
Submitted:
04 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
The short-lived positron-emitter carbon-11 (t1/2 = 20.4 min; b+, 99.8%) is prominent for labeling tracers for use in biomedical research with positron emission tomography (PET). Carbon-11 is produced for this purpose with a cyclotron, nowadays almost exclusively by the 14N(p,a)11C nuclear reaction, either on nitrogen containing a low concentration of oxygen (0.1–0.5%) or hydrogen (~ 5%) to produce [11C]carbon dioxide or [11C]methane, respectively. These primary radioactive products can be produced in high yields and with high molar activities. However, only [11C]carbon dioxide has some utility for directly labeling PET tracers. Primary products are required to be converted rapidly and efficiently into secondary labeling synthons to provide versatile radiochemistry for labeling diverse tracer chemotypes at molecular positions of choice. This review surveys known gas phase transformations of carbon-11 and summarizes the important roles that many of these transformations now play for producing a broad range of labeling synthons in carbon-11 chemistry.
Keywords:
Carbon-11
; Gas phase
; Catalysts
; On-line
; Radiotracer
; Radiochemistry
; PET
1. Introduction
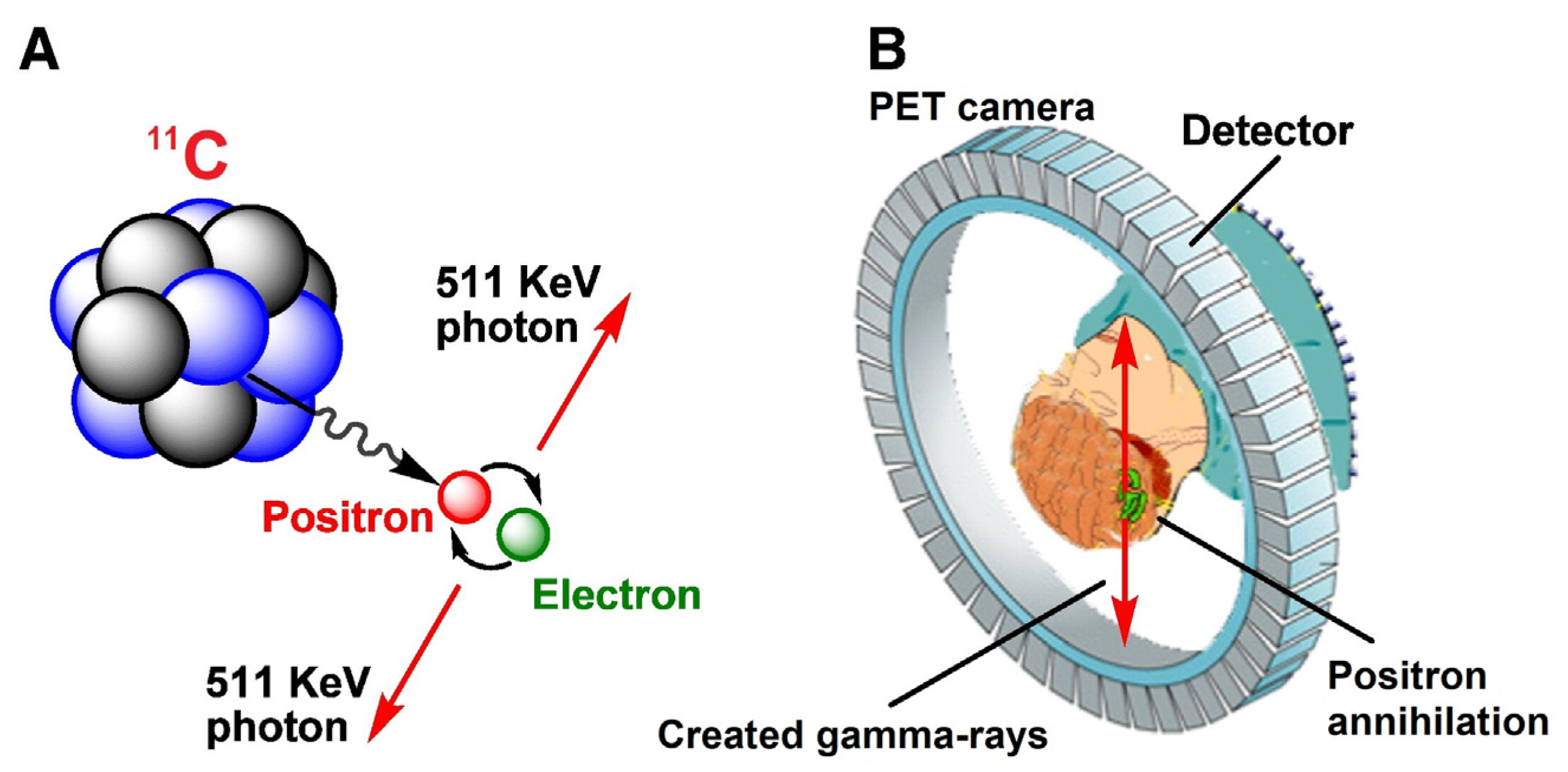
The short-lived positron-emitter carbon-11 (t1/2 = 20.4 min) has prominent application for labeling organic tracers that are used in biomedical research, drug development, and disease diagnosis with the molecular imaging technique of positron emission tomography (PET) [1]. This radionuclide decays almost purely by positron emission (β+, 99.8%) with the remainder by electron capture, each to give stable boron-11. The emitted positron has average and maximum energies of 0.3856 and 0.980 MeV, respectively, giving it a short range in dense tissue before being annihilated by combining with an electron. Positron annihilation results in emission of a pair of antiparallel photons, each with an energy of 511 keV. The photons can readily escape from deep tissue. Their coincident detection provides the basis for locating the administered tracer (or any arising radiometabolite) responsible for the positron emission within a living subject, either animal or human (Scheme 1). Nowadays, there are highly sophisticated ‘PET cameras’ that can record the distribution of positron emissions with time in the whole animal or human body or in major organs such as brain or heart. PET scans with a physical resolution of about a millimeter can be achieved every few seconds or minutes [2]. The biomedical information gained depends on the design of the tracer. Most tracers are designed to provide information on the distributions of a particular low density protein (e.g., neuroreceptor, transporter, enzyme, or pathological plaque), or on the protein interaction with either an endogenous compound (e.g., neurotransmitter) or a known or experimental drug. PET is highly sensitive, being able to detect proteins at sub-nanomolar concentrations. Because of its short half-life, carbon-11 must be rapidly produced and transformed into a PET tracer for time of need. This review surveys gas phase transformations of carbon-11 and highlights the especially strong enabling role that they now play in achieving the aims of rapid, efficient, and reliable PET tracer production.
2. Carbon-11 Production
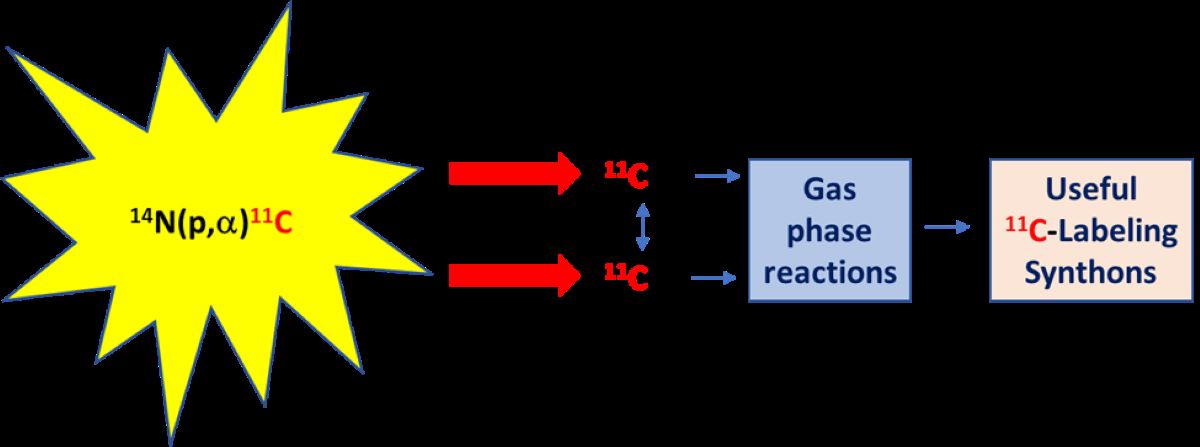
Carbon-11 may be produced for PET by many different cyclotron-promoted nuclear reactions [4,5]. Early methods used irradiations of solid targets, such as the 10B(d,n)11C, 11B(d,2n)11C, and 11B(p,n)11C reactions on boron trioxide [6]. Nowadays, however, carbon-11 is produced almost exclusively by the much higher yielding [7,8] and more easily manageable 14N(p,α)11C nuclear reaction on high pressure nitrogen gas [9]. Typically, the nitrogen has an initial pressure of about 225 psi and irradiations are performed with moderate to high beam currents (typically 40–150 μA) of moderate energy protons (~16.5 MeV). Many compact biomedical cyclotrons are commercially available for this purpose [10,11]. For example, in our laboratory, over 3 Curies of carbon-11 can be produced from a 40-minute irradiation of a nitrogen-oxygen mixture with a 45 μA beam of 16.5 MeV protons from a PETtrace cyclotron. By contrast with this scale of carbon-11 production, only 10 to 20 mCi of a tracer is typically required to be administered to a human subject for a PET experiment.
The chemical form of the retrieved carbon-11 depends on both cyclotron target gas composition and irradiation dose (Scheme 2) [9,12]. Initially, for proton irradiation of nitrogen, [11C]cyano radicals and [11C]carbon monoxide are formed by recoil reactions of nucleogenic 11C atoms with nitrogen and traces of oxygen, respectively. Even at low radiation dose (10-3 eV molecule-1s-1), a trace of oxygen (1–10 ppm) radiolytically oxidizes the [11C]cyano radical to [11C]carbon dioxide. At high irradiation doses, [11C]carbon monoxide also oxidizes radiolytically to [11C]carbon dioxide, leaving only a trace of [11C]carbon monoxide. Thus, relatively intense proton irradiation of high purity nitrogen that contains trace oxygen is an effective means for producing [11C]carbon dioxide in high radiochemical purity and high activity (Scheme 2A). The product is free of chemical impurities, provided that only a low concentration of oxygen (1–10 ppm) is present. Normally, however, the cyclotron target body is made of aluminum which may consume traces of oxygen and lead to diminishing radioactivity recovery over many successive irradiations. Therefore, oxygen (0.1–0.5% v/v) is normally added to the nitrogen target gas to ensure reliable and consistently high recovery of carbon-11 radioactivity. Higher concentrations of oxygen however generate potentially troublesome nitrogen oxides by radiolysis. These can be removed by on-line passage of the recovered irradiated gas into a trap filled with a mixture of chromium trioxide, copper sulfate, and 2 M sulfuric acid that has been dried onto a silica gel support [13]. Alternatively, the recovered [11C]carbon dioxide can be concentrated in a trap composed of a small coil of stainless steel tube immersed in a cryogen, either liquid nitrogen or liquid argon. Liquid argon is preferred because its higher temperature (–185 °C) avoids co-trapping of nitrogen (b.pt. –196 °C) and oxygen (b.pt. –183 °C). Nitrogen trapped by liquid nitrogen cooling may cause a rapid and poorly controllable gas expansion upon the release of the [11C]carbon dioxide at room temperature into a flowing inert gas stream. Alternatively, [11C]carbon dioxide can be trapped without any of the added oxygen on molecular sieves (3, 4, or 5 Å) at room temperature and then released in concentrated form by a flush with an inert gas at higher temperature [14]. This method is overall efficient and reliable, and therefore widely used.
If hydrogen (~5% v/v) is present in the irradiated nitrogen target gas, nucleogenic carbon-11 atoms either react with nitrogen to generate [11C]cyano radicals, which then react with hydrogen to form [11C]hydrogen cyanide, or they sequentially abstract hydrogen to produce [11C]methane (Scheme 2B). At low dose rate (10-4 eV molecule-1 s-1), a significant proportion of the radioactivity is recovered as [11C]hydrogen cyanide. However, at high dose rate (>0.1 eV molecule-1 s-1), radiolytic reduction of [11C]hydrogen cyanide becomes very significant and nearly all the carbon-11 (>95%) is retrieved as [11C]methane. Thus, high activities of [11C]methane can be produced. [11C]Methane is readily recovered and concentrated from irradiated target gas by passage into a Porapak N trap at room temperature or a Porapak Q trap at –186 °C [15]. The [11C]methane can then be released at a higher temperature.
The experience of many laboratories is that recovered activities of [11C]methane are substantially lower than recovered yields of [11C]carbon dioxide for the same energy and beam current in high-intensity production irradiations. For example, Buckley et al. [16]. reported that the recovery of [11C]methane was only 65% of that for [11C]carbon dioxide from the same irradiation conditions on the same target chamber. Some laboratories however report appreciably higher molar activity for [11C]methane than [11C]carbon dioxide [15,17].
The potential to produce predominantly [11C]hydrogen cyanide by the proton irradiation of nitrogen-hydrogen gas mixtures has been well explored but has not delivered a really practical method for high level production. For example, a yield of 0.67 Ci of [11C]hydrogen cyanide has been produced from an irradiation of a flowing nitrogen-(1%) hydrogen mixture (60 psi) in a heated (200 °C) quartz lined target with 30 μA of 15 MeV protons for 30 to 45 minutes [18,19]. This yield however represents only about 20% of that expected for carbon-11 from such an irradiation.
In PET imaging experiments on animals and human subjects, the mass of administered tracer must be limited to avoid possible toxicity and to comply with the tracer principle, which is to avoid perturbation of the biochemical system being studied. In this regard, an extremely important consideration in the production of carbon-11 is the molar activity (Am) of the chemical species that is produced. The molar activity is defined as the ratio of radioactivity (e.g., in Ci or GBq) to the total mass of all isotopologues (e.g., in μmol) of the chemical species in question at a specific time, such as the end of the radionuclide production (ERP). Am values decrease with the decay of the radionuclide. For a 11C-labeled product, the isotopologues to be considered are the corresponding natural abundance 12C and 13C isotopologues. As mentioned above, a cyclotron irradiation may produce a few Curies of carbon-11. One Curie of carbon-11 corresponds to about 0.1 nmol, an extremely small amount of substance with a theoretical molar activity of 9,200 Ci/μmol. However, cyclotron irradiations typically produce carbon-11 with much lower molar activities of 20 to 100 Ci/μmol. This is because sources of trace non-radioactive isotopologues, known as carrier, may enter the cyclotron target or the radioactive product recovery system. For example, during [11C]carbon dioxide production, carbon dioxide may be produced from organic materials during irradiation or a trace of atmospheric carbon dioxide may contaminate the target or product recovery apparatus. Therefore, scrupulous measures are required to minimize ingress or production of trace carrier [20,21]. This includes, for example, using ultra-pure nitrogen, as free as possible of any hydrocarbons and carbon dioxide, as the target gas. Application of careful measures to eliminate potential sources of carrier in post-irradiation chemical processing can lead to good conservation of molar activity [22,23]. Labeled products are described as no-carrier-added (NCA) where such measures are reasonably taken. If carrier is deliberately added, they are described as carrier-added (CA). In the remainder of this review, the discussed products are NCA, unless mentioned as being CA. Herein, cited Am values are for the end of radiosynthesis (EOS) and radiochemical yields are decay-corrected, unless otherwise stated.
