Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
One of the major obstacles confronting the formulation of a mechanistic understanding for Alzheimer’s disease (AD) is its immense complexity – a complexity that traverses the full structural and phenomenological spectrum, including molecular, macromolecular, cellular, neurological and behavioural processes. This complexity is reflected by the equally complex diversity of risk factors associated with AD. However, more than merely mirroring disease complexity, risk factors also provide fundamental insights into the aetiology and pathogenesis of AD as a neurodegenerative disorder since they are central to disease initiation and subsequent propagation. Based on a systematic literature assessment, this review has identified 30 risk factors for AD and then extended the analysis to further identify neuroinflammation as a unifying mechanism present in all 30 risk factors. Although other mechanisms (e.g. vasculopathy) were present in multiple risk factors, dysfunction of the neuroimmune-neuroinflammation axis was uniquely central to all 30 identified risk factors. Though the nature of the neuroinflammatory involvement varied, activation of microglia and the release of pro-inflammatory cytokines was a common pathway shared by all risk factors. This observation provides further evidence for the importance of immunopathic mechanisms in the aetiopathogenesis of AD.
Keywords:
Alzheimer’s disease
; dementia
; neurodegeneration
; neuroinflammation
; neuroimmune
; microglia
; cytokine
1. Introduction
The brain is the most complex organ in the human body, and Alzheimer’s disease (AD) is arguably the most complex disease of the brain. One of the major hurdles encountered when formulating a mechanistic understanding with which to facilitate management strategies for AD is its immense complexity – a complexity that traverses the full structural and phenomenological spectrum, including molecular, macromolecular, cellular, behavioural and neurological processes [1,2]. AD risk factors are excellent examples of this immense complexity; these risk factors include such bewilderingly diverse conditions as medical diseases (diabetes), psychiatric disorders (depression), personal injuries (head trauma), societal factors (social isolation) and environmental issues (air pollution).
To identify a harmonizing mechanistic explanation with which to unify the many and varied risk factors for AD, a comprehensive literature review was initially completed (in PubMed, Web of Science, Scopus and Google Scholar databases including articles published up to November 2023) and identified 30 “risk” factors for AD, employing a broad definition of “risk factor”: some are modifiable risk factors connected in a causative manner with AD (e.g. smoking, alcohol abuse, obesity); others are concomitant disorders occurring as co-morbidities (e.g. glaucoma; people with glaucoma are at risk for also developing AD); others are bidirectional risk factors (e.g. chronic pain causes neuroinflammation which is a risk factor for AD, yet the neuroinflammation is a positive feedback risk factor for continuing pain). This list of 30 risk factors includes the 12 modifiable risk factors identified in the 2020 Lancet commission (less education, hypertension, hearing impairment, smoking, obesity, depression, physical inactivity, diabetes, low social contact, excessive alcohol consumption, traumatic brain injury, and air pollution) as well as 18 others based on our literature review [3].
Next, all literature sources discussing the 30 identified risk factors were searched for common terms providing mechanistic explanations. The term uniting all 30 risk factors was “neuroinflammation”, where neuroinflammation is defined as an activation of the brain's innate immune system in response to varied external (infection, trauma, toxin) and internal (ischaemia) challenges, as characterized by a host of cellular (especially microglial) and molecular (especially cytokine: e.g. Interleukin(IL)-1β, IL-6 and Tumour Necrosis Factor(TNF)-α) changes within the brain [4,5]. Since many studies provide data strongly implicating neuroinflammation as a major contributor to the aetiopathogenesis of AD, a shared neuroimmune-neuroinflammation mechanism clearly emerges as a unifying thread providing harmonization within the rich tapestry of diverse risk factors associated with AD.
Herein, the 30 risk factors for AD are presented in conjunction with a description of their neuroinflammatory mechanisms (Figure 1).
2. Thirty Risk Factors
2.1. Age
Although AD is not a normal consequence of aging, age is accepted as the most important known risk factor for the disease. The number of people with AD doubles every five years beyond age 65; 40% of people age 90 years and older have AD [6]. For the majority of people with AD, symptoms first appear in their mid-60s or later. When the disease develops before age 65, it is regarded as uncommon.
The links between aging and AD are many and complex; however, neuroinflammation is a key component of this link with aging being associated with neuroinflammation and neuroinflammation being associated with AD. D’Avila et al. have shown that aged mice exhibit dystrophic activated microglia in the hippocampus and entorhinal cortex [7]. Aged mice also produce higher levels of pro-inflammatory (IL-1β and IL-6) cytokines in the brain and higher levels of NADPH oxidase 2 (Nox2) expression compared to younger animals [8].
In humans, aging and a chronic inflammatory state frequently co-exist in the periphery and in the brain. Aging impairs functional interactions between brain and the immune system; microglia and astrocytes, functioning in their capacity as innate immune cells, become more pro-inflammatory during aging [9]. This age-associated increase in innate immune reactivity sets the stage for an exaggerated inflammatory cytokine brain response after activation of the innate immune system during the initiation and progression of AD, leading to more severe long-lasting behavioural and cognitive deficits.
2.2. Sex
After age, sex is the other most commonly cited risk factor for AD. Women have a greater risk of developing dementia during their lifetime (even after greater longevity is considered); twice as many women have AD compared to men. A Swedish study by Beam et al. followed 16,926 people and found that, beginning at age 80 years, women were more likely to be diagnosed with AD than men of the same age [10]. An analogous Taiwanese study by Liu et al. concluded that the likelihood of developing AD over seven years was greater in women than men [11]. Finally a meta-analysis by Niu et al. studying the European incidence of AD calculated that annually 13 women out of 1,000 developed AD, compared to only seven men [12].
Immune mediated neuroinflammatory responses are different between men and women. Women are more susceptible to the pathological effects of inflammation than men via neuroimmune alterations including microglial activation, pro-inflammatory cytokine expression, and synaptic plasticity within neural circuitry [13]. In a study involving injecting volunteers with immunogenic lipopolysaccharides (LPS), Engler et al. showed that women exhibited a more profound pro-inflammatory response with significantly higher increases in TNFα and IL-6; in contrast, the LPS-induced increase in anti-inflammatory IL-10 was significantly higher in men [14]. Finally, women represent >80% of all cases of autoimmunity, particularly as demonstrated by differences in the incidence for Sjögren syndrome, systemic lupus erythematosus, Hashimoto thyroiditis, Graves disease, scleroderma, and myasthenia gravis [15]; Meier-Stephenson et al. have argued that AD is an autoimmune disease. Such sex-based neuroimmune differences provide a possible explanation for the corresponding sex differences in the incidence and prevalence of AD [16].
2.3. Arterial Hypertension
Hypertension is an established and accepted risk factor for AD. Numerous studies have demonstrated a relationship between decline in cognitive function and arterial hypertension across different age groups [17,18]. Systemic arterial hypertension, particularly midlife high blood pressure (BP), has been related to a higher risk of dementia, including AD. In the middle age of life (age 40–64 years) there is a positive association between the elevation of BP and the presence of cognitive impairment and AD risk, whilst in elderly population (age ≥ 65 years) this relationship is more controversial.
Not surprisingly, the link between hypertension and AD is multifactorial with vascular factors playing a major contributing role. However, neuroinflammation is another major mechanism linking hypertension and AD [19]. Animal studies have demonstrated that chronically elevated blood pressure leads to deleterious glial activation and increased brain inflammatory mediators, specifically pro-inflammatory cytokines such as TNFα and IL-1β. Solé-Guardia et al. observed that individuals with hypertension had a greater neuroinflammatory response, as illustrated by microglial activation and astrogliosis, and more severe perivascular inflammation compared to normotensives [20]. Carnevale et al. has shown that hypertension induced microglial activation and interleukin IL-1β upregulation triggers neuroinflammation before Aβ deposition [21].
2.4. Hypercholesterolemia
Cholesterol dysregulation serves as a risk factor for multiple diseases, including AD. Experimental studies have shown that a high cholesterol diet (HCD) promotes AD development. In rats and mice, HCD also induces significant cognitive impairment and AD-like disease [22,23]; in Japanese white rabbits on HCD, changes in brain metabolism and structure similar to those of human AD were seen [24]. Epidemiological investigations have also suggested a relationship between hypercholesterolemia and AD [25]. Xu et al. have suggested that high cholesterol levels were associated with increased AD pathology severity, and that the mechanism for this enhanced pathology is not entirely mediated by cerebrovascular conditions [26]. Thus, mounting evidence indicates that excessive cholesterol accumulates in AD, driving AD-associated pathological changes, and that hypercholesterolemia promotes AD development as a risk factor, especially with elevated cholesterol levels in midlife.
As with hypertension, the link between hypercholesterolemia and AD is multifactorial with vascular factors playing a major contributing role. However, neuroinflammation is another major mechanism linking hypercholesterolemia and AD [27]. For example, Thirumangalakudi et al. demonstrated that hyperlipidemic mice showed increased expression of pro-inflammatory microglia and cytokines/mediators including TNFα, IL-1β, IL-6, NOS2 (Nitric oxide synthase 2) and COX2 (Cyclooxygenase-2) [28]. Chen et al. also showed in mice that a high cholesterol diet not only enhanced NLRP3 (NLR family pyrin domain containing 3) inflammasome activation and IL-1β expression [29].
2.5. Smoking
Based on a comprehensive review, Durazzo et al. concluded that smoking is related to a significantly increased risk for AD [30]. Cigarette smoke/smoking is associated with AD neuropathology in both preclinical models and human studies. Jeong et al. have shown that smoking cessation was associated with a lower risk of dementia [31].
The negative consequences of smoking are numerous providing multiple mechanisms by which smoking contributes to pathology. However, immune-based inflammation is a significant contributor to this pathology. Alrouji et al. have concluded that smoking inflicts complex immunological effects, which include enhancement of inflammatory responses (activated microglia with increased pro-inflammatory cytokine responses) with a parallel reduction of some immune defences, resulting in an increased susceptibility to a persistent pro-inflammatory environment [32]. In a case-control study, Liu et al. found that cigarette smoking was associated with increased at-risk biomarkers for AD, as indicated by higher neuroinflammation biomarkers in the cerebrospinal fluid of participants in the active smoking group [33].
2.6. Physical Inactivity
Based on a comprehensive literature review, Meng et al. concluded that physical inactivity is one of the most common preventable risk factors for developing AD and that higher physical activity levels are associated with a reduced risk of AD development [34]. Physical exercise is also effective in improving behavioural and neuropsychiatric symptoms of AD, notably cognitive function. Chen et al. have likewise concluded that physical exercise is important in the prevention of AD, providing non-pharmacological treatment options [35].
The case correlating physical inactivity with AD via a neuroinflammatory mechanism is strong. Recently, Wang et al. demonstrated that exercise ameliorates AD by directly and indirectly regulating brain immune responses and promoting hippocampal neurogenesis [36]. Similarly, Seo et al. have shown that neuroinflammation-mediated microglia activation with pro-inflammatory cytokine release is enhanced by physical inactivity and downregulated by exercise [37]. Svensson and co-workers have likewise shown that exercise leads to elevated expression of anti-inflammatory cytokines, and reduced levels of pro-inflammatory cytokines and activated microglia [38].
2.7. Obesity
Obese individuals are at greater risk of developing age-related cognitive decline, mild cognitive impairment and AD [39,40]. An association between body mass index (BMI) and AD has been described with multiple groups studying the relationship between elevated BMI and AD. Obesity is thus a recognized risk factor for AD [41,42,43,44].
Miller and Spencer have suggested that neuroinflammation is the link that unites obesity to AD; obesity (and high fat feeding) leads to systemic inflammation and excess circulating free fatty acids and inflammatory mediators; these circulating cytokines and activated immune cells reach the brain and initiate local neuroinflammation, including microglial proliferation and causing synaptic remodelling and neurodegeneration [45]. Similarly, Henn et al. also suggest that immune dysregulation, including inflammaging (e.g., increased circulating cytokines) and immunosenescence (declining immune system function), commonly occur in obesity and aging and may impact cognitive impairment, thereby promoting AD [46].
