Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
This case report describes a 71-year-old female with multiple comorbidities who presented with symptoms suggestive of stroke and was subsequently found to have severe acute kidney injury (AKI) due to acute interstitial nephritis (AIN). The patient has normal kidney function at baseline. She has been on hydrochlorothiazide and metoprolol succinate for hypertension for several years. Due to severe acute kidney injury, a kidney biopsy was performed, revealing severe Acute Interstitial Nephritis secondary and multiple intratubular oxalate crystals. The patient's renal function improved after withdrawing HCTZ, volume repletion and initiating prolonged prednisone taper. We attempt to provide some insights through our paper into the mechanism of acute renal failure from crystal nephropathy and its management.
Keywords:
oxalate crystal nephrpathy
; acute renal failure
Introduction
Acute renal failure due to interstitial nephritis is a well-known entity. Interstitial nephritis can be acute or chronic leading to acute renal failure or chronic kidney disease depending on the duration of exposure to the offending agent and severity of insult [1,2]. Interstitial nephritis is usually due to exposure to various drugs. List of the drugs than can cause interstitial nephritis is quite large. But interstitial nephritis due to causes other than medications is uncommon [2]. We here describe a case of severe acute renal failure due to interstitial nephritis which was triggered by intratubular oxalate crystal deposition. We treated this patient with prednisone taper and had significant improvement in renal function.
Case presentation
A 71-year- old female with obstructive sleep apnea, peripheral vascular disease s/p common iliac artery stenting, Gillian Barre syndrome, hypertension, hyperlipidemia was admitted to hospital with symptoms concerning for stroke. She had progressive weakness-in lower extremities (reports left more than right), intermittent slurring of speech, memory difficulties, decreased appetite and severe fatigue. She denied any dysuria, urinary frequency, urgency or decreasing urine output. She reports having diarrhea for a week, 2 weeks prior to admission, which has been resolved without any medical intervention. She takes Hydrochlorothiazide 25 mg daily (has been on it for more than 2 decades) and Metoprolol succinate 100 mg daily for blood pressure. Her vitals on admission are as follows, afebrile, heart rate is around 70 per minute, respiratory rate is 16 per minute, blood pressure is 153/128 mmHg and weight is 76 kg.
Evaluation in the emergency room has shown severe acute kidney injury. She has normal kidney function at baseline with creatinine around 0.7 mg/dL and estimated glomerular filtration rate (EGFR) greater than 60 mL/minute. Basic metabolic panel (BMP) on admission showed creatinine 10 mg/dL and EGFR less than 15 mL/minute. She had anion gap acidosis with anion gap of 17, serum bicarbonate was 24 millimoles per L, sodium was 136 millimoles per L, chloride was 95 millimoles per L and BUN (Blood Urea Nitrogen) was 69 mg/dL. CT (Computed Tomogram) abdomen pelvis did not show any hydronephrosis but showed a 3 mm (about 0.12 in) stone in the distal left ureter. Renal artery Doppler study has shown normal-sized kidneys bilaterally with normal velocities in both renal arteries and no occlusion in the renal arteries. Urinalysis on admission has shown large blood, protein 30 mg/dL, 4-10 WBC per high-power field, no RBC, or granular casts. Creatinine kinase was 862 U/L. Complete blood count (CBC) showed hemoglobin 9.8 grams/deciliter, platelets were 243 and leukocytes were 6. She had severe hypokalemia, potassium was 2.7 millimoles per L on admission and received IV and oral Potassium chloride (KCl) supplements. Magnesium was 2.3 mg/dL.
She underwent extensive investigation with serologies checking for cryoglobulins, Anca vasculitis, anti-GBM antibody disease, antinuclear antibody screen, monoclonal protein study to detect paraproteinemia, serum free light chains, viral hepatitis panel and complements-which have all been negative.
