
Submitted:
06 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Deeper understanding of biological processes, disease mechanisms, and prospective therapeutic targets can be attained by Gene Expression Network Analysis (GENA), which offers a potent framework for revealing the intricate regulatory mechanisms controlling gene expression. GENA is a computational method used to understand the intricate interactions and relationships between the genes in a biological system. It entails using network theory, statistical analysis, and gene expression data to pinpoint functional modules, regulatory linkages, and important genes or pathways involved in a particular biological process or illness. This chapter broadly outlines the principles and practice of GENA, to an novice reader and outlines a simple method of performing a GENA using online Rice gene expression datasets available in various websites.
Keywords:
network analysis
; genetics
; gene expression network analysis
; plants
Introduction
A computational method called gene expression network analysis is used to understand the intricate interactions and relationships between the genes in a biological system. It entails using network theory, statistical analysis, and gene expression data to pinpoint functional modules, regulatory linkages, and important genes or pathways involved in a particular biological process or illness.
Obtaining Gene Expression Data
Obtaining gene expression data, which can be produced using methods like microarrays or RNA sequencing, is the initial stage in gene expression network analysis. The expression levels of thousands of genes across various situations or samples are revealed by these data.
Next, noise is removed from the gene expression data, experimental biases are adjusted for,
Co-Expression Databases for Information
Resources that contain data on the co-expression of genes are called co-expression databases. Co-expression is the occurrence of coordinated gene expression of two or more genes. This can occur when genes are controlled by the same transcription factor or when they are involved in the same biological activity.
The identification of genes that are probably functionally linked can be done using co-expression databases. This can be helpful for a number of things, including:
Co-expression databases can be used to find genes that have not yet been implicated in a given biological process. Co-expression databases can be used to annotate genes with functional data, including the biological process in which they take part.
- Identification of illness genes: Co-expression databases can be used to find genes that are probably implicated in a given disease.
- Drug target identification: Genes that may be prospective drug targets can be found using co-expression databases.
Online internet gene information databases like, Coexpedia, Ingenuity Pathway, GeneFriends, COXPRESdb, and AnalysisDAVID, provide data on the gene co-expression in various cell types, tissues, or organisms. They might also keep records of the genes that are linked together in certain disorders or the functional interactions between different genes.
For deeper understanding of genes functionality and regulation, online co-expression databases represent an invaluable tool, The information is accurate based on experimental or computational data and expression values are normalized (Zhang & Horvath, 2005).
DNA or RNA Microarray
A collection of microscopic DNA or RNA spots adhered to a solid surface is known as a DNA or RNA microarray. DNA or RNA microarrays are used by scientists to genotype different parts of a genome or to evaluate the expression levels of several genes at once. Picomoles (10–12 moles) of a particular DNA or RNA sequence, known as probes (or reporters or oligos), are present in each DNA or RNA location. These can be a brief segment of a gene or other DNA element that is used to hybridize a target sample of cDNA or cRNA (also known as anti-sense RNA/ complementary RNA) under stringent conditions.
The following is the procedure for using a DNA or RNA microarray:
- A thin layer of DNA or RNA probes is placed on top of the microarray.
- A fluorescent dye has been used to label the target sample.
- The microarray is hybridized with the target sample.
- To find the labeled target molecules' fluorescence, a laser is used to scan the microarray.
- The degree of expression of each gene or DNA region is determined by measuring the fluorescence intensity at each site.
Figure 1.
Flowchart of RNA Microarray process (original illustration of authors).