2.8. Dietary Factors
Many studies suggest that diet affects the aging brain’s cognitive abilities and susceptibility to AD. In a systematic search of randomized clinical trials, reviews, and meta-analyses investigating the association between diet and AD, Xu Lou et al. analysed 38 studies concluding that the western diet pattern is a risk factor for developing AD, whereas the Mediterranean diet, ketogenic diet, and supplementation with omega-3 fatty acids and probiotics are protective factors [47]. The Mediterranean diet, the related MIND diet (which includes elements designed to lower blood pressure), and other healthy eating patterns have been associated with cognitive benefits in studies and a decreased likelihood of AD [48,49,50,51].
Kip and Parr-Brownlie have noted that many dietary risks factors are now known to be linked to AD-promoting neuroinflammation, particularly a high saturated and trans fat diet; indeed, dietary modifications in mice can influence levels of pro-inflammatory microglia and cytokines [52]. Conversely, dietary restriction (DR) has been shown to attenuate age-related pro-inflammatory activation of astrocytes and microglia, astrocytes and cytokines while extending lifespan in various organisms [53,54].
The microbiome also plays an essential role in the link between diet and AD. Dietary changes (either deleterious or beneficial) influence the microbiome composition, thereby affecting the gut-brain axis and the subsequent risk for AD progression [55].
2.9. Cerebrovascular Disease
Cerebrovascular disease, manifesting as cerebral atherosclerosis and arteriolosclerosis, is a risk factor associated with AD; thus, cerebral vessel pathology is a pervasive risk factor for both vascular dementia and AD [56]. A number of midlife vascular risk factors are significantly associated with AD – findings consistent with a role of vascular disease in the development of AD [57]. Stroke is a common pathology associated with AD among elderly individuals, a co-morbid relationship strongest in the presence of known vascular risk factors [58].
Beyond the obvious vascular contributions (ischaemia, hypoxemia) to dementia, neurotoxic brain inflammation (pro-inflammatory microglia and cytokines) accompanies the ischaemic conditions of cerebrovascular disease, thereby contributing to AD pathogenesis [59,60,61]. Jurcau and Simion have shown that neuroinflammatory mechanisms significantly contribute to neuronal injury during cerebral ischemia, ultimately increasing the magnitude of cerebral damage and neurological deficit in AD [62].
2.10. Diabetes Mellitus
Many studies indicate that people with diabetes, especially Type 2 Diabetes, are at higher risk to develop AD; indeed, AD has even been referred to as Type 3 diabetes [63,64,65]. Among people with diabetes, the risk for developing AD has been found to be 65% higher than among non-diabetic controls. In people with AD, the rate of diabetes is very high, measured in one large community study to be 35%. An even higher number of people with AD (46%) have glucose intolerance, which can be a precursor to diabetes. Even without overt diabetes, impaired regulation of glucose levels has been associated with cognitive impairment and AD risk.
Given the complexity of diabetes, the possible mechanistic links between diabetes and AD are multi-fold and include Aβ aggregation, tau hyperphosphorylation, neuroinflammation, oxidative stress and mitochondrial dysfunction. Amongst these, Van Dyken and Lacoste have argued that neuroinflammation is one of the key mechanistic connectors between diabetes and AD [66]. Similarly, based on a systematic review of in vitro, preclinical and clinical studies, Vargas-Soria et al. concluded that diabetes triggers specific responses that include upregulation of activated microglia and secretion of a wide variety of pro-inflammatory cytokines and chemokines [67]. Pathways commonly activated by diabetes-related conditions include the NLRP3 inflammasome.
2.11. Oral Hygiene (Porphyromonas gingivalis)
Bacteria and their associated inflammatory molecules are able to travel from regions of mouth infections to the brain via the bloodstream [68]. Researchers at the School of Dentistry, University of Central Lancashire, were the first to report the link between gum disease and AD, concluding that periodontitis is a risk factor for AD [69]. The mouth contains 700 bacterial species, including ones that cause periodontal gingivitis; Porphyromonas gingivalis, a non-motile, Gram-negative, rod-shaped, anaerobic, pathogenic bacterium from the phylum Bacteroidota, is the most common culprit of gum disease. Recent studies indicate that Aβ oligomerization and its associated neuroinflammatory responses may be triggered in response to this infection. Porphyromonas gingivalis and the gingipains enzyme which it produces have been identified in AD brains. Thus, periodontitis is a anatomically specific infection and risk factor for AD [70,71].
Neuroinflammatory processes constitute the connection between chronic, inflammatory disease of the oropharyngeal cavity and gums (periodontitis) and AD [72]. This neuroinflammation may occur by two basic processes: a. local (oral) and/or its associated systemic inflammation triggering a neuroinflammatory reaction within the brain via the distribution of pro-inflammatory mediators; or b. direct entry of bacteria into the cranial space eliciting a protective innate immune response manifesting as neuroinflammation. Also, dysbiotic oral bacteria release both bacterial products and inflammatory molecules into the bloodstream, ultimately crossing the brain-blood barrier (BBB); these bacteria can cause alterations to gut microbiota, enhancing inflammation and affecting brain function via the gut-brain axis. The trigeminal nerve has been suggested as another route for connecting oral bacterial products to the brain. Whatever the mechanism, periodontitis leads to microglial activation and pro-inflammatory cytokine release in brain thereby triggering and promoting AD pathogenesis [73,74].
2.12. Peptic Ulcer Disease (Helicobacter pylori)
There is also an association between peptic ulcer disease and AD – analogous to the connection between oral bacteria and AD, but with the peptic ulcer bacterium (Helicobacter pylori) being further down the gastrointestinal tract [75,76]. Studies have shown that peptic ulcer disease increases the risk of AD via the mechanisms of systemic inflammation and altered gut microbiota [77]. In a population-based study, Chang et al. showed that eradication of Helicobacter pylori is associated with decreased progression of dementia [78]. Thus, periodontitis and peptic ulcer disease are two anatomically specific infections implicated as risk factors for AD.
Noori et al. have shown that Helicobacter pylori infection contributes to the expression of AD-associated risk factors and neuroinflammation, particularly enhanced concentrations of activated microglia and pro-inflammatory cytokines [79]. In a rat model of peptic ulcer disease, increased levels of circulating pro-inflammatory cytokines such as IL-1β have been documented [80].
2.13. Systemic Infection
The relationship between systemic non-CNS infections and AD is complex but a preponderance of evidence supports the supposition that systemic infection is a risk factor for AD [81]. Giridharan et al. showed that sepsis-induced peripheral infection accelerates cognitive decline and AD pathology in the AD mouse model [82]. Based on a systematic review and meta-analysis of human studies, Lei et al. showed that surviving sepsis was associated with an increased risk of dementia (OR = 1.62, 95% CI = 1.23–2.15, I2 = 96.4%, p = 0.001) and that septicaemia is associated with increased risk for dementia and AD [83]. Though many microorganisms have been implicated, Herpes simplex virus 1, Chlamydia pneumonia and Borrelia burgdorferi have been discussed as infectious agents which are possible specific microbiological risk factors for AD. Conversely, systemic infection exacerbates pre-existing AD, accelerating cognitive decline and disease progression.
Systemic infections provoke a systemic inflammatory response which in turn elicits neuroinflammation. In a prospective pilot study in people with AD, Holmes et al. showed that cognitive function can be impaired for at least two months after the resolution of a systemic infection and that cognitive impairment is preceded by raised serum levels of IL-1β [84]. In a post-mortem study, Asby et al. provide evidence that systemic infection raises the levels of multiple cytokines (TNFα, IL-1β, IL-6, IL-8 and IL-15) in brain [85].
2.14. Systemic Inflammation
Acute and chronic systemic inflammation is characterized by the systemic production of pro-inflammatory cytokines (e.g. TNFα) that play a role in immune to brain communication; systemic inflammation increases pro-inflammatory cytokine secretion in brain, which in turn causes an increase in cognitive decline and disease progression in AD [86]. Walker et al. have discussed how systemic pro-inflammatory cytokines can regionally promote a pro-inflammatory environment in the central nervous system (CNS) by crossing the blood-brain barrier, by signalling through endothelial cells, and/or by stimulating the vagus nerve [87]. Through each of these routes, systemic inflammation induces reactive, pro-inflammatory microglia and astrocytic phenotypes which can promote β-amyloid oligomerization, tau hyperphosphorylation, and complement activation. Similarly, Xie et al. likewise discuss how peripheral inflammation is a risk factor contributing to AD by means of neuroinflammation [88]. Finally, diseases typically associated with chronic systemic inflammation, such as rheumatoid arthritis, are regarded as risk factors for AD [89,90].
2.15. Allergies
Joh et al. studied 6,785,948 adults aged ≥40 years who participated in a national health examination without any history of dementia before baseline; during 8.1 years of follow-up, 260,705 dementia cases (195,739 AD) were identified and three allergic diseases (asthma, atopic dermatitis, allergic rhinitis) were positively associated with dementia risk [91]. Compared with individuals without allergies, those with all three allergic diseases had substantially increased risk of AD (multivariable hazard ratios 1.46; 95% CI 1.25-1.70). Bożek et al. have also noted a similar correlation between allergies and AD [92]. Conversely, allergies can exacerbate existing health issues for older adults with AD.
Not surprisingly, there is a relationship between allergies and inflammation [93]. Kabata and Artis have described how allergies affect a variety of cytokines, inflammatory mediators and neuropeptides to yield an enhanced neuroinflammatory response [94]. Similarly, Mirotti et al. have extensively reviewed the relationship between allergies and brain inflammation particularly microglial activation and pro-inflammatory cytokine release [95].
2.16. Migraine Headache
In a nationwide (South Korea) cohort study, Kim et al. showed that the overall incidence of AD was higher in individuals with a history of migraine than in those with no migraine history (8.0 per 1,000 person-years vs. 4.1 per 1,000 person-years) [96]. Similarly, in a population-based cohort study involving 88,390 participants, Hurh et al. concluded that migraine is associated with an increased risk of subsequent AD [97]. Multiple other epidemiological studies support the observation that migraine is a risk factor for AD [98,99].
Migraine is a neuroinflammatory disorder [100]. The implications of neurogenic inflammation and neuroinflammation in the pathophysiology of migraine have been clearly demonstrated in preclinical migraine models involving the trigemino-vascular system, including dural vessels and trigeminal endings. Neuroinflammatory pathways, specifically those involving inflammasome proteins, are regarded as clinical biomarkers and promising druggable targets for migraine [101].
2.17. Chronic Pain
In a nationwide propensity-matched cohort using administrative data, Bornier et al. noted that among 64,496 French individuals, the incidence of AD was higher in the chronic pain population than in a control group (1.13% vs. 0.95%, p <0.001); chronic pain increases the risk of AD [102]. Supportively, in a systematic review, Innes and Sambamoorthi documented the potential contribution of chronic pain and common chronic pain conditions to subsequent cognitive decline, new onset cognitive impairment, and incident dementia including AD [103]. Also, Cao et al. provide evidence that supports a risk factor link between chronic pain and AD [104].
In mechanistic considerations, Vergne-Salle and Bertin discuss how afferent nerve fibres carrying pain information are responsible for peripheral sensitization linked to inflammation molecules; these afferent fibres release neurotransmitters in the dorsal root ganglion and dorsal horn of the spinal cord, activating microglia and producing pro-inflammatory cytokines and chemokines throughout the CNS [105]. Moreover, as with many of these risk factors, the relationship is bidirectional, self-sustaining and mutually triggering, as evidenced by the fact that neuroinflammation enhances chronic pain perception [106].
2.18. Head Trauma
Young adults who experience a moderate or severe head injury have more than double the risk of developing AD and other forms of dementia later in life [107]. In a study based on a population-based prospective historical cohort design, Plassman et al. showed that both moderate head injury (hazard ratio [HR] = 2.32; CI = 1.04 to 5.17) and severe head injury (HR = 4.51; CI = 1.77 to 11.47) were associated with increased risk of AD [108]. Thus, there is evidence for an association between a history of previous head injury and the risk of developing AD.
Schimmel et al. have shown that neuroinflammation following traumatic brain injury is a chronic response to an acute injury [109]. Simon et al. have demonstrated that some individuals with traumatic brain injury develop chronic neuroinflammation, which can last for years after the injury, and is associated with activated microglia and the release of pro-inflammatory cytokines – a conclusion also supported by Xiong et al. and Zheng et al. [110,111,112].
