
Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Microbial community succession in raw milk determines its quality and storage period. In this study, carbon dioxide (CO2) at 2000 ppm was used to treat raw milk to investigate the mechanism of extending the shelf life of raw milk by CO2 treatment from the viewpoint of microbial colonies and metabolites. The results showed that the shelf life of CO2-treated raw milk was extended to 16 days at 4 ℃, while that of the control raw milk was only 6 days. Microbiomics analysis identified 221 Amplicon Sequence Variants (ASVs) in raw milk, and the alpha diversity of microbial communities increased (P < 0.05) with the extension of storage time. Among them, Pseudomonas, Actinobacteria and Serratia were the major microbial genus responsible for the deterioration of raw milk, with a percentage of 85.7%. A combined metagenomics and metabolomics analysis revealed that microorganisms altered the levels of metabolites, such as pyruvic acid, glutamic acid, 5'- cmp, arginine, 2-propenoic acid, and phenylalanine in the raw milk through metabolic activities, such as ABC transporters, pyrimidine metabolism, arginine and proline metabolism and phenylalanine metabolism, and reduced the shelf life of the raw milk. CO2 treatment prolonged the shelf life of the raw milk by inhibiting the abundance of Gram-negative aerobic bacteria, such as Acinetobacter guillouiae, Pseudomonas fluorescens, Serratia liquefaciens and Pseudomonas simiae.
Keywords:
Raw milk
; CO2 treatment
; Microbiomics
; Metabolomics
1. Introduction
Raw milk is commonly produced intensively due to its collection from dispersed farms and subsequent transportation to processing plants for uniform treatment. Throughout the stages of collection, transportation, and waiting for processing, raw milk undergoes a significant duration (several hours), imposing heightened demands on its quality [1,2,3]. Microorganisms play a pivotal role in influencing the quality of raw milk. Given its nutrient-rich composition, raw milk provides an optimal environment and nutrient matrix that facilitate rapid microbial reproduction [4]. Consequently, this can result in undesirable outcomes, such as off-flavors, sourness, discoloration, and a loss of the characteristic freshness associated with milk [1]. Additionally, microorganisms break down and consume essential nutrients present in milk, including fats, proteins and vitamins. This process may lead to a reduction in the nutrient content of raw milk, thereby impacting the nutritional value of the final dairy product [5]. To ensure the provision of high-quality and safe dairy products while prioritizing consumer health and satisfaction, it is imperative to implement effective control measures aimed at mitigating or eliminating microbial hazards inherent in raw milk [2,4].
The sources of microorganisms in raw milk are much more varied; microorganisms can be present in the skin of the cow's udder and teats, and are passed on to the milk when skin microorganisms come into contact with the milk during the milking process [4,5]. In addition, the environment, insects and rodents, milking equipment, and collection devices are all potential sources of microorganisms in raw milk [6]. Notably, these sources of microbial contamination highlight the need to inhibit microbial colonisation through effective technological means [7]. Researchers have explored the use of high pressure to inactivate microorganisms in raw milk while preserving its nutritional quality. However, due to the complexity of high-pressure equipment and limited treatment efficiency, it is not suitable for large-scale implementation [8]. Non-thermal plasma treatment offers an alternative approach to generating reactive oxygen and nitrogen species that can penetrate microorganisms and inhibit or kill them. However, these oxides can also cause oxidation of raw milk, leading to a decline in its quality as a complex emulsion system [9]. Conventional sterilization methods may destabilize raw milk by affecting its composition and structure. To overcome these challenges, alternative technologies, such as gas conditioning or non-thermal treatments, are being investigated to minimize adverse effects on raw milk while preserving its nutritional value and sensory properties [10].
CO2 has been used for fresh milk storage and preservation because it does not affect the nutritional content or flavor compared to conventional heat treatments. Also, CO2 is a natural compound that does not leave harmful residues in the product or the environment [11,12]. CO2 has been used for food preservation. When CO2 is dissolved in milk, it forms carbonic acid, which lowers the pH of the milk. Many bacteria, including those that cause spoilage, cannot survive or grow effectively in this more acidic environment [13,14]. In addition, by replacing oxygen with CO2 in the storage container, packaging or food systems (a technique known as Modified Atmosphere Packaging), the growth of aerobic bacteria and fungi is inhibited. These microorganisms require oxygen to grow, thus removing or reducing oxygen levels can help extend the shelf life of raw milk [15].
In view of the complexity of the raw milk system and microbial community succession, the metabolic pattern of CO2 treatment on raw milk and microorganisms was analyzed by using macro-genome and metabolome, and then the molecular mechanism by which CO2 prolongs the shelf life of raw milk was clarified. The results of the study provide a theoretical basis for the application of CO2 in raw milk.
2. Materials and methods
2.1. Sample collection
Fresh raw milk (the somatic cells < 2×105/mL, total bacteria count < 103 CFU/mL)was collected aseptically from milk tanks at Helan Mountain Dairy Farm, Ningxia, Yinchuan, China in June 2022, refrigerated at 4 ℃ and transported to the laboratory within one hour. Then, a known mass of solid CO2 (dry ice, food grade) was added to 500 mL PET bottles (Servicebio, Wuhan, China) containing 300 mL of raw milk to achieve a CO2 concentration of 2000 ppm in a sterile room immediately [16]. Raw milk without CO2 treatment was used as a control group. Each group of samples was divided into 100 ml aliquots in sterile sealed PET bottles and stored at 4 ℃. One aliquot was used for analysis each day. The day of sample collection was defined as day 0.
2.2. Bacterial growth studies
The total bacterial count (TBC) was measured by the spread plate method. Briefly, 1 mL sample was taken and serially diluted 10 times with 0.85% sterile saline. Then the 200 uL dilutions were spread onto Plate Count Agar (PCA) and cultured at 37 ℃ for 48 h. Analysis of a sample was terminated when it was spoiled, i.e. the TBC reached a threshold of 6 lg(cfu/mL) [17].
2.3. pH measurement
Raw milk pH was determined using a pH acidimeter (PHS-3C, Shanghai Yidian Scientific Instrument Co. Ltd., Shanghai, China) with a three-point calibration standard prior to testing.
2.4. Metagenomics analysis of raw milk
2.4.1. Raw milk DNA extraction and processing
After centrifugation of raw cow's milk, all microbial DNA in raw cow's milk was extracted by DNeasy PowerFood Microbial DNA Isolation Kit (QIAGEN, Dusseldorf, Germany). DNA integrity was determined by 1.2% agarose gel and gel Doc EZ imager (Bio-Rad, California, USA), and DNA concentration was measured by Qubit 4.0 Fluorescence Quantification Instrument (Invitrogen, Carlsbad, California, US). Qubit™ dsDNA HS Assay Kit (Invitrogen, Carlsbad, California, US) was used to accurately quantify the concentration of the genome and determine the total amount of DNA required for library construction. The DNA was fragmented using Covaris 220 ultrasonic crusher (Woburn, MA., US), concentrated and recovered, and then purified and its concentration determined using Qubit 4.0.
2.4.2. Library construction and sequencing
Library construction was carried out according to the instructions of the library construction kit (ONT, Oxford, UK) for Hieff NGS® high-throughput sequencing platform. Briefly, the purified DNA was subjected to end modification using NEBNext FFPE DNA Repair kit (New England Biolabs, Ipswich, MA, USA), junction ligation and magnetic bead sorting to purify the ligated product, and the purified product was subjected to PCR amplification and enrichment, and the PCR amplification product was purified, and the size of library was detected by 2% agarose gel electrophoresis, and the concentration of library was measured by Qubit 4.0, and the qualified library was used. After the libraries were qualified, the Illumina NovaSeq platform was used to analyze the libraries by end-pair sequencing.
2.4.3. Data analysis
In order to ensure the quality of information, the raw data were filtered, and the clean data were obtained by removing the sequences with junctions and low-quality sequences. After removing the junctions in the reads, global cropping and window quality cropping, and the host contamination, the quality of the sequenced raw data were evaluated by Fastp-0.36, and the validated reads were analyzed statistically after the data filtering.
2.5. Metabolomics analysis of raw milk
2.5.1. Metabolite extraction
Raw cow's milk was vortexed at a low speed, 100 μL of sample was pipetted into a centrifuge tube, then 400 μL of pre-cooled pure methanol solution was added, vortexed and mixed, then sonicated in an ice bath for 20 min, and then allowed to stand at -20℃ for 1 h. Frozen samples were centrifuged at 16,000 × g, 4 ℃ for 20 min, and the supernatant was dried in a high-speed vacuum concentrator centrifuge. The supernatant was dried in a high-speed vacuum concentration centrifuge. The dried sample was raw milk metabolites, and 100 μL of methanol-water solution (1:1, v/v) was added to dissolve the metabolites for spectrometry, and then centrifuged at 20,000 × g, at 4 ℃ for 15 min, and then the supernatant was taken as the sample for analysis.
2.5.2. LC-MS/MS analysis
2.5.2.1. Chromatographic separation
The samples were analyzed in an autosampler at 4°C. The samples were separated on a SHIMADZU-LC30 ultra-high-performance liquid chromatography (UHPLC) system using an ACQUITY UPLC® HSS T3 column (2.1×100 mm, 1.8 µm) (Waters, Milford, MA, USA). The flow rate was 0.3 mL/min at 40 ℃ with an injection volume of 4 μL. The chromatographic mobile phases were A: 0.1% formic acid aqueous solution, and B: acetonitrile. The chromatographic gradient elution program was as follows: from 0 to 2 min, 100% A; from 2 to 6 min, A varied linearly from 100% to 52%; from 6 to 10 min, A varied linearly from 52% to 0%; from 10 to 12 min, A was maintained at 0%; from 12 to 12.1 min, A was maintained at 0%; from 12 to 12.1 min, A was maintained at 0%; and from 12 to 12.1 min, A was maintained at 0%. 0%; from 12 to 12.1min, A varied linearly from 0% to 100%, and from 12.1 to 15min, A was maintained at 100%.
2.5.2.2. Mass spectrometry acquisition
Each sample was collected by electrospray ionization (ESI) in positive (+) and negative (-) modes. The samples were separated by UPLC and analyzed by mass spectrometry using a QE Plus mass spectrometer (Thermo Scientific). The ionization conditions were as follows: Spray Voltage: 3.8 kv (+) and 3.2 kv (-); Capillary Temperature: 320 (±); Spray Voltage: 3.8kv (+) and 3.2kv (-); Capillary Temperature: 320 (±); Sheath Gas: 30 (±); Aux Gas: 5 (±); Probe Heater Temp: 350 (±); S-Lens RF Level: 50.
