
Submitted:
06 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
On the thirty-fifth anniversary of the first description of O2-sensitive K+ channels in the carotid body chemoreceptors [1], O2 sensing remains a salient issue in the literature. Whereas much has been learned about this subject, important questions such as the identity of the specific K+ channel subtype(s) responsible for O2 sensing by chemoreceptors and the mechanism(s) by which their activities are altered by hypoxia have not yet been definitively answered. O2 sensing is a fundamental biological process necessary for the acute and chronic responses to varying environmental O2 levels which allow organisms to adapt to hypoxia. Whereas chronic responses depend on the modulation of hypoxia-inducible transcription factors which determine the expression of numerous genes encoding enzymes, transporters and growth factors, acute responses rely mainly on the dynamic modulation of ion channels by hypoxia, causing adaptive changes in cell excitability, contractility, and secretory activity in specialized tissues. The most widely studied oxygen-sensitive ion channels are potassium channels but oxygen sensing by members of both the calcium and sodium channel families has also been demonstrated. Given the explosion of information on this topic, in this review we will focus on the mechanisms of physiological oxygen chemotransduction by PO2-dependent K+ channels, with particular emphasis on their function in carotid body chemoreceptor cells (CBCC) and pulmonary artery smooth muscle cells (PASMC), highlighting areas of consensus and controversy within the field. We will first describe the most well-established concepts, those reproduced in multiple laboratories, and then discuss selected observations or questions that remain unresolved, and that limit our progress in this field.
Keywords:
O2 sensing
; carotid body
; hypoxia
; hypoxic pulmonary vasoconstriction
; BKCa channel
; KV channel
; TASK channel
; mitochondria
; reactive oxygen species
; redox
1. Physiological Oxygen Sensing: The Carotid Body as an O2 sensor
Complex aerobic organisms, such as mammals, must maintain adequate circulating O2 levels (PaO2) to ensure a sufficient supply of oxygen for their metabolic needs. To fulfill this homeostatic function, organisms possess strategically located specialized structures to monitor PaO2 and activate compensatory mechanisms which enable its stabilization. These mechanisms act mainly on the respiratory and circulatory apparatus to mediate enhanced ventilation and pulmonary O2 exchange, improved blood transport at the systemic level, and optimal distribution at the tissue level. The structures responsible for these responses include: i) the chemoreceptor cells of the carotid body (and to a lesser degree the aortic bodies) which in situations of hypoxia reflexively increase pulmonary ventilation and cardiac activity [2], ii) the vascular smooth muscle cells of the pulmonary arteries (PASMC), in which a vasoconstriction to hypoxia (hypoxic pulmonary vasoconstriction or HPV) in poorly ventilated areas produces a redistribution of pulmonary blood flow essential to match perfusion to ventilation, thus ensuring an improvement in pulmonary blood oxygenation and therefore PaO2 [3], iii) the neuroepithelial bodies of the pulmonary airways, which reflexively and together with the above contribute to optimal ventilation-perfusion adjustments [4], and iv) neonatal adrenal medullary chromaffin cells which play a key role in the respiratory adaptation of the newborn to hypoxia associated with delivery and extrauterine life [5].
All these systems share a high sensitivity to hypoxia with a low threshold and high gain (i.e. they respond to moderate hypoxia), with a level of metabolic activity that progressively increases with the intensity of hypoxia. Each, through changes in excitability, contractility, or secretory activity, promotes acute physiological responses, such as HPV and the hypoxic ventilatory response (HVR), which occur within seconds or minutes and enhance the supply of O2 to tissues.
Hyperventilation as a response to high-altitude hypoxia was appreciated by early high-altitude physiologist-expeditioners, although it was Gay-Lussac who first recorded the symptoms he suffered during his balloon flights to verify various aspects of the gas laws (1804). In 1868, the German physiologist Pflüger described the hyperventilation produced by breathing pure nitrogen [6], and in 1908, Boycott and Haldane described in detail the ventilatory effects of hypoxia of different intensities as hyperventilation with increased frequency and depth of respiratory movements, although it was not then known how the decrease in PaO2 could produce these effects [7].
In 1928, Fernando de Castro, a disciple of Cajal, discovered that the carotid body (CB), until then considered a sympathetic ganglion (ganglium minutum) or a gland (glandula carotica) was a sensory organ innervated by a sensory nerve: the carotid sinus nerve (CSN) or Hering's nerve. De Castro also noticed the close relationship between the epithelioid cells of the CB (today called type I, glomus or chemoreceptor cells) and the blood capillaries on one hand, and sensory nerve endings on the other. He proposed that the organ is specialized to detect changes in the chemical composition of the blood, and that the epithelioid cells are the sensors [8]. In 1930, Corneille Heymans discovered that the hyperventilation caused by hypoxia was a reflex originating in the carotid sinus region. This discovery led to his being awarded the Nobel Prize in Physiology and Medicine in 1938, even though until at least 1936 he confused the carotid sinus with the carotid body [9].
CB chemoreceptors have an adaptive function since they detect the PaO2 and, upon its decrease, initiate reflexes aimed at increasing the pumping of air rich in O2 from the atmosphere to the alveoli. However, the mechanisms of CB function at the cellular level have been an enigma for many years. A multitude of hypotheses have arisen, including the “Metabolic hypothesis” of the Russian School of Physiology and the “Acid hypothesis” of Winder and the Oxford School, which emerged initially.
Of these, the one that achieved the widest dissemination was the metabolic hypothesis [10], which linked the chemoreception mechanism to the process of cellular respiration. Hypoxia, like metabolic poisons which act on the mitochondria and are strong stimulants of the CB, would act by reducing ATP levels in chemoreceptor cells. The fall in [ATP] would trigger neurotransmitter release by chemoreceptor cells, although the theory did not explain how this would occur. In support of this hypothesis, there is extensive evidence that the mitochondria in CBCC are specifically adapted for O2 sensing. Whereas in most tissues PO2 must be reduced to very low levels before O2 availability significantly limits respiration, the oxygen sensitivity of mitochondrial function in chemoreceptor cells, an intrinsic property of the Complex 4, appears to be abnormally high. Thus, even at relatively high pO2 electron flux through the respiratory chain would be diminished, thereby decreasing respiration and ATP synthesis in the CBCC. In these cells, mitochondrial depolarization and accumulation of NADH may occur with a relatively modest decline in PO2 to below 40–20 Torr, levels undetected in most other cell types, as first shown by Mills and Jobsis [11,12] who by spectrophotometric analysis correlated changes of NADH/NAD+ ratio with afferent nerve activity over a wide range of O2 levels describing the presence of both a high affinity and a low affinity cytochrome a3. These results were confirmed by Duchen and Biscoe [13,14] who found that the NADH/NAD ratio changed at a PO2 of nearly 60 mmHg and increased as hypoxic challenges became more severe [15]. In rat CBCC, Buckler & Turner obtained P50 values for the effects of oxygen on NADH, electron transport and cytochrome oxidase activity of 40, 5.4 and 2.6 mmHg, respectively [16]. These values are at least one order of magnitude greater than those reported in other tissues and are compatible with a role for mitochondrial metabolism in oxygen sensing. Furthermore, recent evidence suggests that chemoreceptor cells have a different mitochondrial gene expression signature that confers the intrinsically diminished oxygen sensitivity compared to that of other cells. Three atypical mitochondrial electron transport chain subunits, Ndufa4l2, Cox4i2 and Cox8b, are among the most specifically expressed genes in CBCC, highlighting their potential roles in mitochondria-mediated oxygen sensing [17,18]. Along these lines, conditional deletion of Cox4i2 in tyrosine hydroxylase positive cells (TH+) diminishes both the chemoreceptor cell and ventilatory response to hypoxia in the mouse [19].
In the late 1980s, several observations, including those from Constancio González’s grup, led to the proposal known as the “Membrane model” as an alternative hypothesis. By means of biochemical and pharmacological studies, Almaraz et al. [20] demonstrated that depolarization of CB using high extracellular K+ induced Ca2+-dependent release of dopamine. Rocher et al. [21], found that veratridine, a voltage-dependent Na+ channel activator, induced dopamine release in a tetrodotoxin (TTX)-sensitive and Ca2+ dependent manner, and that hypoxia-induced release is inhibited by dihydropyridines, revealing for the first time the excitability of the chemoreceptor cells.
At the same time, in a collaboration between laboratories of Prof. González in Valladolid and Prof. López-Barneo in Sevilla, the excitable nature of rabbit CBCC in primary culture was confirmed, and the first PO2-regulated ion channel was discovered: low PO2 produced a reversible inhibition of a transient K+ current of type IA [1,22]. The existence of hypoxia-sensitive K+ currents in the CB was quickly extended to all animal species studied including cat, rat, and mouse. In the rat, the hypoxia-sensitive K+ current was ascribed to the high-conductance Ca2+ -dependent K+ channel (BKCa)[23] and a K+ ‘leak’ current shown to be due to TASK-1 channels [24,25]. These findings led to the “Membrane hypothesis” of chemotransduction [2] which proposes that the transduction process relies on K+ channels that are reversibly inhibited by hypoxia, thus triggering depolarization, activation of Na+ [26] and Ca2+ channels [27,28], and a resulting increase in intracellular Ca2+ which triggers the release of neurotransmitters to activate afferent chemosensory fibers of the CSN [29,30,31].
Although this general mechanism seems to be present in all O2-sensitive tissues [32,33], there are apparently species-, tissue- and developmental stage-based differences in the identities of oxygen K+ sensitive channels (KO2). More than one type of KO2 may coexist in the same O2-sensing cells [4,34,35,36,37], suggesting that the full hypoxic response may require the concerted action of several types of channels [38,39]. Thus, the molecular identity of the hypoxia-sensitive K+ channels that are responsible for chemotransduction remains a subject of debate. In addition, the nature of the O2 sensor and the mechanisms it employs to modulate CBCC ion channel activities are not well defined.
2. Oxygen-Sensitive K+ Channels in the Carotid Body
Since the first report of the reversible inhibition by hypoxia of K+ channels in rabbit CBCC [1], identifying the hypoxia-sensitive K+ channels has been of paramount interest for the understanding of hypoxic transduction. K+ channels constitute the largest and most diverse family of ion channels. They are ubiquitously expressed in both excitable and non-excitable cells in all organisms, and their open probabilities can be regulated by voltage, Ca2+ and various neurotransmitters and second messengers. Studies of cloned K+ channels have revealed that they are hydrophobic protein complexes which generally consist of tetramers of transmembrane pore-forming α-subunits, each of which is bound to an accessory β-subunit which regulates channel kinetics and membrane trafficking [40,41].
Based on the topology and structure of the α-subunits, we can speak of three groups of O2 sensitive K+ channels (KO2) found in the CB. The first group, voltage-gated K+ channels (KV), are tetramers of 4 α-subunits, each having 6 α-helical transmembrane spanning (TMS) units and a P-loop (6TM/1P), with the latter contributing to the formation of the channel pore in the assembled tetramer. The second group, to which the BKCa channel belongs, is a tetramer of subunits with 7 transmembrane segments and a P-loop (7TM/1P). A third group, which includes leak channels, posses 4 transmembrane segments and two pore-forming domains (4TM/2P). Unlike the other groups that form tetramers, these channels are formed of dimers which are not known to associate with specific β-subunits [42].
2.1. Kv Channels and Oxygen-Sensitivity in the Carotid Body
Twelve families (KV1.x - KV12.x) of KV channels have been identified in mammalian cells, each consisting of several different sub-families with multiple isoforms (e.g., KV1.1–1.9, KV2.1–2.2…etc; see [43]). KV5-6 and KV8-9 family subunits are electrically silent; they cannot assemble into channels by themselves but can form functional heteromultimers with KV2.x subunits, thereby altering the kinetics of the KV2.x channel current [44]. Channel opening occurs when membrane depolarization causes a change in the configuration of the α-subunits due to the outward movement within the membrane of the S4 TMS units, which contain several positively charged arginine residues. KV α-subunits are associated with auxiliary subunits that protrude into the cytoplasm and modulate their activity; the first ones described, termed Kvβ subunits [45,46], associate with KV1.x and KV4.x channels.
When talking about KV channels in hypoxic transduction we cannot forget the regulatory β- subunits as they may be necessary for its coupling to the putative O2 sensor and could play a very important role in regulating the response [47,48]. The kinetic properties of the channel when only the α-subunit is expressed in a model system can be quite different from those observed in the cell in its natural state, which however are seen when the α and the β subunits are co-expressed [40,41].
As mentioned above, the first hypoxia-sensitive K+ channel, a KV channel which is reversibly inhibited by decreasing PO2, was described in rabbit CBCC. It is a voltage-dependent inactivating current with kinetic properties typical of a delayed rectifier, with a macroscopic activation threshold close to -40 mV [1]. Detailed kinetic and pharmacological characterizations of KV currents in rabbit CB have been reported [49,50].
It can be a significant challenge to match native KV currents in intact cells with the KVα and β subunits forming the channels which mediate them. However, several K+ channel genes expressed in chemoreceptor cells have been identified, and some of them have been shown to behave as KO2 in heterologous expression systems. In rabbit CB, using dominant-negative constructs to block the expression of KV1 (shaker) and KV4 (shal) family channels, Perez-Garcia et al. [51] found that only adenoviral infections with Kv4.xDN suppressed the transient KV current in CBCC and prevented hypoxia-induced depolarization, suggesting that native KO2 currents are carried mainly by channels of the KV4 subfamily. Immunological blockade of K+ channels with anti-KV4.3 further supported this conclusion [52]. In a detailed molecular study [53], the subunits identified in rabbit CBCC were KV3.4, KV4.1 and KV4.3 whereas in mouse CB, the subunits described belonged to the KV2 and KV3 families, specifically KV2.2, KV3.1, KV3.2 and KV3.3 [54]. However, it seems that in both species not all the isoforms found represent the molecular correlate of the KO2 currents but may have other physiological functions [55]. Other KV α subunits which are reversibly inhibited by hypoxia when expressed in PASMCs or in heterologous systems are KV1.2, KV1.5, KV2.1, KV3.1b, KV4.2 and KV2.1/9.3 [56,57]. Of these, channels incorporating KV1.5 seem to be of predominant importance in O2 sensing in PASMCs (see Section 6).
The role of the KV channels as triggers of CB activity has been questioned. If the membrane potential (Em) is close to -55mV [28] and the KV activation threshold is close to -40 mV, this implies that the channel is not active at rest (in normoxia) and that its inhibition by hypoxia cannot initiate depolarization. However, it can be argued that: i) at an Em of -55 mV, the probability of opening may be too low to be readily apparent, but nonetheless sufficient to depolarize the cells when further decreased due to the high membrane input resistance, or ii) the cells possess spontaneous electrogenesis [58], making it possible for the K+ channel to be recruited and inhibited. Accordingly, there are data indicating that an O2-sensitive KV current in rabbit CBCC participates in setting the membrane potential and that, therefore, its inhibition could depolarize the cell [22,59]. In contrast, Wang & Kim [60] showed that KV in isolated rat CBCC were mostly closed at rest but became active when cells were depolarized in response to hypoxia and that this strongly limited the hypoxia-induced rise in [Ca2+]i. These findings suggest that KV channels, rather than mediating hypoxic activation of CBCC, provide a hyperpolarizing force which limits the magnitude of the hypoxic response.
2.2. BKCa Channels and Oxygen sensitivity in the Carotid Body
In the rat CBCC, the first PO2-sensitive current to be described was found to be due to the high conductance BKCa channel [23], which is activated by both depolarization and a rise in [Ca2+]i. These have been described in a wide variety of tissues and cells, both excitable and non-excitable, including nerve cells, muscle cells, pituitary cells, chromaffin cells, and renal tubules among others [61,62].
BKca channels consist of α-subunit tetramers with seven transmembrane domains (S0-S6) [63,64]. Segment S4 acts as a voltage sensor and a region between segments S5 and S6 forms the selectivity filter and the channel pore [65]. In addition to the body or core formed by the transmembrane segments, BKCa channels have a very long carboxyl-terminal tail, which constitutes two thirds of the protein and contains four hydrophobic segments (S7-S10) and several alternative splicing sites. There is a region between segments S9 and S10 with numerous negatively charged residues, mostly aspartate, which is highly conserved between species. This region is known as the "calcium bowl" and appears to be responsible for Ca2+ binding and channel activation by Ca2+ [66,67]. Regulatory β-subunits are also essential for BKCa activity, since their co-expression with the channel α-subunit significantly alters the functional properties of the channel [63]. There are four types of β-subunits (β1-β4) with differential expression depending on the tissue [40]; the channel has been found to be much more sensitive to voltage and Ca2+ when α and β subunit types are transfected together than when only the one is transfected [68].
As shown by Peers' group, BKCa channels in the rat CBCC are active at the resting Em and their specific inhibition by hypoxia leads to membrane depolarization. The role of BKCa in transduction of the hypoxic stimulus is supported on the basis that the BKCa blocker charybdotoxin (CHTX) depolarizes type I cells [69,70]. It has also been shown that the blockers iberiotoxin (IBTX) and tetraethylammonium (TEA), like hypoxia, evoke dopamine release in rat or mouse CB slice preparations [71]. The O2-modulation of BKCa in rat CBCC at the single channel level was characterized by Riesco-Fagundo et al [72]. Its activity seems to be very much dependent on the recording configuration used; channel activity is low in inside-out patches (free of cytosolic factors) and high in vesicles studied using the perforated patch technique, which retain a small volume of cytosol [69].
However, the possibility that BKCa channels are involved in depolarization of CBCC seems counterintuitive, since in numerous cell types they are involved in processes in which Ca2+ signaling is coupled to hyperpolarization, rather than depolarization. In fact, BKCa channels play a particularly important role in neuronal signaling by facilitating repolarization after the action potential, thus contributing to the regulation of neurotransmitter release at the synaptic terminal [61]. In other cases, such as during smooth muscle contraction or exocytosis in secretory cells, the hyperpolarization induced by the activation of these channels is a negative feedback mechanism that contributes to terminate these processes by determining the closure of voltage dependent Ca2+ channels [73].
In addition, many authors question whether a voltage- and calcium-dependent channel could be the trigger of CB activity. Its high conductance does not seem well suited to providing a background K+ flux for setting the membrane potential and its re-activation by [Ca2+]i during hypoxic stimulation would tend to oppose depolarization. Furthermore, a large body of publications and experimental data from several laboratories argue against such a role. For example, Buckler [24,25] demonstrated that neither TEA nor CHTX modify membrane potential, intracellular Ca2+ or the response to hypoxia. Lahiri et al [74] and Donelly et al [75] similarly found that BKCa does not participate in either the intracellular Ca2+ increase produced by hypoxia or in the secretory response of CBCC. Gonzalez’s group also reported that neither TEA nor IBTX alter basal catecholamine release, intracellular Ca2+, or the hypoxia-induced secretory response [76]. The conclusions of these studies were that BKCa was not relevant for hypoxia transduction and/or that it possessed other undisclosed functions.
The contradictory findings and different proposed roles for BKCa and KV led to Wang & Kim [60] to re-examine the role of these voltage-dependent K+ channels in basal excitability and in the generation of the hypoxic response in rat CBCC. They showed that TEA/4-AP had no effect on basal [Ca2+]i, suggesting that BKCa and KV were insufficiently active at rest to participate in initiating the hypoxic response. However, TEA/4-AP caused an increase in [Ca2+]i in chemoreceptor cells previously depolarized by high extracellular K+, indicating that BKCa and KV were basally open in these cells. These channels became active when cells depolarized in response to hypoxia, and strongly limited the rise in [Ca2+]i. Furthermore, hypoxia had no effect on BKCa in cell-attached patches of fully depolarized cells with a resting Em of 0 mV, showing that activation of BKCa observed under hypoxia was due to cell depolarization. The authors proposed that the role of KV and BKCa channels is to limit the hypoxia response by opposing membrane depolarization and the opening of voltage gated Ca2+ channels [60]. Since these findings contradicted previous observations in the literature [72,77] the authors argued that the BKCa current present in CBCC of various rat strains (i.e. Wistar vs Sprague-Dale) might be modulated differently by hypoxia, possibly due to the expression of different BKCa isoforms or splice variants [60].
Nothwithstanding these observations, as argued by Peers & Wyatt [78] and Lopez-Barneo [79] in support of a role for BKCa channels in O2 sensing in CBCC, there is a danger that the contribution of BKCa or KV channels to the physiological behavior of chemoreceptor cells may be underestimated by studies carried out using isolated cells or excised patches, since in the intact CB these cells are closely packed with their neighbors within the glomus and may be electrically coupled [80] or subjected to autocrine and paracrine interactions, which could exert depolarizing influences and modify the cell resting potential or [Ca2+]i such that a fraction of KV or BKCa channels are open. Also, regardless of whether hypoxic inhibition of BKCa channels initiates depolarization in chemoreceptor cells, there can be no doubt that their inhibition would delay repolarization and thereby amplify and sustain the hypoxic response.
