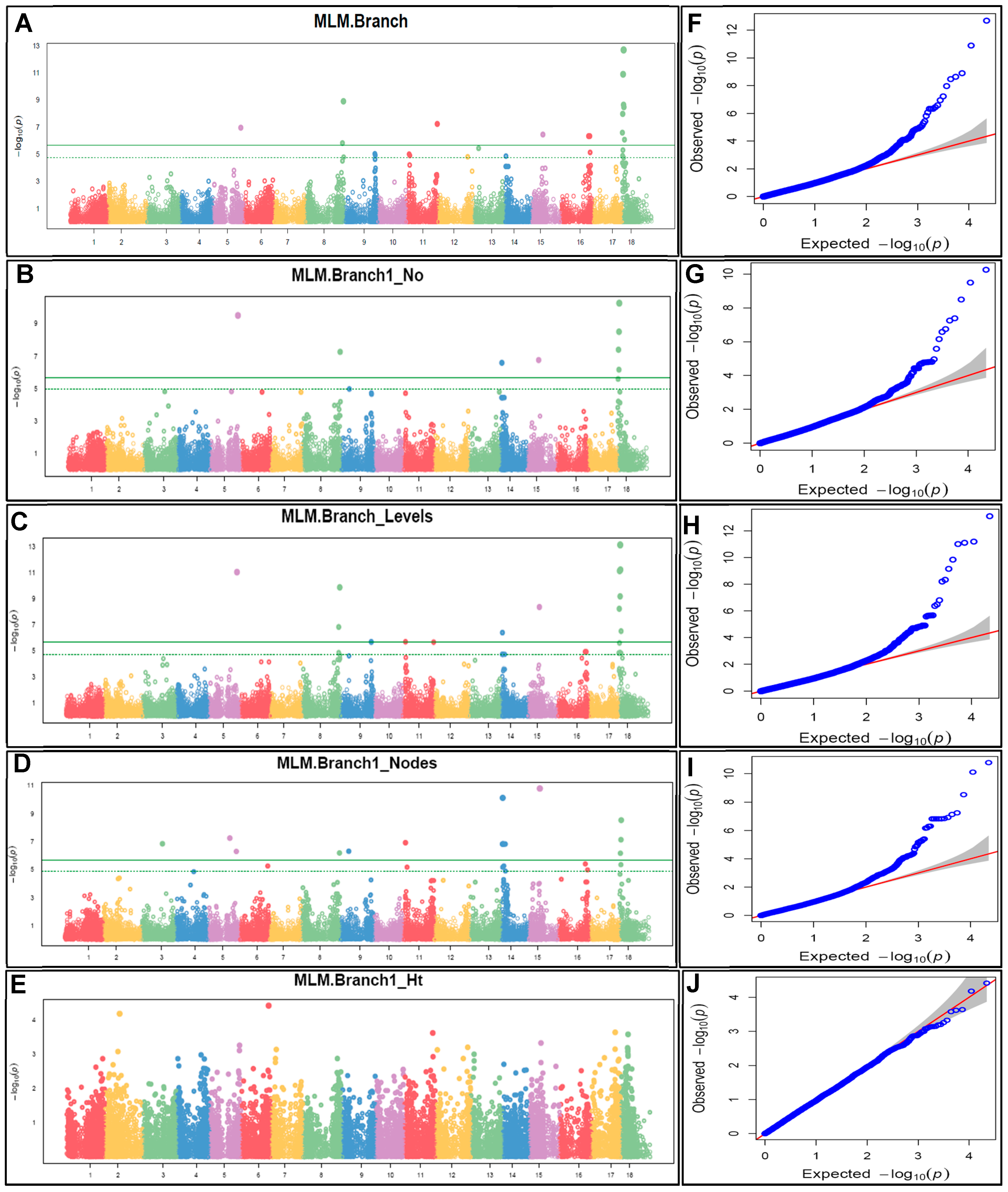

Table 1.
Phenotype variation and heritability estimates of five flowering traits assessed in the cassava GWAS panel.
Table 1.
