
Submitted:
01 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
Visceral leishmaniasis (VL) is a chronic systemic disease. In Brazil this infection is caused by Leishmania (Leishmania) infantum. Extracellular vesicles (EVs) released by Leishmania species have different functions like modulation of host immune system, inflammatory response, among others. This study evaluated the participation of EVs from L. (L.) infantum (Leish-EVs) in stimulation of the humoral and cellular immune responses of hosts with VL. Promastigotes were cultivated in 199 medium and, in the log phase of growth, they were centrifuged, washed, resuspended in RPMI medium and incubated for 2 to 24h, at 25ºC or 37°C to release Leish-EVs. This dynamic was evaluated by transmission (TEM) and scanning (SEM) electron microscopies, as well as nanoparticle tracking analysis (NTA). The results suggested that parasite penetration in mammal macrophages requires more Leish-EVs than those living in insect vectors, since promastigotes incubated at 37°C released more Leish-EVs than those incubated at 25ºC. In comparison to the control group, infected THP-1 cells produced a significantly higher concentration of EVs (THP-1 cells-EVs). Similar results were obtained when THP-1 cells were treated with Leish-EVs or a crude Leishmania antigen. These data suggest that the concentration of host-EVs-Can distinguish between infected and uninfected cells. THP-1 cells treated with Leish-EVs expressed more IL-12 than control THP-1 cells, but were unable to produce IFN-γ. These same cells produced IL-10, which inhibited TNF and IL-6. Equally, THP-1 cells treated with Leish-EVs up-expressed miR-21-5p and miR-146a-5p. These findings indicate that the increased levels of miR-21-5p and miR-146a-5p observed in Leish-EVs-treated-THP-1 cells lead to immunosuppression and the secretion of pro-inflammatory cytokines.
Keywords:
Extracellular vesicles
; Leishmania (Leishmania) infantum
; THP-1 cells
; Cytokines
; miRNAs
1. Introduction
Visceral leishmaniasis (VL) is caused by protozoans classified as Leishmania (Leishmania) donovani complex, whose transmission occurs by infected female sandflies. VL is included in the neglected diseases, since it is an infectious disease lacking effective, affordable treatments [1,2]. This infection is the most severe form of leishmaniasis, affecting humans and dogs in peridomestic and domestic transmission focus. If adequate treatment is not initiated in a timely manner, the condition can progress to death in 90% of cases in both humans and animals [3,4,5,6,7].
The global prevalence of leishmaniasis renders it among the top ten neglected tropical diseases, with over 12 million individuals’ affected around 1 billion people at risk of infection [7]. VL has spread to several regions worldwide and can be found in Africa, Southern Europe, Americas and Asia, with Bangladesh, Sudan, Brazil, and Nepal being the most affected countries [1,3,7,8]. Brazil alone accounts for approximately 90% of the cases in Latina America. Presently, the disease poses a threat to both humans and canine, in wild, rural, and urban environments.
In Brazil, VL is caused by Leishmania (Leishmania) infantum [9,10], which is transmitted through the primary vector, Lutzomyia longipalpis carrying extracellular promastigotes. Following the infection, amastigotes multiply through binary division within cells of the host’s mononuclear phagocytic system [8,11].
One of the crucial mechanisms employed by pathogens to spread effector molecules to guarantee the invasion in the host cells is by the extracellular vesicles (EVs), which are naturally released from all cell types. However, they are enclosed by a lipid bilayer due to the absence of a nucleus; they cannot replicate [12,13,14]. These vesicles are classified into different types based on their size and composition. The largest, apoptotic bodies arise from the outward fragments of apoptotic cells, resulting in a 500–5000 nm in diameter phosphatidylserine-rich vesicles. Microvesicles are particles of 100–1000 nm in diameter that are formed from the plasma membrane and are enriched with phosphatidylserine and cholesterol. Exosomes, the littlest EVs (30–150 nm) are formed by the exocytosis of multivesicular bodies (MVBs). This process liberates intraluminal vesicles upon fusion with the plasma membrane [15,16,17,18]. Studies have shown that different cell types use EVs as mediators of intracellular communication, facilitating the transport of modulatory proteins, lipids, and nucleic acids to short– and long–distance cells. Furthermore they potentiate organ cross-talk, thus playing a critical role in various biological processes [18,19]. Recent research suggests that the identification and characterization of EVs may have significant implications in the diagnosis and treatment of various diseases. To this end, a better understanding of the biology of EVs is imperative [18,19,25].
