
Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Although acute heart failure (AHF) is a common disease associated with significant symptoms, morbidity and mortality, the diagnosis, risk stratification and treatment of patients with hypertensive acute heart failure (H-AHF) still remain a challenge in modern medicine. Despite great progress in diagnostic and therapeutic modalities, this disease is still accompanied by a high rate of both in-hospital (from 3.8% to 11%) and one-year mortality (from 20% to 36%). Considering the high rate of re-hospitalization (22% to 30% in the first three months), the treatment of this disease represents a major financial blow to the health system of each country. This disease is characterized by heterogeneity in precipitating factors, clinical presentation, therapeutic modalities and prognosis. Since heart decompensation usually occurs quickly (within a few hours) in patients with H-AHF, establishing a rapid diagnosis is of vital importance. In addition to establishing the diagnosis of heart failure itself, it is necessary to see the underlying cause that led to it, especially if it is de novo heart failure. Given that hypertension is a precipitating factor of AHF and in up to 11% of AHF patients, strict control of arterial blood pressure is necessary until target values are reached in order to prevent the occurrence of H-AHF, which is still accompanied by a high rate of both early and long-term mortality.
Keywords:
hypertension
; acute heart failure
; congestion
; diagnosis
; modern therapy
1. Introduction
Acute heart failure is defined as the rapid or gradual appearance of pronounced signs and/or symptoms of heart failure, which often require the patient to seek emergency medical help, and which in most cases leads to unplanned hospitalization [1]. AHF does not include heart failure with moderate symptoms and signs, which can be treated on an outpatient basis by changing lifestyle habits and intensifying the medicinal therapy regimen.
Although the classification of heart failure has changed over the years, it is clearly accepted that the classification of chronic heart failure is based on the left ventricular ejection fraction (LVEF) value [2]. So far, different divisions of AHF have been attempted with the aim of clearly defining this entity and easier application of an adequate therapeutic regimen for each phenotype. The initial opinion that every acute heart failure is a consequence of fluid volume overload was rejected by analyzing the pathophysiological mechanisms involved in its occurrence. The 2008 guideline of the European Association of Cardiology for the treatment of heart failure proposed six different clinical forms of AHF: Worsening or decompensated chronic HF; Pulmonary edema; Hypertensive HF; Cardiogenic shock; Isolated right HF and HF in acute coronary syndrome [3]. In the same year, a modification of the AHF categories derived from the Forrester classification for heart failure after myocardial infarction was proposed, which is based on the presence or absence of tissue congestion and perfusion [4]. The heart failure guide from 2012 proposed the classification of AHF based on the level of systolic blood pressure (SBP) at the initial presentation of the patient [5,6]. All of the aforementioned divisions of AHF were analyzed within the ESC Heart Failure Long-Term (HF-LT) registry and the study published by Chioncel et al. In that study were analyzed 6,629 hospitalized patients with AHF and it was shown that there are significant differences in early mortality and adverse events depending on the clinical profile of the patient or the value of arterial blood pressure at the initial examination. This difference was especially registered in patients in the first 6 months of follow-up, while after that time period the one-year outcome of the patient was less influenced by the clinical profile or SBP value at admission [6]. According to the 2016 ESC guide, AHF classification was mainly based on phenotypes resulting from the combined ratio of congestion and hypoperfusion (wet and dry, cold and warm) [7]. By combining them, 4 different phenotypes were created. In the study by Javaloyes P. and associates, this division was shown to be very simple in clinical practice, and the authors emphasized the importance of clinical assessment of congestion and perfusion at the initial presentation right next to the patient's bed. According to their data, the clinical classification into four phenotypic profiles correlated with the early outcome of these patients and was helpful to doctors in making a more precise decision when administering a therapeutic regimen to patients with AHF [2,8]. Other studies conducted on large registries of patients with heart failure showed similar results [4,9,10]. On the other hand, Masip J. and colleagues also mentioned the negative aspects of this division of AHF. The incidence of the mentioned phenotypes is very unbalanced, patients belonging to the "cold and dry" group occurred with a frequency of less than 1%, while patients with the "wet and warm" phenotype made up about 80% of treated patients. Also, with the concept of congestion, no distinction was made between systemic and pulmonary congestion. The third remark according to those authors is that the patient cannot have AHF that requires urgent hospital treatment without congestion or hypoperfusion, so the "warm and dry" phenotype is debatable [11]. In accordance with the current knowledge, the latest division from 2021 focused to the greatest extent on the pathophysiological mechanisms that lead to AHF. According to this classification, AHF is divided into: Acute decompensated heart failure, Acute pulmonary edema, Isolated right ventricular failure and Cardiogenic shock [1].
Despite all efforts to find the best clinical classification of AHF that would make it easier for doctors to choose an individual therapeutic regimen for each patient, mortality from AHF is still very high [12]. We authors think that the main goal is to see the pathophysiological changes in AHF that are responsible for the emergence of difficult clinical scenarios. Patients with H-AHF present a clinical phenotype dominated by an increase in afterload and a decrease in venous capacitance, as well as a consequent increase in ventricular filling pressures. In this paper, we will consider the pathophysiological mechanisms underlying H-AHF, as well as the current data on the best options for its treatment.
2. Epidemiology
Most epidemiological data related to AHF are obtained from large heart failure registries [13,14,15,16,17,18]. In different registries, the frequency of certain phenotypic forms of AHF is different. According to the ALARM-HF registry, the frequency of acutely decompensated congestive HF was 38.6%, pulmonary edema 36.7% and cardiogenic shock 11.7%. Cardiogenic shock was less prevalent according to other registries of about 2-5% [6,19]. A study examining patient outcomes during the first hospitalization due to AHF showed that in-hospital mortality was up to 7.5% [20]. The lowest in-hospital mortality in patients with AHF was shown in the ESC-HF Pilot registry (3.8%), while the highest was recorded in the ALARM HF registry (up to 11%) [14]. Postdischarge mortality up to 3 months was 7% to 11% [13,14,20]. One-year mortality in patients with AHF according to the ESC HF Pilot registry is 17.4%. On the other hand, within the ADHERE registry, one-year mortality was estimated at as much as 36% [18]. A recent study by Lombardy C. and colleagues showed similar data, where the thirty-day mortality from AHF was about 8%, while the one-year mortality was 20% [12]. Registries have shown that the incidence of rehospitalization ranges between 22 and 30% at 1–3 months and reaches 65% at 1 year of the index AHF hospitalization [21,22]. The importance of hospital readmissions due to AHF is reflected in the fact that each new hospitalization has been shown to correlate with worsening cardiac function, reduced quality of life, and a higher incidence of death over a longer follow-up period.
In relation to the geographical distribution, the highest mortality from AHF in the three-month follow-up period is experienced by patients living in South America (17.3%), followed by Western Europe (15.1%), North America (13.3%), Asia and Pacific (11.6%), and the lowest in Central Europe (9.3%) [23].
Analyzing the results of the registers, it was shown that the largest number of patients with AHF already have known heart failure (about two thirds of patients), while the number of patients with de novo AHF is much smaller. About 50% of patients with already known chronic heart failure have preserved EF. The gender ratio is mostly symmetrical with a slight male predisposition. Patients with AHF have hypertension in about 70% [14,16]. Arterial hypertension is more common in HF patients with preserved LVEF (76%) compared to those with reduced LVEF (66%) [24]. Data from the STAT register, which monitors patients hospitalized for hypertensive crisis, indicate that about 25.2% of patients with hypertensive crisis have AHF. Chiocel O. et al. point out that H-AHF constitutes 4.8% of all forms of AHF [6]. However, other authors point out that the share of H-AHF within the total AHF is much higher, up to 11%. [25]. Al-Lawati J.A. et al showed that low SBP values on admission are an independent predictor of mortality in patients with AHF. The higher the SBP on admission, the better the prognosis of AHF patients, regardless of age or estimated LVEF [26]. The OPTIMIZE-HF investigators reported ~50% of patients with AHF had SBP of >140 mm Hg at presentation. According to their study, higher SBP at admission was associated with lower in-hospital mortality rates: 7.2% (<120 mmHg), 3.6% (120-139 mmHg), 2.5% (140-161 mmHg), and 1.7% (>161 mmHg) ) (P<.001 for overall difference). Postdischarge mortality rates in the follow-up cohort by SBP at admission were 14.0%, 8.4%, 6.0%, and 5.4%, respectively (P<.001 for overall difference) [27]. Other studies also showed that SBP values one of the most important predictors of mortality in patients with AHF in intensive care units [28,29].
3. Pathophysiological mechanisms of H-AHF
Hypertensive acute heart failure is defined as the rapid onset of pulmonary congestion in the setting of a systolic blood pressure >140 mm Hg, and often >160 mm Hg [29]. Most patients with H-AHF have previously known heart failure (usually with preserved EF) and long-standing hypertension [24,30]. Maintaining blood pressure is strictly regulated by means of baroreceptors, primarily in the aorta and carotid arteries [31]. A significant role in maintaining normal values of arterial blood pressure is also played by renal regulation mechanisms (the influence of the renin-angiotensin-aldosterone system in the regulation of arterial blood pressure, affecting the volume of circulating blood as well as vascular resistance and tone). The influence of renal mechanisms is reflected in the fact that approximately 20-30% of patients with HF have some degree of renal weakness [32].
