
Submitted:
05 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
In a recent effort to mitigate harm from human pathogens, many biosynthetic pathways are extensively evaluated for their ability to inhibit pathogen growth and determine drug targets. Among those, one of the important products/targets of such pathways is isopentenyl diphosphate, the universal precursor of isoprenoids, which are essential for the normal functioning of microorganisms. In general, two biosynthetic pathways lead to the formation of isopentenyl diphosphate: 1) the mevalonate pathway in animals and 2) the non-mevalonate or methylerythritol phosphate (MEP) in many bacteria, some protozoa, and plants. Because the MEP pathway is not found in mammalian cells, it is considered an attractive target for the development of antimicrobials against a variety of human pathogens, including Mycobacterium tuberculosis (M.tb). In the MEP pathway, 4-diphosphocytidyl-2-c-methyl-d-erythritol kinase (IspE) phosphorylates 4-diphosphocytidyl-2-C-methyl-D-erythritol (CDPME) to form 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate (CDPME2P), followed by cyclization to 2-C-methyl-D-erythritol 2,4-cyclodiphosphate (MECPP) by 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (IspF). A virtual high throughput screening was done by docking IspE protein with commercially available compounds and identified an active heterotricyclic compound. Hence, we designed, synthesized, and tested similar new heterotricyclic compounds. This study will provide the critical insight necessary for the ability to develop novel antimicrobials that target the MEP pathways in pathogens.
Keywords:
pathogens
; IspE inhibitor
; carboline
; synthesis
; minimum inhibitory concentration
1. Introduction
Pulmonary tuberculosis (TB), caused by Mycobacterium tuberculosis (M.tb), is a worldwide health problem with over 2 billion persons infected with latent TB. About 450,000 persons a year are infected with multi-drug-resistant M.tb (MDR-TB), which is resistant to the main first-line drugs isoniazid and rifampicin. Additionally, extensively drug-resistant M.tb (XDR-TB), which is resistant to isoniazid, rifampin, all fluoroquinolones, and at least one of three injectable second-line drugs (i.e., amikacin, kanamycin, or capreomycin), has been reported in 37 countries in all regions of the world since 2006. Human immunodeficiency virus – tuberculosis (HIV-TB) co-infection has contributed in part to the development of this growing pattern of drug resistance. Thus, designing and developing new anti-TB drugs is critical.(1)
The methyl erythritol phosphate (MEP) pathway (Figure 1) has historically been believed to be the pathway whereby microorganisms, including M.tb, synthesize isoprenoids, which are essential for the growth of this and many other prokaryotes. The MEP pathway's absence in humans, coupled with its presence in various human pathogens such as Mycobacterium, Pseudomonas, Klebsiella, Toxoplasma, and Plasmodium species, renders its enzymes attractive targets for the design and development of novel antimicrobials. Hence, intense research worldwide has been spurred by this MEP pathway. Thus, inhibitors of the MEP pathway are being explored as potential anti-infective agents. Most importantly, Targeting IspE would be a better target to determine a critical anti-TB drug. However, recent work(2-5) has identified that parallel and overlapping steps may be present in M.tb MEP pathway. We have also shown that IspC,(6) IspD,(7, 8) IspE,(9, 10) IspF(11) are able to synthesize methyl erythritol cyclo-diphosphate (MECPP)(12) (Figure 1). Recently, our team has conducted a detailed study of the MEP pathway and published the purification and kinetic study of IspC,(13) IspD,(9) IspE(10) and IspF.(11) The second enzyme in the MEP pathway, IspC, catalyzes a two-step reaction, the Mg2+-triggered rearrangement of DXP into a non-isolable aldehyde and NADPH-dependent reduction of the aldehyde to MEP (Figure 1). There is numerous IspC protein crystal structures available in literature from various organisms, including important pathogens such as E. coli, M. tuberculosis, and Y. pestis. Moreover, its active site has been identified as a particularly druggable pocket, indicating that IspC is a potential target for drug development. For example, fosmidomycin was found to exhibit antibacterial activity against a broad-spectrum Gram-negative bacteria, and antimalarial activity against Plasmodium species by targeting IspC. However, the enzyme's significant conformational changes upon ligand binding challenge the development of novel IspC inhibitors.(9, 11, 12, 14).
The third enzyme in the MEP pathway, IspD, serves as a cytidyl transferase, catalyzing the transfer of the cytidyl phosphate group from cytidine triphosphate (CTP) to the phosphate of MEP (Figure 1). This process results in the formation of 4-diphosphocytidyl-2-C-methyl-ᴅ-erythritol (CDP-ME) and inorganic diphosphate. Developing inhibitors that are substrate-competitive for IspD presents a challenge due to its active site being predominantly solvent-exposed and the least lipophilic among all the enzymes within the MEP pathway. Additionally, the existence of a homologous cytidyl transferase in human cells raises concerns about the potential application of discovered inhibitors in therapy. Existing literature on IspD inhibition primarily focuses on herbicides and antimalarials.(9, 11, 12, 14)
The IspD product, CDPME, undergoes ATP-dependent phosphorylation, resulting in the formation of 4-diphosphocytidyl-MEP (CDP-ME2P). This phosphorylation reaction is catalyzed by a cytoplasmic magnesium ion-dependent kinase IspE.(Figure 1) It has been proposed that distinctive features of the IspE active site could facilitate selective targeting. The high conservation of amino acids in the active site provides an opportunity for the design of broad-spectrum antibacterials.(9, 11, 12, 14)
IspF, the fifth enzyme in the MEP pathway, catalyzes the formation of 2-C-methyl-ᴅ-erythritol-2,4 cyclodiphosphate (MEcPP) (Figure 1). The catalytic mechanism of IspF involves the coordination of two metal cations (Zn2+ and Mg2+ or Mn2+), presenting good opportunities for targeted drug design. The Zn2+ ion plays a crucial role by coordinating the phosphate group, thereby enhancing its electrophilicity and facilitating a nucleophilic attack. The intermediate undergoes cyclization, resulting in the formation of MEcPP and cytidine monophosphate (CMP). There are two potential pockets where inhibitors can bind. The first is the active site of IspF, characterized by an unusually high proportion of apolar amino acids. The second is allosteric sites, which opens up another dimension for drug development. The allosteric sites, where larger substrate analog inhibitors can bind to, offer an alternative strategy for inhibiting the activity of IspF. Allosteric inhibition may lead to the development of potent inhibitors with increased specificity.(9, 11, 12, 14).
