
Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Blood-derived products (BP) like citrate-platelet-rich plasma (CPRP) or hyperacute serum (HAS) are known to contain many growth factors. Combined mitogenic and DNA damaging stimuli might lead to increased senescent cell burden and altered senescence-associated secretory pheno-type (SASP). Therefore, the senescent state was extensively tested by γH2AX staining, p21 Q-PCR and western blot, growth curves and senescence associated ß-galactosidase staining. Two main treatments with BP were performed early (immediately after etoposide) and late (after addition-al 11days). Effects of the BP treatment was evaluated by IL-6 and IL-8 measurement as well as collagen (COL1) and p21 mRNA expression. Additionally, XTT assays, cell size measurements, viability assays, and cell number calculations were performed. HAS treated cells in early treat-ment had the lowest observed IL-6 and IL-8. In contrast, there was a clear inflammatory response for IL-8 on HAS and CPRP treated cells in late treatment. For COL1 expression an upregulation in early treatment could be shown, meanwhile cells in the late treatment group remained unaf-fected. In CPRP treated cells, the COL1 expression decreased. All in all, BP treatment seems to have slightly positive effects regarding skin rejuvenation.
Keywords:
blood derived products
; cellular senescence
; fibroblast
; skin
1. Introduction
In the last years, cellular senescence has proven to be one major factor contributing to age-associated degenerative diseases [1,2]. Cellular senescence is a defense mechanism against unlimited cellular proliferation discovered by Leonard Hayflick and Paul Moorhead [3]. This phenomenon of irreversible growth arrest of human diploid cells after serial passaging in culture was causally linked to telomere attrition (replicative senescence). However, cellular senescence is not only induced by telomere shortening but also by activated oncogenes or DNA damage (stress-induced premature senescence (SIPS [4])) [5]. Cellular senescence is like apoptosis a major process of cell fate. In contrast to apoptotic cells, senescent cells stay metabolically active. Therefore, senescent cells accumulate with age although they do not divide anymore. Furthermore, senescent cells are involved in fundamental physiological processes like wound healing [6].
Cellular senescence is a double-bladed mechanism. On one hand, preventing cancer and activating the immune system by establishing the senescence-associated secretory phenotype (SASP) [7]. By this mechanism, inflammatory factors are secreted in the surrounding tissue. The SASP cancer and cellular senescence are strongly interwoven, and you can't look at one without thinking about the other two. Factors of the SASP are attracting as example immune cells like M1-type macrophages that are able to prevent transformation of epithelial cells in carcinoma [8]. M1-type macrophages are characterized by a strong inflammatory phenotype; this subtype plays an important role in elimination of cancer cells. Furthermore, inflammation is part of the normal repair response for healing, and essential in keeping us safe from bacterial and viral infections and noxious environmental agents, but not all inflammation is good. When inflammation becomes prolonged and persists, it can become damaging and destructive [9]. Especially by excreting inflammatory cytokines, this phenotype is contributing to systemic inflammation in older organisms and might subsidize to malignant transformation. Among the SASP factors, IL-6 and IL-8 are of particular interest. They are responsible for inflammatory responses and age-related pathologies and also cancer [10]. Inhibition of SASP is considered an alternative to senolytic therapy to target the deleterious effects of senescent cells [11,12]. But also, activation of immune system to remove senescent cells could be advantageous. Otherwise, by excretion of IL-6 by the SASP, STAT3 could be activated which is an activator of bcl-2 (B-cell lymphoma 2) pro-survival family members which could make cancer cells more resistant to treatment. Besides, STAT3 is well known to restrain anti-tumor activity of immune cells. However, like already mentioned, STAT1 pathway can activate immune cell populations like NK of CD8-T-cells, which eliminate senescent cells [13].
The SASP is induced by severe or irreparable DNA damage, leading to senescence in normal cells by activation of Ataxia Telangiectasia Mutated (ATM) and subsequent activation of the histone H2AX by phosphorylation (γH2AX) forming characteristic foci. Phosphorylation of p53 at serine 15 is upregulating p21, a cell cycle inhibitor [14]. If DNA damage (double-strand breaks) cannot be repaired constitutive DDR signaling and p53-dependent stop of growth can lead to irreversible senescence [10,15].
Etoposide is one of the most important chemotherapeutic agents. Etoposide is used to treat a wide range of human cancers. For more than 20 years, it has been used clinically as a cancer drug. It is one of the most commonly prescribed anticancer drugs in the world. The target of etoposide is topoisomerase II. This ubiquitous enzyme causes transient double-strand breaks in the double helix during repair processes that it catalyzes [16] [17]. Apart from that with regard to research on cellular senescence the induction of fibroblast senescence by etoposide is associated with p53 activation [18]. Consequently, etoposide induced senescence is an optimal model to study SASP in skin fibroblast cells [19].
Rejuvenation of skin is one major factor of research because of unwelcome aesthetic aspects and skin weakening with age. Nevertheless, skin aging is not only a cosmetic problem. There are numerous dermatological disorders, especially in older people with a significant impact on quality of life ranging from disorders like pruritus to the more threatening melanomas [20]. Fibroblasts and surrounding matrix are the main components of dermal stroma. This post-mitotic tissue is prone to accumulation of dysfunctional cells over time [21,22]. Investigations in fibroblast senescence is a major task in studying skin aging, considering the evidence for the SASP and "SASP-like" inflammation in driving skin carcinogenesis, emphasizing how further understanding of both the roles and mechanisms of SASP expression may offer new targets for skin cancer prevention and therapy [23].
The use of blood-derived products is becoming more and more important in regenerative medicine and plastic surgery it is often applied and especially to old skin [24] and there are numerous clinical studies performed related to skin aging [25]. In the literature, many different types of blood-derived products are described like platelet-rich plasma (PRP) (when prepared from citrate supplemented blood CPRP) and hyperacute serum (HAS) [26]. PRP is the most prominent type of blood product and already tested in skin photoaging [27]. In general, these products are known to have a high content of growth factors. HAS has more pronounced effects on proliferation than PRP in human mesenchymal stem cells. Additionally, upregulation of BCL2 mRNA by treatment with 10 % HAS was observed [28]. HAS is also stimulating growth in chondrocytes [29]. Several growth factors have been shown to have positive effects also on skin rejuvenation [30,31] by stimulation of fibroblast proliferation. The rejuvenation strategy is based on overgrowth of damaged and old cells by proliferation of the young and healthy fibroblasts. Nevertheless, old cells remain in the tissue and can produce SASP-Factors. This leads to the question: How are these senescent cells influenced by mitogenic stimuli of BP?
Copp and colleagues showed that a combination of mitogenic stimulation and DNA damage even lead to increased cellular senescent cell number (chondrocytes) [32]. It is of major interest to show how the SASP is influenced by these combined stimuli BP and DNA-Damage in the onset of senescence and in already senescent cells!
Additionally, blood derived products contain a variety of different ingredients like the above-mentioned growth factors but also micro vesicles [33] containing for example miRNAs [34]. Describing the effects of blood derived products in first approximation will lead to a deeper understanding of this complex formulation giving hints for further studies.
Effects of blood-derived products on the proliferation of fibroblast cells and the complex balance between apoptosis and senescence may contribute to skin rejuvenation. However, even more multifaceted effects on onset of cellular senescence and on cellular senescent fibroblasts and their secretory phenotype are still unknown.
2. Materials and Methods
Fibroblast cells were obtained from ATCC and were grown in growth medium (GIBCO DMEM/F12 GlutaMAX-I; Invitrogen, LifeTech Austria, Vienna, Austria) containing antibiotics (penicillin 200 U/mL; streptomycin 0.2 mg/mL) and amphotericin B (2.5 μg/mL) (Sigma-Aldrich Chemie GmbH, Steinheim, Germany) supplemented with 10 % FCS [GIBCO, Invitrogen, LifeTech Austria]. Viability was determined via trypan blue (Sigma-Aldrich Chemie GmbH) dye exclusion and cells were counted using with LUNA cell counter. Cells were passaged every 3 to 4 days.
hypACT was prepared as described in Kardos et al. Briefly, whole blood was drawn from 8 male and female healthy donors (25–45 years) with the hypACT device (catalogue number: 700194, OrthoSera, Krems an der Donau, Austria) according to the manufacturer's protocol. The sample was immediately centrifuged at 1710xg for 5 min at room temperature. After centrifugation a top (platelet-rich fibrin clot) and a bottom layer were formed. The bottom layer, primarily containing red blood cells, was removed.
Whole blood from same 8 donors (woman and men) as in case of hypACT preparation, was drawn in VACUETTE 9ml 9NC Trinatriumcitrat 3,2% blood collection tubes (catalogue number: 455322; Greiner Bio-one, Kremsmünster, Austria) and centrifuged at 440xg for 10 min at room temperature. Three layers were formed in the tube: (i) bottom layer (red blood cells), (ii) middle layer (with the buffy coat) and (iii) top layer (plasma). Leukocytes-poor PRP was obtained by plasma layer aspiration without the middle layer (buffy coat). The aspirate was transferred into a 15 mL falcon tube and centrifuged again at 1700xg for 10 min. The formed platelet pellet was resuspended in 50% of the remaining platelet-poor-plasma supernatant. Blood derived products were prepared after Neubauer et al. [35].
Blood-derived products were pooled and immediately used for experiments or stored at -80°C for later use. Due to the use of citrate as anticoagulant the PRP product, the final product was called CPRP to comply with the nomenclature used in our previous publications.
Growth curves were completed by serial passaging of cells. Cells were seeded 1:3 or 1:6 and passaged when they reached confluence. The control curve was done till population doubling (PD) above 40 just to show that replicative potential was not exhausted in PD where experiments were done.