Changes in cardiac output and systemic vascular resistance trigger sympathetic nervous system and neurohumoral activation [33]. A heart with normal contractility is able to respond promptly to an increase in systemic arterial resistance and to maintain an adequate cardiac output. When a pressure load occurs, the left ventricle undergoes hypertrophic structural remodeling [34]. As the end result of these changes, there is a slow relaxation of the left ventricle with significant diastolic dysfunction. As the functional ventricular-vascular relationship becomes uncoupled, the LV has insufficient cardiac reserve to compensate for the increases in afterload and preload that accompany hypertensive episodes and physical exertion [31,35]. As a result of all this, the cardiac output is unable to increase in response to increased systemic vascular resistance, which leads to increased volume and pressure in the left ventricle and impaired blood flow from the pulmonary veins to the heart, which predisposes to pulmonary congestion. Such increases in filling volume also trigger the Frank-Starling mechanism in the right ventricle (RV), which combines a catecholamine-mediated increase in RV contractile force to drive up pulmonary artery and capillary wedge pressures [31]. The end result of all these processes is the appearance of pulmonary congestion. According to the study by Chiolance J. and colleagues, within the framework of the H-AHF phenotype, the largest number of patients presents with pulmonary congestion - as much as 66% [6]. About 10% of patients have peripheral hypoperfusion with varying degrees of pulmonary congestion, while hypoperfusion without pulmonary congestion is rarely present among H-AHF patients. In one half of all patients with AHF, several different precipitating factors are involved that lead to decompensation in AHF patients [36].
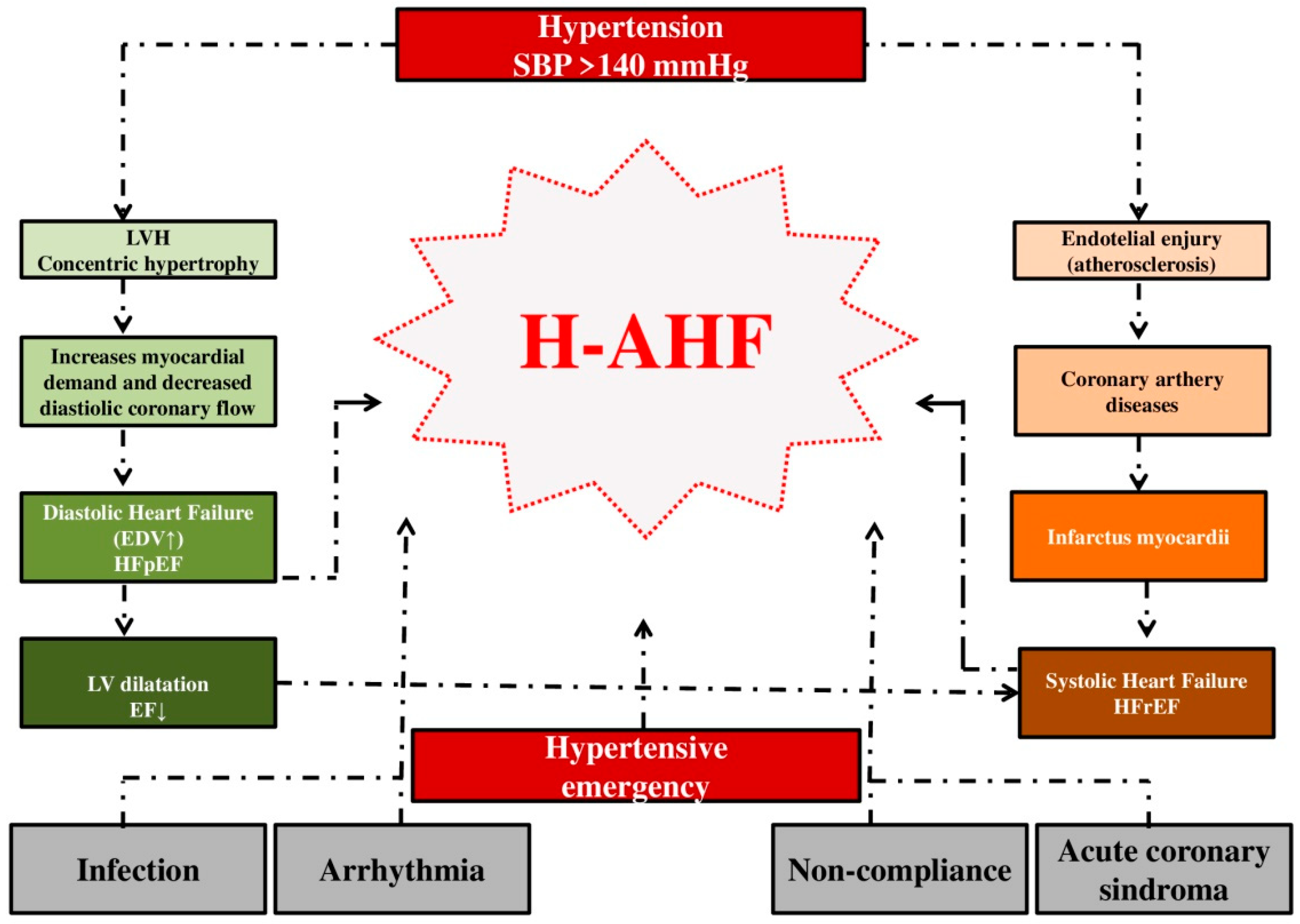
In patients with arterial hypertension, maladaptive changes occur in the myocardium and vasculature as a response to chronic hypertension, creating a system extremely sensitive to changes in pressure, fluid volume, and sympathetic tone. Peripheral and central baroreceptors become tolerant to higher pressures, in fact the aortic baroreflex becomes blunted [34]. Structural disorders in peripheral arterioles and central arteries functionally separate the vascular system from the ventricular system in hypertensive patients [37,38]. Chronically elevated values of arterial blood pressure resulting in increased stiffness of LV and reduced compliance across the cardiovascular system [31,39]. The leading pathophysiological mechanism that in H-AHF leads to decompensation in patients with chronic heart failure is a sudden increase in afterload with significant peripheral vasoconstriction [40]. There is a redistribution of volume with the movement of fluid from the splanchnic and peripheral vascular beds into the pulmonary circulation. Namely, vascular redistribution rather than volume overload may be the primary determinant of elevated cardiac filling pressure and subsequent pulmonary congestion in H-AHF patients [34]. However, to a certain extent, even with this AHF phenotype, fluid volume retention can be an additional significant factor that leads to acute HF [41] (Figure 1).
4. Clinical picture of H-AHF
In patients with H-AHF, cardiac decompensation usually occurs quickly, within a few hours to a few days [25,42,43]. Often the only precipitating factor for AHF is arterial hypertension. Patients usually do not complain of weight gain, nor do they have swelling on the lower legs that occurs due to peripheral congestion. At first examination, they have severely elevated SBP (≥ 160–180 mmHg) with auscultatory signs of pulmonary congestion [25]. The clinical picture is dominated by dyspnea. The patient takes a sitting position because it is easier to breathe.
The most extreme presentation of H-AHF is pulmonary edema. Clinical criteria for acute pulmonary edema diagnosis include dyspnoea with orthopnoea, respiratory failure, tachypnea (>25 breaths/min), and increased work of breathing [44]. Accumulation of fluid in the lungs leads to impaired gas exchange and arterial hypoxemia. In patients with pulmonary congestion, juxtapulmonary capillary (J-type) receptors are stimulated, which leads to tachypnea [45]. Thus, most AHF patients with pulmonary edema hyperventilate due to J-type receptor stimulation before a significant pathologic change in gas exchange occurs [45]. Since CO2 has a better diffusing capacity than O2, CO2 is less likely to increase until the later stages of diaphragmatic fatigue [45,46,47]. Symptoms that correlate with elevated arterial blood pressure are often present: headache, visual disturbances, chest pain, dizziness [48]. Patients may also have some neurological deficits [49].
Patients with H-AHF may have central cyanosis. On auscultation over the lungs, the presence of a weakened respiratory sound on both sides may be a consequence of the presence of pleural effusions, while the finding of inspiratory cracks is in favor of fluid transudation into the pulmonary alveoli [50,51]. Auscultation of the heart often registers the presence of a third heart sound.
5. Diagnostic modalities
In patients with AHF, in addition to establishing the diagnosis of HF itself, it is necessary to see the cause if it is de novo heart failure or to detect all potential precipitating factors and influence them. Physical examination is a fundamental component in the evaluation, risk stratification, and outcome prediction of patients with AHF [54]. Patients with physical examination findings consistent with volume overload (such as elevated jugular venous pressure and presence of peripheral edema) have higher body mass index, higher biomarker values, more precipitating factors for AHF, and lower LVEF compared to those without these findings.
Electrocardiography is a method that does not have sufficient specificity or sensitivity to diagnose AHF. It is very important in diagnosing various rhythm disorders that can be both a triggering factor for the appearance of AHF and a consequence of it. Its greatest importance is reflected in the exclusion of ischemic causes of AHF [55,56]. In addition to the ECG, the measurement of cardiospecific enzymes such as troponin can serve this purpose. Studies have shown that troponin values can be moderately elevated in patients with acute heart failure, and positive troponin values have prognostic significance for the outcome of patients with AHF [57,58]. Analysis of data from the ADHERE observational registry for heart failure showed that elevated TnT values at admission in patients with heart failure are correlated with lower LVEF. Patients with positive TnT values have a higher in-hospital mortality compared to those patients who had negative TnT values (8% vs 2.7%), with an adjusted OR of 2.55 for risk of death [59]. Similar data were shown by the EFECT study conducted on patients with AHF. A peak cTnI >0.5 ug/l measured in the first 48 hours of hospitalization was an independent predictor of all-cause mortality at 1 year with an HR of 1.49 [60]. In AHF that is accompanied or caused by a hypertensive crisis, elevated troponin values are registered [61]. In order to rule out ischemic causes of AHF, it is necessary to correlate the patient's clinical picture, changes in the ECG and dynamics in troponin values.