She received normal saline and potassium chloride supplementation. She had good urine output. But her renal function did not show any significant improvement. HCTZ was discontinued due to hypokalemia and acute kidney injury. She was started on Nifedipine and continued Metoprolol. After 48 hours (about 2 days) of continuous IV fluid infusions her creatinine remained around 9 mg/dL and EGFR remained below 15 mL/minute. A kidney biopsy was done. Kidney biopsy has shown findings of severe acute interstitial nephritis with deposits of oxalate crystals. There was no evidence of any immune complex deposits on immunofluorescence study. Light microscopy has shown mild tubular interstitial scarring but had severe diffuse interstitial edema involving cortex, medulla and severe tubular epithelial cell injury with tubular lumens containing hyper eosinophilic casts and multiple oxalate crystals. Interstitial tissue contained dense infiltrates of lymphocytes and eosinophils. Electron microscopy has shown normal cellularity and mildly expanded mesangial regions with no immune complex or paraprotein related deposits. Glomeruli are Non proliferative and tubular interstitial compartment showed interstitial edema with severe inflammation. There was glomerular basement membrane wrinkling on several segments and no tubular basement membrane deposits.
She denied any exposure to nonprescription medication which can cause interstitial nephritis like proton pump inhibitors or NSAIDs. She has been on hydrochlorothiazide and metoprolol for several years for hypertension. (she recollects being on them for more than 20 years). Because of her severe hypokalemia and AKI, HCTZ was discontinued. She started Prednisolone 60 mg daily for acute interstitial nephritis. She was discharged with Bactrim for Pneumocystis pneumonia (PCP) prophylaxis, calcium and vitamin-D supplements for preventing osteoporosis from high-dose steroids.
Repeat BMP after a month has shown creatinine improving to 1.5 mg/dL and EGFR was close to 35 mL/minute. Her prednisolone dose slowly tapered over 6 weeks (about 1 and a half months). Her potassium has stabilized, and she was taken off potassium supplements at the follow-up visit. Repeat urinalysis did not show any WBC, RBC, or protein. Blood pressure has normalized and was hovering around 120 to 130/70 to 80 mmHg during the clinic visits.
The etiology of her acute interstitial nephritis as per pathologist is due to severe inflammatory reaction in the interstitial tissue surrounding the multiple oxalate crystal deposits in the tubules. She does not have any history of renal calculi. But CT abdomen pelvis shows ureteral calculi and biopsy also has oxalate crystals in multiple tubules. This is a unique case of crystal nephropathy.

Table 1.
Laboratory Test Results.
| Diagnostic Test | Normal Value | At Presentation | Hospital Day 2 | Hospital Day 6 | On Discharge | Follow- Up |
|---|---|---|---|---|---|---|
| Hemoglobin, g/dL | 11.6-15.0 | 9.0 | 7.6 | 9.1 | 7.3 | |
| WBC count, ×10(9) /L | 3.4-9.6 | 6.0 | 4.3 | 16.8 | 9.2 | |
| Platelet count, ×10(9) /L | 157-371 | 243 | 207 | 168 | 124 | |
| Serum Urea Nitrogen, mg/dL | 6-21 | 69 | 62 | 55 | 34 | 23 |
| Serum Creatinine, mg/dL | 0.59-1.04 | 10.15 | 9.10 | 8.03 | 5.03 | 1.6 |
| Serum Sodium | 135-145 mmol/l | 136 | 143 | 143 | 144 | 137 |
| Serum Potassium | 3.6-5.2 mmol/l | 2.7 | 3.6 | 3.9 | 3.4 | 3.9 |
| Serum Chloride | 98-107 mmol/l | 95 | 106 | 108 | 109 | 98 |
| Serum Calcium |
8.8-10.2 mg/dl | 9.4 | 8.8 | 8.6 | 8.4 | 9.7 |
| Serum Phosphorus | 2.5 - 4.5 mg/dl | 5.5 | 5.5 | 5.2 | 3.8 | |
| Serum Magnesium | 1.7- 2.3 mg/dl | 2.3 | 2.0 | 1.6 | ||
| Serum Bicarbonate | 22-29 mmol/l | 24 | 21 | 21 | 22 | 26 |
| eGFR | >=60 mL/min/BSA | < 15 | <15 | <15 | <15 | 34 |
| Anion Gap | 7-15 | 17 | 16 | 14 | 13 | 13 |
| Serum Albumin | 3.5-5.0 g/dl | 3.7 | 4.1 |
Table 2.