2.19. Domestic Violence
Intimate partner violence (IPV; also termed spousal abuse or domestic violence) forms a sub-group of head trauma scenarios uniquely correlated with AD [113]. However, IPV is more than a focussed sub-type of head trauma. Unlike the head trauma typically seen during accidents or in professional athletes, IPV also comprehensively encompasses psychological, sexual, and financial abuse and not infrequently is accompanied by alcohol or substance abuse; the nature of the physical violence in IPV is also different, frequently involving manual or ligature partial strangulation.
A 1990 case report by Roberts et al. describing a 76-year-old woman with dementia connects IPV and AD [114]. A woman was found unconscious with head contusions; her relatives disclosed that her husband had been abusive for years. A post-mortem brain examination revealed morphological and immunological characteristics showing that the woman’s IPV-associated brain trauma contributed significantly to the development and progression of her dementia. The consequences of traumatic brain injuries (TBI) are significant, with evidence suggesting a single TBI may double one’s likelihood of developing dementia. Traumatic brain injuries are highly prevalent amongst victims of IPV, arguably leaving hundreds of millions of women worldwide at increased risk for developing dementia.
The connection between IPV and AD is clear, and involves multiple mechanisms including neuroinflammation. Newton et al. have shown that IPV histories are associated with biologic mediators of inflammation, particularly elevated levels of IL-6 [115]. Similarly, Madison et al. have shown that IPV is associated with augmented pro-inflammatory cytokine response including IL-6 and TNFα [116].
2.20. Depression
Various studies support that depression is a risk factor for AD. Growing evidence implies that timing of the depression may be important to defining the nature of its association with AD. In particular, earlier-life depression or depressive symptoms have been consistently shown to be associated with a 2-fold or greater increase in risk of dementia; in contrast, studies of late-life depression have been less definitive but the majority support an association [117]. A variety of studies support these conclusions that depression is a risk factor for AD [118,119,120,121,122,123].
Multiple studies suggest that neuroinflammation is the key process linking depression to dementia [124]. In depression, chronic activation of innate immunity accelerates central inflammation leading to higher levels of inflammatory cytokines, most consistently IL-1β, IL-6, and TNFα, which in turn correlates with greater depressive symptomatology [125]. Neuroinflammation is involved in the pathophysiology of depression by increasing pro-inflammatory cytokines, activating the hypothalamus–pituitary–adrenal axis, increasing glucocorticoid resistance, and affecting serotonin synthesis and metabolism, neuronal apoptosis and neurogenesis, and neuroplasticity [126].
2.21. Anxiety
Based on a comprehensive literature review, Becker et al. concluded that anxiety is a risk factor for AD (n = 26193, hazard ratio 1.53, 95% CI 1.16-2.01, P < 0.01) [127]. Similarly, based on a meta-analysis of prospective cohort studies listed on PubMed or Web of Science from January 2018 to January 2020, Santabárbara et al. evaluated nine prospective cohorts representing 29,608 participants and identified an overall relative risk of dementia of 1.24 (95% CI: 1.06–1.46) and a population fraction of dementia attributable to anxiety of 3.9%; they concluded that anxiety is significantly associated with an increased risk of AD [128].
The relationship between anxiety, neuroinflammation and AD is complex and bi-directional: anxiety causes neuroinflammation and neuroinflammation causes anxiety. Studies by Won and Kim suggest that anxiety leads to hypothalamic–pituitary–adrenal axis and autonomic nervous system disruption, which in turn induces neuroinflammatory conditions manifesting as enhanced pro-inflammatory cytokine levels from activated microglia particularly in prefrontal and limbic brain structures – this enhances AD progression [129]. Conversely, based on animal and clinical studies, Zheng et al. and Guo et al. have concluded that neuroinflammation induces anxiety by modulating neuronal plasticity in multiple brain regions, but particularly the basolateral amygdala [130,131]. Thus, anxiety triggers neuroinflammation central to the pathogenesis of AD.
2.22. Insomnia
Insomnia and sleep disorders constitute a well-documented risk factor for AD [132]. The majority of neurodegenerative diseases are known to cause sleep disruption, but insomnia is itself a risk factor for neurodegenerative diseases, including AD. Sleep has important roles in learning and memory consolidation and sleep deprivation affects not only the symptoms but also the pathogenesis of AD. Sleep contributes to the removal of Aβ from neural tissue: Kang et al. showed in transgenic mice that chronic insomnia leads to Aβ accumulation and thus AD progression [133]. Many studies have now shown that sleep deprivation is a risk factor for AD [134,135,136,137,138,139].
Neuroinflammation is central to the linkage between insomnia and AD. Zhu et al. showed that sleep disturbance increased IL-6 pro-inflammatory cytokine levels and induced microglia activation in mouse hippocampus, impairing hippocampus-dependent learning and memory [140]. Zielinski and Gibbons have described the altered role of the IL-1β and TNFα inflammatory cytokines and the NLRP3 inflammasome during dysregulated sleep [141]. Sleep impairment leads to neuroinflammation through increasing levels of pro-inflammatory cytokines and inflammatory agents and enzymes such as TNFα, IL-6, and IL-1, and COX levels. Chronic insomnia has also been associated with compromised BBB integrity resulting in an increased entry of peripheral immune cells and cytokines into the CNS, thus further contributing to the neuroinflammation implicated in AD pathogenesis [142].
2.23. Ethanol Abuse
Alcohol abuse has been identified as a risk factor for cognitive decline, AD and dementia and [143]. Alcoholism is associated with extensive and varied cognitive problems, including alcoholic dementia and AD. Because alcohol’s effects on cognition, brain disorders, and brain chemistry share some features with AD’s effects on these three areas, it is postulated that alcohol abuse increases the risk of developing AD [144,145,146,147,148].
Neuroinflammation is a major histochemical component of alcohol-induced neural damage [149]. Alcohol abuse is characterized by induction of peripheral inflammation and neuroinflammation, a hallmark of which is the activation of innate immunity TLR4 (Toll-Like Receptor 4) with associated microglial and inflammatory cytokine involvement. Based on mouse studies, Lowe et al. established that chronic alcohol consumption promotes the recruitment of peripheral macrophages into the CNS and microglia alterations through the CCR2/5 (C-C chemokine receptor types 2 and 5) axis [150].
2.24. Social Isolation
Loneliness and social isolation are widespread and significant public health risks affecting many people and placing them at risk for AD and other related dementias. In a study of 502,506 UK Biobank participants and 30,097 participants from the Canadian Longitudinal Study of Aging, Shafighi et al. analysed risk factors for developing AD and related dementias in the context of loneliness and lack of social support, identifying strong links between social isolation and multiple indicators of AD [151]. Similarly, in a study to establish Cox proportional hazard models with social isolation and loneliness as separate exposures, Shen et al. concluded that social isolation is a risk factor for AD that is independent of loneliness and many other covariates and that social isolation is an early indicator of and a risk factor for an increased risk of dementia [152].
Neuroinflammation is a definite immunological concomitant of the psychosocial problems inflicted by social isolation. In a study on eight-week-old male C57BL/6 mice, Al Omran et al. showed that social isolation resulted in microglial activation and the release of pro-inflammatory cytokines [153]. Analogously, in a study with BALB/c mice, Ayilara and Owoyele demonstrated evidence of neuroinflammation manifesting as increased activated microglial count and elevated IL-1β and TNFα cytokine levels in a social isolation rearing model [154]. Also, Vu et al. have shown that social isolation produces brain region-specific activation of microglia state in C57Bl/6 mice [155].
2.25. Glaucoma
Glaucoma refers to a group of diseases characterized by optic neuropathies that are commonly associated with degeneration of the retinal ganglion cells. Evidence of a link between AD and glaucoma has emerged from studies showing that patients with AD have a significantly increased rate of glaucoma occurrence [156]. Cesareo et al. studied 51 AD subjects and 67 sex-matched controls: subjects underwent measurements of intraocular pressure, visual field testing, and retinal nerve fibre layer thickness assessment by slit-lamp biomicroscopy – patients with AD had a higher frequency of glaucoma-like alterations [157]. Crump et al. studied 324,730 persons diagnosed with glaucoma from 1995-2017 in Sweden and 3,247,300 age- and sex-matched population-based controls without prior dementia: in 16 million person-years of follow-up, 32,339 (10%) persons with glaucoma and 226,896 (7%) controls were diagnosed with dementia [158]. Persons with glaucoma had increased risks for AD (adjusted HR, 1.39; 95% CI, 1.35-1.43); among glaucoma subtypes, both primary open-angle and normal-tension glaucoma were associated with increased risk for AD. Thus, people with glaucoma have an increased risk of developing AD [159,160].
Preclinical and clinical evidence supports the notion that glaucoma is a widespread neurodegenerative condition whose shared pathogenic mechanism with AD is neuroinflammation. Williams et al. have shown that the neuropathology of glaucoma extends beyond the visual pathways and involves pro-inflammatory neuroinflammation at both a cellular (microglia, astrocyte) and molecular (cytokine) level in other CNS locations [161]. Studies by Rolle et al., Rutigliani et al. and Soto & Howell have reached similar conclusions [162,163,164].
2.26. Hearing Loss
Hearing loss in ages 45-65 is a significant risk factor for dementia, possibly accounting for 8 percent of all dementia cases; the 2020 Lancet report determined that hearing loss across a wide variety of types and aetiologies approximately doubles the risk of dementia with even subclinical hearing loss enhancing AD risk [3]. Extensive studies by Lin et al. have concluded that hearing loss is associated with increased cognitive decline and incident AD and other dementias in older adults [165]. Based on an analysis of a UK biobank cohort, Jiang et al. concluded that in people with hearing loss, restorative hearing aid use is associated with a reduced risk of dementia of a similar level to that of people without hearing loss, thereby highlighting the urgent need to take measures to address hearing loss as a remediable risk factor for AD [166].
Seicol et al. have shown that age-related hearing loss is accompanied by chronic inflammation in neural structures with elevated expression of pro-inflammatory cytokines and microglial activation [167]. Similarly, Frye et al. have demonstrated that pro-inflammatory cytokines including TNFα and IL-1β, and chemokines including CCL2, are induced by hearing loss [168].
2.27. Noise Pollution
Despite their obvious interconnection, hearing loss and exposure to noise pollution are regarded as separate risk factors. Hearing loss caused by factors other than noise exposure is a risk factor for AD; chronic noise exposure of insufficient magnitude to cause obvious hearing loss is likewise a risk factor for AD. Epidemiological studies are increasingly identifying the association between external noise exposure (via noise pollution) and dementia [169]. Weuve et al. for example, showed that an increment of 10 A-weighted decibels (dBA) in noise corresponded to 36% and 29% higher odds of prevalent mild cognitive impairment (MCI; odds ratio [OR] = 1.36; 95% confidence interval [CI], 1.15 to 1.62) and AD (OR = 1.29, 95% CI, 1.08 to 1.55) [170]. Cantuaria et al. estimated that as many as 1,216 out of the 8,475 cases of dementia registered in Denmark in 2017 could be attributed to noise exposures, indicating a great potential for dementia prevention through reduction in ambient noise such as that arising from roadway traffic [171].
As with hearing loss, neuroinflammation is a central mechanistic player in the pathogenesis of noise-induced AD. Wang et al. showed that noise exposure is associated with elevated expression of pro-inflammatory cytokines and microglial activation in the primary auditory cortex; genetic knockout of TNFα or pharmacologically blocking TNFα expression prevented neuroinflammation [172]. Similarly, Cui et al. have shown that chronic noise exposure acts cumulatively to exacerbate AD pathology and neuroinflammation in rat hippocampus [173].
2.28. Air pollution
Based on a systematic literature review, Peters et al. concluded that greater exposure to PM2.5, NO2/NOx, and CO were all associated with increased risk of dementia, where PM2.5 is airborne particulate matter ≤2.5 μ in size [174]. Subsequently, Peters and Li have reaffirmed this observation, claiming that constituents of PM2.5, namely black carbon, organic matter, sulphates (SO42−), and ammonium (NH4+) from traffic and fossil fuel combustion are significantly associated with the development of AD [175]. Also, a national cohort study (2000–2018) of long-term air pollution exposure and incident dementia in older adults in the United States showed that exposures to PM2.5 and NO2 are associated with an increased incidence of AD [176,177].
