
Submitted:
06 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
O2 is essential for the life of eukaryotic cells. The ability to sense oxygen availability and initiate a response to adapt the cell to changes in O2 levels is a fundamental achievement of evolution. The key switch for adaptation consists of the transcription factors HIF1A, HIF2A and HIF3A. Their levels are tightly controlled by O2 through the involvement of the oxygen-dependent prolyl hydroxylase domain-containing enzymes (PHDs/EGNLs), the von Hippel-Lindau tumour suppressor protein (pVHL) and the ubiquitin-proteasome system. Furthermore, HIF1A and HIF2A are also under the control of additional post-translational modifications (PTMs) that positively or negatively regulate the activities of these transcription factors. This review focuses mainly on two PTMs of HIF1A and HIF2A: phosphorylation and acetylation.
Keywords:
Hypoxia
; phosphorylation
; HIF-1 alpha
; HIF-2 alpha
; HIF-3 alpha
; acetylation
; HDACs
; KATs
1. Introduction
The hypoxia response
Human cells need O2 to regenerate ATP, to multiply and to survive. When O2 availability decreases, complex adaptive responses are initiated that conserve oxygen consumption by reducing oxidative phosphorylation in the mitochondria and support glycolysis. Angiogenesis is stimulated and cell proliferation is reduced. The transcription factors HIF1A/HIF-1α and EPAS1/HIF2A/HIF-2α are the key players of the adaptive response to O2 depletion [1–4].
HIF-1 is a heterodimer composed of the O2 sensitive subunit HIF1A and a constitutively expressed subunit HIF1B/HIF-1β, also known as aryl hydrocarbon-receptor nuclear translocator (ARNT) [5]. In selected vertebrate cell types or in cancer cells, another O2-sensitive subunit, EPAS1/HIF2A is also expressed (Figure 1). The two subunits can have opposing effects in cancer cells by controlling the transcription of different target genes [6,7].
HIF-3α/HIF3A is the third member of the O2-inducible HIF-TFs. Similar to EPAS1/HIF2A, it is specifically expressed in certain tissues [8]. Structurally, HIF3A differs from HIF1A and EPAS1/HIF2A mainly in the carboxy term, where a leucine zipper domain (LZIP) is present, which is involved in protein-protein interactions, while the TAD is absent [9,10]. Different HIF3A splice variants have been reported (HIF-3α1-10), which originate from alternative initiation transcription sites. HIF3A may act as an inhibitor of transcription mediated by HIF1A and EPAS1/HIF2A by competing for binding to HIF1B [10,11].
O2 controls the stability of HIF1A and EPAS1/HIF2A through the action of 2-oxoglutarate (2-OG) dependent dioxygenases (2-OGDDs), the prolyl hydroxylase domain (PHD1/2/3) also known as EGLN1/2/3 (Figure 2). These enzymes hydroxylate key prolines of HIF1A (Pro-402 and Pro-564) and of EPAS1/HIF2A (Pro-405 and Pro-531) using O2 and α-oxoglutarate [1,12,13]. When HIF1A and EPAS1/HIF2A are hydroxylated, they are recognized by VHL, a tumor suppressor gene responsible for von Hippel-Lindau disease. VHL is the substrate recognition subunit of an ubiquitin E3-ligase complex that directs HIF1A and EPAS1/HIF2A to proteasomal degradation [4]. This multiprotein complex also includes Cullin-2 (Cul-2), Elongin-1, Elongin-2 and Ring-Box 1 (RBX1). It polyubiquitylates HIF1A at K532, K538, K567 or EPAS1/HIF2A at residues K497, K503, K512 [14].
When the O2 content decreases, hydroxylation cannot take place and HIF1A/EPAS1/HIF2A rapidly accumulate. After dimerization, the mature TFs migrate into the nucleus and bind to E-box-like hypoxia response elements (HREs) in the promoter region of hundreds of target genes (Figure 2). These genes represent the hypoxia response and HIFs support their transcription and adaptation to the altered environment [15]. Control by the ubiquitin-proteasome system (UPS) is a key mechanism for cellular adaptation to varying O2 levels.
2. HIF1A and HIF2A: not only protein hydroxylation
Regulating the hydroxylation and stability of HIF1A and EPAS1/HIF2A in response to O2 is a rapid and efficient strategy to link changes in environmental conditions to cell adaptations. However, other PTMs are also used to modulate HIF1A and EPAS1/HIF2A activities and the hypoxia response, mainly by controlling protein stability or enhancing transcriptional activity.
3. Phosphorylation
The regulation of kinase activities in response to hypoxia and the control of phosphorylation of HIFs-TFs in normoxia or hypoxia have been intensively studied. Several kinases have been reported to have HIF1A and EPAS1/HIF2A as substrates. This implies that alternative signaling pathways can be used to modulate the activity of these TFs. Several residues have been reported as targets of phosphorylation (Figure 3A and C) [16]. The possibility that phosphorylation could affect the synthesis, stability, and activity of HIF1A was already observed in early studies using various inhibitors, including tyrosine phosphatase inhibitors [17]. Several studies have subsequently confirmed that receptor tyrosine kinase signaling, or cytoplasmic/nuclear tyrosine kinases can indirectly influence HIF1A [18,19]. However, there is no experimental evidence that tyrosine kinases can directly phosphorylate HIF1A.
3.1. GSK3β: another route to degradation
The serine-threonine kinase GSK3β can phosphorylate HIF1A at multiple residues within the oxygen-dependent-degradation (ODD) domain. This leads to VHL independent polyubiquitylation and degradation of HIF1A [20–22]. FBW7-E3 ubiquitin ligase is involved in GSK3β-dependent degradation of HIF1A in both normoxia and hypoxia. Deubiquitylases are often part of the E3-ligase multiprotein complexes to fine-tune the degradation option [23]. In the case of HIF1A the deubiquitylase USP28 can antagonize the FBW7-E3 ligase to preserve HIF1A from degradation [14,22,24].
3.2. Cell cycle kinases
Hypoxia reduces cell proliferation in a variety of cell types including cancer cells. HIF1A can directly control cell cycle progression [2]. It is therefore not surprising that various kinases that are active during the cell cycle can phosphorylate HIF1A. Polo-like kinase 3 (PLK3), a family of serine-threonine kinases that contribute to the control of mitosis but also has additional non-mitotic functions, regulates the stability of HIF1A in normoxia [25]. Serine 576, within the ODD domain, and serine 657, next to the nuclear export sequence (NES), are the sites phosphorylated by PLK3 [26]. By phosphorylating these sites, PLK3 exerts a negative influence on the stability and activity of HIF1A [27].
During the cell cycle under normoxia, HIF1A is also a target of regulation by the CDKs (cyclin-dependent kinases), the main regulators of cell cycle progression. Opposite effects on the stability of HIF1A have been described for CDK1 and CDK2 [28]. According to one study, the lysosomal degradation pathway is involved in the control of HIF1A levels. More specifically, CDK2 favors the lysosomal degradation of HIF1A, while CDK1 hinders it [28]. However, another group of researchers confirmed that CDK1-dependent phosphorylation of HIF1A can underpin its activity, but by suppressing proteasome-mediated degradation [29]. Finally, it should be mentioned that CDK2 can promote HIF1A activity in certain cancer cells [28]. It can therefore be concluded that the normal progression of the cell cycle requires strict regulation of HIF1A levels.
In endothelial cells, CDK5, which is not involved in cell cycle control but regulates neuronal development, can phosphorylate HIF1A at serine 687 and thus stabilize it [30].
3.3. PKA
A role of PKA in the regulation of HIF1A phosphorylation was first reported in endothelial cells exposed to intermittent hypoxia [31]. Subsequently PKA was shown to phosphorylate T63 and S692 on HIF1A inhibiting its proteasome-mediated degradation and promoting its transcriptional activation. PKA also stimulates the binding of KAT (lysine acetyl-transferase) p300/KAT3B to the carboxy-terminal TAD domain of HIF1A to sustain transcriptional activation [32]. In this way phosphorylation can coordinate acetylation.
3.4. HIF1A phosphorylation and the DNA damage response (DDR)
In proliferating cells hypoxia can also engage elements of the DNA damage response (DDR) due to the induction of replication stress [33–35]. Members of the phosphoinositide 3-kinase-related kinases (PIKKs) family DNA-PK, ataxia telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3 related kinase (ATR) can modulate HIF1A levels and activities [33–35]. ATM has been shown to phosphorylate HIF1A at S696, a process that has been associated with downregulation of mTORC1 signaling [33]. Instead, ATR is required for efficient translation of HIF1A mRNA, by a yet undetermined mechanism [34,35].
An alternative indirect mechanism has been proposed to explain the link between hypoxia, DDR and HIF1A. The histone variant H2AX interacts with HIF1A and stabilizes it, protecting it from nuclear export and degradation. Monoubiquitylation and phosphorylation of H2AX, which are strictly mediated by hypoxia-induced E3 ligase activity of TRAF6 and ATM, can activate HIF1A signaling and promote tumorigenesis [36]. Another indirect mechanism used by ATM/ATR to maintain HIF1A involves seryl-tRNA synthetase (SerRS). SerRS can regulate blood vessel formation by repressing the transcription of VEGFA, independent of its aminoacylation activity. In the proposed mechanism, when SerRS is not phosphorylated by ATM/ATR, it can compete with HIF1A for binding to DNA and turn off the hypoxic genetic program [37].
3.5. Other kinases
ERK1/2 can also phosphorylate both HIF1A and EPAS1/HIF2A. Phosphorylation enhances transcriptional activity by inhibiting nuclear export. In fact, the phosphorylated residues (Ser641/643 in HIF1A and S672 in EPAS1/HIF2A) are located near a non-canonical NES and affect binding to the exportin CRM1 [38,39].
Casein kinase 1 (CK1) has also been described as an upstream regulator of HIF1A activities. Casein kinase 1δ (CK1δ) controls S247 phosphorylation within the N-terminal heterodimerization domain of HIF1A. The result is an impairment of the formation of the HIF1A/HIF1B complex and a reduction in the response to hypoxia [40]. CK1δ is also involved in the phosphorylation of EPAS1/HIF2A, but unlike HIF1A, this phosphorylation enhances transcriptional activity. S383 and T528 are the residues phosphorylated in vitro. As with ERK1/2, the proposed mechanism involves the regulation of CRM1-dependent nuclear export under hypoxia [41].