The main stimulus for increased brain natriuretic peptides (BNP) and N terminal pro brain natriuretic peptide (NT-proBNP) synthesis and secretion is myocardial wall stress. The physiological effects of BNP are manifold and include natriuresis/diuresis, peripheral vasodilatation, and inhibition of the renin–angiotensin–aldosterone system and the sympathetic nervous system [62]. In a large number of studies, BNP and NT-proBNP were consistently found to be elevated in patients with AHF, and the values were found to be related to the severity of the disease (they are higher in patients with a more severe clinical picture, lower LVEF and a more severe degree of diastolic dysfunction) [63,64,65]. During the evaluation of patients with acute dyspnea, it is recommended to measure the value of natriuretic peptides [66]. According to the PRIDE study, NT-proBNP measurement is a valuable addition to standard clinical assessment for the identification and exclusion of AHF in the emergency department setting [67]. Studies have also shown the importance of negative BNP values for excluding AHF in patients who presented with dyspnea in the emergency department [68]. In general, HF is unlikely at BNP values < 100 pg/ml and is very likely at BNP values > 500 pg/ml and, similarly, unlikely at NT-proBNP values < 300 pg/ml and very likely at NT-proBNP values > 450 pg/ml (> 900 pg/ml in patients over 50 years of age) [62]. It is important to note that natriuretic peptides can be elevated both in AHF and in patients with chronic HF, so the measurement of natriuretic peptides is more important in distinguishing between causes of cardiac and non-cardiac origin of AHF than in distinguishing between AHF and chronic HF [69]. Low concentrations of BNP can sometimes be registered in patients with advanced decompensated end-stage HF, in obese patients, in patients with flash pulmonary edema or right-sided AHF [44]. A more recent study by Dal Bianco J.P. and colleagues who examined BNP values in patients with Flash edema and different degrees of cardiac function showed that BNP levels were elevated in every patient, even when BNP was assayed early after dyspnea onset [70]. In addition to BNP, studies have demonstrated that soluble suppressor of tumorigenicity 2 (sST2), GDF-15, cystatin C, galectin-3, serum uric acid, microRNAs and low serum chloride are predictors of outcomes in AHF [71,72,73]. In patients with cardiorenal syndrome, it is important to monitor markers related to renal function. Deterioration of renal function accompanied by a rise in plasma urea and creatinine is associated with a risk of increased mortality and new hospitalizations [74,75]. Proenkephalin levels, a novel marker of renal function, are also associated with worsening renal function, in-hospital and follow-up mortality in patients with AHF [76].
Transthoracic echocardiography should be performed in all de novo AHF or in patients with decompensated chronic HF when a cardiac pathology is suspected, in order to evaluate the function of the left and right ventricles, the presence of segmental outbursts in the kinetics of the left ventricle, valve function and the presence of fluid in the pericardium [77]. All patients who come to the emergency department due to dyspnea must undergo teleradiography of the heart and lungs [78]. Its importance in the differential diagnosis of various lung diseases has been clearly demonstrated [79]. On the other hand, the presence of congestion on the X-ray of the heart and lungs can largely confirm the suspicion that it is AHF. Lung ultrasound proved to be a valid instrument to detect an increase in the superficial density and air space distribution of the lung [80]. Acute pulmonary edema of cardiogenic origin and acute respiratory distress syndrome (ARDS) are diseases that increase the density of the superficial lung and the full/empty ratio of the subpleural lung tissue, but in different ways [80,81]. Therefore, lung ultrasonography is generally considered a useful clinical tool among physicians. An inhomogeneous bilateral pattern of multiple coalescent B-lines and white lung, often with scattered spared areas, clearly characterizes ARDS, whereas the relatively regular presence of discrete B-lines characterizes the initial stages of pulmonary cardiogenic edema [82]. The criteria for the ultrasound differential diagnosis between ARDS and cardiogenic pulmonary edema were proposed in 2008 and revised in 2017 [82,83]. The importance of this technique is that it can be performed in a cheap, quick and simple way at the patient's bedside.
Every patient with AHF should have arterial blood gas analyses. The most common gas exchange disorders in AHF are normoxemia or hypoxemia with hypocapnia [84]. The results of the study that examined gas exchange in arterial blood in patients with HF indicate that about 19% of patients with AHF had acidosis, 37% had normal pH, and 44% had alkalosis. The most common type of acidosis was mixed-type (42%) followed by metabolic (40%), whereas the most common type of alkalosis was respiratory (58%). Acidosis proven in gas analyzes was a significant predictor of mortality (hazard ratio 1.93; 95% confidence intervals 1.27–2.93) [45]. In contrast, alkalosis was not associated with increased mortality. In this study, 19% of the patients had acidosis and most patients had metabolic or mixed-type acidosis, whereas a pure respiratory acidosis was not common, suggesting that tissue hypoperfusion was the main cause of the acidosis, not CO2 retention as a result of impaired gas exchange [45].
In all patients with H-AHF, we must also determine inflammatory markers because only an infectious cause as a precipitating factor for AHF can be ruled out by measuring inflammatory markers (CRP, Procalcitonin) [85].
6. Treatment of patients with H-AHF
Recent data have shown that timely initiation of therapy may be a key factor in the treatment of H-AHF [86]. Reducing ventricular filling pressure is a key component of management, especially when AHF is accompanied by hypertension [25]. In patients with H-AHF, it is recommended to use vasodilators that optimize preload and afterload by decreasing venous and arterial tone and consequently lower SBP and increase stroke volume [87]. As the weakened heart is sensitive to afterload in some patients, pulmonary edema can occur even at SBP values of up to 150 mmHg [31].
The 2016 ESC guideline recommendations for the treatment of HF advise in the treatment of AHF followed by a hypertensive crisis to rapidly reduce SBP (in the range of 25% during the first few hours and cautiously thereafter) [7]. It is necessary to consider the use of vasodilators in all patients with SBP values above 110 mmHg. Kitai T. et al. analyzed data from the REALITY-AHF registry in which AHF patients were analyzed [87]. Patients who received vasodilator therapy with consequent SBP reduction ≤ 25%, vasodilator therapy with >25% SBP reduction, and no vasodilators (mean arrival SBP 149+/−37 mmHg) were compared. Patients treated with vasodilators and with ≤ 25% SBP reduction at 6 h after initial presentation had a greater diuretic response and lower 1-year mortality (HR 0.74; 95%CI 0.57–0.96) compared to patients with ≥ 25% reduction or no vasodilator.
6.1. Vasodilators
6.1.1. Nitrates
Nitrates are primarily venodilators, and their application leads to a decrease in venous inflow to the heart, which reduces the possibility of congestion, lower afterload, increased stroke volume and consequent relief of symptoms. The use of nitroglycerin in the treatment of H-AHF is accompanied by a rapid onset of action of the drug, and the reduction of overload and afterload depends on the applied dose [31]. Nitrates are generally administered with an initial bolus followed by continuous infusion. Nitroglycerin can be given as a 1-2 mg bolus in severely hypertensive patients with acute pulmonary edema [44]. Levy et al. reported aggressive BP control with very high-dose nitroglycerin was associated with fewer intensive care unit (ICU) admissions and less endotracheal intubation compared to historical controls [88]. The Vasodilation in the Management of Acute CHF (VMAC) trial failed to demonstrate statistically significant improvement of pulmonary capillary wedge pressure or self-reported dyspnea scores 3 hours after the initiation of nitroglycerin infusion compared with placebo [89]. It is significant that a significantly lower dose of nitrates than is used in clinical practice was used in the mentioned study. Although nitrates are used very often, no study has shown a reduction in mortality due to the use of nitrates [90,91,92]. One of the potential limitations of using nitrates is their propensity for tachyphylaxis [93,94].
6.1.2. Natriuretic Peptide Vasodilators
Nesiritide is a recombinant human b-type natriuretic peptide (BNP) whose effects mimic those of endogenous hormones [95]. Several smaller studies have shown the benefit of adding nesiritide to standard therapy for the relief of dyspnea in patients with H-AHF, however, on the other hand, the study by O Conor et al. did not show an effect of the use of nesiritide in reducing dyspnea in patients with H-AHF [96,97,98]. It was not associated with a worsening of renal function, but it was associated with an increase in rates of hypotension. Fu S. et al. explored the efficacy and safety of a modified dosage regimen of nesiritide in patients (≥75 years) with AHF [99]. Nesiritide resulted in improvements in dyspnea and edema, and similar adverse effects compared with conventional treatment, but did not show a reduction in short-term mortality. The VMAC study showed a statistically significant improvement in dyspnea at three hours post infusion in patients treated with nesiritide over those treated with NTG or placebo [89]. According to all the above, the use of nesiritide does not pose a risk to kidney function in AHF, however, its use is associated with the appearance of hypotension, so it should be used with caution.