Diverse endogenous molecules, including heme, carbon monoxide (CO), and reactive oxygen/nitrogen species (ROS/RNS), have been reported to activate BKCa channels [81,82]. The association between BKCa and the heme provides an explanation for the direct modulation of the channel by both hypoxia and gaseous second messengers including CO and NO. Although these gases can use intracellular signaling pathways, they can also modulate BKCa channels in isolated membrane patches [72,83]. A study by Jaggar et al [84] proposed the interesting hypothesis that CO can stimulate BKCa channels by altering its binding to the heme. In the absence of CO, the heme binds to BKCa, inhibiting its activity, whereas in the presence of CO the interaction between the channel and the heme is weakened, and the tonic inhibition produced by the latter disappears, resulting in current activation.
The involvement of the interaction between CO, heme and BKCa in O2 sensing was proposed by Kemp’s group [85], who used two-dimensional electrophoresis and mass spectrometry studies to show that heme oxygenase-2 (HO-2), which is expressed in CBCC [86], co-precipitates with the BKCa α-subunit expressed in heterologous systems. The authors proposed that the association of BKCa with the heme group and HO-2 facilitates the production of CO in close proximity to the channel which favors channel modulation [85].
In the presence of O2 and using NADPH as a cofactor, HO catalyzes the degradation of the heme producing biliverdin, iron and CO. In hypoxia, CO production is slower than under normoxia because the breakdown of hemoglobin by HO which it depends upon requires O2. Given that CO (as well as O2) activates K+ channels, it was proposed that the response to hypoxia would be partly the result of a decrease in this CO-mediated activation [85]. This effect of hypoxia was verified in HEK-293 cells stably expressing BKCa channels. However, this does not explain the observation that hypoxia inhibits BKCa in isolated inside-out patches, because HO-2 needs the substrate (heme) and both O2 and NADPH as co-substrates to inhibit the channel, and none of these would be present in excised inside out patches. Furthermore, hypoxia was able to cause a modest inhibition of BKCa channel activity even when HO-2 was inactive suggesting the involvement of an additional mechanism. Additionally, Lopez-Barneo’s group demonstrated that O2 sensing by the mouse CB is largely unaffected by HO-2 knockouts [87]. This suggests that a major role for BKCa channels in CB chemotransduction may be restricted to the rat.
Interesting also was the observation made by McCartney et al [88], who, in a heterologous system, described a highly conserved sequence within a cysteine-rich domain at the carboxy-terminal end of BKCa, that they termed STREX (stress-regulated exon), directly inhibited by hypoxia in excised membrane patches. They suggested that this motif is what confers the channel's sensitivity to hypoxia, and that variability in its expression due to alternative splicing is what gives rise to the plasticity of cellular responses to hypoxia in different tissues. The mechanism by which STREX renders the channel sensitive to hypoxia was not defined, but was dependent upon the presence of a Cys-Ser-Cys motif and did not involve CO. The proposition was attractive, since STREX was the sensor and therefore a membrane-limited mechanism in addition to HO-2 could contribute to hypoxic inhibition of these channels. However, Ross et al [89] found that rat CBCC, a species in which BKCa is sensitive to hypoxia, do not express this variant of the channel.
Mark Evans and colleagues presented evidence for an additional mechanism for oxygen sensing and BKCa channels in rat chemoreceptor cells, involving AMP-activated protein kinase (AMPK) [90,91,92]. AMPK is activated by a rise in the cytosolic AMP/ATP ratio, acting as a tuned metabolic sensor. Given the low concentration of AMP under normal conditions relative to ATP concentration (very low AMP/ATP), a slight predominance of ATP degradation over ATP regeneration could markedly increase the AMP concentration and activate the enzyme. AMPK can phosphorylate a multitude of proteins, including the BKCa channel, which is inhibited by phosphorylation. However, subsequent work by Evans and co-workers [93,94] showed that whereas global knockout of AMPK α1 + α2 abolished the ventilatory response to hypoxia, the CB itself was still fully sensitive to hypoxia. This indicated that AMPK plays a crucial role in the response to hypoxia at sites downstream of the CB, probably in the brainstem, rather than in CBCC. Interestingly, they also presented evidence that knocking out Liver Kinase B1 (LKB1), which phosphorylates AMPK, severely depresses the hypoxic response in CBCC, but via an AMPK-independent mechanism, and speculated that LKB1 may act on the mitochondria in these cells to maintain ATP synthesis, a lack of which could impede O2 sensing through several mechanisms [94].
The activity of BKCa channels is regulated, not only by O2 and CO, but also by hydrogen sulfide (H2S), which acts as an inhibitor of these channels, suggesting that integration of these signals may be crucial to the physiological response of the CB. The intracellular concentration of H2S depends on the balance between its enzymatic generation and its mitochondrial breakdown. Kenneth Olson, the originator of the concept that H2S is involved in O2 sensing, showed that hypoxia increased sulfide production by cell homogenates, and suggests that a rise in cellular [sulfide] may act as an O2-sensing mechanism which occurs ubiquitously because hypoxia depresses the breakdown of H2S, without interfering with its ongoing synthesis [95,96]. Conversely, it has been proposed that hypoxia increases cellular sulfide production in CBCC through a mechanism involving CO [97]. According to this model, during normoxia the enzyme HO-2 generates CO, but hypoxia inhibits its production. The decrease in CO leads to a decrease in protein kinase G-dependent phosphorylation of the enzyme cystathionine γ-lyase (CSE), which causes an increased production of H2S. Prabhakar’s group knocked out the enzyme responsible for generating H2S in mice, cystathionine-γ-lyase (CSE), and observed that this interfered with oxygen sensing in the CB [98], and, based on evidence that application of H2S blocks BKCa currents in CBCC [99,100] proposed that an increased sulfide synthesis by CSE could account for hypoxia-induced inhibition of the BKCa current and CBCC activation.
This mechanism is brought into question by studies which have indicated that the pharmacological block of CSE and cystathionine-β-synthase (CBS), another important sulfide generating enzyme, did not prevent the hypoxia-induced inhibition of the TASK current or the rise in [Ca2+]i in CBCC [101] and that knockout of HO-2 [87] and CSE [102] has no effect on acute oxygen sensing in the CB.
Importantly, however, H2S, which is thought to be present at very low concentrations within cells, exists in equilibrium with substantial amounts of other intracellular sulfur-containing species, including polysulfides and persulfides [103], which are mainly synthesized by sulfurtransferases such as cysteinyl tRNA synthetases (CARS) [104]. In a process analogous to that mediated by ROS, these reactive sulfur species (RSS) can alter protein function via cysteine sulfuration. This has led to the suggestion [105] that RSS rather than H2S may mediate O2 sensing. Moreover, sulfurtransferases can also directly sulfurate proteins, and could therefore also have a signaling function [104]. Whereas it has been argued [106] that H2S is unlikely to be an O2 sensor in CBCC since its high membrane permeability would prevent it from reaching a cellular concentration sufficient to inhibit mitochondrial function, this would not be an issue if hypoxia was acting by increasing [RSS] or promoting direct enzyme-mediated sulfuration. Likewise, evidence that O2 sensing persists in CSE knockouts does not rule out a role for RSS, since they are generated by multiple enzymatic and non-enzymatic pathways [103]. Nevertheless, although it is not known whether RSS affect K+ channels in CBCC, both H2S and the polysulfide Na2S4 have been reported to greatly slow the inactivation of KV3.4 channels expressed in HEK 293T cells [107]. This suggests that H2S/RSS would be likely to depress rather than increase responsiveness to hypoxia if KV channels expressed in CBCC are similarly regulated.
2.3. K2P Channels and Oxygen-Sensitivity in the Carotid Body
In 1997, Buckler presented evidence that TASK channels, which belong to the K2P channel family, play an important role in O2 sensing in CBCC [24,25]. K2P is another K+ channel family in which the α-subunits are made up of four transmembrane domains. They have two pores instead of the single pore formed by the other K+ channel families (hence the designation K2Pore) and exist as dimers in the membrane. They are voltage- and ligand-independent channels which are constitutively open, allowing them to generate leak or background currents. Eight different types of K2P have been cloned in rodents and humans, and can be grouped according to their sensitivity to different stimuli, including TWIK-1 and TWIK-2 (Tandem of P domains in Weak Inward rectifier K+ channels), TREK-1, TREK-2 (TWIK Related K+ channels), TRAAK (TWIK Related Arachidonic Acid stimulated K+ channels, which are activated by polyunsaturated fatty acids and by distension), KCNK6 and KCNK7 (silent subunits), and finally the TASKs: TWIK related Acid-Sensitive K+ channels [108]. In addition to the two best known, TASK-1 and TASK-2, functional TASK-3 and TASK-4 channels and a fifth non-functional type, TASK-5, have been described in different tissues [109,110,111,112,113]; each K2P channel has its own expression pattern and each tissue its own combination of channels [108,114].
Pharmacologically, TASK-1, TASK-2 and TASK-3 are insensitive to TEA and 4-AP, and although many drugs modify TASK activity, there is no specific activator or inhibitor of any of these channels. This complicates their functional analysis, although information about their roles in regulating the membrane potential can be gleaned by using the variety of semi-selective drugs available. For example, TASK-2 is inhibited by quinine, quinidine, and clofilium. Zn2+ is a better inhibitor of TASK-1, which also appears to be blocked by quinidine, Ba2+ and low concentrations of the endocannabinoid anandamide [115]. TASK-1, TASK-2 and TASK-3 are inhibited by the local anesthetic lidocaine and bupivacaine [116] and are opened by volatile anesthetics such as halothane and isoflurane [117].
As previously mentioned, the voltage-dependence of KV and BKCa currents creates a certain degree of uncertainty regarding their functionality: if the activation threshold of hypoxia-sensitive channels is close to -40mV and the resting membrane potential (Em) varies between -50 and -60mV, in normoxia these channels would be closed and, therefore, hypoxia would not be able to reduce their Po. In rat CBCC, Buckler’s experiments revealed the existence of a low conductance O2-sensitive K+ leak current which has very little voltage dependence, is reversibly inhibited by hypoxia, and was responsible for the initial depolarization of the CBCC [24]. Although Buckler identified the channel that carries this current as TASK-1 by its biophysical properties [25,118], other channels of the same family, such as TASK-3 or TASK-1/TASK-3 dimers seem to contribute to the current. Patch-clamp recordings with activators and inhibitors of TASK type channels showed that pharmacologically the O2-sensitive background current of CBCC more closely resembles TASK-3, whereas the conductance and biophysical properties are closer to those of TASK-1 [119,120]. There is evidence that TASK-1 and TASK-3 channels can form heterodimers and can associate with other proteins that could alter their characteristics [121,122]. TASK-1/TASK-3 currents are sensitive to pH, which would also explain the CB response to acidosis. TASK-1/TASK-3 is also sensitive to metabolic poisons that activate CB such as cyanide and DNP [25,118] but resistant to the classical K+-channel inhibitors TEA and 4-AP [24]. In addition to TASK-1 and TASK-3, TASK-2, TASK-5, TREK-2 and TRAAK have been found in CB sections using immunohistochemical techniques [123].
Although gene deletion of TASK-3 in mice was reported to have no effect on the ventilatory response to hypoxia (HVR), deletion of the TASK-1 gene reduced the HVR and depressed the chemoafferent (CSN) response to hypoxia in vitro [124]. Double knockout of both TASK-1 and TASK-3 similarly reduced the ventilatory and chemoreceptor nerve responses to hypoxia but did not completely abolish them [124]. However, in studies performed on CB slices, constitutive deletion of TASK-3 and/or TASK-1 appeared to have little effect on Ca2+ signaling or neurosecretion in response to hypoxia [125]. One explanation for this apparent discrepancy is that the chronic loss of TASK1 and/or TASK3 is largely compensated for in some way. Interestingly, enzymatically dispersed Task1/Task3-null chemoreceptor cells have a relatively depolarized resting potential relative to controls, which could promote a compensatory involvement of KV or other ion channels in the hypoxic response [79,125]. However, the nature of any compensatory response that could maintain O2 sensing following TASK channel knockout in the glomus cells has not yet been defined.
It is important to note that the hypoxia sensitivity of these leak channels, which is evident in cell-attached recordings, is absent in inside-out excised patches, implying that their O2-sensitivity depends on some soluble cytoplasmic factor [72,118]. One cellular constituent that probably plays a key role in maintaining TASK channel activity is MgATP, because addition of millimolar levels of ATP to the intracellular side of excised patches results in a rapid increase of channel activity [119,126]. Thus, although they are often referred to as oxygen-sensitive ion channels, there is no evidence that TASK channels are directly sensitive to oxygen.
Buckler proposed that TASK-type channels are constitutively open, and close with hypoxia and acidosis and that these channels, and not KV or BKCa, are responsible for the CB depolarization and neurosecretion [120,127]. Buckler's proposal provides a compelling explanation for how hypoxia activates CBCC, but other aspects such as the intrinsic sensitivity of the cell membrane to hypoxia remain to be resolved.
Finally, it is important to appreciate that depolarization of any cell reflects a shift in the balance between Na+ influx and K+ efflux. In glomus cells, hypoxic inhibition of K+ efflux tilts the balance in favor of Na+ influx. It is widely thought that Na+ influx is via a voltage-independent, background channel. A calcium-activated non-selective cation channel with a conductance of 20-pS has been described in isolated rat glomus cells and exhibits properties like those of Ca2+ activated monovalent cation channels such as TRPM4 and TRPM5 [128]. It is activated only during moderate to severe hypoxia (<5% O2) due to the necessary intracellular calcium elevation (via voltage gated Ca2+ channels) and thus would contribute to positive feedback driving further membrane depolarization and Ca2+ influx. Thus, O2 sensing would involve both inhibition of an outward K+ current and activation of an inward Na+ current.
3. What we Know and What we Don’t About the Oxygen Sensors in Chemoreceptor Cells.
Although it seems clear that O2-sensitive K+ channels are present in oxygen-sensing cells and explain the activation of CBCC, the question of paramount interest that has not yet been satisfactorily resolved is whether these channels act as direct sensors as opposed to effectors, that is, whether their inhibition by hypoxia requires intracellular element(s) that detect PO2 and transmit this information to the channel. Multiple oxygen sensors that link altered oxygen levels to the opening/closure of potassium channels have been proposed, yet the nature of this link remains controversial.
Most proposals for O2 sensing in the CB can be described by a conceptual framework in which the response of CBCC to hypoxia requires one or more O2 sensor(s) which act(s) through certain mediators to alter the function of specific effectors; these then cause CB depolarization and transmitter release. In the following sections, we discuss the O2 sensors and mediators proposed to link the K+ channel inhibition to a fall in PO2. This information is summarized in a simplified form in Figure 1.
As previously mentioned, there are two main groups of proposals. In one, oxygen sensing would be a process or property intrinsic to the plasma membrane of chemoreceptor cells that would require some cytoplasmic component intimately associated with it. In the other, the PO2 sensor would be some cytoplasmic or mitochondrial pathway enzyme whose product or products would act on certain K+ channels to inhibit them.
With regard to the first group of proposals, in the late 1980s, several researchers, including Constancio Gonzalez’s group, based on experimental observations and on theoretical considerations, proposed that the oxygen sensing function resided in a hypothetical hemoprotein, located in the plasma membrane of chemoreceptor cells, which would detect PO2 as a degree of saturation, changing its conformation and allosterically transmitting this conformational change to certain K+ channel to regulate its probability of opening. This was based on their finding that CO, which they used as a tool since it binds specifically to reduced hemoproteins with accessible iron sites, could prevent and reverse the effect of hypoxia on the K+ channels. Supporting this argument, Ganfornina and Lopez-Barneo [129] demonstrated that hypoxia reduced the probability of K+ channel opening in isolated patches of glomus cells, and in whole cell recordings of glomus cells, Lopez-Lopez & Gonzalez [50] showed that CO prevented or reversed hypoxia-induced inhibition of the K+ current by replacing O2 at the level of the sensing molecule. There are other observations in isolated inside-out patches where hypoxia decreased the opening probability of BKCa and CO reversed the effect [72,130]. The same occurs with O2-sensitive currents obtained in HEK cells by co-transfection of Kvβ1.2 and Kvα4.2 subunits [47], reaffirming the previous conclusion. These researchers concluded that CO binding to the O2 site would lead the sensor to adopt a configuration like that existing in normal PO2 situations with the channel open, preventing the inhibition of chemoreceptor discharge produced by hypoxia.
However, the fact that the channels that are inhibited by hypoxia in isolated patches are distinct from each other and that none of these channels possess consensus domains for O2 and CO binding [131] suggests that the channels are not themselves the sensors. Likewise, a hemoprotein which acts as PO2 sensor and regulates channel activity has never been identified. On the other hand, evidence that heme itself can stimulate BKCa channel activity via a pathway dependent on CO (Section 2.2) suggests that O2 could indirectly regulate channel opening by influencing the activity of HO-2, although the contribution of this pathway to CBCC O2 sensing remains unclear.
Another proposed signaling mechanism linking hypoxia to ion channel function that has received much attention involves reactive oxygen species (ROS) as regulators of the probability of K+ channel opening. ROS are produced from a cascade of reactions that begin with superoxide production, which can be generated by mitochondrial respiration, xanthine oxidase, uncoupled NO synthase or via NADPH oxidase (NOX). Of all these mechanisms, the evidence suggests that changes in ROS production by NOX and/or the mitochondria are likely to be most important during hypoxia.
According to a hypothesis developed by Acker, the sensor would be a NOX analogous to that of phagocytic cells. This reduces molecular O2 with a single electron to form superoxide O2.- which is dismutated to H2O2, which modifies the activity of K+ channels by altering the ratio of -SH/-S-S- groups in the channels [132]. This hypothesis was based on the observation that an inhibitor of NOX, diphenyleneiodonium (DPI), activates CSN discharge and prevents the effect of hypoxia [133]. A decrease in PO2 would inhibit the enzyme, decreasing the production of O2.- and H2O2, shifting the GSH/GSSG ratio, the main determinant of the redox potential of the cell, to a higher value and thereby changing the ratio of free sulfhydryl groups to disulfide bridges (ProtSH/ProtSSG) of the K+ channels and modifying their opening probability [134]. However, this model fails to explain what happens in the CB response to hypoxia. DPI, used at the same concentration as in [133] completely inhibited NADPH oxidase, but produced an effect less than one third of that caused by hypoxia and totally abolished the effect of hypoxia. If DPI totally inhibited NADPH oxidase it should produce a maximal response, and if it was not completely inhibited the response to hypoxia should not be 100% blocked [135]. Moreover, an important problem with Acker’s study is that DPI has been shown to antagonize other flavin-containing enzymes, including Complex 1 and NOS, and also blocks K+ and Ca2+ channels [136].
Another experimental approach, carried out in the labs of Fidone in Salt Lake City and Gonzalez in Valladolid, involved the use of knockout (KO) for the p47phox subunit present in NOX-1 and 2. Previous experiments had used KO for the gp91phox subunit of the NOX-2 isoform present in macrophages, and no effect on chemoreceptor activity had been observed. Whereas the hypoxia induced increase in ROS was absent in cells from mice lacking the p47phox subunit, these cells exhibited increased hypoxia-evoked changes in [Ca2+]i and K+ channel activity [137]. This suggest that, firstly, a NADPH oxidase is not the O2 sensor, since mice lacking p47phox were still responsive to hypoxia; and secondly, that hypoxia not only does not inhibit NADPH oxidase but activates it, and finally, that ROS derived from NADPH oxidase act as negative modulators of the hypoxic transduction cascade, since the response to hypoxia is enhanced in the knockouts [137,138]. Along similar lines, Gonzalez’s group has shown that neither oxidants nor reducing agents altered CB function [139,140], and O2-sensitive BKCa channels of rat glomus cells respond to PO2 changes independently of redox modification [72]. The issue of involvement of NOX in O2 sensing in the CB is further clouded by uncertainties regarding which NOX isoforms are expressed in mouse CBCC. Both Mkrtchian et al [141] and Zhou et al [17] have reported that mRNA for NOX4, but not NOX1 or NOX 2, is present in CBCC from the widely used 657BL/6 mouse strain. In contrast, Yuan et al [142] reported the presence of mRNA for NOX2 in mouse CB, although whether it was expressed in chemoreceptor cells was not determined.