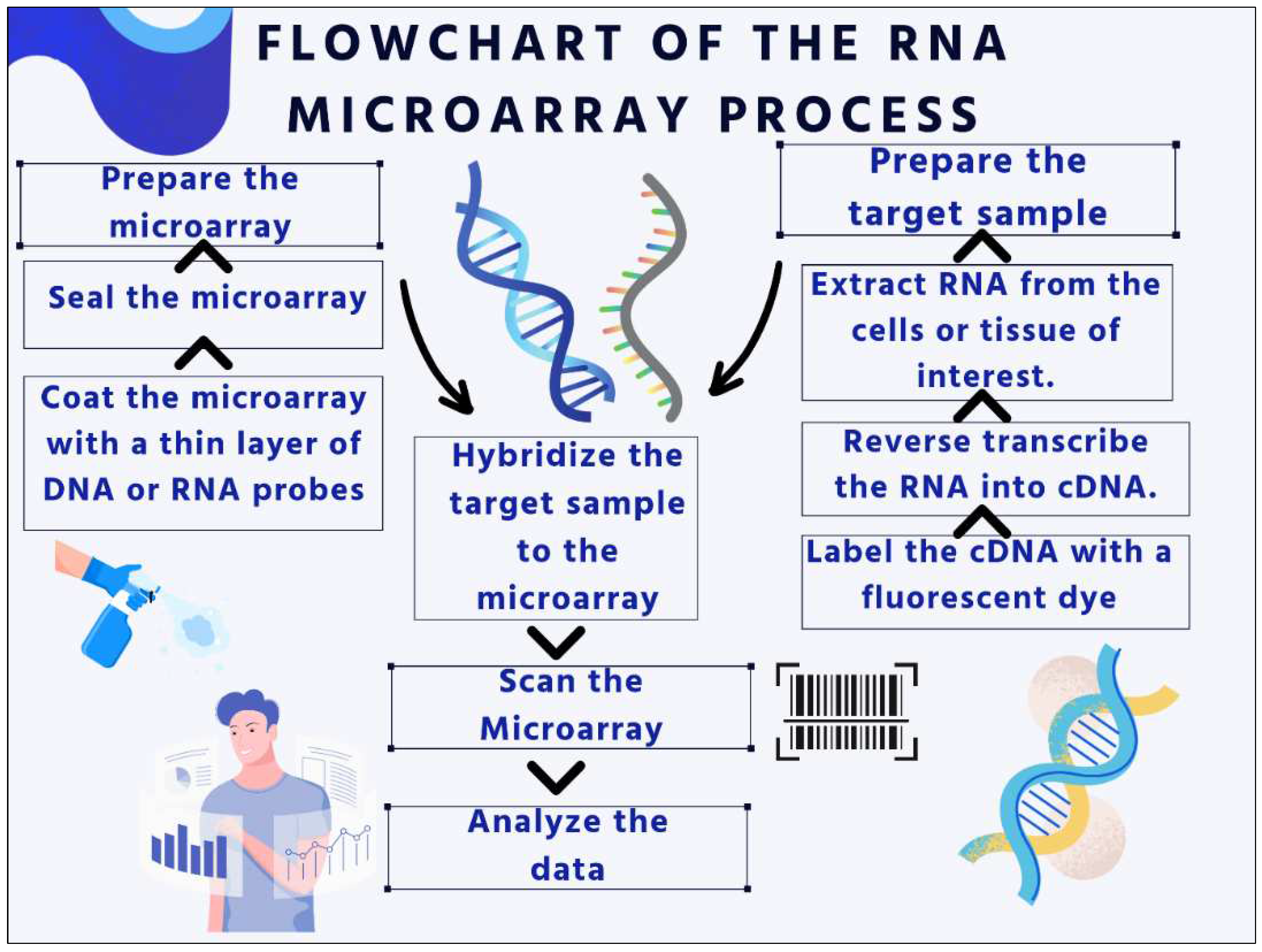
There are numerous uses for DNA or RNA microarrays, including:
- In a single experiment, thousands of genes' levels of expression are measured by a process called gene expression profiling. This can be used to investigate how various therapies affect gene expression or to find genes that are expressed differently in various cell types or tissues.
- Genotype analysis entails detecting whether a certain DNA mutation is present or absent. This can be used to determine who is at risk for contracting particular diseases or to diagnose hereditary diseases.
- In comparative genomic hybridization, the DNA composition of two or more samples is compared. Chromosome abnormalities, such as deletions or duplications, can be found with this method.
Obtaining gene expression data, which can be produced using methods like microarrays or RNA sequencing, is the initial stage in gene expression network analysis. The expression levels of thousands of genes across various situations or samples are revealed by these data.
Next, noise is removed from the gene expression data, experimental biases are adjusted for, and the expression values are normalized.
This helps to ensure sure that the data is organized properly for more analysis.
After the data has been prepared, network inference algorithms are used to build the gene expression network. Based on the patterns of gene expression, these algorithms use statistical and computational techniques to infer the regulatory links between genes. Common strategies include model-based strategies like Bayesian networks or ordinary differential equations as well as correlation-based strategies like Pearson correlation or mutual information (Nacu et al., 2007).
Network Analysis of Genetic Data
After the network has been constructed, numerous network analysis approaches are used to acquire understanding of the dynamics and functional architecture of the gene regulatory network. Detecting densely connected gene groups known as modules or clusters, as well as finding critical regulatory genes, may be necessary to uncover highly connected genes or "hubs" that provide important functions in the network (Zimmermann et al., 2005).