An ability to produce tracers for PET imaging depends on being able to convert a primary cyclotron-produced product, either [11C]carbon dioxide or [11C]methane, into the tracer by rapid and high-yielding post-irradiation chemical means. In practice only 2 or 3 physical half-lives can be allowed for a full tracer production, including the separation and formulation of tracer for intravenous administration. Because of its reactivity, [11C]carbon dioxide has some direct but limited utility (e.g., for 11C-carboxylation reactions). However, [11C]methane must be transformed into some other labeling agent to be useful. Likewise chemical transformations of [11C]carbon dioxide can produce more useful labeling agents.
Whereas the preponderance of carbon-11 chemistry is done in solution, methods for performing 11C-chemistry in the gas phase are highly attractive. They can often be performed on-line and very rapidly in a flow of suitably inert carrier gas (e.g., nitrogen or helium) and they allow easy product isolation, often in a solvent of choice. Catalysts and reactants can often be used repeatedly, and the apparatus can be readily automated for protection of personnel from radioactivity. We now discuss known and important post-irradiation gas phase transformation methods in carbon-11 chemistry, indicating how they have been successful. Opportunities for improvement and expansion will also become apparent.
3. Conversions of Cyclotron-produced 11C-Labeled Materials
3.1. Conversions of [11C]Carbon Dioxide
3.1.1. Into [11C]Methane
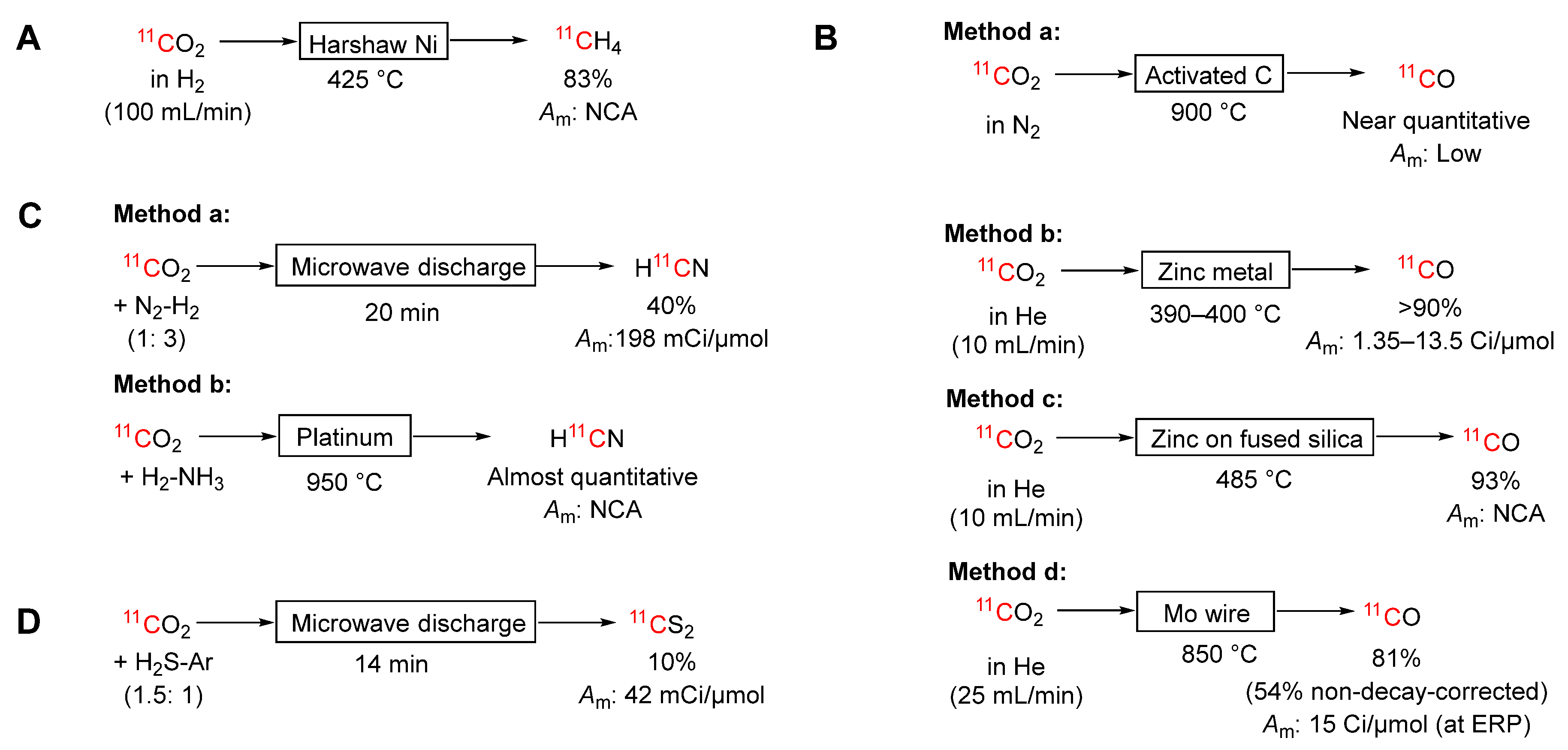
[11C]Methane is a useful precursor for other labeling agents, as discussed below (Section 3.2). Because [11C]carbon dioxide can be obtained in higher activity than [11C]methane, it can be advantageous to convert cyclotron-produced [11C]carbon dioxide into [11C]methane with an efficient on-line process. This can be achieved simply by passing [11C]carbon dioxide with hydrogen over a nickel catalyst at temperatures between 370 and 450 °C (Scheme 3A) [9,24,25,26,27,28]. Various physical forms of the nickel catalyst have been used. Kniess et al. [28] compared Harshaw-nickel, Shimalite-nickel, nickel on silica-alumina (65 wt%), and nanosize (90 nm) nickel (99.99%). High radiochemical yields (83%) were obtained by use of Harshaw nickel at 425 °C. In our laboratory, this method routinely gives very high molar activity for derived PET tracers (40 ± 16 Ci/μmol. mean ± SD at ERP).
3.1.2. Into [11C]Carbon Monoxide
[11C]Carbon monoxide has gained major interest for labeling carbonyl groups in compounds such as ketones, amides, ureas, and carboxylic acids through palladium and other transition metal mediated processes. These methods have been reviewed extensively [29,30,31]. Whereas solution phase methods for producing [11C]carbon monoxide are now known, on-line gas phase reduction of cyclotron-produced [11C]carbon dioxide is still a popular approach because of its simplicity and convenience. Several methods have been described (Scheme 3B).
The earliest gas phase method for producing [11C]carbon monoxide was based on reduction of [11C]carbon dioxide over activated charcoal at 800–900 °C (Method a, Scheme 3B) [32,33]. This method gives near quantitative yield but results in low molar activity, presumably because of unavoidable low-level carbon oxidation. This method is virtually obsolete for radiosynthesis applications because PET tracers usually need to be produced at high NCA molar activity.
[11C]Carbon dioxide can be reduced efficiently to [11C]carbon monoxide by passage over zinc metal heated between 390 and 400 °C (Method b, Scheme 3B) [34,35]. A single pass can give high yield (~70%) [35]. Almost quantitative yield has been achieved by preconcentration and recirculation of the [11C]carbon dioxide over the heated zinc [36]. High molar activities (1.35 to 13.5 Ci/μmol) are repetitively attainable [36]. However, success with this method depends very much on the quality of the zinc. Yields may become irreproducible if oxides form on the zinc over successive heating cycles. Performance may also vary unpredictably from one batch of zinc to another. Furthermore, the required temperature is not far below the melting point of zinc (420 °C). Therefore, overheating of the zinc column must be carefully avoided.
The use of solid-supported zinc has been proposed for improving the gas phase production of [11C]carbon monoxide [37]. Molecular sieves, fused silica (prepared from silica gel), and molybdenum have been investigated as solid supports with fused silica proving to be preferred. The use of a column of zinc supported on fused silica at 485 °C gave impressive yields (93 ± 3%; mean ± SD, n = 20) (Method c, Scheme 3B). This approach overcame the main limitations of fast deactivation and potential zinc metal melting that are experienced with the traditional heated zinc column.
Molybdenum wire heated to 850 °C within a quartz tube is an alternative reductant to zinc [38]. [11C]Carbon monoxide is obtained in up to 81% yield and with high molar activity (up to 15 Ci/μmol) (Method d, Scheme 3B). The process can be completed within 15 minutes and can be operated for a large number of runs without need to replace the molybdenum. It is considered that molybdenum(IV) oxide, generated on the metal surface from carbon dioxide or contaminating gaseous water or oxygen, also accomplishes the reduction. This may be serving to underpin the reliability of this method. In a comparative study, a molybdenum column, although needing to be heated to a much higher temperature, required less and easier maintenance than a zinc column, and gave acceptably high and more reproducible yields (up to 71%) [36]. Therefore, this method has been widely adopted [31,39,40].
Notwithstanding these advances, another method for producing [11C]carbon monoxide from [11C]carbon dioxide has been proposed. This method is based on decomposition of carbon dioxide at room temperature and atmospheric pressure by non-thermal dielectric barrage discharge [41]. The proof of principle for this method was shown with small macro amounts of non-radioactive carbon dioxide. The generated carbon monoxide was directly used as it was formed in microfluidic carbonylation reactions. However, this proposed method has not been adapted and demonstrated with NCA [11C]carbon dioxide.
3.1.3. Into [11C]Hydrogen Cyanide
Niisawa et al. [42] produced about 200 mCi of [11C]hydrogen cyanide with a molar activity of 198 mCi/μmol by microwave discharge through a nitrogen-hydrogen-[11C]carbon dioxide mixture giving 40% yield within 20 minutes of ERP (Method a, Scheme 3C). Hara & Iio [43] produced [11C]hydrogen cyanide from [11C]carbon dioxide (150 to 160 mCi) that had been generated from a low dose irradiation of nitrogen (9.4 MeV protons at 10 μA for 30 min). The [11C]carbon dioxide was mixed with hydrogen and ammonia and passed over platinum at 950 °C giving [11C]hydrogen cyanide in almost quantitative yield (Method b, Scheme 3C). These methods have not been adapted to high level production.
3.1.4. Into [11C]Carbon Disulfide
3.2. Conversions of [11C]Methane
3.2.1. Into [11C]Carbon Dioxide
[11C]Methane is easily converted into [11C]carbon dioxide by passage over cobalt(II-III) oxide (Co3O4) powder heated to 500 °C in a stream of nitrogen containing oxygen (2%) (Scheme 4A) [44]. The yield is 85% but 60% isotopic dilution occurs, giving a low molar activity (400 to 500 mCi/μmol, decay corrected to ERP in the original study). This process does allow a single cyclotron target to be used as a source of both [11C]methane and [11C]carbon dioxide.
3.2.2. Into [11C]Hydrogen Cyanide
[11C]Methane is quantitatively converted into [11C]hydrogen cyanide by passage with ammonia over platinum at 1000 °C [9,45,46]. Ample ammonia for this conversion is produced within a nitrogen-hydrogen gas target under an intense proton irradiation for [11C]methane production [9]. If the [11C]methane is produced by reduction of cyclotron-produced [11C]carbon dioxide, ammonia needs to be added to the [11C]methane. Iwata et al. [47] achieved over 95% yield by passing [11C]methane with ammonia (5 vol %) in carrier nitrogen (200 mL/min) over platinum wire (0.2 mm o.d × 2 cm; 5.4 g) at 920 °C. Prior removal of oxygen and water impurities in the [11C]methane feed with Oxisorb gave an optimal yield of [11C]hydrogen cyanide. This method underpins commercially available ‘boxes’ for enacting this conversion [48,49]. Labaree et al. slightly modified the column configuration housing the platinum catalyst in the commercial device and achieved increased production yields of 1.65 ± 0.45 Ci (mean ± SD, n = 73) over a 2-year period (Scheme 4B) [49]. With this source of [11C]hydrogen cyanide, they labeled a compound with a molar activity of 9.5 Ci/μmol (corrected to ERP).
3.2.3. Into CA [1-11C]Acetylene
Pyrolysis of [11C]methane with added carrier methane in an inductive argon plasma has produced [11C]acetylene in 60% yield within 10 minutes (Scheme 4C) [51]. Only a very low yield was obtained in the absence of added carrier. Notably, carrier-added [11C]acetylene, that had been produced by proton irradiation of calcium carbide followed by hydrolysis, had earlier found application for labeling two steroids, 17α-ethynylestradiol [52] and moxestrol [53].
3.2.4. Into [11C]Fluoroform
[11C]Methane may be converted into [11C]fluoroform in 53 ± 4% yield (mean ± SD; n = 11) by passage in a stream of helium (20 mL/min) over cobalt(III) fluoride (19 g) in a stainless steel tube at 270 °C (Scheme 5A) [54]. The whole process takes only 7 minutes from ERP. A single column was shown to operate consistently for over 80 runs without a need to change the cobalt(III) fluoride. [11C]Fluoroform so formed is readily converted into its reactive copper(I) derivative ([11C]CuCF3) which has a rich and developing chemistry for introducing [11C]trifluoromethyl groups into organic compounds [55,56,57,58]. Such compounds could be labeled with high molar activity (15 Ci/μmol). Ramos-Torres et al. [57] modified the procedure to increase the contact time between [11C]fluoroform and the cobalt(III) fluoride. This increased the purity of the [11C]fluoroform effluent from 88 to about 94%. [11C]Fluoromethane and [11C]difluoromethane are the main byproducts.
3.2.5. Into [11C]Chloromethanes
Passage of purified and concentrated [11C]methane with chlorine in helium over copper(II) chloride on pumice stone (3 g) produces [11C]chloroform at 330 °C in 45 to 50% yield (Method a, Scheme 5B) [59]. [11C]Chloroform prepared in this manner has been used to prepare [11C]diazomethane in solution [59]. At higher temperature (380 °C), the same catalytic system converts [11C]methane mainly into [11C]carbon tetrachloride. This chlorination is an important first step in a method for preparing [11C]phosgene [15] (see Section 3.3.7). Below 300 °C, [11C]chloromethane and [11C]dichloromethane are produced in moderate yields (20–25%) [60]. Crouzel & Hinnen [60] also explored other catalysts. In a meeting abstract, they reported 30 to 35% yield for [11C]dichloromethane (accompanied by 20% yield of [11C]chloroform) by use of 10% zirconium tetrafluoride on alumina (700 mg) at 310 °C with a helium carrier gas flow rate of 30 mL/min (including added chlorine gas) (Method b, Scheme 5B). The ability of [11C]dichloromethane to effect ring closure on catechol was demonstrated.
Later Solbach & Machulla [61] showed that [11C]methane could be chlorinated without catalyst (Method c, Scheme 5B). Using helium at 50 mL/min to carry a mixture of [11C]methane and chlorine through a quartz glass column at 400 °C, [11C]chloroform was obtained in 31% yield with [11C]dichloromethane and [11C]tetrachloromethane as byproducts. [11C]Chloroform from this method is also readily converted into [11C]diazomethane [61]. Higher temperature chlorination (500 °C) gives predominantly [11C]tetrachloromethane (~63%).