Campbell et al. have shown that particulate matter in polluted air increases biomarkers of inflammation in mouse brain, including activated microglia, and levels of pro-inflammatory cytokines such as IL-1β and TNFα [178]. Tin-Tin-Win-Shwe et al. have likewise shown changes in pro-inflammatory cytokine mRNA expressions in mice following nanoparticle air pollution exposure [179]. These data and others have led Block and Calderón-Garcidueñas to conclude that the emerging evidence implicates air pollution as a chronic source of neuroinflammation instigating AD with activation of microglia as key to this process [180].
2.29. Global Warming
In 2021, the World Health Organization (WHO) announced that climate change is the biggest global health threat to humanity. A 1.5°C ambient temperature increase may not influence what clothing people wear but it does induce acclimatization effects on neural pathways and mechanisms that underlie normal brain functioning; these pathways, including oxidative stress and neuroinflammation, are implicated in neurodegeneration [181]. Thus, it is possible that global warming secondary to climate change will emerge as a risk factor for AD. In addition, climate warming puts people with AD at risk for symptom worsening and disease progression [182,183,184]. Gong et al. predict a 4.5% increase in risk of dementia hospital admission per 1°C increase above 17°C and a 300% increase in hospital admissions for AD by 2040 because of climate change [185].
Given the relationship between ambient temperatures and inflammation, it is probable that neuroinflammation is part of the pathological spectrum response to global warming [186,187]. In mice subjected to heat exposure, Lee et al. found: 1) an increased number of glial fibrillary acid protein (GFAP)- and macrophage-1 antigen (Mac-1)-positive cells, 2) up-regulated nuclear factor (NF)-κB, a master regulator of inflammation, and 3) marked increases in COX-2, inducible nitric oxide synthase (iNOS), and cytokine IL-1β and TNFα in the mouse hippocampus [188].
2.30. Educational Level
Lower education is associated with a greater risk for AD and related dementias [189]. The 2020 Lancet Commission that examined dementia risk factors found 7% of worldwide dementia cases could be prevented by increasing early-life education [3]. The study found higher childhood education levels and higher lifelong educational attainment could reduce AD and dementia risk. A focussed sub-type of educational attainment is the ability to speak multiple languages; multiple studies indicate that bilingualism or multilingualism offer a degree of protective delay against the development of AD [190,191,192].
The correlation of educational level with neuroinflammation is not as immediately apparent as for other risk factors, such as head trauma. Nonetheless, there are data clearly supporting a relationship between education and brain inflammatory markers. Steinvil et al. found a statistically significant inverse association between number of school years and high-sensitivity C-reactive protein (CRP), fibrinogen, and erythrocyte sedimentation rate (ESR) concluding that level of education was inversely associated with inflammatory biomarkers even within highly educated populations [193]. Similarly, Maurel et al. found a relationship between educational attainment and five inflammatory biomarkers (CRP, fibrinogen, IL-1β, IL-6 and TNFα) whereby a low educational attainment was associated with higher inflammation even after adjusting for health behaviours and body mass index [194]. A 2015 study by Okonkwo and co-workers showed that older adults who completed at least 16 years of education had less evidence of AD biomarkers in their cerebrospinal fluid (CSF) than people with fewer years of education [195].
Another theory suggests that education is associated with a higher socioeconomic status and quality of life that helps keep people healthy and lowers AD risk. Education can impact the type of job a person has, the environment they live in, the quality of food they can eat and the health care they receive – all of which are directly related to levels of neuroinflammation. Higher levels of education were associated with lower prevalence of diabetes and smoking in both genders and lower prevalence of hypertension and dyslipidemia in women. A combination of these factors over time add up to increased neuroinflammation and a higher AD risk.
3. Conclusions
The development of effective diagnostics and therapeutics for AD is one of humankind’s pressing neuropharmacologic priorities. A hurdle in the successful attainment of these priorities is the immense cellular and molecular complexity of AD. This complexity is reflected by the equally complex diversity of risk factors associated with AD. However, more than merely mirroring disease complexity, risk factors also provide fundamental insights into the aetiology and pathogenesis of AD as a neurodegenerative disorder since they are central to disease initiation and subsequent propagation. Based on a systematic literature review, this analysis has identified 30 risk factors for AD and then extended the analysis to further identify neuroinflammation as a unifying mechanism present in all 30 risk factors. Although other mechanisms (e.g. vasculopathy) were present in multiple risk factors, dysfunction of the neuroimmune-neuroinflammation axis was key to all 30 identified risk factors. Though the nature of the neuroinflammatory involvement varied, activation of microglia and the release of pro-inflammatory cytokines was a common pathway shared by all risk factors. This observation provides further evidence for the importance of immunopathic mechanisms to aetiopathogenesis of AD.
Author Contributions
Conceptualization and writing all drafts by DFW.
Funding
This work was funded by grants from the Krembil Foundation, the Weston Foundation and a Harrington Scholar-Innovator Award.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
DFW acknowledges salary support from the Krembil Foundation.
Conflicts of Interest
The author declares no conflict of interest。
References
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer's Disease: Causes and Treatment. Molecules. 2020, 25, 5789. [CrossRef]
- Knopman, D. S.; Amieva, H.; Petersen, R. C.; Chételat, G.; Holtzman, D. M.; Hyman, B. T.; Nixon, R. A.; Jones, D. T. Alzheimer disease. Nat. Rev. Dis. Primers. 2021, 7, 33. [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; Costafreda, S. G.; Dias, A.; Fox, N.; Gitlin, L. N.; Howard, R.; Kales, H. C.; Kivimäki, M.; Larson, E. B.; Ogunniyi, A.; Orgeta, V.; … Mukadam, N. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020, 396, 413–446. [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D. H. Neuroinflammation in Alzheimer's Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation. 2023, 46, 1–17; [CrossRef]
- Heneka, M. T.; Carson, M. J.; El Khoury, J.; Landreth, G. E.; Brosseron, F.; Feinstein, D. L.; Jacobs, A. H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R. M.; Herrup, K.; Frautschy, S. A.; Finsen, B.; Brown, G. C.; Verkhratsky, A.; Yamanaka, K.; Koistinaho, J.; Latz, E.; Halle, A.; Petzold, G. C.; … Kummer, M. P. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015, 14, 388–405. [CrossRef]
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P. M.; Ferretti, M. T.; Lanman, L.; Chadha, A. S.; & van der Flier, W. M. Global estimates on the number of persons across the Alzheimer's disease continuum. Alzheimers Dement. 2023;19(2):658-670. [CrossRef]
- d'Avila, J. C.; Siqueira, L. D.; Mazeraud, A.; Azevedo, E. P.; Foguel, D.; Castro-Faria-Neto, H. C.; Sharshar, T.; Chrétien, F.; Bozza, F. A. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J. Neuroinflamm. 2018, 15, 28. [CrossRef]
- Andronie-Cioara, F. L.; Ardelean, A. I.; Nistor-Cseppento, C. D.; Jurcau, A.; Jurcau, M. C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and A.D. Progression. Internat. J. Mol. Sci. 2023, 24, 1869. [CrossRef]
- Godbout, J. P.; Johnson, R. W. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol. Allergy Clin. North Amer. 2009, 29, 321–337. [CrossRef]
- Beam, C. R.; Kaneshiro, C.; Jang, J. Y.; Reynolds, C. A.; Pedersen, N. L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer's Disease. J. Alz. Dis. 2018, 64, 1077–1083. [CrossRef]
- Liu, C. C.; Li, C. Y.; Sun, Y.; Hu, S. C. Gender and Age Differences and the Trend in the Incidence and Prevalence of Dementia and Alzheimer's Disease in Taiwan: A 7-Year National Population-Based Study. BioMed. Res. Internat. 2019, 2019, 5378540. [CrossRef]
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer's disease in Europe: A meta-analysis. Prevalencia e incidencia de la enfermedad de Alzheimer en Europa: metaanálisis. Neurologia. 2017, 32, 523–532. [CrossRef]
- Bekhbat, M.; Neigh, G. N. Sex differences in the neuro-immune consequences of stress: Focus on depression and anxiety. Brain Behav. Immun. 2018, 67, 1–12. [CrossRef]
- Engler, H.; Benson, S.; Wegner, A.; Spreitzer, I.; Schedlowski, M.; Elsenbruch, S. Men and women differ in inflammatory and neuroendocrine responses to endotoxin but not in the severity of sickness symptoms. Brain Behav. Immun. 2016, 52, 18-26;. [CrossRef]
- Jacobson, D. L.; Gange, S. J.; Rose, N. R.; Graham, N. M. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997, 84, 223-243;. [CrossRef]
- Meier-Stephenson, F. S.; Meier-Stephenson, V. C.; Carter, M. D.; Meek, A. R.; Wang, Y.; Pan, L.; Chen, Q.; Jacobo, S.; Wu, F.; Lu, E.; Simms, G. A.; Fisher, L.; McGrath, A. J.; Fermo, V.; Barden, C. J.; Clair, H. D. S.; Galloway, T. N.; Yadav, A.; Campágna-Slater, V.; Hadden, M.; … Weaver, D. F. Alzheimer's disease as an autoimmune disorder of innate immunity endogenously modulated by tryptophan metabolites. Alzheimers Dement (N Y). 2022, 8, e12283;. [CrossRef]
- Sierra C. Hypertension and the Risk of Dementia. Front. Cardiovasc. Med. 2020, 7, 5;. [CrossRef]
- Gabin, J. M.; Tambs, K.; Saltvedt, I.; Sund, E.; Holmen, J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: the HUNT Study. Alzheimers Res. Ther. 2017, 9, 37;. [CrossRef]
- Bajwa, E.; Klegeris, A. Neuroinflammation as a mechanism linking hypertension with the increased risk of Alzheimer's disease. Neural. Regen. Res. 2022, 17, 2342-2346;. [CrossRef]
- Solé-Guardia, G.; Custers, E.; de Lange, A.; Clijncke, E.; Geenen, B.; Gutierrez, J.; Küsters, B.; Claassen, J. A. H. R.; de Leeuw, F. E.; Wiesmann, M.; Kiliaan, A. J. Association between hypertension and neurovascular inflammation in both normal-appearing white matter and white matter hyperintensities. Acta Neuropathol. Commun. 2023, 11, 2;. [CrossRef]