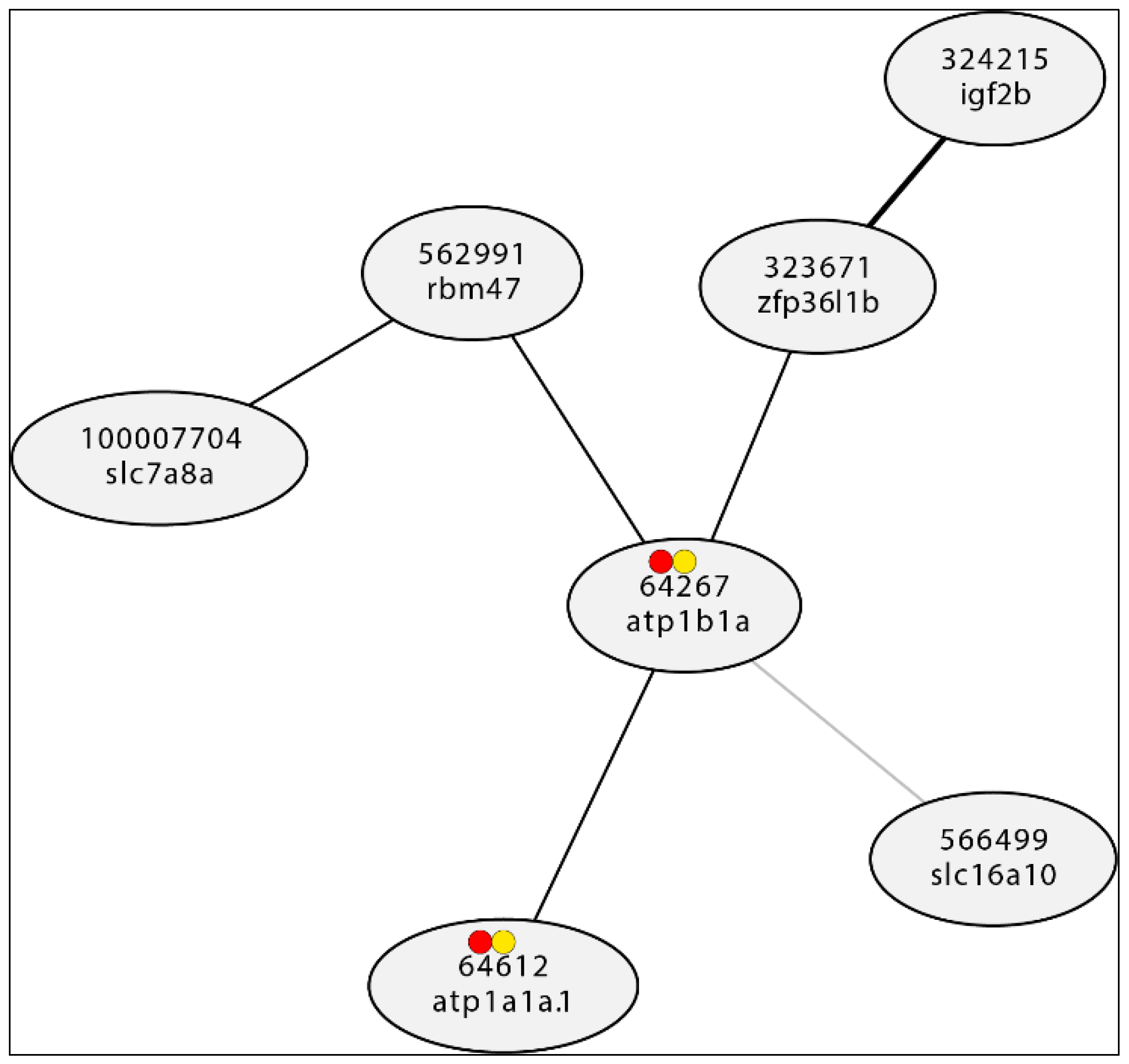
Figure 2.
Example of actual Gene Expression network drawn from expression information available at RiceFriend (RiceFREND is a japanese rice gene coexpression database consisting of a large online collection of microarray Data of rice https://ricefrend.dna.affrc.go.jp/)(Edwards, 2007 and Sato et al., 2012)).
Figure 2.