2.5.3. Data pre-processing
The raw data were subjected to peak alignment, retention time correction and peak area extraction using MSDIAL software. The structural identification of metabolites was performed by exact mass number matching (mass tolerance < 10 ppm) and secondary spectra matching (mass tolerance < 0.01 Da), and the public databases, such as HMDB, MassBank, GNPS, etc., as well as the self-constructed metabolite standard library of Dubai Spectrum (BP-DB), were searched. Ion peaks with > 50% missing values were removed from the extracted data and were not included in the subsequent statistical analysis.
2.6. Statistical analysis
To identify the perturbed biological pathways, the differential metabolite data were subjected to KEGG pathway analysis using KEGG database (http://www.kegg.jp). KEGG enrichment analyses were carried out with the Fisher’s exact test, and an FDR correction was applied to account for multiple testing. Enriched KEGG pathways were considered statistically significant at the P < 0.05 level. All physicochemical data were analysed by one way ANOVA and t-test using SPSS 25 program (SPSS Inc. NY, USA) and significant difference was considered as P < 0.05. Plotting was done using Origin 2023 (Origin Lab., Northampton, USA).
3. Results
3.1. Physicochemical characteristics of raw milk
Total bacterial counts were utilized to assess the storage period of raw milk (Figure 1). The control group was identified as spoilage on day 6, with the TBC greater than 6 lg(cfu/mL). In contrast, the TBC of the CO2-treated group was only 4.03 lg(cfu/mL) on day 6, and did not exceed 6 lg(cfu/mL) until day 16, indicating that the CO2 treatment prolonged the storage time of raw milk by significantly inhibiting the growth of the bacteria. It is noteworthy that the TBC of the control raw milk increased steadily during the first 3 days, and showed a logarithmic increase on day 4, suggesting that day 4 of refrigeration may be a critical time point for changes in the quality of raw milk. Therefore, this study selected samples from days 0, 4 and 6 for metagenomic and metabolomic analysis to explore the mechanism of CO2 prolonging the cold storage period of raw milk. In addition, CO2 at 2000 ppm of significantly reduced the pH of raw milk (P < 0.05).
3.2. Microbial diversity of raw milk
Analysis of the composition of the biome in the raw milk using the Illumina sequencing platform showed that there were 221 ASVs in the raw milk. The control group contained a total of 189 ASVs, 136 of which were unique to that group. Of these, group A0 had 62 ASVs, 27 of which were unique to that group; group A4 had 67 ASVs, 3 of which were unique to that group; group A6 had 176 ASVs, 114 of which were unique to that group; and the CO2-treated group contained a total of 85 ASVs, 5 of which were unique to that group. Of these, the day 4 group had 41 ASVs, 1 of which was unique to that group; and the day 6 group had 48 ASVs, 4 of which were unique to that group (Figure 2A). These findings suggested that CO2 treatment reduced the microbial diversity compared to the control group. The community richness of raw milk microbiota (Simpson diversity index and Chao1 richness estimator) also showed that CO2 treatment reduced raw milk microbial α-diversity (Figure 2B and D). Principal Component Analysis (PCA) showed that the samples from day 0 clustered in the third quadrant. With the extension of refrigeration time, the control group samples continued to move towards the fourth quadrant along the x-axis direction, while the CO2 treated samples continued to move towards the second quadrant along the y-axis direction. There was a significant separation between the two groups of samples, confirming a significant difference in microbial β-diversity between the CO2 treated raw milk and the control group (Figure 2C).
3.3. Microbiological composition of raw milk
The differences in microbial community composition between the two groups were analyzed by nonparametric Kruskal-Wallis and Wilcoxon rank-sum tests, combined with linear discriminant analysis (LDA) and effect size. A total of 100 microbial markers with LDA > 2 and P < 0.05 were identified at different levels. Notably, Figure 2A and 3A showed consistent differences in α- and β-diversity indices, especially in the A6 microbial community, the number of biomarkers was much higher than the D6 group, suggesting that the CO2 treatment inhibited microbial reproduction. The LDA scores for microbial markers were shown in Figure 3B, and the initial microbial communities significantly enriched in raw milk included s_Stenotrophomonas_maltophilia, g_Stenotrophomonas, f_Xanthomonadaceae, s_Lactobacillus_helveticus, g_Lactobacillus_helveticus and g_Lactobacillus. When raw milk spoiled, the microorganisms significantly enriched in group A6 were mainly k_Bacteria, p_Proteobacteria, c_Gammaproteobacteria, f_Pseudomonadaceae, o_Pseudomonas and g_Pseudomonas, suggesting that these may be the key microorganisms responsible for the deterioration of raw milk quality. The significantly enriched in the D6 group treated with CO2 were p_Firmicutes, c_Bacilli,o_Lactobacillales, f_Lactobacillaceae, g_Leuconostoc and s_Leuconostoc_mesenteroides. The sequences were annotated at the genus level for classification, and the results were shown in Figure 3C. Stenotrophomonas accounted for 70.2% of the initial flora of raw milk (A0/D0), and the abundance gradually decreased with the extension of the cold storage time, but still accounted for the highest proportion in groups A4, D4 and D6 (45.7%, 62.1% and 57.9%, respectively). CO2 treatment had a significant effect on the microbial species at the genus level, with Pseudomonas, Actinobacteria and Serratia significantly increasing to 49.2%, 20.9% and 14.6% in the control group at day 6, but only to 0.09%, 3.0% and 0.03% in the D6 group.
3.4. Metabolites
3.4.1. Data quality control
Microorganisms use raw milk as a substrate for reproduction and, in addition to changes in microbial numbers and species, alter the raw milk metabolite system through their own metabolic activities. The study aimed to assess how CO2 treatment affects raw milk metabolite evolution by influencing microbial succession and, consequently, raw milk quality. LC-MS-based untargeted metabolomics identified a total of 1234 metabolites in six batches of raw milk. PCA was initially used as a way to explore the structure of the data and assess its distribution and metabolite variability in PC space. Six raw milk samples and four QC samples were used as the dataset. First, according to Figure 4 A and B, the distributions of the four QC samples in the PCA model overlapped almost completely, and the correlation coefficients were all greater than 0.998, suggesting that mass spectrometry detection was reliable. As shown in Figure 4A, A0 and D0 sample metabolites had good similarity, indicating that carbon dioxide did not affect the quality of raw milk. A4, A6, D4 and D6 were distinguished from the 0-day raw milk group. However, the A4, D4 and D6 metabolites partially overlaped in the whole, indicating that it is not feasible to use PCA unsupervised modeling alone to distinguish the raw milk metabolite groups.
3.4.2. Overview of differences between samples
Orthogonal partial least squares data analysis (OPLS-DA) was further applied to develop discriminative models that could differentiate the metabolites of the control group and CO2-treated group with different storage times. The results of the OPLS-DA models developed showed that R2X = 32.8%, R2Y = 99.7%, and Q2 = 95.2 in the control group and R2X = 35%, R2Y = 98.3%, and Q2 = 89.7% in the CO2-treated group, and the Q2 was greater than 0.5 based on the results of 500 tests of cross-validated alignments (Figure 5) with Q2s of 0.8988 and 0.8735 both being greater than 0.5, indicating that these models are reliable, and different treatments and storage time have significant effects on raw milk metabolites.
3.4.3. Differential metabolites (DMs)
OPLS-DA analysis was used to obtain the variable importance for the projection (VIP) of metabolites to measure the strength of the influence of metabolite expression patterns on the categorical discrimination of each group of samples and the ability to explain, so as to assist in the screening of the marker metabolites (VIP > 1.0 was used as the screening criteria). DMs were indicated by VIP >1 and p < 0.05, as shown in Figure 6 A and C, 402 and 377 DMs were screened in the control and CO2-treated groups, respectively, where red color in the clustering heatmap indicates up-regulation of metabolites and blue color indicates down-regulation.
The structure and function of the metabolites and the differential metabolites in each comparison group were categorized and counted by the Class and Ontology substance classification method of HMDB. As shown in Figure 6B and D, the main DMs in the raw milk were carboxylic acids and derivatives (A-14.32%, D-14.75%), fatty Acyls (A-12.24%, D-7.77%), benzene and substituted derivatives (A-9.11%, D-12.06%), organooxygen compounds (A-5.47%, D-6.7%), and so on. Notably, the Class classification of metabolites in the control and CO2-treated groups showed significant differences, mainly in the increase of 10 benzene and substituted derivatives and the decrease of 18 fatty acyl groups in the CO2-treated group.
3.5. Combined microbiomics and metabolomics analysis
Correlation analysis and elemental screening were performed to investigate the relationship between key microbial community structures and metabolite level parameters. As shown in Figure 7 A and B, some microbes with metabolite changes were identified. Notably, the control microbial community showed a stronger positive correlation with metabolites, indicating that CO2 treatment was effective in reducing the metabolic activities of the raw milk system. The relationship between microbes and metabolites was shown by correlation hierarchical clustering heatmaps (Figure 7 C and D). In the control group, the top 28 microorganisms (Mycoplasmopsis, Aequoribacter, Aeromonas, Carnobacterium, Citrobacter, Cupriavidus, etc.) and metabolites (delta-tridecalactone, cytidine, alpha-aminoorcein, D-glucosamine, 3-hydroxycapric acid, etc.) were correlated. Only five microorganisms, Stenotrophomonas, Anaerobutyricum, Blautia, Leuconostoc and Propionibacterium were correlated with the metabolites in the CO2-treated group compared to the control group.
3.6. Metabolic pathway of microbiomics and metabolomics
Based on Illumina NovaSeq/HiSeq high-throughput sequencing platform, Whole Genome Shotgun (WGS) strategy was used to extract the total DNA of the macro-genome of the obtained colonies, or the cDNA double strand of the macro-transcriptome synthesized by using mRNA as template. The coding regions of prokaryotic microbial and macrogenomic gene sequences were predicted to obtain the corresponding gene sequence files, protein sequence files for protein sequence species and functional annotation. As shown in Figure 8A and Table S1, CO2 treatment significantly reduced metabolic processes, such as metabolism (limonene and pinene degradation, energy metabolism, xenobiotics biodegradation and metabolism, C5-branched dibasic acid metabolism, β-alanine metabolism, monobactam biosynthesis, caprolactam and styrene degradation ), cellular processes (bacterial chemotaxis and biofilm formation - pseudomonas aeruginosa), environmental information processing (ABC transporters), and genetic information processing (sulfur relay system).