- Carnevale, D.; Mascio, G.; Ajmone-Cat, M. A.; D'Andrea, I.; Cifelli, G.; Madonna, M.; Cocozza, G.; Frati, A.; Carullo, P.; Carnevale, L.; Alleva, E.; Branchi, I.; Lembo, G.; Minghetti, L. Role of neuroinflammation in hypertension-induced brain amyloid pathology. Neurobiol. Aging. 2012, 33, 205.e19-205.e2.05E29;. [CrossRef]
- Mancini, G.; Dias, C.; Lourenço, C. F.; Laranjinha, J.; de Bem, A.; Ledo, A. A High Fat/Cholesterol Diet Recapitulates Some Alzheimer's Disease-Like Features in Mice: Focus on Hippocampal Mitochondrial Dysfunction. J. Alzheimers Dis. 2021, 82, 1619-1633;. [CrossRef]
- Ledreux, A.; Wang, X.; Schultzberg, M.; Granholm, A. C.; Freeman, L. R. Detrimental effects of a high fat/high cholesterol diet on memory and hippocampal markers in aged rats. Behav. Brain Res. 2016, 312, 294-304;. [CrossRef]
- Jin, P.; Pan, Y.; Pan, Z.; Xu, J.; Lin, M.; Sun, Z.; Chen, M.; Xu, M. Alzheimer-like brain metabolic and structural features in cholesterol-fed rabbit detected by magnetic resonance imaging [published correction appears in Lipids Health Dis. 2018 Aug 29;17(1):204]. Lipids Health Dis. 2018, 17, 61;. [CrossRef]
- Wu, M.; Zhai, Y.; Liang, X.; Chen, W.; Lin, R.; Ma, L.; Huang, Y.; Zhao, D.; Liang, Y.; Zhao, W.; Fang, J.; Fang, S.; Chen, Y.; Wang, Q.; Li, W. Connecting the Dots Between Hypercholesterolemia and A.D.: A Potential Mechanism Based on 27-Hydroxycholesterol. Front. Neurosci. 2022, 16, 842814: . [CrossRef]
- Xu, C.; Apostolova, L. G.; Oblak, A. L.; Gao, S. Association of Hypercholesterolemia with Alzheimer's Disease Pathology and Cerebral Amyloid Angiopathy. J. Alzheimers Dis. 2020, 73, 1305-1311;. [CrossRef]
- Feringa, F. M.; van der Kant, R. Cholesterol and Alzheimer's Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372;. [CrossRef]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M. S.; Bhat, N. R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 06, 475-485;. [CrossRef]
- Chen, Y.; Yin, M.; Cao, X.; , Hu, G.; Xiao, M. Pro- and Anti-inflammatory Effects of High Cholesterol Diet on Aged Brain. Aging Dis. 2018, 9, 374-390;. [CrossRef]
- Durazzo, T. C.; Mattsson, N.; Weiner, M. W. Smoking and increased Alzheimer's disease risk: a review of potential mechanisms. Alzheimers Dement. 2014, 10, S122-S145;. [CrossRef]
- Jeong, S.; Park, J.; Han, K. Association of Changes in Smoking Intensity with Risk of Dementia in Korea. JAMA Netw. Open. 2023, 6, e2251506;. [CrossRef]
- Alrouji, M.; Manouchehrinia, A.; Gran, B.; Constantinescu, C. S. Effects of cigarette smoke on immunity, neuroinflammation and multiple sclerosis. J. Neuroimmunol. 2019, 329, 24-34;. [CrossRef]
- Liu, Y.; Li, H.; Wang, J. Association of Cigarette Smoking With Cerebrospinal Fluid Biomarkers of Neurodegeneration, Neuroinflammation, and Oxidation. JAMA Netw. Open. 2020, 3, e2018777;. [CrossRef]
- Meng, Q.; Lin, M. S.; Tzeng, I. S. Relationship Between Exercise and Alzheimer's Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131;. [CrossRef]
- Chen, W. W.; Zhang, X.; Huang, W. J. Role of physical exercise in AD. Biomed. Rep. 2016, 4, 403–407. [CrossRef]
- Wang, M.; Zhang, H.; Liang, J.; Huang, J.; Chen, N. Exercise suppresses neuroinflammation for alleviating AD. J. Neuroinflamm. 2023, 20, 76. [CrossRef]
- Seo, D. Y.; Heo, J. W.; Ko, J. R.; Kwak, H. B. Exercise and Neuroinflammation in Health and Disease. Int. Neurourol. J. 2019, 23(Suppl 2), S82-S92;. [CrossRef]
- Svensson, M.; Lexell, J.; Deierborg, T. Effects of Physical Exercise on Neuroinflammation, Neuroplasticity, Neurodegeneration, and Behavior: What We Can Learn From Animal Models in Clinical Settings. Neurorehabil. Neural Repair. 2015, 29, 577-589;. [CrossRef]
- Forny-Germano, L.; De Felice, F. G.; Vieira, M. N. D. N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and A.D. Front. Neurosci. 2019, 12, 1027. [CrossRef]
- Flores-Cordero, J. A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and A.D.: The Role of Leptin. Internat. J. Mol. Sci. 2022, 23, 5202. [CrossRef]
- Hayes, J. P.; Moody, J. N.; Roca, J. G.; Hayes, S. M. Body mass index is associated with smaller medial temporal lobe volume in those at risk for A.D. NeuroImage Clinical. 2020, 25, 102156. [CrossRef]
- Nordestgaard, L. T.; Tybjærg-Hansen, A.; Nordestgaard, B. G.; Frikke-Schmidt, R. Body Mass Index and Risk of A.D.: A Mendelian Randomization Study of 399,536 Individuals. J. Clin. Endo. Metab. 2017, 102, 2310–2320. [CrossRef]
- Emmerzaal, T. L.; Kiliaan, A. J.; Gustafson, D. R. 2003-2013: A decade of body mass index, A.D., and dementia. J. Alzheimers Dis. 2015, 43, 739–755. [CrossRef]
- García-Ptacek, S.; Faxén-Irving, G.; Cermáková, P.; Eriksdotter, M.; Religa, D. Body mass index in dementia. Eur. J. Clin. Nutr. 2014, 68, 1204–1209. [CrossRef]
- Miller, A. A.; Spencer, S. J. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain, Behav. Immun. 2014, 42, 10–21. [CrossRef]
- Henn, R. E.; Elzinga, S. E.; Glass, E.; Parent, R.; Guo, K.; Allouch, A. A.; Mendelson, F. E.; Hayes, J.; Webber-Davis, I.; Murphy, G. G.; Hur, J.; Feldman, E. L. Obesity-induced neuroinflammation and cognitive impairment in young adult versus middle-aged mice. Immunity & Ageing. 2022, 19, 67. [CrossRef]
- Xu Lou, I.; Ali, K.; Chen, Q. Effect of nutrition in A.D.: A systematic review. Front. Neurosci. 2023, 17, 1147177. [CrossRef]
- Tosatti, J. A. G.; Fontes, A. F. D. S.; Caramelli, P.; Gomes, K. B. Effects of Resveratrol Supplementation on the Cognitive Function of Patients with A.D.: A Systematic Review of Randomized Controlled Trials. Drugs & Aging. 2022, 39, 285–295. [CrossRef]
- Samadi, M.; Moradi, S.; Moradinazar, M.; Mostafai, R.; Pasdar, Y. Dietary pattern in relation to the risk of A.D.: a systematic review. Neurological Sciences. 2019, 40, 2031–2043. [CrossRef]
- Pistollato, F.; Iglesias, R. C.; Ruiz, R.; Aparicio, S.; Crespo, J.; Lopez, L. D.; Manna, P. P.; Giampieri, F.; Battino, M. Nutritional patterns associated with the maintenance of neurocognitive functions and the risk of dementia and A.D.: A focus on human studies. Pharmacological Research. 2018, 131, 32–43. [CrossRef]
- Abduljawad, A. A.; Elawad, M. A.; Elkhalifa, M. E. M.; Ahmed, A.; Hamdoon, A. A. E.; Salim, L. H. M.; Ashraf, M.; Ayaz, M.; Hassan, S. S. U.; Bungau, S. A.D. as a Major Public Health Concern: Role of Dietary Saponins in Mitigating Neurodegenerative Disorders and Their Underlying Mechanisms. Molecules. 2022, 27, 6804. [CrossRef]
- Kip, E.; Parr-Brownlie, L. C. Healthy lifestyles and wellbeing reduce neuroinflammation and prevent neurodegenerative and psychiatric disorders. Front. Neurosci. 2023, 17, 1092537. [CrossRef]
- Bok, E.; Jo, M.; Lee, S.; Lee, B. R.; Kim, J.; Kim, H. J. Dietary Restriction and Neuroinflammation: A Potential Mechanistic Link. Internat. J. Mol. Sci. 2019, 20, 464. [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; Monda, M.; Messina, G.; Zammit, C.; Crispino, M.; Mollica, M. P. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [CrossRef]
- Romanenko, M.; Kholin, V.; Koliada, A.; Vaiserman, A. Nutrition, Gut Microbiota, and Alzheimer's Disease. Front. Psych. 2021, 12, 712673. [CrossRef]
- Arvanitakis, Z.; Capuano, A. W.; Leurgans, S. E.; Bennett, D. A.; Schneider, J. A. Relation of cerebral vessel disease to A.D. dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [CrossRef]
- Gottesman, R. F.; Schneider, A. L.; Zhou, Y.; Coresh, J.; Green, E.; Gupta, N.; Knopman, D. S.; Mintz, A.; Rahmim, A.; Sharrett, A. R.; Wagenknecht, L. E.; Wong, D. F.; Mosley, T. H. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. JAMA. 2017, 317, 1443–1450. [CrossRef]
- Honig, L. S.; Tang, M. X.; Albert, S.; Costa, R.; Luchsinger, J.; Manly, J.; Stern, Y.; Mayeux, R. Stroke and the risk of Alzheimer disease. Arch. Neurol. 2003, 60, 1707–1712. [CrossRef]
- Wong, C. H.; Crack, P. J. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr. Med. Chem. 2008, 15, 1–14. [CrossRef]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front. Mol. Neurosci. 2020, 13, 28. [CrossRef]
- Drake, C.; Boutin, H.; Jones, M. S.; Denes, A.; McColl, B. W.; Selvarajah, J. R.; Hulme, S.; Georgiou, R. F.; Hinz, R.; Gerhard, A.; Vail, A.; Prenant, C.; Julyan, P.; Maroy, R.; Brown, G.; Smigova, A.; Herholz, K.; Kassiou, M.; Crossman, D.; Francis, S.; … Allan, S. M. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain, Behav. Immun. 2011, 25, 1113–1122. [CrossRef]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Internat. J. Mol. Sci. 2021, 23, 14. [CrossRef]
- Jayaraj, R. L.; Azimullah, S.; Beiram, R. Diabetes as a risk factor for A.D. in the Middle East and its shared pathological mediators. Saudi J. Biol. Sci. 2020, 27, 736–750. [CrossRef]
- Barbagallo, M.; Dominguez, L. J. Type 2 diabetes mellitus and A.D. World J. Diabet. 2014, 5, 889–893. [CrossRef]
- Whitmer R. A. Type 2 diabetes and risk of cognitive impairment and dementia. Curr. Neurol. Neurosci. Rep. 2007, 7, 373–380. [CrossRef]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [CrossRef]
- Vargas-Soria, M.; García-Alloza, M.; Corraliza-Gómez, M. Effects of diabetes on microglial physiology: a systematic review of in vitro, preclinical and clinical studies. J. Neuroinflamm. 2023, 20, 57. [CrossRef]
- Kanagasingam, S.; von Ruhland, C.; Welbury, R.; Chukkapalli, S. S.; Singhrao, S. K. Porphyromonas gingivalis Conditioned Medium Induces Amyloidogenic Processing of the Amyloid-β Protein Precursor upon in vitro Infection of SH-SY5Y Cells. J. A.D. Rep. 2022, 6, 577–587. [CrossRef]
- Bouziane, A.; Lattaf, S.; Abdallaoui Maan, L. Effect of Periodontal Disease on A.D.: A Systematic Review. Cureus. 2023, 15, e46311. [CrossRef]
- Gao, C.; Larvin, H.; Bishop, D. T.; Bunce, D.; Pavitt, S.; Wu, J.; Kang, J. Oral diseases are associated with cognitive function in adults over 60 years old. Oral Dis. 2023, 10.1111/odi.14757. [CrossRef]
- Bello-Corral, L.; Alves-Gomes, L.; Fernández-Fernández, J. A.; Fernández-García, D.; Casado-Verdejo, I.; Sánchez-Valdeón, L. Implications of gut and oral microbiota in neuroinflammatory responses in A.D. Life Sci. 2023, 333, 122132. [CrossRef]
- Li, X.; Kiprowska, M.; Kansara, T.; Kansara, P.; Li, P. Neuroinflammation: A Distal Consequence of Periodontitis. J. Dent. Res. 2022, 101, 1441–1449. [CrossRef]
- Luo, H.; Wu, B.; Kamer, A. R.; Adhikari, S.; Sloan, F.; Plassman, B. L.; Tan, C.; Qi, X.; Schwartz, M. D. Oral Health, Diabetes, and Inflammation: Effects of Oral Hygiene Behaviour. Internat. Dent. J. 2022, 72, 484–490. [CrossRef]
- Almarhoumi, R.; Alvarez, C.; Harris, T.; Tognoni, C. M.; Paster, B. J.; Carreras, I.; Dedeoglu, A.; Kantarci, A. Microglial cell response to experimental periodontal disease. J. Neuroinflamm. 2023, 20, 142. [CrossRef]
- Hsu, C. C.; Hsu, Y. C.; Chang, K. H.; Lee, C. Y.; Chong, L. W.; Lin, C. L.; Kao, C. H. Association of Dementia and Peptic Ulcer Disease: A Nationwide Population-Based Study. American Journal of A.D. and Other Dementias. 2016, 31, 389–394. [CrossRef]