Summary of key diagnostic tests.
| Diagnostic Test | Normal Value | Result |
|---|---|---|
| HCV Ab screen, S | Negative | Negative |
| HBs antibody, S | Negative | Negative |
| HBs antigen, S | Non-reactive | Non-reactive |
| HBc total ab w/reflex, S | Negative | Negative |
| Complement, C3 | 75-175 mg/dL | 143 mg/dl |
| Complement, C4 | 14-40 mg/dL | 27 mg/dl |
| Cryoglobulins | Negative | Negative |
| GBM, IgG Ab | <0.1(Negative) U | <0.2 U |
| MPO, Ab | <0.4(Negative) U | <0.2 |
| PR 3, Ab | <0.4(Negative) U | <0.2 |
| SPEP | No monoclonal protein | |
| Haptoglobin, S | 30-200 mg/dl | 297 |
| Kappa Free light Chain, S | 0.3300 - 1.94 mg/dL | 13.7 |
| Lambda free light chain, S | 0.5700 - 2.63 mg/dL | 8.08 |
| Kappa/Lambda FLC Ratio | 0.2600 – 1.65 | 1.70 |
| DNA double stranded Ab, IgG, S | <=4 (Negative) IU/mL | 2 |
| Sm. Ab, IgG, S | <1.0(Negative) U | <0.2 |
| CRP | <=8.0 mg/L | < 3.0 mg/l |
| Bilirubin, Total | <=1.2 mg/L | 0.8 |
| ALT, S | 7 - 45 U/L | 11 |
| AST, S | 8 - 43 U/L | 17 |