3.2.6. Into [11C]Bromomethane
Prenant & Crouzel passed [11C]methane over copper(II) bromide-pumice stone at 600 °C and produced [11C]bromomethane and [11C]dibromomethane in 15–20% and 10% yields, respectively (Method a, Scheme 5C) [62]. Mock et al. [63] showed that [11C]methane may be converted into [11C]bromomethane by reaction with heated bromine gas without catalyst. They performed this conversion in two modes, namely i) by single pass through a tube reactor (Method b, Scheme 5C), and ii) by rapid recirculation of [11C]methane through a similar tube reactor (Method c. Scheme 5C) In single pass mode, the [11C]methane is passed with bromine though a narrow borosilicate glass column (2 mm i.d.) held at 500 °C giving 18% conversion into [11C]bromomethane. These results questioned whether the copper(II) bromide-pumice stone was functional in the Prenant & Crouzel method [62]. Mock et al. introduced recirculation of the [11C]methane with the aim of enhancing the overall conversion into [11C]bromomethane. A carbon molecular sieve trap at room temperature was placed in the recirculation path to trap and accumulate the generated [11C]bromomethane. The optimal temperature for bromination was found to be between 525 and 550 °C, with transit times of 0.2 s at 250 mL/min, giving 70 to 75% [11C]bromomethane after only 4 to 5 minutes of recirculation. The relatively stable and highly volatile [11C]bromomethane (b.pt. 3.56 °C) can be efficiently recovered and carried over considerable distances through thin tubing for remote on-line synthesis of highly reactive [11C]methyl triflate (see Section 3.3.8), or itself used as a 11C-methylation agent [63].
3.2.7. Into [11C]Iodomethane
For several decades, [11C]iodomethane has been the most widely used 11C-labeling agent. This is mainly because of its reactivity towards heteroatom nucleophiles, such as amino groups, phenoxides, amides and thiolates, thereby allowing introduction of a 11C-methyl group into a broad swathe of potential PET tracers. For a long period, starting in the late 1970s, [11C]iodomethane was routinely produced by reduction of [11C]carbon dioxide to [11C]methanol followed by iodination with hydroiodic acid [64]. However, such methods were not so ‘user friendly’ because of the need to use vulnerable and aggressive reagents (i.e., lithium aluminum hydride, hydroiodic acid) and the complexity of the required automation. For such reasons, various PET centers explored the direct gas phase iodination of [11C]methane for [11C]iodomethane synthesis (Scheme 5D) [25,65].
Prenant & Crouzel [62] reported a 10 to 15% yield of [11C]iodomethane by passing [11C]methane in nitrogen (15 mL/min) over copper(I) iodide on pumice stone at 600 °C (Method a, Scheme 5D). Later, Link et al. studied reaction of [11C]methane with iodine vapor [65]. Iodine was heated to high temperature (720 to 730 °C) in a quartz tube to generate free iodine radicals. These could extract hydrogen from [11C]methane in a stream of helium (35 to 40 mL/min) to give [11C]methyl radicals that are capable of reacting with iodine (I2) to produce [11C]iodomethane. [11C]Iodomethane was obtained in more than 45 ± 3% yield (mean ± S D, n = 7) with a molar activity of 12 Ci/μmol after only 4 minutes (Method b, Scheme 5D) [25]. The synthesis could be repeated without any apparatus clean-up. Zhang & Suzuki [22] investigated sources of carrier in this single pass method. They suggested that traces of organic solvents and non-volatile oils (e.g., from fingerprints) could react with iodine vapor to yield carrier iodomethane in the heated quartz tube. Therefore, great care is needed to avoid such contamination.
Larsen et al. developed an apparatus for recirculating unreacted [11C]methane in helium (~ 500 mL/min) through iodine vapor at 725 °C to produce [11C]iodomethane continuously. The [11C]iodomethane was separated from the reactants on each cycle by entrapment on Porapak N, from which it could be later released at 190 °C in helium, either to a solution reaction medium or to a ‘loop reactor’ [66]. Under optimal conditions [11C]iodomethane was obtained in high yield (83%) with a molar activity exceeding 15 Ci/μmol at 7 minutes after collecting the [11C]methane [24,65]. The simplicity and reliability of this method has led to widespread use. An automated synthesizer (TRACERlabFXC) offered by GE incorporates this method for [11C]iodomethane synthesis. In this apparatus, cyclotron-produced [11C]carbon dioxide is first reduced to [11C]methane, as described in Section 3.1.1.
3.3. Gas phase Transformations of Other 11C-Labeled Compounds
3.3.1. [11C]Carbon Monoxide into [11C]Phosgene
For several decades, [11C]phosgene has been an important labeling synthon for introducing [11C]carbonyl groups into PET tracers [67]. The production of [11C]phosgene in high molar activity and yield has however been challenging, giving rise to many diverse apparatuses and methods [67].
[11C]Carbon monoxide served as an intermediate in early gas phase routes to [11C]phosgene. The first of these routes used ultraviolet light to promote reaction of [11C]carbon monoxide with chlorine, giving [11C]phosgene in 75 to 90% yield (Method a, Scheme 6A) [68,69]. The second used platinum tetrachloride to achieve the same reaction (Method b, Scheme 6A) [70,71]. A 30-minute irradiation with a 30 μA proton beam consistently gave 200 to 300 mCi of [11C]phosgene. However, the molar activity achieved with either method was very low due to carbon monoxide contamination of the chlorine in the first method (40 mCi/μmol) [69] and of platinum tetrachloride in the second (400 to 500 mCi/μmol at 20 min from ERP) [71]. These methods were quickly superseded by methods from [11C]methane via [11C]carbon tetrachloride [15] (see Section 3.3.7).
3.3.2. [11C]Carbon Monoxide into [11C]Carbonyl Difluoride
Passage of [11C]carbon monoxide in helium (5 mL/min) over a column of silver(II) fluoride (0.4 g) at room temperature provides [11C]carbonyl difluoride (b.pt. –85 °C) in quantitative yield within 7 minutes of ERP (Scheme 6B) [72]. This product is readily trapped in organic solvents and has been shown to label a broad range of cyclic substrates, for example, imidazolidin-2-ones, thiazolidin-2-ones, and oxazolidin-2-ones [72], and unsymmetrical acyclic ureas [73]. High molar activity (> 5.9 ± 1.6 Ci/μmol at end of synthesis) was measured on a labeled product obtained from [11C]carbonyl fluoride. A silver(II) fluoride column could be used reliably for at least ten runs. The reactivity of [11C]carbonyl difluoride is very similar to the more difficult to produce [11C]phosgene and may be set to supersede the latter in many applications.
3.3.3. [11C]Carbon Monoxide into [11C]Methanol
A practical on-line route to [11C]methanol has not been reported. However, a reactor has been designed to allow trace amounts of carbon monoxide to react with hydrogen to form methanol on a copper-zinc oxide catalyst [74]. Reduced and passivated copper-zinc oxide catalyzes the reaction of 50 ppm carbon monoxide with hydrogen to form methanol at 180–240 °C, 55 bar, and with a gas flow of 26–935 mL/min. A kinetic model was fitted to the experimental data with a commercially available process simulator (Hysys). Because the methanol synthesis is exothermic, as the temperature increases the equilibrium conversion of carbon monoxide to methanol decreases. Also, for a given temperature, as the pressure increases, the equilibrium conversion of carbon monoxide to methanol increases. At 50 bar, equilibrium conversion of carbon monoxide to methanol is quantitative below 180 °C and decreases to 90% at 240 °C.
The model predicted optimal operating conditions for practical quantities of [11C]methanol. It was predicted that over 60% conversion of [11C]carbon monoxide into [11C]methanol could be achieved at 224 °C and 55 bar with a gas flow of 20 mL/min (Scheme 6C). The proposed reactor was considered suitable for incorporation into a process that might require [11C]methanol, such as the production of [11C]iodomethane with high molar activity. The experimental results indicated that carbon monoxide and/or carbon dioxide can adsorb on the copper-zinc oxide surface at ambient temperature but desorb at about 200 °C. Accordingly, care would need to be taken to remove any traces of carbon dioxide or carbon monoxide before use of the catalyst to ensure high molar activity for the final [11C]methanol. This might be accomplished by flushing the reactor with pure helium before use, with the reactor heated slightly above operating temperature. The reactor would also need to remain under inert gas while not in use, preferably slightly under pressure, to avoid contamination. The advantage of the proposed gas phase synthesis over the traditional liquid phase method [75] would be the potential to obtain 11C-labeled tracers in high molar activity. However, there is as yet no report for the implementation of this proposal, probably because of the requirement for hydrogen at high pressure.
3.3.4. [11C]Methanol into [11C]Formaldehyde
[11C]Formaldehyde can be produced through silver wool-catalyzed high temperature (270‒520 °C) oxidation of [11C]methanol in nitrogen containing a low percentage of oxygen (Method a, Scheme 7A) [75,76,77]. However, the yield depends strongly on the activity of the catalyst, is poorly reproducible, and seldom exceeds 60%. The catalytic activity of the silver depends on its prior use. With only a low concentration of oxygen in the carrier gas (ppm levels), the catalyst gradually deactivates. At higher oxygen concentration (e.g., 2%) the catalyst increases in activity, tending to further oxidize the [11C]methanol. For optimal molar activity, it is important to prevent non-radioactive contaminants in the [11C]methanol from accessing the silver catalyst, for example, by passing the [11C]methanol through a Porapak P (60–80 mesh) trap [78]. Pretreatment of the catalyst with methanol at 370 °C increases yield but reduces molar activity. More recently, Roeda & Dollé [79] described the use of an Ag+ ion-containing kaolin catalyst (300 mg; 20% silver content) in a 5 mm long column in place of silver wool to achieve the oxidation of [11C]methanol at 500 °C (Method b, Scheme 7A). This method gave [11C]formaldehyde in 67% yield with a molar activity up to 1.6 Ci/μmol.
As an alternative to silver or silver ion catalysis, [11C]methanol may be oxidized to [11C]formaldehyde in about 70% yield by passage with oxygen over a ferric-molybdenum oxide preparation supported on stainless steel balls at 375 °C (Method c, Scheme 7A) [76,80]. The production of [11C]formaldehyde requires about 10 minutes.
As discussed above, there is no practical on-line gas phase method for producing the starting [11C]methanol for these methods. The required [11C]methanol may be produced from cyclotron-produced [11C]carbon dioxide by passage into lithium aluminum hydride in tetrahydrofuran followed by hydrolysis [77]. Even with the need for this prior step, [11C]formaldehyde can normally be produced in about 10 minutes. Molar activities up to 1 Ci/μmol have been reported [77].
[11C]Formaldehyde finds utility mainly for 11C-methylation of secondary amines by reductive alkylation, such as in the synthesis of [11C]erythromycin A [74].
3.3.5. [11C]Methanol into [11C]Iodomethane
[11C]Iodomethane has also been produced on-line in amounts up to 800 mCi by passage of [11C]methanol in a stream of helium (50 mL/min) over alumina-supported triphenylphosphine diiodide (1 g) held within a glass column at 160 °C. The [11C]iodomethane was obtained in 90 ± 2% yield (mean ± SD, n= 25) with a radiochemical purity of > 99.5% and a mean molar activity of 6 Ci/μmol within an overall synthesis times of 11 minutes (Scheme 7B) [81].
3.3.6. [1-11C]Ethanol into [1-11C]Ethylene
[1-11C]Ethylene may be prepared by passing [1-11C]ethanol over quartz glass held entirely within a heated (500 °C) stainless steel tube (Scheme 8) [82]. The [1-11C]ethanol is ideally prepared from cyclotron-produced [11C]carbon dioxide by 11C-carboxylation of methylmagnesium bromide that has been freshly prepared in dibutyl ether, followed by reduction of the generated adduct with lithium aluminum hydride in diglyme. The use of involatile solvents avoids the undesirable formation of carrier ethylene and radioactive and stable diethyl ether by trace solvent cracking over the heated catalyst. Placing the quartz glass entirely within the heated zone of the stainless steel tube avoided adsorptive radioactivity losses. This method was preferred to catalytic dehydration over alumina or pyrolysis at 700 °C. The radiosynthesis takes 21 minutes and has a radiochemical yield of 44%, from [11C]carbon dioxide. [1-11C]Ethylene may be converted quantitatively into [1-11C]l,2-dibromoethane when collected in a solution of bromine in carbon tetrachloride. The NCA [1-l1C]ethylene and [1-11C]l,2-dibromoethane have potential to serve as new and useful 2-carbon labeling agents.
3.3.7. [11C]Carbon Tetrachloride into [11C]Phosgene
[11C]Phosgene may be produced from [11C]carbon tetrachloride by passage in helium over iron filings (1.5 g) at 300 °C (Method a, Scheme 9) [15]. Surface oxide on the filings presumably provides the required oxygen. The generated [11C]phosgene is passed through a trap (3 mm i.d.) of antimony (400 mg) mixed with glass beads (1 mm o.d.) to remove traces of chlorine before collection in a desired solvent. This conversion normally follows the on-line production of [11C]carbon tetrachloride from cyclotron-produced [11C]methane, as described earlier (Section 3.2.5,). The coupled two-step process takes about 10 to 12 minutes. A 30-minute cyclotron irradiation with a 30 μA beam of 20 MeV protons was reported to give 375 to 500 mCi of [11C]phosgene with a molar activity of 1.4 to 1.6 Ci/μmol, corresponding to 2.5 carrier dilution with respect to the molar activity of starting [11C]methane.
Nishijima et al. [83] sought to improve the synthesis of [11C]phosgene from [11C]carbon tetrachloride. They examined two oxidizing agents, iron(III) oxide and copper(II) oxide. The yield of [11C]phosgene, based on labeled derivatives, was significantly increased using iron(III) oxide powder mixed with iron granules, whereas the use of copper(II) oxide alone, or copper(II) oxide powder mixed with iron granules resulted in insignificant yield. The yield and molar activity of the β-adrenoceptor radioligand, [11C](S)-CGP-12177, from [11C]phosgene that had been prepared using iron(III) oxide powder mixed with iron granules (Method b, Scheme 9) were markedly higher than by previous methods using iron granules alone or iron granules mixed with iron powder.
Bramoullé et al. described a simplified on-line method for the production of [11C]phosgene [84]. In this method, cyclotron-produced [11C]methane is mixed with chlorine and converted into [11C]carbon tetrachloride by passage through an empty quartz tube at 510 °C. The outflow is then directed through an antimony filled guard column that takes out chlorine and then, without intentional oxygen addition, through a second empty quartz tube at 750 °C, giving [11C]phosgene in 30 to 35% yield in a total radiosynthesis time of about 12 minutes (Method c, Scheme 9).
Ogawa et al. [85] developed a room temperature method for synthesizing [11C]phosgene. This method is based on utilizing part of a carbon tetrachloride detection tube kit intended for environmental air analysis (Method d, Scheme 9). This used part is a pretreatment tube that is filled with a support material containing iodine pentoxide (I2O5) and fuming sulfuric acid. Optimal flow of [11C]carbon tetrachloride through the tube was 50 mL/min. [11C]Phosgene was obtained in 80.8 ± 3.1% yield (n = 5) from [11C]carbon tetrachloride after 10 minutes of total radiosynthesis time and gave a molar activity of 2.3 ± 1.4 Ci/μmol (n = 3) on a labeled derivative. The synthesis apparatus was automated for preparing [11C]phosgene up to 4 times per week [86].
3.3.8. [11C]Iodomethane into [11C]Methyl Triflate
[11C]Methyl triflate is a more powerful methylation agent than [11C]iodomethane [87,88]. This labeling agent can be prepared in almost quantitative yield and within 7 minutes simply by passing [11C]iodomethane in a stream of nitrogen gas (30–100 mL/min) over graphitized carbon (~ 600 mg) impregnated with 50% by weight of silver(I) triflate held within a small Pyrex tube (6 mm o.d. × 23 cm) at 150‒200 °C (Scheme 10A) [89]. The [11C]methyl triflate (b.pt., 100 °C) is readily trapped in organic solvents at room temperature. The molar activity of the [11C]methyl triflate is dictated by the molar activity of the [11C]iodomethane feed; the process does not add carrier. The very extensive utility that [11C]methyl triflate has gained in the PET radiochemistry field for labeling tracers at heteroatoms has recently been summarized [90].
