
Submitted:
06 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
‘Inner mitochondrial membrane peptidase 2 like’ (IMMP2L) is a nuclear encoded mitochondrial peptidase that has been conserved through evolutionary history as has its target enzyme ‘mitochondrial glycerol phosphate dehydrogenase 2’ (GPD2). IMMP2L is known to cleave the mitochondrial transit peptide from GPD2 and another nuclear encoded mitochondrial respiratory related protein cytochrome C1 (CYC1). However, it is not known whether IMMP2L peptidase activates or alters the activity or respiratory related functions of GPD2 or CYC1. Previous investigations found compelling evidence of behavioural change in the Immp2lKD-/- KO mouse and in this study EchoMRI analysis found that many organs of the Immp2lKD-/- KO mouse were smaller and that the KO mouse had significantly less lean mass and overall body weight compared with wildtype littermates (p<0.05). Moreover, all of the organs analysed from the Immp2lKD-/- KO had lower relative levels of mitochondrial reactive oxygen species (mitoROS). The kidneys of the Immp2lKD-/- KO mouse displayed the greatest decrease in mitoROS levels that were over 50% less compared with wildtype litter mates. Mitochondrial respiration was also lower in the kidney of the Immp2lKD-/- KO mouse compared with other tissues when using succinate as the respiratory substrate, whereas respiration was similar to wildtype when glutamate was used as substrate. When glycerol-3-phosphate (G3P) was used as the substrate for Gpd2 we observed ~20% and ~7% respective decreases in respiration in female and male Immp2lKD-/- KO mice over time. Together these findings suggest that the respiratory related functions of mGpd2 and Cyc1 have been compromised to different degrees in different tissues in the Immp2lKD-/- KO mouse. Structural analyses using AlphaFold2-Multimer further predicted that the interaction between Cyc1 and mitochondrial encoded cytochrome b (Cyb) in Complex III had been altered as had the homodimeric structure of the mGpd2 enzyme within the inner mitochondrial membrane of the Immp2lKD-/- KO mouse. mGpd2 functions as an integral component of the glycerol phosphate shuttle (GPS) which positively regulates both mitochondrial respiration and glycolysis. Interestingly, we found that nonmitochondrial respiration (NMR) was also dramatically lowered in the Immp2lKD-/- KO mouse. Primary mouse embryonic fibroblast (MEF) cell lines derived from the Immp2lKD-/- KO mouse displayed a ~27% decrease in total respiration comprised of a ~50% decrease in NMR and a ~12% decrease in total mitochondrial respiration where the latter was consistent with the decreases in substrate specific mitochondrial respiration reported here. This study is the first to report the role of Immp2l in enhancing Gpd2 structure and function, mitochondrial respiration, nonmitochondrial respiration, organ size and homeostasis.
Keywords:
enzyme structure
; mitochondrial dynamics
; mitochondrial size
; NAD+ biosynthesis
; organ mass
; behaviour
; autism
; mitochondrial respiration
; non-mitochondrial respiration
; lean body mass
; glutathione
1. Introduction
IMMP2L and GPD2 have been conserved throughout evolutionary history from E coli, yeast, mouse and human [1, 2]. IMMP2L is a nuclear encoded mitochondrial peptidase that cleaves the signal transit peptides from other nuclear encoded mitochondrial proteins including two respiratory related proteins, cytochrome c1 (CYC1) and mitochondrial glycerol phosphate dehydrogenase 2 (GPD2) (Figure 1). CYC1 is an integral component of the mitochondrial respiratory (electron transport) chain (ETC) and GPD2 forms an integral component of the glycerol phosphate shuttle (GPS) that transfers electrons from the NADH generated in glycolysis to the mitochondrial ETC to mediate mitochondrial respiration, generate ATP and regenerate NAD+ for another round of glycolysis (Figure 1) [3]. However, prior to this study it was not known whether IMMP2L had any effect on the function and respiratory related roles of CYC1 or GPD2 within the mitochondria.
Prior investigations of a mouse model expressing a truncated form of Immp2l returned conflicting results with respect to Immp2l’s role in cellular respiration [4-7]. These discrepancies were possibly due to gain-of-function interference effects in that prior mouse model arising from the formation of heterodimers between Immp1l and the truncated form of the Immp2l enzyme [3]. While other investigators had erroneously assumed that loss of Immp2L peptidase activity would result in the complete loss of Gpd2 enzyme activity [8]. The present study represents the first respiratory analysis of Immp2l using a ‘clean’ Immp2l KO mouse model (Immp2lKD-/- KO mouse) that is devoid of any Immp2l peptidase activity, as confirmed by its failure to cleave either Cyc1 and Gpd2 [3].
Of great interest in the present study was the finding that all of the organs of the Immp2lKD-/- KO mouse were smaller compared with their wildtype litter mates. This was consistent with an earlier study that had found lower levels of mitochondrial ROS (mitoROS) in all of the organs of the Immp2lKD-/- KO mouse tested where the greatest decrease in mitoROS >50% was found in the kidney [3]. In this study we observed the greatest decrease in mitochondrial respiration was also in the kidney of the Immp2lKD-/- KO mouse when using succinate as the respiratory substrate. This was consistent with the function deficit arising from variant molecular connections between the ‘uncleaved’ Cyc1 and the Cyb respiratory subunit of Complex III in the Immp2lKD-/- KO mouse. When using glycerol-3-phosphate (G3P) as the substrate for Gpd2, we observed further decreases in mitochondrial respiration. We observed ~20% and 7% respective decreases in Gpd2 mediated mitochondrial respiration in female and male Immp2lKD-/- KO mice which were found associated with structural changes that affected the homodimerisation of the Gpd2 dehydrogenase. Perhaps most surprising of all was our finding of a ~50% decrease in nonmitochondrial respiration in primary embryonic fibroblast cell lines derived from the Immp2lKD-/- KO mouse.
2. Results
2.1. Immp2lKD-/- KO is Associated with Lower Lean Body Mass
Male and female Immp2lKD-/- KO mice have decreased body weight compared with wildtype mice. Male Immp2lKD-/- KO mice displayed decreased body mass compared with wildtype mice at 22-29 weeks of age (t(30) = 2.234, p = 0.033; Figure 2A). Using EchoMRI we observed no difference in fat mass between the Immp2lKD-/- KO mouse and wildtype litter mates (Figure 2B), but rather lower lean body mass (Figure 2C). At the time of sacrifice at 25-32 weeks of age, body mass in Immp2lKD-/- KO mice was decreased compared to their wildtype littermates (t(30) = 2.234, p = 0.033; Figure 2), with lower mass observed in many organs from the Immp2lKD-/- KO mice (Figure 2E-L), reaching significance in heart, quadriceps and kidney (Figure 2E-G). When normalised to body mass, there was no significant differences in fat mass, lean mass, organ weights or adipose tissue depots, indicating that the lower body mass of Immp2lKD-/- KO mice is likely due to the proportionally lower mass of many organs, rather than a decrease in size of any specific organ or a disproportionate decrease in fat mass.
2.2. Oxygen Consumption Rates in Mitochondria Isolated from Heart, Brain and Kidney
Respiration rates were determined for mitochondria isolated from kidney, heart and brain using a Clark-type oxygen electrode and different substrates including glutamate that feeds electrons into Complex I and succinate which feeds electrons into Complex II. When using glutamate as the substrate we detected no significant difference in oxygen consumption between the mitochondria isolated from the Immp2lKD-/- KO mouse and their wildtype litter mates (Figure 3). When using succinate as the substrate we detected a decrease in oxygen consumption in mitochondria isolated from the kidney of the Immp2lKD-/- KO mouse when compared with wildtype litter mates but not in mitochondria from heart or brain (Figure 3). Oxygen consumption was significantly lower in kidney mitochondria from the Immp2lKD-/- KO mouse compared to wildtype litter mates (p=0.03) under non-phosphorylating state 2 respiration, when there is substrate present, but no ADP. Under state 3 respiration conditions (ADP present) and state 4 (ATP synthesis inhibited by oligomycin) respiration was still trending lower in the Immp2lKD-/- KO mice, although the differences did not reach statistical significance. Overall, there was an indication that mitochondrial respiration with succinate may be lower in the Immp2lKD-/- KO kidney but not the other organs tested.
2.3. Oxygen Consumption Rates in Mitochondria Isolated from Mouse BAT Tissue
Seahorse XF96 analysis was used to test oxygen consumption rates in mitochondria isolated from brain and brown adipose tissue. Glycerol-3-phosphate (G3P) was used as the substrate for the Gpd2 enzyme. Gpd2 dependent mitochondrial respiration was too low in the brain of wildtype mice for comparative analysis. Given that Gpd2 is expressed at much higher levels in brown adipose tissue (BAT) [9] we isolated BAT mitochondria from mice and used G3P as the substrate for Gpd2 to determine differences in G3P/Gpd2 dependent mitochondrial respiration (Figure 4).
Seahorse analysis of oxygen consumption rates (OCR) in mouse BAT tissue mitochondria indicated that female and male Immp2lKD-/- KO mice had a ~20% and ~7% decrease, respectively, in G3P/Gpd2 enzyme dependent mitochondrial respiration in Immp2lKD-/- KO mice compared with wildtype (Figure 4). Notwithstanding, G3P/Gpd2 dependent mitochondrial respiration was over twice as high in females compared with males (Figure 4).