The metabolic activities of environmental information processing (ABC transporters), metabolism (biosynthesis of cofactors, pyrimidine metabolism, arginine and proline metabolism, biosynthesis of amino acids, carbon metabolism, phenylalanine metabolism, 2-oxocarboxylic acid metabolism, and pantothenate and coa biosynthesis), and organismal systems (protein digestion and absorption) were significantly stronger in the control group than in the CO2-treated group, and microbial metabolism and cellular processes altered raw material metabolites (Figure 8B and Table S1). As shown in Table 1 of Figure 8C, shared metabolic pathways identified by the macrogenome and metabolome, mainly included Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism, and Organismal Systems. A total of 61 shared metabolic pathways involving 53 DMs were screened. Among them, ABC transporters, pyrimidine metabolism, arginine and proline metabolism, and phenylalanine metabolism metabolic processes had the most significant effects on metabolites, involving metabolites, such as pyruvic acid (C00022), glutamic acid (C00025), 5'-CMP (C00055), arginine (C00062), 2-propenoic acid (C00074), and phenylalanine(C00079).
4. Discussion
The diversity of microbial communities in raw milk is usually considered as one of the key factors affecting milk quality, because udder surfaces, milking instruments, containers and environment are potential sources of microorganisms, which leads to microorganisms being inevitable in raw milk [18]. Milk being a nutrient rich liquid is rich in water, proteins, fats, lactose, etc. which makes it a good medium for microbial growth and multiplication [19]. These microorganisms include bacteria, molds and yeasts, among others. Firstly, it was found that Stenotrophomonas maltophilia, Lactococcus lactis and Chryseobacterium sp. NEB161 were the dominant strains of raw milk, with a high percentage of 81.7%. The cold storage process altered the microbial colony diversity of raw milk, and the dominant strains at 6 days were Acinetobacter guillouiae, Pseudomonas fluorescens, Serratia liquefaciens, Pseudomonas simiae and Leuconostoc mesenteroides, with a percentage of 56.2%. These results also verified that the microbial community is a key factor in the spoilage of raw milk. Therefore, it is necessary to use appropriate techniques to inhibit the growth and multiplication of microorganisms in raw milk. Several researchers have shown that adding CO2 to food reduced the rate of food spoilage and the growth of disease-causing microorganisms [19]. We found that CO2 treatment significantly inhibited the propagation of the dominant colonies Acinetobacter guillouiae, Pseudomonas fluorescens, Serratia liquefaciens and Pseudomonas simiae in the CO2-treated group as compared with the control group. The percentage in the CO2 treated group was only 0.2%. CO2 treatment significantly improved the storage period of raw milk (Figure 1), indicating that CO2 improved the quality of raw milk by suppressing microbial levels and diversity.
Through microbiomics analysis, CO2 was found to prolong the storage period of raw milk by inhibiting microbial reproduction, but the exact mechanism of its antimicrobial effect is still unclear [12]. CO2 is highly soluble in water and lipids, and can form carbonic acid in aqueous solutions, which can lead to changes in pH in the suspension medium, and a decrease in pH inhibits the growth and metabolism of certain microorganisms [20]. CO2 significantly lowered the pH of raw milk (Figure 1). Another view is that oxygen is a necessary electron acceptor in the metabolism of aerobic bacteria and is a specific electron acceptor for parthenogenetic anaerobes. Supplementation of raw milk with CO2 reduced the oxygen content of the system, which in turn inhibited bacterial metabolism and reduced growth rates [21]. CO2 treatment significantly inhibited the levels of Acinetobacter guillouiae, Pseudomonas fluorescens, Serratia liquefaciens and Pseudomonas simiae (Figure 3), which inhibited Gram-negative aerobic bacteria, suggesting that CO2 inhibited the growth and reproduction of aerobic microorganisms and prolonged the shelf life of raw milk by reducing the oxygen content of the system.
Microorganisms in raw milk systems can utilize the nutrients in milk for growth and reproduction while breaking down substances, such as proteins, fats and lactose. For example, the pH of milk decreases when lactose is broken down to lactic acid [22,23]. Proteins are broken down, producing toxic substances, such as indole, sulfur and fecal deodorant, as well as foul odors. To be precise, the growth and reproduction of microorganisms is the metabolic process, which leads to changes in the metabolites of the raw milk system through a variety of metabolic processes, reducing the stability of the raw milk system and even spoilage [23,24]. It is worth noting that microbial effects on metabolites are mainly metabolic, including pyrimidine metabolism, arginine and proline metabolism, phenylalanine metabolism, aminoacyl tRNA biosynthesis, glycine, serine and threonine metabolism, lysine degradation, pantothenate and coenzyme A biosynthesis, and pyruvate metabolism (Table 1).
Pyrimidine metabolism refers to the process of pyrimidine nucleotide synthesis and degradation in the cell and is an important component of nucleic acid synthesis. Through pyrimidine metabolism, cells are able to synthesize and maintain sufficient levels of pyrimidine nucleotides to support DNA and RNA synthesis and the stability of genetic material [25,26]. Microbial reproduction requires replication of genetic material, and reduced levels of metabolites, such as cytidylic acid, orotate and uridine, inhibit microbial growth and reproduction (Table S2). In addition, pyrimidine nucleotide degradation produces uracil, which in turn is converted to β-aminobutyric acid, which is involved in amino acid and energy metabolic pathways. The inhibition of energy production results from reduced levels of orotate [27]. The accumulation of N-carbamoyl-L-aspartate, a key compound in the metabolism of alanine, aspartic acid and glutamic acid, suggested that the processes involved in the synthesis of amino acids and the metabolism of other nitrogen compounds were inhibited.
Arginine and proline metabolism can generate nitric oxide (NO) and polyamine compounds, which play important roles in physiological processes, such as cell signaling, immunomodulation and blood flow regulation [28]. In addition, proline metabolism produces polyamines and cofactors required for post-translational modification of proteins, which play key roles in cell growth and development [29]. Phenylalanine metabolism produces tyrosine, and other important bioactive substances, which play important roles in neurotransmitter, hormone and pigment synthesis. Glycine, serine, and threonine metabolism involves a variety of enzymes and intermediates that have multiple effects on metabolites. For example, glycine metabolism produces alanine, which is involved in glucose and fatty acid metabolic pathways. Serine metabolism produces tryptophan. Threonine metabolism produces threonine and methionine, the compounds that play important roles in methionine metabolism and thionine metabolism [30]. Amino acid metabolic processes are attenuated, but accumulation of betaine occurs in the glycerophospholipid metabolic pathway of glycine, serine, and threonine metabolism, which may be due to enhanced upstream glycerophospholipid metabolic activity. Lysine degradation generates intermediates, such as malondialdehyde, acetaldehyde and aminobutyric acid, which can be further involved in oxidation that can alter the stability of the raw milk system [31]. Reduction in glutarate and N6, N6, N6-trimethyl-L-lysine levels were found in the lysine degradation pathway. Generally speaking, it was found that CO2 treatment significantly inhibited amino acid metabolism and reduced microbial biological activity and acid accumulation. In addition, metabolic processes, such as carbohydrate metabolism, lipid metabolism, cell growth and death, and energy metabolism were also inhibited. Overall, CO2 treatment effectively reduced the metabolism of microorganisms and thus inhibited their growth and reproduction.
5. Conclusions
In this study, it was found that CO2 treatment extended the shelf life of raw milk from 6 days to 16 days. This is valuable for the storage and freshness preservation and quality control of raw milk. Carbon dioxide mainly inhibited Gram-negative aerobic bacteria, such as Acinetobacter guillouiae, Pseudomonas fluorescens, Serratia liquefaciens and Pseudomonas simiae, and their growth and multiplication accelerated ABC transporters, pyrimidine metabolism, amino acid metabolism, carbohydrate and lipid metabolism, and metabolism-produced metabolites can lead to raw milk quality deterioration.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Dun-Hua Liu; Data curation, An-Ran Zheng; Formal analysis, An-Ran Zheng; Funding acquisition, Chao-Kun Wei; Investigation, An-Ran Zheng and Ning-Xia Bu; Methodology, An-Ran Zheng; Project administration, Chao-Kun Wei and Dun-Hua Liu; Resources, Dun-Hua Liu; Software, Jun Liu; Supervision, Chao-Kun Wei; Writing – original draft, An-Ran Zheng and Ning-Xia Bu; Writing – review & editing, Jun Liu.
Funding
This work was supported by the National Natural Science Foundation of Ningxia Province (2021AAC02019, 2021AAC03050), the Key research and development projects in Ningxia Province (2021BEB04049).
Data Availability Statement
The data used to support the findings of this study can be made available by the corresponding author upon request.