Example of actual Gene Expression network drawn from expression information available at RiceFriend (RiceFREND is a japanese rice gene coexpression database consisting of a large online collection of microarray Data of rice https://ricefrend.dna.affrc.go.jp/)(Edwards, 2007 and Sato et al., 2012)).
Scale-Free Gene Interaction Network
When a network is scale-free, its degree distribution has a power-law shape, which indicates that only a small number of nodes—known as "hubs" or "high-degree nodes"—have a lot of connections, compared to the vast majority of nodes. In contrast to this distribution, the degree distribution in a random network follows a bell-shaped normal distribution.
Scale-free gene interaction network analysis can provide details about the structure, modularity, and functional characteristics of biological systems. It can aid in locating key genes that are essential for preserving the stability and resilience of a network. The structure and characteristics of the network can also be studied to discover prospective therapeutic targets, understand disease causes, and forecast the consequences of perturbations or genetic variants on the network and cellular behavior.
A scale-free network with hubs indicates that a small number of nodes are crucial in tying together different portions of the network. The effectiveness and connectivity of the network can be greatly impacted by removing or interrupting these hubs. Scale-free networks are strengthened against random failures by this characteristic, but they become more susceptible to deliberate attacks on the high-degree nodes. Scale-free gene interaction networks are analyzed to reveal their biological importance using a variety of computational and statistical techniques, including network creation algorithms, network clustering, centrality measures, and module discovery methods.
Preferential attachment is a common theory to explain how scale-free networks arise. This mechanism states that new nodes entering the network are more likely to link to nodes that already have a large number of connections. As a result, well-connected nodes continue to gain connections over time, causing the power-law degree distribution. This phenomena is known as the "rich-get-richer" phenomenon (Rajamani & Iyer, 2023a)..
The World Wide Web, biological networks, social networks, and many other real-world systems all contain scale-free networks. Due to their special characteristics and implications for network dynamics, information dissemination, and resilience, they have received a great deal of attention in the field of network science (Sato et al., 2012).
The 'barabasi_albert_graph' function from Python NetworkX module is used in the code above to create a scale-free network using the Barabasi-Albert model. The 'n' parameter indicates the number of nodes, whereas the 'm' parameter indicates the number of edges to attach from a new node to existing nodes.
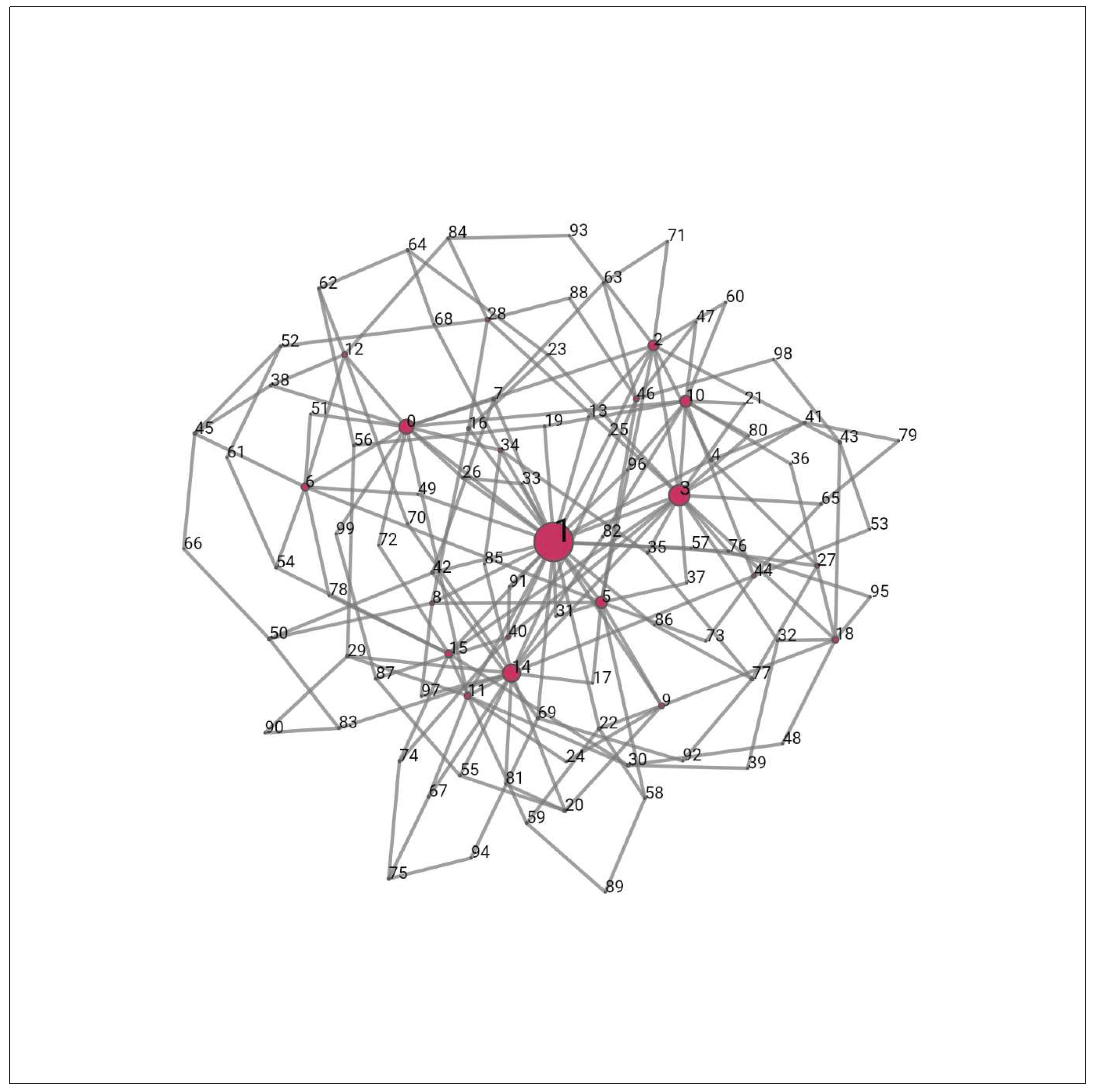
Figure 3.