2.4. Predictive Structural Analysis of Mouse mGpd2 Variant Homodimers Using AlphaFold2-Multimer
The lower Gpd2 dependent respiratory levels in mitochondria isolated from the Immp2lKD−/− KO mouse suggested that there was a decrease in the activity of the ‘uncleaved Gpd2’ enzyme and/or interference of its role within the glycerol phosphate shuttle. We used AlphaFold2-Multimer predictive alignment error (PAE) plots visualised through ChimeraX [10] to interrogate the homodimeric structure and associated inter-subunit connections between the mouse Gpd2 dehydrogenase homodimer variants. AlphFold2-Multimer analysis [10-12] predicted with high confidence that the ‘cleaved Gpd2’ dehydrogenase in wildtype mice and the ‘uncleaved Gpd2’ enzyme in Immp2lKD−/− KO mice both form homodimers, albeit the latter ‘uncleaved mGpd2’ homodimer in mouse forms 6% more inter-subunit connections (Figure 5). This indicated a fundamental change in the structure of the Gpd2 dehydrogenase homodimer within the inner mitochondrial membrane where it is cleaved and where it functions within the glycerol phosphate shuttle (GPS). The GPS shuttle facilitates the transfer of electrons from NADH (from glycolysis) to FAD (embedded within Gpd2) and onto Complex II of the mitochondrial respiratory chain. The AlphaFold2-Multimer analysis also predicted that the signal transit peptides of the ‘uncleaved Gpd2’ enzyme homodimer protrude further into the surrounding inner mitochondrial membrane from where the Gpd2 dehydrogenase is normally embedded (Figure 6) and from where electrons are transferred between the FADH2 generated by Gpd2 in the GPS and Complex II of the electron transport chain (ETC).
2.5. Predictive Structural Analysis of Mouse Cyc1 Variants in Complex with Cyb Using AlphaFold2-Multimer
Three respiratory subunits in Complex III of the electron transport chain interact, namely Cyc1, Cyb and UQCRFS1. AphaFold2-Multimer was used to predict the orientation of these 3 subunits relative to each other. The orientation of Cyc1 with respect to the other 2 subunits is such that the ‘uncleaved’ signal transit peptide of Cyc1 (as found in the Immp2lKD-/- KO mouse) was observed to align in closer proximity to Cyb than to UQCRFS1. To investigate this further, it was queried whether the ‘uncleaved’ transit peptide of Cyc1 established any ‘new’ intermolecular connections with Cyb. Alpha-Fold2-Multimer predicted 56 ‘new’ molecular contacts between the signal transit peptide (Orange) of Cyc1 (Blue chain) and Cyb (Pink chain) (Figure 7). These contacts involved the following residues: Transit Peptide of Cyc1: Met4, Leu8, Leu11, Ala19, Lue22, His23, Val26. Normal Cyb: Gln322, Trp326, Ile327, Trp337, Gln341, Pro346, Phe347, Ile350 (Figure 7D).
2.6. Immp2l KO Mediated Decrease in Mitochondrial and Nonmitochondrial Respiration
Seahorse analysis was used to test for differences in respiration rates between living primary mouse embryonic fibroblast cell lines (MEFs) derived from Immp2lKD-/- KO and wildtype day 15 embryos. MEFs were grown in DMEM using standard cell culture conditions and harvested during the growth phase at ~80% confluence. The total level of cellular respiration was ~27% lower in MEFs derived from Immp2lKD-/- KO mice (H3 and H10) when compared with wildtype (WT) (Figure 8, Table 1). This deficit in respiration in Immp2lKD-/- KO MEFs comprised a dramatic ~50% drop in nonmitochondrial respiration and a modest ~12% decrease in mitochondrial respiration (Figure 8, Table 1). This observation was somewhat surprising given that Immp2l is a mitochondrial peptidase yet nonmitochondrial respiration was more greatly affected than mitochondrial respiration in MEFs derived from Immp2lKD-/- KO mice.
3. Discussion
Our previous investigations found compelling evidence of behavioural change in the Immp2lKD-/- KO mouse that was more pronounced in male mice [3]. In this study, we observed that male and female Immp2lKD-/- KO mice had lower body mass compared with wildtype litter mates. Using EchoMRI, male Immp2lKD-/- KO mice were shown to have a lower lean body mass compared with wildtype litter mates, while fat mass was unaffected by genotype. What was most interesting in this analysis was that all of the organs examined were smaller in size in the Immp2lKD-/- KO mouse compared with wildtype litter mates, with the drop in total lean body mass appearing to represent the cumulative sum of the lower masses of the different organs. Furthermore, we found no marked difference in the mass of two separate fat pads, the epididymal fat pad (EpiWAT) and the inguinal fat pad (IngWAT)) of the Immp2lKD-/- KO mouse compared with wildtype litter mates (Figure 2). This finding was in contrast with the lower fat mass reported for a C-terminal truncated Immp2l mouse model [3, 4] and the lower weight loss reported for a Gpd2 KO mouse line [14]. These results identify a key role for Immp2l in regulating the homeostasis of all the organs of the body.
In an earlier study, we demonstrated that the loss of Immp2l peptidase activity in the Immp2lKD-/- KO mouse resulted in the failure of Immp2l to cleave and remove the mitochondrial transit peptides from two nuclear encoded respiratory associated mitochondrial proteins Cyc1 and Gpd2 [3]. Cyc1 forms part of Complex III of the mitochondrial respiratory chain and the Gpd2 enzyme forms an integral component of the glycerol phosphate shuttle (GPS) which funnels electrons from the NADH generated during glycolysis in the cytosol to the mitochondrial respiratory chain and in doing so regenerates NAD+ for another round of glycolysis (Figure 1). This loss of Immp2l peptidase activity was also associated with a decrease in the level of mitochondrial ROS (mitoROS) to different degrees in different tissues [3]. However, prior to the present study it was not known whether Immp2l had any effect on cellular or mitochondrial respiration rates or the activity or function of Cyc1 or Gpd2 [3-7].
In the present study we used the Immp2lKD-/- KO mouse model to investigate whether Immp2l has a role in the regulation of respiration. In this endeavour, we used a comprehensive approach by testing for differences in respiration between mitochondria isolated from different organs and tissues (kidney, heart, brain and brown adipose tissue) using different respiratory substrates (glutamate, succinate and glycerol-3-phosphate (G3P)). Notwithstanding, all of the respiratory pathways tested feed electrons into the mitochondrial electron transport chain (ETC) upstream of Cyc1/Complex III. Glutamate generates NADH that feeds electrons directly into Complex I and succinate feeds electrons directly into Complex II of the ETC whereas glycerol-3-phosphate (G3P) is a cytosolic substrate for Gpd2 within the GPS. G3P is converted by Gpd2 into dihydroxyacetone phosphate (DHAP) in the GPS which mediates the transfer of electrons from the cytosolic NADH produced during glycolysis to FAD2+ from where the electrons are transferred through the inner mitochondrial membrane to Complex II of the mitochondrial respiratory chain (see Figure 1). We also tested for differences in whole cell respiration using living fibroblast cell lines derived from the Immp2lKD-/- KO mouse.
When using glutamate as the respiratory substrate we found no difference in the level of mitochondrial respiration in brain, heart or kidney from the Immp2lKD-/- KO mouse compared with wildtype littermates. When using succinate as the respiratory substrate we observed a ~31% lower level of mitochondrial respiration in the kidney but not in brain or heart. This result was consistent with the ~50% decrease in mitoROS found in the kidney of the Immp2lKD-/- KO mouse [3] and the much lower decreases in mitoROS observed in brain and heart. These differences between substrates and tissues are difficult to explain, notwithstanding Complex III is comprised of numerous different subunits which may differ proportionally under different conditions and affect respiration accordingly between substrates and tissues. We investigated the structure of Cyc1 and its connections with another closely associated respiratory subunit Cyb (Figure 6). This analysis predicted with confidence that the failure to remove the signal transit peptide from Cyc1 in the Immp2lKD-/- KO mouse had altered its molecular connections with the Cyb respiratory subunit of Complex III. Alpha-Fold2-Multimer predicted 56 ‘new’ molecular interactions between the ‘uncleaved’ signal transit peptide of Cyc1 and Cyb (Figure 7D). How these ‘new’ molecular contacts between the 2 respiratory subunits might affect respiration is uncertain. However, together these results indicated the likelihood of a deficit in mitochondrial respiration in the kidney of the Immp2lKD-/- KO mouse suggestive of a partial/conditional deficit in the function of Cyc1-Cyb in Complex III, however, this needs further investigation.
We used G3P as the substrate for Gpd2 in the glycerol phosphate shuttle (GPS) using mitochondria isolated from brown adipose tissue (BAT) which is known to express high levels of Gpd2 compared with other tissues [9]. Using this approach, we identified a ~20% and ~7% lower level of Gpd2 dependent mitochondrial respiration over time in female and male Immp2lKD-/- KO mice, respectively, compared with wildtype. Results indicated over a 2 fold higher level of Gpd2 dependent mitochondrial respiration in females compared with males. As a result, the levels of Gpd2 dependent mitochondrial respiration were always higher in female mice compared with male mice regardless of genotype. This finding was most interesting given that heterozygous deletions of Gpd2 are relatively common in male autism and a deleterious hemizygous mutation of Gpd2 has been found segregating with autism in a multi-generation autism family [15, 16]. Secondly, autism occurs at a much higher incidence in males compared with females [16]. Thirdly, behavioural change is more pronounced in male compared with the female Immp2lKD-/- KO mice [3].
The above finding further indicated that failure to remove the signal transit peptide from Gpd2 by Immp2l had compromised the activity of the Gpd2 dehydrogenase and/or the role of Gpd2 in the GPS wherein electrons are transferred from NADH to FAD2+ and onto Complex II. To determine if there was any molecular basis for this deficiency in Gpd2 function we searched for structural changes in Gpd2 using AlphaFold2-Multimer predictive structural analysis and found that failure to remove the transit peptide from Gpd2 in the Immp2lKD-/- KO mouse had altered the structure of the Gpd2 enzyme in a number of ways that could affect respiration. Firstly, this structural change altered the homodimerization of Gpd2 by increasing the number of inter subunit connections in the Gpd2 homodimer. Secondly, the transit peptides that remained attached to the Gpd2 homodimer in the Immp2lKD-/- KO mouse extended further into the surrounding inner mitochondrial membrane where they could feasibly interfere with the Gpd2/FADH2 mediated transfer of electrons to Complex II of the ETC.