The serine-threonine kinase PIM (Proviral Integration site for Moloney murine leukemia virus) family are pro-oncogenic factors that control cell cycle progression, proliferation, and survival [42]. They also promote tumor angiogenesis by controlling the phosphorylation of HIF1A and EPAS1/HIF2A. HIF1A is phosphorylated at threonine 455 and HIF2A at serine S435. In both cases, protein stability is increased, even under normoxic conditions. Phosphorylation impairs the binding of prolyl hydroxylases and the canonical pathway of proteasomal-mediated degradation [43].
Using LC/MS/MS-based analysis, Ser451 was identified within the ODD of HIF1A as a target of phosphorylation under hypoxic conditions. This phosphorylation is important for the maintenance of HIF1A levels by inhibiting its interaction with PHD and pVHL. In this way, tumor growth is supported [44]. Protection of HIF1A degradation may depend on binding of the chaperone HSP90, but the kinase involved is unknown.
Finally, it was recently reported that the PKB/AKT kinase phosphorylates HIF3A1 directly at serine 524 in the ODD domain to regulate its stability. Mutagenesis at this site impairs cell proliferation and survival, leading to defects in proliferation, colony formation and cell adherence [45].
4. Acetylation
Lysine acetylation is a widespread and conserved PTM that regulates virtually all cellular processes, from bacteria to human cells [46]. Although much attention has been paid to the acetylation of histones in the context of chromatin organization and epigenetics, it is now clear that hundreds of different proteins can be acetylated in different contexts and cellular compartments [47].
This PTM is reversible, and two enzyme families control it antagonistically [48,49]. The KATs (lysine acetyl transferases) catalyze the transfer of an acetyl group from acetyl-CoA to the ε−amino group of certain lysine residues. Their action is counteracted by KDACs (lysine deacetylases), which are mostly known as HDACs (histone deacetylases).
KATs can be divided into three main families based on their homology to yeast orthologs and their catalytic mechanism. The three families are: i) the p300/CREB-binding proteins (p300/CBP); ii) the GCN5-related N-acetyltransferases (GNAT) and iii) the MOZ, Ybf2, Sas2 and Tip60 (MYST) family [50,51]. In addition, several protein complexes have been reported to possess lysine acetyltransferase activity. They are referred to as non-canonical -KATs [48].
Similarly, the 18 human KDACs/HDACs can be divided into 5 different subfamilies based on their homology to yeast orthologs and their catalysis mechanism. Class I includes HDAC1, HDAC2, HDAC3 and HDAC8, which share homology with Rpd3. Class IIa includes HDAC4, HDAC5, HDAC7 and HDAC9. Class IIb includes HDAC6 and HDAC10. Class IIa and class IIb have a common homology with Hda1. Class III groups, Sirt1, 2, 3, 4, 5, 6 and 7, also known as Silent Information Regulators (SIR), are homologous to Sir2 in yeast. Class IV contains only HDAC11, as it has sequence similarities with the family members of classes I and II. Classes I, II and IV members are zinc-dependent enzymes, while class III members are NAD-dependent [49,52,53].
Importantly, a Tyr/His substitution in the catalytic site that drastically reduces (almost eliminates) the catalytic activity against acetyl-lysine, characterizes class IIa HDACs in vertebrates. However, by assembling with class I HDACs and particularly the NCOR1-NCOR2-HDAC3 complex via the deacetylase domain, they can monitor lysine deacetylation [54,55].
4.1. HIF1A and EPAS1/HIF2A acetylation
Various residues of HIF1A and EPAS1/HIF2A have been experimentally described as acetylation sites experimentally or identified by mass spectrometry analysis. Figure 3D and C summarize these data as reported by the PhosphoSitePlus database for HIF1A and EPAS1/HIF2A [56]. The same lysine is also frequently subjected to ubiquitylation, suggesting a possible link to the regulation of protein stability. In general, the presence of lysine residues targeted for both acetylation and ubiquitylation is frequently observed in different proteins. This is an evolutionarily conserved competition for a more sophisticated control of protein stability [57–59]. In principle, the acetylation of lysines, which are directed to the proteasome via K48-linked poly-ubiquitylation, can be regarded as a protein stabilization factor. This is the case for acetylation at K709 of HIF1A, which increases protein stability by reducing poly-ubiquitylation under both normoxia and hypoxia conditions. This acetylation is mediated by p300/KAT3B and antagonized by HDAC1 but not HDAC3 [60]. However, the consequences of lysine acetylation could be different depending on which residues are modified, especially if the region is under the control of some E3 ligases.
KATs and HDACs can act upstream of HIF1A and EPAS1/HIF2A to control the acetylation status of selected lysines. In addition, KATs and HDACs can also be partners and downstream effectors of these TFs to locally modify chromatin (histone acetylation) to promote or repress gene transcription [61]. In this review, KAT and KDACs are discussed as upstream regulators of HIF1A and EPAS1/HIF2A.
The p300/CBP-associated factor (PCAF/KAT2B) was originally identified as part of a multiprotein complex with HIF1A that controls the transcription of hypoxia-responsive genes [62]. PCAF/KAT2B acetylates HIF1A at K532 and possibly other sites to regulate its stability and selectively directs HIF1A to a subset of target genes [63].
EPAS1/HIF2A is similarly regulated by acetylation. The CREB-binding protein (CREBBP/CBP/KAT3A) can interact via an enzyme/substrate interaction and contributes to HIF2A-mediated EPO transcription. Acetylation mediated by KAT3A is reversed by SIRT1, and this cycle enhances (in an undetermined manner) HIF2A activity [64].
A general increase in HIF1A acetylation was observed in response to DNA damage and LPS treatment, which correlates with increased protein stability in macrophages. The KAT Tip60/KAT5 was involved in the regulation of acetylation and protein stability in response to LPS. Tip60 binds to HIF1A and together with the CDK8 mediator complex is required for efficient expression of most genes under HIF1A control during hypoxia [65].
In a comparative study, K674 of HIF1A and K741 of HIF2A were found to be acetylated by PCAF/KAT2B and CBP/KAT3A, respectively. These residues are deacetylated by SIRT1, which leads to a reduction in the transactivation activity of both TFs, but in the case of EPAS1/HIF2A depending on the cell line [66].
In general, several studies show a positive effect of lysine acetylation on the stability and transcriptional activity of HIF1A and EPAS1/HIF2A. As a logical consequence, one could argue that HDACs/KDACs should act as negative regulators of HIF1A and EPAS1/HIF2A. However, the available literature suggests a more complex scenario.
4.2. HDACs and hypoxia
The involvement of HDACs in the regulation of the cellular response to hypoxia soon attracted interest. Initial studies suggested that HDAC1 exerts a pro-hypoxia function by regulating the expression of TP53 and von Hippel-Lindau [67]. Non-selective HDAC inhibitors (HDACIs) hindered the hypoxia response and the neovascularization process [67,68]. However, since HDACs can act as both regulators and effectors, it soon became apparent that HDACIs act at multiple levels [69–71]. A negative influence of HDACs on HIF1A-dependent transcription was demonstrated by studies with the viral oncoprotein E7 of human papillomaviruses (HPV). E7 was able to displace the binding of class I and class IIa HDACs to HIF1A, while it did not affect the binding of p300/KAT3B and of PCAF/KAT2B [72]. However, contrary results have also been reported with a positive effect of HDACs on HIF1A activities under hypoxia (see below).
HDACs can also be a partner of HIF1A to modulate a repressive genetic program [73,74]. To add complexity, HDACs can also act independently of HIF1A to modulate the hypoxia response. Elongation is a key process that determines efficient transcription and gene expression. Phosphorylation of the carboxy-terminus of RNA polymerase II (RNAPII) by transcription elongation factor b (P-TEFb) is an important regulatory step to promote productive elongation. Active P-TEFb is produced by CDK9, the catalytic subunit, and cyclin T1 or cyclin T2, two distinct regulatory subunits. When associated with HEXIM1/2 in the 7SK snRNP, P-TEFb is kept inactive [75]. Under hypoxia, HDAC3/NcoR complexes colocalize in the nuclei with CDK9 cyclin T1, which are deacetylated, and the formation of the inactive complex with HEXIM1 is favored. As a result, approximately 70% of differentially expressed genes are repressed after 1 hour of hypoxia [76].
5. Class I HDACs
Studies on the involvement of class I HDACs as upstream regulators of HIF1A and EPAS1/HIF2A activities suggest a possible role of HDAC1 and HDAC2 and exclude a contribution of HDAC3. However, it is worth considering that HDAC3 may contribute to the deacetylase activities of class IIa HDACs, as we will discuss in the next section.
HDAC1 and HDAC2 are often partners in various multiprotein complexes containing scaffolding factors that are required both to enhance their enzymatic activity and to mediate interaction with selected TFs or epigenetic readers. In addition, either HDAC1 or HDAC2 have been reported to act independently as part of different complexes [53].
Hypoxia could affect HDAC1 and HDAC2 activities via protein kinase CK2-driven phosphorylation, contributing to the downregulation of pVHL and stabilization of HIF1A [77]. The involvement of HDAC1 in the control of HIF1A acetylation has also been suggested when in complex with Metastasis-associated protein 1 (MTA1), a subunit of the nucleosome remodeling and histone deacetylation complex (NuRD) [78].
PTMs can often be coordinated by the activities of multiprotein complexes. Lysine demethylases can be part of the NuRD complex and influence the activity of HIF1A. LSD1 can indirectly regulate the expression of MTA1 through the control of the MYC oncogene, which increases the interaction with HIF1A and in this way favors the deacetylation of HIF1A. In addition, LSD1 autonomously demethylates HIF1A at K391, a PTM that counteracts its ubiquitylation. Finally, LSD1 can also suppress PHD2-induced HIF1A hydroxylation by reversing Set9-mediated HIF1A methylation [79].
In hepatitis B virus (HBV)-associated hepatocarcinogenesis, the HBV core protein and a regulatory X protein (HBx) enhance the expression of the MTA1 and HDAC1 genes. The MTA1 and HDAC1/2 complex can bind to HIF1A in vivo in the presence of HBx. Deacetylation of the oxygen-dependent degradation domain of HIF1A and dissociation of the prolyl hydroxylases and the von Hippel–Lindau bond resulted in stabilization of the protein. Although no data were available on the specific lysine residues that are deacetylated, the stability of the K532R-HIF1A mutant was not affected even after treatment with HDACIs [80]. Since this lysine may also be subject to ubiquitylation, it is difficult to extrapolate a clear contribution of acetylation to this residue.