6.2. Diuretics
Although intravenous diuretics are the cornerstone of AHF treatment, especially in patients with fluid overload, they are not the basis of treatment in patients with the H-AHF phenotype [25,94]. Their effect is realized in the increased excretion of salt and water through the kidneys, thus reducing the circulating volume of liquid. As already noted, not all patients with H-AHF are volume-loaded, so routine use of diuretics is not necessary when treating this phenotypic variant. Despite vasodilator therapy, diuretics can be used to control blood pressure in patients with H-AHF [94,100]. However, if patients with H-AHF have volume overload (chronic hypertension leads to activation of the renin-angiotensin-aldosterone system, which leads to fluid retention), then diuretics are used to reduce fluid volume [101]. Furosemide has been used most commonly, but alternatives include bumetanide (1 mg equivalent to 40 mg furosemide) and torsemide (20 mg equivalent to 40 mg). More recent studies such as the ADVOR and CLOROTIC studies favor the addition of acetazolamide and hydrochlorothiazide to standard diuretic therapy, although further studies are needed to prove their real effect on the outcome of patients with AHF [102,103].
6.3. ACE-inhibitors
While the use of ACE inhibitors is widely accepted for the treatment of hypertension and chronic HF, the benefit of intravenous ACE inhibitors in patients with AHF has been little studied. To the greatest extent, this is a consequence of the fear of side effects in the direction of hypotension, damage to kidney function and electrolyte imbalance. The main effect of enalaprilat in AHF is reflected in the reduction of arterial blood pressure as well as the effect on splanchnic and arterial circulation [104]. Also, in hypertensive patients with AHF, we often have excessive activation of the renin-angiotensin-aldosterone system, which is why the use of ACE-inhibitors could play a significant role [105,106]. The results of the retrospective cohort study by Ayaz et al., which examined the use of bolus IV enalaprilat in hypertensive patients with AHF, showed a good tolerance to the use of enalaprilat, the absence of negative effects on renal function [107].
6.4. Serelaxin
Serelaxin is a recombinant form of human relaxin-2, a naturally occurring peptide. By binding to the LGR7 and LGR8 receptors, it activates and enhances the vascular endothelin B receptor, vascular endothelial growth factor (VEGF) and leads to the production of nitric oxide [108]. Its influence on the inhibition of angiotensin II and endothelin has also been proven [109]. Serelaxin reduces systemic vascular resistance, increases cardiac output, increases renal blood flow, and increases glomerular filtration rate [108,110]. It is administered as a continuous IV infusion. Within the framework of the RELAX-AHF study, the therapeutic effect of serelaxin in the treatment of AHF was examined. Administration of serelaxin led to relief of dyspnea and improvement in other clinical outcomes, but had no effect on hospital readmissions. Although no differences were observed in the composite outcome at 60 days, a statistically significant improvement in cardiovascular and all-cause mortality was observed at 180 days [111].
6.5. Calcium channel blockers
Although effective antihypertensives, calcium channel blockers have been poorly studied in AHF. Clevidipine is a rapidly acting, arterial selective intravenous calcium-channel blocker. In the PRONTO pilot study, clevidipine safely and rapidly reduced blood pressure and improved dyspnea [112]. According to the study by Koroki T. and associates, which compared the effectiveness of nicardipine and nitroglycerin in patients with H-AHF, the nicardipine group had a shorter length of hospital stay (17.5 [10.0–33.0] days vs. 9.0 [5.0–15.0] days ) than the nitroglycerin group [113].
6.6. Urapidil
Urapidil acts as an α1-adrenoceptor antagonist and as an 5-HT1A receptor agonist [114]. According to a study by Yang W. et al., intravenous urapidil compared with nitroglycerin was associated with better blood pressure control and preserved cardiac function [115]. A meta-analysis of seven smaller studies showed similar data [116].
6.7. Beta blockers
Beta blockers and other non-vasodilator antihypertensives are currently not indicated in the treatment of H-AHF for acute blood pressure reduction [25,117]. However, there are still conflicting opinions when it comes to the use of beta blockers in patients with decompensated HF, who use these drugs in chronic therapy [118]. Namely, sudden discontinuation of beta blocker therapy can lead to a rebound phenomenon, i.e. increased sensitivity to beta-adrenergic agonists in patients undergoing long-term therapy with certain beta-blockers after the blocker is abruptly withdrawn [119]. The results of a meta-analysis published by Prins et al. indicate that in patients treated for AHF, both hospital mortality and the rate of rehospitalization are lower when beta-blockers are not excluded from therapy [120].
6.8. Respiratory support
In patients with H-AHF who present with hypoxia, oxygen supplementation is recommended [44]. In the absence of hypoxia, oxygen supplementation should be avoided given the evidence that high concentrations of inhaled oxygen can have adverse hemodynamic effects (decreased cardiac output, increased systemic vascular resistance) in patients with HFrEF [121]. The use of non-invasive positive pressure ventilation (NPPV) is also useful in patients with H-AHF who have significant work of breathing. It has also been shown that in pulmonary edema, NPPV can affect a faster reduction of dyspnea and correction of metabolic disorders compared to oxygen therapy alone [122,123]. Vital F.M. et al showed in a meta-analysis of 32 studies that the use of NPPV leads to a reduction in respiratory distress and the need for intubation in patients with pulmonary edema [124]. Noninvasive positive pressure ventilation should be started as soon as possible in patients with respiratory distress (respiratory rate >25 breaths/min, SpO2 <90%) [44]. In all patients on NPPV, continuous monitoring of vital functions is necessary with periodic checks of gas exchange. In case of further worsening of respiratory failure, intubation is advised.
7. Conclusions
Acute heart failure caused by arterial hypertension is a specific syndrome that requires urgent diagnosis and treatment. It is defined as a sudden onset of pulmonary congestion in the setting of a systolic blood pressure >140 mm Hg, and often >160 mm Hg. Most patients with H-AHF have previously known heart failure (usually with preserved LVEF), which is why it is necessary to improve diagnostic screening in order to detect both diastolic and systolic dysfunction of the left ventricle. Also, a large number of patients with H-AHF have long-term hypertension, so in order to reduce the risk of H-AHF, it is necessary to detect patients with arterial hypertension more often, quickly achieve target values of arterial blood pressure and maintain adherence. The diagnosis of H-AHF patients must be quick and precise, both due to the rapid establishment of the diagnosis and due to the adequate selection of the therapeutic regimen. New biomarkers such as soluble suppressor of tumorgenicity 2 (sST2), GDF-15, cystatin C, galectin-3, serum uric acid, microRNAs and low serum chloride, which are predictors of outcomes in AHF, must be used in diagnostics. It is also necessary to monitor the values of markers of renal function (urea, creatinine, proenkephalin), the increase of which is associated with the occurrence of renal weakness and increased mortality. Timely initiation of therapy may be a key factor in the successful treatment of H-AHF, with a positive association between a short time from admission to administration of diuretics and vasodilators resulting in reduced in-hospital mortality. However, it is known that the use of diuretics and vasodilators helps control symptoms in hospital conditions, but their effectiveness in reducing H-AHF recurrence, rehospitalization and distant mortality has not been demonstrated. A possible reason for the poor long-term outcome of these patients is the therapy applied in H-AHF, which was most often tested on patients with HF who have reduced LVEF and that in the phase when they had no episodes of AHF, and not on patients with preserved LVEF, which are most common in H- AHF. For now, the main goal is to improve the long-term outcome of these patients. Therefore, further research is needed in the future both in the improvement of diagnostic and therapeutic modalities in order to reduce the mortality of this life-threatening disease.
Author Contributions
Conceptualization, methodology, investigation, data curation, writing, and original draft preparation, R.L. and L.Dj.; original draft preparation and investigation, J.V.; M.Z. and A.R.; visualization and supervision, M.A. and D.S.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021, 42, 3599–3726. [CrossRef]
- Javaloyes, P.; Miró, Ò.; Gil, V.; Martín-Sánchez, F.J.; Jacob, J.; Herrero, P.; Takagi, K.; Alquézar-Arbé, A.; López Díez, M.P.; Martín, E.; Bibiano, C.; et al. ICA-SEMES Research Group. Clinical phenotypes of acute heart failure based on signs and symptoms of perfusion and congestion at emergency department presentation and their relationship with patient management and outcomes. Eur J Heart Fail. 2019, 21, 1353–1365. [CrossRef]
- Dickstein, K.; Cohen-Solal, A.; Filippatos, G.; McMurray, J.J.; Ponikowski, P.; Poole-Wilson, P.A.; Strömberg, A.; van Veldhuisen, D.J.; Atar, D.; Hoes, A.W.; et al. ESC Committee for Practice Guidelines (CPG). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail. 2008, 10, 933–989. [CrossRef]
- Nohria, A.; Tsang, S.W.; Fang, J.C.; Lewis, E.F.; Jarcho, J.A.; Mudge, G.H.; Stevenson, L.W. Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol. 2003, 41, 1797–1804. [Google Scholar] [CrossRef]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A, et al. ESC Committee for Practice Guidelines. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail, 2012, 14, 803–869. [CrossRef]
- Chioncel, O.; Mebazaa, A.; Harjola, V.P.; Coats, A.J.; Piepoli, M.F.; Crespo-Leiro, M.G.; Laroche, C.; Seferovic, P.M.; Anker, S.D.; Ferrari, R. ESC Heart Failure Long-Term Registry Investigators. Clinical phenotypes and outcome of patients hospitalized for acute heart failure: the ESC Heart Failure Long-Term Registry. Eur J Heart Fail. 2017, 19, 1242–1254. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. ESC Scientific Document Group. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016, 37, 2129–2200.