The role of mitochondria in oxygen sensing has been extensively investigated, leading to a revival of the old “Metabolic hypothesis” of Anichkov and Belen'kii [10] by proposing mitochondrial function as a mechanism for oxygen sensing through changes in oxidative phosphorylation and energy metabolism. As oxidative phosphorylation is totally dependent on oxygen, mitochondria would be the true oxygen sensor. As above described, the unique sensitivity of respiration to a small fall in PO2 exhibited by CBCC makes it feasible that the mitochondria in these cells can act as the O2 sensor, and this possibility is supported by evidence that their level of O2-sensitivity is comparable to that of background K+ currents, calcium signaling and neurosecretion in isolated chemoreceptor cells [28,59,119,126].
As a whole, the mitochondrial hypothesis seems remarkably robust. Observations supporting this hypothesis were that almost all inhibitors of oxidative phosphorylation, including uncouplers (DNP and FCCP), numerous electron transport inhibitors at Complex 1 (rotenone), Complex 3 (antimycin A, myxothiazol) and Complex 4 (cyanide, CO) and inhibitors of ATP synthase (oligomycin), are potent stimulants of the CB glomus cells [10,143,144,145,146]. The mechanisms of chemoreceptor activation by mitochondrial inhibitors appear to be identical to those of other chemostimuli, i.e., inhibition of background/TASK channel current, membrane depolarization, voltage-gated calcium entry and neurosecretion [146,147,148].
In a similar way, under conditions of severe hypoxia, the main physiological stimulus of the CB, mitochondrial oxidative phosphorylation, is reduced due to the limited availability of O2, the final electron acceptor of the mitochondrial electron transport chain (ETC). Varas et al [126] presented evidence that the TASK current in CBCC is activated by MgATP, the concentration of which fell when cells were treated with metabolic inhibitors or exposed to hypoxia, presumably due to a decrease in mitochondrial ATP production. Thus, when hypoxia inhibits ATP production, the TASK channel closes and the cell depolarizes, leading to NT release. However, an ATP sensor or MgATP binding domain has not been identified in either TASK-1 or TASK-3, and how MgATP activates these channels is currently unknown. It has further been observed that the effects of metabolic inhibitors and hypoxia upon the background/TASK current are mutually exclusive, suggesting that both signaling pathways converge at the level of mitochondrial metabolism [148].
In addition to changes in cell energy status, other mitochondrial signaling mechanisms linking altered mitochondrial metabolism in hypoxia and CB activation have been proposed, including the local generation of mitochondrial ROS (mitoROS) [149,150], changes in redox status [16], and increases in lactate production [151]. All of them are supported by several key observations; for a recent and comprehensive review, see [152].
Chandel and Schumacker reported that hypoxia increased the production of ROS from O2 and H2O2 at the mitochondrial level (mitoROS model) proposing that the sensor would be the mitochondria and that the signal capable of modifying the activities of effectors such as HIF1α and phospholipase C would be this increase of ROS, and not their decrease as previously proposed by Acker and others [153,154,155,156]. The idea that ROS can be generated in large quantities at Complex 1, following excessive production of succinate in Complex 2 and reverse electron transport (RET), has received significant attention due to its similarity to what happens in cardiac myocytes following ischemia-reperfusion [157], although the degree to which succinate accumulates during hypoxia (as opposed to ischemia) seems not to represent a high concentration in comparison with brain and adrenal medulla [149]. Results from Holmes’ laboratory suggest that succinate metabolism is important for ROS generation and CB stimulation during hypoxia but is not the sole mechanism. They propose an alternative mode of mitochondrial ROS production that is independent of succinate metabolism [158].
Gene expression analysis of mouse CBCC revealed atypical mitochondrial Complex 1 (MC1) and Complex 4 (MC4) subunits with relatively high mRNA levels of Ndufa4I2, Cox4i2 and Cox8b, the latter two coding for important cytochrome c oxidase proteins which have been proposed as the most likely oxygen sensor candidates [17,18]. Mice lacking NDUFS2 in tyrosine hydroxylase positive cells (TH+), one of the three essential subunits that contribute to the ubiquinone binding site in MC1, showed an absence of breathing stimulation by hypoxia and of hypoxia-evoked exocytosis and K+ channel inhibition in chemoreceptor cells [149]. Rotenone binds with high affinity to this site and prevents ubiquinione reduction. Similar results were obtained on conditional deletion of Cox4i2 in TH+ CBCC [19]. It has been proposed that compartmentalized, rapid, and reversible ROS production and NADH accumulation in MC1 acting on ion channels confer chemoreceptor cell response to hypoxia [150]. The existence of O2 sensing microdomains could explain the hypoxic modulation of ion channel activity recorded in excised membrane patches, which may contain attached cytosolic organelles [159].
However, other evidence suggests that modulation of K+ channels by ROS does not occur in CBCC. It has been reported that the CB can still be excited under conditions of complete anoxia, when the generation of ROS would theoretically be zero [16,160]. These experiments instead point towards the existence of a signaling pathway activated upon inhibition of cytochrome c oxidase, which is independent of Complex 1, succinate metabolism and ROS generation [158]. Likewise, as described above, there is evidence that altering the oxidation potential of the GSH/GSSG redox couple, thought to reflect the overall cytoplasmic redox state, does not consistently affect CB activity [139,140].
An alternative hypothesis is based in the recent finding by Chang’s group which suggests that hypoxia inhibits electron transport in CBCC mitochondria ultimately causing glucose to form lactate. Lactate accumulates and activates the G-protein-coupled receptor Olfr 78, which by a yet undetermined mechanism causes the chemoreceptor cells to depolarize and release neurotransmitter [151]. For Olfr 78 to be involved in oxygen sensing it needs to be placed in a microenvironment where it can rapidly signal to appropriate ion channels. Murine chemoreceptor cells express a high abundance of the gene encoding olfactory receptor 78 (Olfr78) and Chang et al. [151] reported impaired carotid sensory nerve (CSN) and breathing response to hypoxia in Olfr78-null mice. However, Torres-Torrelo et al. [161] reported that Olfr78-null mice manifest unaltered breathing responses, as well as [Ca2+]i and transmitter secretion from CBCC in response to hypoxia and lactate. These findings questioned the role of Olfr78-lactate signaling in CB activation by hypoxia.
It has been proposed that the strong dependence of pO2 on aerobic glycolysis, unique to CBCC, may cause rapid lactate production in hypoxia and a consequent increase in its extracellular concentration and an acidic pH [162]. Because of this acidification, Bernadini et al [163] proposed an additional lactate effect inhibiting pH-sensitive TASK channels. Although some studies have shown that lactate can depolarize chemoreceptor cells [164] to date no one has investigated the effect of lactate on any type of K+ channels in CBCC. Bernardini's proposal seems unlikely because inhibition of the TASK current by hypoxia was observed in a recording chamber with solution flowing through it, in which a change in extracellular pH would have been unlikely [24].
A recent study from Prabhakar’s lab [165] shows that hypoxia increases persulfidation of Cys240 of the Olfr78 in the CBCC, and this effect was absent in mice lacking Cth, which encodes CSE, a major H2S synthesizing enzyme in the CB. These results suggest that H2S through redox modification of Olfr78 participates in CB activation by hypoxia to regulate breathing. Since the hypoxia-induced rise in cellular [H2S] is proposed to be caused, at least in part, by inhibition of the mitochondrial electron transport chain [105], this mechanism represents another variant of the mitochondrial hypothesis.
In summary, we do not know the precise nature of the O2 sensor in the carotid body. Most of the mechanisms proposed focus on oxygen sensors that are not in the membrane, but which ultimately act through membrane-located ion channels. Of the proposed O2 sensors within the CB, only mitochondria have been identified as having a unique phenotype compared to other O2-insensitive cell types [166] so potential interactions between mitochondria and the plasma membrane become critical for hypoxic signal transduction [167]. These authors argue that the physical arrangement of glomus cell organelles with their large nuclei favors the positioning of mitochondria proximal to the plasma membrane, an optimal position for interacting with K+ channels. If the mitochondria are indeed the O2 sensor, this raises the question of how hypoxia-induced changes in their activity are coupled to the inhibition of K+ channels. This subject is discussed in the next section.
4. Coupling Mechanisms Between the Sensor and the K+ Channels.
The lack of knowledge of the nature of the O2 sensor makes it difficult to search for the coupling mechanism between the sensor and the O2-sensitive K+ channels, which presumably constitute the first effectors of hypoxic detection-activation. It has been proposed that if the sensor was a hemoprotein, the coupling mechanism could be an allosteric transmission of the conformational change from the sensor to the K+ channel upon saturation and desaturation. In the proposed models in which the sensor is an ROS-producing mechanism, the mitochondria or a NADPH oxidase, changes in the cytosolic levels of ROS could modulate channel activity through oxidative modification of thiol groups on α-subunits [168]. Alternatively, ROS could act indirectly by affecting the potential of the NAD(P)+/NAD(P)H redox couples, thereby altering KV channel kinetics via their regulatory β-subunits, which regulate the α-subunit by virtue of their oxidoreductase activity [169]. The latter concept is supported by evidence that K+ currents are only sensitive to hypoxia if they are expressed together with the β-regulatory subunits or if transfection is done to cell types already expressing β-subunits [47,48]. Although a role for β-subunits in acute O2 sensing in native CBCC has not been established, differences in the mRNA levels of α and β subunits of the BKCa channel have been observed in CB from normoventilating and hypoventilating animals [170], and Hartness et al [171] found that chronic hypoxia, which sensitizes the response to acute hypoxia, results in increased expression and colocalization of specific hypoxia-sensitive K+ channel subunits.
Does O2 sensing in glomus cells involve mitochondrial ROS? The work of Chandel & Schumacker [172] consistently demonstrated in various cell types that acute hypoxia increases the production of ROS at the mitochondrial level. Lopez-Barneo's group proposed a full mechanism of acute oxygen sensing that relies on the production of ROS by the CBCC mitochondria, specifically by Complex 1 of the ETC [146]. Recently, this group has provided more detail about these mechanisms, showing that hypoxic inhibition of mitochondrial electron transport resulted in increased ROS production and a reduction of mitochondrial Complex 1 by pyridine nucleotides [18,149]. According to their hypothesis, increased ROS production would change the redox state of membrane ion channels and modify their opening probability. This general mechanism fits with the idea that multiple ion channels are inhibited by hypoxia depending on the species analyzed and with the concept that when one oxygen-sensitive channel is eliminated (as in knockout mice), others become oxygen-sensitive [125]; ROS would allow this to happen.
However, there is substantial evidence that contradicts the role of increased ROS production in acute oxygen sensing: i) inhibitors and uncouplers which interfere with mitochondrial function, some increase ROS production and some reduce ROS production but all examined consistently raised [Ca2+]i in isolated CBCC [148]; ii) H2O2 applied at a high concentration did not raise [Ca2+]i in isolated CBCC, nor did it interfere with hypoxic rises of [Ca2+]i in CBCC [148]; iii) Acute hyperoxia, which leads to ROS formation in most cells [173], essentially silences CB output [2,174]; iv) Neither oxidizing or reducing agents appear to modulate the ability of intact CB to release catecholamines in response to acute hypoxia [140,175]; v) Although hypoxia has been shown to produce a rise of ROS in CB slices, this effect was modest after exposure of slices to severe hypoxia for 1hr [176]; vi) In anoxia, when the generation of ROS would be zero, CB can still be activated [16,160]; vii) levels of hypoxia that strongly activate CB do not modify the GSH/GSSG ratio in the CB [175].
H2O2, the most important ROS with regard to cellular signaling probably does not act directly on reactive cysteines to modify protein function [177]. Instead, it works indirectly by oxidizing peroxiredoxins, thioredoxins and glutathione [178,179] which then cause the oxidative modifications of protein thiols (e.g. s-glutathionylation) which affect protein activity. In view of the importance of the GSH/GSSG ratio in determining the oxidation of reactive thiols on proteins, further evidence against a role for changes in cellular ROS production is that Sanz-Alfayate et al [175] found that hypoxia had no effect on the redox potential of GSH/GSSG in CB from calf.
Along the same lines, in HEK cells transfected with K+ channel subunits of the Shaker, Shaker/1.2 and Kv4.2/1.2 families, dithiothreitol (DTT) decreased the current of the three channel types, but hypoxia only inhibited the last one [47]. Furthermore, Riesco-Fagundo et al [72] showed that DTT increases the opening probability of BKCa but hypoxia decreases it. Thus, the redox status of these channels does not seem to be involved in the initiation of the transduction cascade, although this does not exclude that it may play a modulatory role. Therefore, although it seems plausible that changes in ROS production and resulting effects on cellular redox networks could modulate K+ channel kinetics, as these can be modified by reducing and oxidizing agents, the observations described above argue against this mechanism as being the primary determinant of K+ channel activity during hypoxia.
One point to consider is that much of this work has made use of oxidizing/reducing agents and ROS scavenging agents [148,180,181] and the extracellular application of these compounds may not accurately mimic/inhibit the rapid signaling that occurs in subcellular microenvironments.
Recently, submicron distance measurements between TASK channels and mitochondria have confirmed the existence of oxygen-sensing micro-domains that are conducive for rapid diffusion of mitochondrial products (i.e., ROS, ATP) and propagation of heat [182]. These results have led to a new oxygen-sensing hypothesis suggesting that CBCC hypoxic signaling may be mediated by mitochondria-generated thermal transients in TASK-channel containing micro-domains. By thermal imaging experiments, Wyatt’s group have demonstrated that mitochondrial thermogenesis is oxygen dependent and mitochondrial inhibition significantly decrease intracellular temperatures in isolated rat CBCC. Perforated patch-clamp electrophysiological recordings of whole-cell resting membrane potentials demonstrated lowering bath temperature to induce consistent and reversible depolarization [182], compatible with TASK channel inhibition opening a new field for future research in CB oxygen sensing.
5. Concluding Remarks in O2 Sensing in the Carotid Body
Electrophysiological techniques helped to characterize the profile of O2-sensitive K+ channels present in CBCC, but it remains to be determined precisely which channels regulate the membrane potential, their possible role in the process of hypoxic transduction and the mechanisms by which hypoxia regulates the activity of these channels. Current models are based on either a heme protein capable of reversibly binding O2 or the production of ROS by NAD(P)H oxidases and/or mitochondria. We cannot exclude that multiple mechanisms, and perhaps other mechanisms not yet considered, can operate simultaneously. It is plausible that a sensor, or any other element of the transduction cascade, could be modified by changes in the levels of ROS in restricted compartments of the cells. Such a modification could enhance the effect of hypoxia or, on the contrary, slow down that effect through the opening/closure modification of K+ channels. A deeper understanding of the cellular mechanisms of O2 sensing will facilitate the development of new pharmacological tools effective in the treatment of diseases caused by localized deficits of O2.
In all studies mentioned here there is an added difficulty that there is only one cell line that partially simulates the behavior of CBCC, and that investigation in the organ is complex due to its location and small size. Pheochromocytoma-derived cells (PC12) share the same embryonic origin, the neural crest, and several structural and neurochemical features with CBCC. In their response to hypoxia, PC12 cells depolarize by inhibition of a voltage-dependent K+ current [183]. Other cells that can be maintained in culture and respond to hypoxia are pulmonary artery smooth muscle cells (PASMC), responsible for hypoxic pulmonary vasoconstriction. In this context, it seems interesting to compare the mechanisms involved in this hypoxia-sensitive cell type, which are discussed in the following sections.
6. O2-Sensitive K+ Channels in the Pulmonary Vasculature
Hypoxic pulmonary vasoconstriction (HPV) is the acute and reversible constriction of the pulmonary vasculature, especially at the level of the small pulmonary arteries, caused by alveolar hypoxia. HPV is an adaptive response which acts to divert pulmonary blood flow from poorly ventilated regions of the lung to those which are more highly oxygenated, thereby optimizing the ventilation/perfusion ratio so as to minimize the effect of hypoxia on PaO2. HPV begins to develop when the PO2 falls below ~80mmHg, and increases as hypoxia deepens, reaching a maximum amplitude at a PO2 of ~20mmHg [57]. Constriction begins within seconds, and is maintained indefinitely if re-oxygenation does not occur, eventually contributing to the development of pulmonary hypertension if alveolar hypoxia is sustained and widespread.
As described in the first part of this review, the response of CBCC to hypoxia depends mainly on K+ current inhibition, membrane depolarization, and Ca2+ influx through voltage gated Ca2+ channels. Although a similar pathway contributes to HPV, the response of PASMC to hypoxia is a much more complex affair which involves a multiplicity of effectors. In addition to K+ channels, these include non-voltage gated Ca2+ channels residing in the plasmalemma (TRP channels, Orai) or the membrane of the sarcoplasmic reticulum sarcoplasmic (RyR, IP3R), and also protein kinases (several PKC isoforms, src kinase, rho kinase) [57].
HPV, especially when evoked in vitro, often exhibits two phases. Phase 1 is typically a transient constriction which peaks within several minutes. This is followed by or superimposed upon a slowly developing constriction (Phase 2) which plateaus within several hours. Although the mechanisms underlying these two phases have not been systematically compared, the evidence available suggests that they are, at least in part, different [184,185]. This concept arises mainly from experiments carried out in isolated PA, in which the two phases are usually readily apparent (especially in rats and mice, which have been used for the bulk of investigations). With some exceptions (e.g. [186]), studies of HPV in isolated perfused lungs or in vivo have typically utilized shorter periods of hypoxia (5-15 minutes), so the information they provide is probably more relevant to Phase 1. A great deal of mechanistic insight has also been gleaned from the use of isolated (usually cultured) PASMC, but it is difficult to ascertain whether this information applies to Phase 1 or Phase 2.
Two further factors which complicate the interpretation of results are the use of multiple species and different levels of hypoxia in experiments. Also, HPV in isolated arteries or perfused lungs has usually been studied in the presence of a small pre-existing stimulus (‘pre-tone’). Angiotensin 2 is generally used to create pre-tone in isolated perfused lung, whereas vasoconstrictors such as PGF2a, U46619, phenylephrine or high K+ PSS have been used for this purpose in isolated PA. Pre-tone is used to amplify the response to hypoxia but is problematic because experimental interventions used to characterize the response to hypoxia may also affect the pre-tone stimulus, which then indirectly affects HPV. On the other hand, it seems likely that some kind of pre-tone exists in vivo, although its nature is unknown. The apparent complexity of HPV, plus these and other issues relating to variability in experimental design, helps to explain why, whereas work to date has established that hypoxia has numerous effects on PASMC which could potentially contribute to HPV, there is little agreement about the extent to which any of these effects actually does shape HPV.
This lack of consensus is embodied by the variety of proposals which have been put forth to explain how HPV arises. As with the response to hypoxia in the CB, the proposed pathways linking a fall in PO2 to HPV can be described according to the sensor/mediator/effector paradigm. Figure 2 presents a simplified overview of these schemes. The first of these, the Redox theory put forward by Stephen Archer, Kenneth Weir and colleagues in the early 1990’s [33], proposes that hypoxia causes a fall in mitochondrial ROS production leading to the closure of KV channels, cell depolarization, and Ca2+ influx through L type voltage-gated Ca2+ channels (L-VGCC). In this case, HPV represents the loss of an ongoing normoxic vasodilating influence exerted by KV channels. Along similar lines, Michael Wolin and co-workers have developed a model (not shown in Figure 2) in which hypoxia causes a fall in ROS production by NOX, resulting ultimately in the decreased activity of protein kinase G that removes tonic normoxic vasodilation (187).
Paul Schumacker’s lab presented a contrasting scheme, often referred to as the Mitochondrial ROS theory, in which hypoxia increases mitochondrial ROS production [156]. It has also been suggested that the increase in ROS from the mitochondria may trigger additional ROS production by NOX, providing a stronger cytoplasmic oxidizing signal [188]. Shumacker and colleagues have not specified any downstream effectors linking the resulting increase in cytoplasmic [ROS] to contraction, but subsequent work by other groups has presented evidence that multiple pathways causing contraction in PASMC, some of which are shown in the Figure 2, can be activated by ROS (see review by [57]).
A variety of other schemes have also been put forward which are somewhat less comprehensive. These include proposals that HPV is caused or promoted by activation of AMPK [91], the pentose phosphate pathway (PPP) [189], or by a rise in the intracellular concentration of hydrogen sulfide and/or reactive sulfur species (RSS) [95].
Regarding the role of K+ channels in O2 sensing, it can be seen that several of these proposals have incorporated K+ channels as effectors. The Redox theory has consistently placed a particular emphasis on the involvement of KV channels in HPV, and observations by Cogolludo et al [190] and Sommer et al [191] also link the increase in cytoplasmic H2O2 predicted by the Mitochondrial ROS theory to hypoxic regulation of KV channel opening. Evidence from the labs of Alison Gurney and Andrea Olschewski also indicates that KV7 (KCNQ) and TASK-1 channels, respectively, may play a role in O2 sensing in PASMC, although the O2-sensors and/or mediators these would link to remain to be fully defined [192,193,194].