3.3.9. [11C]Bromomethane into [11C]Methyl triflate
[11C]Bromomethane may be converted into [11C]methyl triflate in similar manner to [11C]iodomethane (Scheme 10B) [63]. Thus, passage of [11C]bromomethane in a helium stream (30–50 mL/min) through a column of graphitized carbon impregnated with silver(I) triflate held between 280 and 300 °C produces [11C]methyl triflate in over 90% yield. Compounds labeled with [11C]methyl triflate from this method have shown molar activities of about 2.5 Ci/μmol.
3.3.10. Lower [1-11C]Iodoalkanes into Lower [1-11C]Nitroalkanes
[11C]Nitromethane can be prepared simply and efficiently by passing [11C]iodomethane in nitrogen or helium at 20 to 30 mL/min through a soda glass column (3 mm i.d. × 4 cm) packed with silver(I) nitrite (0.4 g) at 80 °C (Scheme 10C) [91]. Higher [11C]nitroalkanes can be prepared similarly from volatile [1-11C]iodoalkanes (R11CH2I, R = Me, Et) [91], produced as described by Långström et al. [92]. The [1-11C]nitroalkanes are readily trapped in solvents of choice for further reaction (e.g., DMSO). The yields are 70, 50, and 65% for R = H, Me, and Et respectively, based on the corresponding [1-11C]alkyl iodides and 55, 30 and 40% overall from starting [11C]carbon dioxide, in radiosynthesis times of 8 to 15 minutes. The on-line method for producing [1-11C]nitromethane overcame many limitations of the earlier solution method based on direct reaction with silver nitrite in an organic solvent at 80 °C [93], such as handling difficulties, poor reproducibility, and limitations on solvents for further reactions. The heated sodium nitrite can produce nitrogen oxides, which can interfere in some labeling reactions with [11C]nitromethane. The volatile [11C]nitromethane product can however be purified on-line by placing a sodium carbonate plug in the heated column after the sodium nitrite [94]. The helium-[11C]nitromethane should then be dried by passage through Sicapent. [11C]Nitromethane reacts readily with aldehydes and found early application for labeling D-glucose and D-mannose at C-1 positions [95].
3.3.11. [11C]Iodomethane into [11C]Methanethiol and then [11C]Mesyl Chloride
[11C]Methanethiol is of interest for introducing [11C]thiomethyl groups into organic structures, such as the amio acid L-methionine. [11C]Alkanethiols have been prepared in solution from [1-11C]iodoalkanes and commercially available sodium hydrosulfide (NaSH) [96,97]. Thus, [11C]methanethiol may be prepared almost instantaneously and quantitatively by dispensing [11C]iodomethane into a solution of sodium hydrosulfide in anhydrous DMF. Because [11C]methanethiol is gaseous (b.pt., 6 °C), simple nitrogen purge of the reaction mixture enables transfer of this product to a collection vessel. However, when McCarron & Pike [98] tried to implement this method only about 60% of the initial [11C]iodomethane radioactivity was successfully transferred over a period of 10 minutes. This experience prompted development of an on-line process for the preparation (Scheme 10D). Passage of [11C]iodomethane in nitrogen (50 mL/min) over sodium hydrosulfide (100 mg) in a silica tube (2 mm i.d. × 30 cm) at 285 °C generated [11C]methanethiol, which was collected in chlorine-saturated water and instantly converted to [11C]mesyl chloride. The yield from [11C]iodomethane was 77%.
Oxidative chlorination of [11C]methanethiol to [11C]mesyl chloride could be achieved on-line by passage over manganese(IV) oxide (40 mg) in a silica tube (2 mm i.d. × 30 cm) at 255 °C and then over a short plug of calcium hypochlorite (20 mg) at room temperature (Scheme 10E). [11C]Mesyl chloride has potential as a labeling agent, as was shown by its ability to mesylate tetrahydroisoquinoline [98].
These on-line procedures are attractive for ease of future radiosynthesis automation and operation with high radioactivity levels, but require further optimization.
3.3.12. [11C]Iodomethane to [methyl-11C]Methyl Isocyanate
[methyl-11C]Methyl isocyanate has been prepared by passing [11C]iodomethane in nitrogen (10 mL/min) for 1 minute over silver(I) cyanate (250 mg) at 180 °C (Scheme 10F) [99]. Typically, the conversion of [11C]iodomethane to [methyl-11C]methyl isocyanate was 70–75% with a total radiosynthesis time of 13 minutes, including 12 minutes for [11C]iodomethane production. About 25% of the radioactivity was retained on the silver(I) cyanate column. Higher temperature failed to drive off this radioactivity. Notably, [methyl-11C]methyl isocyanate has been used to label the antitumor drug temozolomide in an N-methyl group [99].
3.3.13. [11C]Iodomethane into [11C]Hydrogen Cyanide via [11C]Formaldehyde.
Kikuchi et al. [100] have described the conversion of [11C]iodomethane into [11C]hydrogen cyanide by passage in nitrogen through a small two-layered reaction column heated to 170 °C (Scheme 11A). The first layer contains an N-oxide (oxymatrine) and diphenyl sulfoxide for conversion of [11C]iodomethane into [11C]formaldehyde. The generated [11C]formaldehyde is subsequently converted into [11C]hydrogen cyanide in a second layer containing hydroxylamine-O-sulfonic acid (HOSA). The yield of [11C]hydrogen cyanide from this method (~ 52%, corrected to ERP) is similar to that of [11C]hydrogen cyanide produced by the traditional method (51%). The molar activity of the [11C]hydrogen cyanide, determined by derivatization, was estimated to be 9.43 Ci/μmol and almost the same as that of the starting [11C]iodomethane (9.32 Ci/μmol). The first layer of the column can be used to produce gaseous [11C]formaldehyde with a yield estimated from derivatization of about 83%. The authors provided a video on the assembly of the required bi-layer column.
3.3.14. [11C]Iodomethane into [11C]Carbon Disulfide
[11C]Carbon disulfide (b.pt. 46 °C) has been produced by a gas phase reaction from [11C]iodomethane (Scheme 11B) [101]. In this procedure, a stream [11C]iodomethane, is passed through a small glass column packed with a mixture of phosphorus pentasulfide and sand in 1: 2 ratio, at 380 °C. The gases are vented into a vial containing acetonitrile at room temperature, which conveniently traps the [11C]carbon disulfide. The conversion of [11C]iodomethane into [11C]carbon disulfide is instantaneous giving high yields (> 85%) and high molar activities (2.7 Ci/μmol). The only major contaminant is a small amount of unreacted [11C]iodomethane. The flow rate of gas carrying iodomethane through the phosphorus pentasulfide column had a significant impact on conversions. A flow of 5–6 mL/min gave good conversions with an acceptable processing time of less than 10 minutes from the end of [11C]iodomethane production. Higher flow rates gave decreased yields. [11C]Carbon disulfide was reactive towards aniline, benzylamine, and diethylamine, thereby showing considerable potential for labeling prospective sulfur-containing tracers.
3.3.15. [11C]Hydrogen Cyanide into [11C]Cyanogen Bromide
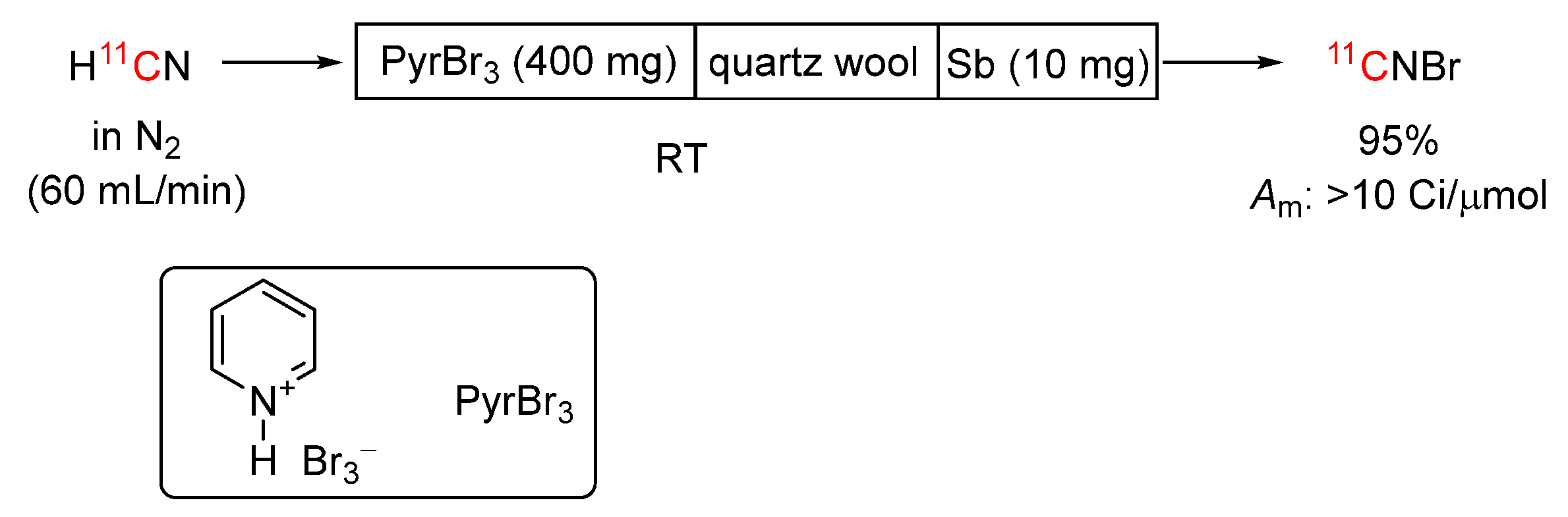
[11C]Cyanogen bromide, an electrophilic labeling agent, has been produced in 95% yield from [11C]hydrogen cyanide within 3 minutes from ERP using a simple and convenient on-line procedure (Scheme 12) [102]. In this procedure, [11C]hydrogen cyanide is passed through a Sicapent tower (10 mm i.d. × 10 cm), and then through a quartz tube (6.25 mm i.d. × 15 cm) contacting first pyridinium perbromide (400 mg) and then antimony powder (10 mg) separated by a quartz wool plug. The generated [11C]cyanogen bromide can be trapped in a suitable solvent for further use. The molar activity of the [11C]cyanogen bromide was difficult to determine directly and was therefore estimated from the molar activity of derivatives, giving values between 10 and 12 Ci/μmol at the end of synthesis. The procedure has been automated [103]. This on-line method for [11C]cyanogen bromide synthesis is shorter, technically simple, and more reproducible than the earlier reported solution method [104]. [11C]Cyanogen bromide has been used to label several compounds, such as the antiviral compound GR121167X [105], albumin [106], and hyaluronic acid [107].
3.3.16. [1-11C]Butyric Acid into [1-11C]Propylketene
Fujii et al. developed a method for preparing [1-11C]but-1-en-1-one ([1-11C]propylketene) in 32% yield based on the pyrolysis of [1-11C]butyric acid (b.pt. 164 °C) at 550 °C in a quartz tube (7 mm i.d. × 24 cm) containing glass beads (1 mm o.d.) [108]. The [1-11C]butyic acid was prepared by 11C-carboxylation of propyl lithium with cyclotron-produced [11C]carbon dioxide followed by acidification with hydrogen chloride gas (Scheme 13). The [1-11C]butyric acid was carried through the quartz tube with helium (70 mL/min) containing a low concentration of dry hydrogen chloride. The whole radiosynthesis required 25 minutes giving an overall yield of 22%. Yields were measured after derivatization. Forskolin, and several phorbol esters and diacylglycerols were successfully labeled with [1-11C]propylketene [109,110].
4. Conclusion and Outlook
Gas phase transformations play a major part in carbon-11 chemistry and in PET tracer utilization and development, largely because of their simplicity, reliability, re-usability, and amenability for automation. Major advances have been made through appropriate selections and optimization of reagents and catalysts, and in the engineering of suitable reactors and their energy supplies. Further advances can be anticipated in all aspects.
Author Contributions
Conceptualization, VWP; methodology, SL, ST, FGS, LC, and VWP; data curation, SL; writing—original draft preparation, VWP; writing—review and editing, SL, ST, FGS, LC, and VWP; supervision, VWP; project administration, VWP; funding acquisition, VWP. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Intramural Research Program of the National Institutes of Health (National Institute of Mental Health; ZIA-MH002793).
Data Availability Statement
All data used in this work were derived from publications. Summaries of extracted data are included in this work.
Acknowledgments
The authors acknowledge the intramural research program of the National Institutes of Health (National Institute of Mental Health; ZIA-MH002793) for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Phelps, M.E. Positron emission tomography provides molecular imaging of biological processes. Proc. Natl. Acad. Sci. 2000, 97, 9226–9233. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.R.; Badawi, R.D.; Karp, J.S.; Moses, W.W.; Price, P.; Jones, T. Total-body imaging: Transforming the role of positron emission tomography. Sci. Transl. Med. 2017, 9, Article ID eaaf6169. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Conti, P.S. Radiopharmaceutical chemistry for positron emission tomography. Adv. Drug Delivery. Rev. 2010, 62, 1031–1051. [Google Scholar] [CrossRef] [PubMed]
- Ferrieri, R.A.; Wolf, A.P. The chemistry of positron emitting nucleogenic (hot) atoms with regard to preparation of labelled compounds of practical utility. Radiochim. Acta 1983, 34, 69–84. [Google Scholar] [CrossRef]
- Ferrieri, R.A. Production and Application of Synthetic Precursors Labeled with Carbon-11 and Fluorine-18. In Handbook of Radiopharmaceuticals - Radiochemistry and Applications; 2002; pp. 229-282. [CrossRef]
- Clark, J.C.; Buckingham, P.D. Short-lived Radioactive Gases for Clinical Use; Butterworths: London, 1975. [Google Scholar]
- Casella, V.R.; Christman, D.R.; Ido, T.; Wolf, A.P. Excitation-function for 14N(p,α)11C reaction up to 15-MeV. Radiochim. Acta 1978, 25, 17–20. [Google Scholar] [CrossRef]
- Bida, G.T.; Ruth, T.J.; Wolf, A.P. Experimentally determined thick target yields for the 14N (p,α)11C reaction. Radiochim. Acta 1980, 27, 181–186. [Google Scholar] [CrossRef]
- Christman, D.R.; Finn, R.D.; Karlstrom, K.I.; Wolf, A.P. Production of ultra high activity 11C-labeled hydrogen cyanide, carbon dioxide, carbon monoxide and methane via 14N(p,α)11C reaction (XV). Int. J. Appl. Radiat. Isot. 1975, 26, 435–442. [Google Scholar] [CrossRef]
- IAEA. Appendix 1: PET Cyclotron Comparison. In Cyclotron Produced Radionuclides: Principles and Practice, Technical Reports Series No. 465, IAEA: Vienna, 2008. https://www.iaea.org/publications/7849/cyclotron-produced-radionuclides-principles-and-practice.