| Alkaline phosphatase | 35 – 104 U/L | 60 |
| Protein, total | 6.3 – 7.9 g/dL | 6.9 g/dl |
| Albumin | 3.5-5.0 g/dL | 3.7 g/dl |
Figure 1.
There is edema and marked diffuse mononuclear tubulointerstitial inflammation. The tubules have simplified epithelium with lost brush border and reactive nuclear changes. There is no significant tubular vacuolization. The glomeruli appear spared.
Figure 1.
There is edema and marked diffuse mononuclear tubulointerstitial inflammation. The tubules have simplified epithelium with lost brush border and reactive nuclear changes. There is no significant tubular vacuolization. The glomeruli appear spared.
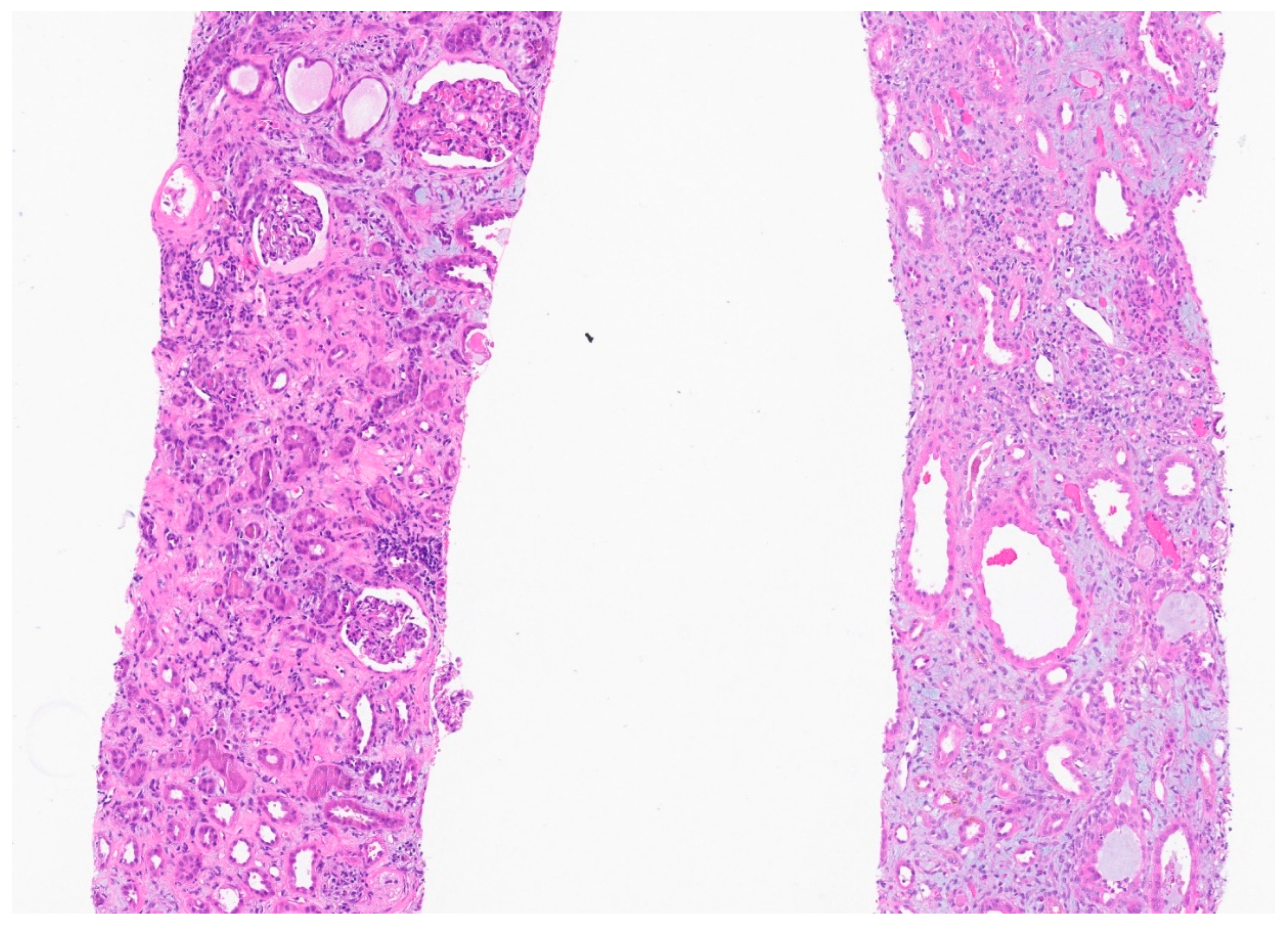
Figure 2.
There is necrotic debris in two tubular lumens (lower left and upper right) and inspissated Tamm-Horsfall protein (center)on severe inflammatory background.
Figure 2.
There is necrotic debris in two tubular lumens (lower left and upper right) and inspissated Tamm-Horsfall protein (center)on severe inflammatory background.