It is unlikely, however, that the inhibition of BKCa channels plays a significant role in acute HPV, at least in adults. BKCa channels do not contribute to the K+ current at potentials close to the resting Em in these cells [195], and blockers of this current have no effect on the resting Em or vascular tone in pulmonary arteries [196,197,198,199]. In contrast, the BKCa current helps to set the resting Em and is inhibited by hypoxia in PASMCs from fetal rats, so could potentially contribute to HPV in the fetus [200].
6.1. Kv Channels and O2 Sensing in PASMC
Initial evidence that acute hypoxia caused membrane depolarization and depressed the K+ current in freshly isolated PASMC was presented by Joseph Hume’s laboratory [201]. Their study also showed that several K+ channel blockers caused PA constriction, consistent with the possibility that hypoxia-induced depression of the current and a resulting membrane depolarization leading to the opening of L-VGCC could cause HPV. This fit with earlier observations that blockers of L-VGCC inhibited HPV recorded in dog lungs [202] and that hypoxia depolarized SMC in isolated cat PA [203]. The K+ current in dog PASMCs was attenuated by the L-VGCC antagonist nisoldipine and the residual current in the presence of this blocker was not affected by hypoxia, suggesting that hypoxia was acting on a Ca2+- activated K+ current. However, subsequent work by Yuan et al [204,205] showed that the Ca2+- activated K+ current in rat PASMC is small, and that hypoxia was instead inhibiting a voltage-gated K+ channel current. Hume’s group later showed that the hypoxia-sensitive K+ current in dog PASMC was also a KV current, which was depressed by increases in [Ca2+]i [206].
Based on the findings of Post et al [201] and on their earlier observations that HPV was associated with a fall in ROS levels in PASMC and was mimicked by application of proximally-acting mitochondrial blockers and exogenous antioxidants, Weir & Archer [33] proposed the Redox theory (Figure 2), which views HPV as being caused by an hypoxia-induced fall in mitochondrial ROS production; this suppresses an ongoing activation of KV channels by ROS produced under normoxic conditions, resulting in channel inhibition, membrane depolarization, and Ca2+ influx via L-VGCC. The importance of K+ channels as HPV effectors was supported by evidence that hypoxia inhibited the Kv current in smooth muscle cells isolated from pulmonary but not systemic arteries, which tend to relax to hypoxia [201,205].
Evans et al [207] soon reported that in addition to displaying typical delayed rectifier and rapidly inactivating voltage gated K+ currents (IKV and IKA, respectively) rabbit PASMCs exhibited a non-inactivating K+ current with an activation threshold negative of -65 mV, indicating that it could contribute to setting the resting Em (which was ~-50mV in these cells). This current, which they christened IKN, could be distinguished from IKV and IKA because it was uniquely resistant to block by 10 mM quinine. Osipenko et al [198] then showed that the depolarization of these cells by hypoxia (14mm Hg) was associated with inhibition of IKN but not IKV or IKA.
At about the same time, several laboratories began to carry out studies designed to establish the molecular identities of the hypoxia sensitive KV channels in PASMC. Patel et al [208] found that cultured rat PASMCs expressed several KV channel α-subunits, including KV2.1. Expression of KV2.1 in COS cells generated a hypoxia-sensitive current in a ~20% of the cells. They also presented evidence that the PASMCs expressed an electrically silent α-subunit, KV9.3, which could form hetero-multimers with KV2.1. Co-expression of KV2.1 and KV9.3 generated a current which activated close to the PASMC resting potential and was hypoxia-sensitive in ~55% of COS cells. The authors proposed that the hypoxia-sensitivity of these KV2.1-containing channels required the action of an endogenous kinase, accounting for the variable responsiveness. Based on the kinetic properties of the current in COS cells resulting from KV2.1/9.3 expression, they suggested that these channels might account for IKN (although this possibility was never confirmed).
Attention shifted to the role of KV1.5 in PASMC O2 sensing when Archer et al [209] reported that antibodies against KV1.5 significantly depressed the KV current and abolished the attenuation of this current by hypoxia. Antibodies against KV2.1 also blocked the KV current and depolarized the cells. The authors suggested that HPV required the hypoxic inhibition of both channels.
The importance of channels incorporating KV1.5 α-subunits for O2 sensing in the pulmonary vasculature was additionally supported by the observation of Archer et al [210] that HPV was significantly smaller in ‘swap’ mice in which the KV1.5 subunit was replaced by KV1.1 [211] compared to wild type controls. Archer et al [186] presented evidence that antibodies to both KV1.5 and KV2.1 caused depolarization of isolated rat distal PA; these also demonstrated a much higher level of KV1.5 protein expression and a larger hypoxia inhibitable KV current than conduit PA, which were less responsive to hypoxia.
Further evidence for the importance of KV1.5 in PASMC O2 sensing was provided by several papers from Jason Yuan’s laboratory. Platoshyn et al [212] showed that overexpression of KV1.5 in PASMCs led to the expression of a 4-AP-sensitive KV current which was strongly suppressed by hypoxia. Intriguingly, the similar 4-AP-sensitive KV current which developed in COS and mesenteric artery smooth muscle cells (MASMC) in which KV1.5 was overexpressed was not diminished by hypoxia. This observation echoed a similar finding by Hulme et al [213], who had found that the current caused by overexpressing KV1.5 in mouse L cells was also not affected by hypoxia. These results implied that the presence of some other co-factor or mediator unique to PASMCs was necessary to confer hypoxia sensitivity on KV1.5. This laboratory [214] later reported that the mRNA expression of KV1.5 varied between individual rat PASMC. The minority of cells had a relatively high expression of KV1.5, and this was associated with a KV current which was sensitive to hypoxia, whereas other cells had a lower expression of KV1.5 and KV currents which were not affected by hypoxia. The authors suggested that the gap junctions between the PASMCs could spread the depolarization induced by hypoxia in the KV1.5-rich PASMCs more widely through the arterial wall.
As described in Section 2.1, the role of KV channels in O2 sensing in CBCC has been questioned because the apparent threshold of activation of these channels is positive of the resting potential of these cells. Similar criticisms of the concept that such channels make a primary contributing to O2 sensing in PASMC have been raised [57], despite the substantial evidence that channels incorporating KV1.5 and possibly KV2.1 are involved in HPV. It is possible, for example, that inhibition of KV channels by hypoxia has evolved to prevent them from resisting the depolarization caused by the hypoxia-induced suppression of another K+ current which is responsible for setting the resting potential (i.e. IKN). Indeed, Gurney et al [215] demonstrated that the K+ leak channel TASK-1 is expressed in rabbit PASMC and provided extensive evidence that it is partly responsible for IKN. Olschewski et al [192] then reported that TASK-1 was expressed in cultured human PASMC and showed that it was inhibited by moderate hypoxia (30 mmHg). In addition, they found that siRNA knockdown of TASK-1 expression caused a significant depolarization of the resting potential, as well as decreasing the amplitude of IKN, and rendering it hypoxia-insensitive.
Gurney’s lab has also presented pharmacological evidence that both the resting potential in rat PASMC and the basal vascular tone of PA in isolated perfused lung are regulated by KV7 (KCNQ) channels [194,216], with the KV7.4 isoform being the most important in this respect. The properties of these channels (very negative activation threshold, lack of inactivation) suggest that they also contribute to IKN and that they could be involved in O2 sensing, although this remains to be established.
7. How Does PO2 Regulate K+ Channels in PASMC?
7.1. Evidence for KV channel regulation by cytoplasmic redox state
The concept that hypoxia depresses plasmalemmal K+ channel opening by altering the production of ROS is consistent with findings that the mitochondria in PASMCs are located close to the cell membrane and that agents which affect the electron transport chain can affect K+ currents [217,218]. However, the effects of hypoxia on mitochondrial production of ROS, and of ROS on KV channels, are disputed.
According to the Redox theory as proposed by Weir and Archer [33], hypoxia inhibits PASMC KV channels by causing a fall in mitochondrial ROS production. This diminishes the cytoplasmic [ROS] (and/or shifts cytoplasmic redox couples to a more reduced state), thereby lessening KV channel opening. Recently, Archer’s group has presented evidence that the basal normoxic production of ROS which is depressed by hypoxia is dependent on the Complex 1 subunit Ndufs2 [219]. The mechanism for KV channel regulation proposed by the Redox theory is supported by reports showing that amplitude of the KV current in PASMC was increased by oxidants such as diamide and H2O2 and decreased by reductants such as DTT and GSH [196,220,221,222,223,224,225].
In contrast, the Mitochondrial ROS theory developed by Paul Schumacker’s laboratory [156] proposes that hypoxia causes an increase in the production of ROS by PASMC mitochondria. The resulting rise in cytoplasmic [ROS] activates one or more effector pathways to cause HPV. Whereas Schumacker’s group has devoted little effort to investigating how the increase in ROS causes PASMC to contract, other labs which have accepted the premise that hypoxia increases cytoplasmic oxidant signaling have sought to determine which effectors are stimulated by ROS to cause HPV. Multiple effectors have been identified (see, e.g. [57,226]), including KV channels. For example, Perez-Vizcaino and co-workers reported that application of H2O2 and tert-butyl hydroperoxide (t-BOOH) depressed the KV current in freshly isolated rat PASMC [227], and, based on this and other observations [190,228,229,230], developed a model in which an increased production of mitochondrial ROS due to hypoxia activates neutral sphingomyelinase, which they showed is more highly expressed in PASMC than in MASMC. This leads to the production of ceramide which inhibits KV channel opening through a pathway involving protein kinase Cζ and the linker protein p62, which they propose is inhibiting the opening of KV1.5 channels by targeting KVb2.1 [231]. The authors showed that the protein expression of nSMase2 is high in rat PASMC but almost absent in smooth muscle cells from mesenteric arteries, thereby providing a potential explanation for why hypoxia-induced inhibition of the KV current is restricted to the pulmonary vasculature.
Sommer et al [191] also found that the KV current in cultured mouse PASMC was similarly diminished by hypoxia and exogenous H2O2, and that this effect of hypoxia (but not H2O2) was smaller in a mouse model in which mitochondrial ROS production was lessened due to knockout of Cox4i2, a subunit of Complex 4. Corresponding differences in the effect of hypoxia and H2O2 on the resting Em in the wild type and the Cox4i2-/- mice were noted.
In addition to these conflicting findings relating to KV channels, divergent effects of hypoxia on cellular ROS levels in PASMC have also been reported by different laboratories [191,219,232]. Other approaches to examining the role of ROS in HPV, including studies of the effects of electron transport chain blockers, antioxidants and pro-oxidants on HPV and pulmonary vascular tone, have similarly yielded inconsistent results (see [57,226]). Although Archer and colleagues have argued, with some justification, that their investigations supporting the Redox theory have generally been carried out under conditions which are more physiologically relevant than those often employed by advocates of the mitochondrial ROS theory [233], this has not always been the case, and the evidence supporting the mitochondrial ROS theory is currently more extensive than that favoring the Redox theory.
In general, the specific molecular mechanisms by which ROS and cell redox mechanisms regulate KV channels remain somewhat enigmatic. The stimulation of KV1.5 channels by oxidants observed in PASMCs in many studies is consistent with, for example, the increase in the current induced by H2O2 in cells overexpressing human α-KV1.5 reported by [234,235], although Duprat et al [236] did not observe this effect with tert-butylhydroperoxide. Park et al [237] showed H2O2 increased the amplitude of the KV current in rat MASMC, and this was associated with the S-glutathionylation of KV2.1, but not KV1.2 or KV1.5. On the other hand, Svoboda et al [238] showed that diamide oxidized a reactive cysteine (C581) on KV1.5 and that this was associated with internalization of the channel in atrial cells, leading to a decrease in the amplitude of the IKur (KV1.5-mediated) current. This effect of diamide on the current amplitude is consistent with the Mitochondrial ROS model. Nonetheless, there is presently no evidence that channel internalization plays a role in the hypoxia-induced effects on KV currents in PASMC, although KV1.5 has been shown to undergo rapid endocytosis when these cells are treated with 5-HT [229]. Also, Gamper et al [239] found that several KV7 isoforms, including KV7.4, are activated by H2O2, an effect that was due to the oxidation of three contiguous cysteine residues in the S2-S3 linker region of the channel protein.
Multiple KVβ subunit isoforms are expressed in PASMC [48,240]. These subunits have been shown to act as oxidoreductases which bind NAD(P)H, using it to reduce various substrates, and thereby converting it to NAD(P)+. This changes the configuration of the β subunit and therefore its interaction with α-subunit, increasing the channel current. Thus, channel activity is sensitive to the redox state of the NAD(P)+: NAD(P)H couple [241].
For this reason, KVβ subunits initially seemed to provide an attractive potential mechanism for redox regulation of KV channels during HPV [48], especially with the demonstration by [47] that the co-expression of KV4.2 α-subunits and KVβ1.2 in HEK293 cells led to a development of a hypoxia-sensitive current, whereas the current due to expression of KV4.2 α-subunits in isolation was unaffected by hypoxia. Recent work from Nystoriak’s laboratory has demonstrated that both H2O2 and NAD(P)H increase the opening KV1.x channels in systemic vascular SMC [242,243]. They presented evidence that NADH was having this effect via an action on KVβ2 subunits, whereas H2O2 stimulated the current through a KVβ2-independent mechanism. Intriguingly, however, the response to H2O2 was potentiated by NADH, and this effect was KVβ2-dependent. Evidence that PASMC express KVβ2 [244] implies that they could also be regulated by the redox state of the NAD(P)H/NAD(P)+ couple. However, PASMC also express KVβ1, which have been proposed to block the KVβ2 -mediated effect of NAD(P)H [245].
Despite the early interest in β subunits as regulators of KV currents in PASMCs, no detailed investigations of this mechanism were subsequently reported. However, Platoshyn et al [212] showed that the expression of KVβ1.1, KVβ2.1, and KVβ3.1 was similar in pulmonary and mesenteric arteries, leading them to suggest that the specific hypoxia-sensitivity of PASMCs was not due to an effect of KVβ subunits. This conclusion was also consistent with their finding that mRNA for the KVβ2 subunit was abundant in HEK293 but negligibly expressed in COS cells, whereas the KV current due to the overexpression of KV1.5α was completely O2-insensitive in both types of cells. Nonetheless, this type of evidence is indirect at best, and the role of KVβ subunits in HPV remains unresolved.
Studies such as those carried by Svoboda et al [238] and Raph et al [243] indicate that redox regulation of KV1.5 channels can be exerted through multiple mechanisms targeting both α and β subunits and may include changes in channel function and membrane expression. This complexity could help explain observations showing that the effects of ROS on K+ current amplitude during changes in PO2 can vary in different types of cells [246,247]. Thus, it is apparent that an in-depth examination of the effect of cell redox on both α- and β - subunit – dependent regulation of channels incorporating KV1.5 in native PASMCs, including an investigation of their membrane trafficking, may be required to resolve the question of how their inhibition during HPV is being orchestrated by changes in ROS production.
7.2. Evidence for TASK-1 channel regulation by cytoplasmic redox state
There is conflicting evidence with regard to whether TASK currents are sensitive to ROS. TASK-1 channels expressed in CHO cells were found to be insensitive to the extracellular application of 5 mM H2O2 [248], a concentration which should raise the intracellular concentration into the micromolar range [249]. On the other hand, Lee et al [250] demonstrated that NOX4, which was endogenously expressed in HEK293 cells, co-localized with TASK-1 when this was expressed in these cells. Hypoxia depressed the TASK-1 current, an effect which was reversed by knockdown of endogenous NOX4 and potentiated by NOX4 overexpression. These results, which suggest that NOX4 is causing an inhibition of TASK-1 which is potentiated by hypoxia, are consistent with the idea that ROS release by NOX4 increases during hypoxia and suppresses the TASK-1 current. NOX4 is expressed in PASMC, so a similar mechanism might also be operative during HPV. However, this has not been investigated, and arguing against this concept is the finding that NOX4 has a KM(O2) of ~135mm Hg [251,252], such that its ability to generate H2O2 is likely to have been severely compromised at the PO2 level (8 mmHg) at which its presence was seen by [250] to most strongly promote TASK-1 inhibition.
7.3. Other factors regulating PASMC K+ currents during hypoxia
Several mechanisms by which hypoxia can regulate PASMC K+ channels independently of ROS have also been described. For example, soon after their discovery that hypoxia depressed the K+ current in canine PASMCs, Hume’s laboratory presented evidence that this was due to the decreased opening of a 4-AP sensitive (i.e. KV) channel with a conductance of 25 pS [206]. Their data indicated that this effect was caused by an increase in the intracellular Ca2+ concentration, associated with the release of Ca2+ from the sarcoplasmic reticulum. If this is indeed the mechanism by which the KV current is inhibited by hypoxia, it would imply that SR Ca2+ release, rather than KV channel opening, may be the key event in the effector pathway causing a rise in [Ca2+]i during HPV. The concept that SR Ca2+ release, probably associated with store operated Ca2+ entry [253], plays an important role in HPV is well established [254]. These processes have been proposed to be due to increases in cellular ROS production [255,256,257], in which case this mechanism is potentially an effector pathway of the Mitochondrial ROS theory.
Mark Evans and colleagues have proposed that HPV is triggered by an increase in the PASMC [AMP]/[ATP] ratio. This results in the stimulation of AMPK and the resulting phosphorylation of HPV effector pathways. Although these remain to be fully defined, Moral-Sanz et al [258] demonstrated that the inhibition of the KV1.5 current caused by hypoxia (~34 mmHg) in freshly isolated rat PASMCs was mimicked by applying three drugs which activate AMPK and also by dialyzing the cells with active AMPK heterotrimer. They also showed that AMPK purified from rat liver phosphorylated human recombinant KV1.5 protein. These results supported the idea that AMPK hypoxia-induced inhibition of KV1.5 occurs during hypoxia, although the kinetic effects on the current of blocking AMPK suggested that this might not be relevant to HPV. However, a later publication from this group which established that AMPK was phosphorylating KV1.5 on serine 592 and 559, [259] concluded that KV1.5 inhibition by AMPK was in fact crucial for HPV.
Jason Yuan and colleagues have published evidence that the plasmalemmal protein Notch, a key regulator of cell differentiation [260] which has been implicated in the development of pulmonary hypertension [261], may also play a role in HPV. This possibility is supported by evidence that the Notch antagonist N-[N-(3,5-diflurophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) depressed HPV in perfused mouse lung [261,262], whereas the Notch agonist Jag-1 increased pulmonary artery pressure. Another paper from this laboratory [263] reported that Jag-1 also attenuated the currents which resulted from expression of KV1.5 and also KV1.2 in HEK293 cells. Taken together, these results suggested that hypoxia might also reduce the KV current in PASMC through a Notch-dependent mechanism, although this has yet to be demonstrated.
With regard to TASK channels, Nagaraj et al [193] presented extensive evidence that the inhibition of the TASK-1 current in cultured human PASMC is dependent on its regulation by Src family tyrosine kinase (SrcTK). They showed that SrcTK is associated with and phosphorylates the TASK-1 channel under normoxic conditions, causing its activation and a corresponding increase in IKN. Hypoxia (~40 mmHg) caused the dephosphorylation of TASK-1 after about 15 minutes, and this was accompanied by the dissociation of TASK-1 from SrcTK, a decrease in IKN, and cell depolarization. The SrcTK inhibitor PP2 also increased the pulmonary artery pressure in isolated perfused mouse lung, thus mimicking the effect of hypoxia. Interestingly, SrcTK similarly regulated the KV and BKCa currents in PASMC. The authors [193] did not investigate the involvement of cell redox in the regulation of SrcTK or its effect on TASK-1. However, work by Greg Knock and co-workers has shown that hypoxia causes the phosphorylation and activation of SrcTK in rat PA [264] and that SrcTK in rat PASMC is activated by superoxide and H2O2 [265], findings which appear to conflict with those of Nagaraj et al [193].
In summary, there is good evidence that both leak and voltage-gated K+ currents in PASMCs are diminished by hypoxia. The common inhibitory effect of hypoxia on these channels suggests that attenuation of both currents are required for PASMC depolarization during HPV, although it may be that their relative contributions to hypoxia-induced depolarization are species-dependent. The observations of Platoshyn et al [214] provide strong evidence for the notion that channels incorporating KV1.5 channels are predominantly responsible for the PO2-dependent KV current, but the mechanism(s) by which their activity is depressed by hypoxia remain controversial. Nonetheless, each of the three schemes proposed to explain this (cytoplasmic oxidation, cytoplasmic reduction, increased AMP/ATP ratio) incorporate a common O2 sensor: the mitochondria. The mechanism by which hypoxia would inhibit SrcTK to attenuate the TASK-1 current has not been defined, and possible alternative pathways by which hypoxia could depress TASK-1 channel opening have not been identified in PASMCs. Observations that SrcTK in PASMC is stimulated rather than inhibited by hypoxia [264] also appear to conflict with the model proposed by Nagaraj et al [193], although this might reflect species differences.