To better understand the effect of Immp2l on whole cell respiration we generated and tested primary fibroblast cell lines (MEFs) derived from Immp2lKD-/- KO mouse embryos. The total level of respiration was decreased ~27% in primary MEFs derived from Immp2lKD-/- KO mice when compared with wildtype (Figure 8, Table 1). This ~27% deficit in total cellular respiration in Immp2lKD-/- KO MEFs comprised a dramatic ~50% decrease in nonmitochondrial respiration concurrent with a more modest ~12% decrease in total mitochondrial respiration (Figure 9, Table 1). This ~12% decrease in total mitochondrial respiration in Immp2l KO MEFs was consistent with the cumulative decreases we had observed in substrate specific mediated mitochondrial respiration in tissues from the Immp2lKD-/- KO mouse (Figure 3 & Figure 4). Notwithstanding evidence suggests that substrate specific mitochondrial respiration rates vary widely between substrates and tissues. However, the source and impact of the ~50% decrease in non-mitochondrial respiration/oxygen consumption remains unexplained.
Together these results identify key roles for Immp2l in enhancing and maximising mitochondrial respiration, glycolysis and nonmitochondrial respiration (NMR). The Immp2l peptidase enhances mitochondrial respiration by cleaving and thereby enhancing the structure and function of two respiratory related mitochondrial proteins Gpd2 and Cyc1. In this respect, the enhanced structure of the Gpd2 dehydrogenase homodimer induced by the Immp2l peptidase was of particular interest given that both Immp2l and Gpd2 have been conserved through evolutionary history from E coli to human. We report that the cleavage of Gpd2 by Immp2l increased the number of molecular connections between the subunits of the Gpd2 homodimer notwithstanding the precise effect of this change on homodimerization requires further research.
This study yielded a number of unexpected results that limit our interpretation of the findings and which require further investigation. It is not certain why the level of substrate mediated mitochondrial respiration differed between tissues. In this respect, the expression level of many of the subunits of complex III may vary between tissues and under different conditions, however, further investigations are required to understand this and the full impact of Immp2l on mitochondrial and cellular homeostasis. This line of research will require expansion of the number of MEFs derived from the Immp2lKD-/- KO mouse so as to better understand the full range and effect that Immp2l has on mitochondrial and nonmitochondrial respiration under different conditions. In this study we did not quantify the impact of the Immp2l KO on glycolysis and changes in metabolism or characterize the nature of the disproportionately large decrease in NMR all of which are the subject of our ongoing studies into the role of Immp2l and its effects on mitochondrial dynamics, glycolysis and metabolism in particular its effect on NAD+ metabolism (Figure 1). In this respect, the Immp2lKD-/- KO mouse and derived MEFs provide an ideal model system to better understand the role of Immp2l in cellular homeostasis, mitochondrial respiration, glycolysis, metabolism, mitoROS production, organ size and behaviour and how this may affect reprograming of cellular metabolism and its impact on NAD+ metabolism, gene expression and mitochondrial dynamics.
4. Conclusion
The Immp2l peptidase cleaves the signal transit peptide from Gpd2 and Cyc1 thereby optimizing and enhancing their structure and function in mitochondrial respiration. As a positive regulator of Gpd2 we can assume that Immp2l also positively regulates glycolysis. The combined positive regulation of mitochondrial respiration and glycolysis may help explain the major effect that Immp2l has on nonmitochondrial respiration. The Immp2lKD-/- KO mouse and derived MEF cell lines represent a valuable model resource to further investigate the role of Immp2l in cellular homeostasis.
5. Materials and Methods
5.1. Animals and Ethics
Mice were bred and group housed at the Ingham Institute Biological Resource Unit (Liverpool, Australia) in individually ventilated cages (GM500 Green, Techniplast Australia Pty Ltd, Rydalmere, Australia) with corn cob bedding, crinkle cut cardboard nesting material and a red igloo (Bioserv, Frenchtown, NJ, USA). At 6 months of age mice were transported to the University of New South Wales, (Sydney, NSW, Australia) for cellular respiration analyses, body composition, mitochondrial and gene expression analyses. Mice were allowed to acclimate to the new facility for one month before testing and were housed in standard holding cages in a temperature and humidity-controlled environment with a 12/12-h modified dark-light cycle (lights on at 0700). Two mice were housed individually to avoid fighting. Food and water or MitoQ-treated water were available ad libitum. Mice were randomly allocated to experimental groups. Mice were euthanized with CO2 or cervical dislocation. Tissues were dissected after perfusion with sterile PBS. All procedures involving mice were carried out under protocols approved by the University of New South Wales Animal Ethics Committee (protocol number 19/6B, 15/48B and 18/78A) and in accordance with National Health and Medical Research Council guidelines. Animals were monitored daily. No unexplained mortality occurred in these studies.
5.2. Mouse Maintenance
Male Immp2lKD−/− KO mice and wildtype litter mates were obtained from the Ingham Institute Biological Resource Unit (Sydney, NSW, Australia). Mice were housed at 22 ± 1°C in a 12:12 hour light/ dark cycle. Mice had ad libitum access to water and a standard low fat rodent diet (Gordon’s Specialty Stock Feeds, Yanderra, NSW, Australia). Physiological assessments were approved by the UNSW animal care and ethics committee (ACEC 18/78A), and followed guidelines issued by the National Health and Medical Research Council of Australia.
5.3. Tissue Preparation and Mitochondrial Isolation
Frozen tissues were thawed from -80°C in ice-cold PBS, minced, and homogenized in MAS buffer (70 mM sucrose, 220 mM mannitol, 5 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2 mM HEPES pH 7.4). Heart and WAT were mechanically homogenized with 20 strokes in a glass–Dounce homogenizer. Liver, BAT, brain, kidney, lung, and muscle were mechanically homogenized with 10–20 strokes (depending on the tissue: lung and muscle require larger number of strokes) in Teflon-glass homogenizer. All homogenates were centrifuged at 1,000 g for 10 min at 4°C; then, the supernatant was collected. Protein concentration was determined by BCA method (ThermoFisher). Isolation of mitochondria followed methods previously described [32-34]. Immediately after excision, kidney and brain tissue were prepared by finely dicing in ice-cold isolation buffer A (250 mM sucrose, 10 mM Tris-HCl, 1 mM EGTA, 1% fatty-acid free BSA, pH 7.4). The tissue was rinsed with buffer A before homogenisation using a glass-Teflon Dounce homogeniser. Heart tissue was similarly prepared in ice-cold isolation buffer B (100 mM sucrose, 100 mM KCl, 50 mM Tris-HCl, 1 mM KH2PO4, 1 mM EGTA, 0.2% fatty-acid free BSA, pH 7.0). The heart tissue was incubated for 2 min in 1 mg/mL of proteinase, and rapidly rinsed in buffer B to stop digestion. The heart tissue was then homogenised using a Polytron homogeniser (ThomasScientific, Swedesboro, NJ, USA). Homogenates were centrifuged at 1,000 g for 5 min at 4°C. The supernatant was collected and centrifuged at 10,000 g for 10 min at 4°C. Pellets were washed in the respective isolation buffers and centrifuged again at 10,000 g for 10 min at 4°C. The pelleted mitochondrial isolate was resuspended in BSA-free ice-cold isolation buffer A and assayed for protein quantification using the bicinchoninic acid (BCA) assay (ThermoFisher Scientific, Rockford, IL, USA).
5.4. Mitochondrial Respiration
Kidney, brain and heart mitochondrial isolates were resuspended in respiration buffer (225 mM mannitol, 75 mM sucrose, 10 mM Tris-HCl, 10 mM KH2PO4, 10 mM KCl, 0.1 mM EDTA, 0.8 mM MgCl2, 0.3% fatty-acid free BSA, pH 7.0) warmed to 37°C. A Clark-type oxygen electrode (Rank Brothers, Cambridge, UK) was used to measure the oxygen consumption of mitochondrial isolates when provided with either succinate (5 mM) + rotenone (2 μM) or glutamate (5 mM) + malate (2 mM). Oxygen consumption of mitochondrial isolates was measured in the presence of the substrates alone (State 2), in the presence of 200 μM ADP (State 3) or in the presence of 2.5 μM oligomycin.
5.5. Mouse BAT Tissue Mitochondria Preparation and Respirometry
Brown adipose tissue (BAT) was processed using a derived protocol to that reported by Acin-Perez et al. [35]. Briefly, frozen samples were thawed from -80°C in ice-cold PBS, minced and homogenized in 1 mL of MAS buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2 mM HEPES; pH 7.2 with KOH). BAT tissues were mechanically homogenized using a Potter-Elvehjem homogenizer (8-10 strokes) and homogenates were centrifuged twice at 1,000 g for 10 min at 4°C. Then, supernatants were centrifuged at 10,000 g for 10 min at 4°C, and mitochondrial pellets were washed twice in wash buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EGTA, 0.5% BSA; pH 7.2 with KOH). The final mitochondrial pellet was re-suspended in a small volume of ice-cold MAS buffer (75ul starting from 30-60mg of wet tissue) with no BSA and quantified (BCA, ThermoFisher). BSA was omitted from the final buffer to prevent interference with the protein assay kit and with the respirometry assay. Mitochondria were loaded 3 mg/well into Seahorse XF96 microplate in 50 ml of MAS. The loaded plate was centrifuged at 2,000 g for 20 min at 4°C (no brake), and an additional 130 microlitres of warm MAS (37°C) was added to each well. To avoid disrupting mitochondrial adherence to the bottom of the plate, MAS was added using a multichannel pipette pointed at a 45° angle to the top of well chamber. Mitochondria were equilibrated for 5 min at 37°C without CO2 before starting the respirometry assay. Glycerol-3-phosphate (G3P, #94124 Sigma) was injected at port A (5mM final concentration, in MAS), and Antimycin A (A8674, Sigma) was injected at port B (4mM final concentration, in MAS). Mix and measure times were 0.5 and 4 min, respectively. Oxygen consumption rate (OCR) was measured using an Agilent Seahorse XFe96 Analyzer (Agilent Technologies, Santa Clara, CA) and Wave software was used to export OCR rates to GraphPad Prism v.9.0.1. OCR rates were normalized to background noise measured in wells with no mitochondria. Note: MAS buffer is diluted 3x with water.