Cells were seeded in a density of 3500 cells /cm2 in 24 well plates [36]. Low seeding densities are important for the development of senescent state cells must not be contact inhibited. Treatments were done in triplicates. The next day (16 h), adhered cells were treated by adding concentrations of 50 µM or 25 µM etoposide (data in supplementary figure) to culture medium and cells were incubated in etoposide for 48h. Medium change and further incubation with fresh medium or blood-derived products for 3 days was done. Another 8-day incubation with normal growth medium in all cases (FCS, CPRP and HAS treated cells), medium change after 4 days was performed for establishment of SIPS. Control samples were performed without etoposide treatment, (growing) cells and (young) cells with the same seeding density after one day. SA-ß-Gal staining was performed after the protocol of Dimri et al. [37]. Briefly, cell culture medium was aspirated, and cells were washed with PBS twice. After the last rinse, PBS 4% PFA for fixation was replaced. Cells were incubated at room temperature for 5 min. 4% PFA were aspirated, and cells washed two times for 5 min at room temperature with gentle shaking (PBS). SA-β-gal staining solution was added (containing 0.1% X-gal, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM Sodium chloride, and 2 mM Magnesium chloride in 40 mM citric acid/sodium phosphate solution, pH 6.0). Cells were incubated in the dark in a 37 °C incubator.
Cells were seeded in chamber slides for 16 h and incubated in 50 µM etoposide for two days. Blood-derived products were added for one day (FCS, CPRP, and HAS), as control, cells were incubated in normal growth medium without damaging treatment. Briefly, cells were fixed in 4% formaldehyde in PBS for 15 min at room temperature. Fixative was aspirated and cells were rinsed three times in PBS for 5 minutes. Blocking was performed for 60 min with PBS supplemented with 5% normal serum and 0.3% Triton™ X-100. After aspiration of blocking agent primary antibody Phospho-Histone H2A.X (Ser139) Antibody (#2577 cell signaling) was added overnight at 4°C. After rinsing three times in PBS for 5 minutes, incubation of specimen in fluorochrome-conjugated secondary antibody diluted in goat-anti-rabbit polyclonal Fab fragment Ab labeled with AF-488 (3 µg/ml, Jackson Laboratories, Bar Harbor, MN) antibody dilution buffer (1 % BSA in PBS, 0.3% Triton™ X-100) for 1 hour at room temperature in dark and then rinsed in PBS as above. Nuclei were stained with DAPI Sigma Aldrich (D9542) 1:1000 in antibody dilution buffer 10 min at 37°C. Slides were washed with PBS and mounting was achieved by adding 20µl Fluoromount-GTM (Southern Biotechnology, Thermo Fisher Scientific), they were covered with high precision coverslip No. 1.5H.
For western-blot cells were lysed in 1 x Cell Lysis Buffer #9803 (Cell Signaling) and protein content was determined by DC assay (Biorad). Equal amounts of total Protein were separated by electrophoresis. Separated proteins were plotted to a 0.2m PVDF membrane (Biorad) with a trans blot turbo system (Biorad). Blot was blocked with 4% dry milk in TBS-Tween20 (0.1%) for one hour and 1:1000 p21 Waf1/Cip1 (12D1) Rabbit mAb #2947 (Cell Signaling) in blocking buffer was added consecutively and incubated over night at 4°C. Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP (Thermo Fisher scientific) was added after washing for 3 times with TBS-Tween (0.1%) for 1h and detected with Novex™ HRP Chromogenic Substrate (TMB) and images were taken with ChemiCoc imaging system (Biorad).
XTT assay: HDFs were seeded in standard growth medium in a 96-well plate (3500 cells/cm2). The CPRP-supplemented medium contained 2 U/mL heparin (Clexane, Sanofi Aventis, Paris, France) in order to prevent clotting in the culture medium. Cell-free wells were used as a technical background. Cell viability was determined using Cell Proliferation Kit II (XTT; Roche, Mannheim, Germany) according to the manufacturer's instructions at different time points. Absorbance was measured after 4 hours incubation in the staining solution using a PowerWave microplate spectrophotometer (BioTek, Winooski, VT, USA) at 480 nm with a reference wavelength at 650 nm. Relevant experiments were done in triplicates. Experiments, which were done only two times are found in the supplemental materials part.
For every approach one vial of cells got thawed at passage 7. Cells were expanded one or two passages (max passage 10). Supernatants were collected at every media change.
Etoposide treatment: Cells got seeded at a density of 3500 cells/cm2 in T75 culture flasks and 15ml media was added. After adherence of cells for at least 16h, cells were treated. To 15ml of media, 15µl of etoposide in DMSO (50mM) were added and cells were incubated for 48h.
BP-treatment for early exposure group: Treatment one was with 10% HAS (1,5ml HAS + 13,5ml media w/o FCS), treatment two was with 10% CPRP (1,5ml CPRP + 3µl Heparin + 13,5ml media w/o FCS) and the third treatment was with 10% FCS (this preparation was also used for the late exposure to BP experiment). Cells were exposed to BP for three days.
Establishment of cellular senescence: After BP pulse cells got normal growth media which was changed every 4 days. On day 8 after BP-treatment, three flasks were harvested.
Harvesting of cells: One flask of each treatment was harvested by accutase treatment (750µl). The reaction was stopped by adding 4.25ml of growth media. Cells were centrifuged at 500g for 10min and resuspended in 250µl Media. 100µl of this suspension were centrifuged twice. One Pellet was lysed in 250 µl RIPA buffer (+ 1:100 Protease inhibitor). The other Pellet was stored at -80°C for RNA extraction, 20µl were used for counting with LUNA cell counter, the residual suspension was mixed with 1ml ice-cold 70% Ethanol and stored at -20°C for flow cytometric analyses (unfortunately we harvested to little cells for reliable results).
For late exposure to BP cells were treated with BP equally to the early exposure group (three and eight), in sum eleven days after Etoposide exposure. (meanwhile fed with FCS containing medium). A three-day pulse with blood-derived products was performed. Thereafter media was changed in all residual groups and the cells were washed two times with PBS. At day 21 (16h+2d+3d+8d+3d+4d) all the residual groups were harvested like described above.
RNA Extraction: After differentiation, cells were collected and pooled group-wise in 200 µl PBS. The RNA was isolated using the High Pure RNA Isolation kit (catalogue number: 11828665001, Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s protocol. RNA was eluted and stored at -80 ̊C until cDNA synthesis.
Gene Expression Analysis: cDNA-Synthesis was performed using Transcriptor First Strand cDNA Synthesis Kit (catalogue number: 04379012001, Roche, Basel, Switzerland). RT-qPCR was performed using FastStart Essential DNA Probes Master kit (catalogue number: 06402682001, Roche, Basel, Switzerland) in triplicates using the LightCycler® 96 from Roche. 1 µl of the cDNA product, FastStart Probe Master 2X, hydrolysis probe (final concentration 250 nM) and primers (final concentration 900 nM) were used for PCR amplification. Primers for each gene were designed to span introns to exclude genomic contamination in PCR products and were designed using the Universal ProbeLibrary System Assay Design. GAPDH was used as a housekeeping gene. PCR conditions were optimized for annealing temperature and limited cycle number to ensure that product formation was in the responsive range.
- GAPDH: (left: 5' CTCTGCTCCTCCTGT 3' right: 5' ACGACCAAATCCGTT 3' universal primer library #60) COL1: (left: 5' GGGATTCCCTGGACC 3' right: 5' GGAACACCTCGCTCT 3' universal primer library #67) p21: (left: 5' TCACTGTCTTGTACCCTTGTGC 3' right: 5' GGC GTT TGG AGT GGT AGA AA 3' universal primer library #32)
Supernatants (15 ml) were harvested from treated samples at indicated time points and stored at -80°C. Cytokines and chemokines including tumor necrosis factor-alpha (TNFα), monocyte chemoattractant protein 1 (MCP1), Interferon-gamma (IFNγ), Interleukin 10 (IL-10), Interleukin 8 (IL-8), Interleukin 6 (IL-6), Interleukin 1ß (IL-1ß), and interleukin- 1 receptor antagonist (IL1RA) were measured with Bio-Plex Pro Assays (BIO-RAD Laboratories, Inc., Hercules, CA, USA). Analyses were done strictly following the instructions of the supplier.
Cells were treated the same way as for the XTT assay and assay procedure of Apo-ONE® Caspase-3/7 (Promega) assay according to manufacturer’s instructions, Briefly, 100μl of Apo-ONE® Caspase-3/7 Reagent were added to each well of a black 96-well plate containing 100μl of blank, control or cells in culture. Contents of wells was gently mixed using a plate shaker at 300–500rpm from 30 seconds up to read time Plates were covered and incubated for 4 hours. fluorescence was measured at 499nm with an emission maximum at a wavelength of 521nm (PowerWave microplate spectrophotometer (BioTek, Winooski, VT, USA)).
Normality testing was done by the Shapiro-Wilk test. Differences between groups were determined by simple ANOVA. Multiple comparison testing was performed by Tukey´s test. When normality was not confirmed, significances were calculated by the Kruskal Wallis test, multiple comparisons were done in this case by Dunn’s multiple comparison test.
3. Results
3.1. Definition of reagents:
Blood-derived products:
Blood-derived products were tested with a common panel of cytokines and chemokines (tumor necrosis factor-alpha (TNFα), monocyte chemoattractant protein 1 (MCP1), Interferon-gamma (IFNγ), Interleukin 10 (IL-10), Interleukin 8 (IL-8), Interleukin 6 (IL-6), Interleukin 1ß (IL-1ß), interleukin- 1 receptor antagonist (IL1RA) All tested substances were below or near the control.
3.2. Induction of senescent state
In all experiments, the cells were used below 20 population doublings (PD) (green area in Figure 1a), control cells were passaged above PD 40 to show that replicative potential of cells was not exhausted at the time point of the experiments (gray line with crosses). Blue vertical line indicates etoposide treatment for two days and the red vertical line indicates the blood product treatment for 3 days, following growth arrest of cells shown by black line with x. Detection of p21 protein by western blot shows typical bands at molecular weight 21 kD (Figure 1b) and confirmed senescent state, young cells were analyzed twice (Y), compared to replicative senescent control (RS) and etoposide induced senescent cells (ES). Furthermore, induction of senescent state was tested by p21-expression on mRNA level, which was positive after 11- and 18-days post-treatment (FCS-group). Significance was confirmed by simple ANOVA and Tukey´s multiple comparison test with a p-value of 0.0127 and 0.0088 respectively (Figure 1c).