Earlier, our group has also synthesized enantiomerically pure DXP, MEP,(9) CDPME,(10) CDPME2P(11) and MECPP(12) for their respective protein characterizations, assay development, and to determine inhibitors. Based on this in-depth study, we initiated a study to determine an IspE inhibitor. Recently 6H-1,3 thiazine(15) compounds have been synthesized using nitro acetophenone, chlorobenzaldehyde, and thiourea and reported as novel IspE inhibitors.(16) Thiazine showed the best activity against P. vulgaris (3.1 µg/ml), E. coli (1.5 µg/ml), P. aeruginosa (3.1 µg/ml). However, the synthesized compounds are racemic mixtures, and it was not tested against IspE enzymes, and its toxicity studies were not conducted in detail. Similarly, recently reported IspE inhibitors(17, 18) were not active against mycobacterium, and they followed nine steps to synthesize the final product with low yield, and they were not chiral pure compounds.(19) Hence, the role of MEP in the biosynthesis of isopentenyl diphosphate and its impact on the development of antimicrobials targeting the synthesis of this critical substrate in M.tb and other pathogens is of high potential impact.
2. Materials and Methods
2.1. Materials and Strains
Tetrahydro-beta-carboline was purchased from ACROS (New Jersey, USA). Morpholine-4-sulfonyl chloride was purchased from Oakwood Chemical (Estill, SC, USA). Mycobacterium tuberculosis H37Ra (M.tb) and Mycobacteroides abscessus 19977 (M.ab) were purchased from the American Type Culture Collection (Manassas, VA, USA). Mycobacterium avium (M. avium) was obtained from the Clinical Pathology/Microbiology Laboratory at Nebraska Medicine, Omaha, NE. 7H9 medium was purchased from BD (Sparks, MD). NMR spectroscopies were recorded on a Varian Unity/Inova-500 NB (500 MHz, Varian Medical Systems Inc. Palo Alto, CA, USA). Chemical shifts are reported in parts per million (ppm) downfield from TMS. Molecular weight is determined using MALD/TOF (Applied Biosystems, CA, USA).
2.2. General Synthesis
To the mixture of tetrahydro-beta-carboline (1 eq.) and K2CO3 (3 eq.) in DMF was added sulfonyl chloride (1.5 eq.) at room temperature. KI (0.3 eq.) was added the reaction mixture after 1 hour. The mixture was stirred overnight at room temperature. The mixture was washed with water, extracted with CH2Cl2, and dried over MgSO4. Flash column chromatography was performed to purify the crude products.
4-((1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)sulfonyl)morpholine (1). The reaction was stirred overnight at room temperature. The mixture was washed with water, extracted with CH2Cl2, and dried over MgSO4. Flash column chromatography was performed to purify the crude products.
The residue was purified by column chromatography (silica, ethyl acetate/hexane) to give 1 as a white solid (40%). 1H NMR (500 MHz, CDCl3): δ 8.04 (s, 1H), 7.48 (d, J = 8.0, 1H), 7.31 (d, J = 8.0, 1H), 7.18 (t, J = 8.0, 1H), 7.12 (t, J = 8.0, 1H), 4.51 (s, 2H), 3.70 (t, J = 5.0, 4H), 3.64 (t, J = 5.0, 2H), 3.25 (t, J = 5.0, 4H), 2.87 (t, J = 6.0, 2H). 13C NMR δ:136.1, 128.9, 126.6, 122.0, 119.6, 117.9, 110.9, 108.0, 66.2, 46.2, 44.3, 43.9, 21.4. MALDI TOF m/z (M + Na)+; Calcd. for C15H19N3O3SNa: 344, found: 344.
4-((1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)sulfonyl)benzo[c][1,2,5]thiadiazole (2). The reaction was stirred overnight at room temperature. The mixture was washed with water, extracted with CH2Cl2, and dried over MgSO4. The residue was purified by column chromatography (silica, ethyl acetate/hexane) to give 2 as a white solid (95%). 1H NMR (500 MHz, DMSO-d6): δ 10.7 (s, NH), 8.30 (d, J = 8.5, 1H), 8.29 (d, J = 6.0, 1H), 7.85 (t, J = 7.5, 1H), 7.24 (d, J = 8.0, 1H), 7.20 (d, J = 8.0, 1H), 6.98 (t, J = 8.0, 1H), 6.86 (t, J = 8.0, 1H), 4.71 (s, 2H), 3.68 (t, J = 6.0, 2H), 2.52 (t, J = 5.0, 2H). 13C NMR δ 155.0, 149.0, 135.9, 131.9, 130.8, 130.2, 129.0, 126.6, 126.3, 121.0, 118.6, 117.5, 111.1, 106.1, 44.1, 43.6, 20.8. MALDI TOF m/z (M + Na)+; Calcd. for C17H14N4O2S2Na: 393, found: 393.
2-((4-bromo-2,5-dichlorothiophen-3-yl)sulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (3). The reaction was stirred overnight at room temperature. The mixture was washed with water, extracted with CH2Cl2, and dried over MgSO4.The residue was purified by column chromatography (silica, ethyl acetate/hexane) to give 3 as a white solid (58%). 1H NMR (500 MHz, DMSO-d6): δ 10.89 (s, NH), 7.36 (d, J = 7.5, 1H), 7.29 (d, J = 8.0, 1H), 7.04 (t, J = 7.0, 1H), 6.95 (d, J = 7.5, 1H), 4.60 (s, 2H), 3.71 (t, J = 5.5, 2H), 2.75 (t, J = 5.5, 2H). 13C NMR δ 136.0, 132.9, 132.7, 129.4, 126.4, 125.2, 121.2, 118.8, 117.8, 111.3, 109.6, 106.3, 43.9, 43.3, 21.1. MALDI TOF m/z (M + H)+. Calcd. for C15H12BrCl2N2O2S2: 464, found: 464.