Acknowledgments
We are very grateful for the support from the Key research and development projects in Ningxia Province and National Natural Science Foundation of Ningxia Province. We thank Shanghai Bioprofile Technology Company for technical support in metagenomics and metabolomics. Meanwhile, we sincerely thank the anonymous reviewers for their critical comments and suggestions for improving the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ndahetuye, J.B.; Artursson, K.; Bage, R.; Ingabire, A.; Karege, C.; Djangwani, J.; Nyman, A.; Ongol, M.P.; Tukei, M.; Persson, Y. MILK Symposium review: Microbiological quality and safe of milk from farm to milk collection centers in Rwanda. J. Dairy Sci. 2020, 103, 9730–9739. [Google Scholar] [CrossRef] [PubMed]
- Mense, L.; Roessler, S.; Hanusch, R.; Rossberg, C.; Ruediger, M. Bacterial contamination of mechanically extracted breast milk. Am. J. Perinatol. 2014, 31, 293–298. [Google Scholar] [CrossRef]
- Pistocchini, E.; Stella, S.; Belli, P.; Cantafora, A.F.A.; Turini, J.; Zecchini, M.; Crimella, C. Dairy production in periurban area of Niamey: milk quality and microbial contamination. Trop. Anim. Health Prod. 2009, 41, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Meunier-Goddik, L.; Waite-Cusic, J.G. Effect of leaving milk trucks empty and idle for 6 h between raw milk loads. J. Dairy Sci. 2018, 101, 1767–1776. [Google Scholar] [CrossRef]
- Peeler, J.T.; Bunning, V.K. Hazard Assessment of Listeria monocytogenes in the Processing of Bovine Milk. J. Food Prot. 1994, 57, 689–697. [Google Scholar] [CrossRef]
- Sharma, N.; Singh, N.K.; Bhadwal, M.S. Relationship of somatic cell count and mastitis: An Overview. Asian Australas. J. Anim. Sci. 2011, 24, 429–438. [Google Scholar] [CrossRef]
- Liang, L.; Wang, P.; Zhao, X.; He, L.; Qu, T.; Chen, Y. Single-molecule real-time sequencing reveals differences in bacterial diversity in raw milk in different regions and seasons in China. J. Dairy Sci. 2022, 105, 5669–5684. [Google Scholar] [CrossRef]
- Viazis, S.; Farkas, B.E.; Allen, J.C. Effects of high-pressure processing on immunoglobulin A and lysozyme activity in human milk. J. Hum. Lact. 2007, 23, 253–261. [Google Scholar] [CrossRef]
- Sriraksha, M.S.; Ayenampudi, S.B.; Noor, M.; Raghavendra, S.N.; Chakka, A.K. Cold plasma technology: An insight on its disinfection efficiency of various food systems. Food Sci. Technol. Int. 2023, 29, 428–441. [Google Scholar]
- Masotti, F.; Cattaneo, S.; Stuknyte, M.; De Noni, I. Current insights into non-thermal preservation technologies alternative to conventional high-temperature short-time pasteurization of drinking milk. Crit. Rev. Food Sci. Nutr. 2023, 63, 5643–5660. [Google Scholar] [CrossRef]
- Li, P.; Mei, J.; Xie, J. The regulation of carbon dioxide on food microorganisms: A review. Food Res. Int. 2023, 172, 113170. [Google Scholar] [CrossRef]
- Singh, P.; Wani, A.A.; Karim, A.A.; Langowski, H. The use of carbon dioxide in the processing and packaging of milk and dairy products: A review. Int. J. Dairy Technol. 2012, 65, 161–177. [Google Scholar] [CrossRef]
- Dixon, N.M.; Kell, D.B. The inhibition by CO2 of the growth and metabolism of micro-organisms. The Journal of Applied Bacteriology. 1989, 67, 109–136. [Google Scholar] [CrossRef]
- Fang, Y.; Franke, C.; Manthei, A.; McMullen, L.; Temelli, F.; Gänzle, M.G. Effects of high-pressure carbon dioxide on microbial quality and germination of cereal grains and beans. The Journal of Supercritical Fluids. 2021, 175, 105272. [Google Scholar] [CrossRef]
- Yang, J.; Yang, X.; Liang, R.; Zhu, L.; Mao, Y.; Dong, P.; Hopkins, D.L.; Luo, X.; Zhang, Y. The response of bacterial communities to carbon dioxide in high-oxygen modified atmosphere packaged beef steaks during chilled storage. Food Res. Int. 2022, 151, 110872. [Google Scholar] [CrossRef]
- Kosasih, L.; Bhandari, B.; Prakash, S.; Bansal, N.; Gaiani, C. Physical and functional properties of whole milk powders prepared from concentrate partially acidified with CO2 at two temperatures. Int. Dairy J. 2016, 56, 4–12. [Google Scholar] [CrossRef]
- Lo, R.; Turner, M.S.; Weeks, M.; Bansal, N. Culture-independent bacterial community profiling of carbon dioxide treated raw milk. Int. J. Food Microbiol. 2016, 233, 81–89. [Google Scholar] [CrossRef]
- Apostolakos, I.; Skarlatoudi, T.; Vatavali, K.; Giannouli, A.; Bosnea, L.; Mataragas, M. Genomic and phenotypic characterization of mastitis-causing Staphylococci and probiotic lactic acid bacteria isolated from raw sheep and rsquo’s Milk. In International Journal of Molecular Sciences, 2023; Vol. 24.
- D'Incecco, P.; Limbo, S.; Hogenboom, J.A.; Pellegrino, L. Novel technologies for extending the shelf life of drinking milk: Concepts, research trends and current applications. Lwt-Food Sci. Technol. 2021, 148.
- DERTLI, E.; SERT, D.; AKIN, N. The effects of carbon dioxide addition to cheese milk on the microbiological properties of Turkish White brined cheese. Int. J. Dairy Technol. 2012, 65, 387–392. [Google Scholar] [CrossRef]
- Couvert, O.; Koullen, L.; Lochardet, A.; Huchet, V.; Thevenot, J.; Le Marc, Y. Effects of carbon dioxide and oxygen on the growth rate of various food spoilage bacteria. Food Microbiol. 2023, 114, 104289. [Google Scholar] [CrossRef]
- Lemos, Á.T.; Casal, S.; Barba, F.J.; Phimolsiripol, Y.; Delgadillo, I.; Saraiva, J.A. Preservation of high pressure pasteurised milk by hyperbaric storage at room temperature versus refrigeration on inoculated microorganisms, fatty acids, volatile compounds and lipid oxidation. Food Chem. 2022, 387, 132887. [Google Scholar] [CrossRef]
- Hotchkiss, J.H.; Werner, B.G.; Lee, E.Y.C. Addition of carbon dioxide to dairy products to improve quality: A comprehensive review. Compr. Rev. Food. Sci. Food Saf. 2006, 5, 158–168. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, Y.; Cheng, Y.; Huang, J.; Huang, M. Monitoring and identification of spoilage-related microorganisms in braised chicken with modified atmosphere packaging during refrigerated storage. Food Sci. Human Wellness. 2023, 12, 28–34. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, Y.; Wu, Y.; Chen, R.; Xu, Y.; Cai, Y.; Chu, M.; Dou, X.; Zhang, Y.; Qin, Y.; et al. Metabonomic analysis of human and 12 kinds of livestock mature milk. Food Chemistry: X. 2023, 17, 100581. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Ziemba, A.; Urasaki, Y.; Hayes, E.; Brotman, S.; Pizzorno, G. Disruption of uridine homeostasis links liver pyrimidine metabolism to lipid accumulation[S]. J. Lipid Res. 2013, 54, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Stretton, S.; Goodman, A.E. Carbon dioxide as a regulator of gene expression in microorganisms. Antonie Van Leeuwenhoek. 1998, 73, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Heng, J.; Tian, M.; Song, H.; Chen, F.; Guan, W.; Zhang, S. Amino acid transportation, sensing and signal transduction in the mammary gland: key molecular signalling pathways in the regulation of milk synthesis. Nutr. Res. Rev. 2020, 33, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Andersen, B.; Arshid, S.; Larsen, M.R.; Albergaria, H.; Lametsch, R.; Arneborg, N. Proteomics insights into the responses of Saccharomyces cerevisiae during mixed-culture alcoholic fermentation with Lachancea thermotolerans. Fems Microbiol. Ecol. 2019, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, W.; Kong, F.; Kang, S.; Liang, X.; Han, H.; Wu, J.; Zheng, Y.; Li, Q.; Yue, X.; et al. Metabolomics methods to analyze full spectrum of amino acids in different domains of bovine colostrum and mature milk. Eur. Food Res. Technol. 2020, 246, 213–224. [Google Scholar] [CrossRef]
- Leandro, J.; Houten, S.M. The lysine degradation pathway: Subcellular compartmentalization and enzyme deficiencies. Mol. Genet. Metab. 2020, 131, 14–22. [Google Scholar] [CrossRef]
Figure 1.
(A) Total bacterial counts of raw milk stored at 4 °C. (B) Effect of 2000 ppm CO2 treatment on raw milk pH.
Figure 1.
(A) Total bacterial counts of raw milk stored at 4 °C. (B) Effect of 2000 ppm CO2 treatment on raw milk pH.
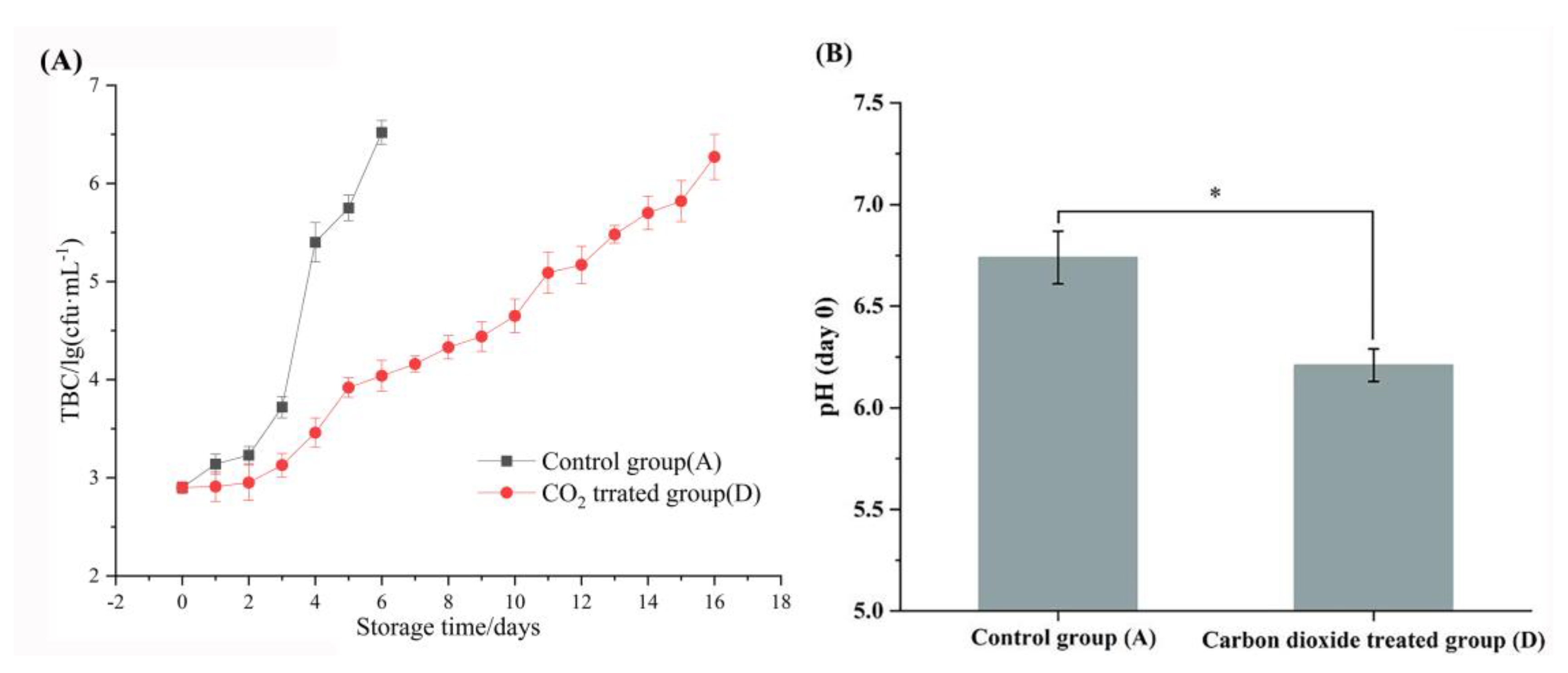
Figure 2.