Previous studies have demonstrated that some Leishmania species secrete compounds involved in pathogenesis. These compounds or virulence factors are transported to host cells by EVs. This, in turn, causes immunosuppression and prepares the host cells for parasite infection [20]. Further, studies conducted on mice and macrophages complement these data and suggest that Leishmania EVs (Leish-EVs) have an impact on the regulation and signaling pathways of the immune system [20,21,22,23,24].
Based on these findings, the main objective of this study was to investigate the role of EVs obtained from L. (L.) infantum in the stimulation of the humoral and cellular immune responses of hosts suffering from VL.
2. Materials and Methods
2.1. Ethical Statements
This study was conducted in compliance with the recommendations of the Ethics Committee for Animal Use of the Adolfo Lutz Institute (Comissão de Ética no Uso de Animais do Instituto Adolfo Lutz, CEUA-IAL) and the National Experimentation Control Council (Conselho Nacional de Controle de Experimentação, CONCEA). Both committees approved this study (Protocol: A-CEUA-003).
2.2. L. (L.) Infantum Cultures and Production of Crude Leishmania Antigen (CLA) and Leish-EVs
The protocols were prepared as described before [25]. Promastigotes of L. (L.) infantum (MHOM/BR/1972/LD) were maintained at 25ºC, in 199 medium, pH 7.2 containing human urine 50μL/mL, gentamicin 30 μg/mL, fetal bovine serum (FBS) 10% and hemin 50μL/mL. For use in experiments, promastigotes were collected from a culture medium in the log phase of growth, counted, washed 3 times, and concentrated by centrifugation (3,000 × g for 10 min) with sterile phosphate-buffered saline (PBS).
For CLA preparation, promastigotes dissolved in sterile in PBS (2 mL) were ruptured in Disruptor (TissueLyser, Qiagen) by agitation by 8 cycles of 4 min (50 oscillations/second) with intervals of 2 min between it cycles and, then, sonicated for 10 min. After optical microscopy for certification of the parasite lysis, the CLA was diluted in 0.3 M NaCl.
To obtain Leish-EVs, promastigotes were first concentrated in a centrifuge (2,000 × g for 15 min) and washed with sterile PBS for 5 times. The parasites were resuspended in RPMI medium (1 mL) and incubated for 2 h at 25ºC for release vesicles in the culture medium. The mixture, which contained both promastigotes and Leish-EVs was filtered using a 0.22 μm filter to remove the parasites.
A BCA protein kit (Pierce) was used to predict the concentrations of protein of the Leish-EVs and CLA according to the manufacturer’s instructions. A NanoDrop spectrophotometer (Thermo Fisher Scientific) at 280 nm also was used for the same purpose. Next, aliquots were treated with 10 μg/mL of a cocktail of protease inhibitors containing per mL, 10 µM EDTA, 1.3 µM Bestatin, 0.14 µM E-64, 10 nM Leupeptin, 20 µM AEBSF (4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride), 3 nM Aprotinin (Sigma-Aldrich). Thereafter, each aliquot was stored at –20°C until analysis.
For immunological reactions, the supernatants containing Leish-EVs were transferred to Ultra-Clear centrifuge tubes (6 mL tube for SW-55 rotor), (Beckman Coulter, Brea, CA, USA) and completed the volume (6 mL) with filtered PBS. Samples were ultracentrifuged at 100,000 × g for 60 minutes at 25ºC in a Beckman® Coulter L8-80M centrifuge. The pellet containing Leish-EV was suspended in 100 µL of filtered PBS. For ultrastructural analyses, promastigotes were analyzed in 5 moments with incubation times of T0, T2, T4, T6, and T24 h.
2.3. Ultrastructural Analyses of Promastigotes Releasing Leish-EVs by Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM)
For ultrastructural analyses in TEM and SEM of promastigotes releasing Leish-EVs, 5 sets of parasites were analyzed based in incubation time for EV release at temperatures of 25°C or 37°C. They were: 0 (in the beginning), 2, 4, 6, and 24 h.
For analysis of Leish-EVs by TEM, supernatants containing Leish-EVs were transferred to Ultra-Clear centrifuge tubes (6 mL tube for SW-55 rotor, Beckman Coulter, Brea, CA, USA), and volume was completed to 6 mL of filtered PBS. Tubes were ultracentrifuged at 100,000 × g for 60 min at 25ºC in a Beckman Coulter L8-80M centrifuge. Next, pellets containing Leish-EVs were suspended in filtered PBS (100 µL).