2-(3-(piperidin-1-yl)propyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (4). The reaction was stirred overnight at room temperature. The mixture was washed with water, extracted with CH2Cl2, and dried over MgSO4.The residue was purified by column chromatography (silica, 5-10 % MeOH in CH2Cl2) to give 4 as a yellow oil (67%). 1H NMR (500 MHz, CDCl3); δ 9.37 (s, NH), 7.36 (t, J = 7.5, 2H), 7.08 (t, J = 7,1H), 7.02 (t, J = 7,1H), 3.82 (s, 2H), 2.95 (m, 8H), 2.74 (m, 4H), 2.04 (p, J = 7.0, 2H), 1.42 (br, 4H), 1.24 (br, 2H). 13C NMR δ:136.1, 130.6, 126.6, 121.3, 119.1, 117.7, 111.3, 106.8, 56.3, 54.8, 53.3, 51.4, 50.5, 49.7, 29.6, 22.9, 21.8, 20.9, 20.7. MALDI TOF m/z (M + H)+: Calcd. for C19H28N3: 298, found: 298.
2.3. CDPME Synthesis
To 10 ml of freshly distilled dry tetrahydrofuran (THF) was added tertiary alcohol (0.54 mmol), and the solution was cooled to 0 °C. Potassium hydride (1.08 mmol) was added to the cooled solution, and the mixture was stirred for 30 min at room temperature by slow warming. The reaction mixture was cooled to 0 °C again, and benzyl bromide (1.35 mmol) was added slowly and warmed to room temperature. Finally, the reaction mixture was stirred for an additional 2 hr to complete the protection reaction. The reaction was quenched by the addition of a saturated ammonium chloride solution and extracted with ethyl acetate to get protected product. The crude mixture was purified by silica gel column chromatography using ethyl acetate: hexane (7:3, v/v) as the solvent, yielding 0.486 mmol benzylated product MEP (90% yield). For MEP and CMP coupling, initially, CMP (0.15 mmol) was dissolved in 1 ml acetonitrile, followed by the addition of 0.6 mmol N,N, dimethyl aniline and 0.15 mmol triethylamine at 0 °C. Then, 0.76 mmol trifluoroacetic anhydride in freshly distilled acetonitrile was slowly added to the mixture. The reaction mixture was stirred for 15 min. Excess TFA and anhydride were removed under reduced pressure at low temperature. To the above reaction mixture, a combination of 0.45 mmol 1-methyl imidazole and 0.76 mmol triethylamine in freshly distilled acetonitrile was added slowly and stirred for 30 more min. The activated CMP was added to the mixture of 0.125 mmol MEP and activated 4 A molecular sieves in freshly distilled acetonitrile at 0 °C and stirred for 2 hr. After the reaction was completed, the mixture was extracted with chloroform, and the aqueous layer was lyophilized to give CDP-ME. CDP-ME was purified by sequential chromatography on a Bio-Gel_ P-2 gel fine column using 100 mM aqueous ammonium bicarbonate solution followed by further purification on a benzyl DEAE cellulose anion exchange column, using a gradient of 0 to 0.5 M aqueous ammonium bicarbonate solution, resulting in a 45% yield of CDP-ME.(10)
2.4. Determination of IC50 assay
To determine the IC50 of compound A1, ADP released from the IspE catalyzed reaction was coupled to commercially available the ADP Quest HS Kinase Assay Kit containing pyruvate kinase, pyruvate oxidase, and horseradish peroxidase to produce a fluorescent dye (resorufin). The reaction mixtures containing 50 mM Tris-Cl (pH 7.0), 100 mM ATP, 100 mM CDP-ME, 5 mM phosphatase inhibitor (Roche Bioscience, Palo Alto, CA), 5 mM MgCl2, and 97.2 pmol Rv1011 I in a 50 ml final reaction volume in 96 well black microplates with clear bottom (Costar, NY), were incubated at 37 °C for 30 min. Followed by, 25 ml reagent A and 50 ml reagent B (ADP Quest HS Kinase Assay Kit) were added sequentially. After shaking and incubating for another 15 min at room temperature, fluorescence was measured using a SynergyTM Multi-Detection Microplate Reader (BioTek instruments) at an excitation wavelength of 530 nm and emission wavelength of 590 nm.(10)
2.4.1. Minimum inhibitory concentration (MIC) against M. avium.
96-well plates were seeded with 5 × 105 CFU/mL in a volume of 200 μL of 7H9-OADC medium per well for M.avium. Compounds were dissolved in DMSO and serially diluted as 1:2 dilutions to give the final concentration ranging from 0.25 to 128 μg/mL per well. The final concentration of DMSO in the assay plates did not exceed 2%. For M.avium, the plates were incubated at 37 °C for 4-5 days, and MIC was determined by reading OD600. MICs were determined using a Tecan microplate reader (OD600).(20-23)
2.4.2. Minimum inhibitory concentration (MIC) against M. abscessus.
96-well plates were seeded with 5 × 105 CFU/mL in a volume of 100 μL of 7H9-OADC medium per well for M.ab. Compounds were dissolved in DMSO and serially diluted as 1:2 dilutions to give the final concentration ranging from 0.25 to 128 μg/mL per well. The final concentration of DMSO in the assay plates did not exceed 2%. The plate was incubated at 37 °C for 3 days. MICs were determined using a Tecan microplate reader (OD600).(20-23)
2.4.3. Minimum inhibitory concentration (MIC) against M. tuberculosis H37Ra.
A microdilution method was used to determine MIC. 96-well plates were seeded with 1 × 106 CFU/mL in a volume of 200 μL of 7H9-OADC medium per well for M.tb. Compounds were dissolved in DMSO and serially diluted as 1:2 dilutions to give the final concentration ranging from 0.25 to 128 μg/mL per well. The final concentration of DMSO in the assay plates did not exceed 2%. The plate was incubated at 37 °C for 2 weeks, and MICs were determined using a Tecan microplate reader (OD600) and confirmed by resazurin reduction assay.(20-23)
3. Results
In contemporary drug discovery research, the quest for effective strategies to combat the deleterious impact of human pathogens remains an area of paramount importance. This pursuit involves a comprehensive exploration of various biosynthetic pathways, evaluating their potential to not only inhibit pathogen growth but also serve as viable targets for drug development. At the heart of these investigations lies a critical focus on isopentenyl diphosphate, a key precursor indispensable for the synthesis of isoprenoids—a class of compounds that plays a pivotal role in the normal functioning of microorganisms.
