
Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
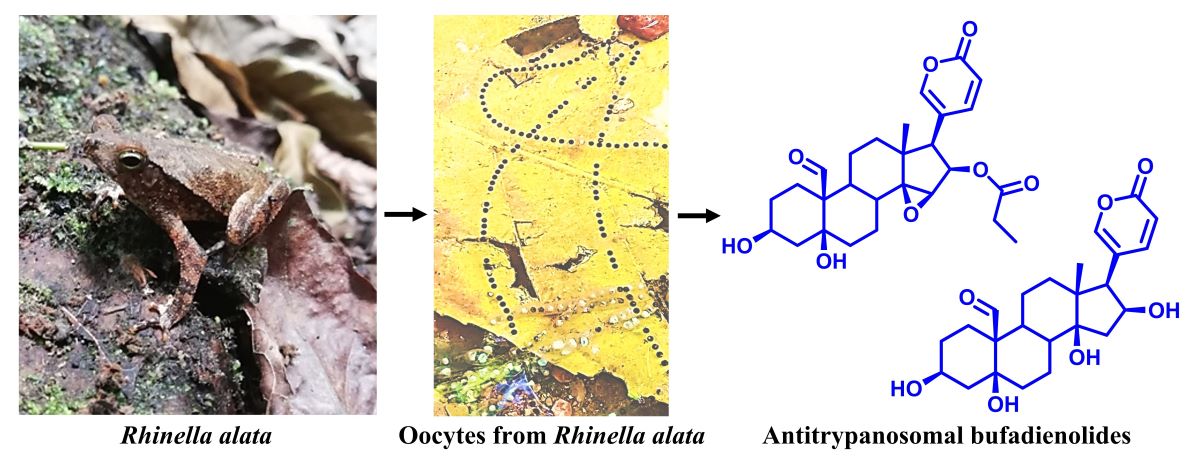
Amphibians are widely known as a prolific source of bioactive metabolites. In this work we isolated and characterized compounds with antiparasitic activity from the oocytes of the toad Rhinella alata collected in Panama. Bio-guided isolation and structural elucidation was carried out using chromatographic and spectroscopic techniques. The organic crude extract was subjected to solid phase extraction followed by HPLC purification of the fraction with in vitro activity against Trypanosoma cruzi trypomastigotes. Seven steroids (1-7) of the bufadienolide family were isolated and their structures determined using NMR and MS analyses; of these 19-formyl-dyscinobufotalin (3) is reported as a new natural product. Compounds 1, 3-7, resulted with a good anti-trypanosomal activity profile, among these 16β-hydroxyl-hellebrigenin (1) and bufalin (7) showed significant selectivity for T. cruzi. Furthermore, molecular docking analysis showed compounds 1, 3 and 7 interact through H-bonds with the amino acid residues GLN-19, ASP-158, HIS-159 and TRP-177 from cruzipain, at the catalytic site. Given the lack of therapeutic options to currently treat the American trypanosomiasis, this work can serve as the basis for further studies that aim the development of bufadienolides or their derivatives as drugs against Chagas disease.
Keywords:
Rhinella alata
; toads
; Bufonidae
; oocytes
; bufadienolides
; anti-trypanosomal activity
; Trypanosoma cruzi
1. Introduction
Chagas disease, or American trypanosomiasis, is a zoonotic disease caused by the parasite Trypanosoma cruzi. This protozoan is transmitted to humans mainly through the feces of insects of the family Reduviidae [1]. Although Chagas disease is recognized as endemic from Latin America, it has become a worldwide issue with 10,000 deaths per year and eight million people at risk of infection [2]. There are no specific symptoms for diagnostics during its acute phase; however, once the disease advances, chronic pathological features such as cardiomyopathy, megacolon, megaesophagus and stroke are developed [3]. The low effectiveness and side effects of the available drugs used for the treatment of this disease, during its chronic phase, highlight the need of more efficacious and safer anti-trypanosomal treatments [4].
Bufadienolides are polyhydroxy steroids that contain an unsaturated 2H-pyran-2-one ring at β-17 position [5]. Because of their wide bioactivity against human cancer cells lines and their great chemical diversity, bufadienolides have attracted a considerable attention in recent years [6]. These metabolites are widely distributed in nature and have been reported in fireflies, mammals, plants, snakes and toads [7]. Bioprospecting research has revealed that bufadienolides isolated from some amphibian species, have promising antibiotic and antiparasitic properties [8].
Herein we report the bio-guided isolation of anti-trypanosomal bufadienolides from the oocytes of the toad Rhinella alata (Thominot, 1884) from Panama [9]. Seven compounds were isolated and their chemical structures were elucidated by spectroscopic techniques. Compounds were tested against trypomastigotes of T. cruzi and evaluated on Vero cells for mammalian cytotoxicity. Furthermore bufadienolides 1, 3 and 7 were analyzed by molecular docking with cruzipain revealing chemical interactions with amino acid residues at the active site.
2. Results and Discussion
2.1. Isolation and structural elucidation of bufadienolides
The crude extract from oocytes of R. alata was prepared by maceration with a mixture of methanol and chloroform and then submitted to C-18 SPE purification. The fraction eluted at 80 % CH3OH showed anti-trypanosomal activity and was further analyzed by semi-preparative RP-HPLC (Figure S1). Compounds 1-7 were isolated and their structures, depicted in Figure 1, were determined based on extensive spectroscopic analysis and comparison of their data with those reported in the literature. While compounds 1, 2 and 4-7 corresponded to known compounds, the bufadienolide 3 is reported as a new natural product.
Compound 3 was isolated as a white solid. The molecular formula was determined as C27H34O8 based on its HR-MALDI-MS data which showed the molecular ion at m/z 487.2283 [M + H]+ (calculated for C27H35O8, 487.2326). Eleven degrees of unsaturation were inferred from the molecular formula. The 1H-NMR and 13C-NMR spectra of compound 3 showed the feature signals for a 2H-pyran-2-one moiety [δH 6.24 (1H, dd, J = 0.8, 9.7 Hz), 7.36 (1H, m), 8.02 (1H, m); δC 114.2, 118.2, 150.9, 153.6, 164.0], a tertiary methyl [δH 0.81 (3H, s); δC 17.3], a secondary methyl [δH 0.94 (3H, t, J = 7.8 Hz); δC 9.3], three oxygenated methines [(δH 3.75, m; δC 60.7), (δH 4.15, m; δC 68.0), (δH 5.47, m; δC 76.3)], two oxygenated quaternary carbons [δC 75.8, 73.1] and two carbonyl carbons characteristic of aldehyde and ester [δC 210.1, 174.9]. Comparison of the NMR signals (Table 1) of compound 3 with those of the reported bufadienolide dyscinobufotalin [10]. revealed the data of the steroidal nucleus of the two compounds were very similar, indicating that compound 3 had the same bufadienolide core with two carbinols at C-3 and C-5, an epoxy group at C-14 and C-15, and the esterification of the hydroxyl group at C-16 with propionic acid. The main difference consisted in the presence of an aldehyde group at C-19 in compound 3 instead of the angular methyl group at C-19 of dyscinobufotalin [10]. HMBC correlations between aldehyde proton at δH 10.00 and the quaternary carbon at C-10 allowed for the assignment of the aldehyde group at C-19 (Figure 1). On the basis of spectral data, the compound 3 was identified as 19-formyl-dyscinobufotalin.