Promastigotes releasing Leish-EVs or only Leish-EVs were fixed with 2.5% glutaraldehyde, 4 % paraformaldehyde in 0.1M sodium cacodylate buffer, pH 7.4 at least 2 h. Then, pellets were rinsed in the same buffer and post-fixed in a solution containing 1% osmium tetroxide, 0.8% ferrocyanide, and 5 mM calcium chloride; washed in 0.1 M sodium cacodylate buffer; dehydrated in graded acetone and embedded in Epon resin. Ultrathin sections (100 nm) were obtained in a Sorvall ultramicrotome; stained with uranyl acetate and lead citrate; and observed under a Transmission Electron Microscope JEOL (model JEM 1011, Peabody, MA, USA) operating at 80 kV. Images were captured on a charge-coupled device camera (model 785 ES1000W, Gatan, Pleasanton, CA, USA) and the Gatan version 1.6 programs.
For high-resolution SEM analysis, after incubations, 1 x 106/mL promastigotes were washed twice in filtered PBS and adhered on coverslips previously coated with 0.1% aqueous poly-L-lysine (Sigma) for at least 2 h at room temperature. Next, coverslips were fixed in 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.2, containing 0.146 mol/L sucrose and 5 mmol/L CaCl2 for 1h at room temperature. The parasites were post-fixed in 1% osmium tetroxide (OsO4) for 15 min and then dehydrated in increasing concentration series of ethanol - 7.5%, 15%, 30%, 50%, 70%, 90%, and 100% (v/v) – for 15 min in each step. Next, the samples were critical point-dried with CO2 and sputter-coated with a thin layer (2 nm) of platinum. The images were recorded at accelerating voltages of 1 Kv on microscopes equipped with the Field Emission Gun (FEG): a Quattro ESEM (ThermoFisher, USA) or an Auriga High-Resolution SEM (Zeiss, Germany).
2.4. THP-1 Cell Line and THP-1 Released EVs (THP-1-EVs) Production
The cell line used in experiments was the THP-1 cell line (ATCC nº TIB 202–The Global Bioresource Center, USA), a human pro-monocyte cell line derived from an acute monocytic leukemia patient. THP-1 cell line was maintained in culture bottles containing RPMI medium, 20% FBS, 2 mM glutamine and 10% gentamicin, at 37°C in 5% CO2. For experiments, THP-1 cells (5 x 105 cells/well) were plated, in duplicate, in 24-well plates containing the same medium, stimulated and differentiated by Phorbol 12-Myristate 13-Acetate (PMA) 160 ng/mL and non-adherent cells were removed after macrophage adhesion.
After the multiplicity of infection (MOI) calculation, macrophages were infected with promastigotes at proportion 5:1. In parallel, THP-1-cells were treated with CLA or Leish-EVs at a concentration of 20 µg/well. For each experiment, a normal control was included, which constituted the same macrophage concentration but without infection or treatment. All experiments were performed in duplicate and THP-1 cells were incubated at 37°C for 24 h, in 5% CO2 to release THP-1-EVs. Following, supernatants (500 μL/well) were collected for the investigation of THP-1-EV concentrations by NTA. Macrophages and supernatants also were collected for the determination of cytokines and miRNAs expression respectively. Both macrophages and supernatants were kept at -70°C until experiments.
2.5. Concentration Determination of Leish-EVs and THP-1-EVs by Nanoparticle Tracking Analysis (NTA)
Each experiment was done twice to confirm results. Leish-EVs and THP-1-EVs were evaluated for concentration/mL by nanoparticle tracking analysis (NTA) on the NanoSight NS300 equipment (Malvern- NanoSight™, NTA 3.0), coupled to an automatic sCMOS camera with a wavelength of 532 nm. NanoSight computes size and particle number based on the measured Brownian motion. The coefficient and hydrodynamic radius were determined using the Stokes–Einstein equation and results were exhibited as particle size distribution. Captures and analyses were done according to the equipment protocol. Data were shown as the average and standard deviation of three video recordings of 30 seconds per sample. Since NTA is accurate between particle concentrations in the range of 2 x 107 to 2 x 109/mL, the aliquots (EVs) were diluted before analysis in filtered PBS that was, also, used as control. Next the relative concentration was calculated according to dilution factor.
2.6. Immunological Investigations
The experiments were carried out to investigate whether Leish-EVs could stimulate the humoral response in infected hosts. For this purpose, five serum samples from dogs with canine visceral leishmaniasis (Can-VL) were tested by Immunoblotting and Dot-Blot, using Leish-EVs as antigen and CLA, as a positive antigen control. Five from healthy dogs were used for negative control. The canine sera have been previously tested for Can-VL by ELISA, Fast test, and Real-Time PCR [25,26].