- Ruth, T.J. Accelerators available for isotope production. In Handbook of Radiopharmaceuticals - Radiochemistry and Applications; 2002; pp. 71-85. [CrossRef]
- Ache, H.J.; Wolf, A.P. Effect of radiation on the reactions of recoil carbon-11 in the nitrogen-oxygen system. J. Phys. Chem. 1968, 72, 1988–1993. [Google Scholar] [CrossRef]
- Tewson, T.J.; Banks, W.; Franceschini, M.; Hoffpauir, J. A trap for the removal of nitrogen oxides from carbon-11 carbon dioxide. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1989, 40, 765–768. [Google Scholar] [CrossRef]
- Mock, B.H.; Vavrek, M.T.; Mulholland, G.K. Solid-phase reversible trap for [11C]carbon dioxide using carbon molecular sieves. Nucl. Med. Biol. 1995, 22, 667–670. [Google Scholar] [CrossRef]
- Landais, P.; Crouzel, C. A new synthesis of carbon-11 labelled phosgene. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1987, 38, 297–300. [Google Scholar] [CrossRef]
- Buckley, K.R.; Huser, J.M.; Jivan, S.; Chun, K.S.; Ruth, T.J. 11C-methane production in small volume, high pressure gas targets. Radiochim. Acta 2000, 88, 201–206. [Google Scholar] [CrossRef]
- Andersson, J.; Truong, P.; Halldin, C. In-target produced [11C]methane: Increased specific radioactivity. Appl. Radiat. Isot. 2009, 67, 106–110. [Google Scholar] [CrossRef]
- Lamb, J.F.; James, R.W.; Winchell, H.S. Recoil synthesis of high specific activity 11C-cyanide. Int. J. Appl. Radiat. Isot. 1971, 22, 475–479. [Google Scholar] [CrossRef]
- Meyer, G.J.; Osterholz, A.; Harms, T. A systematic investigation of [11C]HCN production. Radiochim. Acta 1990, 50, 43–48. [Google Scholar] [CrossRef]
- Suzuki, K.; Yamazaki, T.; Sasaki, M.; Kubodera, A. Specific activity of [11C]CO2 generated in a N2 gas target: effect of irradiation dose, irradiation history, oxygen content and beam energy. Radiochim. Acta 2000, 88, 211–216. [Google Scholar] [CrossRef]
- Gómez-Vallejo, V.; Gaja, V.; Koziorowski, J.; Llop, J. Specific activity of 11C-labelled radiotracers: A big challenge for PET chemists. In Positron Emission Tomography - Current Clinical and Research Aspects, Hsieh, C.-H., Ed.; Intechopen.com: 2012; pp. 183-210. [CrossRef]
- Zhang, M.-R.; Suzuki, K. Sources of carbon which decrease the specific activity of [11C]CH3I synthesized by the single pass I2 method. Appl. Radiat. Isot. 2005, 62, 447–450. [Google Scholar] [CrossRef]
- Pichler, V.; Zenz, T.; Philippe, C.; Vraka, C.; Berrotéran-Infante, N.; Pfaff, S.; Nics, L.; Ozenil, M.; Langer, O.; Willeit, M.; et al. Molar activity – The keystone in 11C-radiochemistry: An explorative study using the gas phase method. Nucl. Med. Biol. 2018, 67, 21–26. [Google Scholar] [CrossRef]
- Larsen, P.; Ulin, J.; Dahlstrom, K.; Jensen, M. Synthesis of [11C]iodomethane by iodination of [11C]methane. Appl. Radiat. Isot. 1997, 48, 153–157. [Google Scholar] [CrossRef]
- Link, J.M.; Krohn, K.A.; Clark, J.C. Production of [11C]CH3I by single pass reaction of [11C]CH4 with I2. Nucl. Med. Biol. 1997, 24, 93–97. [Google Scholar] [CrossRef]
- Buckley, K.R.; Huser, J.; Jivan, S.; McDonald, R.; Ruth, T.J. Methane Production in small volume high pressure gas targets. In Proceedings of the 7th International Workshop on Targetry & Target Chemistry, Heidelberg, Germany, 1997; pp. 75-79. https://wttc.triumf.ca/97-pdf.html.
- Buckley, K.R.; Huser, J.; Jivan, S.; Ruth, T.J. Methane production in small volume, high pressure gas target: Futher studies. In Proceedings of the 8th International Workshop on Targetry & Target Chemistry, St. Louis, Missouri, USA, 1999; pp. 179-181. https://wttc.triumf.ca/99-pdf.html.
- Kniess, T.; Rode, K.; Wuest, F. Practical experiences with the synthesis of [11C]CH3I through gas phase iodination reaction using a TRACERlabFXC synthesis module. Appl. Radiat. Isot. 2008, 66, 482–488. [Google Scholar] [CrossRef]
- Rahman, O. [11C]Carbon monoxide in labeling chemistry and positron emission tomography tracer development: Scope and limitations. J. Label. Compd. Radiopharm. 2015, 58, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Taddei, C.; Pike, V.W. [11C]Carbon monoxide: advances in production and application to PET radiotracer development over the past 15 years. EJNMMI Radiopharm. Chem. 2019, 4, Article ID 25. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Antoni, G.; Långström, B.; Itsenko, O. The development of 11C-carbonylation chemistry: A systematic view. Nucl. Med. Biol. 2021, 92, 115–137. [Google Scholar] [CrossRef]
- Clark, J.C.; Buckingham, P.D. The preparation and storage of carbon-11 labelled gases for clinical use. Int. J. Appl. Radiat. Isot. 1971, 22, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, M.H.; Pike, V.W. Palladium(II)-mediated 11C-carbonylative coupling of diaryliodonium salts with organostannanes - a new, mild and rapid synthesis of aryl [11C]ketones. J. Chem. Soc., Perkin Trans. 1 2000, 1033-1036. [CrossRef]
- Andersson, Y.; Långström, B. Synthesis of 11C-labelled ketones via carbonylative coupling reactions using [11C]carbon monoxide. J. Chem. Soc., Perkin Trans. 1 1995, 287-289. [CrossRef]
- Lidström, P.; Kihlberg, T.; Långström, B. [11C]Carbon monoxide in the palladium-mediated synthesis of 11C-labelled ketones. J. Chem. Soc., Perkin Trans. 1 1997, 2701-2706. [CrossRef]
- Dahl, K.; Itsenko, O.; Rahman, O.; Ulin, J.; Sjöberg, C.-O.; Sandblom, P.; Larsson, L.-A.; Schou, M.; Halldin, C. An evaluation of a high-pressure 11CO carbonylation apparatus. J. Label. Compd. Radiopharm. 2015, 58, 220–225. [Google Scholar] [CrossRef]
- Dahl, K.; Ulin, J.; Schou, M.; Halldin, C. Reduction of [11C]CO2 to [11C]CO using solid supported zinc. J. Label. Compd. Radiopharm. 2017, 60, 624–628. [Google Scholar] [CrossRef]
- Zeisler, S.K.; Nader, M.; Theobald, A.; Oberdorfer, F. Conversion of no-carrier-added [11C]carbon dioxide to [11C]carbon monoxide on molybdenum for the synthesis of 11C-labelled aromatic ketones. Appl. Radiat. Isot. 1997, 48, 1091–1095. [Google Scholar] [CrossRef]
- Lu, S.; Haskali, M.B.; Ruley, K.M.; Dreyfus, N.J.-F.; DuBois, S.L.; Paul, S.; Liow, J.-S.; Morse, C.L.; Kowalski, A.; Gladding, R.L.; et al. PET ligands [18F]LSN3316612 and [11C]LSN3316612 quantify O-linked-β-N-acetyl-glucosamine hydrolase in the brain. Sci. Transl. Med. 2020, 12, Article ID eaau2939. [Google Scholar] [CrossRef]
- Lu, S.Y.; Hong, J.; Itoh, T.; Fujita, M.; Inoue, O.; Innis, R.B.; Pike, V.W. [carbonyl-11C]Benzyl acetate: Automated radiosynthesis via Pd-mediated [11C]carbon monoxide chemistry and PET measurement of brain uptake in monkey. J. Label. Compd. Radiopharm. 2010, 53, 548–551. [Google Scholar] [CrossRef]
- Gaudeau, M.; Zhang, M.; Tatoulian, M.; Lescot, C.; Ognier, S. Fast carbonylation reaction from CO2 using plasma gas/liquid microreactors for radiolabeling applications. React. Chem. Eng. 2020, 5, 1981–1991. [Google Scholar] [CrossRef]
- Niisawa, K.; Ogawa, K.; Saito, J.; Taki, K.; Karasawa, T.; Nozaki, T. Production of no-carrier-added 11C-carbon disulfide and 11C-hydrogen cyanide by microwave discharge. Int. J. Appl. Radiat. Isot. 1984, 35, 29–33. [Google Scholar] [CrossRef]
- Hara, T.; Iio, M. On-line synthesis of [11C]cyanide from cyclotron-produced [11C]carbon dioxide. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1987, 38, 1092–1093. [Google Scholar] [CrossRef]
- Landais, P.; Finn, R. Online preparation of [11C]carbon dioxide from [11C]methane. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1989, 40, 265–266. [Google Scholar] [CrossRef]
- Finn, R.D.; Christman, D.R.; Ache, H.J.; Wolf, A.P. The preparation of cyanide-11C for use in the synthesis of organic radiopharmaceuticals II. Int. J. Appl. Radiat. Isot. 1971, 22, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Christman, D.R.; Fin, R.D.; Karlstrom, K.I.; Wolf, A.P. Production of carrier-free H11CN for medical use and radiopharmaceutical syntheses. IX. J. Nucl. Med. 1973, 14, 864–866. [Google Scholar] [PubMed]
- Iwata, R.; Ido, T.; Takahashi, T.; Nakanishi, H.; Iida, S. Optimization of [11C]HCN production and no-carrier-added [1-11C]amino acid synthesis. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1987, 38, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qu, W. [11C]HCN radiochemistry: Recent progress and future perspectives. Eur. J. Org. Chem. 2021, 2021, 4653–4682. [Google Scholar] [CrossRef]
- Labaree, D.C.; Ropchan, J.R.; Nabulsi, N.; Huang, Y. A modification to improve the reliability of [11C]CN− production in the GE radiochemistry system. J. Label. Compd. Radiopharm. 2017, 60, 592–595. [Google Scholar] [CrossRef]
- Finn, R.D.; Boothe, T.E.; Vora, M.M.; Hildner, J.C.; Emran, A.M.; Kothari, P.J. Syntheses with isotopically labelled carbon. Methyl iodide, formaldehyde and cyanide. Int. J. Appl. Radiat. Isot. 1984, 35, 323–335. [Google Scholar] [CrossRef]
- Crouzel, C.; Sejourne, C.; Comar, D. Production of 11C-acetylene by methane pyrolysis. Int. J. Appl. Radiat. Isot. 1979, 30, 566–568. [Google Scholar] [CrossRef]
- Vaalburg, W.; Reiffers, S.; Beerling, E.; Pratt, J.J.; Woldring, M.G.; Wynberg, H. The preparation of carbon-11 labelled 17α-ethynylestradiol. J. Label. Compd. Radiopharm. 1977, 13, 200–201. [Google Scholar] [CrossRef]
- Vaalburg, W.; Feenstra, A.; Wiegman, T.; Beerling, H.D.; Reiffers, S.; Talma, A.; Woldring, M.G.; Wynberg, H. Carbon-11 labelled Moxestrol and 17α-methylestradiol as receptor binding radiopharmaceuticals. J. Label. Compd. Radiopharm. 1981, 18, 100–101. [Google Scholar] [CrossRef]
- Haskali, M.B.; Pike, V.W. [11C]Fluoroform, a breakthrough for versatile labeling of PET radiotracer trifluoromethyl groups in high molar activity. Chem. Eur. J. 2017, 23, 8156–8160. [Google Scholar] [CrossRef] [PubMed]
- Young, N.J.; Pike, V.W.; Taddei, C. Rapid and efficient synthesis of [11C]trifluoromethylarenes from primary aromatic amines and [11C]CuCF3. ACS Omega 2020, 5, 19557–19564. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Telu, S.; Yang, B.Y.; Haskali, M.B.; Jakobsson, J.E.; Pike, V.W. Rapid syntheses of [11C]arylvinyltrifluoromethanes through treatment of (E)-arylvinyl(phenyl)iodonium tosylates with [11C]trifluoromethyl copper(I). Org. Lett. 2020, 22, 4574–4578. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Torres, K.M.; Zhou, Y.P.; Yang, B.Y.; Guehl, N.J.; Sung-Hyun, M.; Telu, S.; Normandin, M.D.; Pike, V.W.; Brugarolas, P. Syntheses of [11C]2- and [11C]3-trifluoromethyl-4-aminopyridine: potential PET radioligands for demyelinating diseases. RSC Med. Chem. 2020, 11, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Telu, S.; Jana, S.; Haskali, M.B.; Yeun Yang, B.; Jakobsson, J.E.; Zhao, Q.; Ramos-Torres, K.M.; Brugarolas, P.; Pike, V.W. Broad-scope syntheses of [11C/18F]trifluoromethylarenes from aryl(mesityl)iodonium salts. Chem. Eur. J. 2023, 29, Article ID e202204004. [Google Scholar] [CrossRef] [PubMed]
- Crouzel, C.; Amano, R.; Fournier, D. Synthesis of carbon-11 labelled diazomethane. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1987, 38, 669–670. [Google Scholar] [CrossRef]
- Crouzel, C.; Hinnen, F. Synthesis of carbon-11 labelled lower chloromethanes: Application in a methylenation reaction. J. Label. Compd. Radiopharm. 1994, 35, S92–S93. [Google Scholar] [CrossRef]
- Solbach, C.; Machulla, H.J. Production of [11C]chloroform by direct chlorination of [11C]methane without catalyst support for the synthesis of [11C]diazomethane. Appl. Radiat. Isot. 2007, 65, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Prenant, C.; Crouzel, C. A new simple and attractive method of [11C]halogenomethanes production (Br11CH3, I11CH3). J. Label. Compd. Radiopharm. 1991, 30, 125. [Google Scholar] [CrossRef]