Example of Scale-free network with hubs (large nodes) number 1, 14, 3,5, 26,10,2 and 0. The node size is proportional to the number of edges adjacent to it. This is a scale-free network of 100 nodes. Most nodes have only 2 edges, and hubs have multiple edges due to “preferential attachment”. (original illustration of authors) (Aslak & Maier, 2019).
Figure 3.
Example of Scale-free network with hubs (large nodes) number 1, 14, 3,5, 26,10,2 and 0. The node size is proportional to the number of edges adjacent to it. This is a scale-free network of 100 nodes. Most nodes have only 2 edges, and hubs have multiple edges due to “preferential attachment”. (original illustration of authors) (Aslak & Maier, 2019).
Formation of Scale-Free Networks
Hubs and preferential attachment are important ideas in network analysis that have to do with how scale-free networks are formed and structured.
- Hubs: Compared to other nodes in the network, hubs have a disproportionately large number of connections. In other words, they are nodes with lots of connections. Hubs are essential for preserving connectivity and the network's general structure in scale-free networks. They serve as hubs for the network's many nodes, facilitating communication and information exchange. Network connectivity may be adversely affected by the removal or disruption of hubs, leaving them open to deliberate attacks or malfunctions (Sato et al., 2012).
- In gene interaction networks, the edges stand in for interactions or connections between the genes' nodes, which represent genes. Protein-protein interactions are an example of a physical interaction. Regulatory interactions include gene regulation and signaling cascades.
- 2
- Preferential attachment: Preferential attachment is a process that accounts for the expansion of scale-free networks. According to this, new nodes joining a network are more likely to connect to nodes that are already connected to many other nodes. In other words, nodes with more connections and a higher degree are more likely to draw in new connections. The rich-get-richer phenomena, whereby well-connected nodes tend to assemble even more connections over time, is caused by this preferred attachment mechanism. As a result, a power-law degree distribution is formed, with most nodes having just a small number of connections and a select few nodes (hubs) having a huge number of connections(Sato et al., 2011).
- Preferential attachment has been noted in a number of real-world networks, including citation networks, social networks, the World Wide Web, and biological networks. It aids in the development of scale-free networks and provides an explanation for the establishment of hub-like nodes (Rajamani & Iyer, 2023b). Preferential attachment is a notion that has been extensively researched in the field of network science. It has implications for network resilience, information diffusion, and network growth.
- 3
- Power-law degree distribution: A certain structural feature of a complex network known as a power-law degree distribution is present in scale-free networks. The degree distribution of a network, in computing terminology, is the distribution of the number of connections (edges) that each node in the network possesses.