As described above, an opposite scenario was observed in the case of K709. Deacetylation by HDAC1 at this residue promotes ubiquitylation and reduces HIF1A activities in both normoxia and hypoxia, leading to a vigorous debate [60,81].
Recently, a role of HDAC8 in the stability of HIF1A has been proposed. Silencing of HDAC8 increases HIF1A protein levels in normoxia as well as in hypoxia or under cobalt chloride (CoCl2)-induced hypoxic conditions. A similar effect was induced by the HDAC8 inhibitor PCI-34051. This effect appears to play at the level of protein stability and is associated with the general increase in HIF1A acetylation [82]. It would be interesting to clarify on which lysines HDAC8 exerts its effect.
6. Class IIa HDACs and the regulation of the hypoxic response
Several studies have investigated the contribution of class IIa to the acetylation of HIF1A. However, the relationships between hypoxia and HDACs may be reciprocal. For example, ARNT/HIF1B deficiency leads to decreased HDAC activity, increased global histone acetylation, and altered subcellular localization of class IIa HDACs [83]. In the next sections, the contribution of each member of the class IIa family to HIF1A activity will be discussed.
6.1. HDAC4
In the VHL-deficient human renal cell carcinoma cell line UMRC2, HDAC4 and HDAC6 were isolated as part of a complex with HIF1A. Interfering with these HDACs decreased protein expression and transcriptional activity of HIF1A. However, only downregulation of HDAC4 led to increased acetylation of HIF1A, as demonstrated by co-iimmunoprecipitation experiments. [84]. The interaction between HIF1A and HDAC4 was also confirmed in another study using the in-situ proximity ligation assay and fluorescence microscopy [85].
Another study confirmed that HDAC4 can regulate the acetylation and stability of HIF1A. The same authors excluded a contribution of HDAC1 and HDAC3. Based on the differential sensitivity to proteolysis demonstrated by LC-MS/MS analyses, the authors speculated that different lysine residues at the amino terminus of HIF1A appear to be regulated by HDAC4 (lysines 10, 11, 12, 19 and 21). In addition, the silencing of HDAC4 affects the hypoxia-induced increase in glycolysis and resistance to docetaxel chemotherapy [86]. Considering the low/absent enzymatic activity of class IIa HDACs in vertebrates and the exclusion of the involvement of HDAC3, it is unclear which multiprotein complex recruited by HDAC4 is involved in this deacetylation reaction.
In another study, the role of HDAC4 in regulating HIF1A abundance was reconfirmed, but possibly in complex with HDAC3-NCOR1-NCOR2. In this case, a reduction in cell proliferation was only observed in a hypoxic environment when HDAC4 was silenced. Whether this effect is related to HIF1A or to other targets of HDAC4 is unclear [87].
HDAC4 can accumulate in the nucleus, when in complex with nucleus accumbens-associated protein-1 (NAC1), a nuclear factor of the BTB/POZ gene family. The authors hypothesized that HDAC4 is stabilized in the nucleus. Previous studies have indeed shown that HDAC4 is subject to regulation by the UPS [88]. Under hypoxia, higher levels of HDAC4 resulted in reduced HIF1A acetylation and inhibition of UPS-mediated degradation. In this context, the NAC1-HDAC4 axis promotes glycolysis in hypoxic tumor cells [89].
Despite the UPS, the regulation of HDAC4 levels during hypoxia may also be influenced by other factors. A recent study showed that HDAC4 mRNA is strongly regulated under hypoxia conditions. In pancreatic cancer cell lines, hypoxia leads to a reduction of N6-methyladenosine (m6A) modification in mRNAs, possibly because of increased expression of the m6A eraser ALKBH5 [90]. m6A is the most common modification detected in eukaryotic mRNAs, and it is also observed in several other RNA species [91]. This epigenetic modification can affect almost every step of RNA metabolism, including splicing, transport, translation, and stability [92]. By combining MeRIP-seq and RNA-seq obtained from normoxic and hypoxic cells, the authors defined a hypoxia-related m6A modification signature that controls glycolysis and metastasis. Among the most enriched genes was HDAC4, whose expression level was increased under hypoxic conditions and partially in an ALKBH5-dependent manner. A reduction in m6A modification increases the half-life of HDAC4 mRNA, which contributes to increased levels of HDAC4 protein under hypoxia. Similar to previous studies, the authors show that HDAC4 can control HIF1A levels. Further experiments are needed to elucidate the detailed mechanisms, including which lysine residues are specifically involved in this regulation and whether deacetylation is the key event [90]. Nevertheless, it is evident that different mechanisms may contribute to HDAC4 levels and activity under hypoxia.
A peculiar mechanism has been described for the transcription of the coagulation factor VII (FVII) gene in response to hypoxia. In ovarian cancer, HIF2A regulates FVII expression in complex with Sp1, but without ARNT and in an HRE-independent manner. HDAC4 and KAT p300 are also found in this complex. Paradoxically, HDAC4, but not p300, is required for transcriptional activation [93].
Negative influences of HDAC4 on HIF1A activities have also been reported. We have previously discussed the contribution of the viral oncoprotein E7 to HIF1A activities by repressing binding to various HDACs, including HDAC4 [72]. Finally, we should always keep in mind that an epigenetic regulator such as HDAC4 can be part of different multiprotein complexes and is able to regulate different adaptive responses [94]. For example, HDAC4 in combination with RUNX2 can suppress the transcription of vascular endothelial growth factor (VEGF), which is an important HIF1A target gene and the strongest pro-angiogenic factor [95]. In summary, although there are several indications of a contribution of HDAC4 to the cellular response to hypoxia, the mechanisms involved still need to be defined in more detail.
6.2. HDAC5
Among class IIa, HDAC5 has the highest homology to HDAC4, and therefore it is not surprising that it contributes to HIF1A activity. In several cell lines (MCF7, HeLa, Hep3B), silencing HDAC5, but not HDAC1, HDAC3 or HDAC6, leads to a decrease in HIF1A stability. HDAC5 in the cytoplasm contributes to the stabilization of HIF1A in response to hypoxia and to its accumulation in the nucleus [96]. To investigate the involvement of the deacetylase domain, the authors generated a double mutant (C698A/H704A) corresponding to the mutant of HDAC4 (C669A/H675A) in the structural zinc-binding domain. It is important to note that this mutant impairs the binding of HDAC4 to the HDAC3/NCOR1 complex [97]. Therefore, the contribution of HDAC3 in the regulation of HIF1A stability cannot be excluded. As a mechanism, the authors propose that the effect on HIF1A stabilization is mediated by the regulation of HSP70 acetylation [96].
Other studies have reported a repressive effect of HDAC5 on HIF1A expression, although the mechanism was not defined. The experiments included cycles of intermittent hypoxia (IH) as a model to study the manifestations of obstructive sleep apnea (OSA). In this example, down-regulation of HDAC3 and HDAC5 occurred, and it was proposed that this downregulation contributes to the stability of HIF1A by increasing lysine acetylation [98].
The same authors further reported that ROS trigger the degradation of HDAC5 during IH by dephosphorylation of S259. The authors hypothesized that the degradation of HDAC5 is responsible for the increase in HIF1A levels, its acetylation and transcriptional activity [99]. Down-regulation of HDAC5 and HDAC6 expression during hypoxia was also observed in adipocytes from humans and mice. RNAi-mediated silencing of these two HDACs mimicked some of the effects of hypoxia [100].
6.3. HDAC7
The first studies on the possible involvement of class IIa HDACs in the regulation of hypoxia pointed to a role for HDAC7. It was shown that HDAC7 can bind to HIF1A via the carboxy terminus. Under hypoxic conditions, HDAC7 accumulated in the nucleus as a complex with p300/KAT3B and HIF1A and stimulated the transcriptional activity of HIF1A in an undetermined manner [101]. When comparing the subcellular localization of epitope-tagged HDAC4, HDAC5 and HDAC7, only HDAC7 accumulated in the nucleus under hypoxia [101]. An interaction between HDAC7 and HIF1A was also observed in other cell models and under different stimuli. In macrophages, HIF1A expression is induced by LPS. HDAC7 and HIF1A interact and synergistically promote LPS-induced transcription of the End1 gene [102]. In investigating the regulation of the HIF1A-HDAC7 axis in macrophages in response to inflammatory signaling, recent studies have shown that PKM2 may be part of a ternary complex with HIF1A and HDAC7. HDAC7 may control acetylation/dimerization of PKM2, leading to its stabilization and nuclear translocation [103].
6.4. HDAC9
Few studies have investigated the role of HDAC9 in hypoxia [104]. Similar to the other class IIa members of the HDACs family, HDAC9 has been shown to interact with HIF1A, deacetylating it and maintaining its activities [105]. Additional mechanisms have also been proposed. In liver cancer cell lines, HDAC9 can and is required to drive efficient HIF1A translation in an undetermined manner, which in turn is mediated by the eukaryotic translation initiation factor eIF3GA. This mechanism is also employed by SAHA [106]. Finally, mRNA levels of HDAC9 can be dramatically upregulated under 48 hours of hypoxia in renal cell carcinoma cell lines. In this context, HDAC9 acts as a downstream epigenetic regulator of H3K27ac levels and gene expression [107].
7. Class IIb
The enzymes HDAC6 and HDAC10 form class IIb. HDAC6 has attracted much more attention, and more data are available compared to HDAC10. HDAC6 is structurally unique, contains two catalytic domains and is predominantly localized in the cytoplasm. In fact, several substrates of HDAC6 are non-histone proteins with cytosolic activities, including α-tubulin, heat shock protein (HSP90), Cortactin, Peroxiredoxin, etc. [108]. Another distinctive feature of HDAC6 is the presence of the zinc finger ubiquitin-binding domain at the carboxy terminus, which is involved in ubiquitylation and the elimination of misfolded proteins via the aggresome pathway [109].
The link between HDAC6 and HIF1A activity results from the regulation of HSP70/HSP90 chaperones [71,110]. HDAC6 can also deacetylate HIF1A and increase its levels and transcriptional activity under hypoxia. Curiously, both the deacetylase and ubiquitin-binding activities of HDAC6 contributed to the stabilization of HIF1A, but only the deacetylase activity was required for the increase in HIF1A-mediated gene transcription [111].
8. Class III HDACs: Sirtuins
Sirtuins are a family of NAD+-dependent deacetylases that regulate several important cellular processes, responses, and fates. Sirtuins are present in different subcellular compartments, including the mitochondria. Their action is not limited to the removal of acetyl groups, but they can also control other PTMs such as succinylation or glutarylation [112]. Initial studies have focused the attention on Sirt1, but soon after was clear that several Sirtuins can modulate HIF1A and EPAS1/HIF2A activities.