Senescent state was confirmed by gamma-H2AX staining, which was positive in all treatments, except control cells (growing). The treatment with etoposide (50 µM) after 2 days of incubation and subsequent treatment with blood-derived products (FCS, CPRP and HAS (all 10%)) for one day is shown in the upper right of Figure 1b. Gamma H2AX foci were located in the nuclei stained with DAPI (blue) demonstrating DNA damage (double-strand breaks) by etoposide treatment. Cells were used at the beginning of their replicative lifespan (Figure 2a). SA-ß-Gal staining was positive in all treatments, except control cells (growing) as seen in Figure 2b. After treatment and longtime incubation cells were still vital.
3.3. Proliferation and outgrowth of fibroblast cells
Stimulation of outgrowth of fibroblast cells was confirmed by treatment of cells with 25 and 50 µM etoposide and following treatment with blood products for 7 days. In Figure 3 a and b can be seen that growing cells without etoposide treatment (black line with stars) have higher absorption in XTT assay than the ones treated with etoposide. Negative control, serum free medium fed fibroblasts, show the lowest response in XTT assay (black line with circles). FCS supplemented cells (gray line with triangles) which is some kind of positive control show less to similar growth like the other blood products HAS (black box and orange line) and CPRP (black line with triangles). Interestingly HAS shows higher absorbance indicating more cells 24 h post etoposide treatment (25 and 50 µM). Blood products treated with 25 µM etoposide show less outgrowth than 50 µM treatment in all cases (FCS, HAS and CPRP).
Without damaging treatment (Figure 4a, b) and a pulse of just 3 days HAS and CPRP show a higher growth than FCS 10%. When HAS is diluted to 1% and 5% the growth is decreased compared to 10% HAS but still similar to FCS 10%. In Figure 4c it can be seen that with 25 µM etoposide treatment there is still a quite high outgrowth rate compared to 50 µM (Figure 4d) indicating better senescence induction and less cells that are able to proliferate in the higher concentration of etoposide.
Comparing different concentrations of HAS (1%, 5% and 10%) to CPRP and FCS both 10%, treated with 25 µM etoposide (Figure 5a) and 50 µM etoposide (Figure 5b) after 24h of etoposide pulse (48 h) shows a significant dose dependent increase in growth (indicated by higher absorption in XTT assay) which is less pronounced in 50 µM etoposide treated cells but still significant by one way ANOVA with Tukey's multiple comparisons test. This increase in growth is well correlated with a decrease in CASP 3/7 activity shown in Figure 4c and d showing apoptosis inhibition by HAS after 24 h post etoposide treatment.
3.4. Investigations in cellular senescence and SASP
3.4.1. Growth arrest and cell numbers
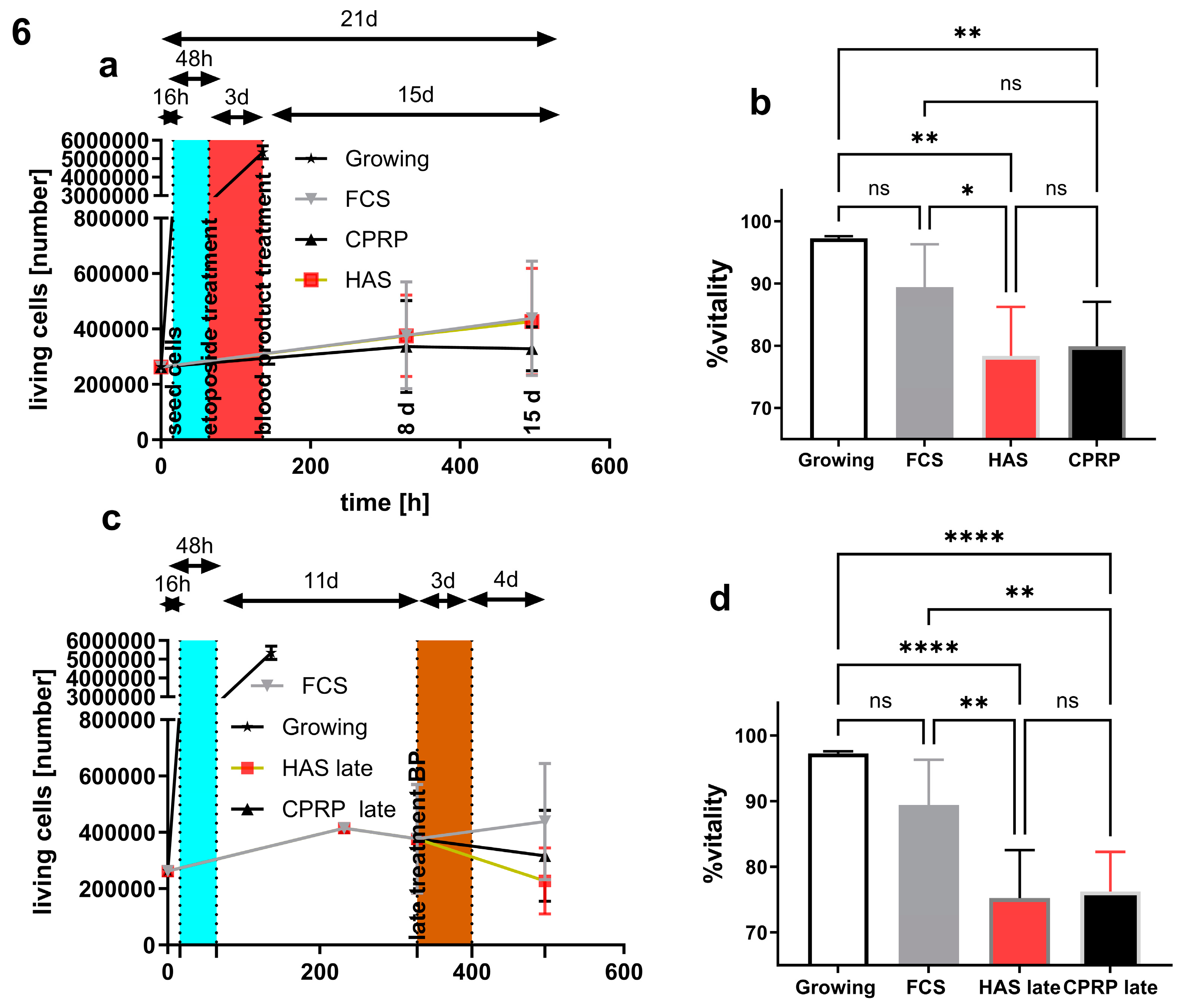
Early treatment (Figure 6a) was performed by seeding the cells and consecutive incubation for 16 h. Etoposide treatment (50 µM) for two days was done (blue area) after incubation with blood-derived products HAS and CPRP or FCS in control group for three days. Sampling time points were 8 days and 15 days after blood product treatment. The number of living cells stayed nearly the same in all three treatment groups in early treatment. Growing cells reach confluency after 4 – 5 days after seeding.
Late treatment (Figure 6c) was performed as follows, after 16 hours incubation after cell seeding, they were treated with 50 µM etoposide (blue area) for 2 days followed by an 11 d (8 d +3 d) period with regular media change (10% FCS) then 3 d blood product pulse (HAS and CPRP or FCS). After this pulse cells were fed again with normal growth medium for 4 d (15d + 3d).
3.4.2. Viability of cells
Viability of early treatment (Figure 6b) at the timepoint after 15 days indicated a more pronounced decrease in vitality in the HAS group compared to FCS treated cells by p = 0.0473. HAS and CPRP groups differ from growing cells by the significances of p = 0.0019 and p = 0.004 respectively.
Viability at the 15 days +3 days timepoint (Figure 6d) in late treatment in HAS and CPRP group decreased compared to growing cells with p >0.0001 in both groups and compared to FCS treatment group in HAS and CPRP group with significances of p = 0,002 and p = 0,0041 respectively.
Figure 6.
early treatment cell numbers (a) and vitality at harvesting timepoint 21 days after start of experiment (b) arrows indicating duration of treatment phases for clearer understanding of treatment conditions. Late treatment, (c) cell numbers and (d) vitalities.
Figure 6.
early treatment cell numbers (a) and vitality at harvesting timepoint 21 days after start of experiment (b) arrows indicating duration of treatment phases for clearer understanding of treatment conditions. Late treatment, (c) cell numbers and (d) vitalities.