5.6. Mouse Embryonic Fibroblast Cell Lines (MEFs)
In this study, we crossed heterozygous Immp2lKD −/+ KO mice and 14.5 days after fertilisation pregnant females were humanely euthanased. Then under sterile conditions individual mouse embryos were removed from the uterus one by one and transferred to individual sterile dishes containing sterile PBS. After the head and all red organs were removed, the remaining tissue was then transferred to individual 1.5 ml tubes containing 0.5 ml PBS. Tissue was minced with scissors before adding 0.5ml digestion solution into each 0.5 ml of PBS in 1.5 ml Eppenorf tubes. Tubes were incubated at 37°C for 1 h, shaking every 10 min. After 1 h, the mixture was removed from the Eppendorf tube and transferred to a 15 mL Falcon tube containing 9 ml of DMEM media and centrifuged at 1000 rpm for 3 min. Supernatant was discarded and the cell pellet resuspended in 10 ml of DMEM (with 10% FBS and Penicillin Streptomycin antibiotics) and transferred to a 10 cm cell culture dish. Cells were incubated and passaged regularly over an 8-week period until cultures were established.
5.7. Mouse Embryonic Fibroblast Cell Line (MEF) Preparation and Seahorse Analysis
MEF cell lines were trypsinised and centrifuged for 5 min at 1000 rpm. Real-time measurements of MEF oxygen consumption rate (OCR) were made using a Seahorse XF-96 analyser following the manufacturer’s instructions (Seahorse Biosciences, North Billerica, MA). MEF cells (H3, WT7 and H10) were seeded into Seahorse 96-well tissue culture plates at a density of 20,000 cells per well and left for 24 h to adhere to the plate. Standard DMEM growth media was removed and replaced with Seahorse media (DMEM + 1 mM Sodium Pyruvate, 4 mM L-glutamine and 25 mM glucose). Cells were incubated at 37℃ in a non-CO2 incubator for 1 h prior to running the assay. Measurements were recorded with a 30 sec mix and a 4 min measurement period [36]. Baseline measurements were followed by the addition of G3P (5mM) and Antimycin A (4uM). BCA assay was performed and readout was normalised to protein content.
5.8. Body Composition Analysis
Whole-body composition data was assessed in 22-29 week old mice yielding lean mass, fat mass, free water mass, and total water was analysed by the EchoMRI-900 (EchoMRI Corporation Pty Ltd, Singapore) in accordance with the manufacturer’s instructions. Organ and fat weight were quantified in the same mice following sacrifice at 25-32 weeks of age yielding mass for whole body, brown adipose tissue (BAT), inguinal white adipose tissue (ingWAT), epididymal white adipose tissue (epiWAT) brain, kidney, liver, heart and quadriceps. Whole-body composition data yielding lean mass, fat mass, free water mass, and total water was analysed by the EchoMRI-900 (EchoMRI Corporation Pty Ltd, Singapore) in accordance with the manufacturer’s instructions.
5.9. Respiratory and Body Composition Statistical Analyses
Mitochondrial respiration data were analysed using unpaired two-tailed t-tests to detect effects of genotype (Immp2lKD −/− KO vs. WT). Body composition data were analysed using unpaired two tailed t-tests to detect effects of genotype (Immp2l-/- vs. WT).
5.10. AlphaFold2-Multimer High-Accuracy Prediction of Complex Structures.
High-accuracy prediction of protein structures and associated oligomerization states were carried out using the AlphaFold2-Multimer program in ChimeraX (v 1.6.1) available at https://www.rbvi.ucsf.edu/chimerax, accessed 1st September 2023 [10] using default settings. Analysis of intermolecular contacts and visualization of structures were performed in ChimeraX [12] with predicted aligned error (PAE) plots interpreted as described [11].
Author Contributions
Conceptualization, R.A.C.; Data curation, N.T, K.S.S. and R.A.C.; Formal analysis, I.C., H.G., M.B., E.O., N.T, K.S.S. and R.A.C.; Investigation, K.H., N.T., K.S.S., W.B., I.C., H.G., M.B., E.O., R.A.C. and A.K.W.; Methodology, N.T., K.S.S., I.C., H.G., M.B., E.O., R.A.C.; Resources, N.T., A.W., W.B., R.A.C., and V.E.; Software, K.S.S., W.B and R.A.C.; Supervision, A.W., N.T., W.B. R.A.C and V.E; Validation, N.T., K.S.S. and R.A.C.; Writing—original draft, W.B, N.T., K.S.S., R.A.C. and V.E.; Writing—review and editing, K.S.S., N.T., R.A.C., A.W., W.B. and V.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded as by a Sydney Partnership for Health Education Research and Enterprise (SPHERE) Grant to V.E. and R.A.C.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Animal Ethics Committee of the University of NSW, Sydney Australia (protocol numbers 19/6B, 15/48B and 18/78A approved 2018) for studies involving animals.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Professor Kyle Hoehn for direction in respiration research. This work was supported by a Sydney Partnership for Health Education Research and Enterprise (SPHERE) Grant to AKW, VE, RC, and TF. AKW is supported by a National Breast Cancer Foundation Australia (PF-15-014), the Schizophrenia Research Institute, UNSW, Australia, and Neuroscience Research Australia, NSW, Australia.
Conflicts of Interest
All authors declare no conflicts of interest.
References
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim Biophys Acta 2002, 1592, 63–77. [Google Scholar] [CrossRef]
- Yeh, J.I.; Chinte, U.; Du, S. Structure of glycerol-3-phosphate dehydrogenase, an essential monotopic membrane enzyme involved in respiration and metabolism. Proc. Natl. Acad. Sci. 2008, 105, 3280–3285. [Google Scholar] [CrossRef] [PubMed]
- Lawther, A.J.; Zieba, J.; Fang, Z.; Furlong, T.M.; Conn, I.; Govindaraju, H.; Choong, L.L.Y.; Turner, N.; Siddiqui, K.S.; Bridge, W.; et al. Antioxidant Behavioural Phenotype in the Immp2l Gene Knock-Out Mouse. Genes 2023, 14, 1717. [Google Scholar] [CrossRef]
- Lu, B.; Poirier, C.; Gaspar, T.; Gratzke, C.; Harrison, W.; Busija, D.; Matzuk, M.M.; Andersson, K.-E.; Overbeek, P.A.; Bishop, C.E. A Mutation in the Inner Mitochondrial Membrane Peptidase 2-Like Gene (Immp2l) Affects Mitochondrial Function and Impairs Fertility in Mice1. Biol. Reprod. 2008, 78, 601–610. [Google Scholar] [CrossRef]
- Ma, Y.; Liang, R.-M.; Ma, N.; Mi, X.-J.; Cheng, Z.-Y.; Zhang, Z.-J.; Lu, B.-S.; Li, P.A. Immp2l Mutation Induces Mitochondrial Membrane Depolarization and Complex III Activity Suppression after Middle Cerebral Artery Occlusion in Mice. Curr. Med Sci. 2023, 43, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, Z.; Chen, Z.; Ma, N.; Sun, S.; Zhang, J.; Ni, X.; Zhang, J.; Li, P.A. Suppression of Inner Mitochondrial Membrane Peptidase 2-Like (IMMP2L) Gene Exacerbates Hypoxia-Induced Neural Death Under High Glucose Condition. Neurochem. Res. 2017, 42, 1504–1514. [Google Scholar] [CrossRef]