- Kamran, H.; Tang, W.H.W. Medical management of acute heart failure. Fac Rev. 2021, 10, 82. [Google Scholar] [CrossRef]
- Chioncel, O.; Mebazaa, A.; Maggioni, A.P.; Harjola, V.P.; Rosano, G.; Laroche, C.; Piepoli, M.F.; Crespo-Leiro, M.G.; Lainscak, M.; Ponikowski, P.; et al. ESC-EORP-HFA Heart Failure Long-Term Registry Investigators. Acute heart failure congestion and perfusion status - impact of the clinical classification on in-hospital and long-term outcomes; insights from the ESC-EORP-HFA Heart Failure Long-Term Registry. Eur J Heart Fail. 2019, 21, 1338–1352. [CrossRef]
- Nieminen, M.S.; Brutsaert, D.; Dickstein. K.; Drexler, H.; Follath, F.; Harjola, V,P.; Hochadel, M.; Komajda, M.; Lassus, J.; Lopez-Sendon, J.L:, et al. EuroHeart Survey Investigators; Heart Failure Association, European Society of Cardiology. EuroHeart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur Heart J. 2006, 27, 2725–2736. [CrossRef]
- Masip, J.; Frank Peacok, W.; Arrigo, M.; Rossello, X.; Platz, E.; Cullen, L.; Mebazaa, A.; Price, S.; Bueno, H.; Di Somma, S.; et al. Acute Heart Failure Study Group of the Association for Acute Cardiovascular Care (ACVC) of the European Society of Cardiology. Acute Heart Failure in the 2021 ESC Heart Failure Guidelines: a scientific statement from the Association for Acute CardioVascular Care (ACVC) of the European Society of Cardiology. Eur Heart J Acute Cardiovasc Care. 2022, 11, 173–185. [CrossRef]
- Lombardi, C.; Peveri, G.; Cani, D.; Latta. F.; Bonelli, A.; Tomasoni, D.; Sbolli, M.; Ravera, A.; Carubelli, V.; Saccani, N.; et al. In-hospital and long-term mortality for acute heart failure: analysis at the time of admission to the emergency department. ESC Heart Fail. 2020, 7, 2650–2661. [CrossRef] [PubMed]
- Follath, F.; Yilmaz, M.B.; Delgado, J.F.; Parissis, J.T.; Porcher, R.; Gayat, E.; Burrows, N.; McLean, A.; Vilas-Boas, F.; Mebazaa, A. Clinical presentation, management and outcomes in the Acute Heart Failure Global Survey of Standard Treatment (ALARM-HF). Intensive Care Med. 2011, 37, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Maggioni, A.P.; Dahlström, U.; Filippatos, G.; Chioncel, O.; Crespo Leiro, M.; Drozdz, J.; Fruhwald, F.; Gullestad, L.; Logeart, D.; Fabbri, G.; et al. Heart Failure Association of the European Society of Cardiology (HFA). EURObservational Research Programme: regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESC-HF Pilot). Eur J Heart Fail. 2013, 15, 808–817. [CrossRef]
- Cleland, J.G.; Swedberg, K.; Follath, F.; Komajda, M.; Cohen-Solal, A.; Aguilar, J.C.; Dietz, R.; Gavazzi, A.; Hobbs, R.; Korewicki, J.; et al. Study Group on Diagnosis of the Working Group on Heart Failure of the European Society of Cardiology. The EuroHeart Failure survey programme-- a survey on the quality of care among patients with heart failure in Europe. Part 1: patient characteristics and diagnosis. Eur Heart J. 2003, 24, 442–463. [CrossRef]
- Komajda, M.; Follath, F.; Swedberg, K.; Cleland, J.; Aguilar, J.C.; Cohen-Solal, A.; Dietz, R.; Gavazzi, A.; Van Gilst, W.H.; Hobbs, R.; et al. Study Group on Diagnosis of the Working Group on Heart Failure of the European Society of Cardiology. The EuroHeart Failure Survey programme--a survey on the quality of care among patients with heart failure in Europe. Part 2: treatment. Eur Heart J. 2003, 24, 464–474. [CrossRef]
- O'Connor, C.M.; Abraham, W.T.; Albert, N.M.; Clare, R.; Gattis Stough, W.; Gheorghiade, M.; Greenberg, B.H.; Yancy, C.W.; Young, J.B.; Fonarow, G.C. Predictors of mortality after discharge in patients hospitalized with heart failure: an analysis from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF). Am Heart J. 2008, 156, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.F. Jr.; Fonarow, G.C.; Emerman, C.L.; LeJemtel, T.H.; Costanzo, M.R.; Abraham, W.T.; Berkowitz, R.L.; Galvao, M.; Horton, D.P; ADHERE Scientific Advisory Committee and Investigators. Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005, 149, 209–216.
- Berg, D.D.; Bohula, E.A.; van Diepen, S.; Katz, J.N.; Alviar, C.L.; Baird-Zars, V.M.; Barnett, C.F.; Barsness, G.W.; Burke, J.A.; Cremer, P.C.; et al. Epidemiology of Shock in Contemporary Cardiac Intensive Care Units. Circ Cardiovasc Qual Outcomes. 2019, 12, e005618. [Google Scholar] [CrossRef]
- Miró, Ò.; García Sarasola, A.; Fuenzalida, C.; Calderón, S.; Jacob, J.; Aguirre, A.; Wu, D.M.; Rizzi, M.A.; Malchair, P.; Haro, A.; et al. ICA-SEMES Research Group. Departments involved during the first episode of acute heart failure and subsequent emergency department revisits and rehospitalisations: an outlook through the NOVICA cohort. Eur J Heart Fail. 2019, 21, 1231–1244. [CrossRef]
- Kociol, R.D.; Hammill, B.G.; Fonarow, G.C.; Klaskala, W.; Mills, R.M.; Hernandez, A.F.; Curtis, L.H. Generalizability and longitudinal outcomes of a national heart failure clinical registry: Comparison of Acute Decompensated Heart Failure National Registry (ADHERE) and non-ADHERE Medicare beneficiaries. Am Heart J. 2010, 160, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.T.; Fonarow, G.C.; Albert, N.M.; Stough, W.G.; Gheorghiade, M.; Greenberg, B.H.; O'Connor, C.M.; Sun, J.L.; Yancy, C.W.; Young, J.B. OPTIMIZE-HF Investigators and Coordinators. Predictors of in-hospital mortality in patients hospitalized for heart failure: insights from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF). J Am Coll Cardiol. 2008, 52, 347–356. [CrossRef]
- Metra, M.; Mentz, R.J.; Hernandez, A.F.; Heizer, G.M.; Armstrong, P.W.; Clausell, N.; Corbalan, R.; Costanzo, M.R.; Dickstein, K.; Dunlap, M.E.; et al. Geographic Differences in Patients in a Global Acute Heart Failure Clinical Trial (from the ASCEND-HF Trial). Am J Cardiol. 2016, 117, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Fonarow, G.C.; Stough, W.G.; Abraham, W.T.; Albert, N.M.; Gheorghiade, M.; Greenberg, B.H.; O'Connor, C.M.; Sun, J.L.; Yancy, C.W.; Young, J.B. OPTIMIZE-HF Investigators and Hospitals. Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: a report from the OPTIMIZE-HF Registry. J Am Coll Cardiol. 2007, 50, 768–777. [CrossRef]
- Harrison, N.; Pang, P.; Collins, S.; Levy, P. Blood Pressure Reduction in Hypertensive Acute Heart Failure. Curr Hypertens Rep. 2021, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Al-Lawati, J.A.; Sulaiman, K.J.; Al-Zakwani, I.; Alsheikh-Ali, A.A.; Panduranga, P.; Al-Habib, K.F.; Al-Suwaidi, J.; Al-Mahmeed, W.; Al-Faleh, H.; El-Asfar. A. et al. Systolic Blood Pressure on Admission and Mortality in Patients Hospitalized With Acute Heart Failure: Observations From the Gulf Acute Heart Failure Registry. Angiology 2017, 68, 584–591. [CrossRef]
- Gheorghiade, M.; Abraham, W.T.; Albert, N.M.; Greenberg, B.H.; O'Connor, C.M.; She, L.; Stough, W.G.; Yancy,C.W.; Young, J.B.; Fonarow, G.C. OPTIMIZE-HF Investigators and Coordinators. Systolic blood pressure at admission, clinical characteristics, and outcomes in patients hospitalized with acute heart failure. JAMA 2006, 296, 2217–2226. [CrossRef]
- Collins, S.P.; Jenkins, C.A.; Harrell, F.E.Jr.; Liu, D.; Miller, K.F.; Lindsell, C.J.; Naftilan, A.J.; McPherson, J.A.; Maron, D.J.; Sawyer, D.B.; et al. Identification of Emergency Department Patients With Acute Heart Failure at Low Risk for 30-Day Adverse Events: The STRATIFY Decision Tool. JACC Heart Fail. 2015, 3, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Rosman, Y.; Kopel, E.; Shlomai, G.; Goldenberg, I.; Grossman, E. The association between admission systolic blood pressure of heart failure patients with preserved systolic function and mortality outcomes. Eur J Intern Med. 2015, 26, 807–812. [Google Scholar] [CrossRef]
- Fonarow, G.C. Epidemiology and risk stratification in acute heart failure. Am Heart J. 2008, 155, 200–207. [Google Scholar] [CrossRef]