Biosynthetically, two principal pathways contribute to the production of isopentenyl diphosphate: the well-established mevalonate pathway and the non-mevalonate or methylerythritol phosphate (MEP) pathway. The emphasis on isopentenyl diphosphate stems from its central role as a universal building block for isoprenoids, essential for processes such as cell membrane structure, electron transport, and the biosynthesis of various biomolecules.
A focal point of intrigue within these investigations is the MEP pathway, notable for its absence in mammalian cells. This unique feature positions the MEP pathway as an attractive and, importantly, a differentially exploitable target for developing antimicrobials. The absence of the MEP pathway in humans opens a strategic avenue for designing selective inhibitors, presenting a promising prospect for therapeutic intervention against a broad spectrum of pathogens.
The development of inhibitors targeting the MEP pathway is a dynamic and evolving area of research. Researchers are delving into the intricacies of this pathway, focusing on enzymes that play pivotal roles in isopentenyl diphosphate production. By scrutinizing these key enzymes, novel compounds are being systematically explored with the goal of disrupting the MEP pathway's functionality. The rationale behind this approach is to impede the synthesis of essential isoprenoids, thereby compromising the normal functioning of microbial organisms.
Strategic targeting of the MEP pathway represents a promising avenue for the development of next-generation antimicrobials. As researchers unravel the complexities of this pathway, they gain insights that inform the design of inhibitors with enhanced efficacy and specificity. The potential impact of such inhibitors extends beyond conventional drug development paradigms, providing a novel approach to combating infections caused by a diverse array of human pathogens.
The absence of the MEP pathway in human cells not only underscores its appeal as a drug target but also accentuates the selectivity achievable in drug design. This selectivity is a key determinant in minimizing potential side effects on human cells while effectively targeting the microbial pathogens. The unique biochemical features of the MEP pathway, such as its reliance on specific enzymes and the absence of counterparts in mammalian cells, offer a compelling case for its exploitation in drug development.
Reported IspE inhibitors showed good IC50 but less MIC activity.(17, 18, 24) Therefore, determining a new IspE inhibitor and its lead optimization is important with high activity. The lack of MIC in the reported compounds may be because of the presence of MEPP pathway (resistance by novel different pathway approach). We discovered a new lead compound, and lead optimization will be carried out with our novel lead IspE inhibitor. These compounds will be designed to overcome potential resistance to MEP pathway and study of new mechanistic changes in the organisms in the future.(25)
A virtual high throughput screening was done by docking IspE protein against 15 million commercially available compounds in Zinc database. The top ten candidates with high docking scores were bought from commercial sources and tested against M.tb IspE protein in the lab. The most active compound, A1 (Figure 2) showed enzymatic activity, IC50 of 6 µg/ml against M.tb IspE and MIC of 12 µg/ml against M. tb (H37Rv) (Table 1) and was chosen as the lead compound for preliminary optimization. The compound also underwent a docking study with M.tb IspE protein and showed high binding with the carbonyl groups and enzyme. Similarly high affinity was observed with the amine groups and IspE (Figure 3).
Since the active compound from the virtual screening was a tricyclic compound and amine groups, we initiated to synthesize similar tricyclic compounds, carboline derivatives. Four different carboline derivatives were synthesized (Scheme 1). Briefly, carboline was alkylated or sulfated using potassium carbonate and potassium iodide at room temperature for 48 hours with good yield. The synthesized, column-purified compounds were characterized by Nuclear Magnetic Resonance (NMR) and mass spectra (MS).
The synthesized compounds were screened against various bacteria. Compounds 2 and 3 showed good effect (5- 10 µg/mL) against M. avium (Table 2). However, several compounds showed little to no effect against M.tb, and medium effect against Mycobacteroides abscessus (M.ab) (Table 2). These results confirm that optimization of heterotricyclic compounds could lead to an effective IspE inhibitor, and hence, more new compounds related to virtual hit compound A1 or compound 2 have to be synthesized and tested against mycobacterium in the future.
4. Discussion
MEP pathway is an essential pathway in mycobacterium and other bacteria. It is crucial for bacterial viability. Isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) serve as precursors for all isoprenoid compounds. There are two pathways for their biosynthesis. The mevalonate (MVA) pathway is present in eukaryotes, algae, archaea, and some gram-positive bacteria. Gram-negative bacteria, plants, and some other gram-positive bacteria utilize the methyl erythritol phosphate (MEP) pathway. The distinct distribution and orthogonal nature of these pathways make the MEP pathway an appealing target for antibiotics and herbicides.
Enzymes involved in MEP pathways have been characterized, and extensive research have been performed to develop inhibitors that block the MEP pathway.(9, 11, 12, 14) Based on this research, we focused on identifying an IspE inhibitor that possesses 1,3-thiazine moiety, which was designed based on a lead molecule A1 from in-silico screening. Virtual high throughput screening is a powerful computational approach employed in drug discovery to identify potential lead structures. In this method, we screened 15 million chemical compounds using molecular docking tool to accelerates the drug discovery process by simulating the interactions between small molecules and biological target, IspE protein, in Zinc database. The virtual screening process generated a ranked list of compounds that exhibit the most favorable interactions with the target. These top-ranked compounds served as potential lead structures for further experimental validation. We determined MIC and IC50 for hit compounds and identified an active heterotricyclic compound, A1 (Figure 4). The identified compound A1 showed high activity against M.tb and enzymatic activity against M.tb IspE. Thus, virtual screening allowed us to design, synthesize, and test similar new heterotricyclic compounds. We synthesized carboline derivatives that possess similar tricyclic moiety. Four different carboline derivatives were synthesized (Scheme 1). Briefly, carboline was alkylated or sulfated using potassium carbonate and potassium iodide with good yield. The synthesized compounds are highly active against M. avium. Especially, compounds 2 and 3 are active IspE inhibitor against M. avium and has the potential to undergo lead optimization and in vivo study to determine a promising drug-like compound.