- Choi, H. G.; Soh, J. S.; Lim, J. S.; Sim, S. Y.; Jung, Y. J.; Lee, S. W. Peptic ulcer does not increase the risk of dementia: A nested case control study using a national sample cohort. Medicine. 2020, 99, e21703. [CrossRef]
- Huang, W. S.; Yang, T. Y.; Shen, W. C.; Lin, C. L.; Lin, M. C.; Kao, C. H. Association between Helicobacter pylori infection and dementia. J. Clin. Neurosci. 2014, 21, 1355–1358. [CrossRef]
- Chang, Y. P.; Chiu, G. F.; Kuo, F. C.; Lai, C. L.; Yang, Y. H.; Hu, H. M.; Chang, P. Y.; Chen, C. Y.; Wu, D. C.; Yu, F. J. Eradication of Helicobacter pylori Is Associated with the Progression of Dementia: A Population-Based Study. Gastroenterology Research and Practice, 2013, 2013, 175729. [CrossRef]
- Noori, M.; Mahboobi, R.; Nabavi-Rad, A.; Jamshidizadeh, S.; Fakharian, F.; Yadegar, A.; Zali, M. R. Helicobacter pylori infection contributes to the expression of A.D.-associated risk factors and neuroinflammation. Heliyon. 2023, 9, e19607. [CrossRef]
- Watanabe, T.; Higuchi, K.; Tanigawa, T. Mechanisms of peptic ulcer recurrence: role of inflammation. Inflammopharmacology. 2002, 10, 291–302. [CrossRef]
- Rakic, S.; Hung, Y. M. A.; Smith, M.; So, D.; Tayler, H. M.; Varney, W.; Wild, J.; Harris, S.; Holmes, C.; Love, S.; Stewart, W.; Nicoll, J. A. R.; Boche, D. Systemic infection modifies the neuroinflammatory response in late stage A.D. Acta Neuropathologica Communications. 2018, 6, 88. [CrossRef]
- Giridharan, V. V.; Catumbela, C. S. G.; Catalão, C. H. R.; Lee, J.; Ganesh, B. P.; Petronilho, F.; Dal-Pizzol, F.; Morales, R.; Barichello, T. Sepsis exacerbates A.D. pathophysiology, modulates the gut microbiome, increases neuroinflammation and amyloid burden. Molecular Psychiatry. 2023, 10.1038/s41380-023-02172-2; [CrossRef]
- Lei, S.; Li, X.; Zhao, H.; Feng, Z.; Chun, L.; Xie, Y.; Li, J. Risk of Dementia or Cognitive Impairment in Sepsis Survivals: A Systematic Review and Meta-Analysis. Frontiers in Aging Neuroscience, 2022, 14, 839472. [CrossRef]
- Holmes, C.; El-Okl, M.; Williams, A. L.; Cunningham, C.; Wilcockson, D.; Perry, V. H. Systemic infection, interleukin 1beta, and cognitive decline in A.D. Journal of Neurology, Neurosurgery, and Psychiatry. 2003, 74, 788–789. [CrossRef]
- Asby, D.; Boche, D.; Allan, S.; Love, S.; Miners, J. S. Systemic infection exacerbates cerebrovascular dysfunction in A.D.. Brain. 2021, 144, 1869–1883. [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V. H. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009, 73, 768–774. [CrossRef]
- Walker, K. A.; Ficek, B. N.; Westbrook, R. Understanding the Role of Systemic Inflammation in A.D. ACS Chem. Neurosci. 2019, 10, 3340–3342. [CrossRef]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R. E. The Impact of Systemic Inflammation on A.D. Pathology. Front. Immunol. 2022, 12, 796867. [CrossRef]
- Sangha, P. S.; Thakur, M.; Akhtar, Z.; Ramani, S.; Gyamfi, R. S. The Link Between Rheumatoid Arthritis and Dementia: A Review. Cureus. 2020, 12, e7855. [CrossRef]
- Trzeciak, P.; Herbet, M.; Dudka, J. Common Factors of A.D. and Rheumatoid Arthritis-Pathomechanism and Treatment. Molecules. 2021, 26, 6038. [CrossRef]
- Joh, H. K.; Kwon, H.; Son, K. Y.; Yun, J. M.; Cho, S. H.; Han, K.; Park, J. H.; Cho, B. Allergic Diseases and Risk of Incident Dementia and A.D. Ann. Neurol. 2023, 93, 384–397. [CrossRef]
- Bożek, A.; Bednarski, P.; Jarzab, J. Allergic rhinitis, bronchial asthma and other allergies in patients with A.D. Postepy Dermatologii i Alergologii, 2016, 33, 353–358. [CrossRef]
- Voisin, T.; Bouvier, A.; Chiu, I. M. Neuro-immune interactions in allergic diseases: novel targets for therapeutics. Internat. Immunol. 2017, 29, 247–261. [CrossRef]
- Kabata, H.; Artis, D. Neuro-immune crosstalk and allergic inflammation. J. Clin. Invest. 2019, 129, 1475–1482. [CrossRef]
- Mirotti, L.; Castro, J.; Costa-Pinto, F. A.; Russo, M. Neural pathways in allergic inflammation. Journal of Allergy, 2010, 2010, 491928. [CrossRef]
- Kim, J.; Ha, W. S.; Park, S. H.; Han, K.; Baek, M. S. Association between migraine and A.D.: a nationwide cohort study. Frontiers in Aging Neuroscience. 2023, 15, 1196185. [CrossRef]
- Hurh, K.; Jeong, S. H.; Kim, S. H.; Jang, S. Y.; Park, E. C.; Jang, S. I. Increased risk of all-cause, Alzheimer's, and vascular dementia in adults with migraine in Korea: a population-based cohort study. Journal of Headache and Pain. 2022, 23, 108. [CrossRef]
- Morton, R. E.; St John, P. D.; Tyas, S. L. Migraine and the risk of all-cause dementia, A.D.; and vascular dementia: A prospective cohort study in community-dwelling older adults. International Journal of Geriatric Psychiatry, 2019, 34, 1667–1676. [CrossRef]
- Yang, F. C.; Lin, T. Y.; Chen, H. J.; Lee, J. T.; Lin, C. C.; Kao, C. H. Increased Risk of Dementia in Patients with Tension-Type Headache: A Nationwide Retrospective Population-Based Cohort Study. PloS one. 2016, 11, e0156097. [CrossRef]
- Biscetti, L.; Cresta, E.; Cupini, L. M.; Calabresi, P.; Sarchielli, P. The putative role of neuroinflammation in the complex pathophysiology of migraine: From bench to bedside. Neurobiology of Disease, 2023, 180, 106072. [CrossRef]
- Kursun, O.; Yemisci, M.; van den Maagdenberg, A. M. J. M.; Karatas, H. Migraine and neuroinflammation: the inflammasome perspective. Journal of Headache and Pain. 2021, 22, 55. [CrossRef]
- Bornier, N.; Mulliez, A.; Chenaf, C.; Elyn, A.; Teixeira, S.; Authier, N.; Bertin, C.; Kerckhove, N. Chronic pain is a risk factor for incident A.D.: A nationwide propensity-matched cohort using administrative data. Frontiers in Aging Neuroscience. 2023, 15, 1193108. [CrossRef]
- Innes, K. E.; Sambamoorthi, U. The Potential Contribution of Chronic Pain and Common Chronic Pain Conditions to Subsequent Cognitive Decline, New Onset Cognitive Impairment, and Incident Dementia: A Systematic Review and Conceptual Model for Future Research. J. Alzheimers Dis. 2020, 78, 1177–1195. [CrossRef]
- Cao, S.; Fisher, D. W.; Yu, T.; Dong, H. The link between chronic pain and A.D. J. Inflamm. 2019, 16, 204. [CrossRef]
- Vergne-Salle, P.; Bertin, P. Chronic pain and neuroinflammation. Joint Bone Spine. 2021, 88, 105222. [CrossRef]
- Ji, R. R.; Xu, Z. Z.; Gao, Y. J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug. Disc. 2014, 13, 533–548. [CrossRef]
- Gottlieb S. Head injury doubles the risk of A.D. Brit. Med. J. 2000, 321, 1100.
- Plassman, B. L.; Havlik, R. J.; Steffens, D. C.; Helms, M. J.; Newman, T. N.; Drosdick, D.; Phillips, C.; Gau, B. A.; Welsh-Bohmer, K. A.; Burke, J. R.; Guralnik, J. M.; Breitner, J. C. Documented head injury in early adulthood and risk of A.D. and other dementias. Neurology. 2000, 55, 1158–1166. [CrossRef]
- Schimmel, S. J.; Acosta, S.; Lozano, D. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circulation. 2017, 3, 135–142. [CrossRef]
- Simon, D. W.; McGeachy, M. J.; Bayır, H.; Clark, R. S.; Loane, D. J.; Kochanek, P. M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chinese Journal of Traumatology. 2018, 21, 137–151. [CrossRef]
- Zheng, R. Z.; Lee, K. Y.; Qi, Z. X.; Wang, Z.; Xu, Z. Y.; Wu, X. H.; Mao, Y. Neuroinflammation Following Traumatic Brain Injury: Take It Seriously or Not. Front. Immunol. 2022, 13, 855701. [CrossRef]
- Mehr, J. B.; Bennett, E. R.; Price, J. L.; de Souza, N. L.; Buckman, J. F.; Wilde, E. A.; Tate, D. F.; Marshall, A. D.; Dams-O'Connor, K.; Esopenko, C. Intimate partner violence, substance use, and health comorbidities among women: A narrative review. Frontiers in Psychology. 2023, 13, 1028375. [CrossRef]
- Roberts, G. W.; Whitwell, H. L.; Acland, P. R.; Bruton, C. J. Dementia in a punch-drunk wife. Lancet. 1990, 335, 918–919. [CrossRef]
- Newton, T. L.; Fernandez-Botran, R.; Miller, J. J.; Lorenz, D. J.; Burns, V. E.; Fleming, K. N. Markers of inflammation in midlife women with intimate partner violence histories. Journal of Women's Health. 2011, 20, 1871–1880. [CrossRef]
- Madison, A. A.; Wilson, S. J.; Shrout, M. R.; Malarkey, W. B.; Kiecolt-Glaser, J. K. Intimate Partner Violence and Inflammaging: Conflict Tactics Predict Inflammation Among Middle-Aged and Older Adults. Psychosomatic medicine. 2023, 10.1097/PSY.0000000000001179; [CrossRef]
- Byers, A. L.; Yaffe, K. Depression and risk of developing dementia. Nat. Rev. Neurol. 2011, 7, 323–331; [CrossRef]
- Gatz, J. L.; Tyas, S. L.; St John, P.; Montgomery, P. Do depressive symptoms predict A.D. and dementia? Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 2005, 60, 744–747. [CrossRef]
- Chen, R.; Hu, Z.; Wei, L.; Qin, X.; McCracken, C.; Copeland, J. R. Severity of depression and risk for subsequent dementia: cohort studies in China and the UK. The British Journal of Psychiatry. 2008, 193, 373–377. [CrossRef]
- Byers, A. L.; Covinsky, K. E.; Barnes, D. E.; Yaffe, K. Dysthymia and depression increase risk of dementia and mortality among older veterans. Amer. J. Geriatric Psych. 2012, 20, 664–672. [CrossRef]
- Wilson, R. S.; Barnes, L. L.; Mendes de Leon, C. F.; Aggarwal, N. T.; Schneider, J. S.; Bach, J.; Pilat, J.; Beckett, L. A.; Arnold, S. E.; Evans, D. A.; Bennett, D. A. Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology. 2002, 59, 364–370. [CrossRef]
- Fuhrer, R.; Dufouil, C.; Dartigues, J. F.; PAQUID Study. Exploring sex differences in the relationship between depressive symptoms and dementia incidence: prospective results from the PAQUID Study. J. Amer. Geriat. Soc. 2003, 51, 1055–1063. [CrossRef]
- Geerlings, M. I.; Schmand, B.; Braam, A. W.; Jonker, C.; Bouter, L. M.; van Tilburg, W. Depressive symptoms and risk of A.D. in more highly educated older people. J. Amer. Geriat. Soc. 2000, 48, 1092–1097. [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; Camus, V. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [CrossRef]
- Hassamal S. Chronic stress, neuroinflammation, and depression: an overview of pathophysiological mechanisms and emerging anti-inflammatories. Frontiers in Psychiatry. 2023, 14, 1130989. [CrossRef]
- Jeon, S. W.; Kim, Y. K. The role of neuroinflammation and neurovascular dysfunction in major depressive disorder. J. Inflamm. Res. 2018, 11, 179–192. [CrossRef]
- Becker, E.; Orellana Rios, C. L.; Lahmann, C.; Rücker, G.; Bauer, J.; & Boeker, M. Anxiety as a risk factor of A.D. and vascular dementia. British Journal of Psychiatry. 2018, 213, 654–660. [CrossRef]
- Santabárbara, J.; Lipnicki, D. M.; Olaya, B.; Villagrasa, B.; Bueno-Notivol, J.; Nuez, L.; López-Antón, R.; Gracia-García, P. Does Anxiety Increase the Risk of All-Cause Dementia? An Updated Meta-Analysis of Prospective Cohort Studies. J. Clin. Med. 2020, 9, 1791. [CrossRef]
- Won, E.; Kim, Y. K. Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders. Internat. J. Mol. Sci. 2020, 21, 6546. [CrossRef]