- Collins, S.P.; Levy, P.D.; Martindale, J.L.; Dunlap, M.E.; Storrow, A.B.; Pang, P.S.; Albert, N.M.; Felker, G.M.; Fermann, G.J.; Fonarow, G.C.; et al. Clinical and Research Considerations for Patients With Hypertensive Acute Heart Failure: A Consensus Statement from the Society of Academic Emergency Medicine and the Heart Failure Society of America Acute Heart Failure Working Group. J Card Fail. 2016, 22, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Emmens, J.E.; Ter Maaten, J.M.; Matsue, Y.; Figarska, S.M.; Sama, I.E.; Cotter, G.; Cleland, J.G.F.; Davison, B.A.; Felker, G.M.; Givertz, M.M.; et al. Worsening renal function in acute heart failure in the context of diuretic response. Eur J Heart Fail. 2022, 24, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.A.; D'Amario, D.; Bozic, J.; Glavas, D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Martindale, J. Optimizing Hypertensive Acute Heart Failure Management with Afterload Reduction. Curr Hypertens Rep. 2018, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Melenovsky, V.; Russell, S.D.; Kessler, K.; Pacak, K.; Becker, L.C.; Kass, D.A. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation. 2006, 114, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Farmakis, D.; Parissis, J.; Lekakis, J.; Filippatos, G. Acute heart failure: Epidemiology, risk factors, and prevention. Rev Esp Cardiol (Engl Ed). 2015, 68, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Viau, D.M.; Sala-Mercado, J.A.; Spranger, M.D.; O'Leary, D.S.; Levy, P.D. The pathophysiology of hypertensive acute heart failure. Heart. 2015, 101, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Ooi, H.; Chung, W.; Biolo, A. Arterial stiffness and vascular load in heart failure. Congest Heart Fail. 2008, 14, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; De Luca, L.; Fonarow, G.C.; Filippatos, G.; Metra, M.; Francis, G.S. Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol. 2005, 96, 11G–17G. [Google Scholar] [CrossRef]
- Levy, P.D.; Bellou, A. Acute Heart Failure Treatment. Curr Emerg Hosp Med Rep. 2013, 1, 10–10.1007/s40138-013-0012-8. [Google Scholar] [CrossRef]
- Oh, G.C.; Cho, H.J. Blood pressure and heart failure. Clin Hypertens. 2020, 26, 1. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, S.F.; Yuzefpolskaya, M.; Allemann, Y.; Messerli, F. Flash pulmonary edema. Prog Cardiovasc Dis. 2009, 52, 249–259. [Google Scholar] [CrossRef] [PubMed]
- López-Rivera, F.; Cintrón Martínez, H.R.; Castillo LaTorre, C.; Rivera González, A.; Rodríguez Vélez, J.G.; Fonseca Ferrer, V.; Méndez Meléndez, O.F.; Vázquez Vargas, E.J.; González Monroig, H.A. Treatment of Hypertensive Cardiogenic Edema with Intravenous High-Dose Nitroglycerin in a Patient Presenting with Signs of Respiratory Failure: A Case Report and Review of the Literature. Am J Case Rep. 2019, 20, 83–90. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2022, 24, 4–131. [CrossRef]
- Park, J.J.; Choi, D.J.; Yoon, C.H.; Oh, I.Y.; Lee, J.H.; Ahn, S.; Yoo, B.S.; Kang, S.M.; Kim, J.J.; et al. KorHF Registry. The prognostic value of arterial blood gas analysis in high-risk acute heart failure patients: an analysis of the Korean Heart Failure (KorHF) registry. Eur J Heart Fail. 2015, 17, 601–611. [CrossRef]
- Aubier, M.; Trippenbach, T.; Roussos, C. Respiratory muscle fatigue during cardiogenic shock. J Appl Physiol Respir Environ Exerc Physiol. 1981, 51, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.; Cooper, J.S. Physiology, Carbon Dioxide Transport. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532988/.
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; et al. ESC Scientific Document Group. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018, 39, 3021–3104. [CrossRef]
- Dhadke, S.V.; Dhadke, V.N.; Batra, D.S. Clinical Profile of Hypertensive Emergencies in an Intensive Care Unit. J Assoc Physicians India. 2017, 65, 18–22. [Google Scholar] [PubMed]
- Andrès, E.; Gass, R.; Charloux, A.; Brandt, C.; Hentzler, A. Respiratory sound analysis in the era of evidence-based medicine and the world of medicine 2.0. J Med Life. 2018, 11, 89–106. [Google Scholar]
- Sarkar, M.; Madabhavi, I.; Niranjan, N.; Dogra, M. Auscultation of the respiratory system. Ann Thorac Med. 2015, 10, 158–168. [Google Scholar] [CrossRef]
- Lee, D.S.; Lee, J.S.; Schull, M.J.; Borgundvaag, B.; Edmonds, M.L.; Ivankovic, M.; McLeod, S.L.; Dreyer, J.F.; Sabbah, S.; Levy, P.D.; et al. Prospective Validation of the Emergency Heart Failure Mortality Risk Grade for Acute Heart Failure. Circulation. 2019, 139, 1146–1156. [Google Scholar] [CrossRef]
- Sokolska, J.M.; Sokolski, M.; Zymliński, R.; Biegus, J.; Siwołowski, P.; Nawrocka-Millward, S.; Jankowska, E.A.; Todd, J.; Banasiak, W. Ponikowski P. Patterns of dyspnoea onset in patients with acute heart failure: clinical and prognostic implications. ESC Heart Fail. 2019, 6, 16–26. [CrossRef]
- Thibodeau, J.T.; Drazner, M.H. Reply: The Role of the Clinical Examination in Patients With Heart Failure. JACC Heart Fail. 2018, 6, 971. [Google Scholar] [CrossRef]
- Herring, N.; Paterson, D.J. ECG diagnosis of acute ischaemia and infarction: past, present and future. QJM. 2006, 99, 219–230. [Google Scholar] [CrossRef]
- Harjola, V.P.; Parissis, J.; Bauersachs, J.; Brunner-La Rocca, H.P.; Bueno, H.; Čelutkienė, J.; Chioncel, O.; Coats, A.J.S.; Collins, S.P, et al.; et al. Acute coronary syndromes and acute heart failure: a diagnostic dilemma and high-risk combination. A statement from the Acute Heart Failure Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2020, 22, 1298–1314. [Google Scholar] [CrossRef] [PubMed]
- Arenja, N.; Reichlin, T.; Drexler, B.; Oshima, S.; Denhaerynck, K.; Haaf, P.; Potocki, M.; Breidthardt, T.; Noveanu, M.; et al. Sensitive cardiac troponin in the diagnosis and risk stratification of acute heart failure. J Intern Med. 2012, 271, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Hasselblad, V.; Tang, W.H.; Hernandez, A.F.; Armstrong, P.W.; Fonarow, G.C.; Voors, A.A.; Metra, M.; McMurray, J.J.; Butler, J.; et al. Troponin I in acute decompensated heart failure: insights from the ASCEND-HF study. Eur J Heart Fail. 2012, 14, 1257–1264. [Google Scholar] [CrossRef]
- Peacock, W.F 4th.; De Marco, T.; Fonarow, G.C.; Diercks, D.; Wynne, J.; Apple, F.S.; Wu, A.H; ADHERE Investigators. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008, 358, 2117–2126. [CrossRef]
- You, J.J.; Austin, P.C.; Alter, D.A.; Ko, D.T.; Tu, J.V. Relation between cardiac troponin I and mortality in acute decompensated heart failure. Am Heart J. 2007, 153, 462–470. [Google Scholar] [CrossRef]
- Kim, W.; Kim, B.S.; Kim, H.J.; Lee, J.H.; Shin, J.; Shin, J.H. Clinical implications of cardiac troponin-I in patients with hypertensive crisis visiting the emergency department. Ann Med. 2022, 54, 507–515. [Google Scholar] [CrossRef]
- Weber, M.; Hamm, C. Role of B-type natriuretic peptide (BNP) and NT-proBNP in clinical routine. Heart. 2006, 92, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L.; van Kimmenade, R.; Lainchbury, J.; Bayes-Genis, A.; Ordonez-Llanos, J.; Santalo-Bel, M.; Pinto, Y.M.; Richards, M. NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart failure: an international pooled analysis of 1256 patients: the International Collaborative of NT-proBNP Study. Eur Heart J. 2006, 27, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Belagavi, A.C.; Rao, M.; Pillai, A.Y.; Srihari, U.S. Correlation between NT proBNP and left ventricular ejection fraction in elderly patients presenting to emergency department with dyspnoea. Indian Heart J. 2012, 64, 302–304. [Google Scholar] [CrossRef]
- Krishnaswamy, P.; Lubien, E.; Clopton, P.; Koon. J.; Kazanegra, R.; Wanner, E.; Gardetto, N.; Garcia, A.; DeMaria, A.; Maisel, A.S. Utility of B-natriuretic peptide levels in identifying patients with left ventricular systolic or diastolic dysfunction. Am J Med. 2001, 111, 274–279. [CrossRef] [PubMed]