2.2. Anti-trypanosomal activity and mammalian cytotoxicity
The trypanosomaticidal activity of the bufadienolides 1-7 was evaluated in vitro against T. cruzi. The ability of these compounds to inhibit growth of the trypomastigote form was evaluated at four different concentrations. The results showed that bufadienolides 1, 3, 4, 5, 6 and 7 reduce the growth of T. cruzi while compound 2 was inactive at the screening concentration of 10 µg/mL. The bufadienolides 3 and 7 presented EC50 values (0.19 and 3.93 µM, respectively) being compound 3 even more active than the positive control benznidazole (Table 2). The cytotoxicity of bufadienolides against mammalian cells was also tested in vitro using healthy epithelial kidney Vero cells. The IC50 values of bufadienolides 1-7 ranged from 0.02 to > 135 µM. As shown below, compounds 1, 2 and 4 presented the lowest cytotoxic effects. Both the antiparasitic and cytotoxic activities exhibited by each bufadienolide assessed are shown in Table 2. Selectivity was analyzed by the cytotoxicity on Vero cells/anti-trypanosomal activity ratio.
2.3. Molecular docking of bufadienolides 1, 3 and 7
A docking protocol was validated and its reproducibility confirmed by the root mean square deviation (RMSD) values obtained. The RMSD for each cruzipain-co-crystallized inhibitor complex were as follows: 2.3586 for 1EWO-VSC, 1.9060 for 2AIM-ZRA, 1.6817 for 1U9Q-186 and 2.9319 for 4XUI-2VC. In addition, relative docking scores for each complex resulted in -7.0484, -6.9388, -8.2353 and -8.1351, respectively. After validation of the protocol accuracy, interactions of anti-trypanosomal bufadienolides 1, 3 and 7 with the active site of proteins were studied via docking calculations. Table 3 shows the calculated parameters for the best pose obtained for each bufadienolide tested. The energy variation for the predictions obtained by simulation ranged from -2.2 to +1.3 kcal/mol. The isolated bufadienolides 1, 3 and 7 displayed interactions with cruzipain. Bufadienolide 1 showed H-bonds with ASP-158 and GLN-19 at a distance of 2.10 Å and 2.32 Å, respectively. On the other hand, bufadienolide 3 presents three H-bonds with amino acid residues GLN-19, HIS-159 and TRP-177. Bufadienolide 7 formed one H-bond with ASP-158 at a distance of 1.95 Å (Figure 2).
Currently, there is no effective chemotherapeutic treatment available for American trypanosomiasis [4]. Poisonous terrestrial vertebrates are rarely considered for natural product research programs; however, it has been reported that bioactive materials derived from animals, have resulted in effective medicines, highlighting the potential of vertebrate animals’ toxins in pharmacology. For instance, teprotide a nonapeptide isolated from the venom of the snake Bothrops jararaca, has served as template to produce captopril and other angiotensin-converted enzyme inhibitors that currently are routinely medicated for hypertension patients [13]. The alkaloid epibatidine obtained from the skin of the dendrobatid frog Epipedobates tricolor presents analgesic properties 200 times more potent than morphine. Epibatidine has served as template for producing anti-nociceptive derivatives such as tebanicline (ABT-594) that reached clinical phase II [14,15].
In this study, through the analysis of the spectroscopic data, we elucidated the chemical structure of the new bufadienolide 3 from the oocytes of R. alata. Moreover, five bufadienolides (2, 4-7) were previously reported in amphibians and one in plants (1). Bufadienolide 2 was isolated from the poison of the toad Duttaphrynus melanostictus as a minor constituent [5]. Compounds 4 and 5 have been obtained from skin and parotoid gland secretions of bufonids, but not in toads of the genus Rhinella [16]. The bufadienolide 6 was isolated from Chan Su, a traditional chinese remedy prepared with the poison of Asian toads [10]. The bufadienolide 7 has been identified in bufonid extracts including oocytes [17].
Due to the marked inhibitory activity against the pump (Na+-K+)ATPase, the antiparasitic potential of bufadienolides against human pathogens has currently received some attention [18]. In this study, bufadienolides 1-7 were evaluated in vitro for inhibition of the growth of the trypomastigote form of T. cruzi. According to our data, substitution of methyl at C-19 by formyl group increases the anti-trypanosomal activity as seen in bufadienolide 3 in comparison with bufadienolide 6. Similarly, in a previous study hellebrigenin showed trypanocidal activity at 91.7 µg/mL while telocinobufagin, which contains a C-19 methyl group instead, was inactive [19]. Likewise, comparing the activities of bufadienolides 2 and 7, it can be inferred that the lack of a hydroxyl group at C-16 is required for the anti-trypanosomal activity, however if this hydroxyl group is esterified, a moderate bioactivity is observed (compounds 4-6, Table 2).
Also, previous evaluations on cancer cell lines with bufadienolides revealed a decrease in growth inhibition by introduction of a hydroxyl group at C-16 position [20]. Bufadienolide 7 presents higher anti-trypanosomal activity than its 19-OH analogue (IC50 = 19.4 µM) [21]. On the basis of these results, we can suggest that the presence of a formyl group at C-19 and the esterification of the hydroxyl group at C-16 are essential elements required for the effective biomolecular interactions with components present in the trypomastigote form of T. cruzi, which led to the anti-protozoal activity observed.
In mammals, bufadienolides block the transport activity of the (Na+-K+)ATPase pump by binding to the α-subunit. This inhibition provokes that intracellular sodium concentration increases until cells get depolarized [22]. In T. cruzi, there is no report on the identification of this protein; maintenance of the ionic steady-state is carried out by K+ channels, Na+ efflux pumps and (H+)ATPases [23]. Kyoichi, I. et al. [24] cloned and characterized a (Na+)ATPase from a gene encoding of T. cruzi. After biochemical evaluations it was evidenced that the enzymatic activity of this pump was not inhibited by the steroid (cardenolide) ouabain. In the search for anti-trypanosomal drugs, the most considered targets for Chagas disease are cruzipain, sterol-14α-demethylase, trans-syalidase, trypanothione reductase and tubulin [4,24]. Currently, the mechanism by which bufadienolides cause inhibition of T. cruzi is unknown, despite of previous findings suggest that bufadienolides inhibit the growth of T. cruzi parasites by a biochemical pathway other than the blockage of the (Na+-K+)ATPase pump. In this study, in silico analysis by molecular docking was carried out, using the T. cruzi protease cruzipain. Redocking with previously reported co-crystallized inhibitors was employed for protocol validation and active site prediction (Figure S9). Molecular docking with the anti-trypanosomal bufadienolides 1, 3 and 7 revealed strong interactions with cruzipains. These interactions correspond to H-bonds between amino acid residues GLN-19, ASP-158, HIS-159 and TRP-177 with hydroxyl groups at C-5 and C-14 position; and formyl at C-19 position of bufadienolides. The most potent anti-trypanosomal bufadienolide (3), presented three interactions with cruzipain while bufadienolides 1 and 7 showed two and one H-bonds, respectively. Bufadienolide 7 interacts with cruzipain by the hydroxyl at C-14, a functional group essential for bioactivity [25]. No interaction with polar groups at C-16 such as hydroxyl and propionyloxy were observed for bufadienolides 1 and 3. Cruzipain contains, at the active site, a catalytic triad formed by GLY-23, CYS-25 and GLY-65 [26]. Furthermore, some amino acid residues have been revealed to be involved in enzymatic catalysis of cruzipain such as residues GLN-19, GLY-66, ASP-158, HIS-159 and TRP-177 that keep peptidyl inhibitors anchored to the active site [27]. A molecular docking analysis with 173 compounds reported to be inhibitors of cruzipain showed that in addition to the catalytic triad, the amino acid residues GLN-19 and ASP-158 are involved in ligand interactions with a frequency of 47 % and 44 %, respectively [26].