For immunoblotting, CLA or Leish-EVs were solubilized in lysis buffer (2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 60 mM Tris-HCl, pH 6.8 and 0.002% bromophenol blue), boiled and run in 12% polyacrylamide-SDS gels. After visualization, by silver staining, Leish-EVs, and CLA proteins were transferred to nitrocellulose membranes, at room temperature, membranes were blocked with 5% skim milk-PBS, and washed for 5 times during 5 min with PBS. Proteins, in membranes, were incubated for 18 h at 4°C with serum samples diluted 1:50 in PBS-5% skimmed milk. After 2 h and more five washes, membranes were incubated for 2h at room temperature, under agitation with a peroxidase-conjugated rabbit anti-dog IgG, diluted 1:1,500 in 5% skim milk-PBS, and washed again.
For Dot blots, strips of nitrocellulose membranes with 0.45 μm in diameter were incubated with CLA or Leish-EVs (1.5 μg/mL). After five washes with PBS during 5 min, the strips were incubated for 18 h at 4°C with each serum diluted 1:50 in 5% skim milk-PBS. After five more washes with PBS during 5 min and drying, at least 2 h, strips were incubated at room temperature, under agitation for 2 h with peroxidase-conjugated rabbit anti-dog IgG (diluted 1:4,000 in 5% skim milk-PBS) and washed again.
Bound antibodies from Dot blots and immunoblotting were visualized after treatment with chemiluminescence western blotting substrate (Pierce ECL Western Solution, Thermo Scientific). The images were captured on documenting gel with a chemiluminescence filter (UVITEC, Cleaver Scientific) in a Blot Scanner (C-DiGit).
2.7. Cytokine and miRNA Analysis by Gene Expression in Quantitative Real Time PCR (qPCR)
To evaluate whether Leish-EVs could stimulate the cellular immune response, THP-1 cells (5×105cells/well) were incubated for 24h with Leish-EVs (20 µg). For each experiment, a normal control was included, which constituted the same cell concentration but without treatment. Before the experimental process and handling samples, all materials and working surfaces were cleaned with RNase decontamination solution (RNaseZap – Thermo Fisher), in order to minimize the RNA degradation.
Reactions for cytokine and miRNA expression were performed as described before [27,28]. For cytokine investigation, RNA molecules were extracted from THP-1 cells using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Isolated RNA molecules were treated with RNA later to remove residual genomic DNA. Next, 10 μL of each sample was reverse-transcripted (RT) using a high-capacity cDNA reverse transcription kit in a Veriti 96-Well Thermal Cycler, according to the manufacturer’s instructions under the following thermal conditions: 10 min at 25°C, 120 min at 37°C followed by 5 min at 85°C. cDNA samples were stored at -70°C until use in qPCR. Each qPCR amplification mixture contained 5 μL of 2X TaqMan Universal PCR Master Mix and 0.5 μL TaqMan Gene Expression Assays were mixture with the following genes: Interferon gamma (IFN-γ), Interleukin 10 (IL-10), Interleukin 12 (IL-12), Interleukin 6 (IL-6), Transforming growth factor beta (TGF-β), Tumor necrosis factor (TNF), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Assay IDs are shown in Table 1. Template cDNA (2 µL) and 2.5 µL of RNAse-free water were added to a total volume of 10 µL. Reactions were prepared in duplicate for each sample and all assays. Samples were amplified and detected using a StepOne Real-Time PCR System (Applied Biosystems) using the following thermal profile: 2 min, 50° C, and 95° C for 10 min, followed by 40 cycles performed at 95°C for 15 sec and 60°C for 1 min. The results with no amplification in qPCR were repeated at least two times.
In parallel miRNA expression profiles were assessed in supernatants of the same samples using the miRNeasy Serum/Plasma Kit (Qiagen, Valencia, CA) and miRNAs were eluted into 30 µL of nuclease-free water. The normalization of miRNA expression started in miRNA extractions, since each sample was spiked with 5 µL (25 fmol) of the spike-in control (Caenorhabditis elegans miRNA, the synthetic molecule cel-miR-39, Ambion). Cel-miR-39-3p was chosen as an exogenous gene. The extractions well performed were confirmed with the amplification of the exogenous miRNA by qPCR. The cDNA synthesis was performed using 2 μL of total RNA in RT-PCR using TaqMan Advanced miRNA cDNA synthesis kit (Applied Biosystems). The protocols was done as the to manufacturer's instructions with four steps and under the following thermal conditions: 45 min at 37° C, 10 min at 65º C for poly (A) tailing reaction; 60 min at 16° C for ligation reaction; 15 min at 42° C, 5 min at 85º C for reverse transcription reaction; 5 min at 95° C, followed by 14 cycles at 95° C for 3 sec and 60º C for 30 sec; stop reaction at 99° C for 10 min for miR-Amp reaction. The cDNA samples were diluted 1:10, according to the manufacturer's instructions. qPCR was performed in a customized assay produced by Applied Biosystems. Each qPCR amplification mix contained TaqMan Fast Advanced Master Mix (5 µL), cDNA (2.5 µL), RNAse-free water (2 µL), and TaqMan Advanced miRNA Assays (0.5 µL) for each gene that were: miR-21-5p, miR-146a-5p, miR-125b-5p and miR-144-3p. Table 2 describes, in detail, each miRNA species. Reactions were performed in duplicate in a final volume of 10 µL. A negative control (amplification mix only) was included in which reaction. The fast mode of the StepOne Real-Time PCR Systems equipment (Applied Biosystems) was used with the program: one cycle of 95° C at 20 sec, 40 cycles of 95 C at 1 sec, and 60° C for 20 sec.