Phenotype variation and heritability estimates of five flowering traits assessed in the cassava GWAS panel.
| Variables | Branching | Branch-type | Branching levels (number) | Nodes (number) | Stem height (cm) | Pistillates (number) | Staminates (number) |
|---|---|---|---|---|---|---|---|
| Observations (n) | 1565 | 1,565 | 1,565 | 1,547 | 1,562 | 801 | 801 |
| Mean | 0.93 | 2.19 | 4.86 | 36.90 | 63.06 | 2.09 | 34.63 |
| Skewedness | - | + | - | + | + | + | + |
| SEM | 0.01 | 0.02 | 0.05 | 0.51 | 0.85 | 0.30 | 8.51 |
| CI (0.95) | 0.01 | 0.04 | 0.11 | 1.01 | 1.67 | 0.59 | 16.72 |
| Variance | 0.06 | 0.51 | 4.60 | 407.04 | 1119.56 | 39.00 | 31506.04 |
| SD | 0.25 | 0.71 | 2.15 | 20.18 | 33.46 | 6.25 | 177.50 |
| CV (%) | 26.87 | 32.72 | 44.16 | 54.66 | 53.08 | 2.98 | 5.13 |
| Heritability, H2 | 0.34 | 0.53 | 0.38 | 0.42 | 0.60 | 0.83 | 0.00 |
| SNP heritability, h2 | 0.25 | 0.00 | 0.19 | 0.01 | 0.35 | - | - |
| Significance | * | *** | *** | *** | * | * | *** |
Table 2.
Analysis of variance for flowering traits assessed in 293 cassava accessions.
| Variable | Sums of squares | Mean squares | F value | Significance | ||||
|---|---|---|---|---|---|---|---|---|
| Acc | Loc | Acc | Loc | Acc | Loc | Acc | Loc | |
| Branching | 57.143 | 0.634 | 0.12841 | 0.63371 | 3.533 | 17.4353 | *** | *** |
| Branch type | 435.27 | 0.96 | 0.9781 | 0.9603 | 3.2537 | 3.1945 | *** | ns |
| Branching levels | 5021.7 | 367.3 | 11.28 | 367.26 | 9.733 | 316.7597 | *** | *** |
| Nodes (at 1st branch) | 280458 | 16954 | 630.2 | 16954 | 2.4511 | 65.9356 | *** | *** |
| Stem height (1st branch) (cm) | 755806 | 4734 | 1698 | 4734 | 2.2902 | 6.3829 | *** | * |
Acc, accession; Loc, location. ANOVA tests of significant main effects are indicated: P ≤ 0.10 (•), P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***), ns not significant
Table 3.
Marker-trait associations for flowering traits of 293 cassava accessions.
| Trait | No. of MTA | Chromosome | Associated SNP marker ID |
|---|---|---|---|
| Branching | 15 | 5 (01) | S5_29309724 |
| 8 (02) | S8_38134897; S8_39184412 | ||
| 11 (01) | S11_32333764 | ||
| 15 (01) | S15_11747301 | ||
| 16 (03) | S16_28288554; S16_28711444; S16_29508150 | ||
| 18 (07) | S18_263746; S18_597957; S18_1002380; S18_1489472; S18_1562744; S18_1832353; S18_2456168 | ||
| Branch type | 08 | 5 (01) | S5_29309724 |
| 8 (01) | S8_39184412 | ||
| 14 (01) | S14_1230231 | ||
| 15 (01) | S15_11747301 | ||
| 18 (04) | S18_1002380; S18_1489472; S18_1562744; S18_1832353 | ||
| Branching levels | 14 | 5 (01) | S5_29309724 |
| 8 (02) | S8_38134897; S8_39184412 | ||
| 9 (02) | S9_31290323; S9_31489987 | ||
| 11 (01) | S11_3127381 | ||
| 14 (01) | S14_1230231 | ||
| 15 (01) | S15_11747301 | ||
| 18 (06) | S18_597957; S18_1002380; S18_1489472; S18_1562744; S18_1832353; S18_2456168 | ||
| Nodes at 1st branch | 16 | 3 (01) | S3_21330906 |
| 5 (02) | S5_22566689; S5_29309724 | ||
| 8 (01) | S8_39184412 | ||
| 9 (01) | S9_8276994 | ||
| 11 (01) | S11_3127381 | ||
| 14 (06) | S14_964612; S14_1209616; S14_1230231; S14_1653289; S14_2247549; S14_3759369 | ||
| 15 (01) | S15_11747301 | ||
| 18 (03) | S18_1002380; S18_1562744; S18_1832353 | ||
| Stem height at 1st branch | - | - | - |
Table 4.