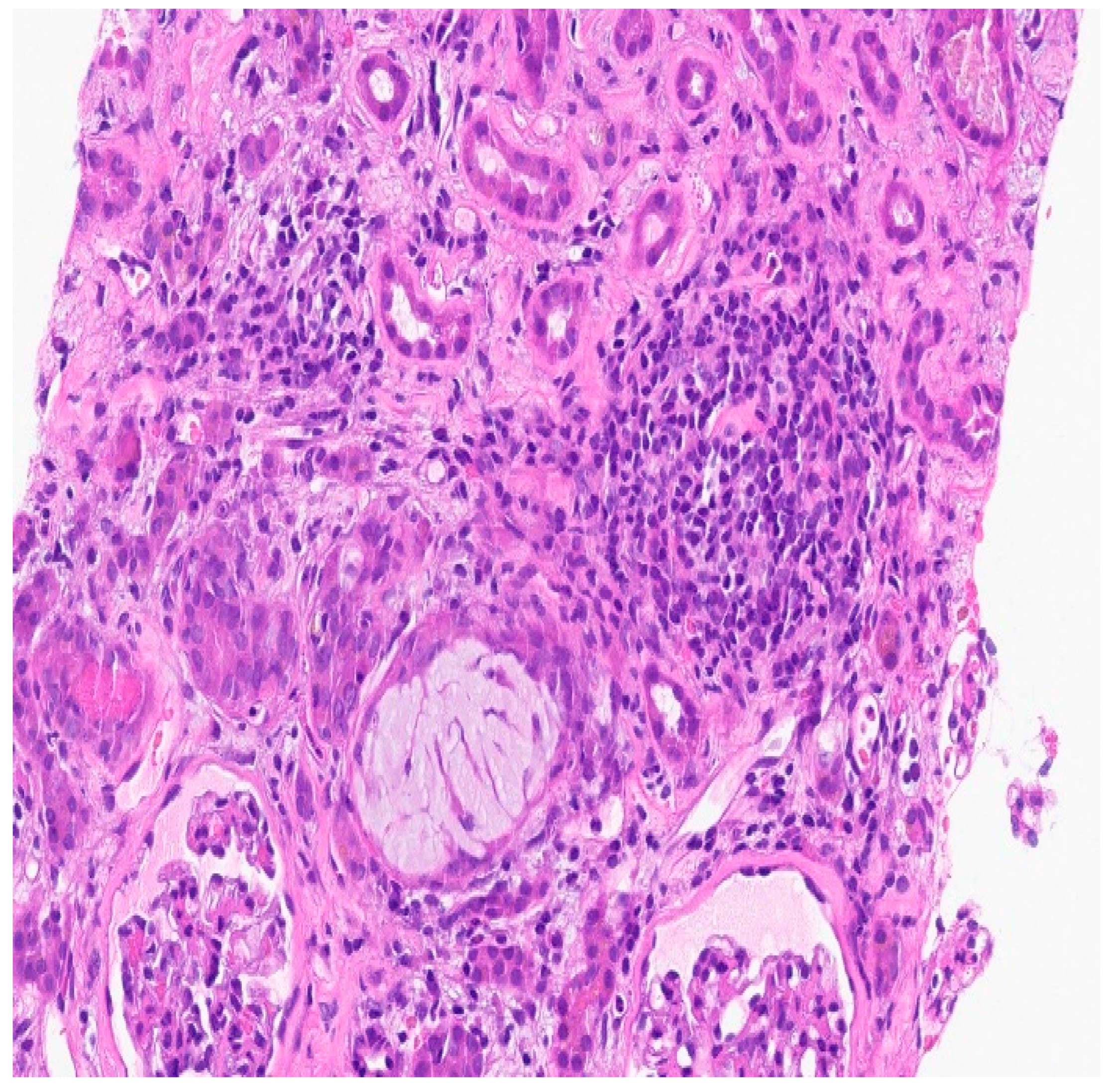
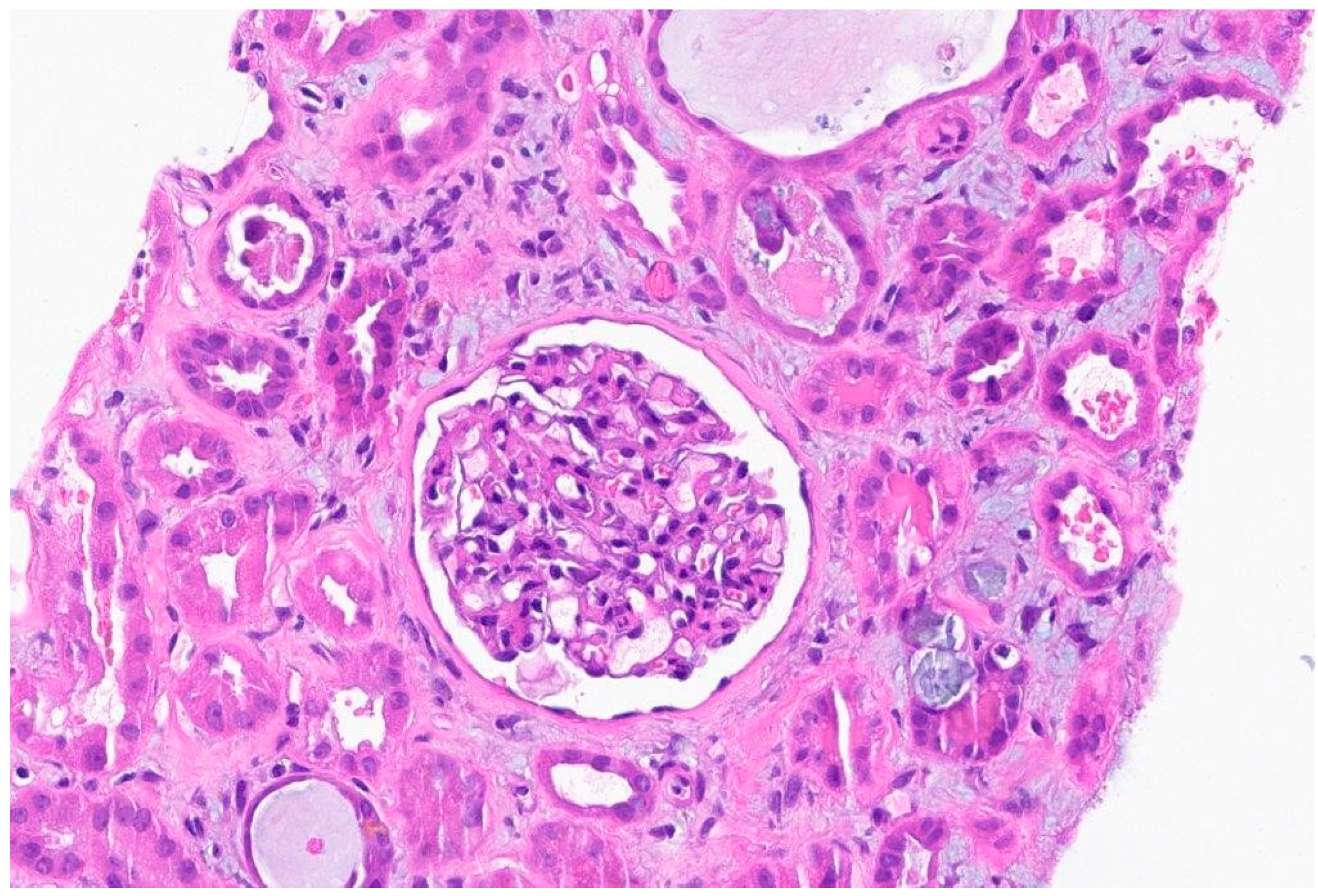
Figure 3.
The background is inflammatory and there is an oxalate crystal in the center right. Necrotic debris in 2-3 tubules.
Figure 3.
The background is inflammatory and there is an oxalate crystal in the center right. Necrotic debris in 2-3 tubules.
Biopsy report:
Final diagnosis: Acute onset of interstitial nephritis, severe.
Light Microscopy: The glomeruli are of normal size with normal mesangial matrix. There is no mesangial or endocapillary hypercellularity. Special stains do not demonstrate spikes, craters, or basement membrane remodeling.
TUBULES AND INTERSTITIUM: There is severe diffuse interstitial edema involving cortex and medulla. There is severe tubular epithelial cell injury with luminal ectasia, fraying of the brush border, and simplification of the lining epithelium. Tubular lumina contain necrotic debris, and some lumina contain hyper eosinophilic ropy casts. The interstitium contains dense infiltrates of lymphocytes. Some areas contain aggregates of eosinophils. Mild tubulitis is seen. There are intratubular oxalate crystals (at least 3).
VESSELS: The visualized arteries show severe intimal fibrosis. No vasculitis, thrombi, or Atheroembolic lesions.