2.8. Data Analysis
Statistical analyses and graphics were performed using Graph Pad Prism software version 8.4.3 (San Diego, CA, USA). Unpaired Test one-tailed test based on a critical value of p ⩽ 0.05 were used to compare concentration and size of Leish-EVs and THP-1-EVs. Values are presented as mean ± SEM (standard error of the mean). The indicated data were representative of at least two individual experiments.
For cytokine and miRNA expressions, the data were based on an amplification plot, which represented the cycle threshold (CT) values for each sample. The values of expression (cytokines and miRNAs) are shown as Relative Quantification (RQ) and were calculated by the comparative CT method (2-ΔΔCT) as described before [29]. THP-1 cells without infection or treatment (control) were considered calibrators, whose values were always 1.0 according to the comparative CT method. To describe how many times the gene under investigation is up expressed, the use of a calibrator is recommended. To perform this formula, the divergence in cDNA concentration between samples (ΔCT value) was corrected by subtracting the CT value obtained from the exogenous gene (cel-miR-39-3p) or gene endogenous for cytokines (GAPDH). Next, the ΔCT values of the tested samples were decreased from the ΔCT value of the calibrator.
3. Results
The initial experiments were performed to define the ideal conditions for the production of Leish-EVs and THP-1-EVs. They included the promastigote number for each cell (MOI), ideal temperature, incubation time, and transformation of THP-1 monocytes in macrophages.
3.1. Biological Characteristics of Leish-EVs Released by L. (L.) Infantum
These experiments were aimed to investigate how L. (L.) infantum promastigotes release EVs over time in different environments. Figure 1 shows images captured by TEM and SEM of promastigotes releasing Leish-EVs, at 25°C (vector temperature), in five periods. Panels 1A and 1F show promastigotes at the onset of Leish-EV releasing (T=0). The images show the Leish-EVs inside the promastigotes and preparing to be released by the plasma membrane. After 2 h (Panels 1B and 1G); 4 h (Panels 1C and 1H); 6 h (Panels 1D and 1I), and 24 h (Panels 1E and 1J) incubation, it was possible to note different moments of Leish-EV release through the plasma membrane and already released to external environment. Figure 2 shows, also, images captured by TEM and SEM, in the same five periods, of promastigotes releasing Leish-EVs, but at 37°C (mammal body temperature). In these images it was possible to note promastigotes releasing more Leish-EVs than those from Figure 1. The image captured by TEM in Figure 3A shows concentrated Leish-EVs released by promastigotes.
The images captured by TEM and SEM in Figure 1 and Figure 2 were confirmed by NTA results, concerning the concentration of Leish-EVs (Figure 3B). However, the mean sizes of Leish-EVs were similar in both incubation periods. (Figure 3C) Promastigotes incubated in RPMI medium at 25°C for 2, 4, 6 and 24 h released the mean concentrations (and sizes) of 2.54 x 108 (150.8 nm) 7.15 x 108 (195.9 nm), 9.45 x 108 (205.5 nm) and 2,45 x 109 (142.3nm) Leish-EVs/mL, respectively. Those incubated at 37°C, the mean concentrations (and sizes) were 1.75 x 109 (142.3), 6.71 x 109 (170.5), 1.71 x 1010 (198.4 nm) and 1.71 x 1010 (150.8 nm) Leish-EVs/mL, respectively.