Selectivity is a well-accepted parameter of the safety and pharmacological activity of a compound [28]. Bufadienolides from oocytes of R. alata were evaluated against kidney monkey Vero cells. The bufadienolides 1 (EC50 = 22.80 µM, S.I. > 5) and 7 (EC50 = 3.93 µM, S.I. = 2.7) presented significant selectivity (Table 2). Although bufadienolide 7 shows a promising S.I., less cytotoxicity is required. On the contrary, the low cytotoxicity showed by the bufadienolide 1 highlights its potential for further studies. Recently, 19-hydroxy-bufalin was evaluated against Vero cells and its low cytotoxicity, in comparison with bufadienolide 7, suggest that hydroxyl at position 19 decrease cytotoxic effects against mammals [21]. Despite of the potent anti-trypanosomal effect shown by compounds 3, 5 and 6, these bufadienolides were toxic to Vero cells, resulting in a reduced selectivity for the assayed parasite. In a previous study, hellebrigenin and telocinobufagin, two bufadienolides with antiprotozoal activity were tested for cytotoxicity on mouse macrophages and erythrocytes, showing no toxic effects [19]. However, it has been found that rodent cells are over 1000-fold more resistant than human cells to cytotoxic effects of cardiotonic sterols [29]. Differential cytotoxicity and water solubility are great attributes for potential anti-trypanosomals. In this context, microbial transformations carried out with bufadienolides 4, 5 and 7 have produced nontoxic derivatives to human cells [30,31,32]. Nanoparticles containing bufadienolide 7 were injected in mice xenografted with human colon cancer cells, and their therapeutic potential was tested. The particles were able to inhibit the growth of tumor cells with efficient tumor targeting and cellular uptake [33].
Some bufadienolides have been found to be active against amphibian pathogens. Arenobufagin, γ-bufotalin and telocinobufagin isolated from the skin secretion of the boreal toad Anaxyrus boreas inhibit the harmful fungus Batrachochytrium dendrobatidis in vitro [34]. Amphibians are also affected by parasitic diseases caused by protozoa [35]. In fact, different families of anurans, including Bufonidae, are hosts of trypanosomes and are frequently parasitized by more than one species [36]. Blood samples from R. alata have not been examined for the presence of trypanosomes; however, congeneric species, as well as syntopic species such as the frog Engystomops pustulosus, are known to be parasitized by trypanosomes [37,38]. Trypanosomiasis in amphibians is characterized by anemia, food refusal, listlessness and localized hemorrhages with swollen lymph glands. Species of leeches from the family Hirudinea are considered to be the aquatic vectors of amphibian trypanosomes, while mosquitos, sand flies and midges, their main terrestrial vectors [37,38,39]. The presence of anti-trypanosomal bufadienolides in oocytes of toads is intriguing. Bufonids biosynthesize bufadienolides, as shown by studies using cholesterol marked isotopically [40]. These compounds have been isolated from parotoid glands and skin of toads, as well as from their plasma, bile, ovaries and oocytes [17,41,42,43]. Because they inhibit (Na+-K+)ATPase pump, bufadienolides are considered as possible regulators of ionic equilibrium in anurans, and the high levels of bufadienolides present in some bufonid species presumably serve as defense against predation [44]. However, a possible role as endogenous antiprotozoal substances should not be discarded.
3. Materials and Methods
3.1. General Experimental Procedures
Optical activity was determined on a digital P-2000 polarimeter (Jasco, Easton, MD, USA) at room temperature. Ultraviolet spectra were registered using a UV-2401 PC recording spectrophotometer (Shimadzu, Columbia, MD, USA). Infrared spectra were measured using a Platinum ATR Alpha instrument (Bruker, Billerica, MA, USA). Reverse-phase high-performance liquid chromatography was carried out on an Agilent 1100 HPLC equipped with a Diode Array detector 1200 series (Agilent, Santa Clara, CA, USA). Nuclear magnetic resonance (NMR) experiments were carried out at 25 °C on an Eclipse+ 400 Fourier transform spectrometer with a field strength of 9.38 T and equipped with a 5 mm TH tunable probe (JEOL, Peabody, MA, USA). Chemical shifts (δ) were expressed in parts per million (ppm) and coupling constants (J) in Hz. NMR spectra were processed using the MestReNova software version 12.0.3-21384 (© Mestrelab Research S.L., Spain) and referenced based on the residual signal of chloroform-d (δH 7.26 and δC 77.00), dimethyl sulfoxide-d (δH 2.50 and δC 39.51) or methanol-d (δH 3.31 and δC 49.00). For High Resolution Mass Spectrometry analysis, compounds were diluted in methanol and mass spectra were acquired on either a micrOTOF-QIII™ spectrometer (Bruker Daltonics, Billerica, MA) or a MALDI-TOF-TOF Ultraflex (Bruker Daltonics, Billerica, MA, USA). For HR-ESI-Q-TOF analysis, compounds were directly infused into the spectrometer and MS spectra were obtained in positive electrospray ionization mode in the range of 50 to 2500 mass to charge ratio (m/z) and capillary voltage set at 4500 V. Nitrogen was used as the nebulizer gas at 2.0 bar, 200 °C and 9.0 L/min. External calibration was performed with Agilent ESI-L Low Concentration Tuning Mix (Agilent Technologies, Santa Clara, CA) prior and throughout data collection. Hexakis (1H, 1H, 2H-di-fluoro-ethoxy-phosphazene; m/z 622.0290 [M+H]+) (Synquest Laboratories, Alachua, FL) was used as internal standard. For HR-MALDI-TOF analysis, the samples were transferred to a MTP plate (Bruker Daltonics, Billerica, MA, USA), and mixed with the α-cyano-4-hydroxy-cinammic acid matrix (Sigma Aldrich). All experiments were carried out in positive mode. Pepcalibstandard was used as external calibrant (Bruker, Daltonics, Billerica, MA, USA).
3.2. Sample collection
Ninety grams of oocytes were obtained from six gravid females of R. alata collected at Parque Nacional Soberanía, Republic of Panama (9.134994° N 79.722597° W). These gravid females spontaneously laid their oocytes when held in isolation inside plastic containers. The toads were collected under the authorization of the Ministry of Environment of Panama (Permit: SE/AQ-2-14) and released at the same collection site. The Ministry of Environment of Panama reviewed and approved the experimental protocols of the study before granting collection permits. All methods were carried out in accordance with relevant guidelines and regulations of the INDICASAT. All methods are reported in accordance with ARRIVE guidelines.