Microbial diversity of raw milk from the control (A) and CO2-treated (D) groups. (A) Number of common and unique microbial taxa in raw milk from both groups. (B) Simpson's index to assess the alpha diversity (genus level) of microbial communities in the two groups. (C) Principal component analysis (PCA) to assess β-diversity (genus level). (D) Chao1 index to assess the α-diversity (genus level) of the two microbial communities.
Figure 2.
Microbial diversity of raw milk from the control (A) and CO2-treated (D) groups. (A) Number of common and unique microbial taxa in raw milk from both groups. (B) Simpson's index to assess the alpha diversity (genus level) of microbial communities in the two groups. (C) Principal component analysis (PCA) to assess β-diversity (genus level). (D) Chao1 index to assess the α-diversity (genus level) of the two microbial communities.
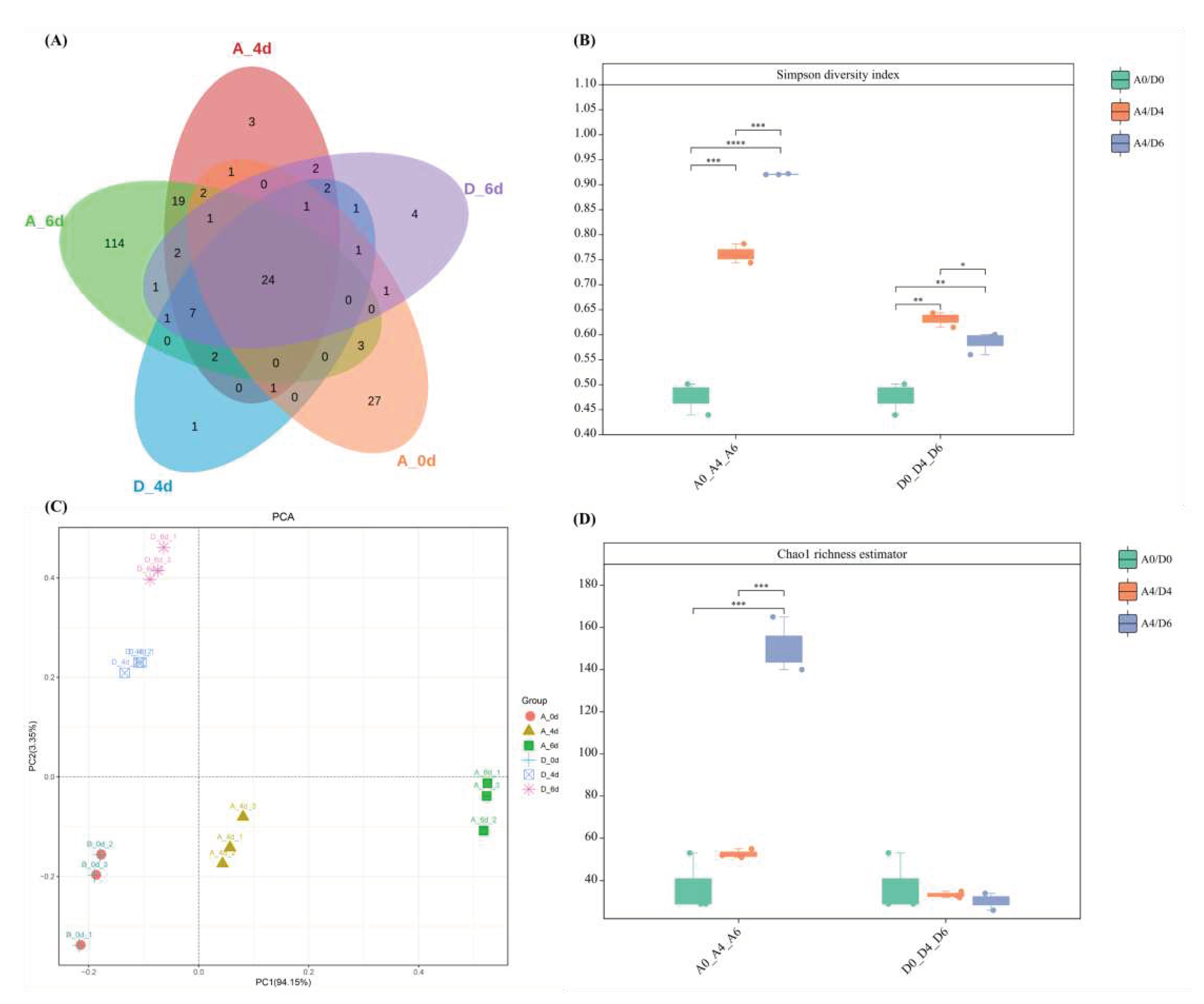
Figure 3.
Microbial community structure of raw milk from the control (A) and CO2 treated (D) groups. (A) Linear discriminant analysis effect size (LEfSe) species branching diagram. The taxonomic branching diagram shows the taxonomic hierarchy of the major taxonomic units in the sample community from phylum to species (from inner circle to outer circle, phylum, order, order, family, genus, and species), with node sizes corresponding to the average relative abundance of the taxonomic unit; hollow nodes represent taxonomic units with insignificant intergroup differences, while nodes of other colors indicate that these taxonomic units exhibit significant intergroup differences and are more abundant in the sample than in the group represented by the color. higher abundance in the sample. Letters identify the names of taxonomic units with significant intergroup differences. (B) Histogram of LEfSe. Vertical coordinates show taxonomic units with significant differences between groups, and horizontal coordinates visualize the logarithmic scores of LDA analysis for each taxonomic unit in a bar chart. Taxonomic units are sorted by the size of the score to characterize their specificity within the sample grouping. Longer lengths indicate more significant differences for that taxonomic unit, and the color of the bar indicates the sample subgroup with the highest abundance corresponding to that taxonomic unit. (C) Taxonomic analysis of bacteria at the genus level. Horizontal coordinates are arranged according to the sample name, each bar represents one sample and is color-coded to distinguish each taxonomic unit, vertical coordinates represent the relative abundance of each taxonomic unit, the longer the bar, the higher the relative abundance of that taxonomic unit in the corresponding sample.
Figure 3.
Microbial community structure of raw milk from the control (A) and CO2 treated (D) groups. (A) Linear discriminant analysis effect size (LEfSe) species branching diagram. The taxonomic branching diagram shows the taxonomic hierarchy of the major taxonomic units in the sample community from phylum to species (from inner circle to outer circle, phylum, order, order, family, genus, and species), with node sizes corresponding to the average relative abundance of the taxonomic unit; hollow nodes represent taxonomic units with insignificant intergroup differences, while nodes of other colors indicate that these taxonomic units exhibit significant intergroup differences and are more abundant in the sample than in the group represented by the color. higher abundance in the sample. Letters identify the names of taxonomic units with significant intergroup differences. (B) Histogram of LEfSe. Vertical coordinates show taxonomic units with significant differences between groups, and horizontal coordinates visualize the logarithmic scores of LDA analysis for each taxonomic unit in a bar chart. Taxonomic units are sorted by the size of the score to characterize their specificity within the sample grouping. Longer lengths indicate more significant differences for that taxonomic unit, and the color of the bar indicates the sample subgroup with the highest abundance corresponding to that taxonomic unit. (C) Taxonomic analysis of bacteria at the genus level. Horizontal coordinates are arranged according to the sample name, each bar represents one sample and is color-coded to distinguish each taxonomic unit, vertical coordinates represent the relative abundance of each taxonomic unit, the longer the bar, the higher the relative abundance of that taxonomic unit in the corresponding sample.
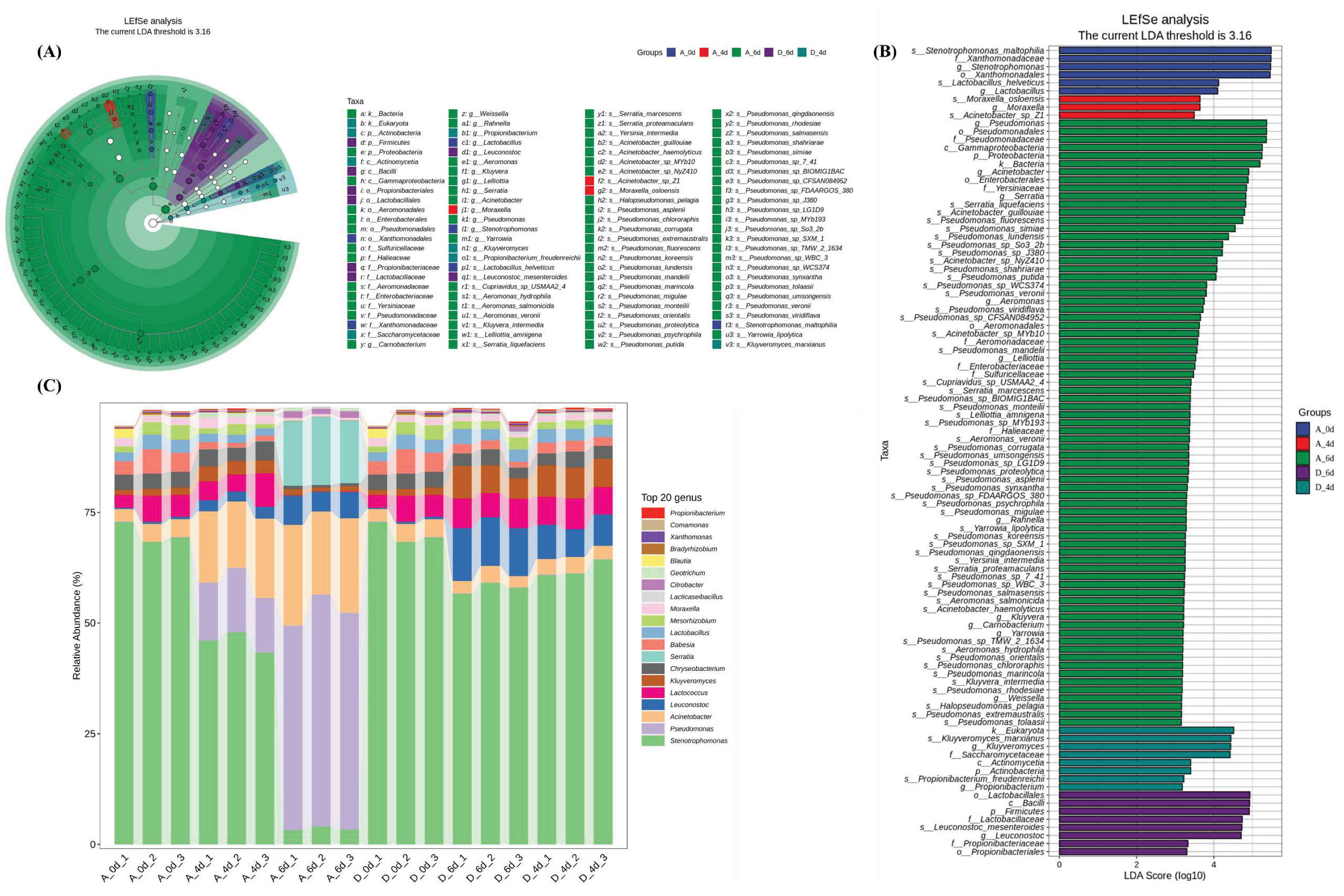
Figure 4.