3.4.3. Cell morphology
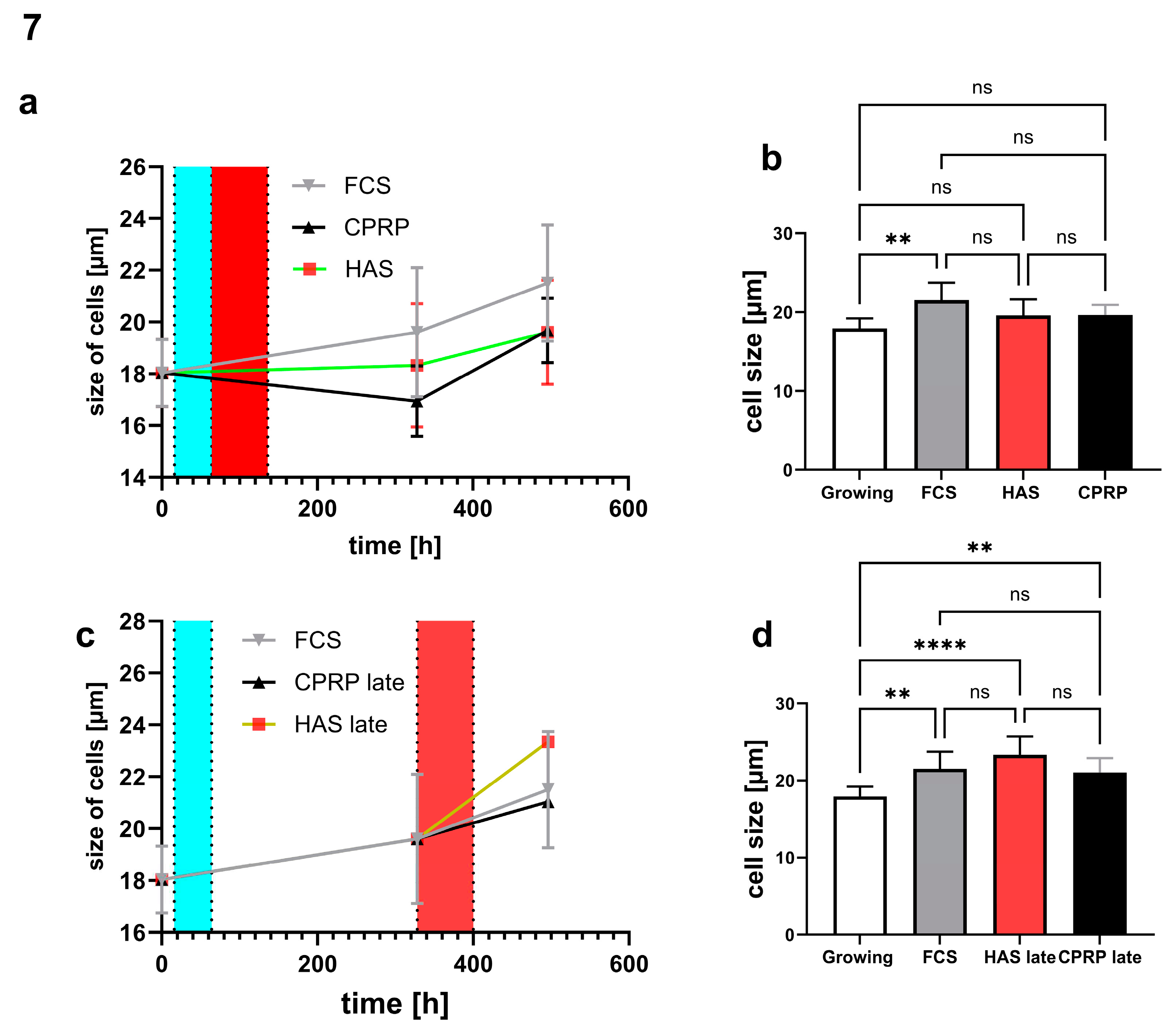
Cell morphology was determined by cell size and was not affected significantly in early treatment setup (Figure 7a and b) in HAS and CPRP treated fibroblast cells. The size of FCS (normal growth medium) treated cells increased significantly with p = 0.0028. Contrary, late treatment setup (Figure 7c and d) resulted in a highly significant increase in the size of HAS treated cells (p < 0.0001) and less but also significant increase in CPRP group (p = 0.0093). Significances were calculated by Kruskal Wallis test, data that did not pass the normality test (Shapiro-Wilk), a multiple comparison by Dunn’s multiple comparison test was performed (n = 3 or 4). Column graphs represent the 21-day timepoint at the end of the experiment.
3.4.4. p21 expression
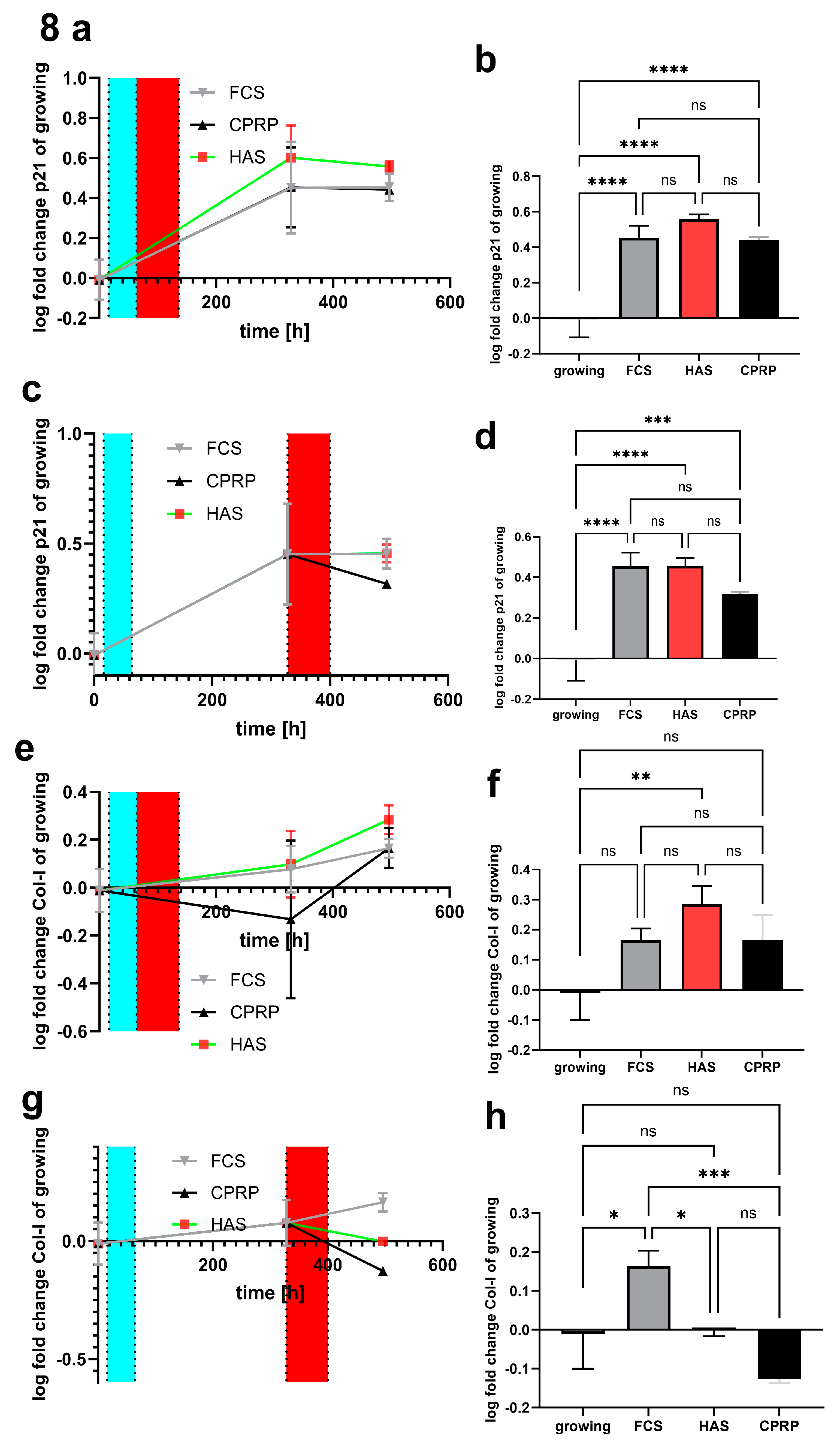
p21 expression (Figure 8a) was increased on day 8 and day 15 after etoposide treatment in early treatment setup. On day 15 FCS, HAS and CPRP the expression of p21 was significant increased (p < 0.0001) in comparison to growing cells (Figure 8b). The most prominent increase in p21 expression compared to growing cells was observed in the HAS group nevertheless, this increase was not significant if compared to FCS a CPRP at the same timepoint.
p21 expression was increased on day 11 days and on day 18 days post etoposide treatment (pt) in late treatment setup (Figure 8c), which is as high as HAS and FCS with p < 0,0001 if compared to growing cells. However, CPRP p21 expression on day 18 pt is decreased and less significant compared to growing cells (p = 0.0008) (Figure 8d). Data were expressed in logarithmical fold change values and passed Shapiro-Wilk normality testing.
Figure 8.
pPCR of mRNA expression of p21 early treatment time course (a) as well as 21d end point column diagram (b). Late treatment also shows log fold change values of different timepoints (c) and the final state after 21 d (d). Similarly, collagen 1 expression early treatment (e and f) and late treatment (g and h).
Figure 8.
pPCR of mRNA expression of p21 early treatment time course (a) as well as 21d end point column diagram (b). Late treatment also shows log fold change values of different timepoints (c) and the final state after 21 d (d). Similarly, collagen 1 expression early treatment (e and f) and late treatment (g and h).
3.4.5. Type I collagen expression
Type I collagen (COL1) a parameter of extracellular matrix (ECM) was investigated by qPCR. In Figure 8e an increase in expression pattern of all three treatment is shown, only HAS COL1 Expression reached significance (p = 0.0041) after 15 days post-treatment in early treatment group (Figure 8f). Nevertheless, by late treatment with blood-derived products HAS kept unaffected compared to zero time points (growing cells) in 18 days pt time point. FCS (normal growth medium) was highly significant increased compared to CPRP treated cells (p = 0.0004) which showed a decreased expression in COL1 compared to growing cells calculated by simple ANOVA with Tukey`s multiple comparison tests (Figure 8g and h). Data are expressed in logarithmical fold change values and passed Shapiro-Wilk normality testing.