3.2. THP-1-EVs Releasing Was Stimulated by L. (L.) Infantum Antigens and Leish-EVs
The objective of these experiments was to determine whether uninfected and promastigotes-infected d macrophages could release the same amount of THP-1-EVs. For that, THP-1 cells were infected with promastigotes at proportion 5:1 and incubated for 6 and 24h. Next, concentrations of THP-1-EVs in supernatants were analyzed by NTA. Values illustrate the mean of three readings in the NanoSight instrument and the mean concentration ± SEM (standard error of the mean) of each aliquot prepared in duplicate. The comparison between aliquots was statistically determined by Unpaired Test T, one-tailed. As shown in Figure 4, the mean concentration released by the control aliquot (THP-1) was 1.6 x 109 ± 1.9 x 108 THP-1-EVs/mL. The mean concentration released by THP-1 aliquot incubated with promastigotes during 6 h increased to 2.7 x109 ± 9.5 x 107 THP-1-EVs/mL. THP-1 cells incubated with promastigotes for 24 h released 3.8 x 109 ± 1.5 x 108 THP-1-EVs/mL. The THP-1 cells without infection (control) were released during 24h, 1.8 x109 ± 3.8 x 108 THP-1-EVs/mL. The increase of THP-1-EV production in aliquots from infected THP-1 cells (incubations with 6 and 24 h) compared with control aliquots was statistically different at *p < 0.018 and *p < 0.042, respectively.
Thus, the next experiments were performed to understand whether the excess of EVs was released by promastigotes or not. In parallel, THP-1 cells were treated with CLA (20 µg/well) and, also, incubated for 6 or 24 h. The mean concentration released by control aliquot and incubated for 6 h was 1.0 x 109 ± 3.4 x 108 THP-1-EVs/mL. The mean concentration released by THP-1 aliquots treated with CLA increased to 5.6 x109 ± 3.8 x 108 THP-1-EVs/mL. THP-1 cells incubated at 24 h released 1.5 x 109 ± 7.0 x 107 THP-1-EVs/mL (control). Those treated with CLA and incubated for 24 h released 8.2 x 109 ± 1.0 x 109 THP-1-EVs/mL. The increase of THP-1-EV production compared with both control aliquots was statistically different at *p < 0.022 and **p < 0.006, respectively (Figure 4).
In the next experiment, THP-1 cells were treated with Leish-EVs at a concentration of 20 µg/well and incubated for 6 h or 24 h. The mean concentration released by the control aliquot, incubated for 6h was 1.2 x 109 ± 9.0 x 107 THP-1-EVs/mL. The mean concentration of THP-1 aliquots treated with Leish-EVs was 3.1 x 109 ± 2.9 x 107 THP-1-EVs/mL. Those without treatment (controls) incubated for 24 h, the mean concentration was 1.5 x 109 ± 4.0 x 108 THP-1-EVs/mL. In THP-1 aliquots treated with Leish-EVs, the mean concentration was 3.4 x 109 ± 6.2 x 108 THP-1-EVs/mL. The increase of THP-1-EV production compared with both control aliquots was statistically different at **p < 0.018 and **p < 0.051, respectively (Figure 4).
3.3. Immunological Experiments
The immunological assays evaluated whether Leish-EVs were reactive with sera from infected hosts. The assays were performed using Leish-EVs as antigen. CLA was used as positive control. Figure 5A shows the electrophoretic profile Leish-EVs on 12% SDS-PAGE after silver nitrate staining (strip 1). Next, Leish-EVs were analyzed as antigen in immunoblot using 2 canine sera. One was positive (strip 2) and the other was negative (strip 3) for Can-VL. As positive antigen control, CLA was used against the same sera used for Leish-EVs, strips 4 and 5 respectively. Leish-EVs and CLA were reactive against positive sera (strips 2 and 4). Negative sera showed no reactivity (strips 3 and 5), for both experiments. Equally, Figure 5B shows the same reactivity against Leish-EVs in Dot-Blot in five positive and five negative canine sera for CanVL. CLA was used as control.
3.4. Leish-EVs Stimulated the THP-1 Cells to Produce Cytokines and miRNAs
The results of the gene expression of cytokines produced by THP-1 cells after stimulation with Leish-EVs are shown in Figure 6A. IL-10 and IL-12 were expressed around 7 and 4 times in the THP-1 cells treated with Leish-EVs when compared with the THP-1 cell controls. Already, TGF-β, TNF and IL-6 also were up expressed, although with low intensity (1.51, 1.20 and 1.11 more expressed than THP-1 aliquot controls). IFN-γ was not expressed in the tested samples.
The relative quantification of the four miRNA species (miR-144-3p, miR-21-5p, miR-125b-5p and miR-146a-5p) was estimated from miRNAs purified from THP-1 cells (5×105 cells/well) treated with Leish-EVs (20 µg) during 24h. THP-1 cells without Leish-EV treatment were considered calibrators with fixed value of 1.0. Thus, the values of treated cells inform how many times they were more expressed. Treated THP-1 cells up-expressed the miR-21-5p and miR-146a-5p, with RQ values of 6.97 and 2.73 respectively. These results represented around 7 and 3 times more expressed than THP-1 cells without Leish-EV treatment. miR-125b-5p and miR-144-3p were slightly expressed, with a RQ mean of 1.40 and 0.42, respectively. Figure 6B shows the RQ mean of each miRNA species.