Given that gene interaction networks are scale-free, it follows that a limited subset of genes (hubs) are essential for tying together various genes or network modules. These hub genes are frequently linked to crucial signaling networks, fundamental cellular processes, or important biological activities. The integrity of the entire network and biological function may be significantly impacted by disruption or dysfunction of these hub genes (RiceFREND, n.d.).
This allows researchers to in locating important transcription factors or regulatory genes that regulate the expression of other genes. Additionally, regulatory patterns and signaling pathways within the network can be found with the aid of network analysis. Additionally, it can be used to compare networks between various samples or situations in order to spot changes in gene regulatory relationships linked to certain biological functions or disease states (Rajamani & Iyer, 2023c). Experimental validation methods like knockouts or overexpression of genes, chromatin immunoprecipitation, or reporter gene assays can be used to verify the results of gene expression network analysis. This allows researchers to in locating important transcription factors or regulatory genes that regulate the expression of other genes.
Additionally, regulatory patterns and signaling pathways within the network can be found with the aid of network analysis. Additionally, it can be used to compare networks between various samples or situations in order to spot changes in gene regulatory relationships linked to certain biological functions or disease states. Experimental validation methods like knockdown or overexpression experiments, chromatin immunoprecipitation, or reporter gene assays can be used to verify the results of gene expression network analysis.
Conclusion
Deeper understanding of biological processes, disease mechanisms, and prospective therapeutic targets can be attained by gene expression network analysis, which offers a potent framework for revealing the intricate regulatory mechanisms controlling gene expression.
References
- Aslak, U., & Maier, B. (2019). Netwulf: Interactive visualization of networks in Python. Journal of Open Source Software, 4(42), 1425. [CrossRef]
- Edwards, D. (2007). Plant bioinformatics : methods and protocols. Humana Press.
- Nacu, S., Critchley-Thorne, R., Lee, P., & Holmes, S. (2007). Gene expression network analysis and applications to immunology. Bioinformatics, 23(7), 850–858. [CrossRef]
- Rajamani, S. K., & Iyer, R. S. (2023a). Machine Learning-Based Mobile Applications Using Python and Scikit-Learn. Advances in Wireless Technologies and Telecommunication Book Series, 282–306. [CrossRef]
- Santhosh Kumar Rajamani & Radha Srinivasan Iyer. (2023b). Declining Freshwater Species Biodiversity. In N. K. Chourasia & K. Chahal (Eds.), Perspectives on Global Biodiversity Scenarios and Environmental Services in the 21st Century (pp. 22–56). IGI Global. [CrossRef]
- Rajamani, S. K., & Iyer, R. S. (2023c). Methods of Complex Network Analysis to Screen for Cyberbullying (pp. 218–242). CRC Press. [CrossRef]
- RiceFREND. (n.d.). Ricefrend.dna.affrc.go.jp. Retrieved July 8, 2023, from https://ricefrend.dna.affrc.go.jp/.
- Sato, Y. S., Antonio, B. A., Nobukazu Namiki, Hinako Takehisa, Minami, H., Kaori Kamatsuki, Sugimoto, K., Shimizu, Y., Hirohiko Hirochika, & Yoshiaki Nagamura. (2011). RiceXPro: a platform for monitoring gene expression in japonica rice grown under natural field conditions. 39(Database), D1141–D1148. [CrossRef]
- Sato, Y. S., Hinako Takehisa, Kaori Kamatsuki, Minami, H., Nobukazu Namiki, Hiroshi Ikawa, Hajime Ohyanagi, Sugimoto, K., Antonio, B. A., & Yoshiaki Nagamura. (2012). RiceXPro Version 3.0: expanding the informatics resource for rice transcriptome. 41(D1), D1206–D1213. [CrossRef]
- Sato, Y., Namiki, N., Takehisa, H., Kamatsuki, K., Minami, H., Ikawa, H., Ohyanagi, H., Sugimoto, K., Itoh, J.-I., Antonio, B. A., & Nagamura, Y. (2012). RiceFREND: a platform for retrieving coexpressed gene networks in rice. Nucleic Acids Research, 41, D1. [CrossRef]
- Zhang, B., & Horvath, S. (2005). A General Framework for Weighted Gene Co-Expression Network Analysis. Statistical Applications in Genetics and Molecular Biology, 4(1). [CrossRef]
- Zimmermann, P., Hennig, L., & Gruissem, W. (2005). Gene-expression analysis and network discovery using Genevestigator. Trends in Plant Science, 10(9), 407–409. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