5. Conclusion
In summary, the quest to combat human pathogens has entered an era where precision and strategic innovation play pivotal roles. The MEP pathway, with its absence in mammalian cells and its central role in isoprenoid biosynthesis, stands out as a promising target for the development of antimicrobials. As research endeavors continue to unfold the intricacies of this pathway, novel inhibitors hold the potential to revolutionize our approach to infectious diseases, offering hope for enhanced therapeutic interventions against a wide spectrum of microbial pathogens. This study provides the critical insight necessary for the ability to develop IspE inhibitors that target the MEP pathway in mycobacteria. Strategic targeting of the MEP pathway represents a promising avenue for the development of next-generation antimicrobials, with enhanced efficacy and specificity. Moreover, virtual screening process enables the exploration of a broader chemical space, considering a vast array of compounds that may not be feasible for experimental screening. This diversity increases the likelihood of discovering novel lead structures with therapeutic potential.
Author Contributions
Conceptualization, P.N.; methodology, S.C.; software, S.C.; validation, P.N.; formal analysis, P.N.; investigation, P.N.; resources, P.N.; data curation, P.N.; writing—original draft preparation, P.N.; writing—review and editing, P.N. and S.C.; supervision, P.N.; project administration, P.N.; funding acquisition, P.N. All authors have read and agreed to the published version of the manuscript.
Funding
We would like to thank Nebraska Research Initiative Faculty Award to P.N. for funding.
Acknowledgments
We would like to thank Dr. Dean Crick and Dr. Hyungjin Eoh from Colorado State University for determining IC50 for the compound A1.
Competing financial interests
The authors declare no competing financial interests.
References
- Brennan PJ, Crick DC. 2007. The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr Top Med Chem 7:475-88. [CrossRef]
- Hunter WN, Bond CS, Gabrielsen M, Kemp LE. 2003. Structure and reactivity in the non-mevalonate pathway of isoprenoid biosynthesis. Biochem Soc Trans 31:537-42. [CrossRef]
- Rodriguez-Concepcion, M. 2004. The MEP pathway: a new target for the development of herbicides, antibiotics and antimalarial drugs. Curr Pharm Des 10:2391-400. [CrossRef]
- Koppisch AT, Fox DT, Blagg BS, Poulter CD. 2002. E. coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry 41:236-43. [CrossRef]
- Rekittke I, Wiesner J, Rohrich R, Demmer U, Warkentin E, Xu W, Troschke K, Hintz M, No JH, Duin EC, Oldfield E, Jomaa H, Ermler U. 2008. Structure of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate reductase, the terminal enzyme of the non-mevalonate pathway. J Am Chem Soc 130:17206-7. [CrossRef]
- Steinbacher S, Kaiser J, Eisenreich W, Huber R, Bacher A, Rohdich F. 2003. Structural basis of fosmidomycin action revealed by the complex with 2-C-methyl-D-erythritol 4-phosphate synthase (IspC). Implications for the catalytic mechanism and anti-malaria drug development. J Biol Chem 278:18401-7. [CrossRef]
- Bjorkelid C, Bergfors T, Henriksson LM, Stern AL, Unge T, Mowbray SL, Jones TA. 2011. Structural and functional studies of mycobacterial IspD enzymes. Acta Crystallogr D Biol Crystallogr 67:403-14. [CrossRef]
- Gao P, Yang Y, Xiao C, Liu Y, Gan M, Guan Y, Hao X, Meng J, Zhou S, Chen X, Cui J. 2012. Identification and validation of a novel lead compound targeting 4-diphosphocytidyl-2-C-methylerythritol synthetase (IspD) of mycobacteria. Eur J Pharmacol 694:45-52. [CrossRef]
- Narayanasamy P, Eoh H, Crick DC. 2008. Chemoenzymatic synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol: A substrate for IspE. Tetrahedron Lett 49:4461-4463. [CrossRef]