Electron Microscopy: Normal cellularity and mildly expanded mesangial regions are confirmed. No immune complex or paraprotein-related deposits are seen, and the glomerular basement membranes show wrinkling of several of the segments, but otherwise show no ultrastructural abnormalities. There is mild foot process effacement present. Examination of the tubulointerstitial compartment shows interstitial edema, severe interstitial inflammation, and multifocal tubulitis. No tubular basement membrane deposits are seen.
Impression: Kidney, needle biopsy: Acute interstitial nephritis.
Immunofluorescence: There is no significant glomerular staining with albumin, IgA, IgG, IgM, C1q, C3, fibrinogen, kappa, and lambda light chains. The cast is seen staining equally with IgA, kappa, and lambda light chains.
Discussion:
Interstitial nephritis is a renal disease characterized by inflammation and scarring of the kidney's tubular and interstitial components. It manifests in three primary types: immune-mediated, infection-mediated and idiopathic. The immune-mediated Interstitial nephritis is caused by drug -reactions or due to immunologic diseases. Many drugs have been linked to interstitial nephritis, comprising of antibiotics, antacids, analgesics, immunotherapies, diuretics (including thiazide diuretics), antivirals, anti-convulsant, lithium, allopurinol, and many more [2]. The mechanism through which drugs induce interstitial nephritis vary [3]. As we mentioned, drug induced interstitial nephritis is well documented entity with thiazide diuretics and our case report adds a complex layer to this well-known entity [1,2,4].
In our patient, the complexity is enhanced when multiple oxalate crystals were identified on kidney biopsy along with interstitial nephritis. Oxalate nephropathy is a rare pathology that can be difficult to diagnose clinically and needs a biopsy. This presentation aligns with crystalline nephropathy, a condition marked by crystal precipitation in kidney tubules [2,3]. Crystalline nephropathy poses risks of both acute and chronic kidney injuries. Various factors contribute to the risk of crystal deposition, encompassing intravascular volume depletion, underlying kidney diseases, and metabolic imbalances altering urinary Ph [2,3]. The intricate interplay of supersaturation, urine pH, and crystallization inhibitors influences intratubular crystal deposition. Drug-induced crystal precipitation, often associated with supersaturation in low urine volume or drug insolubility in acidic or alkaline urine pH, can exacerbate renal complications. Metabolic disturbances, including systemic acidosis or alkalosis and renal tubular acidosis, play a role in worsening intrarenal crystal deposition. Patient characteristics linked to medication intake predispose individuals to intratubular crystal deposition and subsequent tubular obstruction [2,3].
Severe volume depletion, prevalent in conditions such as chronic diarrhea, anorexia, excessive diuresis, febrile illnesses, adrenal insufficiency, and renal salt wasting are crucial factors in the development of acute kidney injury (AKI). Conditions leading to effective intravascular volume depletion such as pancreatitis, ascites, heart failure, pleural effusions, and nephrotic syndrome contribute to renal hypoperfusion, heightening the risk of tubular crystal deposition [2,3]. Urine pH further modulates crystallization, with certain drugs exhibiting varying solubility in acid or alkaline urine. Crystal precipitation within the kidneys obstructs tubular lumens in the distal nephron, involving crystals mixed with cellular debris and proteinaceous material. Gastrointestinal disorders with small bowel dysfunction or defective fat and bile acid absorption contribute to enteric hyperoxaluria, characterized by increased gastrointestinal oxalate absorption and excessive urinary oxalate excretion [2,3].
There are numerous case reports that have described drug- induced oxalate crystallopathy [4,5,6,7,8]. In our case, the patient has been on thiazide diuretic for over two decades without any previous kidney injury or associated complications. The continued exposure to thiazide diuretic through a period where she had diarrhea raises the possibility of severe volume depletion and enteric hyperoxalurias as contributing factors in crystal formation. Prolonged exposure to thiazide diuretic without any adverse events rules out the possibility that the interstitial nephritis was solely due to thiazide diuretic. But it is quite possible that thiazide diuretic use in the setting of diarrhea might have aggravated volume depletion leading to oxalate crystal deposition which has triggered the onset of interstitial nephritis.