SIRT1 is redox-sensing and can stimulate EPAS1/HIF2A transcriptional activity during hypoxia. SIRT1 forms a complex with EPAS1/HIF2A and reverses lysine acetylation [113]. Conversely, SIRT1 can inhibit HIF1A by deacetylating K674, which is acetylated by PCAF. In this way, the binding of p300 and the transcriptional activity of HIF1A is reduced [114].
The results on SIRT1, HIF1A and hypoxia are contradictory and show both neutral, positive and negative effects. A heterogeneity of the results that was explained by the context-dependent effect of SIRT1 on additional targets [113,115–117]. Contrasting results have also been reported for the regulation of SIRT1 expression during hypoxia, with some studies indicating a suppression of SIRT1 transcription during hypoxia-induced epithelial-mesenchymal transition or cancer stem cell-like characteristics [118–120]. A relationship that is also confirmed during aging. In fact, HIF1A is significantly higher in aged mice, while SIRT1 levels decrease. During hypoxia, SIRT1 was downregulated, allowing the acetylation and activation of HIF1A. Chronic activation of HIF1A promoted apoptosis and fibrosis [121]. In contrast, another study showed that SIRT1 expression is increased under hypoxia in a HIF1A-dependent manner. In a kind of positive feedback loop, SIRT1 was able to maintain HIF2A, but not HIF1A-mediated transcriptional activation of the isolated SIRT1 promoter [122]. Finally, DNA damage affects the activity of SIRT1 and the acetylation of H3K27 and HIF1A through the consumption of NAD+ [123]. In general, several reports indicate that a reduction in SIRT1 levels/activities correlates with increased acetylation of HIF1A and increased protein stability.
SIRT2 is a regulator of cellular metabolism and can influence the stability and transcription of HIF1A. Under hypoxia, SIRT2 deacetylates HIF1A at Lys709, but under normoxia it can promote the instability of HIF1A [124]. SIRT2 has an important role in the regulation of neuronal survival [125]. Similar to SIRT1, SIRT2 could also be part of a regulatory feedback loop that is regulated by HIF1A [126].
Mitochondrial and cytoplasmic SIRT3 may regulate HIF1A levels/stability, albeit indirectly. SIRT3 monitors the level of ROS production, which may promote the stabilization of HIF1A [127,128]. The contribution of SIRT3 as a negative regulator of HIF1A stability has also been confirmed in other studies [129–132].
SIRT4 is another, mainly mitochondrial Sirtuin [133]. SIRT4 can negatively regulate aerobic glycolysis and suppresses HIF1A expression in pancreatic cancer [134]. This effect also appears to be mediated by ROS formation. In clear cell renal cell carcinoma, however, preliminary data suggest that SIRT4 interacts directly with HIF1A and can reduce HIF1A protein levels [135].
SIRT6 has attracted attention for its role in the regulation of chromatin structure and DNA repair, as well as for its involvement in aging and longevity [136]. Originally, SIRT6 was characterized as an epigenetic effector of HIF1A that acts as an H3K9 deacetylase. SIRT6 was required for the regulation of nutrient stress responses [137].
Overexpression of SIRT6 in human umbilical vein endothelial cells (HUVECs) can prevent HIF1A degradation by increasing deubiquitylation at K37 and K532. This measure monitors the expression of HIF1A under both normoxia and hypoxia [138]. In another work, deacetylation of HIF1A in response to capsaicin treatment, as driven by SIRT6, led to degradation of HIF1A [139].
SIRT7 has also been identified as a negative regulator of both HIF1A and HIF2A activity. The proposed mechanism is quite peculiar and does not involve enzymatic activity but a physical interaction [140]. This role was recently confirmed in vivo using sirt7-null zebrafish. Here, sirt7 facilitates polyubiquitylation and degradation of hif-1αa, hif-1αb, hif-2αa and hif-2αb. These animals are more resistant to hypoxic conditions and are characterized by an increased expression of hypoxia-responsive genes and an increased number of erythrocytes compared to their wild-type counterparts [141].
9. Inhibitors
The involvement of HDACs in the regulation of hypoxia has been exploited therapeutically, not only in cancer but also in various other diseases, such as ischemia and pulmonary hypertension [142–145]. The effect of HDACIs on HIF1A activities and the hypoxia response were the first evidence for the subsequent investigation of the contribution of different KATs and HDACs to HIF1A and EPAS1/HIF2A activities. The effects of HDACIs could be more indirect and explained at multiple levels. For example, as an adaptive response to treatment with HDACIs, cells reduce the expression of highly expressed genes [146].
HDACIs can also affect the stability of mutant pVHL, which is misfolded and unstable. By inhibiting the HDAC-HSP90 chaperone axis, HDACIs stabilize pVHL, restore its activity and regulate HIF1A-dependent gene expression [147].
In soft tissue sarcomas (STS), SAHA/vorinostat can upregulate EPAS1/HIF2A expression but not HIF1A expression. HIF2A levels are downregulated in STS, and this downregulation supports cell proliferation and mTORC1 signaling. In contrast, its expression inhibits the growth of high-grade STS cells in vivo [148].
During hypoxia, translation of cap-dependent mRNAs is inhibited, while mRNAs encoding HIF1A and proangiogenic, hypoxia and survival factors undergo cap-independent enhanced translation [149]. The investigation of changes in the acetylome after treatment with the HDACI MS-275 revealed that the Y-box-binding protein 1 (YB-1/YBX1) controls translation under stress conditions. YB-1 is an RNA-binding protein (RBP) that binds to 5′- and 3′-untranslated regions (UTRs) of various mRNAs, including HIF1A [150]. Its binding to RNA is inhibited by lysine-81 acetylation. Therefore, HDACIs and acetylation may also indirectly control HIF1A levels. Indeed, a possible contribution of HDACIs to HIF1A translation through an undetermined effect of HDAC9 was originally proposed [106].
These few examples attest to the complexity of the effects that HDACIs could trigger on the response to hypoxia. Therefore, the discussion of HDACIs and hypoxia requires a detailed and comprehensive review that is beyond the scope of this manuscript.
10. Conclusions
HIF1A and HIF2A are two key players in the cellular adaptive response to the reduction in O2 availability. O2 directly controls their activity by targeting to degradation via the UPS. However, HIF1A and HIF2A are subject to further controls to fine-tune their activities. These controls are mainly through PTMs to rapidly adapt cellular activities to environmental changes. Here, we have discussed phosphorylation and acetylation of HIF-1 TFs as important additional options to modulate their transcriptional activities. In some cases, particularly with HDACs and the effects on HIF1A and HIF2A activities, the results are sometimes contradictory. Only by dissecting the mechanisms involved, the possible contribution of additional factors to HDACs regulation (including their epigenetic modulations) and the specific lysine residues involved, will it be possible to clarify the contribution of HDACs to HIF1A and HIF2A activities. Certainly, further experimental work is required to understand the fine regulation of the hypoxia response.
Author Contributions
“Conceptualization, C.B.; writing—original draft preparation, C.B.; writing—review and editing, M.M., E.C. and C.B.; visualization, C.B.; supervision, C.B.; funding acquisition, C.B. All authors have read and agreed to the published version of the manuscript.
Funding
Research activities in C.B. lab a funded by Interreg Italia-Osterreich [ITAT11-018 SENECA] and by AIRC under IG 2021-ID. 26200 project—P.I.
Conflicts of Interest
“The authors declare no conflict of interest.”.
References
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013, 123, 3664–71. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am J Physiol Cell Physiol. 2015, 309, C775-82. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. New horizons in hypoxia signaling pathways. Exp Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef]
- Kaelin, W.G.Jr. Von Hippel-Lindau disease: insights into oxygen sensing, protein degradation, and cancer. J Clin Invest. 2022, 132, e162480. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995, 270, 1230–7. [Google Scholar] [CrossRef] [PubMed]
- Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59(22), 5830–5835.