8. Concluding Remarks: O2 sensing in CBCC versus PASMCs
Although the involvement of K+ channels in O2 sensing in CBCC and PASMCs has been mooted for more than three decades, despite substantial research it is clear that in both cases what we know is far outweighed by what we don’t. To some extent this is because proposed mechanisms have not yet been investigated sufficiently to establish their salience (e.g. the involvement of β-subunits in KV channel inhibition), but often it is difficult to draw firm conclusions because apparently conflicting results have been obtained. It is likely that this has arisen in part because of the use by experimenters of different species, preparations, and experimental conditions. The employment of pharmacological agents with well-known off-target effects (e.g. DPI) has not helped matters.
With regard to K+ channels as effectors in O2 sensing pathways, at this point it seems incontrovertible that depolarization of both CBCC and PASMC by hypoxia is predominantly due to a decrease in the opening probability of K+ channels, although the involvement of non-selective cation channels remains a possibility. In addition, it is widely accepted that both CBCC and PASMC express several types of K+ currents which are inhibited by hypoxia. There is persuasive evidence that channels incorporating the KV1.5α subunit are largely responsible for the voltage-gated K+ channel component of depolarization in PASMCs, at least in mice and rats. In contrast, the identity(ies) of the voltage-gated K+ channel(s) which respond to hypoxia in CBCC are less well characterized and may vary between species; the strongest evidence is for the involvement of KV4.x channels in rabbit. It is also well established that hypoxia also attenuates the activity of BKCa and TASK channels, (most likely TASK1/3 heterodimers) in CBCC, and of TASK-1 channels in PASMC; however, a role for BKCa channels in HPV is unlikely.
As to O2 sensing, there is persuasive evidence that the mitochondria act as O2 sensors linked to K+ channel inhibition in both types of cells. There is an increasing body of evidence [19,150,191] that hypoxia acts on both CBCC and PASMC to cause the reduction of Coenzyme Q10 pool, resulting in an increase in mitochondrial ROS production, probably at complex 3, and that this is enabled by the high expression of the atypical cytochrome C oxidase subunit COX4i2in both types of cell [17,18]. This has been proposed by Pak et al to be a ‘unifying’ mechanism for O2 sensing [266]. However, there is also evidence against this concept for both types of cell [140,175,219], and several other sensor/mediator pathways which could link a decrease in mitochondrial respiration to K+ channel inhibition have been mooted.
In addition, a number of proposals for non-mitochondrial O2 sensors, for example involving the decreased production of CO by HO-2 and resulting effects on BKCa channels mediated through heme and H2S, respectively, have been made for CBCC, and the extent to which these various sensors contribute to the hypoxic response also remains to be established. In the case of PASMC, all of the proposed mechanisms for hypoxia-induced suppression of the KV current incorporate the mitochondria as the O2 sensor; a ‘membrane’ model designed to explain K+ channel inhibition by hypoxia analogous to that proposed for CBCC has never arisen. In contrast, the suppression of TASK-1 channel opening mediated by its the displacement from SrcTK potentially constitutes a mechanism for PASMC depolarization which is independent of the mitochondria, although this remains to be established. Likewise, the relative importance of KV vs TASK channels in causing hypoxic depolarization of these cells is undetermined.
There has been relatively little discussion of the possibility that multiple O2 sensing systems acting via K+ channels may coexist in PASMCs, although Wolin and colleagues have proposed that the response to hypoxia in these cells involves diverse effects on cell redox pathways acting through various effectors such as protein kinase G and Rho kinase [267]. In contrast, it has often been proposed that more than one mechanism of O2 sensing coexists in CBCC, and might be more or less important at different levels of hypoxia [152,167,268] and the recent study by Swiderska et al [158] which presented evidence that increased succinate-dependent mitochondrial ROS production makes a significant but limited contribution to O2 sensing in rat CB represents a promising attempt to begin to establish the relative importance of the component mechanisms mediating the hypoxia response.
Looking ahead, the introduction over the past 10-15 years of greatly improved methods for measuring free pyridine nucleotide concentrations [269] and H2O2 and the redox poise of the GSH/GSSG couple in specific cell compartments [270,271,272], as well for pharmacologically manipulating of mitochondrial ROS production without interfering with respiration [179,273], represents an opportunity for the study of O2 sensing in CBCC and PASMC which currently remains underexploited. The more widespread use of these and other novel experimental approaches will hopefully cause a favorable shift in the balance between what we know and don’t know about acute O2 sensing in the carotid body and the pulmonary circulation over the next decade.
Author Contributions
A.R. and P.I.A. are senior coauthors and have contributed equally at all stages of the manuscript. Both authors have read and accepted the published version of the manuscript.
Funding
Consejería de Educación, Junta de Castilla y León (Spain), grant CCVC848 to IBGM.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lopez-Barneo, J. , Lopez-Lopez, J.R., Urena, J., Gonzalez, C. Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science 1988, 241, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C., Almaraz, L., Obeso, A., Rigual, R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994, 74(4), 829-898. [CrossRef]
- Marshall, B.E.; Marshall, C.; Frasch, F.; Hanson, C.W. (1994). Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. 1. Physiologic concepts. Intensive Care Med 1994, 20, 291-297. [CrossRef]
- Peers, C.; Kemp, P.J. Acute oxygen sensing: diverse but convergent mechanisms in airway and arterial chemoreceptors. Respir Res 2001, 2, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Seidler, F.J. Adrenomedullary catecholamine release in the fetus and newborn: secretory mechanisms and their role in stress and survival. J Dev Physiol. 1988, 10(1),1-16.
- Pflüger, E. Ueber die ursacheder athembewegungen, sowie der dyspnoë und apnoë. Pflugers Arch. Gesamte Physiol. Meschen Tiere. 1868, 1, 61-106.
- Boycott, A.E.; Haldane, J.S. The effects of low atmospheric pressures on respiration. J Physiol 1908, 37, 355–377. [Google Scholar] [CrossRef] [PubMed]
- De Castro, F. Sur la structure et l'innervation du sinus carotidien de l'homme et des mammifères: Nouveaux faits sur l'innervation et la fonction du glomus caroticum. Trab Lab Invest Biol Univ Madrid 1928, 25, 330–380. [Google Scholar]
- Gonzalez, C.; Conde, S.V.; Gallego-Martín, T.; Olea, E.; Gonzalez-Obeso, E.; Ramirez, M.; Yubero, S.; Agapito, M.T.; Gomez-Nino, A.; Obeso, A.; Rigual, R.; Rocher, A. Fernando de Castro and the discovery of the arterial chemoreceptors. Front Neuroanat. 2014, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Anichkov, S.V.; Belen'kii, M.L. Pharmacology of the Carotid Body Chemoreceptors. McMillan, New York, 1963.
- Mills, E.; Jöbsis, F.F. Simultaneous measurement of cytochrome a3 reduction and chemoreceptor afferent activity in the carotid body. Nature 1970, 225, 1147–9. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Jöbsis, F.F. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J Neurophysiol. 1972, 35, 405–28. [Google Scholar] [CrossRef]
- Duchen, M.R.; Biscoe, T.J. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J Physiol. 1992, 450, 13–31. [Google Scholar] [CrossRef]
- Duchen, M.R.; Biscoe, T.J. ; Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J Physiol. 1992, 450, 33–61. [Google Scholar] [CrossRef]
- Lahiri, S.; Rumsey, W.L.; Wilson, D.F.; Iturriaga, R. Contribution of in vivo microvascular PO2 in the cat carotid body chemotransduction. J Appl Physiol (1985), 1993, 75(3), 1035–1043. [Google Scholar] [CrossRef]
- Buckler, K.J.; Turner, P.J. Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J Physiol 2013, 591(14), 3549–63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chien, M.S.; Kaleem, S.; Matsunami, H. Single cell transcriptome analysis of mouse carotid body glomus cells. J Physiol. 2016, 594(15), 4225–4251. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Bonilla-Henao, V.; García-Flores, P.; Arias-Mayenco, I.; Ortega- Sáenz, P.; López-Barneo, J. Gene expression analyses reveal metabolic specifications in acute O2-sensing chemoreceptor cells. J Physiol 2017, 595, 6091–6120. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Domínguez, A.; Ortega-Sáenz, P.; Gao, L.; Colinas, O.; García-Flores, P.; Bonilla-Henao, V.; Aragonés, J.; Hüttemann, M.; Grossman, L.I.; Weissmann, N.; Sommer, N.; López-Barneo, J. Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci Signal. 2020, 13(615), eaay9452. [Google Scholar] [CrossRef] [PubMed]
- Almaraz, L.; Gonzalez, C.; Obeso, A. Effects of high potassium on the release of [3H] dopamine from the cat carotid body in vitro. J Physiol. 1986, 379, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Rocher, A.; Obeso, A.; Herreros, B.; Gonzalez, C. Activation of the release of dopamine in the carotid body by veratridine. Evidence for the presence of voltage-dependent Na+ channels in type I cells. Neurosci Lett 1988, 94, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, J.R.; Gonzalez, C.; Urena, J.; Lopez-Barneo, J. Low pO2 selectively inhibits K channel activity in chemoreceptor cells of the mammalian carotid body. J Gen Physiol 1989, 93, 1001–1015. [Google Scholar] [CrossRef]
- Peers, C. Hypoxic suppression of K+ currents in type I carotid body cells: selective effect on the Ca2(+)-activated K+ current. Neurosci Lett 1990, 119, 253–256. [Google Scholar] [CrossRef]
- Buckler, K.J. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol 1997, 498 (Pt 3), 649–662. [Google Scholar] [CrossRef]
- Buckler, K.J. Background leak K+ currents and oxygen sensing in carotid body type 1 cells. Respir Physiol. 1999, 115(2), 179–87. [Google Scholar] [CrossRef]
- Rocher, A.; Obeso, A.; Cachero, M.T.; Herreros, B.; Gonzalez, C. Participation of Na+ channels in the response of carotid body chemoreceptor cells to hypoxia. Am J Physiol 1994, 267, C738–C744. [Google Scholar] [CrossRef] [PubMed]
- Obeso, A.; Rocher, A.; Fidone, S.; Gonzalez, C. The role of dihydropyridine-sensitive Ca2+ channels in stimulus-evoked catecholamine release from chemoreceptor cells of the carotid body. Neuroscience 1992, 47(2), 463–72. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Vaughan-Jones, R.D. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994, 476(3), 423–8. [Google Scholar] [CrossRef] [PubMed]
- Stea, A.; Nurse C.A. Whole-cell and perforated-patch recordings from O2-sensitive rat carotid body cells grown in short- and long-term culture. Pflugers Arch. 1991, 418(1-2), 93-101. [CrossRef]
- Ureña, J.; Fernández-Chacón, R.; Benot, A.R.; Alvarez de Toledo, G.A.; López-Barneo, J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci U S A 1994, 91(21), 10208–10211. [Google Scholar] [CrossRef]
- Rocher, A.; Geijo-Barrientos, E.; Caceres, A.I.; Rigual, R.; Gonzalez, C.; Almaraz, L. Role of voltage-dependent calcium channels in stimulus-secretion coupling in rabbit carotid body chemoreceptor cells. J Physiol 2005, 562, 407–420. [Google Scholar] [CrossRef]
- Youngson, C.; Nurse, C.; Yeger, H.; Cutz, E. Oxygen sensing in airway chemoreceptors. Nature. 1993, 365(6442), 153–155. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.K.; Archer, S.L. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J 1995, 9(2), 183–189. [Google Scholar] [CrossRef] [PubMed]
- Peers, C.; Buckler, K.J. Transduction of chemostimuli by the type I carotid body cell. J Membr Biol. 1995, 144(1), 1–9. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, M.T.; Lopez-Lopez, J.R. Are Kv channels the essence of O2 sensing? Circ Res. 2000, 86(5), 490–491. [Google Scholar] [CrossRef]
- Patel, A.J.; Honore, E. Molecular physiology of oxygen-sensitive potassium channels. Eur Respir J. 2001, 8(1), 221–227. [Google Scholar] [CrossRef]
- Coppock, E.A.; Martens, J.R.; Tamkun, M.M. Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am J Physiol Lung Cell Mol Physiol. 2001, 281(1), L1–12. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.J. Detecting acute changes in oxygen: will the real sensor please stand up? Exp Physiol. 2006, 91(5), 829–34. [Google Scholar] [CrossRef] [PubMed]
- Peers, C.; Wyatt, C.N.; Evans, A.M. Mechanisms for acute oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2010, 174(3), 292–8. [Google Scholar] [CrossRef] [PubMed]
- Jan, L.Y.; Jan, Y.N. Cloned potassium channels from eukaryotes and procaryotes. Annu Rev Neurosci 1997, 20, 91–123. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, W.A.; Amarillo, Y.; Chiu, J.; Chow, A.; Lau, D.; McCormack, T.; Moreno, H.; Nadal, M.S.; Ozaita, A.; Pountney, D.; Saganich, M.; Vega-Saenz de Miera, E.; Rudy, B. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999, 868, 233–85. [Google Scholar] [CrossRef] [PubMed]
- Lotshaw, D.P. Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem Biophys. 2007, 47(2), 209–256. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Adelman, J.P.; Aiyar, J.; Bayliss, D.A.; Clapham, D.E.; Covarriubias, M.; Desir, G.V.; Furuichi, K.; Ganetzky, B.; Garcia, M.L.; Grissmer, S.; Jan, L.Y.; Karschin, A.; Kim, D.; Kuperschmidt, S.; Kurachi, Y.; Lazdunski, M.; Lesage, F.; Lester, H.A.; McKinnon, D.; Nichols, C.G.; O'Kelly, I.; Robbins, J.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.; Seino, S.; Stuehmer, W.; Tamkun, M.M.; Vandenberg, C.A.; Wei, A.; Wulff, H.; Wymore, R.S. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev. 2003, 55(4), 583-6. [CrossRef]
- Bocksteins, E.; Snyders, D.J. Electrically silent Kv subunits: their molecular and functional characteristics. Physiology (Bethesda), 2012, 27(2), 73-84. [CrossRef]
- Martens, J.R.; Kwak, Y.G.; Tamkun, M.M. Modulation of Kv channel alpha/beta subunit interactions. Trends Cardiovasc Med. 1999, 9(8), 253–258. [Google Scholar] [CrossRef]
- Biggin, P.C.; Roosild, T.; Choe, S. Potassium channel structure: domain by domain. Curr Opin Struct Biol. 2000, 10(4), 456–61. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, M.T.; López-López, J.R.; González, C. Kvbeta1. 2 subunit coexpression in HEK293 cells confers O2 sensitivity to kv4.2 but not to Shaker channels. J Gen Physiol. 1999, 113(6), 897–907. [Google Scholar] [CrossRef]
- Coppock, E.A.; Tamkun, M.M. Differential expression of K(v) channel alpha- and beta-subunits in the bovine pulmonary arterial circulation. Am J Physiol Lung Cell Mol Physiol 2001, 281(6), L1350–1360. [Google Scholar] [CrossRef]
- Ganfornina, M.D.; López-Barneo, J. Gating of O2-sensitive K+ channels of arterial chemoreceptor cells and kinetic modifications induced by low PO2. J Gen Physiol. 1992, 100(3), 427–455. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, J.R.; Gonzalez, C. Time course of K+ current inhibition by low oxygen in chemoreceptor cells of adult rabbit carotid body: effects of carbon monoxide. FEBS Lett. 1992, 299, 251–254. [Google Scholar] [CrossRef]
- Perez-Garcia, M.T.; Lopez-Lopez, J.R.; Riesco, A.M.; Hoppe, U.; Gonzalez, C.; Marban, E.; Johns, D.C. Supression of transient outward K+ currents in chemoreceptor cells of the rabbit carotid body by viral gene transfer of inducible dominant negative Kv4.3 constructs. J Neurosci. 2000, 20, 5689–5695. [Google Scholar] [CrossRef]
- López-López, J.R.; Pérez-García, M.T.; Sanz-Alfayate, G.; Obeso, A.; Gonzalez, C. Functional identification of Kvalpha subunits contributing to the O2-sensitive K+ current in rabbit carotid body chemoreceptor cells. Adv Exp Med Biol. 2003, 536, 33–39. [Google Scholar]
- Sanchez, D.; López-López, J.R.; Pérez-García, M.T.; Sanz-Alfayate, G.; Obeso, A.; Ganfornina, M.D.; Gonzalez, C. Molecular identification of Kvalpha subunits that contribute to the oxygen-sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J Physiol. 2002, 542(Pt 2), 369–382. [Google Scholar] [CrossRef]
- Pérez-García, M.T.; Colinas, O.; Miguel-Velado, E.; Moreno-Domínguez, A.; López-López, J.R. Characterization of the Kv channels of mouse carotid body chemoreceptor cells and their role in oxygen sensing. J Physiol. 2004, 557 Pt 2, 457–471. [Google Scholar] [CrossRef] [PubMed]
- López-López, J.R.; Pérez-García, M.T. Oxygen sensitive Kv channels in the carotid body. Respir Physiol Neurobiol. 2007, 157(1), 65–74. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Yuan, J.X. Hypoxic pulmonary vasoconstriction: role of voltage-gated potassium channels. Respir Res. 2000, 1(1), 40–48. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol Rev 2012, 92(1), 367–520. [Google Scholar] [CrossRef]
- Lopez-Barneo, J.; Benot, A.R.; Urena, J. Oxygen Sensing and the Electrophysiology of Arterial Chemoreceptor Cells. NIPS 1993, 8, 191–195. [Google Scholar] [CrossRef]
- Montoro, R.J.; Urena, J.; Fernandez-Chacon, R.; Alvarez de Toledo, G.; López-Barneo, J. Oxygen sensing by ion channels and chemotransduction in single glomus cells. J. Gen. Physiol 1996, 107, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, D. Activation of voltage-dependent K+ channels strongly limits hypoxia-induced elevation of [Ca2+ ]i in rat carotid body glomus cells. J Physiol. 2018, 596(15), 3119–3136. [Google Scholar] [CrossRef]