3.4.6. IL-6 concentration in supernatant
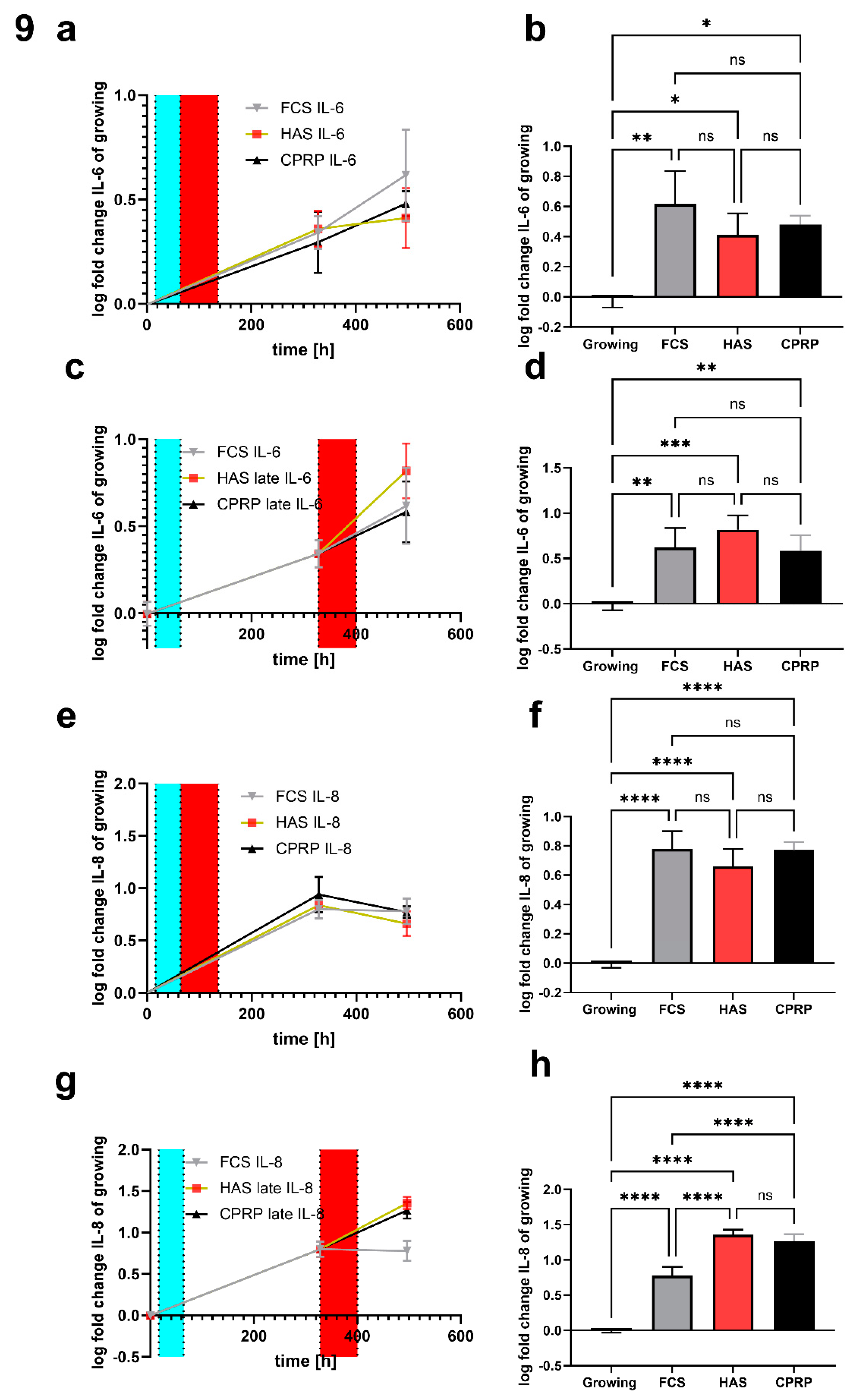
IL-6 in early treatment on day 15 after etoposide treatment is still increased in all groups compared to growing control. FCS (normal growth medium) treatment showed the most pronounced increase with significance p = 0.0019, CPRP and HAS were increased by significance 0.0142 and 0.0328 respectively (Figure 9a and b). In longtime treatment 15 days, + 3 days or rather 18 d pt time point displayed a highly significant increase in IL-6 in all groups FCS, HAS and CPRP (p = 0.0028, 0.0003 and 0.0042) where HAS increased most significantly (Figure 9c and d). Data were expressed in logarithmical fold change values furthermore they were normalized by the mean cell number of the corresponding experiment. Shapiro-Wilk test normality test indicated normality in all treatments and simple ANOVA with Tukey’s multiple comparison test was used to calculate significances.
3.4.7. IL-8 concentration in supernatant
IL-8 increased significantly in early treatment in all treatments compared to growing cells on day 15 (p <0.0001) again determined by simple ANOVA with Holm Tukey’s multiple comparison test (Figure 9e and f). Late treatment (Figure 9g and h) uncovered not only differences between treatments and growing cells (all p < 0.0001) but also significantly increased HAS and CPRP values compared to normal growth medium treatment (FCS) by p < 0.0001 in both cases analyzed by Tukey’s multiple comparison test. Data are expressed in logarithmical fold change values and passed Shapiro-Wilk normality testing furthermore they were normalized by the mean cell number of the corresponding experiment.
Figure 9.
Luminex assay of inflammatory markers. (a) time course of early treatment with different BP and final IL-6 concentration in supernatant normalized to cell count (in all cases) at day 21 (b). Late treatment time course (c) and 21-day timepoint column graph of late treatment (d). Same for IL-8 for early treatment (e and f) and late treatment (g and h).
Figure 9.
Luminex assay of inflammatory markers. (a) time course of early treatment with different BP and final IL-6 concentration in supernatant normalized to cell count (in all cases) at day 21 (b). Late treatment time course (c) and 21-day timepoint column graph of late treatment (d). Same for IL-8 for early treatment (e and f) and late treatment (g and h).
4. Discussion
Autologous blood-derived products like platelet-rich plasma and hyperacute serum are considered to be promising for therapeutic use. Drawbacks are the demand of standardization of their preparation, and no sufficient characterization of their biomolecule composition [38]. Nevertheless, there is constant progress in research regarding blood-derived products [26]. Several important questions relate to the timing of treatment are also still to answer [39]. This becomes more complicated considering different cell types, states of the cells and other factors. In this study, we just focused on states of cellular senescence of skin fibroblast cells.
Cellular senescence (CS) is a multifactorial process. Therefore, not a single marker can confirm the senescent state. SA-ß-gal is one of the most recognized indicators of CS and was positive in all treatments following etoposide exposure (2d) and BP treatment (3d) after further incubation for eight days. Nevertheless, there are some concerns about SA-ß-gal assay performance. SA-ß-gal staining can be false positive for many reasons like cell confluence, radical damage to the cells, and others. Furthermore, SA-ß-gal staining is not easy to quantify [40]. For that reason, we were determining senescent state with a variety of methods like establishment and maintenance of γH2AX foci, which were present after twenty-four hours following etoposide withdrawal and were used as an additional clear indicator of CS [41]. The appearance of γH2AX foci was not influenced by 24 h treatment with different blood-derived products representing persistent DNA damage signaling and development of SASP in all treatments performed. DNA damage response is also confirmed by p21 upregulation. This upregulation of p21 was significant in all treatments demonstrating established CS or at least DNA-damage signaling. Additionally, increased p21 protein expression was also confirmed by western blot. Finally, confirming growth arrest by growth curves is one of the most important items in approving CS. Thus, cells are still alive and metabolic active even 15 days of post-treatment. After all these analyses, we can clearly say that we demonstrated onset of CS for validation of further experiments.
Aging is an extremely complex process and so is the SASP. SASP can differ from cell type to cell type. Furthermore, the type of stimuli that induces cellular senescence is also influencing the resulting SASP. Often oncogene induction or ionizing radiation and oxidative stress are used beside chemotherapeutic drugs to induce cellular senescence. We focused on a reliable and reproducible form of senescence induction and characterized the senescent state in detail. Etoposide is inducing DNA-damage response and CS [17,18] which is one requirement for SASP development [10,15,42].
The main goal of the current study was determining whether cellular senescence in skin fibroblasts is accompanied by the development of SASP, regardless of the type of senescence (replicative, SIPS, or etoposide-induced) like in the study of Odeh et al. [19]. Human fibroblasts were used due to the well-characterized SASP in these cells.
The formation of CS is a process that takes some time. For that reason, cells were incubated for 11 days (8 days post 3 days blood product treatment) after etoposide exposure for two days. One aim of this work was to investigate the influence of blood-derived products on the setup of senescence and on already senescent cells. Hence, a stringent method for induction of CS was used. Methods such as UV-irradiation or radical treatment with regard to skin aging are not the focus of this work. The late and early treatments put that into praxis, late treatment is representing the treatment of already senescent cells. Time point for late treatment was chosen by taking into consideration the results of analyses of extensive definition of senescent state of cells.
Easily quantifiable senescence parameters are not easy to find, therefore it was chosen to use cell size as an easy to analyze parameter for morphological changes in senescent cells [43,44,45]. Senescent morphology indicated by the size of cells did not change significantly by CPRP and HAS treatment compared to FCS (normal growth medium). This is consistent with visual observations of cells in SA-ß-gal staining where cells were blue but did not change morphology. This impression was not quantified or checked for statistical significance. Although, other senescence parameters like p21 and growth arrest indicate the senescent state of cells. Nevertheless, cell size is consistent with IL-6 and IL-8 expression pattern in late treatment (late pulse with blood-derived products) where especially HAS and also CPRP show increased size and inflammatory cytokine and chemokine production. Where in the early treatment there is the opposite effect with FCS treated fibroblasts showing more swelling than HAS and CPRP treatment. This could indicate a dampening effect in early treatment and a stimulating effect in late treatment which is an inflammatory (“healing?”) effect.
With regard to p21 expression, all treatments show a significant increase in p21. The tumor suppressor p21 shows less significant increase in late treatment of CPRP which could be a sign of senolytic activity of this BP.
With regard to SASP, IL-6 levels in culture supernatant in early treatment reveal less and less significant excretion of IL-6 when treating fibroblast cells with CPRP and HAS. Together with the IL-8 results of early treatment, an attenuation of SASP related inflammatory cytokine and chemokine levels was shown, when cells are treated with BP right after exposure to damaging substance etoposide.
Opposing results are obtained with the treatment of already senescent cells. HAS is increasing IL-6 and IL-8 excretion significantly. In case of IL-8 the increase of HAS and CPRP treated cells is even significantly higher than that of FCS treatment. It would be interesting to see if increased cytokine release leads to attraction of immune cells and clearance of senescent cells in vivo. In combination with strong growth signals and subsequent outgrowth of young fibroblast induced by BPs, this could be an optimal outcome for this treatment.
Additionally, a clear anti apoptotic effect of HAS could be observed. This effect resulted in higher cell numbers and XTT absorption after 24 hours but did not lead to differences in the number of cells to other BP 8- and 15-days post treatment.
It is always questionable how useful FCS supplementation could be as a control, taking into account the origin of this serum. FCS is a mixture of growth factors and might be not the best control for experiments with other blood-derived products and could mask some effects or make them seem less pronounced than they would be under normal physiological conditions.
Anyway, the growth of fibroblasts is highly induced by HAS and even more by CPRP. Furthermore, hints for an additional short time anti-apoptotic effect of HAS after damaging treatment could be accomplished.