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013, 123, 3664–71. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman. L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–13. [Google Scholar]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3alpha. Biochem Biophys Res Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Yang, S.L.; Wu, C.; Xiong, Z.F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function. Mol Med Rep. 2015, 12, 2411–6. [Google Scholar] [CrossRef]
- Pasanen, A.; Heikkilä, M.; Rautavuoma, K.; Hirsilä, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3alpha is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int J Biochem Cell Biol. 2010, 42, 1189–200. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999, 399, 271–5. [Google Scholar] [CrossRef] [PubMed]
- Collier, H.; Albanese, A.; Kwok, C.S.; Kou, J.; Rocha, S. Functional crosstalk between chromatin and hypoxia signalling. Cell Signal. 2023, 106, 110660. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Daly, L.A.; Mennerich, D.; Kietzmann, T.; Sée, V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. Int J Mol Sci. 2020, 22, 268. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics, 2014, 13, 1690–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Semenza, G.L. Effect of protein kinase and phosphatase inhibitors on expression of hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1995, 216, 669–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Lee, T.; Lee, N.; Yang, E.G.; Lee, C.; Lee, J.; Moon, E.Y.; Ha, J.; Park, H. Src activates HIF-1α not through direct phosphorylation of HIF-1α specific prolyl-4 hydroxylase 2 but through activation of the NADPH oxidase/Rac pathway. Carcinogenesis. 2011, 32, 703–12. [Google Scholar] [CrossRef] [PubMed]
- Martinengo, C.; Poggio, T.; Menotti, M.; Scalzo, M.S.; Mastini, C.; Ambrogio, C.; Pellegrino, E.; Riera, L.; Piva R, Ribatti D. et al. ALK-dependent control of hypoxia-inducible factors mediates tumor growth and metastasis. Cancer Res. 2014, 74, 6094–106. [Google Scholar] [CrossRef]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem. 2003, 278, 31277–85. [Google Scholar] [CrossRef]
- Flügel, D.; Görlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol. 2007, 27, 3253–65. [Google Scholar] [CrossRef] [PubMed]
- Cassavaugh, J.M.; Hale, S.A.; Wellman, T.L.; Howe, A.K.; Wong, C.; Lounsbury, K.M. Negative regulation of HIF-1α by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem. 2011, 112, 3882–3890. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.P.; MacGurn, J.A. Coupling Conjugation and Deconjugation Activities to Achieve Cellular Ubiquitin Dynamics. Trends Biochem Sci. 2020, 45, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Flügel, D,; Görlach, A. ; Kietzmann, T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood. 2012, 119, 1292–301. [Google Scholar] [CrossRef]
- Raab, C.A.; Raab, M.; Becker, S.; Strebhardt, K. Non-mitotic functions of polo-like kinases in cancer cells. Biochim Biophys Acta Rev Cancer. 2021, 1875, 188467. [Google Scholar] [CrossRef]
- Xu, D.; Yao, Y.; Lu, L. ; Costa, M,; Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J Biol Chem. 2010, 285, 38944–50. [Google Scholar] [CrossRef]
- Yang, Y.; Bai, J.; Shen, R. , Brown, S.A.; Komissarova, E.; Huang, Y.; Jiang, N.; Alberts, G.F.; Costa, M.; Lu, L.. et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008, 68, 4077–85. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz, Ahmed, I. ; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc Natl Acad Sci USA. 2014, 111, E3325–34. [Google Scholar] [CrossRef]
- Warfel, N.A.; Dolloff, N.G.; Dicker, D.T.; Malysz, J.; El-Deiry, W.S. CDK1 stabilizes HIF-1α via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle. 2013, 12, 3689–701. [Google Scholar] [CrossRef]
- Herzog, J.; Ehrlich, S.M.; Pfitzer, L.; Liebl, J.; Fröhlich, T.; Arnold, G.J.; Mikulits, W.; Haider, C.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 stabilizes hypoxia-inducible factor-1α: a novel approach for inhibiting angiogenesis in hepatocellular carcinoma. Oncotarget. 2016, 7, 27108–21. [Google Scholar] [CrossRef]
- Toffoli, S.; Feron, O.; Raes, M.; Michiels, C. Intermittent hypoxia changes HIF-1alpha phosphorylation pattern in endothelial cells: unravelling of a new PKA-dependent regulation of HIF-1alpha. Biochim Biophys Acta 2007, 1773, 1558–1571. [Google Scholar] [CrossRef]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol Cell. 2010, 40, 509–20. [Google Scholar] [CrossRef] [PubMed]
- Fallone, F.; Britton, S.; Nieto, L.; Salles, B.; Muller, C. ATR controls cellular adaptation to hypoxia through positive regulation of hypoxia-inducible factor 1 (HIF-1) expression. Oncogene. 2013, 32, 4387–96. [Google Scholar] [CrossRef]
- Bouquet F, Ousset M, Biard D, Fallone F, Dauvillier S, Frit P, Salles B, Muller C. A DNA-dependent stress response involving DNA-PK occurs in hypoxic cells and contributes to cellular adaptation to hypoxia. J Cell Sci 2011, 124, 1943–1951. [CrossRef] [PubMed]
- Rezaeian, A.H.; Li, C.F.; Wu, C.Y.; Zhang, X.; Delacerda, J.; You, M.J.; Han, F.; Cai, Z.; Jeong, Y.S.; Jin, G.; et al. A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1α activation, tumorigenesis and metastasis. Nat Cell Biol. 2017, 19, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, Z.; Zhang, Q.; Vallee, I.; Mo, Z.; Kishi, S.; Yang, X.L. Phosphorylation of seryl-tRNA synthetase by ATM/ATR is essential for hypoxia-induced angiogenesis. PLoS Biol. 2020, 18, e3000991. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J Biol Chem 2008, 283, 27620–27627. [Google Scholar] [CrossRef] [PubMed]
- Gkotinakou, I.M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 phosphorylates HIF-2α and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J Cell Sci 2019, 132, jcs225698. [Google Scholar] [CrossRef]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J Cell Sci. 2010, 123, 2976–86. [Google Scholar] [CrossRef]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2α phosphorylation by CK1δ promotes erythropoietin secretion in liver cancer cells under hypoxia. J Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef]
- Malone, T.; Schäfer, L.; Simon, N.; Heavey, S.; Cuffe, S.; Finn, S.; Moore, G.; Gately, K. Current perspectives on targeting PIM kinases to overcome mechanisms of drug resistance and immune evasion in cancer. Pharmacol Ther. 2020, 207, 107454. [Google Scholar] [CrossRef] [PubMed]
- Casillas AL, Chauhan SS, Toth RK, Sainz AG, Clements AN, Jensen CC, Langlais PR, Miranti CK, Cress AE, Warfel NA. Direct phosphorylation and stabilization of HIF-1α by PIM1 kinase drives angiogenesis in solid tumors. Oncogene 2021, 40, 5142–5152. [CrossRef] [PubMed]
- Han, H.J.; Saeidi, S.; Kim, S.J.; Piao, J.Y.; Lim, S.; Guillen-Quispe, Y.N.; Choi, B.Y.; Surh, Y.J. Alternative regulation of HIF-1α stability through Phosphorylation on Ser451. Biochem Biophys Res Commun. 2021, 545, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.H.; Bergmann, U.; Kietzmann, T.; Mennerich, D. Protein kinase B/AKT phosphorylates hypoxia-inducible factor-3α1 in response to insulin, promoting cell growth and migration. Front Cell Dev Biol. 2023, 11, 1250000. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Modak, R. Protein Acetyltransferases Mediate Bacterial Adaptation to a Diverse Environment. J Bacteriol. 2021, 203, e0023121. [Google Scholar] [CrossRef] [PubMed]
- Toro, T.B.; Watt, T.J. Critical review of non-histone human substrates of metal-dependent lysine deacetylases. FASEB J. 2020, 34, 13140–13155. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Kumar, S.; Bhatt, R.; Ramachandran, R.; Trivedi, A.K.; Kundu, T.K. Lysine Acetyltransferases (KATs) in Disguise: Diseases Implications. J Biochem. 2023, 173, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp Mol Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007, 8, 284–95. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Lysine Acetyltransferase Inhibitors From Natural Sources. Front Pharmacol. 2020, 11, 1243. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Florean, C.; Brancolini, C. Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J Cell Mol Med. 2011, 15, 1833–46. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, C.; Gagliano, T.; Minisini, M. HDACs and the epigenetic plasticity of cancer cells: Target the complexity. Pharmacol Ther. 2022, 238, 108190. [Google Scholar] [CrossRef] [PubMed]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007, 104, 17335–40. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: it is not only matter of subcellular localization. Epigenomics. 2016, 8, 251–69. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–20. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Chakraborty, P.; Biswas, D. Fine tuning of the transcription juggernaut: A sweet and sour saga of acetylation and ubiquitination. Biochim Biophys Acta Gene Regul Mech. 2023, 1866, 194944. [Google Scholar] [CrossRef] [PubMed]
- Shukri, A.H.; Lukinović, V.; Charih, F.; Biggar, K.K. Unraveling the battle for lysine: A review of the competition among post-translational modifications. Biochim Biophys Acta Gene Regul Mech. 2023, 1866, 194990. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Gi, M.; Suzuki, S.; North, B.J.; Watahiki, A.; Fukumoto, S.; Asara, J.M.; Tokunaga, F.; Wei, W.; Inuzuka, H. Interplay between protein acetylation and ubiquitination controls MCL1 protein stability. Cell Rep. 2021, 37, 109988. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1α protein stability is increased by acetylation at lysine 709. J Biol Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Y. Epigenetic regulators: multifunctional proteins modulating hypoxia-inducible factor-α protein stability and activity. Cell Mol Life Sci. 2018, 75, 1043–1056. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A. 1996, 93, 12969–73. [Google Scholar] [CrossRef] [PubMed]