3.3. Extraction and Isolation of bufadienolides
Freshly collected oocytes were macerated with 100 mL of chloroform-methanol (1:1) three times overnight in a shaker at room temperature. Extractions were pooled and dried under reduced pressure. The pool was suspended with deionized water and extracted with equal volume of chloroform-methanol (2:1). The organic phase was dried to provide 641.2 mg of residue. This extract was loaded onto SupelcleanTM LC-18 SPE cartridges (Supelco, Bellefonte, PA, USA) and fractionated by employing a stepwise gradient of methanol in water at 40, 80 and finally to 100 % of methanol to produce fractions 1-3, respectively. Solvent was removed by rotary evaporation under vacuum and the remaining water was removed by lyophilization. Fraction 3 was diluted in methanol and the compounds were separated by HPLC. We used a semipreparative Synergy Hydro C18 column (250 x 10 mm, 4 µm, 80 Å) (Phenomenex, Torrance, CA, USA). The mobile phase consisted of water with 0.1 % of trifluoroacetic acid (TFA) (Solvent A), and a mixture of methanol-acetonitrile (1:1) acidified with TFA at 0.1 % (Solvent B). Elution detection was performed at λ 300 nm at a flow rate of 1 mL/min. The fractions were collected individually and as result the isolation of compounds 1 (5.7 mg, Rt 42.1 min), 2 (8.3 mg, Rt 63.0 min), 3 (1.7 mg, Rt 65.8 min), 4 (3.4 mg, Rt 67.5 min), 5 (4.4 mg, Rt 70.7 min), 6 (2.1 mg, Rt 75.4 min) and 7 (2.4 mg, Rt 77.1 min) was achieved. The purity of all compounds was found to be higher than 95 % as analyzed by 1H NMR spectroscopy.
3.4. Compounds characterization data
16β-hydroxyl-hellebrigenin (1): yellow solid; for 1H and 13C NMR data see supplementary information file; HR-ESI-MS m/z 433.2222 [M + H]+ (calculated for C24H33O7, 433.2221).
Desacetyl-bufotalin (2): yellow solid; For 1H and 13C-NMR data see supplementary information file. HR-ESI-MS m/z 403.2472 [M + H]+ (calculated for C24H35O5, 403.2479).
19-formyl-dyscinobufotalin (3): white solid; [α]= + 11° (0.001 c, methanol); UV (CH3OH) λmax (log ɛ) 201 nm (4.45) 300 nm (3.90); IR υmax 3589, 2864, 1715 cm-1; for 1H and 13C NMR data see Table 1 and spectra in supporting information (Figures S2–S7); HR-MALDI-MS m/z 487.2283 [M + H]+ (calculated for C27H35O8, 487.2326) (Figure S8).
Bufotalin (4): white solid; For 1H and 13C-NMR data see supplementary information file. HR-ESI-MS m/z 445.2616 [M + H]+ (calculated for C26H37O6, 445.2585).
Cinobufotalin (5): yellow solid; For 1H and 13C-NMR data see supplementary information file. HR-ESI-MS m/z 459.2385 [M + H]+ (calculated for C26H35O7, 459.2377).
Dyscinobufotalin (6): white solid; For 1H and 13C-NMR data see supplementary information file. HR-ESI-MS m/z 473.2518 [M + H]+ (calculated for C27H37O7, 473.2534).
Bufalin (7): white solid; For 1H and 13C-NMR data see supplementary information file. HR-ESI-MS m/z 387.2512 [M + H]+ (calculated for C24H35O4, 387.2530).
3.5. Anti-trypanosomal activity
The evaluation of activity against trypanosomes was carried out using the recombinant strain Tulahuen, clone C4, of T. cruzi trypomastigotes (ATCC, Manassas, VA, USA). This strain expresses the β-galactosidase enzyme [45]. The parasites were maintained at 37 °C under a 5 % CO2 atmosphere in RPMI-1640 culture medium with L-glutamine,4-(2-hydroxyethyl)-piperazine-1-ethanesulfonic acid (HEPES) buffer, NaHCO3, 10 % FBS and 0.05 % gentamicin (50 mg/mL) as supplements. Epithelial kidney monkey Vero cells from the American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured for 24 hours, and one day prior to the addition of the compound, cells were infected with trypomastigotes. After additional 24 hours of infection, bufadienolides (1-7) were dissolved in di-methyl-sulfoxide and tested at 10, 2, 0,4 and 0.08 µg/mL for five days. Benznidazole was used as positive control. To determine the anti-trypanosomal activity, chlorophenol-red-β-D-galactopyranoside (Roche Applied Science) was added to each well and allowed to react with the β-galactosidase of the remaining living parasites for 4.5 hours. Absorbance was calculated at 570 nm using a microtiter plate reader (Sinergy HT, BioTek Instruments Inc., Winooski, VT, USA).
3.6. Mammalian cytotoxicity assay
Epithelial kidney monkey Vero cells were cultivated in 96-well plates at 37 °C under a 5% CO2 atmosphere, using RPMI-1640 medium (Sigma-Aldrich, USA) sterilized with 0.05 % gentamicin (50 mg/mL) and supplemented with 10 % FBS (fetal bovine serum; Gibco, Invitrogen, Carlsbad, CA, USA). Vero cells were allowed to adhere for 24 hours prior to incubation for five days with the samples. During the treatment, dimethyl-sulfoxide was used as negative control and doxorubicin as positive control. After incubation, 3-(4, 5-di-methyl-thiazol-2-yl)-2, 5-di-phenyl-tetra-zolium bromide was added to each well and absorbance was determined 4 hours later at 570 nm, using a color plate reader. Cytotoxicity was evaluated colorimetrically by calculating the ability of the remanent living Vero cells to reduce the pale yellow MTT into the black purple formazan product, as described [46].
3.7. Statistical analysis of bioassays
All anti-trypanosomal and cytotoxicity assays represent independent analysis and were performed in duplicate. The statistical analysis for the 50 % inhibiting (IC50) and cytotoxic (CC50) concentration values were carried out employing the Data Analysis complement Wizard of Excel 2000 by adjusting the dose-response curve to a sigmoidal model (Microsoft, Seattle, WA, USA).
3.8. Molecular docking
Molecular docking analysis was performed with the Molecular Operating Environment (MOE), software version 2018.01 (Chemical Computing Group, Montreal, QC, Canada). Molecular docking was carried out by allowing ligands to move keeping proteins rigid. Cruzipains and ligands were prepared with the standard protocol of MOE. The X-ray crystallographic data of twenty-four proteins were extracted from the Protein Data Bank. The structures of the proteins were corrected on its misallocated amino acid residues, protonated in 3D and the energies were minimized using the MMFF94x force field. Downloaded proteins were aligned before docking simulation. All cruzipains showed protein similarity from 99.1 to 100 %.
Validation of the protocol was carried out by redocking of the cruzipains 1EWO [47], 2AIM [48], 1U9Q [49] and 4XUI [50] (resolution between 2.10 - 2.51 Å); with their respective co-crystallized ligands VSC, ZRA, 186 and 2VC. Co-crystallized ligands were subjected to a simulated auto mode coupling induced fit. The binding pocket was defined as the set of amino acid residues within 4.5 Å from the co-crystallized ligand. Bufadienolides (1, 3, 7) were selected for molecular docking with cruzipain since they present the highest bioactivity and a significant selectivity in vitro. The docking conformations that showed lowest docking scores were selected for analysis.