Wound healing, for example, is a complex multistep process in which senescence plays a role in both positive and negative ways. After an initial hemostatic phase, an inflammatory response begins. In the course of this, a large number of cells migrate in the wounded tissue or differentiate locally to support the healing process. Collagen synthesis and remodeling processes also take place. Wound healing is not optimal especially in old organisms or in people with diabetes; it is known that especially in old skin senescent cells accumulate [46]. Moreover, induction of type I collagen especially by HAS also contributes to the alleviation of negative effects forced by etoposide treatment. Summing up these observations there could be beneficial effects and skin rejuvenation of HAS and CPRP by influencing dermal fibroblasts with regard to senescence (and also induction of proliferation of fibroblast cells).
Further studies should uncover if activated SASP is leading to accumulation of M1-type macrophages in tissue treated with blood derived products. Also, NK cells infiltration could lead to the elimination of malignant or senescent cells [8,47]. Consequently, these NK cells engage explicit regulatory mechanisms to eliminate senescent cells by for example perforin-granzyme exocytosis mediated NK-cell killing of senescent cells [48]. In the end, BP treatment could lead to real rejuvenation of skin by stimulating these factors leading to elimination of senescent cells and stimulating the growth of young and healthy cells, an effect that has to be proved in further experiments.
5. Conclusions
Nevertheless, our experiments demonstrate clearly that aging skin might be a challenging environment for the treatment with blood-derived products taking into account all the states of cells, different cell types and other factors. In general, no detrimental effect was observed BP do have slightly positive effects increased inflammatory markers can also induce healing effects.
Author Contributions
Conceptualization, H.K.; methodology, HK, MP, OK, KK and CB.; investigation, HK, MP, KK, and CB.; resources, SN and ZL.; data curation, HK.; writing—original draft preparation HK.; writing—review and editing, HK, MP, SN.; visualization, HK.; supervision, SN, HK.; project administration, HK SN; funding acquisition, SN, ZL. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by OrthoSera GmbH and The APC was funded _______________
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are thankful for OrthoSera GmbH for the research support Danube Universities. Furthermore, the authors thank Claudia Schildboeck, Eugenia-Paulina Niculescu-Morzsa and Eva Rossmanith for technical assistance.
Conflicts of Interest
Authors Z.L.and O.K. are employees, stockholders, or advisory board members of OrthoSera GmbH, a startup company developing hyperacute serum technology towards clinical applications. “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
References
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4apositive senescent cells delays ageingassociated disorders. Nature. 2011; 479:232. [CrossRef]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016; 530:184–9. [CrossRef]
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Experimental cell research. 1961; 25:585–621. [CrossRef]
- Toussaint O, Dumont P, Remacle J, Dierick J-F, Pascal T, Frippiat C, et al. Stress-induced premature senescence or stress-induced senescence-like phenotype: one in vivo reality, two possible definitions. TheScientificWorldJournal. 2002; 2:230–47. [CrossRef]
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509:439–46. [CrossRef]
- Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nature medicine. 2015; 21:1424–35. [CrossRef]
- Velarde MC, Demaria M, Campisi J. Senescent cells and their secretory phenotype as targets for cancer therapy. Cancer and Aging: From Bench to Clinics. ; 2013. [CrossRef]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell. 2008; 134:657–67. [CrossRef]
- Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front Immunol. 2018; 9. [CrossRef]
- Rodier F, Coppé J-P, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11:973–9. [CrossRef]
- Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer cell. 2018; 34:85-102.e9. [CrossRef]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. 00219738; 2013.
- Guillon J, Petit C, Toutain B, Guette C, Lelièvre E, Coqueret O. Chemotherapy-induced senescence, an adaptive mechanism driving resistance and tumor heterogeneity. Cell cycle (Georgetown, Tex.). 2019; 18:2385–97. [CrossRef]
- Loughery J, Cox M, Smith LM, Meek DW. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic acids research. 2014; 42:7666–80. [CrossRef]
- Rodier F, Kim S-H, Nijjar T, Yaswen P, Campisi J. Cancer and aging: the importance of telomeres in genome maintenance. Int J Biochem Cell Biol. 2005; 37:977–90. [CrossRef]
- Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005; 5:363–72. [CrossRef]
- Petrova NV, Velichko AK, Razin SV, Kantidze OL. Small molecule compounds that induce cellular senescence. Aging Cell. 2016; 15:999–1017. [CrossRef]
- Probin V, Wang Y, Bai A, Zhou D. Busulfan selectively induces cellular senescence but not apoptosis in WI38 fibroblasts via a p53-independent but extracellular signal-regulated kinase-p38 mitogen-activated protein kinase-dependent mechanism. The Journal of pharmacology and experimental therapeutics. 2006; 319:551–60. [CrossRef]
- Odeh A, Dronina M, Domankevich V, Shams I, Manov I. Downregulation of the inflammatory network in senescent fibroblasts and aging tissues of the long-lived and cancer-resistant subterranean wild rodent, Spalax. Aging Cell. 2020; 19:e13045. [CrossRef]
- Farage MA, Miller KW, Elsner P, Maibach HI. Characteristics of the Aging Skin. Adv Wound Care (New Rochelle). 2013; 2:5–10. [CrossRef]
- Boukamp P. Skin aging: a role for telomerase and telomere dynamics. Curr Mol Med. 2005; 5:171–7. [CrossRef]
- Waldera Lupa DM, Kalfalah F, Safferling K, Boukamp P, Poschmann G, Volpi E, et al. Characterization of Skin Aging-Associated Secreted Proteins (SAASP) Produced by Dermal Fibroblasts Isolated from Intrinsically Aged Human Skin. J Invest Dermatol. 2015; 135:1954–68. [CrossRef]
- Ghosh K, Capell BC. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J Invest Dermatol. 2016; 136:2133–9. [CrossRef]
- Charles-de-Sá L, Gontijo-de-Amorim NF, Takiya CM, Borojevic R, Benati D, Bernardi P, et al. Effect of Use of Platelet-Rich Plasma (PRP) in Skin with Intrinsic Aging Process. Aesthet Surg J. 2018; 38:321–8. [CrossRef]
- Maisel-Campbell AL, Ismail A, Reynolds KA, Poon E, Serrano L, Grushchak S, et al. A systematic review of the safety and effectiveness of platelet-rich plasma (PRP) for skin aging. Archives of dermatological research. 2019. [CrossRef]
- Kardos D, Simon M, Vácz G, Hinsenkamp A, Holczer T, Cseh D, et al. The Composition of Hyperacute Serum and Platelet-Rich Plasma Is Markedly Different despite the Similar Production Method. Int J Mol Sci. 2019; 20. [CrossRef]
- Jia C, Lu Y, Bi B, Chen L, Yang Q, Yang P, et al. Platelet-rich plasma ameliorates senescence-like phenotypes in a cellular photoaging model. RSC Adv. 2017; 7:3152–60. [CrossRef]
- Kuten O, Simon M, Hornyák I, Luna-Preitschopf A de, Nehrer S, Lacza Z. The Effects of Hyperacute Serum on Adipogenesis and Cell Proliferation of Mesenchymal Stromal Cells. Tissue Eng Part A. 2018; 24:1011–21. [CrossRef]
- Jeyakumar V, Niculescu-Morzsa E, Bauer C, Lacza Z, Nehrer S. Platelet-Rich Plasma Supports Proliferation and Redifferentiation of Chondrocytes during In Vitro Expansion. Front Bioeng Biotechnol. 2017; 5:75. [CrossRef]
- Fabi S, Sundaram H. The potential of topical and injectable growth factors and cytokines for skin rejuvenation. Facial Plast Surg. 2014; 30:157–71. [CrossRef]
- Passaretti F, Tia M, D'Esposito V, Pascale M de, Del Corso M, Sepulveres R, et al. Growth-promoting action and growth factor release by different platelet derivatives. Platelets. 2014; 25:252–6. [CrossRef]
- Copp ME, Flanders MC, Gagliardi R, Gilbertie JM, Sessions GA, Chubinskaya S, et al. The combination of mitogenic stimulation and DNA damage induces chondrocyte senescence. Osteoarthritis and cartilage. 2020. [CrossRef]
- Otahal A, Kuten-Pella O, Kramer K, Neubauer M, Lacza Z, Nehrer S, et al. Functional repertoire of EV-associated miRNA profiles after lipoprotein depletion via ultracentrifugation and size exclusion chromatography from autologous blood products. Sci Rep. 2021; 11:5823. Epub 2021/03/12. [CrossRef]
- Tao SC, Guo SC, Zhang CQ. Platelet-derived Extracellular Vesicles: An Emerging Therapeutic Approach. Int J Biol Sci. 2017; 13:828–34. [CrossRef]
- Neubauer M, Kuten O, Stotter C, Kramer K, Luna A de, Muellner T, et al. The Effect of Blood-Derived Products on the Chondrogenic and Osteogenic Differentiation Potential of Adipose-Derived Mesenchymal Stem Cells Originated from Three Different Locations. Stem Cells Int. 2019; 2019:1–20. [CrossRef]
- Lammermann I, Terlecki-Zaniewicz L, Weinmullner R, Schosserer M, Dellago H, de Matos Branco, A D, et al. Blocking negative effects of senescence in human skin fibroblasts with a plant extract. NPJ Aging Mech Dis. 2018; 4:4. [CrossRef]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92:9363–7.