- Zheng, Z. H.; Tu, J. L.; Li, X. H.; Hua, Q.; Liu, W. Z.; Liu, Y.; Pan, B. X.; Hu, P.; Zhang, W. H. Neuroinflammation induces anxiety- and depressive-like behavior by modulating neuronal plasticity in the basolateral amygdala. Brain, Behav. Immun. 2021, 91, 505–518. [CrossRef]
- Guo, B.; Zhang, M.; Hao, W.; Wang, Y.; Zhang, T.; Liu, C. Neuroinflammation mechanisms of neuromodulation therapies for anxiety and depression. Translational Psychiatry. 2023, 13, 5. [CrossRef]
- Minakawa, E. N.; Wada, K.; Nagai, Y. Sleep Disturbance as a Potential Modifiable Risk Factor for Alzheimer's Disease. Internat. J. Mol. Sci. 2019, 20, 803. [CrossRef]
- Kang, J. E.; Lim, M. M.; Bateman, R. J.; Lee, J. J.; Smyth, L. P.; Cirrito, J. R.; Fujiki, N.; Nishino, S.; & Holtzman, D. M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009, 326, 1005–1007. [CrossRef]
- Bubu, O. M.; Brannick, M.; Mortimer, J.; Umasabor-Bubu, O.; Sebastião, Y. V.; Wen, Y.; Schwartz, S.; Borenstein, A. R.; Wu, Y.; Morgan, D.; Anderson, W. M. Sleep, Cognitive impairment, and Alzheimer's disease: A Systematic Review and Meta-Analysis. Sleep. 2017, 40, 10.1093/sleep/zsw032. [CrossRef]
- Sadeghmousavi, S.; Eskian, M.; Rahmani, F.; Rezaei, N. The effect of insomnia on development of A.D. J. Neuroinflammation. 2020, 17, 289:. [CrossRef]
- Shamim, S.A.; Warriach, Z.I.; Tariq, M.A.; Rana, K.F.; Malik, B.H. Insomnia: Risk Factor for Neurodegenerative Diseases. Cureus. 2019, 11, e6004;. [CrossRef]
- Johar, H.; Kawan, R.; Emeny, R.T.; Ladwig, K.H. Impaired Sleep Predicts Cognitive Decline in Old People: Findings from the Prospective KORA Age Study. Sleep. 2016, 39, 217-26;. [CrossRef]
- Benito-León, J.; Bermejo-Pareja, F.; Vega, S.; Louis, E.D. Total daily sleep duration and the risk of dementia: a prospective population-based study. Eur. J. Neurol. 2009, 16, 990-7;. [CrossRef]
- Cricco, M.; Simonsick, E.M.; Foley, D.J. The impact of insomnia on cognitive functioning in older adults. J. Am. Geriat. Soc. 2001, 49, 1185-9;. [CrossRef]
- Zhu, B.; Dong, Y.; Xu, Z.; Gompf, H. S.; Ward, S. A.; Xue, Z.; Miao, C.; Zhang, Y.; Chamberlin, N. L.; Xie, Z. Sleep disturbance induces neuroinflammation and impairment of learning and memory. Neurobiol. Dis. 2012, 48, 348–355. [CrossRef]
- Zielinski, M. R.; Gibbons, A. J. Neuroinflammation, Sleep, and Circadian Rhythms. Frontiers in cellular and infection microbiology, 2022, 12, 853096. [CrossRef]
- Herrero Babiloni, A.; Baril, A.-A.; Charlebois-Plante, C.; Jodoin, M.; Sanchez, E.; De Baets, L.; Arbour, C.; Lavigne, G.J.; Gosselin, N.; De Beaumont, L. The Putative Role of Neuroinflammation in the Interaction between Traumatic Brain Injuries, Sleep, Pain and Other Neuropsychiatric Outcomes: A State-of-the-Art Review. J. Clin. Med. 2023, 12, 1793. [CrossRef]
- Rehm, J.; Hasan, O. S. M.; Black, S. E.; Shield, K. D.; Schwarzinger, M. Alcohol use and dementia: a systematic scoping review. Alzheimer's Research & Therapy. 2019, 11, 1. [CrossRef]
- Evert, D. L.; Oscar-Berman, M. Alcohol-Related Cognitive Impairments: An Overview of How Alcoholism May Affect the Workings of the Brain. Alcohol Health and Research World, 1995, 19, 89–96.
- Smith, D. M.; Atkinson, R. M. Alcoholism and dementia. The International Journal of the Addictions, 1995, 30, 1843–1869. [CrossRef]
- Tyas S. L. Are tobacco and alcohol use related to Alzheimer's disease? A critical assessment of the evidence and its implications. Addiction Biology. 1996, 1, 237–254. [CrossRef]
- Tyas S. L. Alcohol use and the risk of developing A.D. Alcohol Research & Health: Journal of the National Institute on Alcohol Abuse and Alcoholism. 2001, 25, 299–306.
- Jeon, K. H.; Han, K.; Jeong, S. M.; Park, J.; Yoo, J. E.; Yoo, J.; Lee, J.; Kim, S.; Shin, D. W. Changes in Alcohol Consumption and Risk of Dementia in a Nationwide Cohort in South Korea. JAMA Network Open. 2023, 6, e2254771. [CrossRef]
- Orio, L.; Alen, F.; Pavón, F. J.; Serrano, A.; García-Bueno, B. Oleoylethanolamide, Neuroinflammation, and Alcohol Abuse. Front. Mol. Neurosci. 2019, 11, 490. [CrossRef]
- Lowe, P. P.; Morel, C.; Ambade, A.; Iracheta-Vellve, A.; Kwiatkowski, E.; Satishchandran, A.; Furi, I.; Cho, Y.; Gyongyosi, B.; Catalano, D.; Lefebvre, E.; Fischer, L.; Seyedkazemi, S.; Schafer, D. P.; Szabo, G. Chronic alcohol-induced neuroinflammation involves CCR2/5-dependent peripheral macrophage infiltration and microglia alterations. J. Neuroinflamm. 2020, 17, 296. [CrossRef]
- Shafighi, K.; Villeneuve, S.; Rosa Neto, P.; Badhwar, A.; Poirier, J.; Sharma, V.; Medina, Y. I.; Silveira, P. P.; Dube, L.; Glahn, D.; Bzdok, D. Social isolation is linked to classical risk factors of A.D.-related dementias. PloS one. 2023, 18, e0280471. [CrossRef]
- Shen, C.; Rolls, E.; Cheng, W.; Kang, J.; Dong, G.; Xie, C.; Zhao, X. M.; Sahakian, B.; Feng, J. Associations of Social Isolation and Loneliness with Later Dementia. Neurology. 2022, 10.1212/WNL.0000000000200583. [CrossRef]
- Al Omran, A. J.; Shao, A. S.; Watanabe, S.; Zhang, Z.; Zhang, J.; Xue, C.; Watanabe, J.; Davies, D. L.; Shao, X. M.; & Liang, J. Social Isolation Induces Neuroinflammation And Microglia Overactivation, While Dihydromyricetin Prevents And Improves Them. Research Square. 2021, rs.3.rs-923871. [CrossRef]
- Ayilara, G. O.; Owoyele, B. V. Neuroinflammation and microglial expression in brains of social-isolation rearing model of schizophrenia. IBRO Neuroscience Reports. 2023, 15, 31–41. [CrossRef]
- Vu, A. P.; Lam, D.; Denney, C.; Lee, K. V.; Plemel, J. R.; Jackson, J. Social isolation produces a sex- and brain region-specific alteration of microglia state. Eur. J. Neurosci. 2023, 57, 1481–1497. [CrossRef]
- Wostyn, P.; Audenaert, K.; De Deyn, P.P. A.D. and glaucoma: Is there a causal relationship? Brit. J. Ophthalmol. 2009, 93, 1557-1559.
- Cesareo, M.; Martucci, A.; Ciuffoletti, E.; Mancino, R.; Cerulli, A.; Sorge, R. P.; Martorana, A.; Sancesario, G.; Nucci, C. Association Between Alzheimer's Disease and Glaucoma: A Study Based on Heidelberg Retinal Tomography and Frequency Doubling Technology Perimetry. Front. Neurosci. 2015, 9, 479;. [CrossRef]
- Crump, C.; Sundquist, J.; Sieh, W.; Sundquist, K. Risk of Alzheimer's Disease and Related Dementias in Persons With Glaucoma: A National Cohort Study. Ophthalmology. 2023, S0161-6420(23)00756-X; [CrossRef]
- Sugiyama, T. Glaucoma and A.D.: Their clinical similarity and future therapeutic strategies for glaucoma. World J. Ophthalmol. 2014, 4, 47-51:. [CrossRef]
- Mancino, R.; Martucci, A.; Cesareo, M.; Giannini, C.; Corasaniti, M. T.; Bagetta, G.; Nucci, C. Glaucoma and Alzheimer Disease: One Age-Related Neurodegenerative Disease of the Brain. Curr. Neuropharmacol. 2018, 16, 971-977;. [CrossRef]
- Williams, P. A.; Marsh-Armstrong, N.; Howell, G. R.; & Lasker/IRRF Initiative on Astrocytes and Glaucomatous Neurodegeneration Participants. Neuroinflammation in glaucoma: A new opportunity. Experimental Eye Research, 2017, 157, 20–27. [CrossRef]
- Rolle, T.; Ponzetto, A.; Malinverni, L. The Role of Neuroinflammation in Glaucoma: An Update on Molecular Mechanisms and New Therapeutic Options. Front. Neurol. 2021, 11, 612422. [CrossRef]
- Soto, I.; Howell, G. R. The complex role of neuroinflammation in glaucoma. Cold Spring Harbor Perspectives in Medicine, 2014, 4, a017269. [CrossRef]
- Rutigliani, C.; Tribble, J. R.; Hagström, A.; Lardner, E.; Jóhannesson, G.; Stålhammar, G.; Williams, P. A. Widespread retina and optic nerve neuroinflammation in enucleated eyes from glaucoma patients. Acta Neuropathologica Communications, 2022, 10, 118. [CrossRef]
- Lin, F. R.; Pike, J. R.; Albert, M. S.; Arnold, M.; Burgard, S.; Chisolm, T.; Couper, D.; Deal, J. A.; Goman, A. M.; Glynn, N. W.; Gmelin, T.; Gravens-Mueller, L.; Hayden, K. M.; Huang, A. R.; Knopman, D.; Mitchell, C. M.; Mosley, T.; Pankow, J. S.; Reed, N. S.; Sanchez, V.; … ACHIEVE Collaborative Research Group. Hearing intervention versus health education control to reduce cognitive decline in older adults with hearing loss in the USA (ACHIEVE): a multicentre, randomised controlled trial. Lancet. 2023, 402, 786–797. [CrossRef]
- Jiang, F.; Mishra, S. R.; Shrestha, N.; Ozaki, A.; Virani, S. S.; Bright, T.; Kuper, H.; Zhou, C.; Zhu, D. Association between hearing aid use and all-cause and cause-specific dementia: an analysis of the UK Biobank cohort. The Lancet. Public Health. 2023, 8, e329–e338. [CrossRef]
- Seicol, B. J.; Lin, S.; Xie, R. Age-Related Hearing Loss Is Accompanied by Chronic Inflammation in the Cochlea and the Cochlear Nucleus. Frontiers in Aging Neuroscience. 2022, 14, 846804; [CrossRef]
- Frye, M. D.; Ryan, A. F.; Kurabi, A. Inflammation associated with noise-induced hearing loss. The Journal of the Acoustical Society of America. 2019, 146, 4020. [CrossRef]
- Huang, L.; Zhang, Y.; Wang, Y.; Lan, Y. Relationship Between Chronic Noise Exposure, Cognitive Impairment, and Degenerative Dementia: Update on the Experimental and Epidemiological Evidence and Prospects for Further Research. J Alzheimers Dis. 2021, 79, 1409-1427;. [CrossRef]
- Weuve, J.; D'Souza, J.; Beck, T, Evans.; DA, Kaufman, J.D.; Rajan, K.B.; de Leon, C.F.M.; Adar, S.D. Long-term community noise exposure in relation to dementia, cognition, and cognitive decline in older adults. Alzheimers Dement. 2021, 17, 525-533:. [CrossRef]
- Cantuaria, M.L.; Waldorff, F.B.; Wermuth, L.; Pedersen, E.R.; Poulsen, A.H.; Thacher, J.D.; Raaschou-Nielsen, O.; Ketze, M, Khan.; J, Valencia V.H,.; Schmid, J.H.; Sørensen, M. Residential exposure to transportation noise in Denmark and incidence of dementia: national cohort study. BMJ. 2021, 374, n1954;. [CrossRef]
- Wang, W.; Zhang, L.S.; Zinsmaier, A.K.; Patterson, G.; Leptich, E.J.; Shoemaker, S.L.; Yatskievych, T.A.; Gibboni, R.; Pace, E.; Luo, H.; Zhang, J.; Yang, S.; Bao, S. Neuroinflammation mediates noise-induced synaptic imbalance and tinnitus in rodent models. PLoS Biol. 2019, 17, e3000307;. [CrossRef]
- Cui, B.; Li, K.; Gai, Z.; She, X.; Zhang, N.; Xu, C.; Chen, X.; An, G.; Ma, Q.; Wang, R. Chronic Noise Exposure Acts Cumulatively to Exacerbate A.D.-Like Amyloid-β Pathology and Neuroinflammation in the Rat Hippocampus. Sci. Rep. 2015, 5, 12943;. [CrossRef]
- Peters, R.; Ee, N.; Peters, J.; Booth, A.; Mudway, I.; Anstey, K. J. Air Pollution and Dementia: A Systematic Review. J. Alzheimers Dis. 2019, 70, S145–S163; [CrossRef]
- Peters A. Ambient air pollution and A.D.: The role of the composition of fine particles. Proc. Natl. Acad. Sci. 2023, 120, e2220028120. [CrossRef]
- Shi, L. Incident dementia and long-term exposure to constituents of fine particle air pollution: A national cohort study in the United States. Proc. Natl. Acad. Sci. U.S.A. 2022, 120, e2211282119.