4. Conclusions
Seven steroids from the bufadienolide class were isolated from the oocytes of the toad R. alata. Among them, 19-formyl-dyscinobufotalin (3) is reported as new natural product. While compound 2 was the only one reported as inactive, new bufadienolide 3 showed the most potent anti-trypanosomal activity, even more active than the control drug benznidazole. The in vitro assays and structural-activity relationship analysis suggest the formyl group at C-19 and esterification of the hydroxy group at C-16 increases anti-trypanosomal activity. 16β-hydroxyl-hellebrigenin showed significant selectivity and a promising therapeutic window. Furthermore, molecular docking analysis revealed bufadienolides 1, 3 and 7 interacts with the main protease from T. cruzi, cruzipain, at the amino acid residues GLN-19, ASP-158, HIS-159 and TRP-177 through H-bonds. Chemical interactions involve the groups hydroxyl at positions C-5, and C-14 and formyl at C-19 at the cruzipain active site. Given the lack of therapeutic options to currently treat the American trypanosomiasis, this work can serve as the basis for further studies that aim the development of bufadienolides or their derivatives as drugs against Chagas disease.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: RP-HPLC chromatogram profile of fraction-3 from oocytes of Rhinella alata; Figure S2: 1H-NMR spetrum of 19-formyl-dyscinobufotalin (3); Figure S3: 13C-NMR spetrum of 19-formyl-dyscinobufotalin (3); Figure S4: DEPT spectrum comparison of 19-formyl-dyscinobufotalin (3); Figure S5: HMBC spetrum of 19-formyl-dyscinobufotalin (3); Figure S6: HSQC spetrum of 19-formyl-dyscinobufotalin (3); Figure S7: COSY spetrum of 19-formyl-dyscinobufotalin (3); Figure S8: HR-MALDI-MS spetrum of 19-formyl-dyscinobufotalin (3); Figure S9: Alignment and superposition of 24 cruzipains and cocrystallized ligands; NMR data for compounds 1, 2, 4-7 isolated from the oocytes of Rhinella alata; Figure S10: 1H NMR spectra for compound 16β-Hydroxyl-hellebrigenin (1); Figure S11: 1H NMR spectra for compound Desacetyl-bufotalin (2); Figure S12: 1H NMR spectra for compound Bufotalin (4); Figure S13: 1H NMR spectra for compound Cinobufotalin (5); Figure S14: 1H NMR spectra for compound Dyscinobufotalin (6); Figure S15: 1H NMR spectra for compound Bufalin (7); Supplementary references.
Author Contributions
Conceptualization, M.G., and C.R.; methodology, M.G., C.R., C.S. A.A.D.-A.; software, D.O.; validation, M.G., C.R., D.O. and C.S.; formal analysis, M.G., C.R., M.N., D.O. and C.S.; investigation, C.R., M.G., C.S. M.N., R.I.; resources, M.G., C.S., R.I.; data curation, C.R., M.G. C.S., D.O.; writing—original draft preparation, C.R., M.G., A.A.D.-A., R.I., D.O.; writing—review and editing, M.G., C.S. A.A.D.-A., R.I., D.O.; visualization, C.R., M.G.,; supervision, M.G., C.S.; project administration, M.G.; funding acquisition, M.G., C.S., R.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the INDICASAT-BID Program (grant number IND-JAL-05). C.R., R.I., A.A.D.-A., C.S. and M.G. were supported by the Sistema Nacional de Investigación (SNI), Panama.
Institutional Review Board Statement
The toads were collected under the authorization of the Ministry of Environment of Panama (Permit: SE/AQ-2-14) and released at the same collection site. The Ministry of Environment of Panama reviewed and approved the experimental protocols of the study before granting collection permits. All methods were carried out in accordance with relevant guidelines and regulations of the INDICASAT. All methods are reported in accordance with ARRIVE guidelines.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available in the main text or the Supplementary Materials.
Acknowledgments
CR was supported by a scholarship of the Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU), Panama and The Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT), Panama (No. 270-2014-007). We gratefully acknowledge the Government of Panama (MiAmbiente) for granting permission to collect the frogs used in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Steverding, D. The history of Chagas disease. Parasites and Vectors, 7. [CrossRef]
- Centers for Disease Control (CDC) - Chagas Disease. Available online: https://www.cdc.gov/parasites/chagas/index.html (accessed on 21 December 2020).
- Pérez-Molina, J.P. and Molina, I. Chagas disease. Lancet, 2017, 391, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Sueth-Santiago, V.; Decote-Ricardo, D.; Morrot, A.; Freire-de-Lima, C.G.; Freire-Lima, M.E. Challenges in the chemotherapy of Chagas disease: Looking for possibilities related to the differences and similarities between the parasite and host. World J. Biol. Chem. 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Verpoorte, R.; Phax-quôc-Kinh; Svendsen, A.B. Chemical constituents of Vietnamese toad venom, collected from Bufo Melanostictus Schneider. Part II. The bufadienolides. J. Nat. Prod. 1980, 43, 347–352. [CrossRef]
- Sousa, L.Q.; Machado, K.C.; Carvalho, O.S.F.; Silva, A. L.; Moncao-Filho, E.S.; Melo-Cavalcante, A.A.; Vieira-Júnior, G.M.; Pinheiro, F.P.M. Bufadienolides from amphibians: a promising source of anticancer prototypes for radical innovation, apoptosis triggering and Na+/K+-ATPase inhibition. Toxicon 2017, 127, 63–76. [Google Scholar] [CrossRef]