- Eoh H, Narayanasamy P, Brown AC, Parish T, Brennan PJ, Crick DC. 2009. Expression and characterization of soluble 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase from bacterial pathogens. Chem Biol 16:1230-9. [CrossRef]
- Narayanasamy P, Eoh H, Brennan PJ, Crick DC. 2010. Synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate and kinetic studies of Mycobacterium tuberculosis IspF. Chem Biol 17:117-22. [CrossRef]
- Narayanasamy P, Crick DC. 2008. Enantiomeric Synthesis of 2-C-Methyl-D-Erythritol 2, 4-Cyclodiphosphate. Heterocycles 76:243-247. [CrossRef]
- Eoh H, Brennan PJ, Crick DC. 2009. The Mycobacterium tuberculosis MEP (2C-methyl-d-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis (Edinb) 89:1-11. [CrossRef]
- Narayanasamy, P. 2015. MEP pathway: A novel Pathway for New antibiotics. Chem Sci J 6. [CrossRef]
- Singh UP, Pathak M, Dubey V, Bhat HR, Gahtori P, Singh RK. 2012. Design, synthesis, antibacterial activity, and molecular docking studies of novel hybrid 1,3-thiazine-1,3,5-triazine derivatives as potential bacterial translation inhibitor. Chem Biol Drug Des 80:572-83. [CrossRef]
- Udaya Pratap Singh, Hans Raj Bhat, Singh RK. 2013. Ceric ammonium nitrate (CAN) catalysed expeditious one-pot synthesis of 1,3-thiazine as IspE kinase inhibitor of Gram-negative bacteria using polyethylene glycol (PEG-400) as an efficient recyclable reaction medium. Comptes Rendus Chimie 16:462-8. [CrossRef]
- Hirsch AK, Alphey MS, Lauw S, Seet M, Barandun L, Eisenreich W, Rohdich F, Hunter WN, Bacher A, Diederich F. 2008. Inhibitors of the kinase IspE: structure-activity relationships and co-crystal structure analysis. Org Biomol Chem 6:2719-30. [CrossRef]
- Hirsch AK, Lauw S, Gersbach P, Schweizer WB, Rohdich F, Eisenreich W, Bacher A, Diederich F. 2007. Nonphosphate inhibitors of IspE protein, a kinase in the non-mevalonate pathway for isoprenoid biosynthesis and a potential target for antimalarial therapy. ChemMedChem 2:806-10. [CrossRef]
- Masini T, Hirsch AK. 2014. Development of inhibitors of the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents. J Med Chem 57:9740-63. [CrossRef]
- Choi SR, Britigan BE, Moran DM, Narayanasamy P. 2017. Gallium nanoparticles facilitate phagosome maturation and inhibit growth of virulent Mycobacterium tuberculosis in macrophages. PLoS ONE 12:e0177987. [CrossRef]
- Choi SR, Britigan BE, Switzer B, Hoke T, Moran D, Narayanasamy P. 2018. In Vitro Efficacy of Free and Nanoparticle Formulations of Gallium(III) meso-Tetraphenylporphyrine against Mycobacterium avium and Mycobacterium abscessus and Gallium Biodistribution in Mice. Mol Pharm 15:1215-1225. [CrossRef]
- Choi SR, Switzer B, Britigan BE, Narayanasamy P. 2020. Gallium Porphyrin and Gallium Nitrate Synergistically Inhibit Mycobacterial Species by Targeting Different Aspects of Iron/Heme Metabolism. ACS Infect Dis 6:2582-2591. [CrossRef]
- Choi SR, Talmon GA, Britigan BE, Narayanasamy P. 2021. Nanoparticulate beta-Cyclodextrin with Gallium Tetraphenylporphyrin Demonstrates in Vitro and in Vivo Antimicrobial Efficacy against Mycobacteroides abscessus and Mycobacterium avium ACS Infect Dis. [CrossRef]
- Tidten-Luksch N, Grimaldi R, Torrie LS, Frearson JA, Hunter WN, Brenk R. 2012. IspE inhibitors identified by a combination of in silico and in vitro high-throughput screening. PLoS One 7:e35792. [CrossRef]
- Zhao X, Vainshtein I, Gellibolian R, Shu Y, Dotimas H, Wang XM, Fung P, Horecka J, Bosano BL, Eglen RM. 2003. Homogeneous assays for cellular protein degradation using beta-galactosidase complementation: NF-kappaB/IkappaB pathway signaling. Assay Drug Dev Technol 1:823-33. [CrossRef]
Figure 1.
MEP pathway.
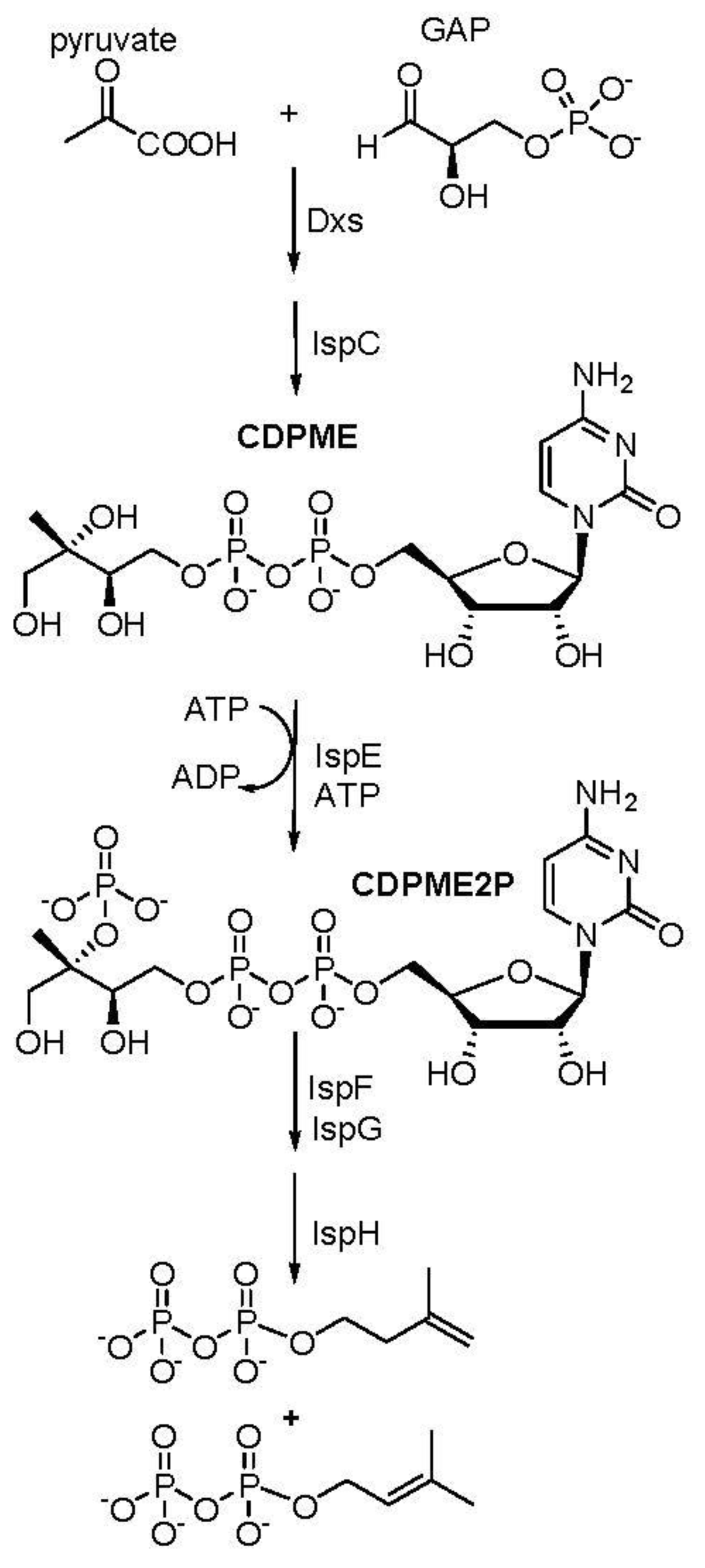
Figure 2.