- Xenaki, G.; Ontikatze, T.; Rajendran, R.; Stratford, I.J.; Dive, C.; Krstic-Demonacos, M.; Demonacos, C. PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene. 2008, 27, 5785–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xu, M.; Hogg, R.T.; Li, J.; Little, B.; Gerard, R.D.; Garcia, J.A. The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J Biol Chem. 2012, 287, 30800–11. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 2016. 16, 37–47. [Google Scholar] [CrossRef]
- Yoon, H.; Shin, S.H.; Shin, D.H.; Chun, Y.S.; Park, J.W. Differential roles of Sirt1 in HIF-1α and HIF-2α mediated hypoxic responses. Biochem Biophys Res Commun. 2014, 444, 36–43. [Google Scholar] [CrossRef]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.E.; Lee, S.W.; Moon, E.J.; Kim, H.S.; Lee, S.K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001, 7, 437–43. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Kim, M.S.; Kim, M.J.; Nakajima, H.; Kim, K.W. Histone deacetylase inhibitor FK228 inhibits tumor angiogenesis. Int J Cancer. 2002, 97, 290–6. [Google Scholar] [CrossRef] [PubMed]
- Mie Lee, Y.; Kim, S.H.; Kim, H.S.; Jin Son, M.; Nakajima, H.; Jeong Kwon, H.; Kim, K.W. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem Biophys Res Commun. 2003, 300, 241–6. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G. Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003, 4, 147–58. [Google Scholar] [CrossRef]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006, 26, 2019–28. [Google Scholar] [CrossRef]
- Bodily, J.M.; Mehta, K.P.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Lee, K.Y.; Lee, Y.M. Downregulation of a tumor suppressor RECK by hypoxia through recruitment of HDAC1 and HIF-1alpha to reverse HRE site in the promoter. Biochim Biophys Acta. 2010, 1803, 608–16. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Ding, J.; Sun, W.; Wu, K.; Ning, B.; Gong, W.; He, G.; Huang, S.; Ding, X.; Yin, P.; et al. Suppression of cyclin D1 by hypoxia-inducible factor-1 via direct mechanism inhibits the proliferation and 5-fluorouracil-induced apoptosis of A549 cells. Cancer Res. 2010, 70, 2010–9. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Huang, F.; Peterlin, B.M. P-TEFb: The master regulator of transcription elongation. Mol Cell. 2023, 83, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Safronova, O.S.; Nakahama, K.; Morita, I. Acute hypoxia affects P-TEFb through HDAC3 and HEXIM1-dependent mechanism to promote gene-specific transcriptional repression. Nucleic Acids Res. 2014, 42, 8954–69. [Google Scholar] [CrossRef] [PubMed]
- Pluemsampant, S.; Safronova, O.S.; Nakahama, K.; Morita, I. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int J Cancer. 2008, 122, 333–41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Deng, S.; He, C.; Liu, M.; Chen, H.; Zeng, Z.; Zhong, J.; Ye, Z.; Deng, S.; Wu, H.; et al. Reciprocal loop of hypoxia-inducible factor-1α (HIF-1α) and metastasis-associated protein 2 (MTA2) contributes to the progression of pancreatic carcinoma by suppressing E-cadherin transcription. J Pathol. 2018, 245, 349–360. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.H.; Choi, H.J.; Won, H.Y.; Joo, H.S.; Shin, D.H.; Park, M.K.; Han, B.; Kim, K.P.; Lee, T.J.; et al. LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 2017, 36(39), 5512–5521. [Google Scholar] [CrossRef]
- Yoo, Y.G.; Na, T.Y.; Seo, H.W.; Seong, J.K.; Park, C.K.; Shin, Y.K.; Lee, M.O. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene. 2008, 27, 3405–13. [Google Scholar] [CrossRef]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; He, Q.; Yu, W. The histone deacetylase HDAC1 activates HIF1α/VEGFA signal pathway in colorectal cancer. Gene. 2020, 754, 144851. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma. Cancers (Basel). 2023, 15, 1123. [Google Scholar] [CrossRef]
- Maltepe, E.; Krampitz, G.W.; Okazaki, K.M.; Red-Horse, K.; Mak, W.; Simon, M.C.; Fisher, S.J. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development. 2005, 132, 3393–403. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–21. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Leithner, K.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1α destabilization. Mol Cancer. 2015, 4, 4. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Antony, L.; Dalrymple, S.L.; Brennen, W.N.; Gerber, S.; Hammers, H.; Wissing, M.; Kachhap, S.; Luo, J.; Xing, L.; et al. Tasquinimod Is an Allosteric Modulator of HDAC4 survival signaling within the compromised cancer microenvironment. Cancer Res. 2013, 73, 1386–99. [Google Scholar] [CrossRef]
- Cernotta, N.; Clocchiatti, A.; Florean, C.; Brancolini, C. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol Biol Cell. 2011, 22, 278–89. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, Y.J.; Guo, L.C.; Ji, C.; Hu, J.; Zhang, H.H.; Xu, Q.H.; Zhu, W.D.; Ming, Z.J.; Yuan, Y.S.; et al. Nucleus accumbens-associated protein-1 promotes glycolysis and survival of hypoxic tumor cells via the HDAC4-HIF-1α axis. Oncogene. 2017, 36, 4171–4181. [Google Scholar] [CrossRef]
- Liu, X.; Feng, M.; Hao, X.; Gao, Z.; Wu, Z.; Wang, Y.; Du, L.; Wang, C. m6A methylation regulates hypoxia-induced pancreatic cancer glycolytic metabolism through ALKBH5-HDAC4-HIF1α positive feedback loop. Oncogene. 2023, 42, 2047–2060. [Google Scholar] [CrossRef]
- Liu, N.; Pan, T. N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol. 2016, 23, 98–102. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Su, H.; Zhang, X.; Chen, H.; Yu, J. RNA N6-Methyladenine Modification, Cellular Reprogramming, and Cancer Stemness. Front Cell Dev Biol. 2022, 10, 935224. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Ito, S.; Miyagi, E.; Hirahara, F.; Nakamura, Y.; Sakuma, Y.; Osaka, H.; Takano, Y.; Ruf, W.; Miyagi, Y. HIF2α-Sp1 interaction mediates a deacetylation-dependent FVII-gene activation under hypoxic conditions in ovarian cancer cells. Nucleic Acids Res. 2012, 40, 5389–401. [Google Scholar] [CrossRef] [PubMed]
- Cuttini, E.; Goi, C.; Pellarin, E.; Vida, R.; Brancolini, C. HDAC4 in cancer: A multitasking platform to drive not only epigenetic modifications. Front Mol Biosci. 2023, 10, 1116660. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wie, L.; Chen, Q.; Terek, R.M. HDAC4 represses vascular endothelial growth factor expression in chondrosarcoma by modulating RUNX2 activity. J Biol Chem. 2009, 284, 21881–21890. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. MPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle. 2015, 14, 2520–36. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Lo Surdo, P.; Di Giovine, P.; Cirillo, A.; Scarpelli, R.; Ferrigno, F.; Jones, P.; Neddermann, P.; De Francesco, R.; Steinkühler, C. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008, 283, 26694–704. [Google Scholar] [CrossRef]
- Wang, N.; Peng, Y.J.; Su, X.; Prabhakar, N.R.; Nanduri, J. Histone Deacetylase 5 Is an Early Epigenetic Regulator of Intermittent Hypoxia Induced Sympathetic Nerve Activation and Blood Pressure. Front Physiol. 2021, 12, 688322. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Prabhakar, N.R.; Nanduri, J. Protein phosphatase 1 regulates reactive oxygen species-dependent degradation of histone deacetylase 5 by intermittent hypoxia. Am J Physiol Cell Physiol. 2022, 323, C423–C431. [Google Scholar] [CrossRef]
- Bricambert, J.; Favre, D.; Brajkovic, S.; Bonnefond, A.; Boutry, R.; Salvi, R.; Plaisance, V.; Chikri, M.; Chinetti-Gbaguidi, G.; Staels, B.; et al. Impaired histone deacetylases 5 and 6 expression mimics the effects of obesity and hypoxia on adipocyte function. Mol Metab. 2016, 5, 1200–1207. [Google Scholar] [CrossRef]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004, 279, 41966–74. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Hohenhaus, D.M.; Kelly, G.M.; Kamal, N.A.; Gupta, P.; Labzin, L.I.; Schroder, K.; Garceau, V.; Barbero, S.; Iyer, A.; et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J Biol Chem. 2013, 288, 25362–25374. [Google Scholar] [CrossRef] [PubMed]
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.B.; Murthy, A.M.V.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone Deacetylases Drive Toll-like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep. 2020, 30, 2712–2728. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, C.; Di Giorgio, E.; Formisano, L.; Gagliano, T. Quis Custodiet Ipsos Custodes (Who Controls the Controllers)? Two Decades of Studies on HDAC9. Life (Basel). 2021, 11, 90. [Google Scholar] [CrossRef]
- Sanguigno, L.; Guida, N.; Anzilotti, S.; Cuomo, O.; Mascolo, L.; Serani, A.; Brancaccio, P.; Pennacchio, G.; Licastro, E.; Pignataro, G.; et al. Stroke by inducing HDAC9-dependent deacetylation of HIF-1 and Sp1, promotes TfR1 transcription and GPX4 reduction, thus determining ferroptotic neuronal death. Int J Biol Sci. 2023, 19, 2695–2710. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS One. 2014, 9, e106224. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Z.; Xu, Q.; Liu, Y.; Chen, L.; Guo, S.; Wang, H.; Zeng, K.; Liu, J.; Zeng, S.; et al. The failure of DAC to induce OCT2 expression and its remission by hemoglobin-based nanocarriers under hypoxia in renal cell carcinoma. Theranostics 2020, 10, 3562–3578. [Google Scholar] [CrossRef] [PubMed]
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol Res. 2021, 163, 105274. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007, 26, 5468–76. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Costa, M.; Gao, J.; Huang, C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res. 2010, 70, 813–23. [Google Scholar] [CrossRef]