- Steyn, P.S.; Van Heerden, F.R. Bufadienolides of plant and animal origin. Nat. Prod. Rep. 1998, 15, 397–413. [Google Scholar] [CrossRef]
- Rodríguez, C.; Rollins-smith, L.; Ibáñez, R.; Durant-Archibold, A.A.; Gutierrez, M. Toxins and pharmacologically active compounds from species of the family Bufonidae (Amphibia, Anura). J. Ethnopharmacol. 2017, 235–254. [Google Scholar] [CrossRef]
- Santos, S.P.; Ibáñez, R.; Ron, S.R. Systematics of the Rhinella margaritifera complex (Anura, Bufonidae) from western Ecuador and Panama with insights in the biogeography of Rhinella alata. Zookeys 2015, 145, 109–145. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhao, X.; Zhong, W.T.; Jiao, F.R.; Wang, X.L.; Ma, L.; Duan, D.Z.; Yang, D.S.; Tang, S.Q. Bufadienolides from the venom of Bufo gargarizans and their enzyme inhibition activities and brine shrimp lethality. Nat. Prod. Commun. 2018, 13, 827–830. [Google Scholar] [CrossRef]
- Krenn, L.; Stapf, V.; Kopp, B. Bufadienolides from Drimia robusta BAK. Scientia Pharmazeutica 2000, 68, 421–427. [Google Scholar] [CrossRef]
- Kamano, Y.; Nogawa, T.; Yamashita, A.; Pettit, G.R. The 1H and 13C NMR chemical shift assignments for thirteen bufadienolides isolated from the traditional Chinese drug Ch’an Su. Collect. Czechoslov. Chem. Commun. 2001, 66, 1841–1848. [Google Scholar] [CrossRef]
- Ondetti, M.A.; Williams, N.J.; Sabo, E.; Josip, P.F.; Weaver, E.R.; Kocy, O. Angiotensin-converting enzyme inhibitors from the venom of Bothrops jararaca. Isolation, elucidation of structure, and synthesis. Biochemistry 1971, 10, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Katsuyama, S.; Orito, T.; Suzuki, T.; Sakurada, S. Antinociceptive effect of tebanicline for various noxious stimuli-induced behaviours in mice. Neurosci. Lett. 2017, 638, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Spande, T.F.; Garrafo, H.M.; Edwards, M.W.; Yeh, H.J.C.; Pannell, L.; Daly, J.W. Epibatidine: a novel (Chloropyridy1)azabicycloheptane with potent analgesic activity from an Ecuadorian poison frog. J. Am. Chem. Soc. 1992, 114, 3475–3478. [Google Scholar] [CrossRef]
- Gao, H.; Zehl, M.; Leitner, A.; Wu, X.; Wang, Z.; Kopp, B. Comparison of toad venoms from different Bufo species by HPLC and LC-DAD-MS/MS. J. Ethnopharmacol. 2010, 131, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.W.; Tian, H.Y.; Nie, Q.L.; Wang, L.; Zhou, S.W.; Ye, W.C.; Zhang, D.M.; Jiang, R.W. Structures and inhibitory activity against breast cancer cells of new bufadienolides from the eggs of toad: Bufo bufo gargarizans. RSC Adv. 2016, 6, 93832–93841. [Google Scholar] [CrossRef]
- Servillon, R.J.T.; Dingal, M.C.; Lusica, M.C.; Yamson, M.K.A.; Balonebro, M.J.B.; Alzate, F.B. Phytochemical screening and in vitro antibacterial evaluation of Persea americana (Avocado) crude peel extract. Optima 2013, 1, 96. [Google Scholar]
- Tempone, A.G.; Pimenta, D.C.; Lebrun, I.; Sartorelli, P.; Taniwaki, N.N.; De Andrade, H.F., Jr.; Antoniazzi, M.M.; Jared, C. Antileishmanial and anti-trypanosomal activity of bufadienolides isolated from the toad Rhinella jimi parotoid macrogland secretion. Toxicon 2008, 52, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, D.; Li, Y.; Chen, W.; Ruan, Z.; Deng, L.; Wang, L.; Tian, H.; Yiu, A.; Fan, C.; Luo, H.; Liu, S.; Wang, Y.; Xiao, G.; Chen, L.; Ye, W. Discovery of bufadienolides as a novel class of ClC-3 chloride channel activators with antitumor activities. J. Med. Chem. 2013, 56, 5734–5743. [Google Scholar] [CrossRef]
- Rodriguez, C.; Ibáñez, R.; NG, M.; Spadafora, C.; Durant-Archibold, A.A.; Gutiérrez, M. 19-Hydroxy-bufalin, a major bufadienolide isolated from the parotoid gland secretions of the Panamanian endemic toad Rhinella centralis (Bufonidae), inhibits the growth of Trypanosoma cruzi. Toxicon 2020, 177, 89–92. [Google Scholar] [CrossRef]
- Córdova, W.H.P.; Leitao, S.G.; Cunha-Filho, G.; Bosch, R.A.; Pascual, I.A.; Pereda-Miranda, R.; Gervou, R.; Arújo, T.N.; Quintas, M.L.E.; Noel, F. Bufadienolides from the parotoid gland secretions of Cuban toad Peltophryne fustiger (Bufonidae): inhibition of human (Na+/K+)-ATPase activity. Toxicon 2016, 110, 27–34. [Google Scholar] [CrossRef]
- Van Der Heyden, N.; Docampo, R. Proton and sodium pumps regulate the plasma membrane potential of different stages of Trypanosoma cruzi. Mol. Biochem. Parasitol. 2002, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kyoichi, I.; Mikami, Y.; Hashimoto, M.; Nara, T.; Hara, Y.; Aoki, T. Molecular cloning and characterization of ouabain-insensitive Na+-ATPase in the parasitic protist, Trypanosoma cruzi. Biochim. Biophys. Acta 2006, 1758, 738–746. [Google Scholar] [CrossRef]
- Kamano, Y.; Kotake, A.; Hashima, H.; Inoue, M.; Morita, H.; Takeya, K.; Itokawa, H.; Nandachi, N.; Segawa, T.; Yukita, A.; Saitou, K.; Katsuyama, M.; Pettit, F.G.R. Structure-cytotoxic activity relationship for the toad poison bufadienolides. Bioorg. Med. Chem. 1998, 6, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Silva-Junior, E.F.; Barcellos, F.P.H.; Ribeiro, F.F.; Bezerra, M.J.; Scotti, L.; Tullius, S.M.; De Aquino, T.M.; De Araujo-Junior, J.X. Molecular docking studies applied to a dataset of cruzain inhibitors. Curr. Comput. Aided. Drug Des. 2017, 14, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Robertson, S.A.; Brinen, L.S.; McKerrow, J.H. Cruzain: the path from target validation to the clinic. Adv. Exp. Med. Biol. 2011, 712, 100–115. [Google Scholar] [CrossRef]