(A) PCA score plots for raw milk metabolite and quality control (QC) sample datasets. (B) Correlation plot for QC samples.
Figure 4.
(A) PCA score plots for raw milk metabolite and quality control (QC) sample datasets. (B) Correlation plot for QC samples.
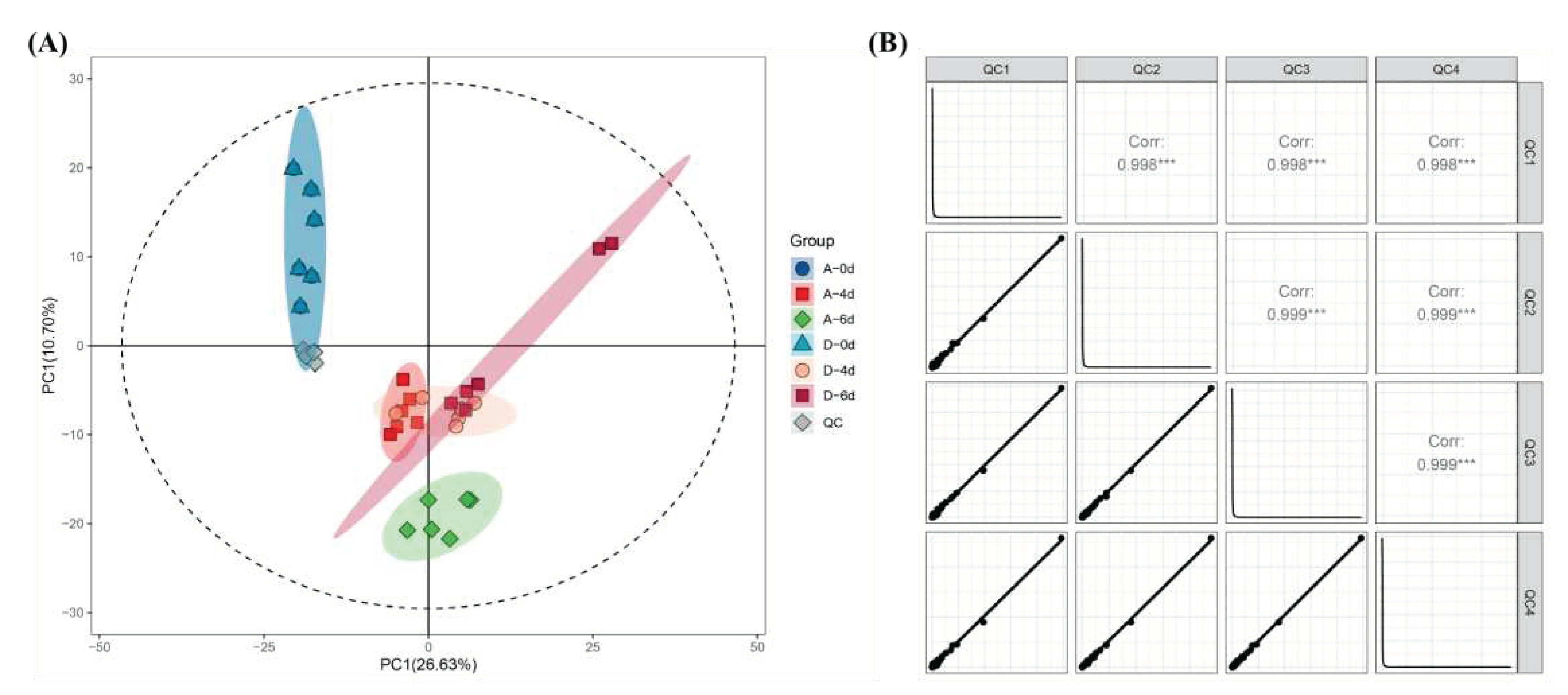
Figure 5.
(A) OPLS-DA of the metabolite dataset of the control group. (B) OPLS-DA of the metabolite dataset of the CO2-treated group. (C) Alignment test results of the OPLS-DA of the control group. (D) Alignment test results of OPLS-DA for the CO2-treated group.
Figure 5.
(A) OPLS-DA of the metabolite dataset of the control group. (B) OPLS-DA of the metabolite dataset of the CO2-treated group. (C) Alignment test results of the OPLS-DA of the control group. (D) Alignment test results of OPLS-DA for the CO2-treated group.
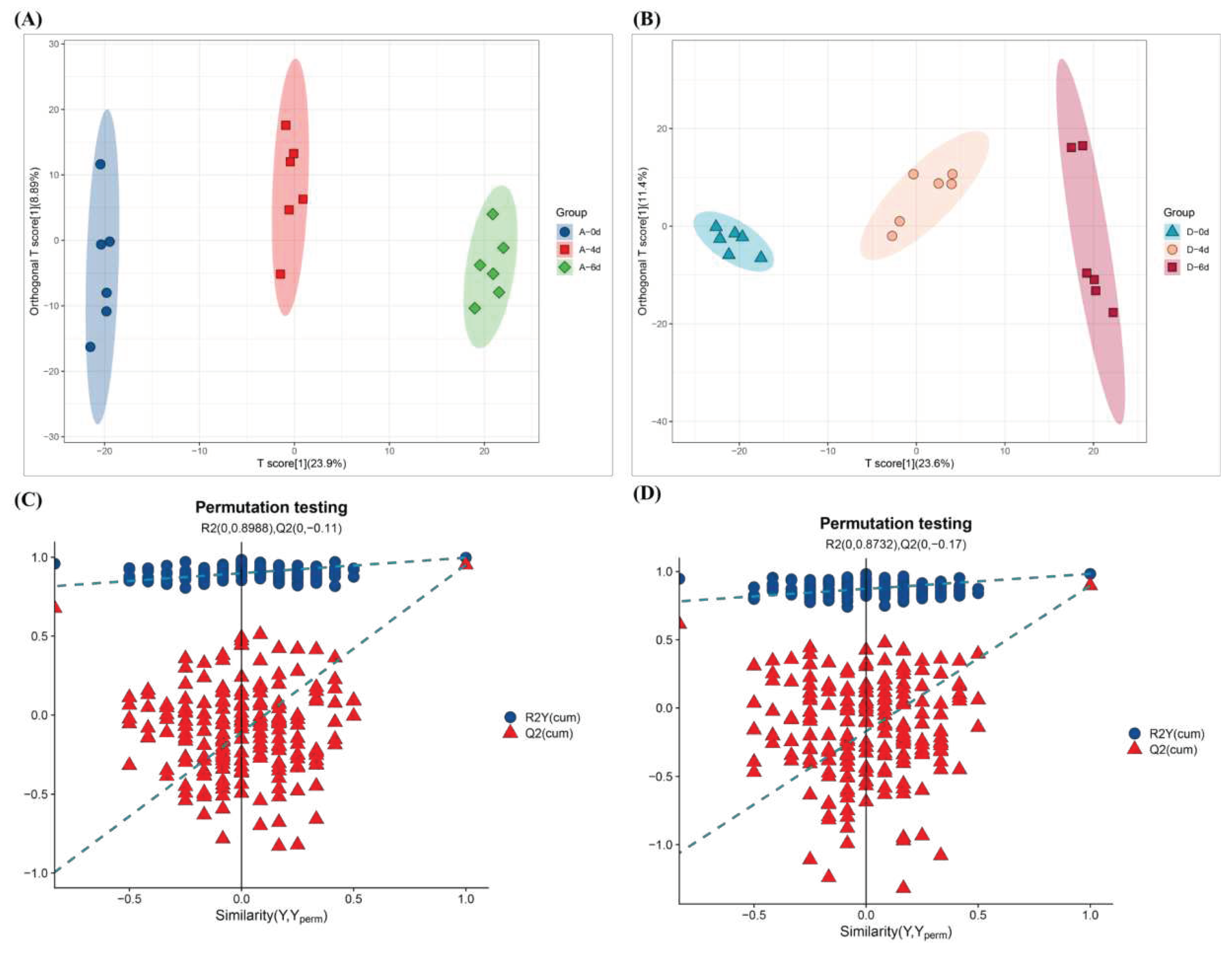
Figure 6.
(A) Heatmap of DMs in the control group. (B) Heat map of DMs in the CO2-treated group. (C) Classification loop diagram (Ontology) of DMs in the control group. (D) Classification loop diagram (Ontology) of DMs in the CO2-treated group.
Figure 6.
(A) Heatmap of DMs in the control group. (B) Heat map of DMs in the CO2-treated group. (C) Classification loop diagram (Ontology) of DMs in the control group. (D) Classification loop diagram (Ontology) of DMs in the CO2-treated group.