- Bharadwaj, M.S.; Zhou, Y.; Molina, A.J.; Criswell, T.; Lu, B. Examination of bioenergetic function in the inner mitochondrial membrane peptidase 2-like (Immp2l) mutant mice. Redox Biol. 2014, 2, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhai, L.; Qian, L.; Huang, D.; Ding, Y.; Xiang, H.; Liu, X.; Thompson, J.W.; Liu, J.; He, Y.-H.; et al. Switching off IMMP2L signaling drives senescence via simultaneous metabolic alteration and blockage of cell death. Cell Res. 2018, 28, 625–643. [Google Scholar] [CrossRef]
- Gong, D.W. Rat mitochondrial glycerol-3-phosphate dehydrogenase gene: multiple promoters, high levels in brown adipose tissue, and tissue-specific regulation by thyroid hormone. l. DNA Cell Bio 1998, 17, 301–309. [Google Scholar] [CrossRef]
- Mirdita, M. ColabFold: making protein folding accessible to all. Nat Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Elfmann, C. and J. Stulke, PAE viewer: a webserver for the interactive visualization of the predicted aligned error for multimer structure predictions and crosslinks. Nucleic Acids Res 2023, 51, W404–W410. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Sullivan, C.R.; Mielnik, C.A.; Funk, A.; O'Donovan, S.M.; Bentea, E.; Pletnikov, M.; Ramsey, A.J.; Wen, Z.; Rowland, L.M.; McCullumsmith, R.E. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.; DosSantos, R.A.; Stepanyan, Z.; Marrif, H.; Silva, J.E. Mice with deletion of the mitochondrial glycerol-3-phosphate dehydrogenase gene exhibit a thrifty phenotype: effect of gender. Am. J. Physiol. Integr. Comp. Physiol. 2004, 287, R147–R156. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.M.A.; Meganathan, K.; Baldridge, D.; Gontarz, P.; Zhang, B.; Bonni, A.; Constantino, J.N.; Kroll, K.L. Cellular and molecular characterization of multiplex autism in human induced pluripotent stem cell-derived neurons. Mol. Autism 2019, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- A Clarke, R.; Lee, S.; Eapen, V. Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including Autism. Transl. Psychiatry 2012, 2, e158. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Inducible Factor-Dependent Extension of the Replicative Life Span during Hypoxia. Mol. Cell. Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and human disease: One highly involved factor. Genes Dev. 2000, 14, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Calvani, M.; Comito, G.; Giannoni, E.; Chiarugi, P. Time-Dependent Stabilization of Hypoxia Inducible Factor-1α by Different Intracellular Sources of Reactive Oxygen Species. PLOS ONE 2012, 7, e38388. [Google Scholar] [CrossRef]
- Willson, J.A.; Arienti, S.; Sadiku, P.; Reyes, L.; Coelho, P.; Morrison, T.; Rinaldi, G.; Dockrell, D.H.; Whyte, M.K.B.; Walmsley, S.R. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood 2022, 139, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, M.M. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep 2012, 2, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Nambu, A.; Jung, J.; Shibata, M.; Aksoylar, H.I.; Lei, J.; Xu, P.; Doan, M.T.; Jiang, H.; MacArthur, M.R.; et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 2019, 20, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef]
- Flashman, E.; Hoffart, L.M.; Hamed, R.B.; Jr, J.M.B.; Krebs, C.; Schofield, C.J. Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 2010, 277, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Leung, B.K.; Merlin, S.; Walker, A.K.; Lawther, A.J.; Paxinos, G.; Eapen, V.; Clarke, R.; Balleine, B.W.; Furlong, T.M. Immp2l knockdown in male mice increases stimulus-driven instrumental behaviour but does not alter goal-directed learning or neuron density in cortico-striatal circuits in a model of Tourette syndrome and autism spectrum disorder. Behav. Brain Res. 2023, 452, 114610. [Google Scholar] [CrossRef]
- Kreilaus, F.; Chesworth, R.; Eapen, V.; Clarke, R.; Karl, T. First behavioural assessment of a novel Immp2l knockdown mouse model with relevance for Gilles de la Tourette syndrome and Autism spectrum disorder. Behav. Brain Res. 2019, 374, 112057. [Google Scholar] [CrossRef] [PubMed]
- Bentley, N.L.; Fiveash, C.E.; Osborne, B.; Quek, L.-E.; Ogura, M.; Inagaki, N.; Cooney, G.J.; Polly, P.; Montgomery, M.K.; Turner, N. Protein hypoacylation induced by Sirt5 overexpression has minimal metabolic effect in mice. Biochem. Biophys. Res. Commun. 2018, 503, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Osborne, B.; Brandon, A.E.; O'Reilly, L.; Fiveash, C.E.; Brown, S.H.J.; Wilkins, B.P.; Samsudeen, A.; Yu, J.; Devanapalli, B.; et al. Regulation of mitochondrial metabolism in murine skeletal muscle by the medium-chain fatty acid receptor Gpr84. FASEB J. 2019, 33, 12264–12276. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Osborne, B.; Brown, S.H.J.; Small, L.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J. Lipid Res. 2013, 54, 3322–3333. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Benador, I.Y.; Petcherski, A.; Veliova, M.; A Benavides, G.; Lagarrigue, S.; Caudal, A.; Vergnes, L.; Murphy, A.N.; Karamanlidis, G.; et al. A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 2020, 39, e104073. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
IMMP2L peptidase association with the Glycerol Phosphate Shuttle that helps maximise mitochondrial respiration and glycolysis.
Figure 1.
IMMP2L peptidase association with the Glycerol Phosphate Shuttle that helps maximise mitochondrial respiration and glycolysis.
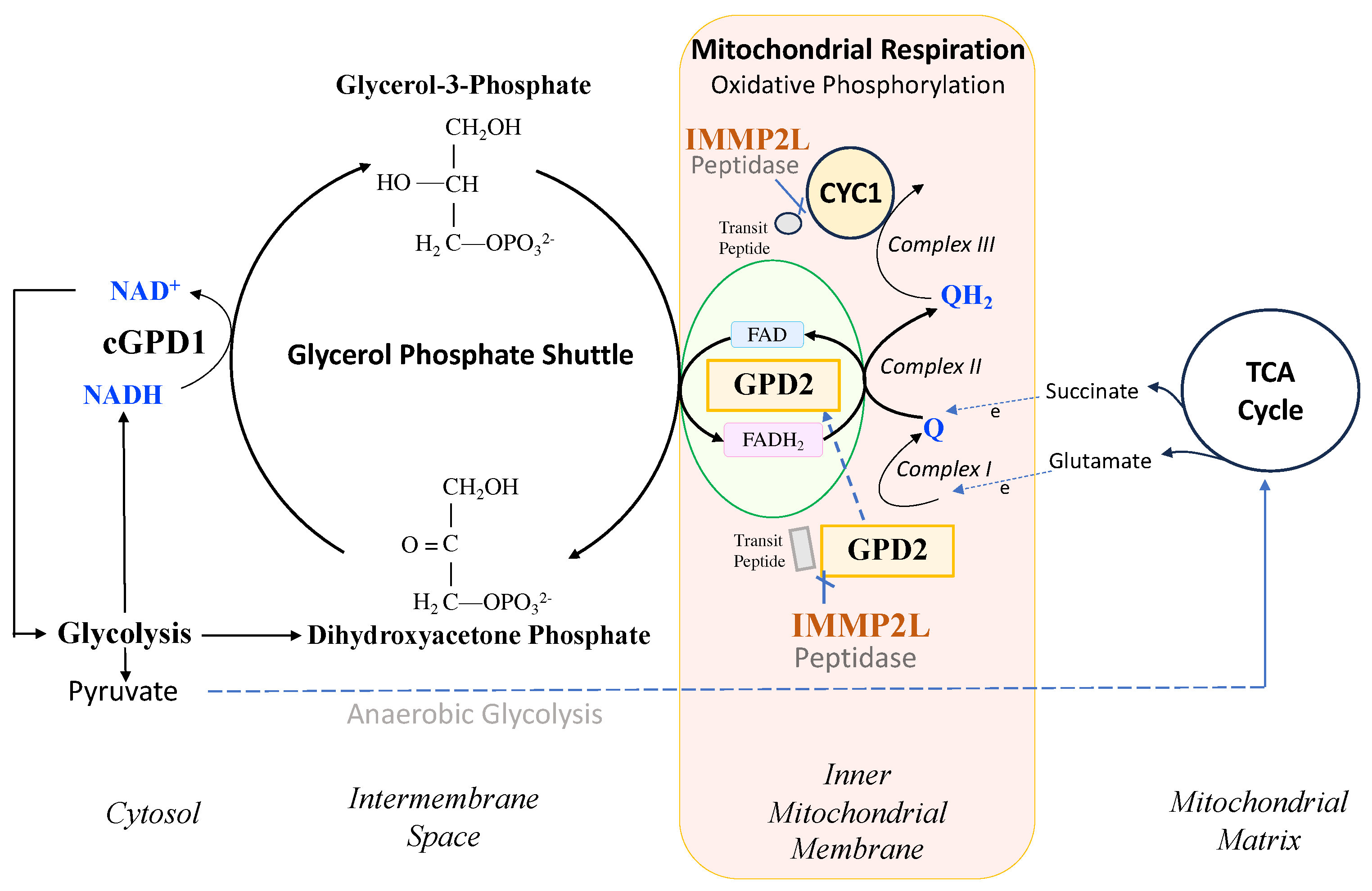
Figure 2.
Immp2lKD-/- KO decreases lean body mass in mice. A-C. Body mass, fat mass and lean mass of Immp2lKD-/- KO and wild-type mice at 22-29 weeks of age. D-L. Body mass and organ weights of Immp2lKD-/- KO and wild-type mice at 25-32 weeks of age. Data represent mean + SE (n = 7-16) and were analyzed by an unpaired t test *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 2.
Immp2lKD-/- KO decreases lean body mass in mice. A-C. Body mass, fat mass and lean mass of Immp2lKD-/- KO and wild-type mice at 22-29 weeks of age. D-L. Body mass and organ weights of Immp2lKD-/- KO and wild-type mice at 25-32 weeks of age. Data represent mean + SE (n = 7-16) and were analyzed by an unpaired t test *p < 0.05, **p < 0.01, ***p < 0.001.