- Mock, B.H.; Mulholland, G.K.; Vavrek, M.T. Convenient gas phase bromination of [11C]methane and production of [11C]methyl triflate. Nucl. Med. Biol. 1999, 26, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Crouzel, C.; Långström, B.; Pike, V.W.; Coenen, H.H. Recommendations for a practical production of [11C]methyl iodide. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1987, 38, 601–603. [Google Scholar] [CrossRef]
- Link, J.M.; Clark, J.C.; Larsen, P.; Krohn, K.A. Production of [11C]methyl iodide by reaction of 11CH4 with I2. J. Label. Compd. Radiopharm. 1995, 37, 76–78. [Google Scholar] [CrossRef]
- Wilson, A.A.; Garcia, A.; Jin, L.; Houle, S. Radiotracer synthesis from [11C]iodomethane: A remarkably simple captive solvent method. Nucl. Med. Biol. 2000, 27, 529–532. [Google Scholar] [CrossRef]
- Roeda, D.; Dollé, F. [11C]Phosgene: A versatile reagent for radioactive carbonyl insertion into medicinal radiotracers for Positron Emission Tomography. Curr. Top. Med. Chem. 2010, 10, 1680–1700. [Google Scholar] [CrossRef]
- Brinkman, G.A.; Hass-Lisewska, I.; Veenboer, J.T.; Lindner, L. Preparation of 11COCl2. Int. J. Appl. Radiat. Isot. 1978, 29, 701–702. [Google Scholar] [CrossRef]
- Diksic, M.; Jolly, D.; Farrokhzad, S. An on-line synthesis of “no-carrier-added” [11C]phosgene. Int. J. Nucl. Med. Biol. 1982, 9, 283–285. [Google Scholar] [CrossRef]
- Roeda, D.; Zanten, B.v.; Crouzel, C. The production of 11C-phosgene without added carrier. Radiochem. Radioanal. Lett. 1978, 33, 175–178. [Google Scholar]
- Crouzel, C.; Roeda, D.; Berridge, M.; Knipper, R.; Comar, D. 11C-labelled phosgene: an improved procedure and synthesis device. Int. J. Appl. Radiat. Isot. 1983, 34, 1558–1559. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, J.E.; Lu, S.Y.; Telu, S.; Pike, V.W. [11C]Carbonyl difluoride-a new and highly efficient [11C]carbonyl group transfer agent. Angew. Chem. Int. Ed. 2020, 59, 7256–7260. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, J.E.; Telu, S.; Lu, S.Y.; Jana, S.; Pike, V.W. Broad scope and high-yield access to unsymmetrical acyclic [11C]ureas for biomedical imaging from [11C]carbonyl difluoride. Chem. Eur. J. 2021, 27, 10369–10376. [Google Scholar] [CrossRef] [PubMed]
- van Lier, E.J.; Posarac, D.; Kwok, K.E.; Lim, C.J. Modeling, simulation and experimental study of methanol synthesis for 11C- radiopharmaceuticals. Chem. Prod. Process Model. 2008, 3, Article ID 39. [Google Scholar] [CrossRef]
- Marazano, C.; Maziere, M.; Berger, G.; Comar, D. Synthesis of methyl iodide-11C and formaldehyde-11C. Int. J. Appl. Radiat. Isot. 1977, 28, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Christman, D.; Crawford, E.J.; Friedkin, M.; Wolf, A.P. Detection of DNA synthesis in intact organisms with positron-emitting [methyl-11C]thymidine. Proc. Natl. Acad. Sci. 1972, 69, 988–992. [Google Scholar] [CrossRef]
- Berger, G.; Maziere, M.; Sastre, J.; Comar, D. Carrier-free 11C formaldehyde - An approach. J. Label. Compd. Radiopharm. 1980, 17, 59–71. [Google Scholar] [CrossRef]
- Pike, V.W.; Palmer, A.J.; Horlock, P.L.; Perun, T.J.; Freiberg, L.A.; Dunnigan, D.A.; Liss, R.H. Semi-automated preparation of a 11C-labelled antibiotic - [N-methyl-11C]erythromycin-A lactobionate. Int. J. Appl. Radiat. Isot. 1984, 35, 103–109. [Google Scholar] [CrossRef]
- Roeda, D.; Dollé, F. Preparation of [11C]formaldehyde using a silver-containing ceramic catalyst. J. Label. Compd. Radiopharm. 2003, 46, 449–458. [Google Scholar] [CrossRef]
- Straatmann, M.G.; Welch, M.J. A general method for labeling proteins with 11C. J. Nucl. Med. 1975, 16, 425–428. [Google Scholar]
- Holschbach, M.; Schüller, M. A new and simple on-line method for the preparation of n.c.a. [11C]methyl iodide. Appl. Radiat. Isot. 1993, 44, 779–780. [Google Scholar] [CrossRef]
- Shah, F.; Pike, V.W.; Dowsett, K. Preparation of no-carrier-added [1-11C]ethylene and [1-11C]1,2-dibromoethane as new labelling agents. Appl. Radiat. Isot. 1997, 48, 931–941. [Google Scholar] [CrossRef]
- Nishijima, K.-i.; Kuge, Y.; Seki, K.-i.; Ohkura, K.; Motoki, N.; Nagatsu, K.; Tanaka, A.; Tsukamoto, E.; Tamaki, N. A simplified and improved synthesis of [11C]phosgene with iron and iron (III) oxide. Nucl. Med. Biol. 2002, 29, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Bramoullé, Y.; Roeda, D.; Dollé, F. A simplified [11C]phosgene synthesis. Tetrahedron Lett. 2010, 51, 313–316. [Google Scholar] [CrossRef]
- Ogawa, M.; Takada, Y.; Suzuki, H.; Nemoto, K.; Fukumura, T. Simple and effective method for producing [11C]phosgene using an environmental CCl4 gas detection tube. Nucl. Med. Biol. 2010, 37, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, T.; Mori, W.; Ogawa, M.; Fujinaga, M.; Zhang, M.R. [11C]phosgene: Synthesis and application for development of PET radiotracers. Nucl. Med. Biol. 2021, 92, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Någren, K.; Müller, L.; Halldin, C.; Swahn, C.-G.; Lehikoinen, P. Improved synthesis of some commonly used PET radioligands by the use of [11C]methyl triflate. Nucl. Med. Biol. 1995, 22, 235–239. [Google Scholar] [CrossRef]
- Någren, K.; Halldin, C.; Muller, L.; Swahn, C.G.; Lehikoinen, P. Comparison of [11C]methyl triflate and [11C]methyl-iodide in the synthesis of PET radioligands such as [11C]β-CIT and [11C]β-CFT. Nucl. Med. Biol. 1995, 22, 965–970. [Google Scholar] [CrossRef]
- Jewett, D.M. A simple synthesis of [11C]methyl triflate. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1992, 43, 1383–1385. [Google Scholar] [CrossRef]
- Jewett, E.M.; Någren, K.; Mock, B.H.; Watkins, G.L. 30 years of [11C]methyl triflate. Appl. Radiat. Isot. 2023, 197, Article ID 110812. [Google Scholar] [CrossRef]
- Schoeps, K.-O.; Stone-Elander, S.; Halldin, C. On-line synthesis of [11C]nitroalkanes. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1989, 40, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Långström, B.; Antoni, G.; Gullberg, P.; Halldin, C.; Någren, K.; Rimland, A.; Svärd, H. The synthesis of 1-11C-labelled ethyl, propyl, butyl and isobutyl iodides and examples of alkylation reactions. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1986, 37, 1141–1145. [Google Scholar] [CrossRef]
- Schoeps, K.-O.; Halidin, C.; Stone-Elander, S.; Långström, B.; Greitz, T. Preparation of 11C-nitromethane and an example of its use as a radiolabeling precursor. J. Label. Compd. Radiopharm. 1988, 25, 749–758. [Google Scholar] [CrossRef]
- Gustavsson, S.A.; Kato, K.; Långström, B. Purification of [11C]nitromethane for use in asymmetric nitroaldol reactions. J. Label. Compd. Radiopharm. 2003, 46, 1279–1285. [Google Scholar] [CrossRef]
- Schoeps, K.O.; Långström, B.; Stone-Elander, S.; Halldin, C. Synthesis of [1-11C]D-glucose and [1-11C]D-mannose from online produced [11C]nitromethane. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1991, 42, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, M.; Shiue, C.Y.; Gonzalez, C.; Dembowski, B. The synthesis of [11C]methanethiol, a precursor for S-[11C]methylations. J. Label. Compd. Radiopharm. 1995, 37, 109–110. [Google Scholar] [CrossRef]
- Kaneko, S.; Ishiwata, K.; Ishii, S.-I.; Omura, H.; Senda, M. Enzymatic synthesis of carbon-11 labeled methionine and its derivatives with immobilized γ-cyano-α-aminobutyric acid synthase. Appl. Radiat. Isot. 1999, 51, 285–291. [Google Scholar] [CrossRef]
- McCarron, J.A.; Pike, V.W. Synthesis of no-carrier-added [11C]methanesulfonyl chloride as a new labeling agent for PET radiopharmaceutical development. J. Label. Compd. Radiopharm. 2003, 46, 1127–1140. [Google Scholar] [CrossRef]
- Brown, G.D.; Luthra, S.K.; Brock, C.S.; Stevens, M.F.G.; Price, P.M.; Brady, F. Antitumor imidazotetrazines. 40. Radiosyntheses of [4-11C-carbonyl]- and [3-N-11C-methyl]-8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (temozolomide) for positron emission tomography (PET) studies. J. Med. Chem. 2002, 45, 5448–5457. [Google Scholar] [CrossRef]
- Kikuchi, T.; Ogawa, M.; Okamura, T.; Gee, A.D.; Zhang, M.-R. Rapid ‘on-column’ preparation of hydrogen [11C]cyanide from [11C]methyl iodide via [11C]formaldehyde. Chem. Sci. 2022, 13, 3556–3562. [Google Scholar] [CrossRef]
- Miller, P.W.; Bender, D. [11C]Carbon disulfide: A versatile reagent for PET radiolabelling. Chem. Eur. J. 2012, 18, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, G.; Långström, B. On-line production of [11C]cyanogen bromide. Appl. Radiat. Isot. 1997, 48, 459–461. [Google Scholar] [CrossRef]
- Bjurling, P.; Reineck, R.; Westerburg, G.; Gee, A.D.; Sutcliffe, J.; Långström, B. Synthia, a compact radiochemistry system for automated production of radiopharmaceuticals. In Proceedings of the 6th International Workshop on Targetry & Target Chemistry, TRIUMF, Vancouver, 1995; pp. 282-285. https://wttc.triumf.ca/95-pdf.html.
- Westerberg, G.; Långström, B. Synthesis of [11C]-and [13C]-cyanogen bromide. Useful electrophilic labelling precursors. Acta Chem. Scand. 1993, 47, 974–978. [Google Scholar] [CrossRef]
- Westerberg, G.; Bamford, M.; Daniel, M.J.; Långström, B.; Sutherland, D.R. Synthesis of 5-acetylamino-4-[11C]guanidino-2,6-anhydro-3,4,5-trideoxy-D-glycero-D-galacto-non-2-enoic acid ([11C]GG167)—an influenza virus neuraminidase inhibitor. J. Label. Compd. Radiopharm. 1996, 38, 585–589. [Google Scholar] [CrossRef]
- Westerberg, G.; Långström, B. Labelling of proteins with 11C in high specific radioactivity: [11C]albumin and [11C]transferrin. Appl. Radiat. Isot. 1994, 45, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, G.; Bergström, M.; Gustafson, S.; Lindqvist, U.; Sundin, A.; Långström, B. Labelling of polysaccharides using [11C]cyanogen bromide. In vivo and in vitro evaluation of 11C-hyaluronan uptake kinetics. Nucl. Med. Biol. 1995, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Imahori, Y.; Fujii, R.; Ido, T.; Hirakawa, K.; Nakahashi, H. Positron labeled phorbol ester: Synthesis method for “non-carrier added” phorbol 13-[1-11C] butyrate using ketene reaction. J. Label. Compd. Radiopharm. 1989, 27, 1025–1034. [Google Scholar] [CrossRef]
- Fujii, R.; Imahori, Y.; Ido, T.; Yagyu, T.; Horii, H.; Wakita, K.; Moriyama, Y.; Ueda, S.; Yamamoto, Y.L.; Nakahashi, H. New synthesis system of (C-11)propyl ketene and its reactons with various alcohols. J. Label. Compd. Radiopharm. 1991, 29, 497–505. [Google Scholar] [CrossRef]
- Fujii, R.; Imahori, Y.; Ido, T.; Yagyu, T.; Horii, H.; Wakita, K.; Ueda, S.; Nakahashi, H. New Synthesis of [C-11]propyl ketene using HCl/He gas mixture and the reaction on various alcohols. J. Label. Compd. Radiopharm. 1991, 30, 127–128. [Google Scholar] [CrossRef]
Scheme 1.
Decay of carbon-11 (A) and principle of PET (B). Reprinted and modified from Li and Conti, 2010 [3] with permission from Elsevier.
Scheme 1.
Decay of carbon-11 (A) and principle of PET (B). Reprinted and modified from Li and Conti, 2010 [3] with permission from Elsevier.
Scheme 2.
Processes leading to [11C]carbon dioxide and [11C]methane by proton irradiation of nitrogen gas with low level oxygen (A) or ~ 5% hydrogen (B), respectively.
Scheme 2.
Processes leading to [11C]carbon dioxide and [11C]methane by proton irradiation of nitrogen gas with low level oxygen (A) or ~ 5% hydrogen (B), respectively.
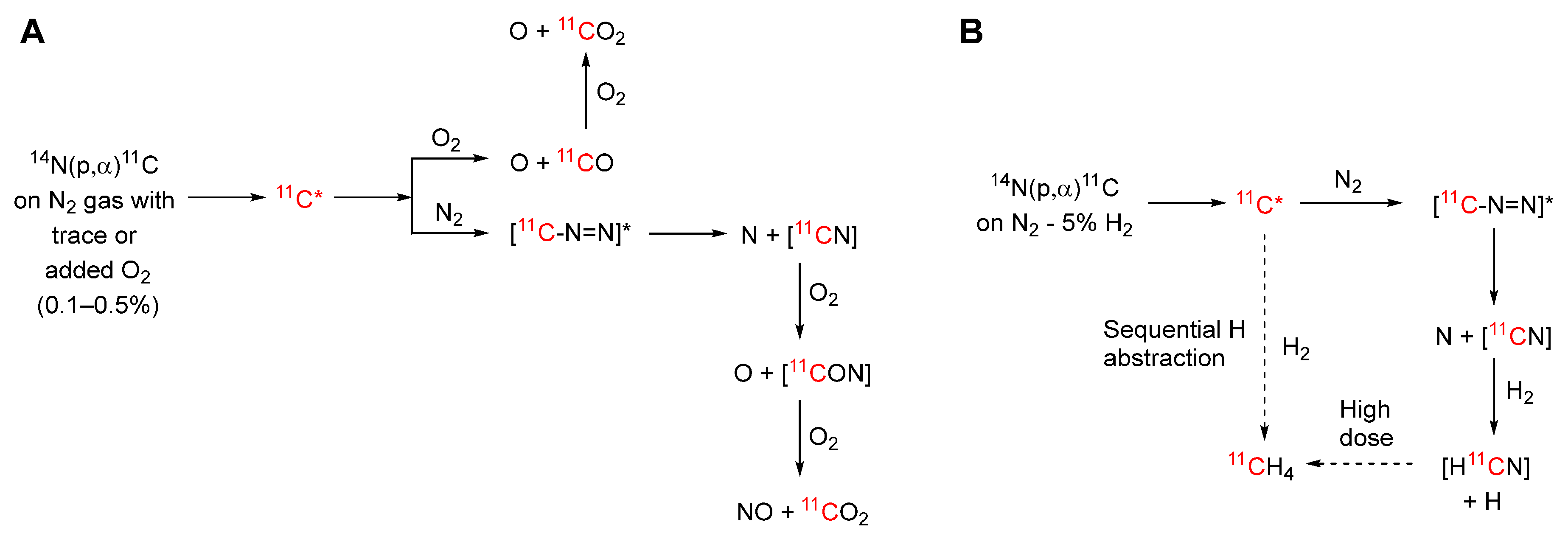
Scheme 3.
Conversions of [11C]carbon dioxide into other 11C-labeling agents: (A) [11C]methane; (B) [11C]carbon monoxide; (C) [11C]hydrogen cyanide; (D) [11C]carbon disulfide.
Scheme 3.
Conversions of [11C]carbon dioxide into other 11C-labeling agents: (A) [11C]methane; (B) [11C]carbon monoxide; (C) [11C]hydrogen cyanide; (D) [11C]carbon disulfide.
Scheme 4.
Conversion of [11C]methane into: (A) [11C]carbon dioxide; (B) [11C]hydrogen cyanide; (C) [11C]acetylene.