- Nogueira, R.C.; Oliveira-Costa, J.F.; De Sá, M.S.; Dos Santos, R.R.; Soares, M.B.P. Early toxicity screening and selection of lead compounds for parasitic diseases. Curr. Drug Targets 2009, 10, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Montaño, J.; Burgos-Morón, E.; López-Lázaro, M. The in vivo antitumor activity of cardiac glycosides in mice xenografted with human cancer cells is probably an experimental artifact. Oncogene 2014, 33, 2947–2948. [Google Scholar] [CrossRef]
- Lu, J.; Deng, S.; Chen, H.; Hou, J.; Zhang, B.; Tian, Y.; Wang, C.; Ma, X. Microbial transformation of cinobufotalin by Alternaria alternate AS 3.4578 and Aspergillus niger AS 3.739. J. Mol. Catal. B Enzym. 2013, 89, 102–107. [Google Scholar] [CrossRef]
- Xiu-Lan, X.; Li-Bin, Z.; Feng-Yun, L.; Xiao-Chi, M.; Ke-Xin, L.; Jian, H.; De-An, G. Microbial transformation of bufotalin by Alternaria alternata AS 3.4578. J. Asian Nat. Prod. Res. 2009, 11, 7–11. [Google Scholar] [CrossRef]
- Ye, M.; Han, J.; Tu, G.; An, D.; Guo, D. Microbial hydroxylation of bufalin by Cunninghamella blakesleana and Mucor spinosus. J. Nat. Prod. 2005, 68, 626–628. [Google Scholar] [CrossRef]
- Yin, P.; Wang, Y.; Qiu, Y.Y.; Hou, L.; Liu, X.; Qin, J.; Duan, Y.; Liu, P.; Qiu, M.; Li, Q. Bufalin-loaded mPEG-PLGA-PLL-cRGD nanoparticles: preparation, cellular uptake, tissue distribution, and anticancer activity. Int. J. Nanomedicine 2012, 7, 3961–3969. [Google Scholar] [CrossRef]
- Barnhart, K.; Forman, M.E.; Umile, T.P.; Kuenemn, J.; McKenzie, V.; Salinas, I.; Minbiole, K.P.C.; Woodhams, D.C. Identification of bufadienolides from the boreal toad, Anaxyrus boreas, active against a fungal pathogen. Microb. Ecol. 2017, 74, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Densmore, C.L.; Green, D.E. Diseases of Amphibians. Inst. fo Lab. Anim. Res. J. 2007, 48, 235–254. [Google Scholar] [CrossRef]
- Gupta, D.K.; Gupta, N.; Gangwar, R. Infectivity of Bufo melanostictus (Amphibia: Bufonidae) to two new species of haematozoan parasites from Rohilkhand, India. Proc. Zool. Soc. 2012, 65, 22–32. [Google Scholar] [CrossRef]
- Ferreira, J.I.G.S.; Da Costa, A.P.; Ramirez, D.; Roldan, J.A.M.; Saraiva, D.; Founier, S.G.F.R.; Sue, A.; Zambelli, E.R.; Minervino, H.A.H.; Verdade, K.V.; Gennari, M.S.; Marcili, A. Anuran trypanosomes: phylogenetic evidence for new clades in Brazil. Syst. Parasitol. 2015, 91, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Bernal, X.E.; Pinto, C.M. Parasites and wildlife sexual differences in prevalence of a new species of trypanosome infecting túngara frogs. Int. J. Parasitol. Parasites Wildl. 2016, 5, 40–47. [Google Scholar] [CrossRef]
- Barta, J.R.; Desser, S.S. Blood parasites of amphibians from Algonquin Park, Ontario. J. Wildl. Dis. 1984, 20, 180–189. [Google Scholar] [CrossRef]
- Garraffo, H.M.; Gras, E.G. Biosynthesis of bufadienolides in toads VI. Experiments with [1,2-3H] cholesterol, [21-14C] coprostanol, and 5-beta-[21-14C] pregnonolone in the toad Bufo arenarum. Steroids 1986, 48, 251–257. [Google Scholar] [CrossRef]
- Akizawa, T.; Mukai, T.; Matsukawa, M.; Yoshioka, M.; Morris, J.F.; Butler, V.P. Structures of novel bufadienolides in the eggs of a toad, Bufo marinus. Chem. Pharm. Bull. 1994, 42, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Lichtstein, D.; Gati, I.; Haver, E.; Katz, U. Digitalis-like compounds in the toad Bufo viridis: tissue and plasma levels and significance in osmotic stress. Life Sci. 1992, 51, 119–128. [Google Scholar] [CrossRef]
- Lee, S.S.; Derguini, F.; Bruening, R.C.; Nakanishi, K.; Wallick, E.T.; Akizawa, T.; Rosenbaum, C.S.; Butler, V.P. Digitalis-like compounds of toad bile: sulfation and reduction of bufadienolides decrease potency of Na+, K+-ATPase inhibition. Heterocycles 1994, 39, 669–686. [Google Scholar]
- Flier, J.; Edwards, M.; Daly, J.W.; Myers, C. Widespread occurrence in frogs and toads of skin compounds interacting with the ouabain site of Na+, K+-ATPase. Science 1980, 208, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Buckner, F.S.; Verlinde, C.L.M.J.; Flamme, A.C.L.A.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Brinen, L.S.; Gillmor, S.A. and Fletterick, R.J. RCSB PDB - 1EWO: The cysteine protease cruzain bound to WRR-204. Available online: https://www.rcsb.org/structure/1EWO (accessed on 7 January 2021).
- Gillmor, S.A.; Craik, C.S. and Fletterick, R.J. Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 1997, 6, 1603–1611. [CrossRef]
- Choe, Y.; et al. Development of alpha-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorganic Med. Chem. 2005, 13, 2141–2156. [Google Scholar] [CrossRef]
- Jones, B.D.; et al. Synthesis and evaluation of oxyguanidine analogues of the cysteine protease inhibitor WRR-483 against cruzain. Med. Chem. Lett, 2016; 7. [Google Scholar]
Figure 1.
Chemical structures of bufadienolides 1-7.
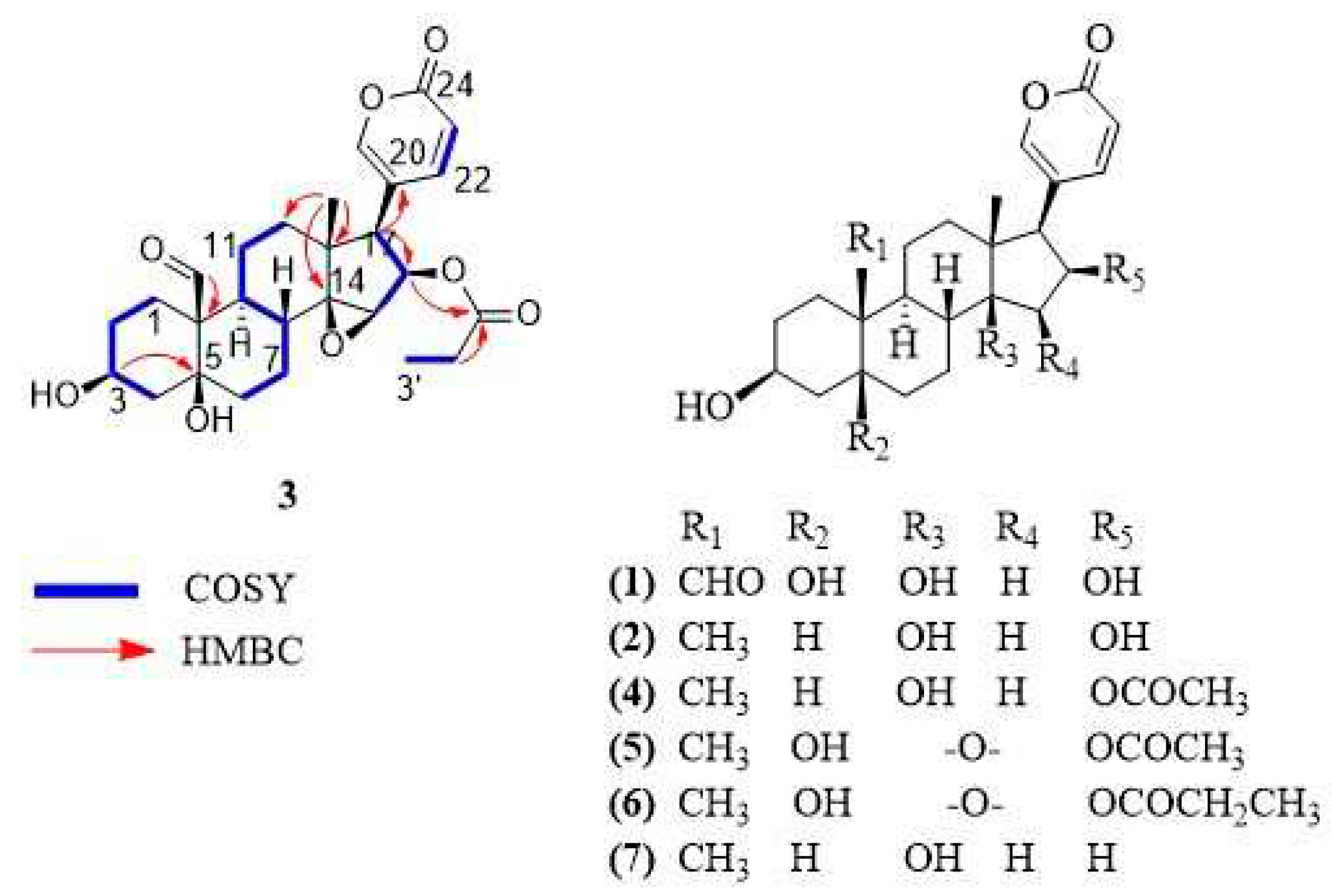
Figure 2.
Representation in 2D format for molecular docking of bufadienolides 1, 3 and 7 with active site amino acid residues of cruzipains. X-ray crystallographic structures of the proteins were extracted from the Protein Data Bank (PDB). Bufadienolide/cruzipain PDB ID= A: 16β-hydroxyl-hellebrigenin (1)/1EWO; B: 19-formyl-dyscinobufotalin (3)/1EWO; and C: bufalin (7)/2AIM.