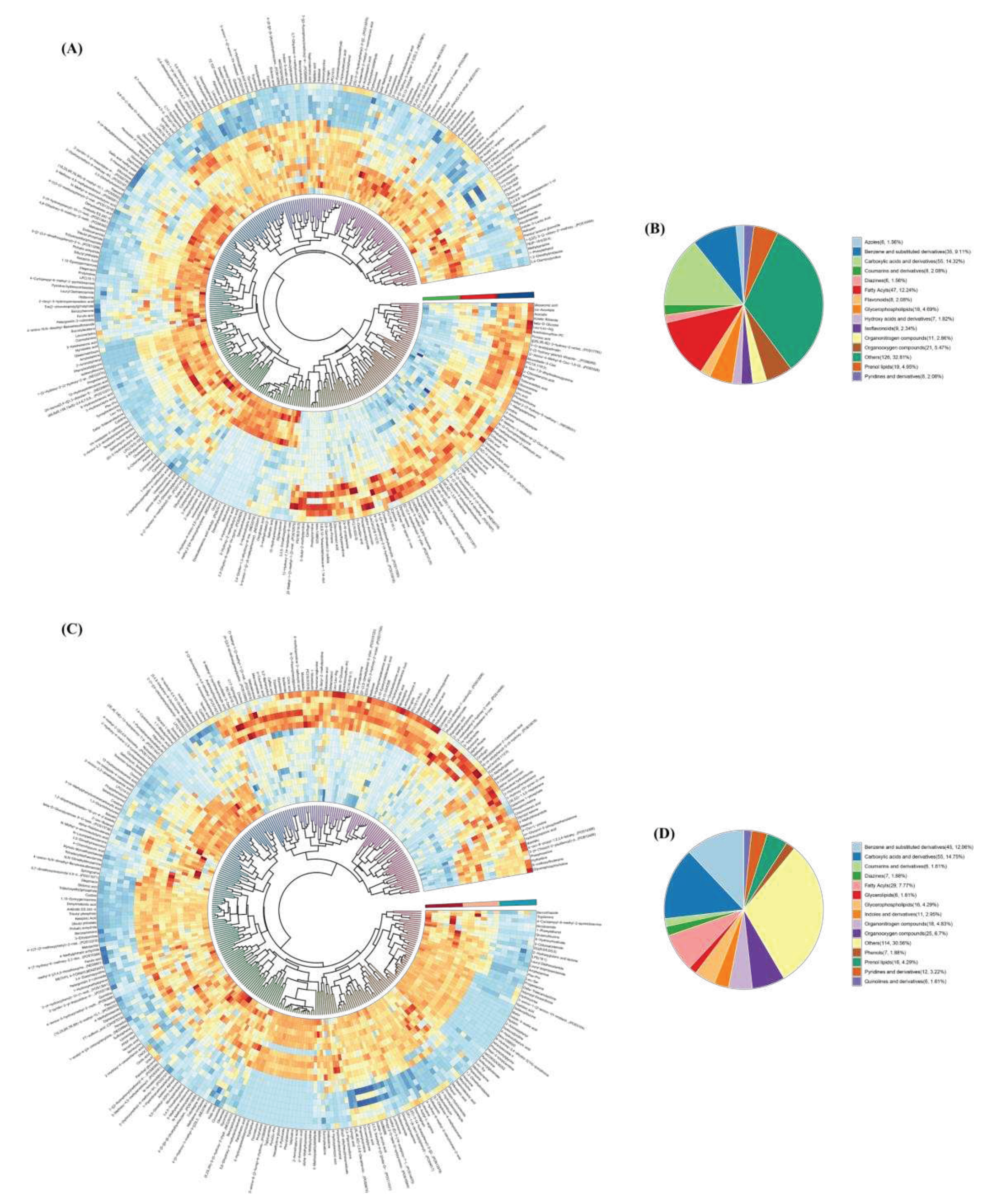
Figure 7.
(A) Histogram of the distribution of microbiomics and metabolomics correlation coefficients (r) in the control group. (B) Histogram of the distribution of microbiomics and metabolomics correlation coefficients (r) in the CO2-treated group. The horizontal coordinate is the distribution of r between the two histologies, and the vertical coordinate is the density of the distribution of the corresponding r. r ≥ 0.7 or ≤ -0.7 indicates that the correlation is very strong, i.e., the blue negative correlation and the yellow positive correlation part of the plot. (C) Hierarchical clustering heat map of microbial and metabolite correlations in the control group (Top 28). (D) Hierarchical clustering heatmap of microbial and metabolite correlations in the CO2-treated group. Rows indicate metabolites and columns indicate genera. *** denotes correlation test P <0.001, ** denotes correlation test P <0.01, * denotes correlation test P <0.05.
Figure 7.
(A) Histogram of the distribution of microbiomics and metabolomics correlation coefficients (r) in the control group. (B) Histogram of the distribution of microbiomics and metabolomics correlation coefficients (r) in the CO2-treated group. The horizontal coordinate is the distribution of r between the two histologies, and the vertical coordinate is the density of the distribution of the corresponding r. r ≥ 0.7 or ≤ -0.7 indicates that the correlation is very strong, i.e., the blue negative correlation and the yellow positive correlation part of the plot. (C) Hierarchical clustering heat map of microbial and metabolite correlations in the control group (Top 28). (D) Hierarchical clustering heatmap of microbial and metabolite correlations in the CO2-treated group. Rows indicate metabolites and columns indicate genera. *** denotes correlation test P <0.001, ** denotes correlation test P <0.01, * denotes correlation test P <0.05.
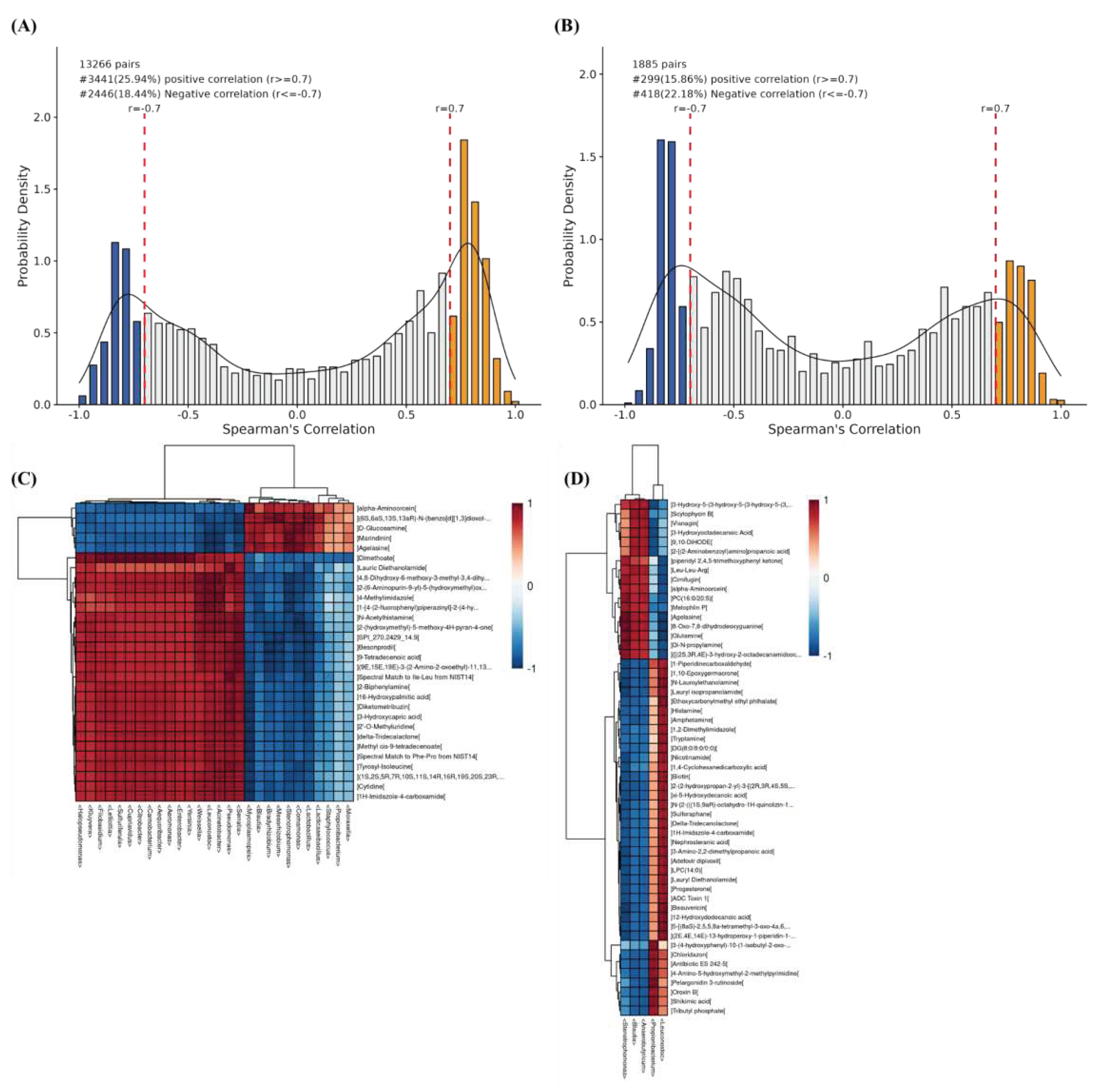
Figure 8.
Microbiomics and untargeted metabolomics KEGG enrichment pathway analysis. (A) Microbiomics and identification of KEGG secondary metabolic pathway composition bar graphs for each sample. The vertical coordinates represent the relative abundance of each functional taxon; the longer the bar, the higher the relative abundance of that functional taxon in the corresponding sample. (B) Comparison group A-6d and D-6d KEGG pathway category bar graphs. G is Genetic Information Processing, M is Metabolism,E is Environmental Information Processing,o is Organismal Systems,H is Human Diseases. (C) Interaction maps of KEGG-enriched pathway-regulated metabolite networks shared by microbiomics and non-targeted metabolomics.
Figure 8.
Microbiomics and untargeted metabolomics KEGG enrichment pathway analysis. (A) Microbiomics and identification of KEGG secondary metabolic pathway composition bar graphs for each sample. The vertical coordinates represent the relative abundance of each functional taxon; the longer the bar, the higher the relative abundance of that functional taxon in the corresponding sample. (B) Comparison group A-6d and D-6d KEGG pathway category bar graphs. G is Genetic Information Processing, M is Metabolism,E is Environmental Information Processing,o is Organismal Systems,H is Human Diseases. (C) Interaction maps of KEGG-enriched pathway-regulated metabolite networks shared by microbiomics and non-targeted metabolomics.