Scheme 4.
Conversion of [11C]methane into: (A) [11C]carbon dioxide; (B) [11C]hydrogen cyanide; (C) [11C]acetylene.
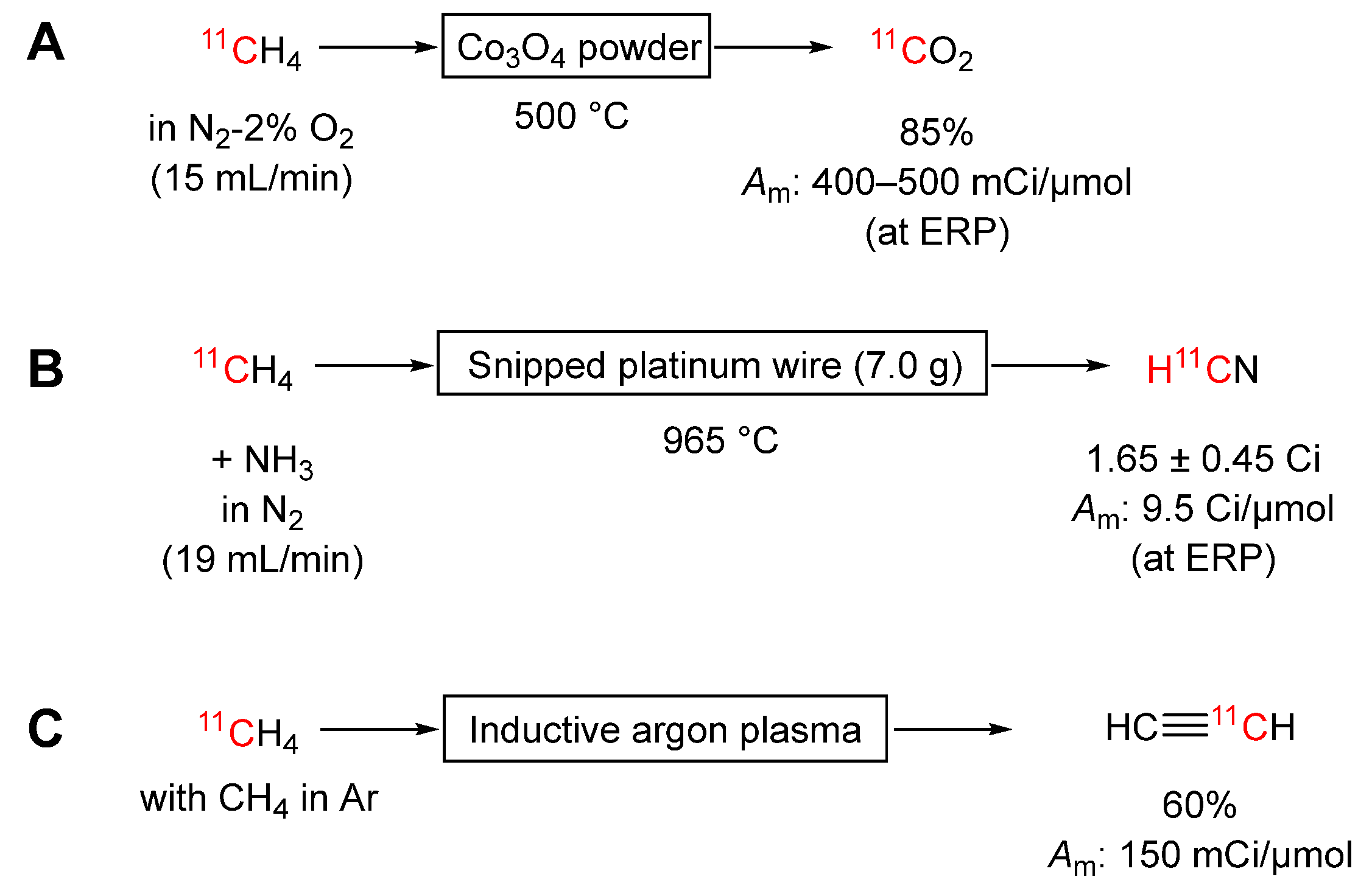
Scheme 5.
Halogenations of [11C]methane to produce useful labeling synthons: (A) fluorination; (B) chlorination; (C) bromination; (D) iodination.
Scheme 5.
Halogenations of [11C]methane to produce useful labeling synthons: (A) fluorination; (B) chlorination; (C) bromination; (D) iodination.
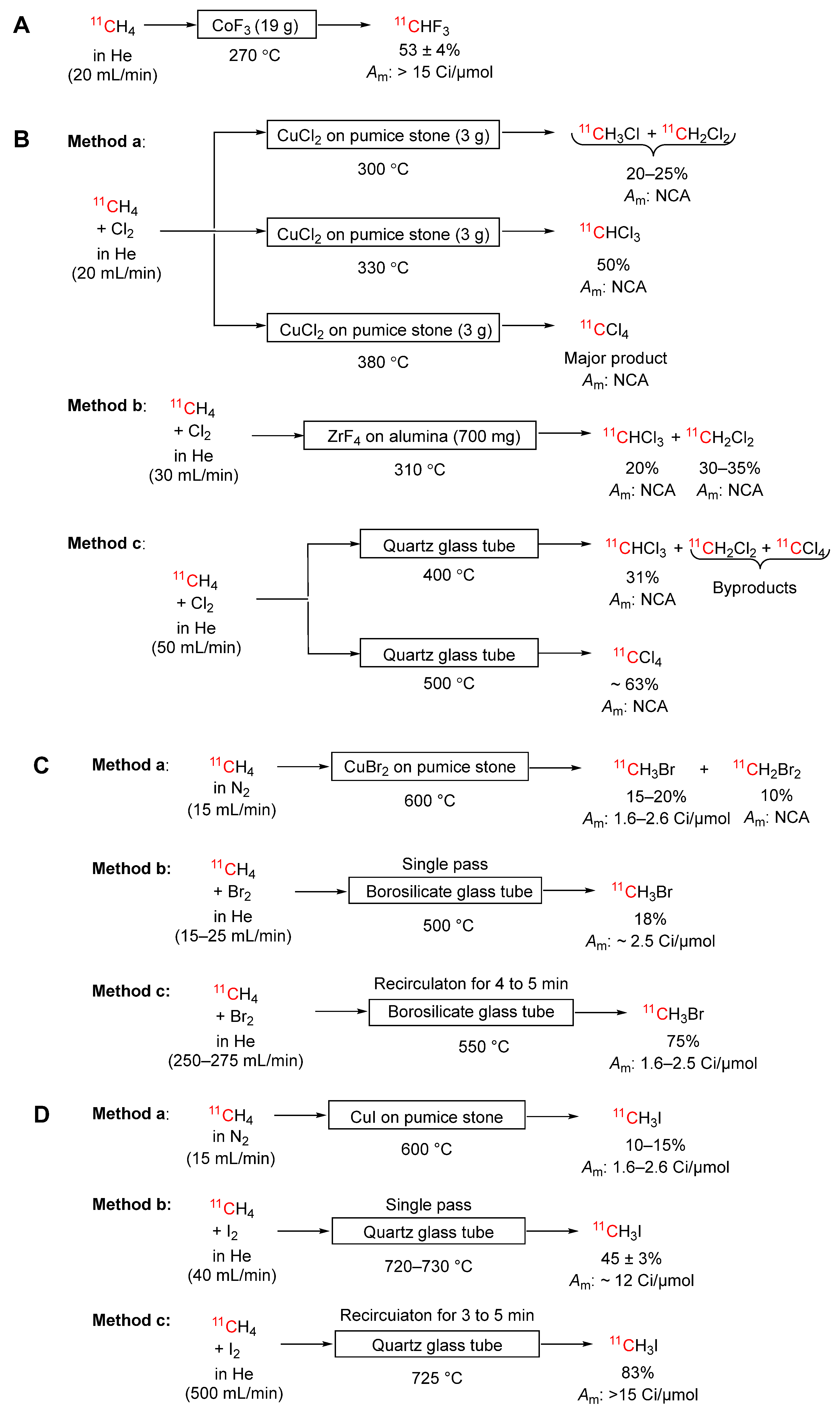
Scheme 6.
Conversions of [11C]carbon monoxide into other labeling synthons and intermediates: (A) [11C]phosgene; (B) [11C]carbonyl difluoride; C) Proposed for [11C]methanol.
Scheme 6.
Conversions of [11C]carbon monoxide into other labeling synthons and intermediates: (A) [11C]phosgene; (B) [11C]carbonyl difluoride; C) Proposed for [11C]methanol.
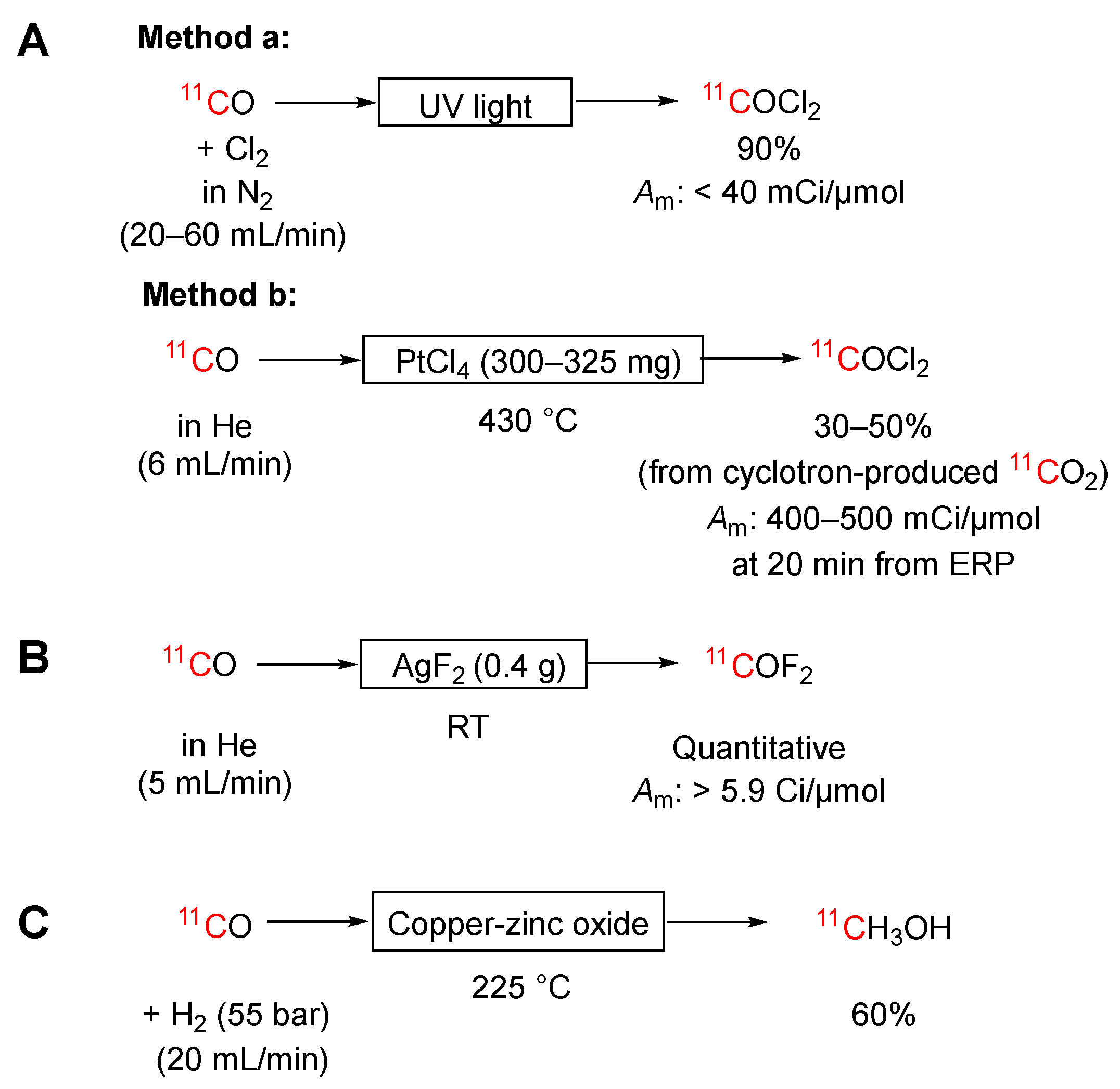
Scheme 7.
Conversions of [11C]methanol into (A) [11C]formaldehyde; (B) [11C]iodomethane.
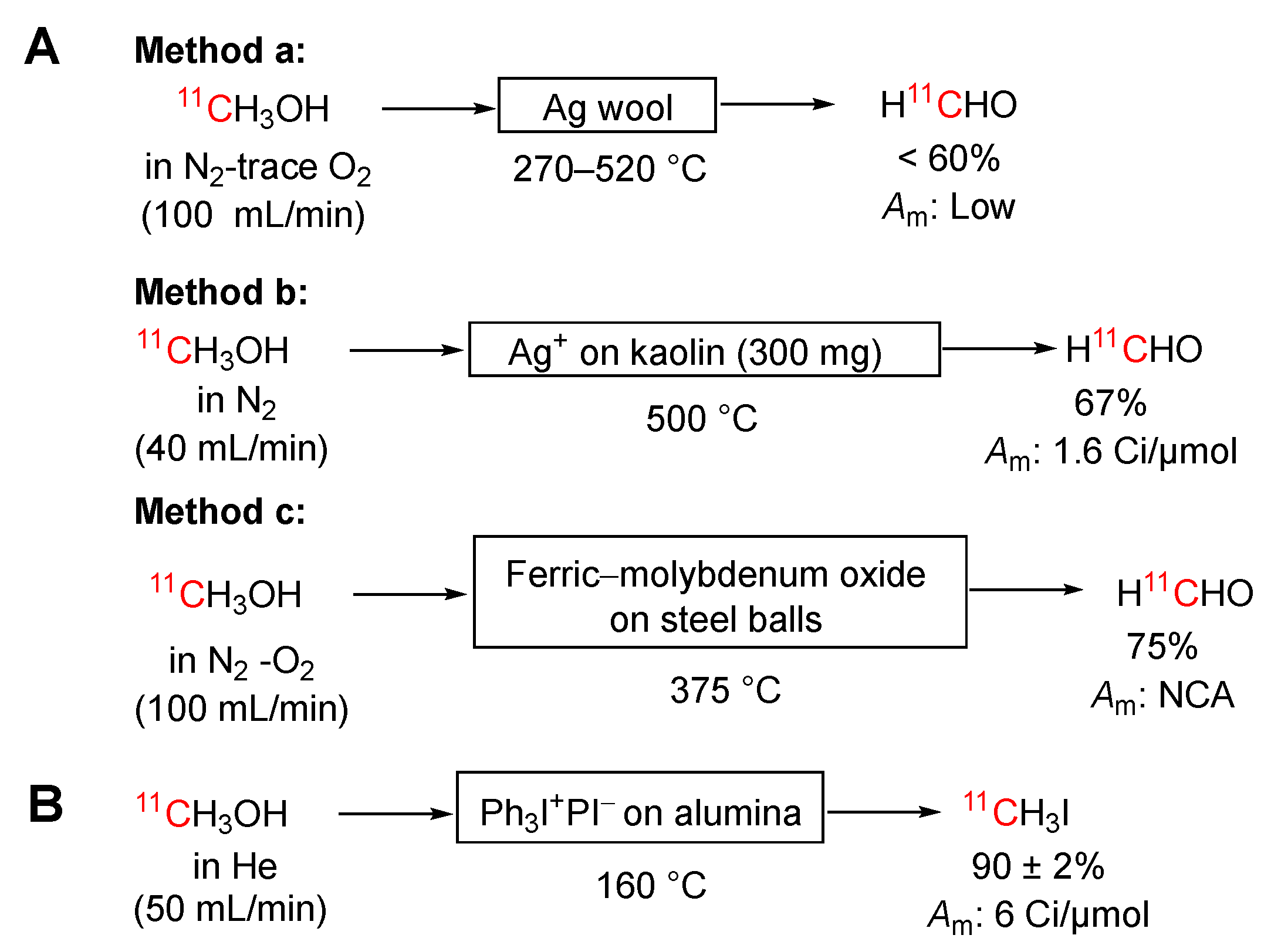
Scheme 8.