- Shi, L.; Steenland, K.; Li, H.; Liu, P.; Zhang, Y.; Lyles, R. H.; Requia, W. J.; Ilango, S. D.; Chang, H. H.; Wingo, T.; Weber, R. J.; Schwartz, J. A national cohort study (2000-2018) of long-term air pollution exposure and incident dementia in older adults in the United States. Nat. Comm. 2021, 12, 6754. [CrossRef]
- Campbell, A.; Oldham, M.; Becaria, A.; Bondy, S. C.; Meacher, D.; Sioutas, C.; Misra, C.; Mendez, L. B.; Kleinman, M. Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology. 2005, 26, 133–140. [CrossRef]
- Tin-Tin-Win-Shwe, Mitsushima, D.; Yamamoto, S.; Fukushima, A.; Funabashi, T.; Kobayashi, T.; Fujimaki, H. Changes in neurotransmitter levels and proinflammatory cytokine mRNA expressions in the mice olfactory bulb following nanoparticle exposure. Toxicology and Applied Pharmacology. 2008, 226, 192–198. [CrossRef]
- Block, M. L.; Calderón-Garcidueñas, L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends in Neurosciences. 2009, 32, 506–516. [CrossRef]
- Bongioanni, P.; Del Carratore, R.; Corbianco, S.; Diana, A.; Cavallini, G.; Masciandaro, S. M.; Dini, M.; Buizza, R. Climate change and neurodegenerative diseases. Environmental Research. 2021, 201, 111511. [CrossRef]
- Zuelsdorff, M.; Limaye, V. S. A Framework for Assessing the Effects of Climate Change on Dementia Risk and Burden. The Gerontologist. 2023, gnad082. [CrossRef]
- Stella, A. B.; Galmonte, A.; Deodato, M.; Ozturk, S.; Reis, J.; & Manganotti, P. Climate Change and Global Warming: Are Individuals with Dementia - Including A.D. - At a Higher Risk? Curr. Alz. Res. 2023, 20, 209–212. [CrossRef]
- Ruszkiewicz, J. A.; Tinkov, A. A.; Skalny, A. V.; Siokas, V.; Dardiotis, E.; Tsatsakis, A.; Bowman, A. B.; da Rocha, J. B. T.; Aschner, M. Brain diseases in changing climate. Environmental Research. 2019, 177, 108637. [CrossRef]
- Gong, J.; Part, C.; Hajat, S. Current and future burdens of heat-related dementia hospital admissions in England. Environment International. 2022, 159, 107027. [CrossRef]
- O'Donnell S. The neurobiology of climate change. Die Naturwissenschaften. 2018, 105, 11: . [CrossRef]
- Habibi, L.; Perry, G.; Mahmoudi, M. Global warming and neurodegenerative disorders: speculations on their linkage. BioImpacts. 2014, 4, 167–170. [CrossRef]
- Lee, W.; Moon, M.; Kim, H. G.; Lee, T. H.; Oh, M. S. Heat stress-induced memory impairment is associated with neuroinflammation in mice. J. Neuroinflamm. 2015, 12, 102. [CrossRef]
- Sharp, E.S.; Gatz, M. Relationship between education and dementia: an updated systematic review. Alzheimer Dis. Assoc. Disord. 2011, 25, 289-304;. [CrossRef]
- Klimova, B.; Valis, M.; Kuca, K. Bilingualism as a strategy to delay the onset of Alzheimer's disease. Clin. Interv. Aging. 2017, 12, 1731-1737;. [CrossRef]
- Craik, F.I.; Bialystok, E.; Freedman, M. Delaying the onset of Alzheimer disease: bilingualism as a form of cognitive reserve. Neurology. 2010, 75, 1726-1729;. [CrossRef]
- Liu, H.; Wu, L. Lifelong Bilingualism Functions as an Alternative Intervention for Cognitive Reserve against Alzheimer's Disease. Front. Psychiatry. 2021, 12, 696015;. [CrossRef]
- Steinvil, A.; Shirom, A.; Melamed, S.; Toker, S.; Justo, D.; Saar, N, Shapira, I.; Berliner, S.; Rogowski, O. Relation of educational level to inflammation-sensitive biomarker level. Am. J. Cardiol. 2008, 102, 1034-9;. [CrossRef]
- Maurel, M.; Castagné, R.; Berger, E.; Bochud, M.; Chadeau-Hyam.; M, Fraga, S.; Gandini, M.; Hutri-Kähönen, N.; Jalkanen, S.; Kivimäki, M.; Marmot, M.; McCrory, C.; Preisig, M.; Raitakari, O.; Ricceri, F.; Salmi, M.; Steptoe, A.; Vineis, P.; Delpierre, C.; Kelly-Irving, M. Patterning of educational attainment across inflammatory markers: Findings from a multi-cohort study. Brain. Behav. Immun. 2020, 90, 303-310;. [CrossRef]
- Almeida, R.P.; Schultz, S.A.; Austin, B.P.; Boots, E.A.; Dowling, N.; Gleason, C.E.; Bendlin, B.B.; Sager, M.A.; Hermann, B.P.; Zetterberg, H.; Carlsson, C.M.; Johnson, S.C.; Asthana, S.; Okonkwo, O.C. Effect of Cognitive Reserve on Age-Related Changes in Cerebrospinal Fluid Biomarkers of Alzheimer Disease. JAMA Neurol. 2015, 72, 699-706;. [CrossRef]
Figure 1.
Thirty risk factors for Alzheimer’s disease are unified by their common ability to elicit neuroinflammation, manifesting as microglial activation and pro-inflammatory cytokine release, ultimately contributing to the pathogenesis of the disease.
Figure 1.
Thirty risk factors for Alzheimer’s disease are unified by their common ability to elicit neuroinflammation, manifesting as microglial activation and pro-inflammatory cytokine release, ultimately contributing to the pathogenesis of the disease.
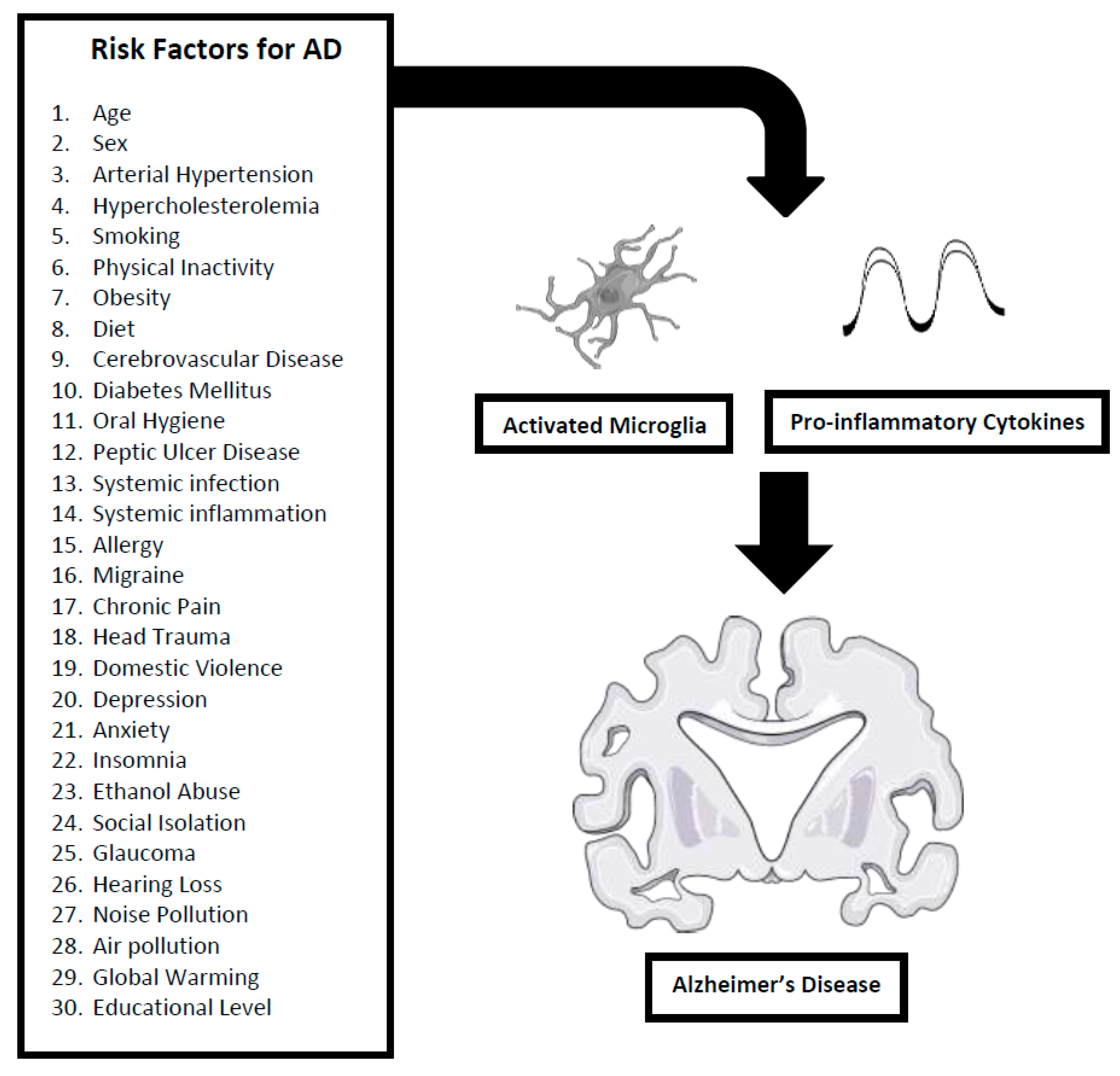
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