Summary of associated markers and number of candidate genes for flowering traits.
| SNP ID | Chromosome | No. of associated genes | Associated traits |
|---|---|---|---|
| S3_21330906 | 3 | 06 | Nodes at 1st branch |
| S5_22566689 | 5 | 04 | Nodes at 1st branch |
| S5_29309724 | 5 | 05 | Branching; Branch type; Branching levels; Nodes at 1st branch |
| S15_11747301 | 15 | 12 | Branching; Branch type; Branching levels; Nodes at 1st branch |
| S18_1832353 | 18 | 10 | Branching; Branch type; Branching levels |
MTA = marker-trait association, number in brackets shows number of MTA on the chromosome
Table 5.
Associated markers and putative candidate genes for cassava flowering traits according to cassava genome annotations.
Table 5.
Associated markers and putative candidate genes for cassava flowering traits according to cassava genome annotations.
| Trait | Associated SNP | Chr. No. | P.value | MAF | R2 | SNP Effect | PVE (%) | Putative candidate gene ID (Phytozome v13; Manihot 7.1) | Gene functional annotation (according to NCBI) |
|---|---|---|---|---|---|---|---|---|---|
| Nodes at 1st branch | S3_21330906 | 3 | 1.45 x10-7 | 0.017 | 0.106 | 0.82 | 12.94 | Manes.03G102000.1 | dol-P-Man:Man(6)GlcNAc(2)-PP-Dol alpha-1,2-mannosyltransferase |
| Manes.03G102100.1 | olee1-like protein | ||||||||
| Manes.03G101900.3 | phosphoenolpyruvate carboxylase | ||||||||
| Manes.03G102600.3 | release factor glutamine methyltransferase | ||||||||
| Manes.03G102400.1 | TPR repeat-containing thioredoxin TTL1 | ||||||||
| Manes.03G102200.1 | ubiquitin-conjugating enzyme E2 36 | ||||||||
| S5_22566689 | 5 | 5.75 x10-8 | 0.010 | 0.113 | -1.07 | 4.85 | Manes.05G131430.1 | mogroside IE synthase | |
| Manes.05G131410.1 Manes.05G131420.1 | mogroside IE synthase-like | ||||||||
| Manes.05G131440.1 | probably inactive leucine-rich repeat receptor-like protein kinase At5g48380 | ||||||||
| Branching; Branch type; Branching levels |
S18_1832353 | 18 | 1.15 x10-9 | 0.017 | 0.177 | -1.26 | 5.95 | Manes.18G016700.1 | aldehyde oxidase GLOX |
| Manes.18G016200.2 | cytochrome P450 83B1 | ||||||||
| Manes.18G016500.2 | histone H3.3 | ||||||||
| Manes.18G016725.1 Manes.18G016800.2 | lysine-specific histone demethylase 1 homolog 3 | ||||||||
| Manes.18G016372.1 | protein IQ-DOMAIN 32 | ||||||||
| Manes.18G016212.1 | protein RCC2 | ||||||||
| Manes.18G016775.1 | vicilin-like seed storage protein At2g28490 | ||||||||
| Branch type; Branching levels; Nodes at 1st branch |
S5_29309724 | 5 | 1.15 x10-9 | 0.010 | 0.147 | 1.36 | 26.86 | Manes.05G186700.1 | protein DETOXIFICATION 48 |
| Branching; Branch type; Branching levels; Nodes at 1st branch |
S5_29309724 | 5 | 1.57 x10-7 | 0.010 | 0.147 | 1.36 | 26.86 | Manes.05G186500.1 | receptor-like protein kinase 7 |
| Manes.05G186300.4 | reticulon-4-interacting protein 1 homolog, mitochondrial | ||||||||
| Manes.05G186600.1 | sucrose transport protein SUC4 | ||||||||
| Manes.05G186400.1 | xylulose kinase 2 | ||||||||
| S15_11747301 | 15 | 1.40 x10-7 | 0.014 | 0.141 | -1.14 | 11.48 | Manes.15G141100.1 Manes.15G141200.1 | endonuclease V | |
| Manes.15G140500.1 | eukaryotic translation initiation factor 3 subunit I | ||||||||
| Manes.15G140900.1 | myb family transcription factor MOF1 | ||||||||
| Manes.15G140700.1 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7-like | ||||||||
| Manes.15G140300.1 | non-specific lipid-transfer protein 4.1 | ||||||||