Recommendations to reduce the risk of developing crystal nephropathy emphasize maintaining adequate volume status and avoiding concurrent use of diuretics, ACE-Is, ARBs, and NSAIDs [2,3]. In cases where crystal nephropathy occurs, sustaining a high urine flow rate is crucial to minimize further crystal precipitation and dislodge obstructing crystals. Early detection and withdrawal of the causative agent can be achieved through timely examination of urine sediment in individuals experiencing AKI which sometimes can demonstrate crystals. Monitoring facilitates the implementation of supportive measures, including volume repletion, urine alkalinization and in severe cases of AKI, rarely, hemodialysis [2,3].
In this case, we treated the patient with prolonged prednisolone taper with a strategy targeting the inflammatory response associated with interstitial nephritis. There was significant improvement in renal function, which provides insight into the role of corticosteroids in treatment of oxalate crystallopathy induced interstitial nephritis.
In addition, research studies indicate that oxalate crystal deposition is associated with kidney epithelial cell damage, suggesting that crystals induce epithelial injury and progressive inflammation leading to interstitial nephritis [9]. Our case provides us with the learning that prolonged exposure to diuretics can lead to volume depletion triggering oxalate crystal deposition which can induce severe interstitial nephritis.
It is important to fully assess the impact of oxalate crystal deposition on renal function to provide comprehensive patient care. The frequency of interstitial nephritis due to oxalate crystal deposition, especially in the context of long-term thiazide diuretic use in the absence of other risk factors for interstitial nephritis, highlights the importance of a comprehensive diagnostic approach. This case contributes to the expanding knowledge of renal pathology, highlighting the clinical importance of identifying and addressing uncommon causes of interstitial nephritis. Further research and case studies will play an important role in improving our understanding of such unusual presentations and optimizing treatment strategies.
Author Contributions
E.P. and L.M. contributed to the investigation, coordination, writing (original and final draft), reviewing, editing and S.R contributed to the reviewing, and editing.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent Statement
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Acknowledgments
The authors are very appreciative of the reviewers for their valuable feedback during review which helped us improve the article and publishers of the journal for publishing our article.
Conflicts of Interest
The author(s) declared no conflicts of interest regarding the research, authorship, and/or publication of this article.
References
- Magil, A.B.; et al. Acute interstitial nephritis associated with thiazide diuretics. Clinical and pathologic observations in three cases. Am J Med 1980, 69, 939–43. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A.; Rosner, M.H. Drug-Induced Acute Kidney Injury. Clin J Am Soc Nephrol 2022, 17, 1220–1233. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, S.G.; Perazella, M.A. Drug-induced crystal nephropathy: an update. Expert Opin Drug Saf 2008, 7, 147–58. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; et al. Triamterene crystalline nephropathy. Am J Kidney Dis 2014, 63, 148–52. [Google Scholar] [CrossRef] [PubMed]
- Farge, D.; et al. Dyazide-induced reversible acute renal failure associated with intracellular crystal deposition. Am J Kidney Dis 1986, 8, 445–9. [Google Scholar] [CrossRef] [PubMed]
- Ansari, F.A.; et al. A Rare Case of Acute Kidney Injury Due to Levofloxacin-induced Crystal Nephropathy. Indian J Nephrol 2019, 29, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Garneau, A.P.; Riopel, J.; Isenring, P. Acute Methotrexate-Induced Crystal Nephropathy. N Engl J Med 2015, 373, 2691–3. [Google Scholar] [CrossRef] [PubMed]
- Goli, R.; et al. Acute Ciprofloxacin-Induced Crystal Nephropathy with Granulomatous Interstitial Nephritis. Indian J Nephrol 2017, 27, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.; Wood, K.; Sayer, J.A. Calcium oxalate crystal deposition in the kidney: identification, causes and consequences. Urolithiasis 2020, 48, 377–384. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