Conversion of [1-11C]ethanol into [1-11C]ethylene.
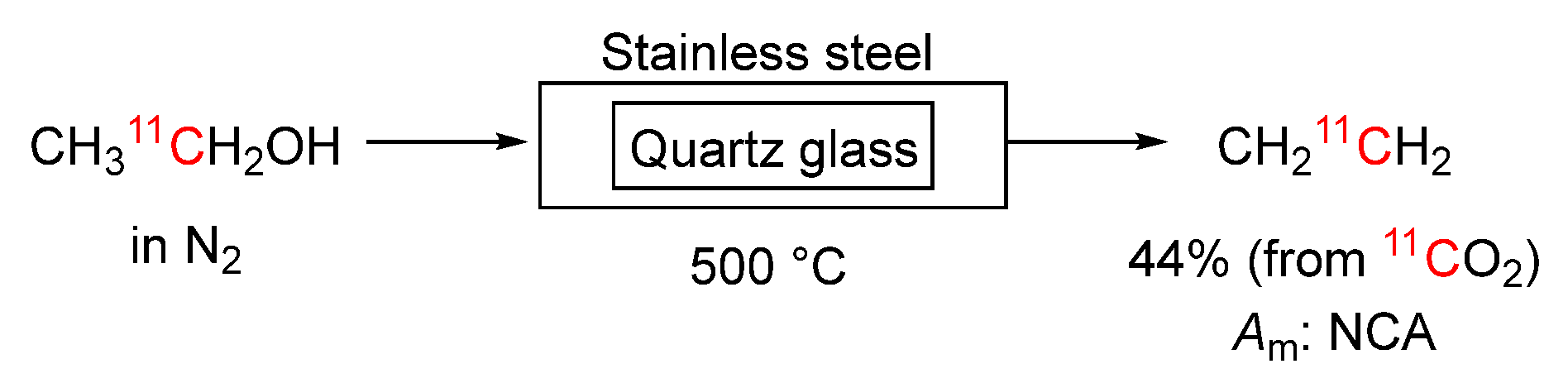
Scheme 9.
Conversions of [11C]carbon tetrachloride into [11C]phosgene.
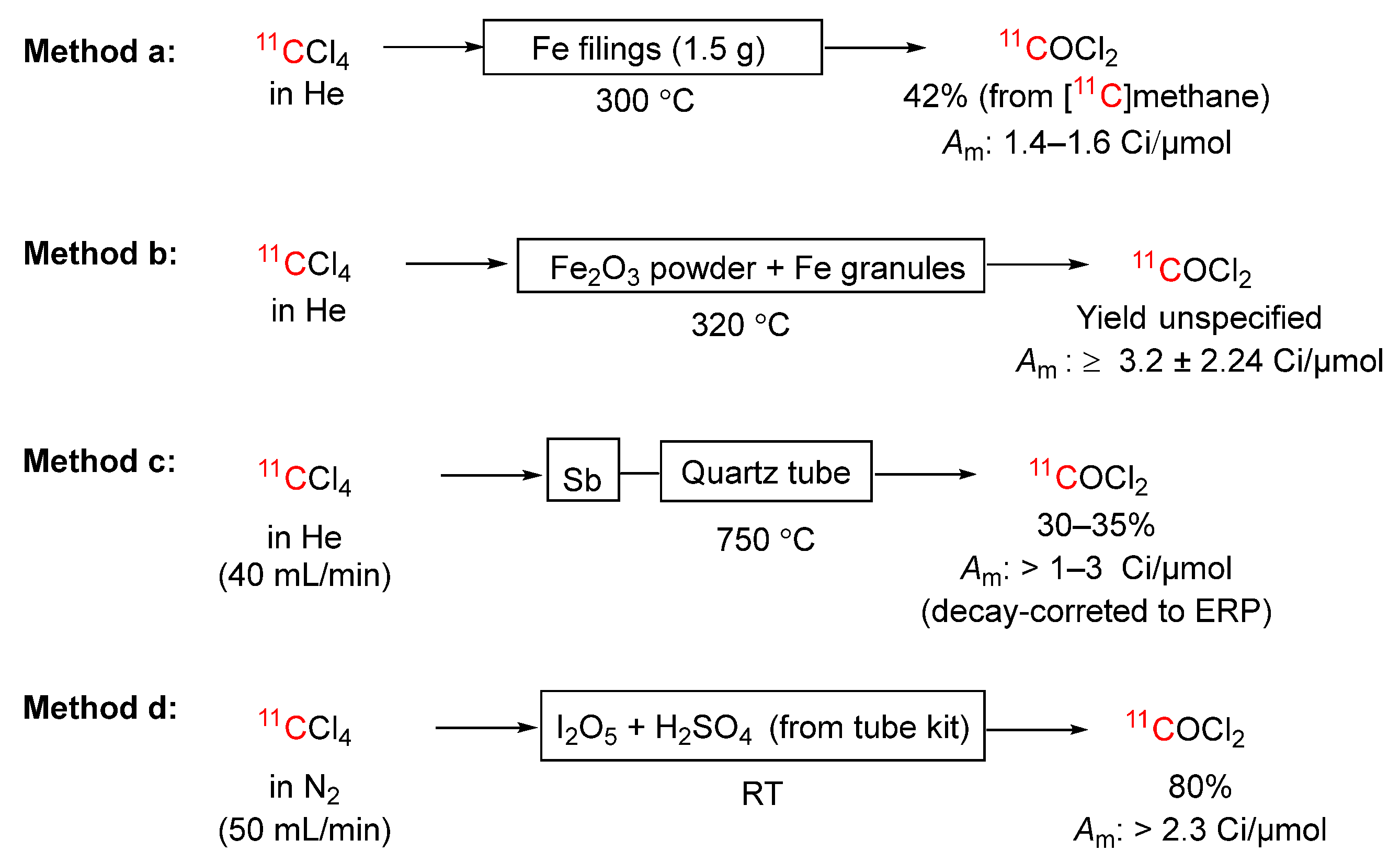
Scheme 10.
Metathetical conversions of [11C]haloalkanes into useful labeling synthons by passage over heated silver(I) or sodium salts into: (A) and (B) [11C]methyl triflate; (C) [11C]nitroalkanes; (D) [11C]methanethiol; (E) [11C]mesyl chloride; (F) [methyl-11C]methyl isocyanate.
Scheme 10.
Metathetical conversions of [11C]haloalkanes into useful labeling synthons by passage over heated silver(I) or sodium salts into: (A) and (B) [11C]methyl triflate; (C) [11C]nitroalkanes; (D) [11C]methanethiol; (E) [11C]mesyl chloride; (F) [methyl-11C]methyl isocyanate.
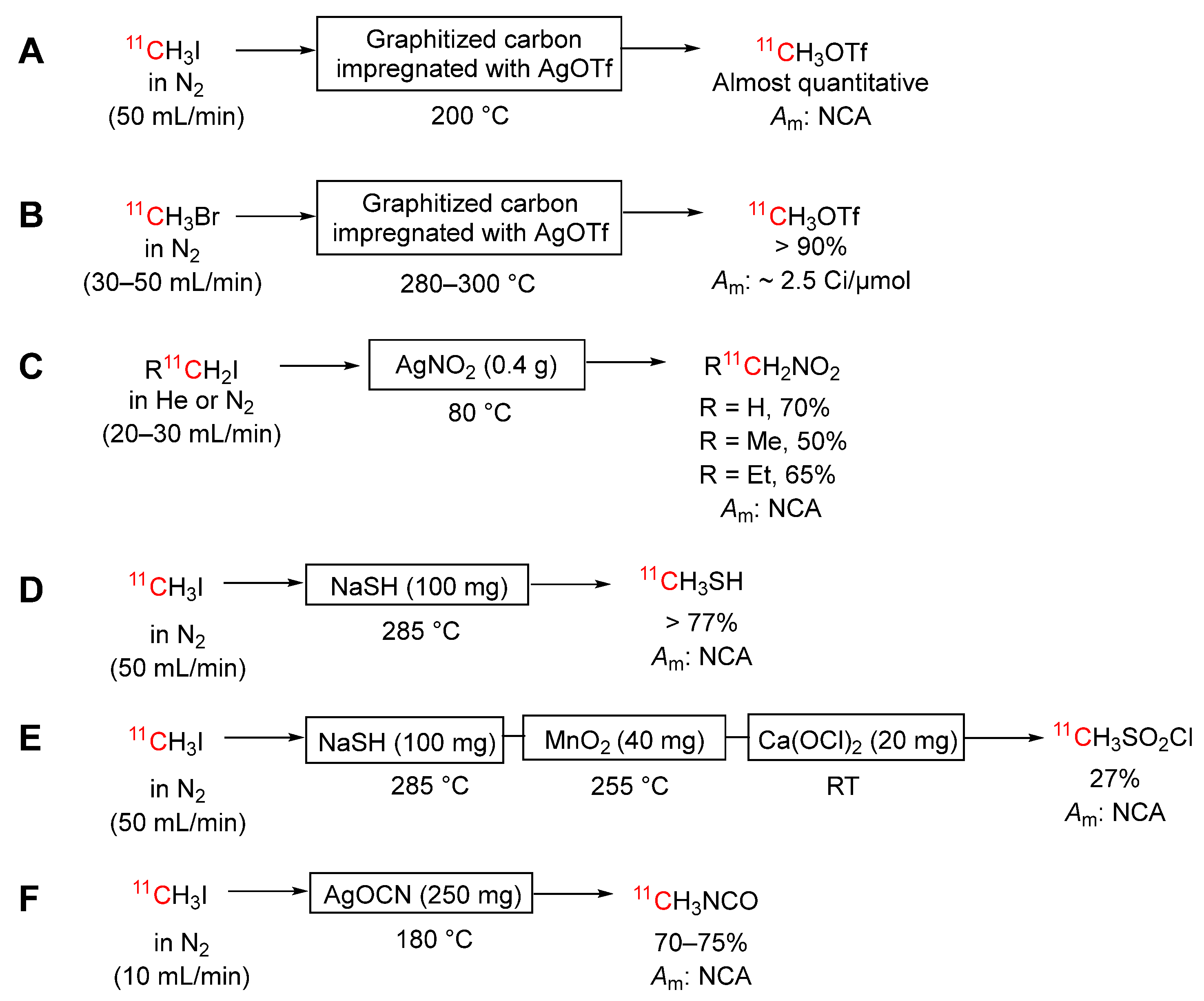
Scheme 11.
Conversions of [11C]iodomethane into: (A) [11C]hydrogen cyanide; (B) [11C]carbon disulfide.
Scheme 11.
Conversions of [11C]iodomethane into: (A) [11C]hydrogen cyanide; (B) [11C]carbon disulfide.
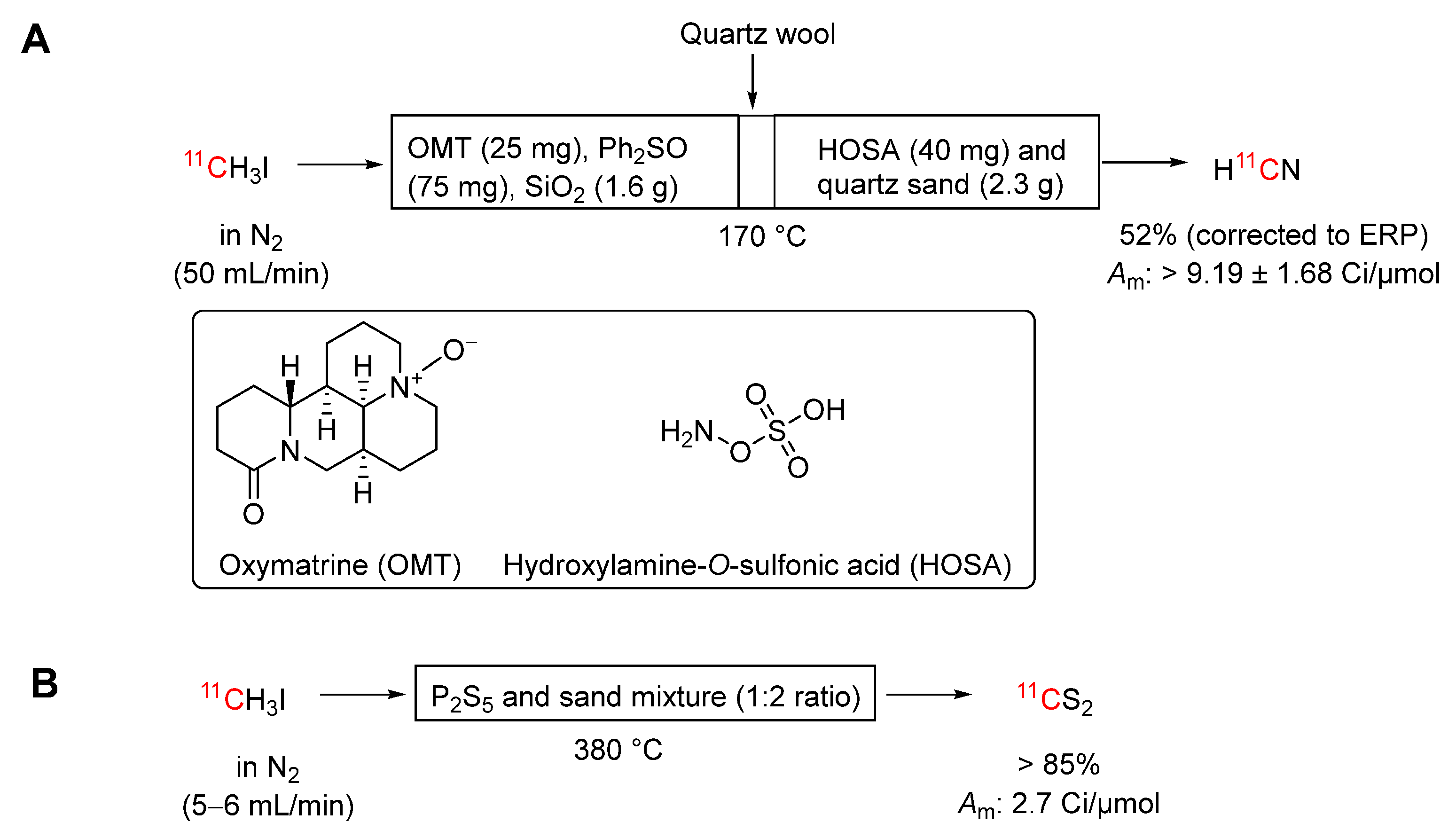
Scheme 12.
Conversion of [11C]hydrogen cyanide into [11C]cyanogen bromide.
Scheme 13.
Conversion of [1-11C]butyric acid into [1-11C]propylketene.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