- Chahla J, Cinque ME, Piuzzi NS, Mannava S, Geeslin AG, Murray IR, et al. A Call for Standardization in Platelet-Rich Plasma Preparation Protocols and Composition Reporting: A Systematic Review of the Clinical Orthopaedic Literature. J Bone Joint Surg Am. 2017; 99:1769–79. [CrossRef]
- Martínez CE, Smith PC, Palma Alvarado VA. The influence of platelet-derived products on angiogenesis and tissue repair: a concise update. Front Physiol. 2015; 6:290. [CrossRef]
- Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017; 9:1867–84. [CrossRef]
- Soubeyrand S, Pope L, Haché RJG. Topoisomerase IIalpha-dependent induction of a persistent DNA damage response in response to transient etoposide exposure. Mol Oncol. 2010; 4:38–51. [CrossRef]
- Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. Journal of cell science. 2011; 124:68–81. [CrossRef]
- Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017; 8:422. [CrossRef]
- Block TJ, Marinkovic M, Tran ON, Gonzalez AO, Marshall A, Dean DD, et al. Restoring the quantity and quality of elderly human mesenchymal stem cells for autologous cell-based therapies. Stem Cell Res Ther. 2017; 8:239. [CrossRef]
- Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, et al. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017; 16:661–71. [CrossRef]
- Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, et al. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers. 2020; 12. [CrossRef]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445:656–60. [CrossRef]
- Adi Sagiv, Valery Krizhanovsky. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013; 14:617–28. [CrossRef]
Figure 1.
definition of senescent state by growth curve (a) and p21 expression on protein level bands at molecular weight 21 kD indicate p21 protein (b) and qPCR of p21 mRNA (c).
Figure 1.
definition of senescent state by growth curve (a) and p21 expression on protein level bands at molecular weight 21 kD indicate p21 protein (b) and qPCR of p21 mRNA (c).

Figure 2.
(a) gamma-H2AX staining of growing, and etoposide treated fibroblasts. All three blood product treatments FCS, CPRP and HAS show blue DAPI stained nuclei with green dots marked by white arrow are indicating DNA lesion by immune fluorescence of phosphorylated histone H2AX. (b) SA-ß-Gal-stained cells blue color indicates establishment of senescent phenotype in etoposide treatment (2 d) and FCS CPRP and HAS pulse for 3 d and additional 8 d of establishment of senescence.
Figure 2.
(a) gamma-H2AX staining of growing, and etoposide treated fibroblasts. All three blood product treatments FCS, CPRP and HAS show blue DAPI stained nuclei with green dots marked by white arrow are indicating DNA lesion by immune fluorescence of phosphorylated histone H2AX. (b) SA-ß-Gal-stained cells blue color indicates establishment of senescent phenotype in etoposide treatment (2 d) and FCS CPRP and HAS pulse for 3 d and additional 8 d of establishment of senescence.
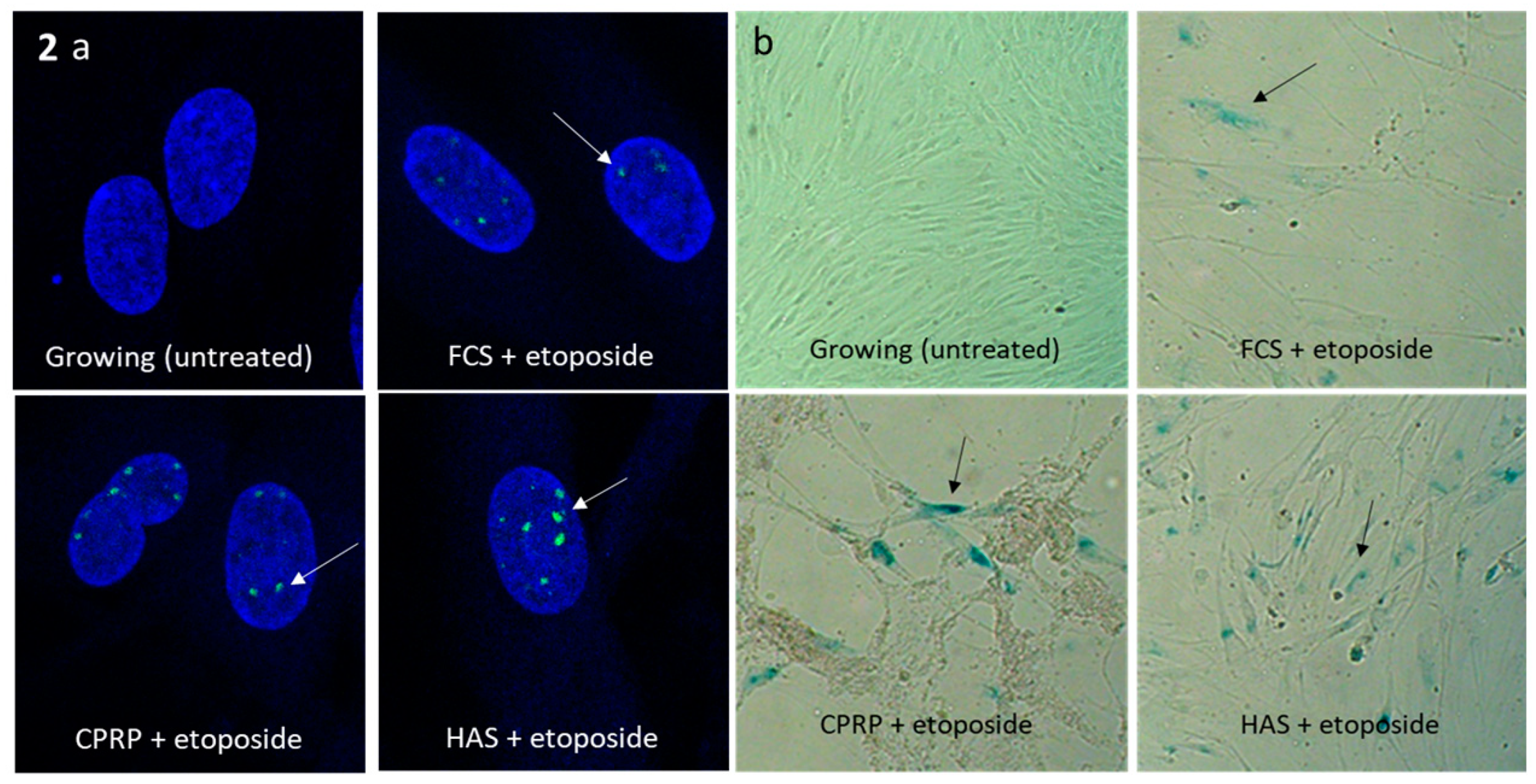
Figure 3.
outgrowth of cells (a) treatment with etoposide (blue area 48 h) leads to a decrease in growth but does not absolutely stop the growth of FCS (gray with triangles) HAS (cubes with orange line) and CPRP (black with triangles) treated for seven days (red area) with 25 µM etoposide not even 50 µM (b) treatment could stop outgrowth absolutely indicating massive growth signal by blood products. Growing and serum free cells in both cases show growth without damaging treatment and without blood products respectively.
Figure 3.
outgrowth of cells (a) treatment with etoposide (blue area 48 h) leads to a decrease in growth but does not absolutely stop the growth of FCS (gray with triangles) HAS (cubes with orange line) and CPRP (black with triangles) treated for seven days (red area) with 25 µM etoposide not even 50 µM (b) treatment could stop outgrowth absolutely indicating massive growth signal by blood products. Growing and serum free cells in both cases show growth without damaging treatment and without blood products respectively.
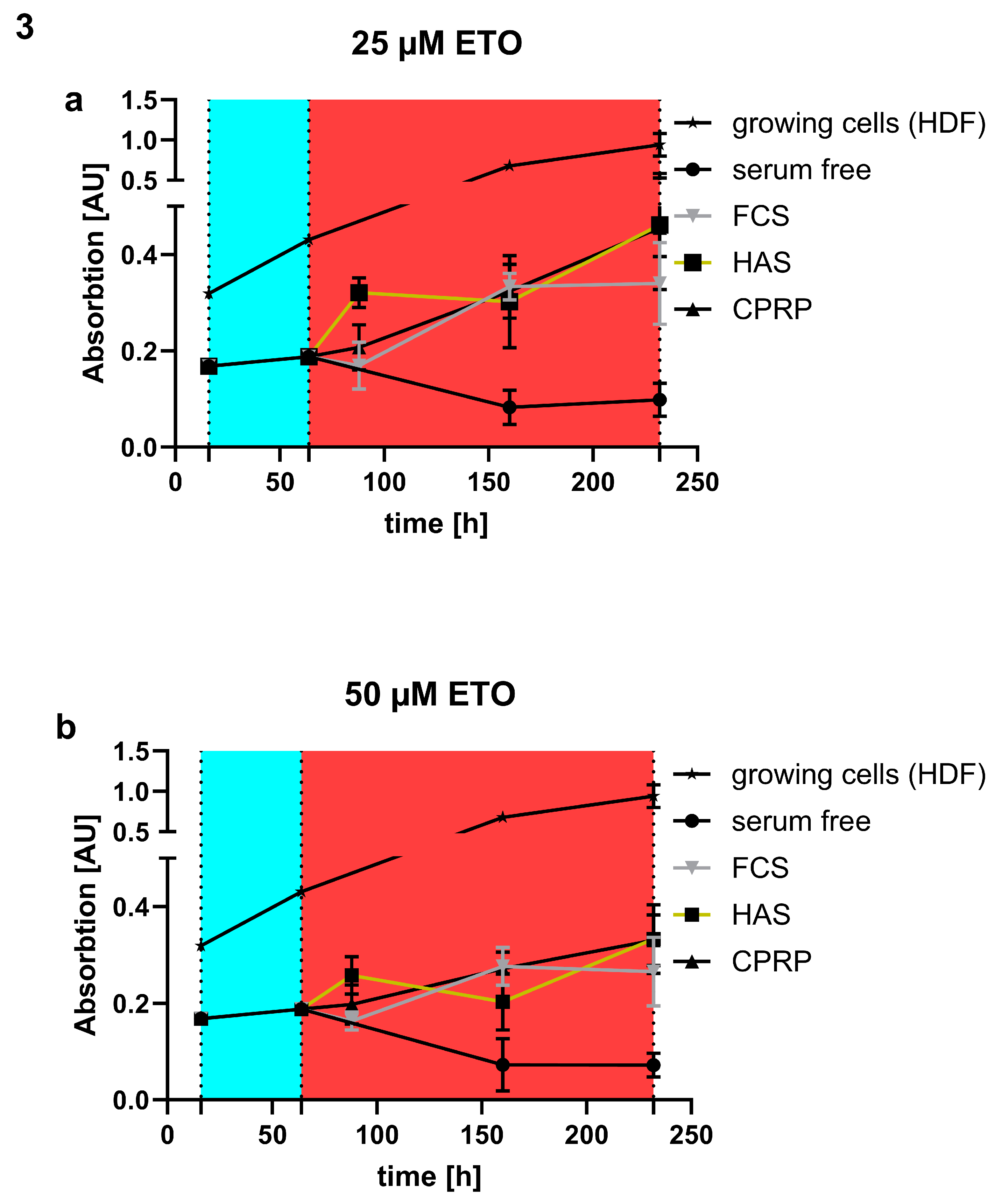
Figure 4.
growth of fibroblasts without etoposide (a) and different concentrations of HAS (b) and (c) reduced outgrowth by 3d treatment with blood products (red area) following etoposide treatment (blue area) final treatment conditions 50 µM (d) indicating only minor outgrowth representing establishment of senescent state.