- Latorre, R.; Oberhauser, A.; Labarca, P.; Alvarez, O. Varieties of calcium-activated potassium channels. Annu Rev Physiol. 1989, 51, 385–399. [Google Scholar] [CrossRef]
- Vergara, C.; Latorre, R.; Marrion, N.V.; Adelman, J.P. Calcium-activated potassium channels. Curr. Opin. Neurobiol. 1998, 8, 321–329. [Google Scholar] [CrossRef]
- Knaus, H.G.; Eberhart, A.; Glossmann, H.; Munujos, P.; Kaczorowski, G.J.; Garcia, M.L. Pharmacology and structure of high conductance calcium-activated potassium channels. Cell Signal. 1994, 6(8), 861–870. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, G.J.; Knaus, H.G.; Leonard, R.J.; McManus, O.B.; Garcia, M.L. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J Bioenerg Biomembr. 1996, 28(3), 255–267. [Google Scholar] [CrossRef]
- Butler, A.; Tsunoda, S.; McCobb, D.P.; Wei, A.; Salkoff, L. mSlo, a complex mouse gene encoding "maxi" calcium-activated potassium channel. Science 1993, 261, 221–224. [Google Scholar] [CrossRef]
- Schreiber, M.; Salkoff, L. A novel calcium-sensing domain in the BK channel. Biophys J. 1997, 73(3), 1355–63. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.H. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem Biophys. 2005, 42(2), 167–95. [Google Scholar] [CrossRef]
- McManus, O.B.; Helms, L.M.; Pallanck, L.; Ganetzky, B.; Swanson, R.; Leonard, R.J. Functional role of the beta subunit of high conductance calcium-activated potassium channels. Neuron 1995, 14(3), 645–650. [Google Scholar] [CrossRef]
- Wyatt, C.N.; Peers, C. Ca(2+)-activated K+ channels in isolated type I cells of the neonatal rat carotid body. J Physiol. 1995, 483, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, C.N.; Wright, C.; Bee, D.; Peers, C. O2-sensitive K+ currents in carotid body chemoreceptor cells from normoxic and chronically hypoxic rats and their roles in hypoxic chemotransduction. Proc Natl Acad Sci U S A. 1995, 92(1), 295–299. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Ludewig, U.; Garcia-Hirschfeld, J.; Lopez-Barneo, J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci U S A. 2000, 97(5), 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Riesco-Fagundo, A.M.; Pérez-García, M.T.; González, C.; López-López, J.R. O(2) modulates large-conductance Ca(2+)-dependent K(+) channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res 2001, 89(5), 430–436. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Castillo, K.; Carrasquel-Ursulaez, W.; Sepulveda, R.V.; Gonzalez-Nilo, F.; Gonzalez, C.; Alvarez, O. Molecular Determinants of BK Channel Functional Diversity and Functioning. Physiol Rev. 2017, 97(1), 39–87. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Roy, A.; Rozanov, C.; Mokashi, A. K current modulated by PO2 in type I cells in rat carotid body is not a chemosensor. Brain Res. 1998, 794(1), 162–165. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.F. Are oxygen dependent K+ channels essential for carotid body chemo-transduction? Respir Physiol. 1997, 110(2-3), 211-8. [CrossRef]
- Gomez-Niño, A.; Obeso, A.; Baranda, J.A; Santo-Domingo, J.; Lopez-Lopez, J.R.; Gonzalez, C. MaxiK potassium channels in the function of chemoreceptor cells of the rat carotid body. Am J Physiol Cell Physiol. 2009, 297(3), C715–722. [Google Scholar] [CrossRef] [PubMed]
- Hatton, C.J.; Carpenter, E.; Pepper, D.R.; Kumar, P.; Peers, C. Developmental changes in isolated rat type I carotid body cell K+ currents and their modulation by hypoxia. J Physiol. 1997, 501 Pt 1, 49–58. [Google Scholar] [CrossRef]
- Peers, C.; Wyatt, C.N. The role of maxiK channels in carotid body chemotransduction. Respir Physiol Neurobiol. 2007, 157(1), 75–82. [Google Scholar] [CrossRef]
- López-Barneo, J. All for one - O2 -sensitive K+ channels that mediate carotid body activation. J Physiol. 2018, 596(15), 2951–2952. [Google Scholar] [CrossRef]
- Eyzaguirre, C. Chemical and electric transmission in the carotid body chemoreceptor complex. Biol Res. 2005, 38(4), 341–345. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Heinemann, S.H.; Hoshi, T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiology (Bethesda). 2009, 24, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kyle, B.D.; Braun, A.P. The regulation of BK channel activity by pre- and post-translational modifications. Front Physiol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef]
- Jaggar, J.H.; Li, A.; Parfenova, H.; Liu, J.; Umstot, E.S.; Dopico, A.M.; Leffler, C.W. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res. 2005, 97(8), 805–812. [Google Scholar] [CrossRef]
- Williams, S.E.; Wootton, P.; Mason, H.S.; Bould, J.; Iles, D.E.; Riccardi, D.; Peers, C.; Kemp, P.J. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004, 306(5704), 2093–2097. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Dinerman, J.L.; Agani, F.H.; Snyder, S.H. Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci U S A 1995, 92(6), 1994–7. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Sáenz, P.; Pascual, A.; Gómez-Díaz, R.; López-Barneo, J. Acute oxygen sensing in heme oxygenase-2 null mice. J Gen Physiol. 2006, 128(4), 405–11. [Google Scholar] [CrossRef]
- McCartney, C.E.; McClafferty, H.; Huibant, J.M.; Rowan, R.G.; Shipston, M.J.; Rowe, I.C.M. A cysteine-rich motif confers hypoxia sensitivity to mammalian large conductance voltage- and Ca-activated K (BK) channel alpha-subunits. Proc Natl Acad Sci U S A, 2005, 102(49), 17870-17876. [CrossRef]
- Ross, F.A.; Rafferty, J.N.; Dallas, M.L.; Ogunbayo, O.; Ikematsu, N.; McClafferty, H.; Tian, L.; Widmer, H.; Rowe, I.C.; Wyatt, C.N., Shipston MJ, Peers C, Hardie DG, Evans AM. Selective expression in carotid body type I cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by AMP-activated protein kinase. J Biol Chem. 2011, 286(14), 11929-11936. [CrossRef]
- Evans, A.M.; Mustard, K.J.; Wyatt, C.N.; Peers, C.; Dipp, M.; Kumar, P.; Kinnear, N.P.; Hardie, D.G. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? J Biol Chem. 2005, 280(50), 41504–11. [Google Scholar] [CrossRef]
- Evans, A.M.; Hardie, D.G.; Peers, C.; Wyatt, C.N.; Viollet, B.; Kumar, P.; Dallas, M.L.; Ross, F.; Ikematsu, N.; Jordan, H.L.; Barr, B.L.; Rafferty, J.N.; Ogunbayo, O. Ion channel regulation by AMPK: the route of hypoxia-response coupling in thecarotid body and pulmonary artery. Ann N Y Acad Sci. 2009, 1177, 89–100. [Google Scholar] [CrossRef]
- Wyatt, C.N.; Mustard, K.J.; Pearson, S.A.; Dallas, M.L.; Atkinson, L.; Kumar, P.; Peers, C.; Hardie, D.G.; Evans, A.M. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007, 282(11), 8092–8. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.D.; Lewis, S.; Juričić, L.; Udoh, U.A.; Hartmann, S.; Jansen, M.A.; Ogunbayo, O.A.; Puggioni, P.; Holmes, A.P.; Kumar, P.; Navarro-Dorado, J.; Foretz, M.; Viollet, B.; Dutia, M.B.; Marshall, I.; Evans, A.M. AMP-activated Protein Kinase Deficiency Blocks the Hypoxic Ventilatory Response and Thus Precipitates Hypoventilation and Apnea. Am J Respir Crit Care Med. 2016, 193(9), 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, S.; Holmes, A.P.; Dallas, M.L.; Mahmoud, A.D.; Shipston, M.J.; Peers, C.; Hardie, D.G.; Kumar, P.; Evans, A.M. LKB1 is the gatekeeper of carotid body chemosensing and the hypoxic ventilatory response. Commun Biol. 2022, 5(1), 642. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol. 2006, 209 Pt 20, 4011–23. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen sulfide and oxygen sensing: implications in cardiorespiratory control. J Exp Biol. 2008, 211(Pt 17), 2727–34. [Google Scholar] [CrossRef]
- Yuan, G.; Vasavda, C.; Peng, Y.J.; Makarenko, V.V.; Raghuraman, G.; Nanduri, J.; Gadalla, M.M.; Semenza, G.L.; Kumar, G.K. Snyder, S.H.; Prabhakar, N.R. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci Signal. 2015, 8(373), ra37. [CrossRef]
- Peng, Y.J.; Nanduri, J.; Raghuraman, G.; Souvannakitti, D.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci USA. 2010, 107(23), 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sun, B.; Wang, X.; Jin, Z.; Zhou, Y.; Dong, L.; Jiang, L.H.; Rong, W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal. 2010, 12(10), 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Telezhkin, V.; Brazier, S.P.; Cayzac, S.H.; Wilkinson, W.J.; Riccardi, D.; Kemp, P.J. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol. 2010, 172(3), 169–78. [Google Scholar] [CrossRef]
- Kim, D.; Kim, I.; Wang, J.; White, C.; Carroll, J.L. Hydrogen sulfide and hypoxia-induced changes in TASK (K2P3/9) activity and intracellular Ca(2+) concentration in rat carotid body glomus cells. Respir Physiol Neurobiol. 2015, 215, 30–38. [Google Scholar] [CrossRef]
- Wang, J.; Hogan, J.O.; Wang, R.; White, C.; Kim, D. Role of cystathionine-γ-lyase in hypoxia-induced changes in TASK activity, intracellular [Ca2+] and ventilation in mice. Respir Physiol Neurobiol. 2017, 246, 98–106. [Google Scholar] [CrossRef]
- Olson, K.R. Are Reactive Sulfur Species the New Reactive Oxygen Species? Antioxid Redox Signal 2020, 33(16), 1125–1142. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; Nishimura, A.; Morita, M.; Tomizawa, K.; Nishimura, A.; Watanabe, S.; Inaba, K.; Shima, H.; Tanuma, N.; Jung, M.; Fujii, S.; Watanabe, Y.; Ohmuraya, M.; Nagy, P.; Feelisch, M.; Fukuto, J.M.; Motohashi, H. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun. 2017, 8(1), 1177. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. A Case for Hydrogen Sulfide Metabolism as an Oxygen Sensing Mechanism. Antioxidants (Basel). 2021, 10(11), 1650. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012, 463(5), 743–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Coburger, I.; Langner, J.M.; Peter, N.; Hoshi, T.; Schonherr, R.; Heinemann, S.H. Modulation of K(+) channel N-type inactivation by sulfhydration through hydrogen sulfide and polysulfides. Pflugers Arch 2019, 471(4), 557–571. [Google Scholar] [CrossRef] [PubMed]
- Lesage, F.; Lazdunski, M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000, 279(5), F793–F801. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Bang, H.; Kim, D. TBAK-1 and TASK-1, two-pore K(+) channel subunits: kinetic properties and expression in rat heart. Am J Physiol. 1999, 277(5), H1669–H1678. [Google Scholar] [CrossRef]
- Kim, Y.; Bang, H.; Kim, D. TASK-3, a new member of the tandem pore K(+) channel family. J Biol Chem. 2000, 275(13), 9340–9347. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Wischmeyer, E.; Xin Liu, G.; Preisig-Müller, R.; Daut, J.; Karschin, A.; Derst, C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J Biol Chem. 2000, 275(22), 16650–7. [Google Scholar] [CrossRef]
- Ashmole, I.; Goodwin, P.A.; Stanfield, P.R. TASK-5, a novel member of the tandem pore K+ channel family. Pflugers Arch. 2001, 442(6), 828–833. [Google Scholar] [CrossRef]
- Decher, N.; Maier, M.; Dittrich, W.; Gassenhuber, J.; Brüggemann, A.; Busch, A.E.; Steinmeyer, K. Characterization of TASK-4, a novel member of the pH-sensitive, two-pore domain potassium channel family. FEBS Lett. 2001, 492(1-2), 84-9. [CrossRef]
- Medhurst, A.D.; Rennie, G.; Chapman, C.G.; Meadows, H.; Duckworth, M.D.; Kelsell, R.E.; Gloger, I.I.; Pangalos, M.N. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res. 2001, 86(1-2), 101-114. [CrossRef]
- Maingret, F.; Patel, A.J.; Lazdunski, M.; Honoré, E. The endocannabinoid anandamide is a direct and selective blocker of the background K(+) channel TASK-1. EMBO J. 2001, 20(1-2), 47-54. [CrossRef]
- Kindler CH, Yost CS, Gray AT. Local anesthetic inhibition of baseline potassium channels with two pore domains in tandem. Anesthesiology 1999, 90(4), 1092-1102. [CrossRef]
- Lesage, F. Pharmacology of neuronal background potassium channels. Neuropharmacology. 2003, 44(1), 1–7. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Williams, B.A.; Honore, E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000, 525 (Pt 1), 135–42. [Google Scholar] [CrossRef]
- Williams, B.A.; Buckler, K.J. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am J Physiol Lung Cell Mol Physiol. 2004, 286(1), L221–230. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Williams, B.A.; Orozco, R.V.; Wyatt, C.N. The role of TASK-like K+ channels in oxygen sensing in the carotid body. Novartis Found Symp. 2006, 272, 73–94. [Google Scholar] [CrossRef]
- Czirják, G.; Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem. 2002, 277(7), 5426–5432. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Han, J.; Talley, E.M.; Bayliss, D.A.; Kim, D. Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J Physiol. 2004, 554(Pt1), 64–77. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kummer, W.; Atoji, Y.; Suzuki, Y. TASK-1, TASK-2, TASK-3 and TRAAK immunoreactivities in the rat carotid body. Brain Res. 2002, 950(1-2), 304-307. [CrossRef]
- Trapp, S.; Aller, M.I.; Wisden, W.; Gourine, A.V. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J Neurosci. 2008, 28(35), 8844–8850. [Google Scholar] [CrossRef]
- Ortega-Sáenz, P.; Levitsky, K.L.; Marcos-Almaraz, M.T.; Bonilla-Henao, V.; Pascual, A.; López-Barneo, J. Carotid body chemosensory responses in mice deficient of TASK channels. J Gen Physiol. 2010, 135(4), 379–92. [Google Scholar] [CrossRef]
- Varas, R.; Wyatt, C.N.; Buckler, K.J. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J Physiol. 2007, 583(Pt 2), 521–536. [Google Scholar] [CrossRef]
- Buckler, K.J. TASK channels in arterial chemoreceptors and their role in oxygen and acid sensing. Pflugers Arch. 2015, 467(5), 1013–25. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Wang, J.; Hogan, J.O.; Vennekens, R.; Freichel, M.; White, C.; Kim, D. Increase in cytosolic Ca2+ produced by hypoxia and other depolarizing stimuli activates a non-selective cation channel in chemoreceptor cells of rat carotid body. J Physiol. 2014, 592(9), 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M.D.; López-Barneo, J. Single K+ channels in membrane patches of arterial chemoreceptor cells are modulated by O2 tension. Proc Natl Acad Sci U S A. 1991, 88(7), 2927–2930. [Google Scholar] [CrossRef]
- Lewis, A.; Peers, C.; Ashford, M.L.; Kemp, P.J. Hypoxia inhibits human recombinant large conductance, Ca(2+)-activated K(+) (maxi-K) channels by a mechanism which is membrane delimited and Ca(2+) sensitive. J Physiol. 2002, 540 Pt 3, 771–780. [Google Scholar] [CrossRef]
- Gonzalez, C.; Vaquero, L.M.; López-López, J.R.; Pérez-García, M.T. Oxygen-sensitive potassium channels in chemoreceptor cell physiology: making a virtue of necessity. Ann N Y Acad Sci. 2009, 1177, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Acker, H. Cellular oxygen sensors. Ann N Y Acad Sci. 1994, 718, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.R.; Henderson, L.; Jamesm, T.G.; Delpiano, A.; Hentschel, J.; Acker, H. Involvement of an NAD(P)H oxidase as a PO2 sensor protein in the rat carotid body. Biochem J. 1990, 272, 743–747. [Google Scholar] [CrossRef]
- Acker, H.; Bölling, B.; Delpiano, M.A.; Dufau, E.; Görlach, A.; Holtermann, G. The meaning of H2O2 generation in carotid body cells for PO2 chemoreception. J Auton Nerv Syst. 1992, 41(1-2), 41-51. [CrossRef]
- Obeso, A.; Gómez-Niño, A.; Gonzalez, C. NADPH oxidase inhibition does not interfere with low PO2 transduction in rat and rabbit CB chemoreceptor cells. Am J Physiol. 1999; 276(3), C593-601. [CrossRef]
- Hutchinson, D.S.; Csikasz, R.I.; Yamamoto, D.L.; Shabalina, I.G.; Wikstrom, P.; Wilcke, M.; Bengtsson, T. Diphenylene iodonium stimulates glucose uptake in skeletal muscle cells through mitochondrial complex I inhibition and activation of AMP-activated protein kinase. Cell Signal 2007, 19(7), 1610–1620. [Google Scholar] [CrossRef]
- He, L.; Dinger, B.; Sanders, K.; Hoidal, J.; Obeso, A.; Stensaas, L.; Fidone, S.; Gonzalez, C. Effect of p47phox gene deletion on ROS production and oxygen sensing in mouse carotid body chemoreceptor cells. Am J Physiol Lung Cell Mol Physiol. 2005, 289(6), L916–L924. [Google Scholar] [CrossRef]
- Dinger, B.; He, L.; Chen, J.; Liu, X.; Gonzalez, C.; Obeso, A.; Sanders, K.; Hoidal, J.; Stensaas, L.; Fidone, S. The role of NADPH oxidase in carotid body arterial chemoreceptors. Respir Physiol Neurobiol. 2007, 157(1), 45–54. [Google Scholar] [CrossRef]
- Gonzalez, C.; Sanz-Alyayate, G.; Agapito, M.T.; Obeso, A. Effects of reducing agents on glutathione metabolism and the function of carotid body chemoreceptor cells. Biol Chem. 2004, 385(3-4), 265-274. [CrossRef]
- Agapito, M.T.; Sanz-Alfayate, G.; Gomez-Niño, A.; Gonzalez C, Obeso A. General redox environment and carotid body chemoreceptor function. Am J Physiol Cell Physiol, 2009, 296(3), C620-631. [CrossRef]
- Mkrtchian, S.; Kahlin, J.; Ebberyd, A.; Gonzalez, C.; Sanchez, D.; Balbir, A.; Kostuk, E.W.; Shirahata, M.; Fagerlund, M. J.; Eriksson, L.I. The human carotid body transcriptome with focus on oxygen sensing and inflammation--a comparative analysis. J Physiol, 2012, 590(16), 3807-3819. [CrossRef]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 2011, 226(11), 2925–2933. [Google Scholar] [CrossRef] [PubMed]