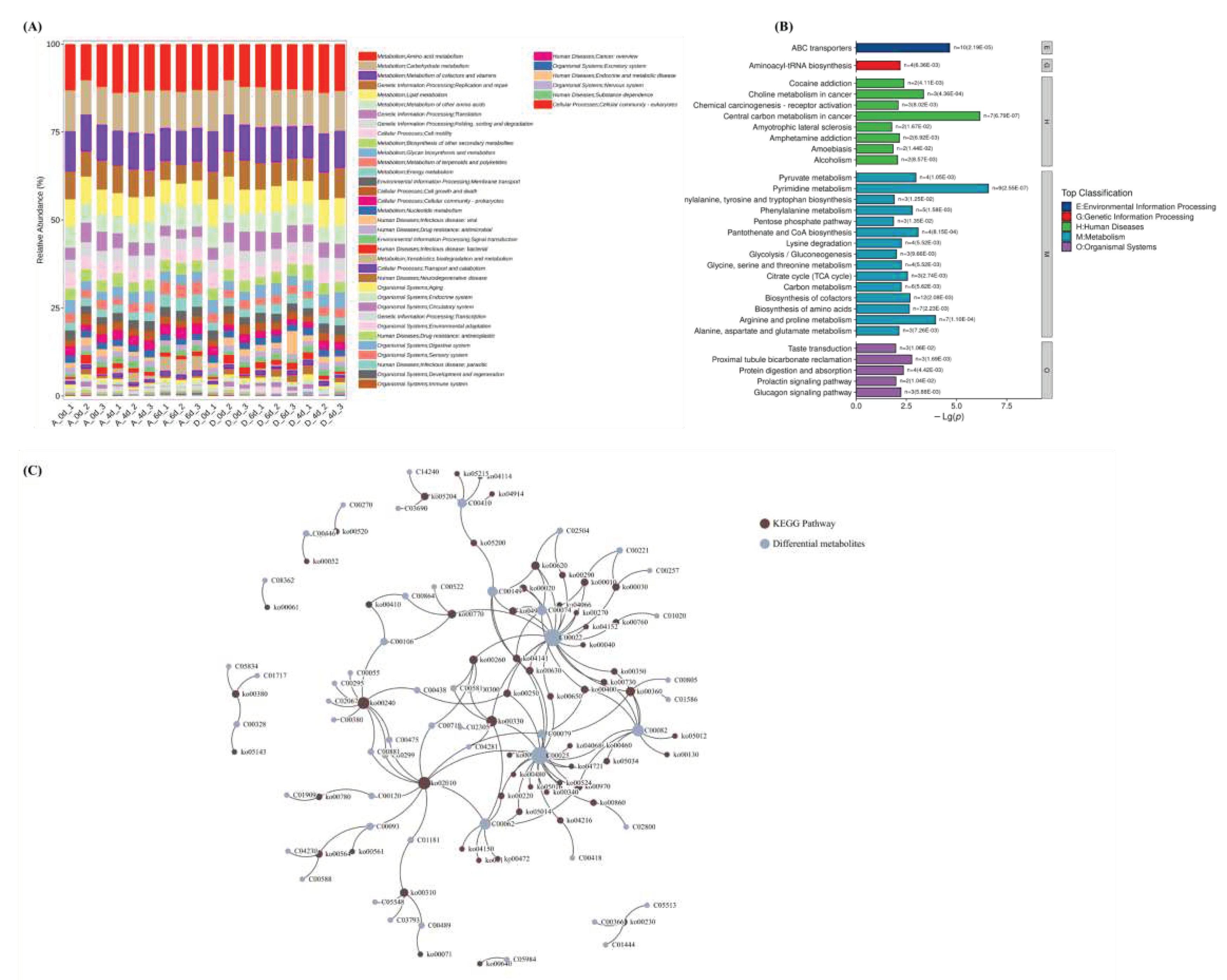

Table 1.
Macrogenomics screened proteins corresponding to KO abundance and metabolomics annotated KEGG shared pathway analysis.
Table 1.
Macrogenomics screened proteins corresponding to KO abundance and metabolomics annotated KEGG shared pathway analysis.
| No. | ID | ID | Description | Count | KEGG ID | Top Class |
|---|---|---|---|---|---|---|
| 1 | ko02010 | bta02010 | ABC transporters | 10 | C00719/C00062/C00025/C00120/C00093/C00299/C00475/C00881/C00079/C01181 | Metabolism |
| 2 | ko00240 | bta00240 | Pyrimidine metabolism | 9 | C00055/C00380/C00299/C00475/C00881/C00106/C00438/C02067/C00295 | Environmental Information Processing |
| 3 | ko00330 | bta00330 | Arginine and proline metabolism | 7 | C00062/C00581/C02305/C00025/C04281/C00022/C00300 | Metabolism |
| 4 | ko00360 | bta00360 | Phenylalanine metabolism | 5 | C00082/C01586/C00805/C00079/C00022 | Metabolism |
| 5 | ko00770 | bta00770 | Pantothenate and CoA biosynthesis | 4 | C00864/C00522/C00106/C00022 | Metabolism |
| 6 | ko00620 | bta00620 | Pyruvate metabolism | 4 | C02504/C00074/C00149/C00022 | Metabolism |
| 7 | ko00260 | bta00260 | Glycine, serine and threonine metabolism | 4 | C00719/C00581/C00022/C00300 | Organismal Systems |
| 8 | ko00310 | bta00310 | Lysine degradation | 4 | C03793/C00489/C01181/C05548 | Metabolism |
| 9 | ko00970 | bta00970 | Aminoacyl-tRNA biosynthesis | 4 | C00062/C00025/C00082/C00079 | Human Diseases |
| 10 | ko04141 | bta04964 | Proximal tubule bicarbonate reclamation | 3 | C00074/C00025/C00149 | Metabolism |
| 11 | ko00020 | bta00020 | Citrate cycle (TCA cycle) | 3 | C00074/C00149/C00022 | Metabolism |
| 12 | ko04922 | bta04922 | Glucagon signaling pathway | 3 | C00074/C00149/C00022 | Organismal Systems |
| 13 | ko00250 | bta00250 | Alanine, aspartate and glutamate metabolism | 3 | C00025/C00438/C00022 | Genetic Information Processing |
| 14 | ko05204 | bta05207 | Chemical carcinogenesis - receptor activation | 3 | C14240/C03690/C00410 | Metabolism |
| 15 | ko00010 | bta00010 | Glycolysis / Gluconeogenesis | 3 | C00074/C00221/C00022 | Human Diseases |
| 16 | ko00400 | bta00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 3 | C00074/C00082/C00079 | Human Diseases |
| 17 | ko00030 | bta00030 | Pentose phosphate pathway | 3 | C00257/C00221/C00022 | Metabolism |
| 18 | ko00564 | bta00564 | Glycerophospholipid metabolism | 3 | C00588/C00093/C04230 | Metabolism |
| 19 | ko00630 | bta00630 | Glyoxylate and dicarboxylate metabolism | 3 | C00025/C00149/C00022 | Metabolism |
| 20 | ko00380 | bta00380 | Tryptophan metabolism | 3 | C01717/C05834/C00328 | Human Diseases |
| 21 | ko00230 | bta00230 | Purine metabolism | 3 | C00366/C01444/C05513 | Metabolism |
| 22 | ko00460 | bta05030 | Cocaine addiction | 2 | C00025/C00082 | Metabolism |
| 23 | ko05034 | bta05034 | Alcoholism | 2 | C00025/C00082 | Metabolism |
| 24 | ko05014 | bta05014 | Amyotrophic lateral sclerosis | 2 | C00062/C00025 | Environmental Information Processing |
| 25 | ko00220 | bta00220 | Arginine biosynthesis | 2 | C00062/C00025 | Cellular Processes |
| 26 | ko00290 | bta00290 | Valine, leucine and isoleucine biosynthesis | 2 | C02504/C00022 | Organismal Systems |
| 27 | ko00780 | bta00780 | Biotin metabolism | 2 | C00120/C01909 | Metabolism |
| 28 | ko04216 | bta04216 | Ferroptosis | 2 | C00025/C00418 | Metabolism |
| 29 | ko05200 | bta05200 | Pathways in cancer | 2 | C00410/C00149 | Cellular Processes |
| 30 | ko00730 | bta00730 | Thiamine metabolism | 2 | C00082/C00022 | Environmental Information Processing |
| 31 | ko00410 | bta00410 | beta-Alanine metabolism | 2 | C00864/C00106 | Human Diseases |
| 32 | ko00650 | bta00650 | Butanoate metabolism | 2 | C00025/C00022 | Metabolism |
| 33 | ko00760 | bta00760 | Nicotinate and nicotinamide metabolism | 2 | C01020/C00022 | Metabolism |
| 34 | ko00350 | bta00350 | Tyrosine metabolism | 2 | C00082/C00022 | Human Diseases |
| 35 | ko00520 | bta00520 | Amino sugar and nucleotide sugar metabolism | 2 | C00270/C00446 | Human Diseases |
| 36 | ko00860 | bta00860 | Porphyrin and chlorophyll metabolism | 2 | C00025/C02800 | Human Diseases |
| 37 | ko04150 | bta04150 | mTOR signaling pathway | 1 | C00062 | Metabolism |
| 38 | ko04114 | bta04114 | Oocyte meiosis | 1 | C00410 | Metabolism |
| 39 | ko04914 | bta04914 | Progesterone-mediated oocyte maturation | 1 | C00410 | Metabolism |
| 40 | ko04068 | bta04068 | FoxO signaling pathway | 1 | C00025 | Human Diseases |
| 41 | ko05142 | bta05142 | Chagas disease | 1 | C00062 | Metabolism |
| 42 | ko05016 | bta05016 | Huntington disease | 1 | C00025 | Organismal Systems |
| 43 | ko05143 | bta05143 | African trypanosomiasis | 1 | C00328 | Metabolism |
| 44 | ko00472 | bta00472 | D-Arginine and D-ornithine metabolism | 1 | C00062 | Environmental Information Processing |
| 45 | ko05215 | bta05215 | Prostate cancer | 1 | C00410 | Metabolism |
| 46 | ko04721 | bta04721 | Synaptic vesicle cycle | 1 | C00025 | Environmental Information Processing |
| 47 | ko04066 | bta04066 | HIF-1 signaling pathway | 1 | C00022 | Metabolism |
| 48 | ko00910 | bta00910 | Nitrogen metabolism | 1 | C00025 | Human Diseases |
| 49 | ko04152 | bta04152 | AMPK signaling pathway | 1 | C00022 | Metabolism |
| 50 | ko05012 | bta05012 | Parkinson disease | 1 | C00082 | Metabolism |
| 51 | ko00480 | bta00480 | Glutathione metabolism | 1 | C00025 | Metabolism |
| 52 | ko00561 | bta00561 | Glycerolipid metabolism | 1 | C00093 | Metabolism |
| 53 | ko00640 | bta00640 | Propanoate metabolism | 1 | C05984 | Metabolism |
| 54 | ko00052 | bta00052 | Galactose metabolism | 1 | C00446 | Metabolism |
| 55 | ko00340 | bta00340 | Histidine metabolism | 1 | C00025 | Metabolism |
| 56 | ko00071 | bta00071 | Fatty acid degradation | 1 | C00489 | Environmental Information Processing |
| 57 | ko00524 | bta04080 | Neuroactive ligand-receptor interaction | 1 | C00025 | Metabolism |
| 58 | ko00061 | bta00061 | Fatty acid biosynthesis | 1 | C08362 | Metabolism |
| 59 | ko00040 | bta00040 | Pentose and glucuronate interconversions | 1 | C00022 | Metabolism |
| 60 | ko00270 | bta00270 | Cysteine and methionine metabolism | 1 | C00022 | Metabolism |
| 61 | ko00130 | bta00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 1 | C00082 | Metabolism |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