Figure 4.
growth of fibroblasts without etoposide (a) and different concentrations of HAS (b) and (c) reduced outgrowth by 3d treatment with blood products (red area) following etoposide treatment (blue area) final treatment conditions 50 µM (d) indicating only minor outgrowth representing establishment of senescent state.
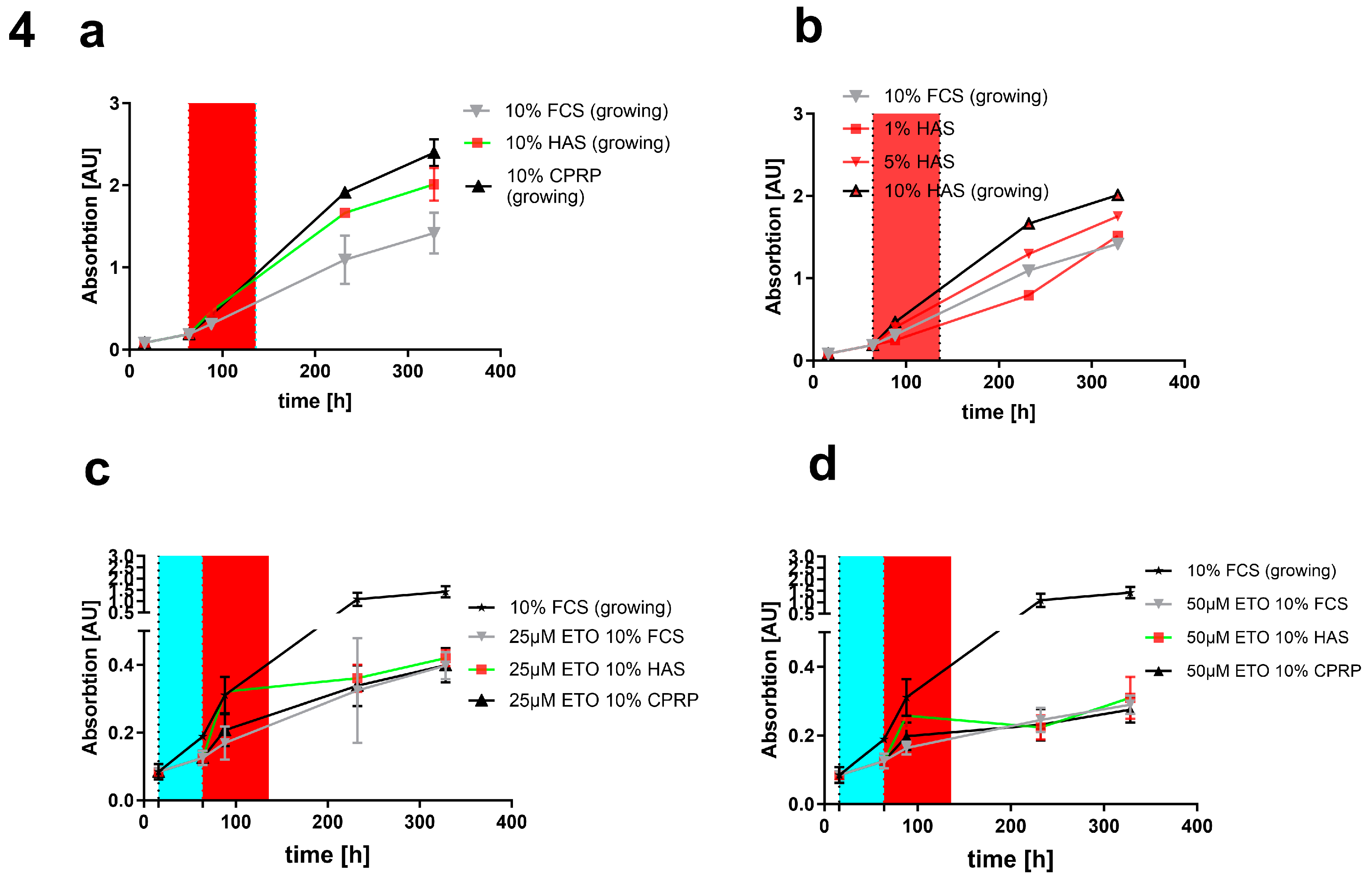
Figure 5.
different HAS condensations (a) 25 µM (b) 50 µM etoposide (48 h) and 24 h of BP treatment showing increased growth of HAS treated cells correlated with decreased CASP3/7 activity indicating prevention of apoptosis by HAS at the same timepoint with 25 (c) and 50 µM (d) etoposide treated fibroblasts. 0.0001 to 0.001Extremely significant *** 0.001 to 0.01 Very significant ** 0.01 to 0.05 Significant * ≥ 0.05 Not significant ns.
Figure 5.
different HAS condensations (a) 25 µM (b) 50 µM etoposide (48 h) and 24 h of BP treatment showing increased growth of HAS treated cells correlated with decreased CASP3/7 activity indicating prevention of apoptosis by HAS at the same timepoint with 25 (c) and 50 µM (d) etoposide treated fibroblasts. 0.0001 to 0.001Extremely significant *** 0.001 to 0.01 Very significant ** 0.01 to 0.05 Significant * ≥ 0.05 Not significant ns.
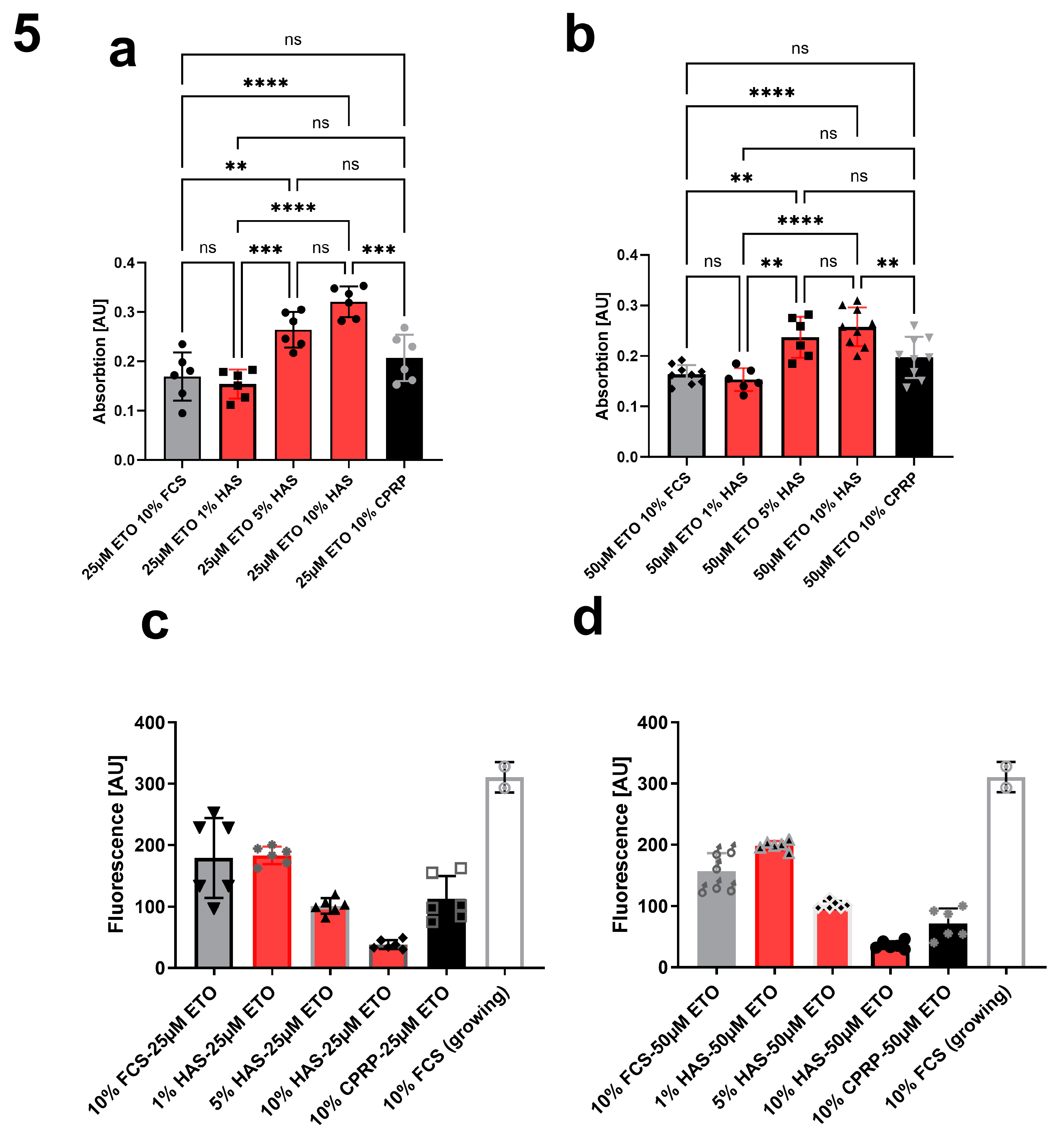
Figure 7.
morphology of cells determined by cell size. (a) time course of early treatment with different BP and final sizes at day 21 (b). Late treatment time course (c) and 21-day timepoint column graph of late treatment (d).
Figure 7.
morphology of cells determined by cell size. (a) time course of early treatment with different BP and final sizes at day 21 (b). Late treatment time course (c) and 21-day timepoint column graph of late treatment (d).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