Lead IspE inhibitor, A1.
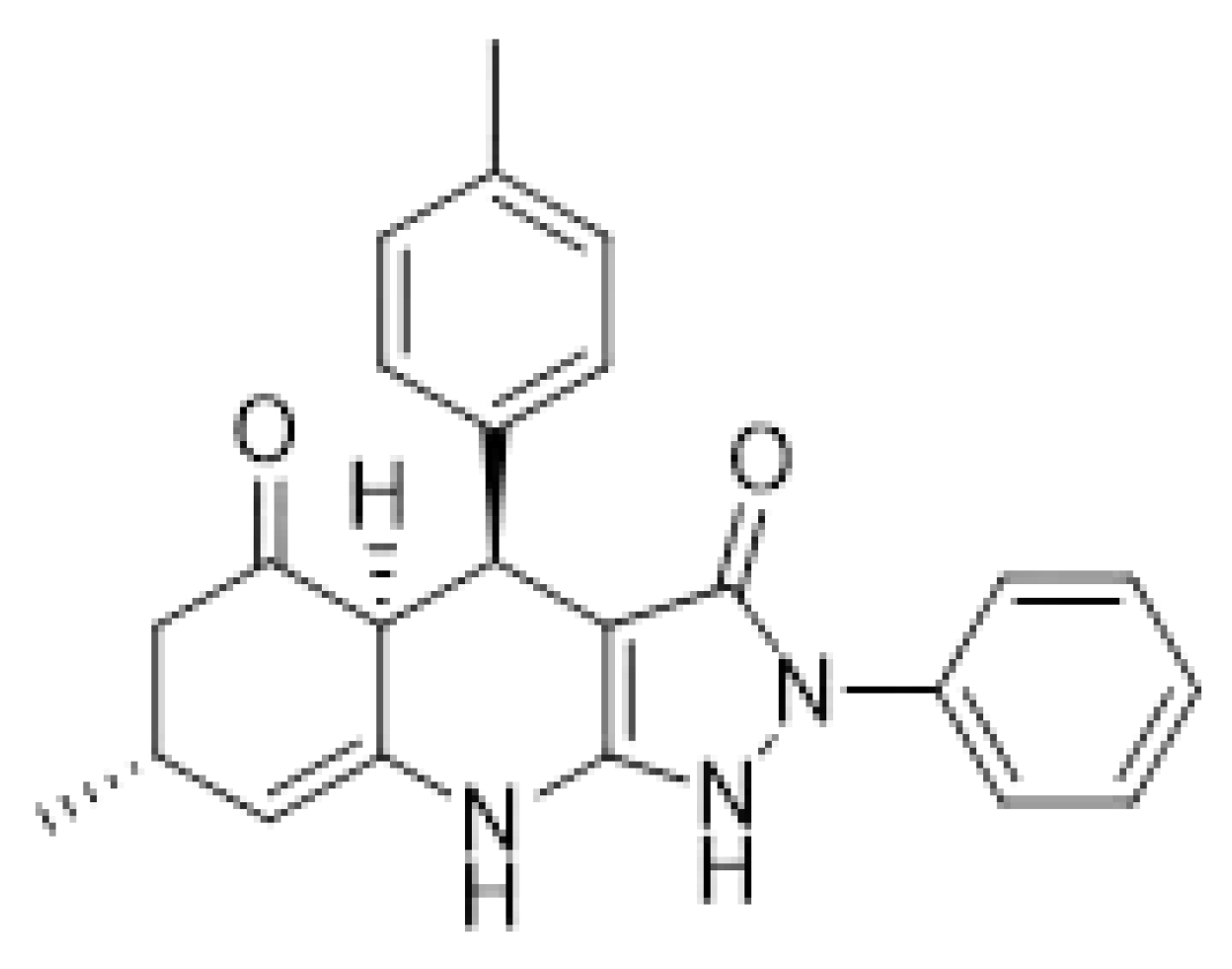
Figure 3.
Autodocking of A1 with M.tb IspE. The docking was predicted using SwissDock. (21, 22) IspE (PDB: 3PYG).
Figure 3.
Autodocking of A1 with M.tb IspE. The docking was predicted using SwissDock. (21, 22) IspE (PDB: 3PYG).
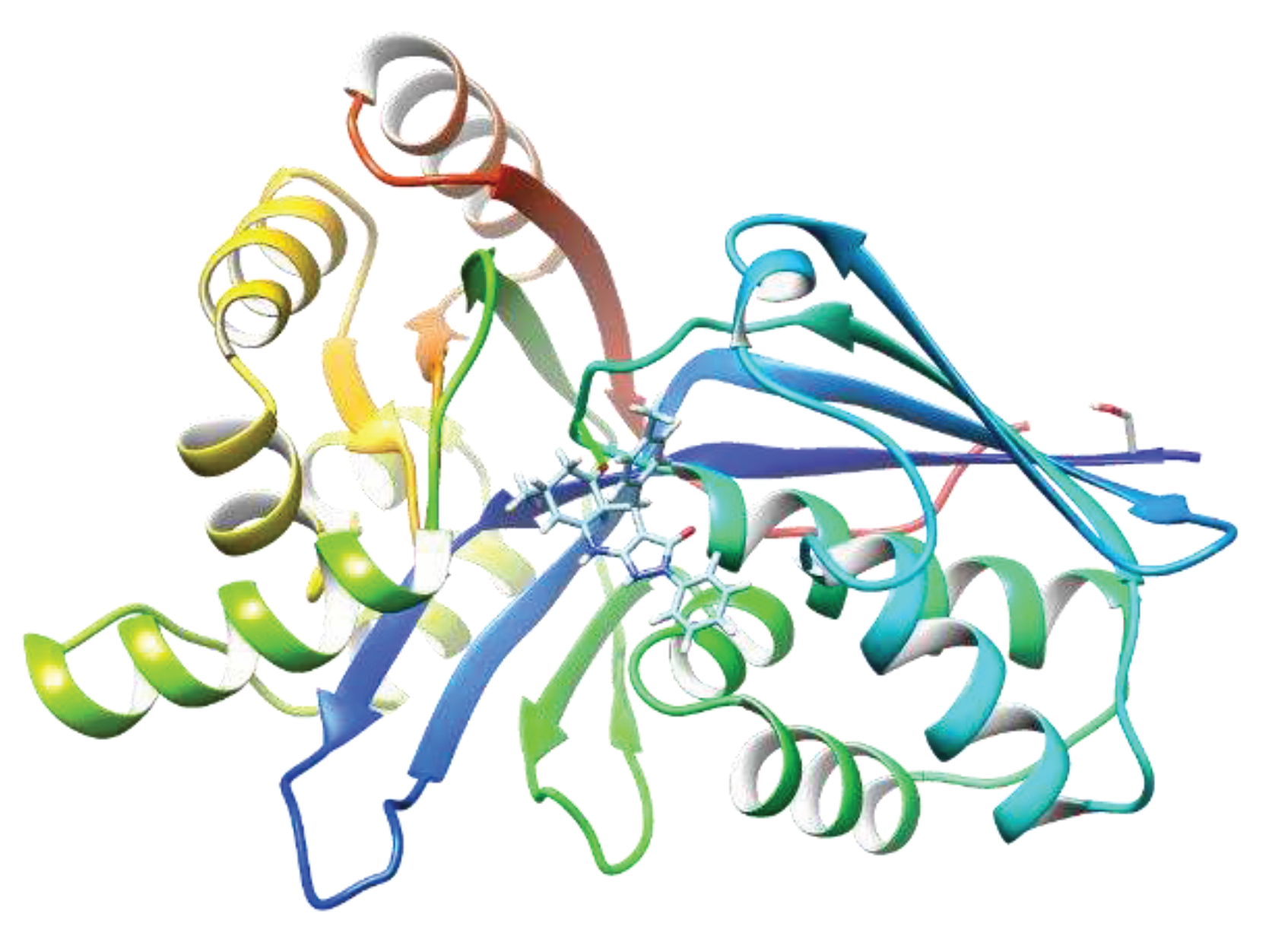
Scheme 1.
Synthesis of carboline derivatives designed from A1 compound.
Figure 4.
Scheme for IspE targeted drug discovery: Hit molecules were obtained from in-silico docking study with IspE protein and 15 million commercially available compounds. Virtual screening with experimental determination of inhibitory activity led to the discovery of compound A1 as a lead structure against mycobacteria, including M. avium, M. abscessus, and M. tuberculosis.
Figure 4.
Scheme for IspE targeted drug discovery: Hit molecules were obtained from in-silico docking study with IspE protein and 15 million commercially available compounds. Virtual screening with experimental determination of inhibitory activity led to the discovery of compound A1 as a lead structure against mycobacteria, including M. avium, M. abscessus, and M. tuberculosis.