- Obeso, A.; Almaraz, L.; Gonzalez, C. Effects of cyanide and uncouplers on chemoreceptor activity and ATP content of the cat carotid body. Brain Res. 1989, 481(2), 250–7. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F.; Mokashi, A.; Chugh, D.; Vinogradov, S.; Osanai, S.; Lahiri, S. The primary oxygen sensor of the cat carotid body is cytochrome a3 of the mitochondrial respiratory chain. FEBS Lett. 1994, 351(3), 370–4. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, M.; Iturriaga, R. Carotid body chemosensory excitation induced by nitric oxide: involvement of oxidative metabolism. Respir Physiol Neurobiol. 2002, 131(3), 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Sáenz, P.; Pardal, R.; García-Fernandez, M.; López-Barneo, J. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J Physiol. 2003, 548(Pt 3), 789–800. [Google Scholar] [CrossRef]
- Buckler, K.J.; Vaughan-Jones, R.D. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol 1998, 513 (Pt 3), 819–833. [Google Scholar] [CrossRef]
- Wyatt, C.N.; Buckler, K.J. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J Physiol. 2004, 556(Pt 1), 175–91. [Google Scholar] [CrossRef]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22(5), 825–37. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mayenco, I.; González-Rodríguez, P.; Torres-Torrelo, H.; Gao, L.; Fernández-Agüera, M.C.; Bonilla-Henao, V.; Ortega-Sáenz, P.; López-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28(1), 145–158. [Google Scholar] [CrossRef]
- Chang, A.J.; Ortega, F.E.; Riegler, J.; Madison, D.V.; Krasnow, M.A. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 2015, 527(7577), 240–244. [Google Scholar] [CrossRef]
- Holmes, A.P.; Swiderska, A.; Nathanael, D.; Aldossary, H.S.; Ray, C.J.; Coney, A.M.; Kumar, P. Are Multiple Mitochondrial Related Signalling Pathways Involved in Carotid Body Oxygen Sensing? Fron. Physiol. 2022, 13, 908617. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998, 95(20), 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Duranteau, J.; Chandel, N.S.; Kulisz, A.; Shao, Z.; Schumacker, P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998, 273(19), 11619–11624. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000, 275(33), 25130–25138. [Google Scholar] [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res 2001, 88(12), 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E., Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; Eyassu, F.; Shirley, R.; Hu, C.H.; Dare, A.J.; James, A.M.; Rogatti, S.; Hartley, R.C.; Eaton, S.; Costa, A.S.H.; Brookes, P.S.; Davidson, S.M.; Duchen, M.R.; Saeb-Parsy, K.; Shattock, M.J.; Robinson, A.J.; Work, L.M.; Frezza, C.; Krieg, T.; Murphy, M.P. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature, 2014, 515(7527), 431-435. [CrossRef]
- Swiderska, A.; Coney, A.M.; Alzahran,i A. A.; Aldossary, H.S.; Batis, N.; Ray, C.J.; Kumar, P.; Holmes, A.P. Mitochondrial Succinate Metabolism and Reactive Oxygen Species Are Important but Not Essential for Eliciting Carotid Body and Ventilatory Responses to Hypoxia in the Rat. Antioxidants (Basel). 2021, 10(6), 840. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Sáenz. P.; Lopez-Barneo, J. Physiology of the Carotid Body: From Molecules to Disease. Annu. Rev. Physiol. 2020, 82, 127–149. [CrossRef]
- Peng, Y.J.; Makarenko, V.V.; Gridina, A.; Chupikova, I.; Zhang, X.; Kumar, G.K.; Fox, A.P.; Prabhakar, N.R. H2S mediates carotid body response to hypoxia but not anoxia. Respir Physiol Neurobiol. 2019, 259, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Torres-Torrelo, H.; Ortega-Sáenz, P.; Macías, D.; Omura, M.; Zhou, T.; Matsunami, H.; Johnson, R.S.; Mombaerts, P.; López-Barneo, J. The role of Olfr78 in the breathing circuit of mice. Nature 2018, 561(7724), E33–E40. [Google Scholar] [CrossRef] [PubMed]
- Delpiano, M.A; Acker, H. Extracellular pH changes in the superfused cat carotid body during hypoxia and hypercapnia. Brain Res. 1985, 342(2), 273–280. [Google Scholar] [CrossRef]
- Bernardini, A.; Wolf, A.; Brockmeier, U.; Riffkin, H.; Metzen, E.; Acker-Palmer, A.; Fandrey, J.; Acker, H. Carotid body type I cells engage flavoprotein and Pin1 for oxygen sensing. Am J Physiol Cell Physiol. 2020, 318(4), C719–C731. [Google Scholar] [CrossRef]
- Monti-Bloch, L.; Abudara, V.; Eyzaguirre C. Electrical communication between glomus cells of the rat carotid body.Brain Res. 1993, 622(1-2), 119-31. [CrossRef]
- Peng, Y.J.; Nanduri, J.; Wang, N.; Kumar, G.K.; Bindokas, V.; Paul, B.D.; Chen, X.; Fox, A.P.; Vignane, T.; Filipovic, M.R.; Prabhakar, N.R. Hypoxia sensing requires H2S-dependent persulfidation of olfactory receptor 78. Sci Adv. 2023, 9(27), eadf3026. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.P.; Ray, C.J.; Coney, A.M.; Kumar, P. Is Carotid Body Physiological O2 Sensitivity Determined by a Unique Mitochondrial Phenotype? Front Physiol. 2018, 9, 562. [Google Scholar] [CrossRef]
- Rakoczy, R.J.; Wyatt, C.N. Acute oxygen sensing by the carotid body: a rattlebag of molecular mechanisms. J Physiol. 2018, 596(15), 2969–2976. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Sanz-Alfayate, G.; Agapito, M.T.; Gomez-Niño, A.; Rocher, A.; Obeso, A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir Physiol Neurobiol. 2002, 132(1), 17–41. [Google Scholar] [CrossRef] [PubMed]
- Kilfoil, P.J.; Tipparaju, S.M.; Barski, O.A.; Bhatnagar, A. Regulation of ion channels by pyridine nucleotides. Circ Res. 2013, 112(4), 721–741. [Google Scholar] [CrossRef]
- Otsubo, T.; Kostuk, E.W.; Balbir, A.; Fujii, K.; Shirahata, M. Differential Expression of Large-Conductance Ca-Activated K Channels in the Carotid Body between DBA/2J and A/J Strains of Mice. Front Cell Neurosci. 2011, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Hartness, M.E.; Brazier, S.P.; Peers, C.; Bateson, A.N.; Ashford, M.L.; Kemp, P.J. Post-transcriptional control of human maxiK potassium channel activity and acute oxygen sensitivity by chronic hypoxia. J Biol Chem. 2003, 278(51), 51422–51432. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol (1985) 2000, 88(5):1880-9. [CrossRef]
- Jamieson, D.; Chance, B.; Cadenas, E.; Boveris, A. The relation of free radical production to hyperoxia. Annu Rev Physiol. 1986, 48, 703–719. [Google Scholar] [CrossRef]
- Kumar, P. Sensing hypoxia in the carotid body: from stimulus to response. Essays Biochem. 2007, 43, 43–60. [Google Scholar] [CrossRef]
- Sanz-Alfayate, G.; Obeso, A.; Agapito, M.T.; González, C. Reduced to oxidized glutathione ratios and oxygen sensing in calf and rabbit carotid body chemoreceptor cells. J Physiol. 2001, 537(Pt 1), 209–20. [Google Scholar] [CrossRef]
- Yamamoto, Y.; König, P.; Henrich, M.; Dedio, J.; Kummer, W. Hypoxia induces production of nitric oxide and reactive oxygen species in glomus cells of rat carotid body. Cell Tissue Res. 2006, 325(1), 3–11. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid Redox Signal. 2018, 29(6), 541–551. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol Cells. 2016, 39(1), 65–71. [Google Scholar] [CrossRef] [PubMed]
- Grayson, C.; Mailloux, R.J. J. Coenzyme Q10 and nicotinamide nucleotide transhydrogenase: Sentinels for mitochondrial hydrogen peroxide signaling. Free Radic Biol Med. 2023, 208, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Agapito, M.T.; Rocher, A.; Gomez-Niño, A.; Rigual, R.; Castañeda, J.; Conde, S.V.; Obeso, A. A revisit to O2 sensing and transduction in the carotid body chemoreceptors in the context of reactive oxygen species biology. Respir Physiol Neurobiol. 2010, 174(3), 317–30. [Google Scholar] [CrossRef] [PubMed]
- Papreck, J.R.; Martin, E.A.; Lazzarini, P.; Kang, D.; Kim, D. Modulation of K2P3.1 (TASK-1), K2P9.1 (TASK-3), and TASK-1/3 heteromer by reactive oxygen species. Pflugers Arch. 2012, 464(5), 471-480. [CrossRef]
- Rakoczy, R.J.; Schiebrel, C.M.; Wyatt, C.N. Acute Oxygen-Sensing via Mitochondria-Generated Temperature Transients in Rat Carotid Body Type I Cells. Front Physiol. 2022, 13, 874039. [Google Scholar] [CrossRef] [PubMed]
- Conrad, P.W.; Conforti, L.; Kobayashi, S.; Beitner-Johnson, D.; Rust, R.T.; Yuan, Y.; Kim, H.W.; Kim, R.H.; Seta, K.; Millhorn, D.E. The molecular basis of O2-sensing and hypoxia tolerance in pheochromocytoma cells. Comp Biochem Physiol B Biochem Mol Biol. 2001, 128(2), 187–204. [Google Scholar] [CrossRef] [PubMed]
- Robertson, T. P.; Hague, D.; Aaronson, P.I.; Ward, J.P. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol 2000, 525 (Pt 3), 669–680. [Google Scholar] [CrossRef]
- Weissmann, N.; Dietrich, A; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Mederos-Schnitzler, M.; Ghofrani, H.A.; Schermuly, R.T.; Pinkenburg, O.; Seeger, W.; Grimminger, F.; Gudermann, T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A 2006, 103(50), 19093-19098. [CrossRef]
- Archer, S.L.; Wu, X.C.; Thebaud, B.; Nsair, A.; Bonnet, S.; Tyrrell, B.; McMurtry, M.S.; Hashimoto, K.; Harry, G.; Michelakis, E.D. Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells. Circ Res 2004, 95(3), 308–318. [Google Scholar] [CrossRef]
- Neo, B.H.; Patel, D.; Kandhi, S.; Wolin, M.S. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of protein kinase G1alpha. Am J Physiol Heart Circ Physiol 2013, 305(3), H330–343. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y. M.; Niu, C. F.; Liu, Q.H.; Korde, A.; Ho, Y. S.; Wang, Y. X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 2008, 45(9), 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.S.; Rawat, D.K.; Chettimada, S.; Cioffi, D.L.; Wolin, M.S.; Gerthoffer, W.T.; McMurtry, I.F.; Gupte, S.A. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 2010, 285(25), 19561–19571. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc Res 2009, 82(2), 296–302. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Huttemann, M.; Pak, O;, Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; Jonas, F.; Veith, C.; Aras, S.; Sydykov, A.; Alebrahimdehkordi, N.; Giehl, K.; Hecker, M.; Brandes, R.P.; Seeger, W.; Grimminger, F.; Ghofrani, H.A.; Schermuly, R.T.; Grossman, L.I.;Weissmann, N. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ Res 2017, 121(4), 424-438. [CrossRef]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; Kwapiszewska, G.; Klepetko, W.; Seeger, W.; Olschewski, H. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res 2006, 98(8), 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Tang, B.; Balint, Z.; Wygrecka, M.; Hrzenjak, A.; Kwapiszewska, G.; Stacher, E.; Lindenmann, J.; Weir, E. K.; Olschewski, H.; Olschewski, A. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J 2013, 41(1), 85–95. [Google Scholar] [CrossRef]
- Sedivy, V.; Joshi, S.; Ghaly, Y.; Mizera, R.; Zaloudikova, M.; Brennan, S.; Novotna, J.; Herget, J.; Gurney, A.M. Role of Kv7 channels in responses of the pulmonary circulation to hypoxia. Am J Physiol Lung Cell Mol Physiol 2015, 308(1), L48–57. [Google Scholar] [CrossRef]
- Peng, W.; Karwande, S.V.; Hoidal, J.R.; Farrukh, I.S. Potassium currents in cultured human pulmonary arterial smooth muscle cells. J Appl Physiol (1985) 1996, 80(4), 1187–1196. [Google Scholar] [CrossRef]
- Yuan, X.J. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 1995, 77(2), 370–378. [Google Scholar] [CrossRef]
- Wiener, C.M.; Banta, M.R.; Dowless, M.S.; Flavahan, N.A.; Sylvester, J.T. Mechanisms of hypoxic vasodilation in ferret pulmonary arteries. Am J Physio 1995, 269(3 Pt 1), L351–357. [Google Scholar] [CrossRef]
- Osipenko, O.N.; Evans, A.M.; Gurney, A.M. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold, O2-sensing potassium current. Br J Pharmacol 1997, 120(8), 1461–1470. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Sylvester, J.T.; Sham, J.S. Chronic hypoxia alters effects of endothelin and angiotensin on K+ currents in pulmonary arterial myocytes. Am J Physiol 1999, 277(3), L431–439. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Li, Y.; Tang, B.; Bordag, N.; Guntur, D.; Enyedi, P.; Olschewski, H; Olschewski, A. Potassium Channels in the Transition from Fetal to the Neonatal Pulmonary Circulation. Int J Mol Sci 2022, 23, 9. [Google Scholar] [CrossRef]
- Post, J.M.; Hume, J.R.; Archer, S.L.; Weir, E.K. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol. 1992, 262(Pt1), C882–890. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, I.F.; Davidson, A.B.; Reeves, J.T.; Grover, R.F. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res 1976, 38(2), 99–104. [Google Scholar] [CrossRef] [PubMed]
- Harder, D.R. , Madden, J.A., Dawson, C. A membrane electrical mechanism for hypoxic vasoconstriction of small pulmonary arteries from cat. Chest 1985, 88(4), 233S–235S. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 1993, 264(2 Pt 1), L116–123. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Ionic currents in rat pulmonary and mesenteric arterial myocytes in primary culture and subculture. Am J Physiol 1993, 264(2 Pt 1), L107–115. [Google Scholar] [CrossRef]
- Post, J.; Gelband, C.H.; Hume, J.R. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 1995, 77(1), 131-139. [CrossRef]
- Evans, A.M.; Osipenko, O.N.; Gurney, A.M. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J Physiol 1996, 496 ( Pt 2), 407–420. [Google Scholar] [CrossRef]
- Patel, A.J.; Lazdunski, M.; Honore, E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J 1997, 16(22), 6615–6625. [Google Scholar] [CrossRef]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998, 101(11), 2319–2330. [Google Scholar] [CrossRef]
- Archer, S.L.; London, B.; Hampl, V.; Wu, X.; Nsair, A.; Puttagunta, L.; Hashimoto, K.; Waite, R.E.; Michelakis, E.D. Impairment of hypoxic pulmonary vasoconstriction in mice lacking the voltage-gated potassium channel Kv1.5. FASEB J 2001, 15(10), 1801–1803. [Google Scholar] [CrossRef]
- London, B.; Guo, W.; Pan, X.; Lee, J.S.; Shusterman, V.; Rocco, C.J.; Logothetis, D.A.; Nerbonne, J.M.; Hill, J.A. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res 2001, 88(9), 940–946. [Google Scholar] [CrossRef]
- Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Yu, Y.; Remillard, C.V.; Yuan, J.X. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 2006, 290(3), C907–916. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; Coppock, E.A; Felipe, A.; Martens, J.R.; Tamkun, M.M. Oxygen sensitivity of cloned voltage-gated K(+) channels expressed in the pulmonary vasculature. Circ Res 1999, 85(6), 489–497. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Yu, Y.; Ko, E.A.; Remillard, C.V.; Yuan, J.X. Heterogeneity of hypoxia-mediated decrease in I(K(V)) and increase in [Ca2+](cyt) in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007, 293(2), L402–416. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A. M; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res 2003, 93(10), 957–964. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Sedivy, V.; Hodyc, D.; Herget, J.; Gurney, A. M. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther 2009, 329(1), 368–376. [Google Scholar] [CrossRef]
- Archer, S.L; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 1993, 73(6), 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Firth, A. L.; Gordienko, D.V.; Yuill, K.H.; Smirnov, S.V. Cellular localization of mitochondria contributes to Kv channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am J Physiol Lung Cell Mol Physiol 2009, 296(3), L347–360. [Google Scholar] [CrossRef]
- Dunham-Snary, K.J.; Wu, D.; Potus, F.; Sykes, E.A.; Mewburn, J.D.; Charles, R.L.; Eaton, P.; Sultanian, R. A; Archer, S.L. Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction. Circ Res 2019, 124(12), 1727–1746. [Google Scholar] [CrossRef]
- Olschewski, A.; Hong, Z.; Peterson, D.A.; Nelson, D.P; Porter, V.A.; Weir, E.K. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am J Physiol Lung Cell Mol Physiol 2004, 286(1), L15–22. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Lee, S.H.; Ho, W.K.; Earm, Y.E. Redox agents as a link between hypoxia and the responses of ionic channels in rabbit pulmonary vascular smooth muscle. Exp Physiol 1995, 80(5), 835–842. [Google Scholar] [CrossRef]
- Park, M. K.; Bae, Y.M.; Lee, S.H.; Ho, W.K.; Earm, Y.E. Modulation of voltage-dependent K+ channel by redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflugers Arch 1997, 434(6), 764–771. [Google Scholar] [CrossRef] [PubMed]
- Reeve, H.L.; Weir, E.K.; Nelson, D.P.; Peterson, D.A.; Archer, S.L. Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue. Exp Physiol 1995, 80(5), 825–834. [Google Scholar] [CrossRef] [PubMed]
- Schach, C.; Xu, M.; Platoshyn, O.; Keller, S.H.; Yuan, J.X. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store operated Ca2+ channels. Am J Physiol Lung Cell Mol Physiol 2007, 292(3), L685–698. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.K.; Hong, Z.; Porter, V.A.; Reeve, H.L. Redox signaling in oxygen sensing by vessels. Respir Physiol Neurobiol 2002, 132(1), 121–130. [Google Scholar] [CrossRef]
- Olschewski, A.; Weir, E.K. Redox regulation of ion channels in the pulmonary circulation. Antioxid Redox Signal 2015, 22(6), 465–485. [Google Scholar] [CrossRef]
- Frazziano, G.; Moreno, L.; Moral-Sanz, J.; Menendez, C.; Escolano, L.; Gonzalez, C.; Villamor, E.; Alvarez-Sala, J.L; Cogolludo, A.L.; Perez-Vizcaino, F. (2011). Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J Cell Physiol 2011, 226(10), 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res 2003, 93(7), 656–663. [Google Scholar] [CrossRef]
- Cogolludo, A.; Frazziano, G.; Cobeno, L.; Moreno, L.; Lodi, F.; Villamor, E.; Tamargo, J.; & Perez-Vizcaino, F.; & Perez-Vizcaino, F. Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann NY Acad Sci 2006, 1091, 41–51. [Google Scholar] [CrossRef]