- Ryu, H.W.; Won, H.R.; Lee, D.H.; Kwon, S.H. HDAC6 regulates sensitivity to cell death in response to stress and post-stress recovery. Cell Stress Chaperones. 2017, 22, 253–261. [Google Scholar] [CrossRef]
- Sun, X.; Yang, X.; Gui, W.; Liu, S.; Gui, Q. Sirtuins and autophagy in lipid metabolism. Cell Biochem Funct. 2023. Sep 27. [Google Scholar] [CrossRef] [PubMed]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009, 324, 1289–93. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010, 38, 864–78. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.Y.; Yun, M.; Jeong, J.; Park, E.R.; Shin, H.J.; Woo, S.R.; Jung, J.K.; Kim, M.; Park, J.J.; Kim, J.; Lee, K.H. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem Biophys Res Commun. 2015, 462, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Laemmle, A.; Lechleiter, A.; Roh, V.; Schwarz, C.; Portmann, S.; Furer, C.; Keogh, A.; Tschan, M.P.; Candinas, D.; Vorburger, S.A.; et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS One. 2012, 7, e33433. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Wang, M.; Zhong, A.; Wang, Y.; Du, J.; Wang, J.; Qi, L.; Bi, Z.; Zhang, P.; Lin, T.; et al. SRT1720 inhibits the growth of bladder cancer in organoids and murine models through the SIRT1-HIF axis. Oncogene. 2021, 40, 6081–6092. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, H.; Chen, J.; Dehennaut, V.; Zhao, Y.; Yang, Y.; Iwasaki, Y.; Kahn-Perles, B.; Leprince, D.; Chen, Q.; et al. A SUMOylation-dependent pathway regulates SIRT1 transcription and lung cancer metastasis. J Natl Cancer Inst. 2013, 105, 887–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, H.; Chen, J.; Iwasaki, Y.; Kubota, T.; Matsuoka, M.; Shen, A.; Chen, Q.; Xu, Y. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J Cell Sci. 2013, 126, 3939–47. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, Y.; Lu, Y.; Liu, M.; Li, M.; Li, J.; Wu, L. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci Rep. 2017, 7, 10592. [Google Scholar] [CrossRef]
- Ryu, D.R.; Yu, M.R.; Kong, K.H.; Kim, H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Noh, H. Sirt1-hypoxia-inducible factor-1α interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell. 2019, 18, e12904. [Google Scholar] [CrossRef]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J Biol Chem. 2011, 286, 13869–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Cui, K.; Zhao, Z.; Xu, X.; Liu, Y.; Shen, Y.; Chen, F.; Mai, K.; Ai, Q. LPS stimulation stabilizes HIF-1α by enhancing HIF-1α acetylation via the PARP1-SIRT1 and ACLY-Tip60 pathways in macrophages. FASEB J. 2022, 36, e22418. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.S.; Park, J.H.; Heo, J.Y.; Jing, K.; Han, J.; Min, K.N.; Kim, C.; Koh, G.Y.; Lim, K.; Kang, G.Y.; et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene. 2015, 34, 1354–62. [Google Scholar] [CrossRef]
- Kaitsuka, T.; Matsushita, M.; Matsushita, N. SIRT2 inhibition activates hypoxia-inducible factor 1α signaling and mediates neuronal survival. Biochem Biophys Res Commun. 2020, 529, 957–962. [Google Scholar] [CrossRef]
- Shu, L.; Xu, C.Q.; Yan, Z.Y.; Yan, Y.; Jiang, S.Z.; Wang, Y.R. Post-Stroke Microglia Induce Sirtuin2 Expression to Suppress the Anti-inflammatory Function of Infiltrating Regulatory T Cells. Inflammation. 2019, 42, 1968–1979. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell. 2011, 19, 416–28. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011, 30, 2986–96. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Ji, S.; Qin, Y.; Yang, J.; Xu, J.; Zhang, B.; Xu, W.; Liu, J.; Shi, S.; Liu, L.; et al. Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1α axis. Mol Cancer. 2014, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhou, Y.; Qiao, C.; Ni, T.; Li, Z.; You, Q.; Guo, Q.; Lu, N. Oroxylin A inhibits glycolysis-dependent proliferation of human breast cancer via promoting SIRT3-mediated SOD2 transcription and HIF1α destabilization. Cell Death Dis. 2015, 6, e1714. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Zhou, L.; Dorfman, R.G.; Liu, L.; Cai, R.; Jiang, C.; Tang, D.; Wang, Y.; Zou, X.; et al. SIRT3 elicited an anti-Warburg effect through HIF1α/PDK1/PDHA1 to inhibit cholangiocarcinoma tumorigenesis. Cancer Med. 2019, 8, 2380–2391. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Geng, K.; Yang, K.; Shao, J.; Xia, W. Sirt3 Protects Against Ischemic Stroke Injury by Regulating HIF-1α/VEGF Signaling and Blood-Brain Barrier Integrity. Cell Mol Neurobiol. 2021, 41, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.; Xudong, Z.; Zhenyu, J.; Saretzki, G. Mitochondrial sirtuins in stem cells and cancer. FEBS J. 2022, 289, 3393–3415. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Qin, Y.; Ji, S.; Xu, W.; Liu, W.; Sun, Q.; Zhang, Z.; Liu, M.; Ni, Q.; Yu, X.; et al. UHRF1 promotes aerobic glycolysis and proliferation via suppression of SIRT4 in pancreatic cancer. Cancer Lett. 2019, 452, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Kai, J.; Wang, S.; Yu, Y.; Xie, S.; Zheng, H.; Wang, Y.; Liu, Y.; Zhu, K.; Guan, X.; et al. VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1α/HO-1 pathway. Cell Death Dis. 2021, 12, 621. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, A.; Seluanov, A.; Gorbunova, V. Sirtuin 6: linking longevity with genome and epigenome stability. Trends Cell Biol. 2021, 31, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D'Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010, 140, 280–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1α and reactive oxygen species. Cell Death Dis. 2021, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Li, W.; Xie, C.; Yin, L.; Su, X.; Chen, J.; Huang, H. Capsaicin Attenuates Arterial Calcification Through Promoting SIRT6-Mediated Deacetylation and Degradation of Hif1α (Hypoxic-Inducible Factor-1 Alpha). Hypertension. 2022, 79, 906–917. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Hu, H.; Kshitiz, Gilkes, D.M.; Semenza, G.L. Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J Biol Chem. 2013, 288(29), 20768–20775. [Google Scholar] [CrossRef]
- Liao, Q.; Zhu, C.; Sun, X.; Wang, Z.; Chen, X.; Deng, H.; Tang, J.; Jia, S.; Liu, W.; Xiao, W.; et al. Disruption of sirtuin 7 in zebrafish facilitates hypoxia tolerance. J Biol Chem. 2023, 299, 105074. [Google Scholar] [CrossRef]
- Verheul, H.M.; Salumbides, B.; Van Erp, K.; Hammers, H.; Qian, D.Z.; Sanni, T.; Atadja, P.; Pili, R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008, 14, 3589–97. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Hammers, H.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009, 280, 145–53. [Google Scholar] [CrossRef] [PubMed]
- Cavasin, M.A.; Demos-Davies, K.; Horn, T.R.; Walker, L.A.; Lemon, D.D.; Birdsey, N.; Weiser-Evans, M.C.; Harral, J.; Irwin, D.C.; Anwar, A.; et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012, 110, 739–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012, 52, 928–36. [Google Scholar] [CrossRef]
- Minisini, M.; Di Giorgio, E.; Kerschbamer, E.; Dalla, E.; Faggiani, M.; Franforte, E.; Meyer-Almes, F.J.; Ragno, R.; Antonini, L.; Mai, A.; et al. Transcriptomic and genomic studies classify NKL54 as a histone deacetylase inhibitor with indirect influence on MEF2-dependent transcription. Nucleic Acids Res. 2022, 50, 2566–2586. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Huntoon, K.; Ksendzovsky, A.; Zhuang, Z.; Lonser, R.R. Proteostasis modulators prolong missense VHL protein activity and halt tumor progression. Cell Rep. 2013, 3, 52–9. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.S.; Eisinger-Mathason, T.S.; Sadri, N.; Ochocki, J.D.; Gade, T.P.; Amin, R.K.; Simon, M.C. Epigenetic re-expression of HIF-2α suppresses soft tissue sarcoma growth. Nat Commun. 2016, 7, 10539. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, S.; Karpisheva, K.; Pola, C.; Goldberg, J.; Hochman, T.; Yee, H.; Cangiarella, J.; Arju, R.; Formenti, S.C.; Schneider, R.J. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell. 2007, 28, 501–12. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, XQ, Delaidelli A, Rafn B, Cran J. ; et al. Huntsman DG, Morin GB, Sorensen PH. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef]
Figure 1.
HIF1A and HIF2A/EPAS1 proteins. A) AlphaFold prediction of HIF1A and HIF2A/EPAS1. Colors indicate the different per-residue confidence score (pLDDT) as indicated. Some regions below 50 pLDDT may be unstructured in isolation. https://alphafold.ebi.ac.uk. B) Schematic representation of HIF1A and HIF2A/EPAS1. Both proteins contain basic helix-loop-helix (bHLH) and Par-Arnt-SIM (PAS) transcription factors domains that facilitates the heterodimerization with ARNT. They also have N- and C-terminal transactivation domains (N/C-TAD), oxygen-dependent degradation domain (ODD) and nuclear localization sequence (NLS). Their sequences show a high degree of similarity.
Figure 1.
HIF1A and HIF2A/EPAS1 proteins. A) AlphaFold prediction of HIF1A and HIF2A/EPAS1. Colors indicate the different per-residue confidence score (pLDDT) as indicated. Some regions below 50 pLDDT may be unstructured in isolation. https://alphafold.ebi.ac.uk. B) Schematic representation of HIF1A and HIF2A/EPAS1. Both proteins contain basic helix-loop-helix (bHLH) and Par-Arnt-SIM (PAS) transcription factors domains that facilitates the heterodimerization with ARNT. They also have N- and C-terminal transactivation domains (N/C-TAD), oxygen-dependent degradation domain (ODD) and nuclear localization sequence (NLS). Their sequences show a high degree of similarity.
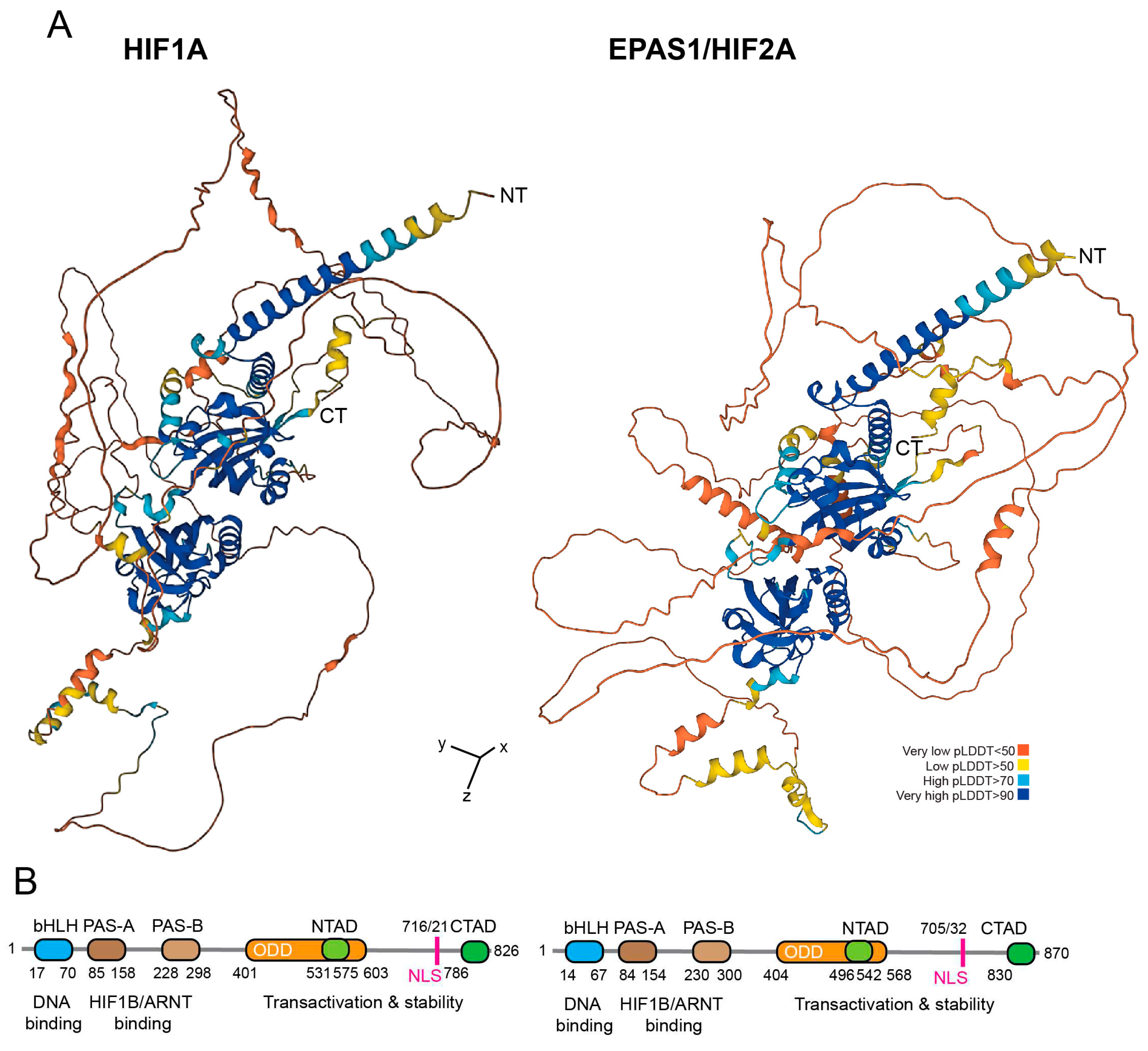
Figure 2.