- Christ, M.; Mueller, C. Use of natriuretic peptide assay in dyspnea. Dtsch Arztebl Int. 2008, 105, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L. Jr.; Camargo, C.A.; Anwaruddin, S.; Baggish, A.L.; Chen, A.A.; Krauser, D.G.; Tung, R.; Cameron, R.; Nagurney, J.T.; et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol. 2005, 95, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.S.; Krishnaswamy, P.; Nowak, R.M.; McCord, J.; Hollander, J.E.; Duc, P.; Omland, T.; Storrow, A.B.; Abraham, W.T.; Wu, A.H, et al. Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med. 2002, 347, 161–167. [CrossRef]
- Arrigo, M.; Jessup, M.; Mullens, W.; Reza, N.; Shah, A.M.; Sliwa, K.; Mebazaa, A. Acute heart failure. Nat Rev Dis Primers. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Dal-Bianco, J.P.; Jaffe, A.S.; Bell, M.R.; Oh, J.K. Cardiac function and brain-type natriuretic peptide in first-time flash pulmonary edema. Mayo Clin Proc. 2008, 83, 289–296. [Google Scholar] [CrossRef]
- Ip, C.; Luk, K.S., Yuen, V.L.C.; Chiang, L.; Chan, C.K.; Ho, K.; Gong, M.; Lee, T.T.L.; Leung, K.S.K.; Roever, L,; Bazoukis, G.; et al; International Health Informatics Study (IHIS) Network. Soluble suppression of tumorigenicity 2 (sST2) for predicting disease severity or mortality outcomes in cardiovascular diseases: A systematic review and meta-analysis. Int J Cardiol Heart Vasc. 2021, 37, 100887. [CrossRef]
- Miró, Ò.; González de la Presa, B.; Herrero-Puente, P.; Fernández Bonifacio, R.; Möckel, M.; Mueller, C.; Casals, G.; Sandalinas, S.; Llorens, P.; Martín-Sánchez, F.J.; et al. The GALA study: relationship between galectin-3 serum levels and short- and long-term outcomes of patients with acute heart failure. Biomarkers. 2017, 22, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, V.; Aimo, A.; Vergaro, G.; Saccaro, L.; Passino, C.; Emdin, M. Biomarkers for the diagnosis and management of heart failure. Heart Fail Rev. 2022, 27, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cheang, I.; Liao, S.; Wang, K.; Yao, W., Yin, T.; Lu, X.; Zhou, Y.; Zhang, H.; Li, X. Blood Urea Nitrogen to Creatinine Ratio and Long-Term Mortality in Patients with Acute Heart Failure: A Prospective Cohort Study and Meta-Analysis. Cardiorenal Med. 2020, 10, 415–428. [CrossRef] [PubMed]
- Qian, H.; Tang, C.; Yan, G. Predictive value of blood urea nitrogen/creatinine ratio in the long-term prognosis of patients with acute myocardial infarction complicated with acute heart failure. Medicine (Baltimore). 2019, 98, e14845. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.L.; Squire, I.B.; Jones, D.J.L.; Cao, T.H.; Chan, D.C.S.; Sandhu, J.K.; Quinn, P.A.; Davies, J.E.; Struck, J.; Hartmann, O.; et al. GREAT Network. Proenkephalin, Renal Dysfunction, and Prognosis in Patients With Acute Heart Failure: A GREAT Network Study. J Am Coll Cardiol. 2017, 69, 56–69. [CrossRef]
- Price, S.; Platz, E.; Cullen, L.; Tavazzi, G.; Christ, M.; Cowie, M.R.; Maisel, A.S.; Masip, J.; Miro, O.; McMurray, J.J.; et al. Acute Heart Failure Study Group of the European Society of Cardiology Acute Cardiovascular Care Association. Expert consensus document: Echocardiography and lung ultrasonography for the assessment and management of acute heart failure. Nat Rev Cardiol. 2017, 14, 427–440. [CrossRef]
- Milos, R.I.; Bartha, C.; Röhrich, S.; Heidinger, B.H.; Prayer, F.; Beer, L.; Wassipaul, C.; Kifjak, D.; Watzenboeck, M.L.; Pochepnia, S. ; Imaging in patients with acute dyspnea when cardiac or pulmonary origin is suspected. BJR Open. 2023, 5, 20220026. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.; Simon, B.; Alter, H.J.; Cheung, P. Ability of physicians to diagnose congestive heart failure based on chest X-ray. J Emerg Med. 2011, 40, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Soldati, G.; Demi, M.; Demi, L. Ultrasound patterns of pulmonary edema. Ann Transl Med. 2019, 7 (Suppl. 1), S16. [Google Scholar] [CrossRef]
- Soldati, G.; Inchingolo, R.; Smargiassi, A.; Sher, S.; Nenna, R.; Inchingolo, C.D.; Valente, S. Ex vivo lung sonography: morphologic-ultrasound relationship. Ultrasound Med Biol. 2012, 38, 1169–1179. [Google Scholar] [CrossRef]
- Soldati, G.; Demi, M. The use of lung ultrasound images for the differential diagnosis of pulmonary and cardiac interstitial pathology. J Ultrasound. 2017, 20, 91–96. [Google Scholar] [CrossRef]
- Copetti, R.; Soldati, G.; Copetti, P. Chest sonography: a useful tool to differentiate acute cardiogenic pulmonary edema from acute respiratory distress syndrome. Cardiovasc Ultrasound. 2008, 6, 16. [Google Scholar] [CrossRef]
- Garus, M.; Zdanowicz, A.; Fudim, M.; Zymliński, R.; Niewiński, P.; Paleczny, B.; Rosiek-Biegus, M.; Iwanek, G.; Ponikowski, P.; Biegus, J. Clinical determinants and prognostic significance of hypocapnia in acute heart failure. Sci Rep. 2022, 12, 16889. [Google Scholar] [CrossRef]
- Bezati, S.; Velliou, M.; Ventoulis, I.; Simitsis, P.; Parissis, J.; Polyzogopoulou, E. Infection as an under-recognized precipitant of acute heart failure: prognostic and therapeutic implications. Heart Fail Rev. 2023, 28, 893–904. [Google Scholar] [CrossRef]
- Abdin, A.; Anker, S.D.; Butler, J.; Coats, A.J.S.; Kindermann, I.; Lainscak, M.; Lund, L.H.; Metra, M.; Mullens, W.; Rosano, G. 'Time is prognosis' in heart failure: time-to-treatment initiation as a modifiable risk factor. ESC Heart Fail. 2021, 8, 4444–4453. [Google Scholar] [CrossRef]
- Kitai, T.; Tang W.H.W.; Xanthopoulos, A.; Murai, R.; Yamane, T.; Kim, K.; Oishi, S.; Akiyama. E.; Suzuki, S.;Yamamoto, M. Impact of early treatment with intravenous vasodilators and blood pressure reduction in acute heart failure. Open Heart. 2018, 5, e000845. [CrossRef] [PubMed]
- Levy, P.; Compton, S.; Welch, R.; Delgado, G.; Jennett, A.; Penugonda, N.; Dunne, R.; Zalenski, R. Treatment of severe decompensated heart failure with high-dose intravenous nitroglycerin: a feasibility and outcome analysis. Ann Emerg Med. 2007, 50, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002, 287, 1531–1540, Erratum in: JAMA 2002, 288(5), 577. [CrossRef]
- Ho, E.C.; Parker, J.D.; Austin, P.C.; Tu, J.V.; Wang, X.; Lee, D.S. Impact of Nitrate Use on Survival in Acute Heart Failure: A Propensity-Matched Analysis. J Am Heart Assoc. 2016, 5, e002531. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Puente, P.; Jacob, J.; Martín-Sánchez, F.J.; Vázquez-Álvarez, J.; Martínez-Camblor, P.; Miró, Ò.; Lucas-Imbernón, F.J.; Martínez-Zapico, A.; Llorens, P. ICA-SEMES group. Influence of Intravenous Nitrate Treatment on Early Mortality Among Patients With Acute Heart Failure. NITRO-EAHFE Study. Rev Esp Cardiol (Engl Ed). 2015, 68, 959–967. [CrossRef]
- Breidthardt, T.; Noveanu, M.; Potocki, M.; Reichlin, T.; Egli, P.; Hartwiger, S.; Socrates, T.; Gayat, E.; Christ, M.; Mebazaa, A.; et al. Impact of a high-dose nitrate strategy on cardiac stress in acute heart failure: a pilot study. J Intern Med. 2010, 267, 322–330. [Google Scholar] [CrossRef]
- Abrams, J. Nitrate tolerance and dependence. A critical assessment. Nouv Presse Med. 1980, 9 (Suppl. 34), 2499–2504. [Google Scholar]
- Liu, J.X.; Uppal, S.; Patel, V. Management of Acute Hypertensive Heart Failure. Heart Fail Clin. 2019, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Strain, W.D. The use of recombinant human B-type natriuretic peptide (nesiritide) in the management of acute decompensated heart failure. Int J Clin Pract. 2004, 58, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Colucci, W.S.; Elkayam, U.; Horton, D.P.; Abraham, W.T.; Bourge, R.C.; Johnson, A.D.; Wagoner, L.E.; Givertz, M.M.; Liang, C.S.; Neibaur, M.; et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000, 343, 246–253. [CrossRef]
- Elkayam, U.; Akhter, M.W.; Singh, H.; Khan, S.; Usman, A. Comparison of effects on left ventricular filling pressure of intravenous nesiritide and high-dose nitroglycerin in patients with decompensated heart failure. Am J Cardiol. 2004, 93, 237–240. [Google Scholar] [CrossRef]