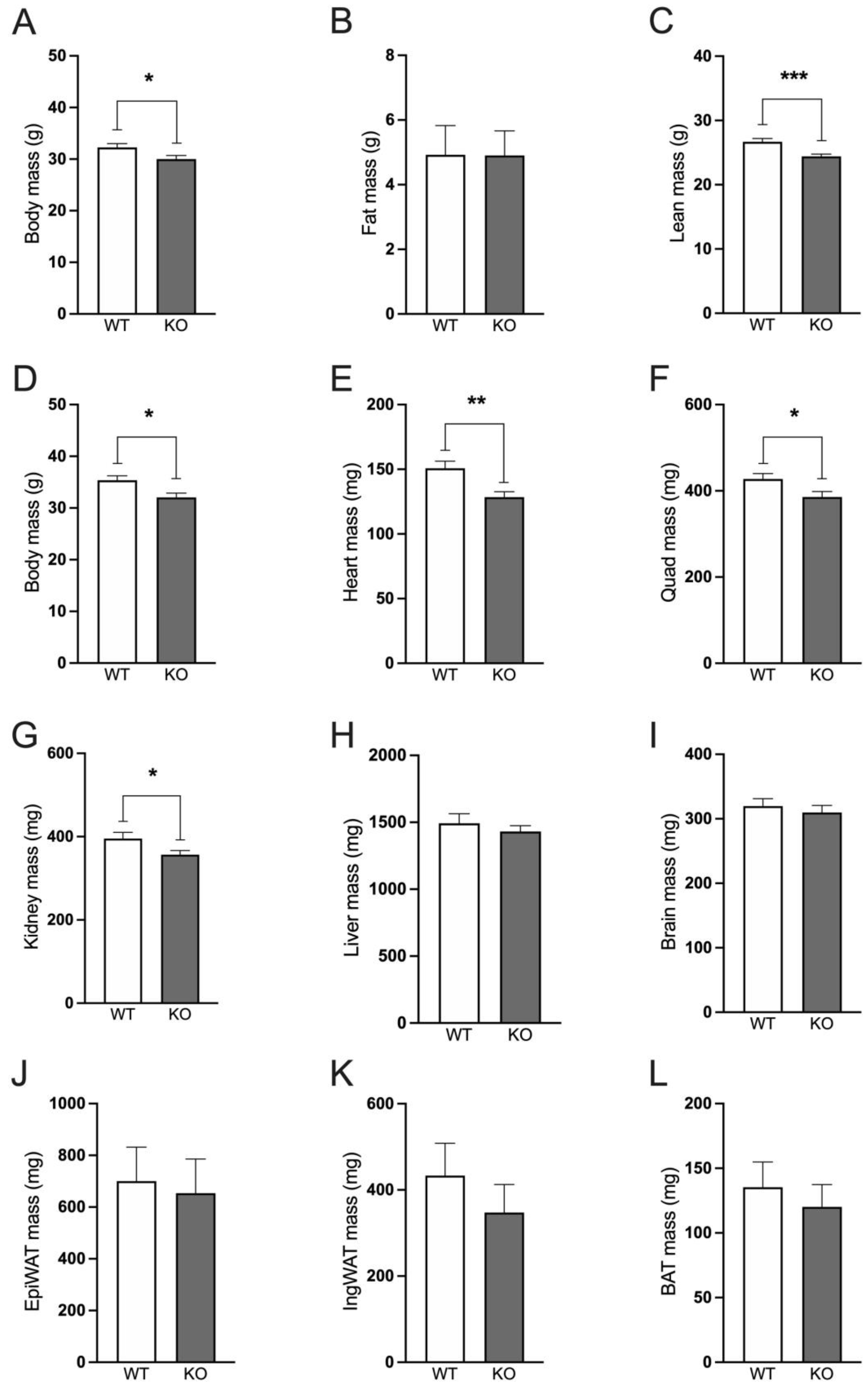
Figure 3.
Immp2lKD-/- KO impact on respiration in mitochondria isolated from mouse organs. Respiration in mitochondria isolated from organs of wild-type mice and Immp2lKD-/- KO mice using glutamate (A.) and succinate (B.) as respiratory substrates. Respiration was measured under State 2 conditions (substrate only), State 3 (ADP present) or State 4 (chemical inhibition of ATP synthesis by oligomycin). Data represent mean + SE (n = 6-8) and were analyzed by an unpaired t test * p < 0.05.
Figure 3.
Immp2lKD-/- KO impact on respiration in mitochondria isolated from mouse organs. Respiration in mitochondria isolated from organs of wild-type mice and Immp2lKD-/- KO mice using glutamate (A.) and succinate (B.) as respiratory substrates. Respiration was measured under State 2 conditions (substrate only), State 3 (ADP present) or State 4 (chemical inhibition of ATP synthesis by oligomycin). Data represent mean + SE (n = 6-8) and were analyzed by an unpaired t test * p < 0.05.
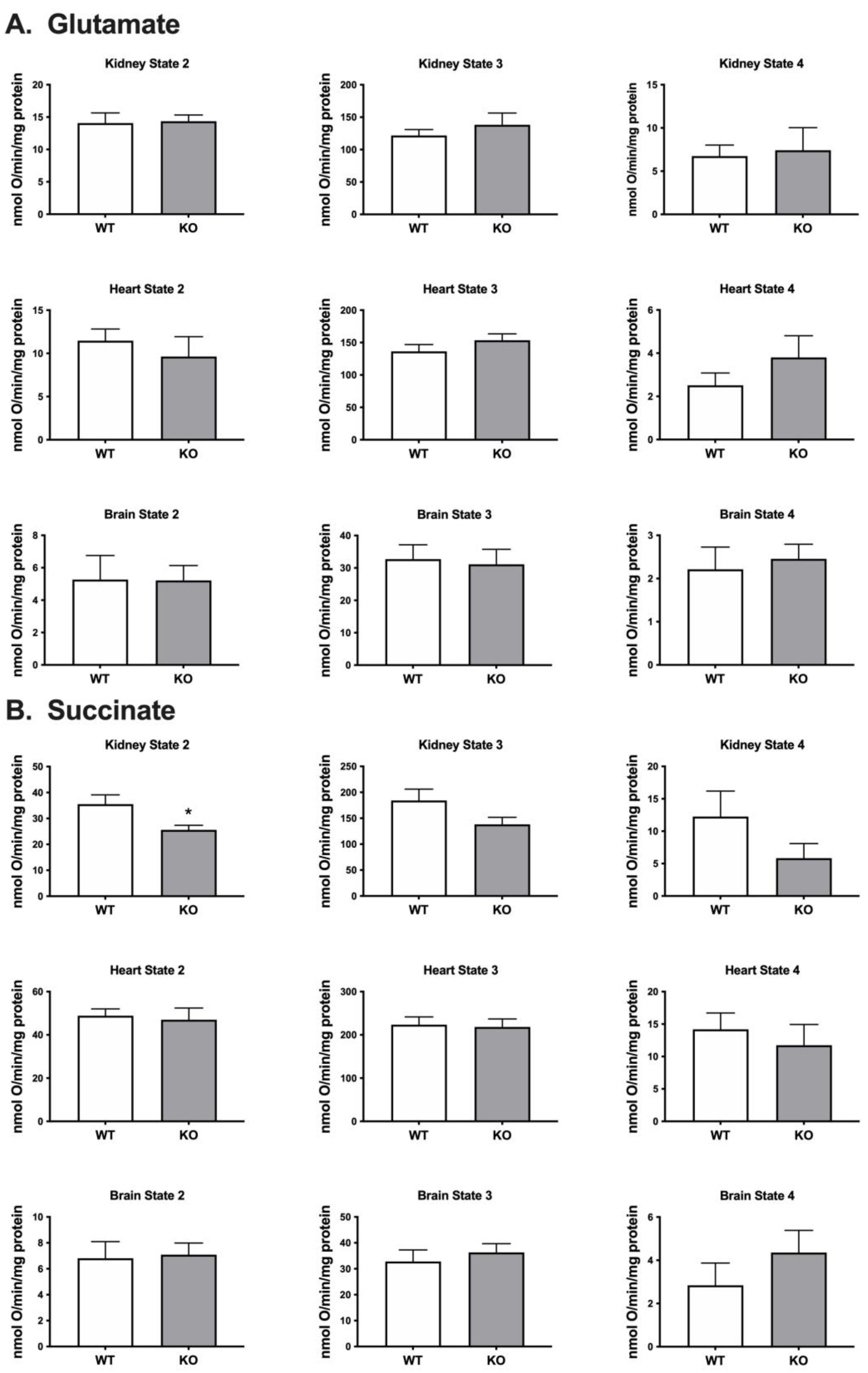
Figure 4.
Immp2lKD-/- KO lowers G3P substrate mediated mitochondrial respiration in brown adipose tissue (BAT). Oxygen consumption rate (OCR) of frozen brown adipose tissue from wild-type (n=3) and Immp2lKD-/- KO (n=3) male and female mice. Glyceraldehyde 3-phosphate (G3P, 5mM) and Antimycin A (AntA, 4 M) were injected at the indicated times. OCR rates are represented as percentage of baseline. n=3 wells per sample; values are represented as mean + SE.
Figure 4.
Immp2lKD-/- KO lowers G3P substrate mediated mitochondrial respiration in brown adipose tissue (BAT). Oxygen consumption rate (OCR) of frozen brown adipose tissue from wild-type (n=3) and Immp2lKD-/- KO (n=3) male and female mice. Glyceraldehyde 3-phosphate (G3P, 5mM) and Antimycin A (AntA, 4 M) were injected at the indicated times. OCR rates are represented as percentage of baseline. n=3 wells per sample; values are represented as mean + SE.