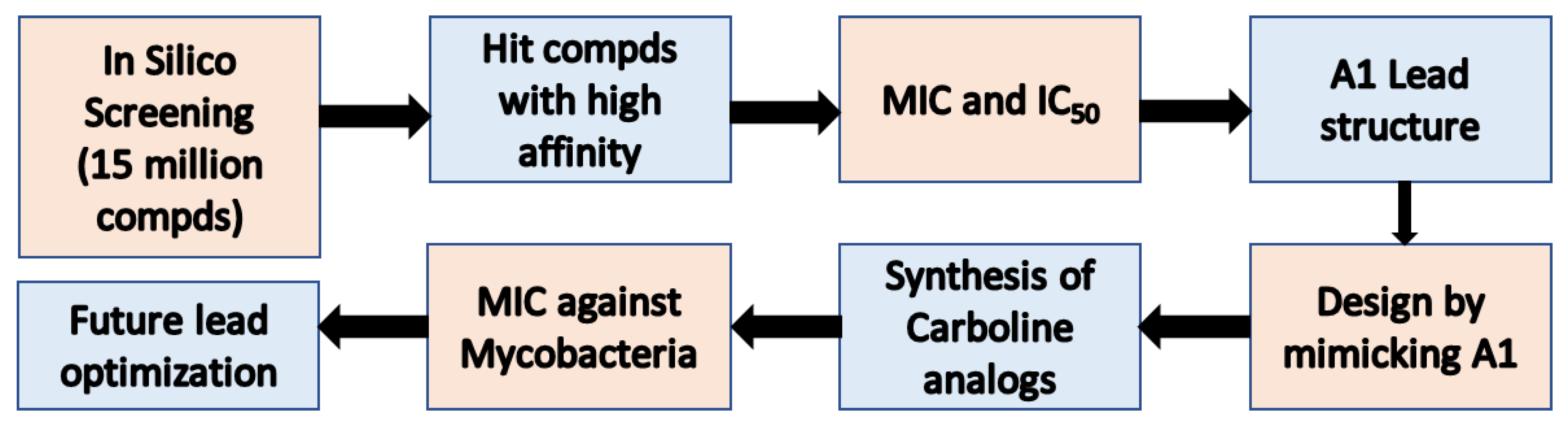

Table 1.
The minimum inhibitory concentration (MIC) against bacteria and Enzymatic activity (IC50) against IspE for lead compound A1.
Table 1.
The minimum inhibitory concentration (MIC) against bacteria and Enzymatic activity (IC50) against IspE for lead compound A1.
| MIC (µg/mL) | IC50 | ||
|---|---|---|---|
| Compound | M.tb | M.tb IspE | |
| A1 | 12 | 6 | |
M.tb–Mycobacterium tuberculosis (H37Rv).
Table 2.
The minimum inhibitory concentration of synthesized carboline derivatives against mycobacteria.
Table 2.
The minimum inhibitory concentration of synthesized carboline derivatives against mycobacteria.
| MIC (µg/mL) | |||
|---|---|---|---|
| Compound | M.avium | M.tb | M.ab |
| 1 | 80 | 128 | 32 |
| 2 | 5 | >128 | 64 |
| 3 | 10 | >128 | 32 |
| 4 | nd | >128 | nd |
Mycobacterium avium (M. avium), Mycobacteroides abscessus (M.ab), Mycobacterium tuberculosis H37Ra (M.tb). MIC was determined using a broth dilution method. nd: not determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