| Manes.15G141500.1 | protein CHROMATIN REMODELING 19 | ||||||||
| Manes.15G141300.1 Manes.15G141400.3 | protein indeterminate-domain 5, chloroplastic | ||||||||
| Manes.15G140400.1 | protein NRT1/ PTR FAMILY 8.3 | ||||||||
| Manes.15G140600.1 | short-chain dehydrogenase reductase ATA1 | ||||||||
| Manes.15G141700.1 | transcription factor bHLH111 | ||||||||
| S18_1832353 | 18 | 1.15 x10-9 | 0.017 | 0.177 | -1.26 | 5.95 | Manes.18G016750.1 Manes.18G017000.2 |
V-type proton ATPase 16 kDa proteolipid subunit |
Chr., chromosome; MAF, minor allele frequency; PVE, phenotype variance explained; NCBI, National Centre for Biotechnology Information
Table 6.
Functional annotation of putative genes for flowering traits in cassava.
| Trait | SNP | Ch | Gene ID | Description | Function | Reference species | References |
|---|---|---|---|---|---|---|---|
| Branch, Branch1_No, Branch_Levels | S18_1832353 | 18 | Manes.18G016700.1 | aldehyde oxidase GLOX | Catalyzes the oxidation of aldehydes to the corresponding carboxylate by coupling the reaction to the reduction of dioxygen to hydrogen peroxide; involved in anther development and play a role in tapetum and pollen development |
Arabidopsis thaliana Vitis pseudoreticulata (Chinese wild grapevine) |
[58,65] |
| Manes.18G016725.1; Manes.18G016800.2 | lysine-specific histone demethylase 1 homolog 3 | Reduces the levels of histone H3 'Lys-4' methylation in chromatin; promotion of floral transition. | Arabidopsis thaliana | [59] | |||
| S5_29309724 | 5 | Manes.05G186700.1 | protein DETOXIFICATION 48 | Functions as a multidrug and toxin extrusion transporter. Contributes to iron homeostasis during stress responses and senescence; Could be involved in specifying the lateral organ initiation rate; May act as a negative regulator of hypocotyl cell elongation in the light | Arabidopsis thaliana | [60,66] | |
| S15_11747301 | 15 | Manes.15G140500.1 | eukaryotic translation initiation factor 3 subunit I | Component of the eukaryotic translation initiation factor 3 (eIF-3) complex, involved in protein synthesis of a specialized repertoire of mRNAs and, together with other initiation factors, stimulates binding of mRNA and methionyl-tRNAi to the 40S ribosome. Regulates negatively translation during flower development | Arabidopsis thaliana | [61] | |
| Manes.15G140900.1 | myb family transcription factor MOF1 | Transcriptional repressor that plays a role in the regulation of organ identity and spikelet meristem determinacy. Interacts with the TPR corepressors to possibly repress the expression of downstream target genes in Rice | Oryza sativa subsp. japonica (Rice) | [62] | |||
| Manes.15G140300.1 | non-specific lipid-transfer protein 4.1 | Plant non-specific lipid-transfer proteins transfer phospholipids as well as galactolipids across membranes. May play a role in wax or cutin deposition in the cell walls of expanding epidermal cells and certain secretory tissues. Lipid transfer protein involved in seed and ovule maturation and development, probably by regulating the fatty acids homeostasis during suberin and sporopollenin biosynthesis or deposition. | Hordeum vulgare (Barley); Arabidopsis thaliana | [63] | |||
| Manes.15G140600.1 | short-chain dehydrogenase reductase ATA1 | May play a role in tapetum development. | Arabidopsis thaliana | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