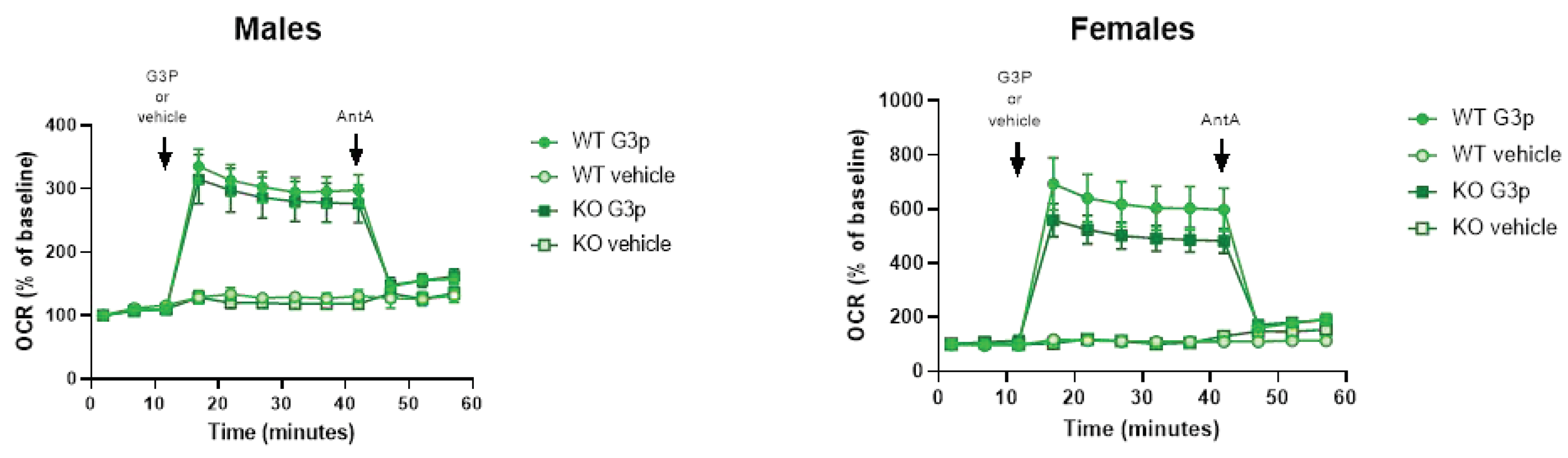
Figure 5.
Mouse Gpd2 dehydrogenase homodimer structural variants. AlphaFold2-Multimer high-accuracy prediction of mouse Gpd2 enzyme homodimeric structures [10-12] with predicted alignment error (PAE) plots visualised through ChimeraX. Rows: A. ‘cleaved Gpd2’ enzyme homodimer B. ‘uncleaved Gpd2’ enzyme homodimer found in the Immp2lKD−/− KO mouse. Columns: (1) Dimer predicted alignment error (PAE) colour comparison; (2) PAE plots where green colour intensity depicts higher confidence. Upper left and lower right quadrants represent the confidence of the predicted structure of the monomeric enzyme whereas lower left and upper right quadrants depict the confidence of the predicted dimerization of the enzyme subunits; (3) Dimer (Chain A, blue; Chain B, wood colour) inter-subunit connections (yellow) with 156 inter-subunit connections predicted for the ‘cleaved Gpd2’ dehydrogenase homodimer in mouse compared with 166 connections predicted for the ‘uncleaved Gpd2’ dehydrogenase homodimer.
Figure 5.
Mouse Gpd2 dehydrogenase homodimer structural variants. AlphaFold2-Multimer high-accuracy prediction of mouse Gpd2 enzyme homodimeric structures [10-12] with predicted alignment error (PAE) plots visualised through ChimeraX. Rows: A. ‘cleaved Gpd2’ enzyme homodimer B. ‘uncleaved Gpd2’ enzyme homodimer found in the Immp2lKD−/− KO mouse. Columns: (1) Dimer predicted alignment error (PAE) colour comparison; (2) PAE plots where green colour intensity depicts higher confidence. Upper left and lower right quadrants represent the confidence of the predicted structure of the monomeric enzyme whereas lower left and upper right quadrants depict the confidence of the predicted dimerization of the enzyme subunits; (3) Dimer (Chain A, blue; Chain B, wood colour) inter-subunit connections (yellow) with 156 inter-subunit connections predicted for the ‘cleaved Gpd2’ dehydrogenase homodimer in mouse compared with 166 connections predicted for the ‘uncleaved Gpd2’ dehydrogenase homodimer.

Figure 6.
Surface structure of ‘uncleaved Gpd2’ dehydrogenase homodimer. AlphaFold2-Multimer [10-12] predicted surface structure of the ‘uncleaved Gpd2’ enzyme homodimer embedded within the inner mitochondrial membrane of the Immp2lKD-/- KO mouse. The two subunits of the Gpd2 enzyme homodimer (different shades of grey), uncleaved N-terminal signal transit peptides (pink) with visible peptide cleavage recognition sites (yellow). Note: Mouse Gpd2 shares 30% homology with E coli GlpD upon which this structural rendition was modelled [2].
Figure 6.
Surface structure of ‘uncleaved Gpd2’ dehydrogenase homodimer. AlphaFold2-Multimer [10-12] predicted surface structure of the ‘uncleaved Gpd2’ enzyme homodimer embedded within the inner mitochondrial membrane of the Immp2lKD-/- KO mouse. The two subunits of the Gpd2 enzyme homodimer (different shades of grey), uncleaved N-terminal signal transit peptides (pink) with visible peptide cleavage recognition sites (yellow). Note: Mouse Gpd2 shares 30% homology with E coli GlpD upon which this structural rendition was modelled [2].
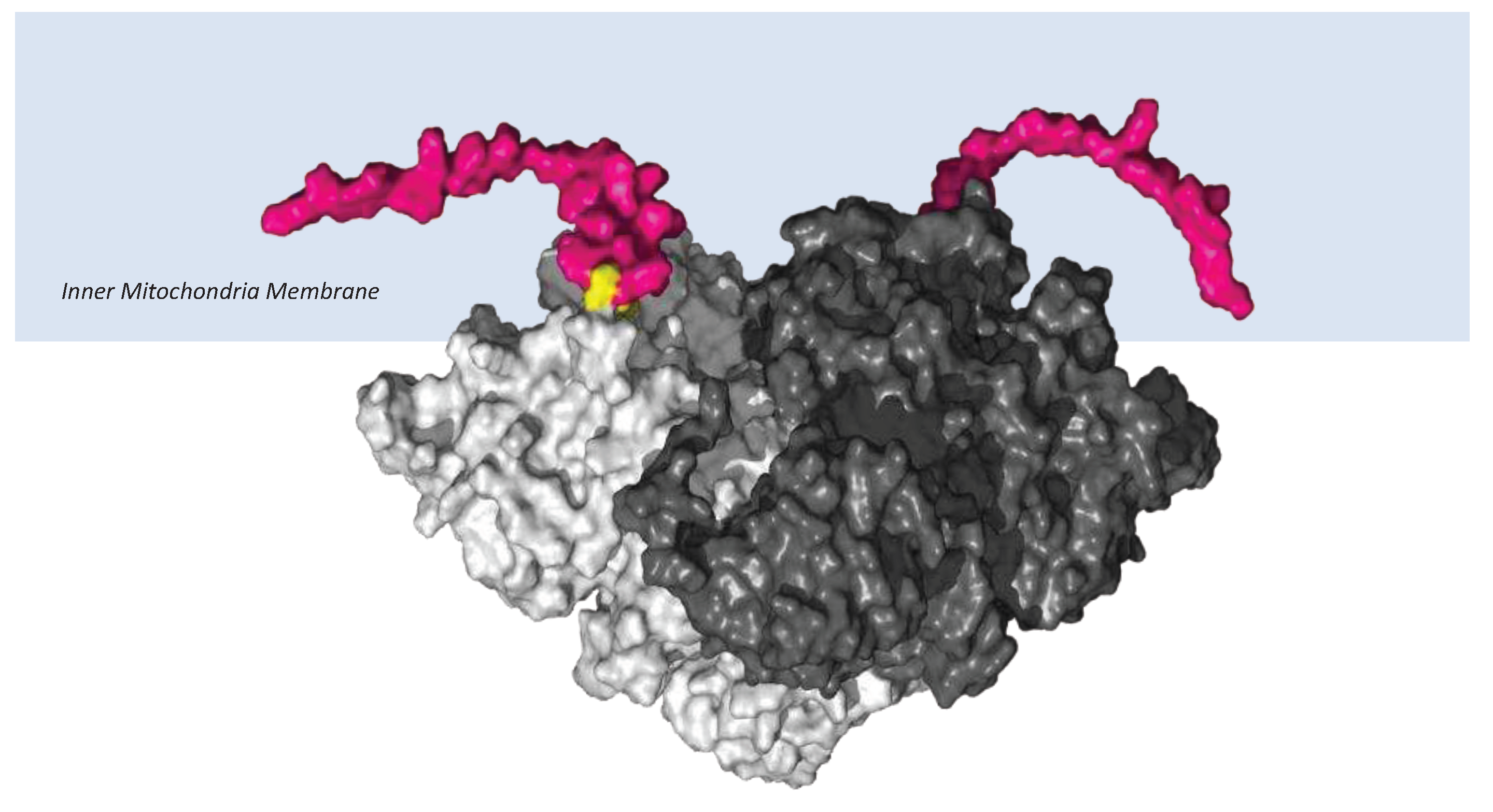
Figure 7.
Cyc1 variant connections with the Cyb respiratory subunit of Complex III. AlphaFold2-Multimer high-accuracy prediction of mouse Cyc1-Cyb of Complex III [10-12] with predicted alignment error (PAE) plots visualised through ChimeraX. A. ‘cleaved Cyc1’ interaction with Cyb. B. ‘uncleaved Cyc1’ interaction with Cyb as found in Immp2lKD-/- KO mouse. C. PAE plot of ‘uncleaved Cyc1’-Cyb where blue colour intensity depicts higher confidence. Upper left and lower right quadrants of PAE plot represent the confidence of the predicted structure of the unitary respiratory subunits whereas the lower left and upper right quadrants depict the confidence of the predicted respiratory subunit complex of Cyc1 (blue), Cyb (wood) and the ‘uncleaved’ signal transit peptide of Cyc1 (orange). D. Magnified lateral view of image B - highlighting the contact forming residues between the ‘uncleaved’ signal transit peptide (Orange) of Cyc1 (blue) and Cyb (wood). The contact forming residues are shown in green and the contacts are shown as yellow dotted lines.
Figure 7.