4. Discussion
Diverse studies focused on the interaction between EVs and pathogenic protozoa have highlighted the important role played by these nanoparticles in parasite survival. They facilitate the infection in experimental models [30] and help the parasite to adapt to the host environment [31]. EVs carry virulence factors that impact on modulation of cellular immune response, as already observed in Leishmania species, such as L. (L.) mexicana [24], L. (L.) major [32], L. (L.) amazonensis [33] and L. (L.) donovani [34].
Similar to other Leishmania species, L. (L.) infantum promastigotes were observed to release a greater number of Leish-EVs at 37º C as compared to that incubated at 25º C. The images capitated by electronic microscopy and NTA analyses showed that these nanoparticles have a size and shape characteristic of microvesicles and exosomes. These results suggest that parasite penetration in mammal macrophages require more Leish-EV than those living in insect vectors, and the virulence factors carry them contribute to parasite adaptation and maintenance in host cells. These data are in line with previous studies [24,35].
Macrophages play a dual role during infections caused by the different Leishmania species. They are responsible for both internalizing and destroying parasites, and, at the same time, provide a safe place for Leishmania multiplication. Therefore, these cells are a key to the failure or success of the infection [36].
The experiments conducted using THP-1 cells were performed to understand the relationship between macrophages, L. (L.) infantum and THP-1-EV release during infection. The initial investigations revealed that THP-1 cells infected with promastigotes were capable of producing an increase in the release of THP-1-EVs. Furthermore, the results also demonstrated an increase in the concentrations of THP-1-EVs released from 6 to 24 hours. Additionally, THP-1 cells treated with the CLA or Leish-EVs exhibited the same results. Similar observations were done with EVs from bone marrow in L. (L.) amazonensis [34]. The comparison between control and infected groups showed statistically significant differences. These findings indicate that the concentration of EVs may be able to distinguish between infected and uninfected hosts [25].
The immunological analyses were conducted to investigate the ability of Leish-EVs to stimulate the host's antibody response. The immunoblots and dot-blots used in this study indicated that Leish-EVs have a high potential to be used as biomarkers in serological diagnosis for VL. These data are consistent with our earlier study [25], which had also shown high reactivity against sera from infected hosts.
In natural CanVL infection, the protective cellular immunity is associated with an increase in the Th1 response mediated by IFN-γ and TNF. As a result, macrophage activation and cytotoxic T lymphocytes display an apparent resistance to the infection. However, in the symptomatic infection, the role of IL-4 and IL-10 becomes more prominent, progressing to reduce the effects of Th1 cytokines. T, in turn, leads to a decrease in the production of nitric oxide in macrophages, thereby, preventing the destruction of the parasite [36].
The Leish-EVs released to the extracellular space target the host cells resulting in immunosuppression that enables the parasite to invade those cells [37]. IL-12 drives the Th1 and Th2 responses inducing IFN-γ production [38]. THP-1 cells treated with Leish-EVs were unable to produce IFN-γ, but expressed 3.89 times more IL-12 than untreated THP-1 cells. At the same time, these same cells produced the potent anti-inflammatory cytokine IL-10, which was produced 6.67 times more than untreated macrophages. The high expression of IL-10 effectively inhibits the pro-inflammatory cytokines TNF and IL-6. Although TGF-β was expressed in low levels, it cooperated with IL-10 to cause immunosuppression [37,38,39].
One of the functions of miRNAs is directly to activate cytokine production, which regulates functions related to cellular immune responses [40,41,42,43,44]. Previous studies have detected miRNAs species such miR-21-5p, mir-146a-5p in hosts with VL [25,39,45,46]. Equally, when THP-1 cells were treated with Leish-EVs, miR-21-5p and miR-146a-5p were up-expressed. These findings suggest that the high expression of the miR-21-5p and miR-146a-5p in THP-1 cells treated with Leish-EVs caused immunosuppression and production of inflammatory cytokines. These results are consistent with our previous study that showed that dogs with CanVL had high production of EVs in serum and high expression of miR21-5p and miR146a-5p, which together might contribute to the evolution of CanVL [25].