Figure 2.
Representation in 2D format for molecular docking of bufadienolides 1, 3 and 7 with active site amino acid residues of cruzipains. X-ray crystallographic structures of the proteins were extracted from the Protein Data Bank (PDB). Bufadienolide/cruzipain PDB ID= A: 16β-hydroxyl-hellebrigenin (1)/1EWO; B: 19-formyl-dyscinobufotalin (3)/1EWO; and C: bufalin (7)/2AIM.
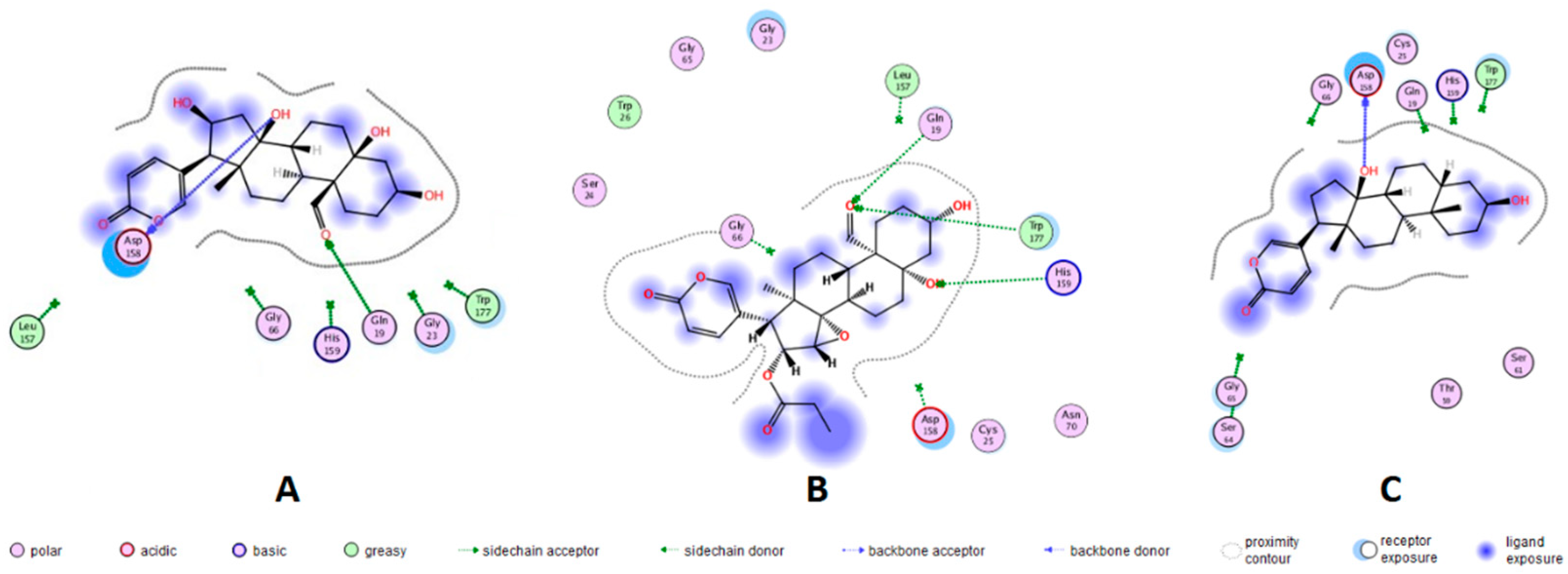

Table 1.
NMR (1H 400 MHz, 13C 100 MHz) data for compound 3 in methanol-d4 (δ in ppm, J, in Hz).
| No. | 13C, Type | 1H, Mult. (J in Hz) |
|---|---|---|
| 1a | 18.6, CH2 | 2.26, m |
| 1b | 1.77, m | |
| 2a | 27.5, CH2 | 1.77, m |
| 2b | 1.77, m | |
| 3 | 68.0, CH | 4.15, m |
| 4a | 36.7, CH2 | 1.99, m |
| 4b | 1.61, m | |
| 5 | 75.8, C | |
| 6a | 38.3, CH2 | 2.23, m |
| 6b | 1.49, m | |
| 7a | 24.1, CH2 | 1.77, m |
| 7b | 1.11, m | |
| 8 | 43.1, CH | 1.77, m |
| 9 | 34.4, CH | 2.47, td (3.8, 12.2) |
| 10 | 56.0, C | |
| 11a | 22.9, CH2 | 1.66, m |
| 11b | 1.39, m | |
| 12a | 40.5, CH2 | 1.77, m |
| 12b | 1.49, m | |
| 13 | 46.0, C | |
| 14 | 73.1, C | |
| 15 | 60.7, CH | 3.75, m |
| 16 | 76.3, CH | 5.47, m |
| 17 | 51.2, CH | 2.93, d (9.2) |
| 18 | 17.3, CH3 | 0.81, s |
| 19 | 210.1, CH | 10.00 s |
| 20 | 118.2, C | |
| 21 | 153.6, CH | 7.36, m |
| 22 | 150.9, CH | 8.02, m |
| 23 | 114.2, CH | 6.24, dd (0.8, 9.7) |
| 24 | 164.0, C=O | |
| 1' | 174.9, C=O | |
| 2'a | 28.1, CH2 | 2.23, m |
| 2'b | 2.11, m | |
| 3' | 9.3, CH3 | 0.94, t (7.8) |
Table 2.
Anti-trypanosomal activity and mammalian cytotoxic of bufadienolides 1-7 isolated from the oocytes of Rhinella alata.
Table 2.
Anti-trypanosomal activity and mammalian cytotoxic of bufadienolides 1-7 isolated from the oocytes of Rhinella alata.
| Compound | T. cruzi | Vero cell | Selectivity (CC50/EC50) |
|---|---|---|---|
| EC50 (µM) | CC50 (µM) | ||
| 1 | 22.80 ± 4.11 | > 135 | > 5 |
| 2 | Inactive | > 28 | ---- |
| 3 | 0.19 ± 0.07 | 0.22 ± 0.09 | 1.16 |
| 4 | 19.60 ± 2 | > 46 | > 2 |
| 5 | 27.80 ± 3.77 | 1.88 ± 0.20 | 0.07 |
| 6 | 12.30 ± 1.87 | 0.02 ± 0.002 | < 0.01 |
| 7 | 3.93 ± 0.57 | 10.61 ± 1.54 | 2.69 |
| benznidazole | 2.73 ± 0.10 | Not tested | ---- |
| doxorubicin | Not tested | 0.23 | ---- |
Table 3.
Calculations for molecular docking of bufadienolides with cruzipain.
| Bufadienolide | RMSD (Å) | Docking Score | Ligand group | Aminoacid residue |
Energy (kcal/mol) | Distance (Å) |
|---|---|---|---|---|---|---|
| 1 | 2.5812 | -4.2777 | CH=O at 19 | GLN-19 | -1.5 | 2.32 |
| OH at 14 | ASP-158 | -1.7 | 2.10 | |||
| 3 | 2.0099 | -5.3984 | CH=O at 19 | GLN-19 | -2.2 | 2.09 |
| CH=O at 19 | TRP-177 | -0.5 | 2.59 | |||
| OH at 5 | HIS-159 | -1.5 | 2.35 | |||
| 7 | 3.0381 | -4.1206 | OH at 14 | ASP-158 | +1.3 | 1.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