- Moreno, L.; Frazziano, G.; Cogolludo, A,; Cobeno, L. ; Tamargo, J.; Perez-Vizcaino, F. Role of protein kinase Czeta and its adaptor protein p62 in voltage-gated potassium channel modulation in pulmonary arteries. Mol Pharmacol 2007, 72(5), 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Moral-Sanz, J.; Gonzalez, T.; Menendez, C.; David, M.; Moreno, L.; Macias, A.; Cortijo, J.; Valenzuela, C.; Perez-Vizcaino, F.; Cogolludo, A. Ceramide inhibits Kv currents and contributes to TP-receptor-induced vasoconstriction in rat and human pulmonary arteries. Am J Physiol Cell Physiol 2011, 301(1), C186–194. [Google Scholar] [CrossRef] [PubMed]
- Desireddi, J.R.; Farrow, K.N.; Marks, J.D.; Waypa, G.B.; Schumacker, P.T. Hypoxia increases ROS signaling and cytosolic Ca(2+) in pulmonary artery smooth muscle cells of mouse lungs slices. Antioxid Redox Signal 2010, 12(5), 595–602. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. Hypoxic pulmonary vasoconstriction. J Appl Physiol (1985). 2005, 98(1), 390–403. [Google Scholar] [CrossRef] [PubMed]
- Caouette, D.; Dongmo, C.; Berube, J.; Fournier, D.; Daleau, P. Hydrogen peroxide modulates the Kv1.5 channel expressed in a mammalian cell line. Naunyn Schmiedebergs Arch Pharmacol 2003, 368(6), 479–486. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ma, J.H.; Zhang, P.H.; Zou, A.R.; Tu, D.N. Quercetin activates human Kv1.5 channels by a residue I502 in the S6 segment. Clin Exp Pharmacol Physiol 2009, 36(2), 154–161. [Google Scholar] [CrossRef] [PubMed]
- Duprat, F.; Guillemare, E.; Romey, G.; Fink, M.; Lesage, F.; Lazdunski, M.; Honore, E. Susceptibility of cloned K+ channels to reactive oxygen species. Proc Natl Acad Sci U S A 1995, 92(25), 11796–11800. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Noh, H.J.; Sung, D.J.; Kim, J.G.; Kim, J.M.; Ryu, S.Y.; Kang, K.; Kim, B.; Bae, Y.M.; Cho, H. Hydrogen peroxide induces vasorelaxation by enhancing 4-aminopyridine-sensitive Kv currents through S-glutathionylation. Pflugers Arch. 2015, 467(2), 285–97. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, L.K.; Reddie, K. G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O'Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; Carroll, K.S.; Martens, J.R. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ Res 2012, 111(7), 842–853. [Google Scholar] [CrossRef]
- Gamper, N.; Zaika, O.; Li, Y.; Martin, P.; Hernandez, C.C.; Perez, M.R.; Wang, A.Y.; Jaffe, D.B.; Shapiro, M.S. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J 2006, 25(20), 4996–5004. [Google Scholar] [CrossRef]
- Yuan, X.J.; Wang, J.; Juhaszova, M.; Golovina, V.A.; Rubin, L.J. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol 1998, 274(4), L621–635. [Google Scholar] [CrossRef] [PubMed]
- Bahring, R.; Milligan, C.J.; Vardanyan, V.; Engeland, B.; Young, B.A.; Dannenberg, J.; Waldschutz, R.; Edwards, J.P.; Wray, D; Pongs, O. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvbeta subunits. J Biol Chem 2001, 276(25), 22923–22929. [Google Scholar] [CrossRef] [PubMed]
- Dwenger, M.M. ; Raph, S.M.; Reyzer, M.L.; Lisa Manier, M.; Riggs, D.W.; Wohl, Z.B.; Ohanyan, V.; Mack, G.; Pucci, T.; Moore, J.B.; Hill, B.G.; Chilian, W.M.; Caprioli, R.M.; Bhatnagar, A.; Nystoriak, M. A. Pyridine nucleotide redox potential in coronary smooth muscle couples myocardial blood flow to cardiac metabolism. Nat Commun, 2022, 13(1), 2051. [CrossRef]
- Raph, S.M.; Dwenger, M.M.; Hu, X.; Nystoriak, M.A. Basal NAD(H) redox state permits hydrogen peroxide-induced mesenteric artery dilatation. J Physiol 2023, 601(13), 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Remillard, C.V.; Fantozzi, I.; Mandegar, M.; Sison, T.T.; Zhang, S.; Burg, E.; Yuan, J.X. Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2004, 287(1), L226–238. [Google Scholar] [CrossRef]
- Dwenger, M.M.; Raph, S.M.; Baba, S.P.; Moore, J.B.t.; Nystoriak, M.A. Diversification of Potassium Currents in Excitable Cells via Kvbeta Proteins. Cells 2022, 11(14). [CrossRef]
- Michelakis, E.D.; Hampl, V.; Nsair, A.; Wu, X.; Harry, G.; Haromy, A.; Gurtu, R.; Archer, S.L. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res 2002, 90(12), 1307–1315. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Rebeyka, I.; Wu, X.; Nsair, A.; Thebaud, B.; Hashimoto, K.; Dyck, J.R., Haromy, A.; Harry, G.; Barr, A.; Archer, S.L. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res 2002, 91(6), 478-486. [CrossRef]
- Kim, Y.; Lee, S.H.; Ho, W.K. Hydrogen peroxide selectively increases TREK-2 currents via myosin light chain kinases. Front Biosci 2007, 12, 1642–1650. [Google Scholar] [CrossRef]
- Lim, J.B.; Langford, T.F.; Huang, B.K.; Deen, W.M.; Sikes, H.D. A reaction-diffusion model of cytosolic hydrogen peroxide. Free Radic Biol Med. 2016, 90, 85–90. [Google Scholar] [CrossRef]
- Lee, Y.M.; Kim, B.J.; Chun, Y.S.; So, I.; Choi, H.; Kim, M.S.; Park, J.W. NOX4 as an oxygen sensor to regulate TASK-1 activity. Cell Signal. 2006, 18(4), 499–507. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014, 53(31), 5111–20. [Google Scholar] [CrossRef]
- Stuart, J.A.; Fonseca, J.; Moradi, F.; Cunningham, C.; Seliman, B.; Worsfold, C.R.; Dolan, S.; Abando, J; Maddalena, L. A. How Supraphysiological Oxygen Levels in Standard Cell Culture Affect Oxygen-Consuming Reactions. Oxid Med Cell Longev. 2018, 2018, 8238459. [Google Scholar] [CrossRef]
- Lu, W; Wang, J.; Shimoda, L.A.; Sylvester, J.T. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 2008, 295(1), L104-113. [CrossRef]
- Wang, J.; Shimoda, L.A.; Sylvester, J.T. Ca2+ responses of pulmonary arterial myocytes to acute hypoxia require release from ryanodine and inositol trisphosphate receptors in sarcoplasmic reticulum. Am J Physiol Lung Cell Mol Physiol 2012, 303(2), L161–168. [Google Scholar] [CrossRef]
- Chen, T.X.; Xu, X.Y.; Zhao, Z.; Zhao, F.Y.; Gao, Y.M.; Yan, X.H.; Wan, Y. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca(2+) entry into rat pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2017, 312(4), L477–L487. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Yang, X.R.; Cao, Y.N.; Sham, J.S. Hydrogen peroxide induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007, 292(6), L1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.R.; Song, T.; Joseph, L.; Mei, L.; Zheng, Y.M.; & Wang, Y.X. X. Important role of PLC-gamma1 in hypoxic increase in intracellular calcium in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2013, 304(3), L143–151. [Google Scholar] [CrossRef] [PubMed]
- Moral-Sanz, J.; Mahmoud, A.D.; Ross, F.A.; Eldstrom, J.; Fedida, D.; Hardie, D.G.; Evans, A.M. AMP-activated protein kinase inhibits Kv1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J Physiol 2016, 594(17), 4901–4915. [Google Scholar] [CrossRef] [PubMed]
- Moral-Sanz, J.; Lewis, S.A.; MacMillan, S.; Ross, F.A.; Thomson, A.; Viollet, B.; Foretz, M.; Moran, C.; Hardie, D.G.; Evans, A.M. The LKB1-AMPK-alpha1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria. Sci Signal 2018, 11(550). [CrossRef]
- Del Gaudio, F.; Liu, D.; Lendahl, U. Notch signalling in healthy and diseased vasculature. Open Biol 2022, 12(4), 220004. [Google Scholar] [CrossRef]
- Smith, K.A.; Voiriot, G.; Tang, H.; Fraidenburg, D.R.; Song, S.; Yamamura, H.; Yamamura, A.; Guo, Q.; Wan, J.; Pohl, N.M.; Tauseef, M.; Bodmer, R.; Ocorr, K.; Thistlethwaite, P.A.; Haddad, G.G.; Powell, F.L.; Makino, A.; Mehta, D; Yuan, J.X. Notch Activation of Ca(2+) Signaling in the Development of Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Am J Respir Cell Mol Biol 2015, 53(3), 355-367. [CrossRef]
- Jain, P.P.; Hosokawa, S.; Xiong, M.; Babicheva, A.; Zhao, T.; Rodriguez, M.; Rahimi, S; Pourhashemi, K.; Balistrieri, F.; Lai, N.; Malhotra, A.; Shyy, J.Y.; Valdez-Jasso, D.; Thistlethwaite, P.A.; Makino, A.; Yuan, J.X. Revisiting the mechanism of hypoxic pulmonary vasoconstriction using isolated perfused/ventilated mouse lung. Pulm Circ 2020, 10(4), 2045894020956592. [CrossRef]
- Song, S.; Babicheva, A.; Zhao, T.; Ayon, R.J.; Rodriguez, M.; Rahimi, S.; Balistrieri, F.; Harrington, A.; Shyy, J.Y.; Thistlethwaite, P.A.; Makino, A.; Yuan, J.X. Notch enhances Ca(2+) entry by activating calcium-sensing receptors and inhibiting voltage-gated K(+) channels. Am J Physiol Cell Physiol 2020, 318(5), C954–C968. [Google Scholar] [CrossRef]
- Knock, G.A.; Snetkov, V.A.; Shaifta, Y.; Drndarski, S.; Ward, J.P.; Aaronson, P.I. Role of src-family kinases in hypoxic vasoconstriction of rat pulmonary artery. Cardiovasc Res 2008, 80(3), 453–462. [Google Scholar] [CrossRef]
- MacKay, C.E.; Shaifta, Y.; Snetkov, V.V.; Francois, A.A.; Ward, J.P.T.; Knock, G.A. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: Role of Src-family kinases and ARHGEF1. Free Radic Biol Med 2017, 110, 316–331. [Google Scholar] [CrossRef]
- Pak, O.; Nolte, A.; Knoepp, F.; Giordano, L.; Pecina, P.; Huttemann, M.; Grossman, L. I., Weissmann, N.; Sommer, N. Mitochondrial oxygen sensing of acute hypoxia in specialized cells - Is there a unifying mechanism? Biochim Biophys Acta Bioenerg, 2022, 1863(8), 148911. [CrossRef]
- Alruwaili, N.; Kandhi, S.; Sun, D.; Wolin, M. S. (2019, Oct 1). Metabolism and Redox in Pulmonary Vascular Physiology and Pathophysiology. Antioxid Redox Signal 2019, 31(10), 752–769. [Google Scholar] [CrossRef]
- Kemp, P.J.; Telezhkin, V. Oxygen sensing by the carotid body: is it all just rotten eggs? Antioxid Redox Signal. 2014, 20(5), 794–804. [Google Scholar] [CrossRef] [PubMed]
- Bilan, D.S.; Belousov, V.V. New tools for redox biology: From imaging to manipulation. Free Radic Biol Med 2017, 109, 167–188. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, K.A.; Belousov, V.V. Genetically encoded fluorescent redox sensors. Biochim Biophys Acta 2014, 1840(2), 745–756. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol J 2014, 9(2), 282–293. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; Gems, D;, Kagan, V.E.; Kalyanaraman, B.; Larsson, N.G.; Milne, G.L.; Nystrom, T.; Poulsen, H.E.; Radi, R.; Van Remmen, H.; Schumacker, P.T.; Thornalley, P. J.; Toyokuni, S.; Winterbourn, C.C.; Yin, H.; Halliwell, B. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab 2022, 4(6), 651-662. [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016, 100, 14–31. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram summarizing the main proposed mechanisms of oxygen sensing and chemotransduction in the carotid body (CB). According to the “Membrane Model,” several oxygen-sensitive potassium channels (O2 sensitive KvO2, large conductance BKCa, and TASK) close in the CB chemoreceptor cells in response to hypoxia, as indicated by the reduction of channel activity by hypoxia in excised patches. This inhibition leads to membrane depolarization and the opening of voltage gated Ca2+ channels. The ensuing influx of Ca2+ leads to neurotransmitter release (NT) which activates afferent fibers and signal propagation to the respiratory center. Another proposal is that hypoxia inhibits an NADPH oxidase, promoting changes in cytosolic ROS that directly affect K+ channel activity. Additionally, hypoxia could inhibit HO-2, which decreases CO and inhibits protein kinase G causing an increase in CSE activity and elevated local production of H2S, which then inhibits K+ channels. According to the “Mitochondrial Hypothesis”, hypoxia inhibits mitochondrial electron transport leading to activation of multiple signaling pathways: decreased ATP production, increased production of mitochondrial ROS, activation of AMP-activated protein kinase (AMPK), a rise in NADH, and a rise in lactate. Some of these pathways seem to directly (or indirectly) modify K+ channel activity and cause membrane depolarization. Hypoxia further leads to a higher lactate/pH level that potentiates the inhibition of TASK channels through acidosis or Olfr78 stimulation. BKCa, large conductance K channel; TASK, TWIK-related acid-sensitive K channel; Kv, voltage dependent K channel(s). AMPK, AMP-activated kinase; HO-2, heme oxygenase-2; NOX, NADPH oxidase; ROS, reactive oxygen species; Olfr78, olfactory receptor 78; CSE, cystathionine-γ-lyase.
Figure 1.
Schematic diagram summarizing the main proposed mechanisms of oxygen sensing and chemotransduction in the carotid body (CB). According to the “Membrane Model,” several oxygen-sensitive potassium channels (O2 sensitive KvO2, large conductance BKCa, and TASK) close in the CB chemoreceptor cells in response to hypoxia, as indicated by the reduction of channel activity by hypoxia in excised patches. This inhibition leads to membrane depolarization and the opening of voltage gated Ca2+ channels. The ensuing influx of Ca2+ leads to neurotransmitter release (NT) which activates afferent fibers and signal propagation to the respiratory center. Another proposal is that hypoxia inhibits an NADPH oxidase, promoting changes in cytosolic ROS that directly affect K+ channel activity. Additionally, hypoxia could inhibit HO-2, which decreases CO and inhibits protein kinase G causing an increase in CSE activity and elevated local production of H2S, which then inhibits K+ channels. According to the “Mitochondrial Hypothesis”, hypoxia inhibits mitochondrial electron transport leading to activation of multiple signaling pathways: decreased ATP production, increased production of mitochondrial ROS, activation of AMP-activated protein kinase (AMPK), a rise in NADH, and a rise in lactate. Some of these pathways seem to directly (or indirectly) modify K+ channel activity and cause membrane depolarization. Hypoxia further leads to a higher lactate/pH level that potentiates the inhibition of TASK channels through acidosis or Olfr78 stimulation. BKCa, large conductance K channel; TASK, TWIK-related acid-sensitive K channel; Kv, voltage dependent K channel(s). AMPK, AMP-activated kinase; HO-2, heme oxygenase-2; NOX, NADPH oxidase; ROS, reactive oxygen species; Olfr78, olfactory receptor 78; CSE, cystathionine-γ-lyase.
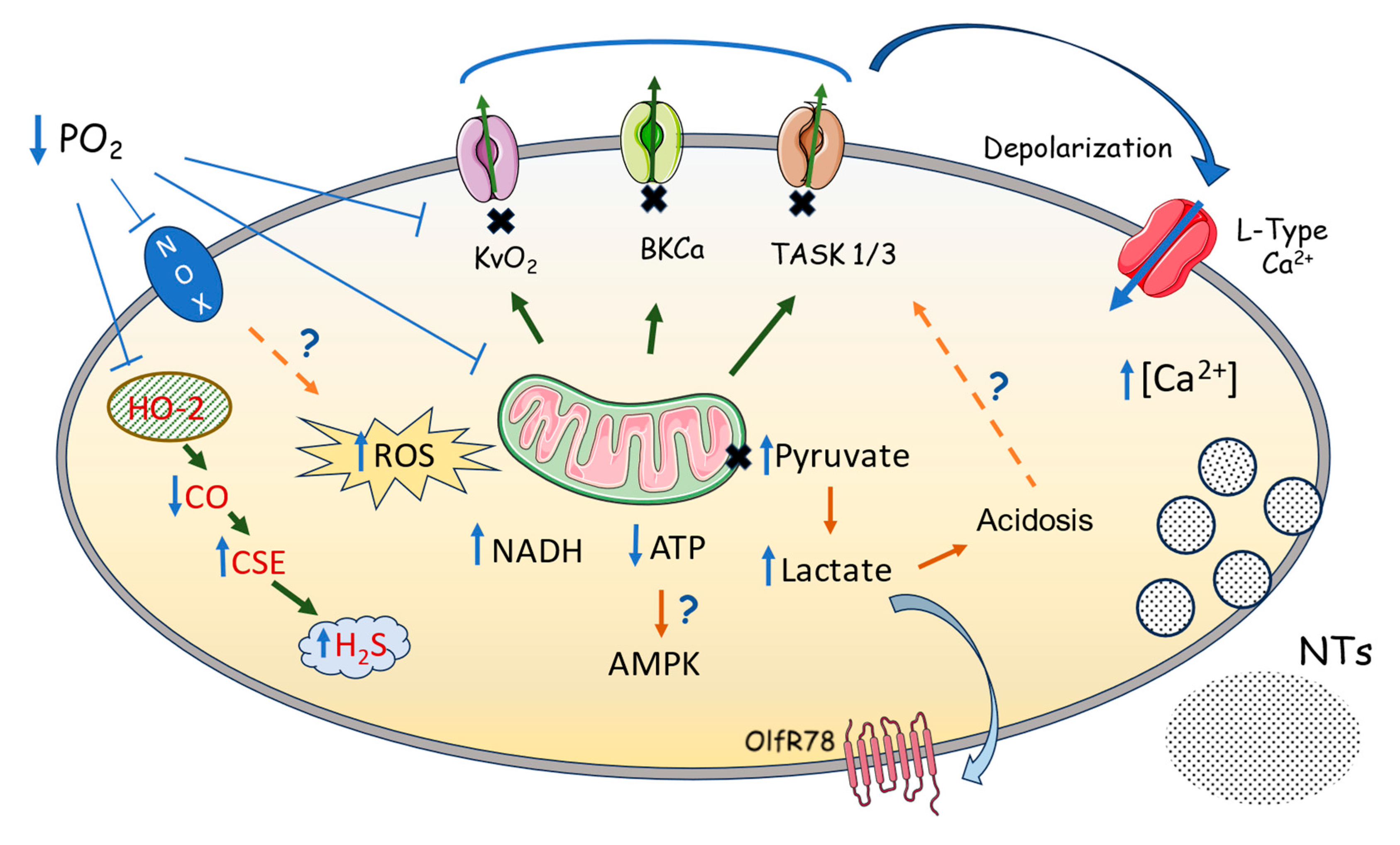
Figure 2.
Flow chart illustrating the proposed involvement of KV channels and other effector mechanisms in O2 sensing during hypoxic pulmonary vasoconstriction. The processes linking hypoxia to pulmonary artery contraction according to the Redox theory are indicated in red, whereas those proposed by, or consistent with, the Mitochondrial ROS theory are shown in blue. Processes shown in black relate to models based on the involvement of AMPK kinase, hydrogen sulfide/RSS, and the pentose phosphate pathway (see references in the text). In the case of the Redox theory, a fall in PO2 causes a fall in mitochondrial ROS production primarily because less O2 is available to form superoxide as it reacts with electrons which are able to 'escape' from the ETC before they reach Complex 4. This occurs during the sojourn of electrons at a dozen or more sites along the ETC, most prominently those within Complexes 1, 2 and 3, pyruvate dehydrogenase and α-ketoglutarate dehydrogenase [179], with the IcoQ site in Complex 1 being proposed by the Redox theory as being of primary importance. According to the Mitochondrial ROS model, hypoxia slows the ETC, effectively increasing the occupancy of electrons at sites within the coenzyme Q pool, from which they flow into the IIICoQ site in Complex 3 to escape and react with O2; superoxide production increases because the increased electron occupancy outweighs the relative lack of O2. Superoxide formed by this process is immediately converted by superoxide dismutase to H2O2, which can access the cytoplasm and directly or indirectly (via intermediates such as peroxiredoxins) cause oxidative modifications of protein thiols. All the effector pathways illustrated in the figure contain proteins which have been shown to be regulated via thiol oxidation, although the extent, nature and importance of such modulation during HPV remains imperfectly understood, and controversial. .
Figure 2.
Flow chart illustrating the proposed involvement of KV channels and other effector mechanisms in O2 sensing during hypoxic pulmonary vasoconstriction. The processes linking hypoxia to pulmonary artery contraction according to the Redox theory are indicated in red, whereas those proposed by, or consistent with, the Mitochondrial ROS theory are shown in blue. Processes shown in black relate to models based on the involvement of AMPK kinase, hydrogen sulfide/RSS, and the pentose phosphate pathway (see references in the text). In the case of the Redox theory, a fall in PO2 causes a fall in mitochondrial ROS production primarily because less O2 is available to form superoxide as it reacts with electrons which are able to 'escape' from the ETC before they reach Complex 4. This occurs during the sojourn of electrons at a dozen or more sites along the ETC, most prominently those within Complexes 1, 2 and 3, pyruvate dehydrogenase and α-ketoglutarate dehydrogenase [179], with the IcoQ site in Complex 1 being proposed by the Redox theory as being of primary importance. According to the Mitochondrial ROS model, hypoxia slows the ETC, effectively increasing the occupancy of electrons at sites within the coenzyme Q pool, from which they flow into the IIICoQ site in Complex 3 to escape and react with O2; superoxide production increases because the increased electron occupancy outweighs the relative lack of O2. Superoxide formed by this process is immediately converted by superoxide dismutase to H2O2, which can access the cytoplasm and directly or indirectly (via intermediates such as peroxiredoxins) cause oxidative modifications of protein thiols. All the effector pathways illustrated in the figure contain proteins which have been shown to be regulated via thiol oxidation, although the extent, nature and importance of such modulation during HPV remains imperfectly understood, and controversial. .
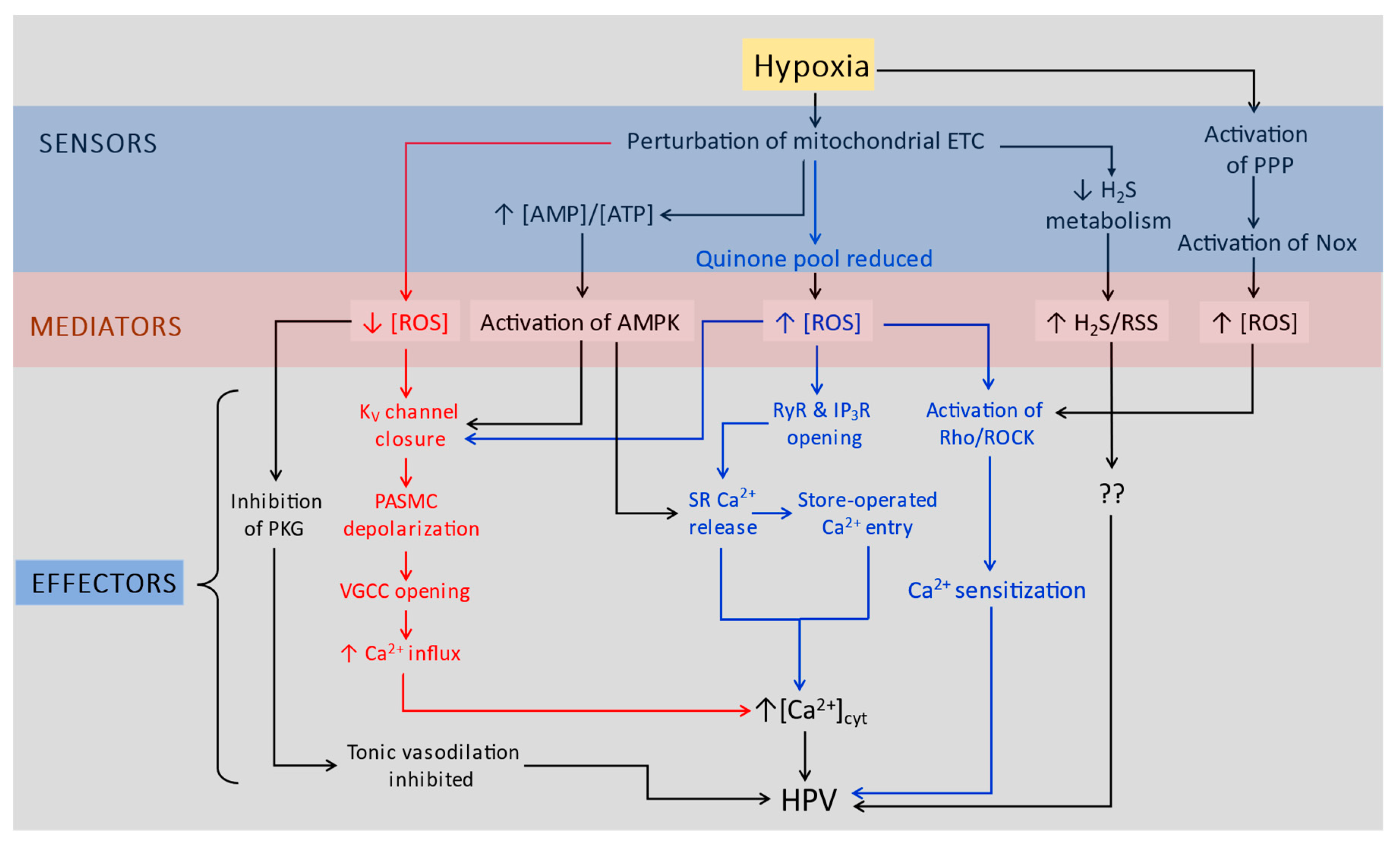
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