The cellular response to hypoxia. Under normoxia key prolines of HIF1A are hydroxylated by PHD/EGLN proteins using O2 and α-oxoglutarate (also known as α-ketoglutarate). When hydroxylated, HIF1A is recognized by VHL, polyubiquitylated and target to proteasomal-mediated degradation. Under hypoxia HIF1A accumulates and associates with ARNT/HIF1B. The heterodimer binds the HRE consensus motif and supervises the transcription of genes of the adaptive response to low oxygen levels.
Figure 2.
The cellular response to hypoxia. Under normoxia key prolines of HIF1A are hydroxylated by PHD/EGLN proteins using O2 and α-oxoglutarate (also known as α-ketoglutarate). When hydroxylated, HIF1A is recognized by VHL, polyubiquitylated and target to proteasomal-mediated degradation. Under hypoxia HIF1A accumulates and associates with ARNT/HIF1B. The heterodimer binds the HRE consensus motif and supervises the transcription of genes of the adaptive response to low oxygen levels.
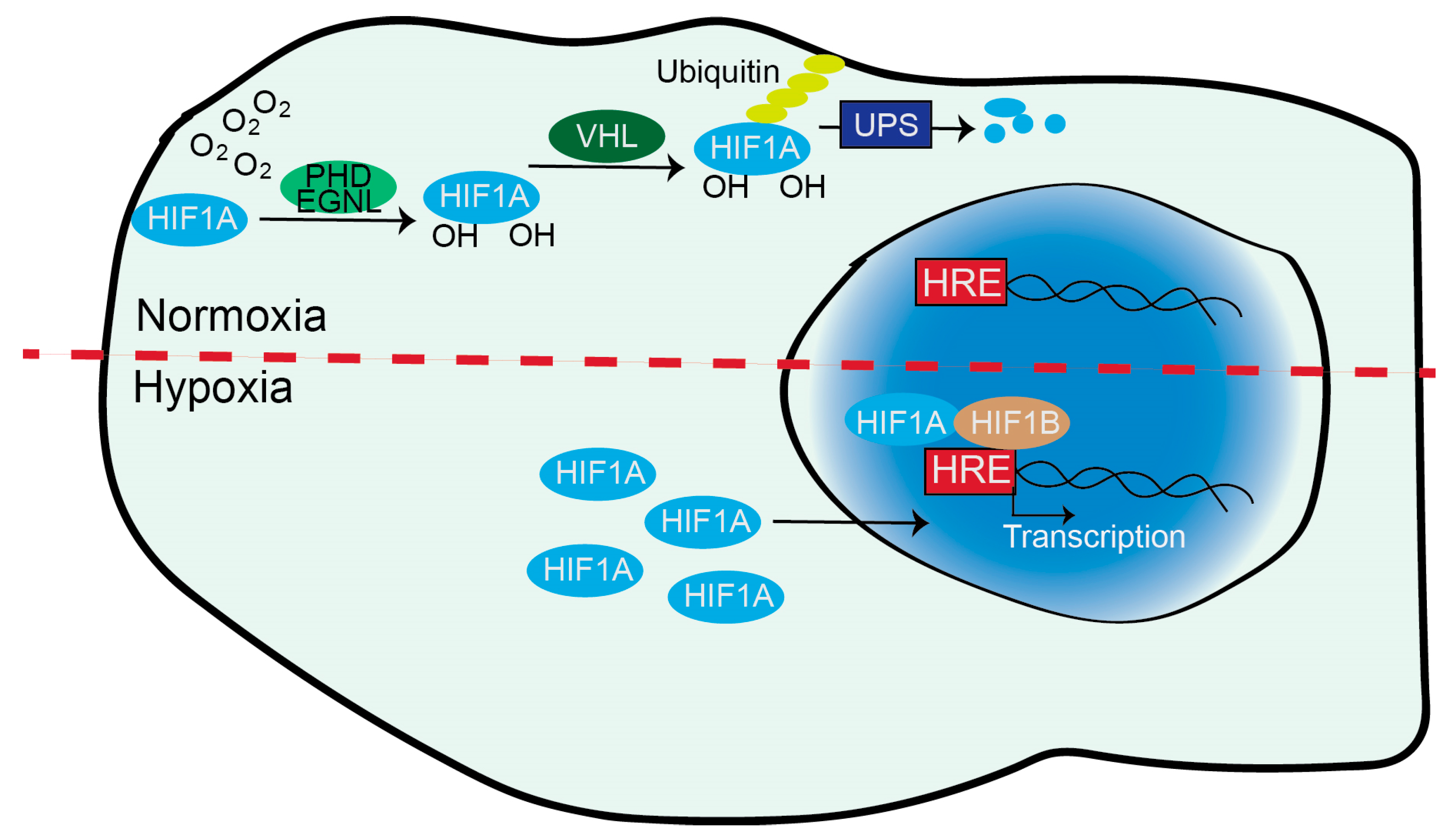
Figure 3.
Phosphorylated, acetylated and ubiquitylated sites in human HIF1A and EPAS1/HIF2A. A) Phosphorylated, acetylated and ubiquitylated sites in human HIF1A as determined by PhosphoSitePlus https://www.phosphosite.org/proteinAction.action?id=4987&showAllSites=true. HTP: number of records where the phosphorylation was demonstrated only with mass-spectrometry. LPT: number of records where the phosphorylation was experimental demonstrated without mass-spec. [56]. The different domains are indicated. B) Phosphorylated, acetylated and ubiquitylated sites in human EPAS1/HIF2A as determined by PhosphoSitePlus https://www.phosphosite.org/proteinAction.action?id=4986&showAllSites=true. HTP: number of records where the phosphorylation was demonstrated only with mass-spectrometry. LPT: number of records where the phosphorylation was experimental demonstrated without mass-spec [56]. The different domains are indicated.
Figure 3.
Phosphorylated, acetylated and ubiquitylated sites in human HIF1A and EPAS1/HIF2A. A) Phosphorylated, acetylated and ubiquitylated sites in human HIF1A as determined by PhosphoSitePlus https://www.phosphosite.org/proteinAction.action?id=4987&showAllSites=true. HTP: number of records where the phosphorylation was demonstrated only with mass-spectrometry. LPT: number of records where the phosphorylation was experimental demonstrated without mass-spec. [56]. The different domains are indicated. B) Phosphorylated, acetylated and ubiquitylated sites in human EPAS1/HIF2A as determined by PhosphoSitePlus https://www.phosphosite.org/proteinAction.action?id=4986&showAllSites=true. HTP: number of records where the phosphorylation was demonstrated only with mass-spectrometry. LPT: number of records where the phosphorylation was experimental demonstrated without mass-spec [56]. The different domains are indicated.
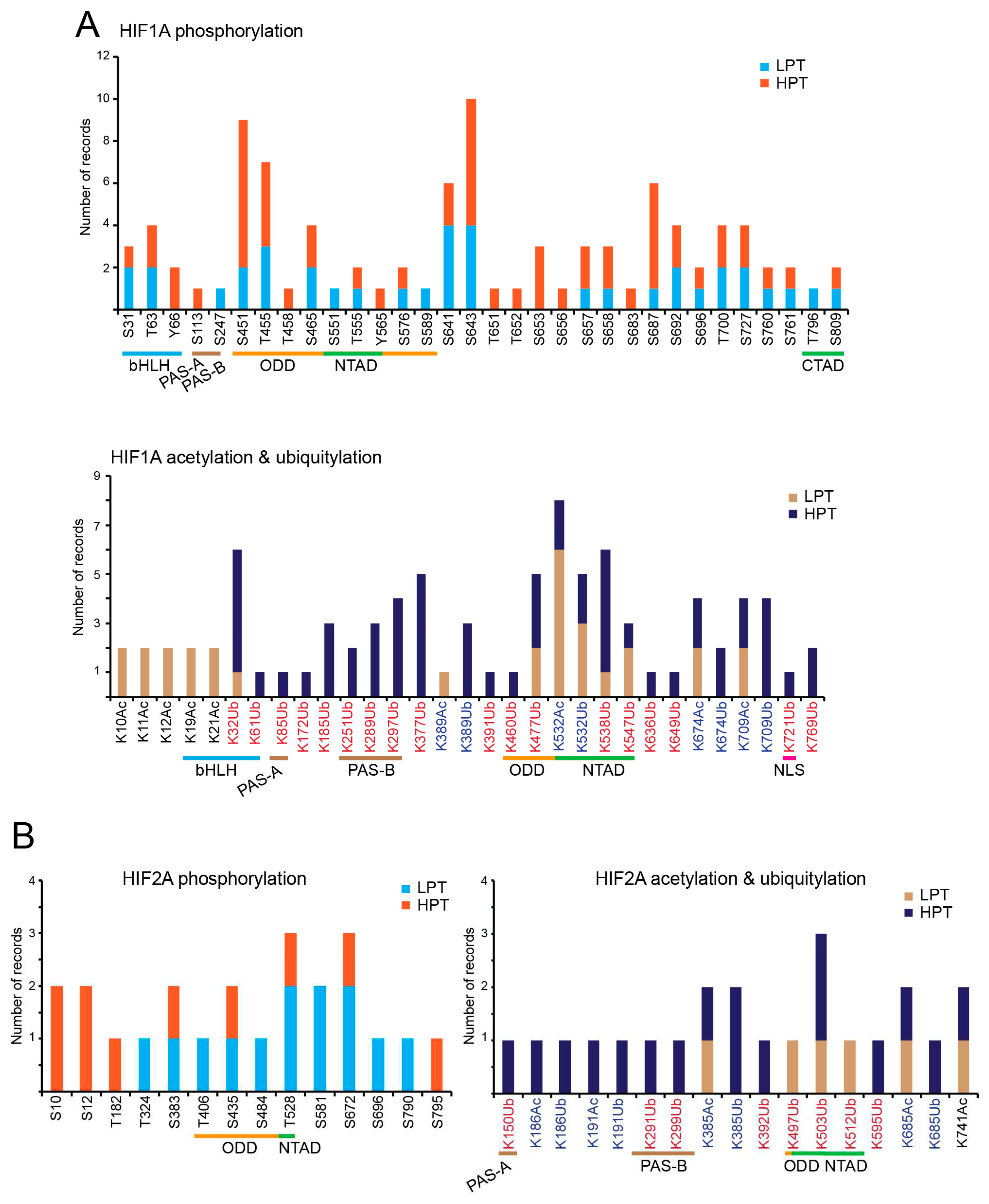
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