Cyc1 variant connections with the Cyb respiratory subunit of Complex III. AlphaFold2-Multimer high-accuracy prediction of mouse Cyc1-Cyb of Complex III [10-12] with predicted alignment error (PAE) plots visualised through ChimeraX. A. ‘cleaved Cyc1’ interaction with Cyb. B. ‘uncleaved Cyc1’ interaction with Cyb as found in Immp2lKD-/- KO mouse. C. PAE plot of ‘uncleaved Cyc1’-Cyb where blue colour intensity depicts higher confidence. Upper left and lower right quadrants of PAE plot represent the confidence of the predicted structure of the unitary respiratory subunits whereas the lower left and upper right quadrants depict the confidence of the predicted respiratory subunit complex of Cyc1 (blue), Cyb (wood) and the ‘uncleaved’ signal transit peptide of Cyc1 (orange). D. Magnified lateral view of image B - highlighting the contact forming residues between the ‘uncleaved’ signal transit peptide (Orange) of Cyc1 (blue) and Cyb (wood). The contact forming residues are shown in green and the contacts are shown as yellow dotted lines.
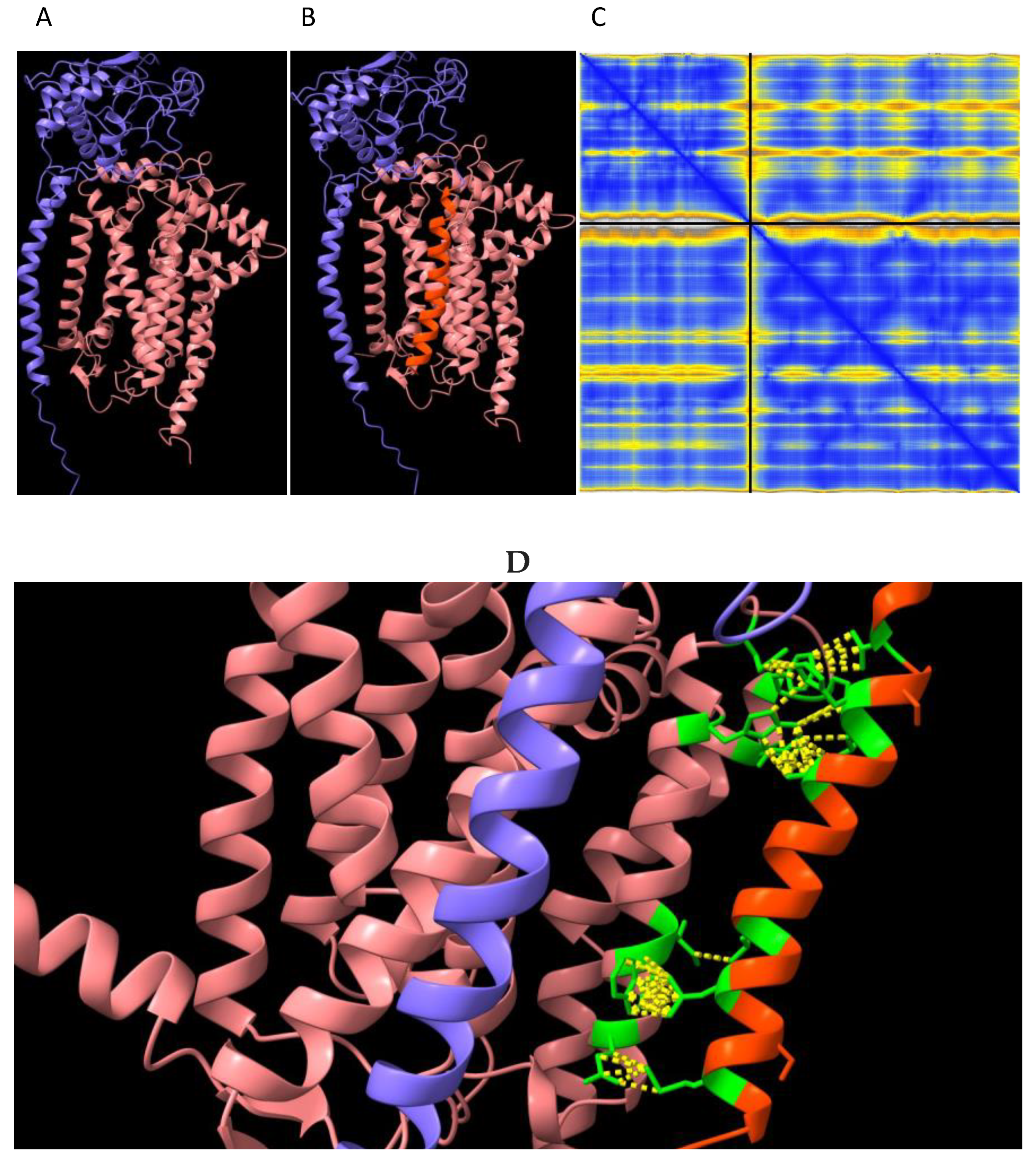
Figure 8.
Immp2lKD-/- KO lowered oxygen consumption rates in primary mouse embryonic fibroblast cell lines (MEFs).Primary MEFs were generated from embryo litter mates and harvested at the same time and grown and analysed under identical conditions in DMEM cell culture media for the determination of oxygen consumption rates (OCR). Two MEFs derived from homozygote male Immp2lKD-/- KO mouse embryos ‘H3’ and ‘H10’ were compared with the OCR of one male wildtype litter mate MEF ‘WT7’.
Figure 8.
Immp2lKD-/- KO lowered oxygen consumption rates in primary mouse embryonic fibroblast cell lines (MEFs).Primary MEFs were generated from embryo litter mates and harvested at the same time and grown and analysed under identical conditions in DMEM cell culture media for the determination of oxygen consumption rates (OCR). Two MEFs derived from homozygote male Immp2lKD-/- KO mouse embryos ‘H3’ and ‘H10’ were compared with the OCR of one male wildtype litter mate MEF ‘WT7’.
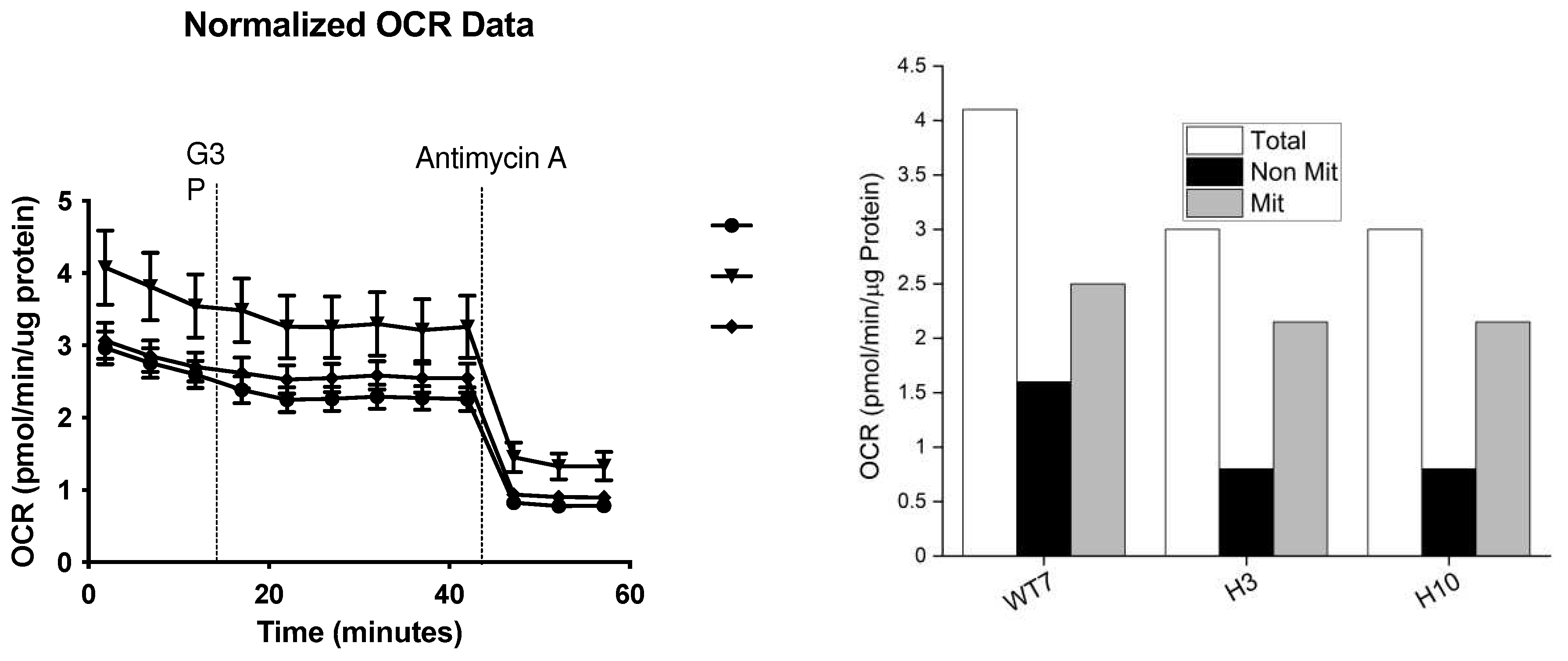

Table 1.
Comparative analysis of respiration rates in mouse embryonic fibroblast cell lines (MEFs).
| MEF Cell Line | TOTAL OCR pmol/min/ug protein |
Non-mitochondrial Respiration |
Mitochondrial Respiration |
| Wildtype - WT7 | 4.1 | 1.6 (39% of Total) | 2.5 (61% of Total) |
| Immp2l KO - H3 | 3.0 (73%) | 0.8 (50%) | 2.2 (88%) |
| Immp2l KO - H10 | 3.0 (73%) | 0.8 (50%) | 2.2 (88%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