- O'Connor, C.M.; Starling, R.C.; Hernandez, A.F.; Armstrong, P.W.; Dickstein, K.; Hasselblad, V.; Heizer, G.M.; Komajda, M.; Massie, B.M.; McMurray, J.J.; et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011, 365, 32–43. [Google Scholar] [CrossRef]
- Fu, S.; Yi, S.; Zhu, B.; Wang, L.; Wang, H., Bai, Y.; Ye, P.; Luo, L. Efficacy and safety of a modified dosage regimen of nesiritide in patients older than 75 years with acute heart failure. Aging Clin Exp Res. 2012, 24, 524–529. [CrossRef]
- Pickkers, P.; Dormans, T.P.; Russel, F.G.; Hughes, A.D.; Thien, T.; Schaper, N.; Smits, P. Direct vascular effects of furosemide in humans. Circulation. 1997, 96, 1847–1852. [Google Scholar] [CrossRef]
- Amatruda, J.G.; Scherzer, R.; Rao, V.S.; Ivey-Miranda, J.B.; Shlipak, M.G.; Estrella, M.M.; Testani, JM. Renin-Angiotensin-Aldosterone System Activation and Diuretic Response in Ambulatory Patients With Heart Failure. Kidney Med. 2022, 4, 100465. [Google Scholar] [CrossRef]
- Mullens, W.; Dauw, J.; Martens, P.; Verbrugge, F.H.; Nijst, P.; Meekers, E.; Tartaglia, K.; Chenot, F.; Moubayed, S.; Dierckx, R.; et al. ADVOR Study Group. Acetazolamide in Acute Decompensated Heart Failure with Volume Overload. N Engl J Med. 2022, 387, 1185–1195. [CrossRef]
- Trullàs, J.C.; Morales-Rull, J.L.; Casado, J.; Carrera-Izquierdo, M.; Sánchez-Marteles, M.; Conde-Martel, A.; Dávila-Ramos, M.F.; Llácer, P.; Salamanca-Bautista, P.; Pérez-Silvestre, J.; et al. CLOROTIC trial investigators. Combining loop with thiazide diuretics for decompensated heart failure: the CLOROTIC trial. Eur Heart J. 2023, 44, 411–421. [CrossRef]
- Wang, S.Y.; Manyari, D.E.; Scott-Douglas, N.; Smiseth, O.A.; Smith, E.R.; Tyberg, J.V. Splanchnic venous pressure-volume relation during experimental acute ischemic heart failure. Differential effects of hydralazine, enalaprilat, and nitroglycerin. Circulation 1995, 91, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- AlHabeeb, W.; Hayajneh, A. Continuation of Angiotensin Converting Enzyme Inhibitors in Acute Heart Failure. Int J Gen Med. 2021, 14, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Mielniczuk, L.; Stevenson, L.W. Angiotensin-converting enzyme inhibitors and angiotensin II type I receptor blockers in the management of congestive heart failure patients: what have we learned from recent clinical trials? Curr Opin Cardiol. 2005, 20, 250–255. [Google Scholar] [CrossRef]
- Ayaz, S.I.; Sharkey, C.M.; Kwiatkowski, G.M.; Wilson, S.S.; John, R.S.; Tolomello, R.; Mahajan, A.; Millis, S.; Levy, P.D. Intravenous enalaprilat for treatment of acute hypertensive heart failure in the emergency department. Int J Emerg Med. 2016, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Miyares, M.A.; Davis, K.A. Serelaxin, a 'breakthrough' investigational intravenous agent for acute heart failure. P T. 2013, 38, 606–611. [Google Scholar]
- Hernandez-Montfort, J.A.; Arora, S.; Slawsky, M.T. Relaxin for treatment of acute heart failure: making the case for treating targeted patient profiles. Curr Heart Fail Rep. 2013, 10, 198–203. [Google Scholar] [CrossRef]
- Dschietzig, T.; Teichman, S.; Unemori, E.; Wood, S.; Boehmer, J.; Richter, C.; Baumann, G.; Stangl, K. First clinical experience with intravenous recombinant human relaxin in compensated heart failure. Ann N Y Acad Sci. 2009, 1160, 387–392. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Ponikowski, P.; Unemori, E.; Voors, A.A.; Adams, K.F. Jr.; et al. RELAXin in Acute Heart Failure (RELAX-AHF) Investigators. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013, 381, 29–39. [CrossRef]
- Peacock, W.F.; Chandra, A.; Char, D.; Collins, S.; Der Sahakian, G.; Ding, L.; Dunbar, L.; Fermann, G.; Fonarow, G.C.; Garrison, N., et al. Clevidipine in acute heart failure: Results of the A Study of Blood Pressure Control in Acute Heart Failure-A Pilot Study (PRONTO). Am Heart J. 2014, 167, 529–536. [CrossRef]
- Koroki, T.; Abe, T.; Ochiai, H. Nicardipine versus nitroglycerin for hypertensive acute heart failure syndrome: a single-center observational study. J Rural Med. 2022, 17, 33–39. [Google Scholar] [CrossRef]
- Bopp, C.; Auger, C.; Diemunsch, P.; Schini-Kerth, V. The effect of urapidil, an alpha-1 adrenoceptor antagonist and a 5-HT1A agonist, on the vascular tone of the porcine coronary and pulmonary arteries, the rat aorta and the human pulmonary artery. Eur J Pharmacol. 2016, 779, 53–58. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, Y.J.; Fu, Y.; Qin, J.; Qin, S.; Chen, X.M.; Guo, J.C,. Wang, Z.; Zhan, H.; Li, J.; He, J.Y.; et al. Efficacy and Safety of Intravenous Urapidil for Older Hypertensive Patients with Acute Heart Failure: A Multicenter Randomized Controlled Trial. Yonsei Med J. 2017, 58, 105–113. [CrossRef]
- Shi, J.; Li, Y.; Xing, C.; Peng, P.; Shi, H.; Ding, H.; Zheng, P., Ning, G.; Feng, S. Urapidil, compared to nitroglycerin, has better clinical safety in the treatment of hypertensive patients with acute heart failure: a meta-analysis. Drug Des Devel Ther. 2018, 13, 161–172. [CrossRef]
- Jondeau, G.; Neuder, Y.; Eicher, J.C.; Jourdain, P.; Fauveau, E.; Galinier, M.; Jegou, A.; Bauer, F.; Trochu, J.N.; Bouzamondo, A. B-CONVINCED Investigators. B-CONVINCED: Beta-blocker CONtinuation Vs. INterruption in patients with Congestive heart failure hospitalizED for a decompensation episode. Eur Heart J. 2009, 30, 2186–2192. [CrossRef]
- Jondeau, G.; Milleron, O. ; Beta-Blockers in Acute Heart Failure: Do They Cause Harm? JACC Heart Fail. 2015, 3, 654–656. [Google Scholar] [CrossRef]
- Koracevic, G.; Micic, S.; Stojanovic, M.; Tomasevic, M.; Kostic, T.; Velickovic Radovanovic, R.; Lovic, D.; Djordjevic, D.; Randjelovic, M.; Koracevic, M. Beta blocker rebound phenomenon is important, but we do not know its definition, incidence or optimal prevention strategies. Hypertens Res. 2020, 43, 591–596. [Google Scholar] [CrossRef]
- Prins, K.W.; Neill, J.M.; Tyler, J.O.; Eckman, P.M.; Duval, S. Effects of Beta-Blocker Withdrawal in Acute Decompensated Heart Failure: A Systematic Review and Meta-Analysis. JACC Heart Fail. 2015, 3, 647–653. [Google Scholar] [CrossRef]
- Mebazaa, A.; Tolppanen, H.; Mueller, C.; Lassus, J.; DiSomma, S.; Baksyte, G.; Cecconi, M.; Choi, D.J.; Cohen Solal, A.; Christ, M. Acute heart failure and cardiogenic shock: a multidisciplinary practical guidance. Intensive Care Med. 2016, 42, 147–163. [Google Scholar] [CrossRef]
- Masip, J.; Roque, M.; Sánchez, B.; Fernández, R.; Subirana, M.; Expósito, J.A. Noninvasive ventilation in acute cardiogenic pulmonary edema: systematic review and meta-analysis. JAMA. 2005, 294, 3124–3130. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.L.; Zhao, Y.T.; Liu, Q.H.; Fu, C.J.; Sun, F.; Ma, Y.L.; Chen, Y.W.; He, Q.Y. Meta-analysis: Noninvasive ventilation in acute cardiogenic pulmonary edema. Ann Intern Med. 2010, 152, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Vital, F.M.; Ladeira, M.T.; Atallah, A.N. Non-invasive positive pressure ventilation (CPAP or bilevel NPPV) for cardiogenic pulmonary oedema. Cochrane Database Syst Rev. 2013, 5, CD005351. [Google Scholar] [CrossRef]
Figure 1.
The influence of hypertension on the development of hypertensive acute heart failure. Legend: SBP = systolic blood pressure; LV = left ventricular; LVH = left ventricular hypertrophy; EDV = end-diastolic volume; EF = ejection fraction; HFpEF = heart failure with preserved ejection fraction; HFrER = heart failure with reduced ejection fraction; H-AHF- hypertensive acute heart failure.
Figure 1.
The influence of hypertension on the development of hypertensive acute heart failure. Legend: SBP = systolic blood pressure; LV = left ventricular; LVH = left ventricular hypertrophy; EDV = end-diastolic volume; EF = ejection fraction; HFpEF = heart failure with preserved ejection fraction; HFrER = heart failure with reduced ejection fraction; H-AHF- hypertensive acute heart failure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