Author Contributions
VLP-C coordinated the experiments and designed the study. VLP-C, FMC and ABC wrote the manuscript. RG and RMN performed the serological and molecular diagnoses for visceral leishmaniasis. RMN performed the selection of dog samples. VLP-C, ABC, FMC and MMM performed EV isolation and quantified the particles using the NanoSight-NTA; SDS-PAGE, and immunoblot. FMC and ISP performed the genic expression of cytokines and miRNAs. NNT and GMN performed experiments using transmission electron microscopy. RMM, VM and BV performed the experiments using high-resolution scanning electron microscopy. All authors contributed substantially to the data and manuscript writing. They, also, approved the final version submitted for publication and agreed to be accountable for all aspects of the work to ensure that questions about the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This study was supported by grants from the FAPESP (Fundação de Amparo à Pesquisa do Estado de Sao Paulo, Brazil) 2021/02217-3. VLP-C was supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) 303566/2021-3. ABC, FMC, and MMM were supported by scholarship from CAPES (Coordination for the Improvement of Higher Education Personnel (Process SCBA nº88881.689557/2022-0, Program PROAP/ CAPES - AUXPE nº 115/2022.
Acknowledgments
We thank the Graduate Program in Science, Coordinator for Disease Control, Ministry of Health (PPG-CCD-SES/SP), and Coordination for the Improvement of Higher Education Personnel (CAPES). Furthermore, the authors would like to thank the teams from the Rudolf Barth Electron Microscopy Platform (Oswaldo Cruz Institute, Fiocruz, Brazil) and the Advanced Microscopy Unit of the National Center for Structural Biology and Bioimaging (CENABIO, UFRJ) for their technical support.
Data Availability Statement
Data supporting reported results can be requested from the authors.
Conflicts of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entire with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript or financial relationships that could be construed as a potential conflict of interest.
References
- World Health Organization. Leishmaniasis, 2022. Available online: https://www.who.int/health-topics/leishmaniasis#tab=tab (accessed on 2 October 2023).
- Matsumoto, P.S.S; Taniguchi, H.H.; Pereira, V.B.R.; Hiramoto, R.M.; Seviero Rampazzi, K.L.; de Raeffray Barbosa, J.E.; Puci Neto, R.A.; Camprigher, V.M.; de Barros Cortez, L.R.P.; Rahaman, K.R.; Novak, M.; Tolezano, J.E. Efficacies of insecticide dog collars against visceral leishmaniasis in low and high-income areas and the effects for non-collared neighbor dogs. Acta Trop. 2022, 235, 106626. [Google Scholar] [CrossRef] [PubMed]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J. ; den Boer M; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. 2012, 7: e35671. PLoS ONE. [CrossRef]
- Alvar, J.; Yactayo, S.; Bern, C. Leishmaniasis and poverty. Trends Parasitol. 2006, 22, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Dantas-Torres, F. The role of dogs as reservoirs of Leishmania parasites, with emphasis on Leishmania (Leishmania) infantum and Leishmania (Viannia) braziliensis. Vet. Parasitol. 2007, 149, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Dantas-Torres, F.; Otranto, D. Best practices for preventing vector-borne diseases in dogs and humans. Trends Parasitol. 2016, 32, 43–55. [Google Scholar] [CrossRef] [PubMed]
- PAHO–Pan American Health Organization, Visceral leishmaniasis, 2022. Available online: https://www.paho.org/en/topics/leishmaniasis/visceral-leishmaniasis and https://www.paho.org/en/topics/leishmaniasis (accessed on 10 October 2023).
- Grimaldi, G. Jr.; Tesh, R. B. Leishmaniases of the New World: Current concepts and implications for future research. Clin. Microbiol. Rev. 1993, 6, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Degrave, W.; Fernandes, O.; Campbell, D.; Bozza, M.; Lopes, U. Use of molecular probes and PCR for detection and typing of Leishmania-a mini-review. Mem. Inst. Oswaldo Cruz. 1994, 89, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Lainson, R. ; Shaw, J, J. New world leishmaniasis. The neotropical Leishmania species. In: Collier L, Balows A, Sussman M (Eds) Microbiology and Microbial Infectious Diseases, 1998. Volume 5 Topley & Wilson’s 9th, London.
- Lainson, R.; Rangel, E.F. Lutzomyia longipalpis and the eco-epidemiology of American visceral leishmaniasis, with particular reference to Brazil: A review. Mem. Inst. Oswaldo Cruz. 2005, 100, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracellular Vesicles. 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Torrecilhas, A.C.; Soares, R.P.; Schenkman, S.; Fernandez-Prada, C.; Olivier, M. Extracellular vesicles in Trypanosomatids: Host cell communication. Front. Cell. Infect. Microbiol. 2020, 10, 602502. [Google Scholar] [CrossRef] [PubMed]
- Marti, M.; Johnson, P.J. Emerging roles for extracellular vesicles in parasitic infections. Curr. Opin. Microbiol. 2016, 32, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Sampey, G.; Chung, M.C.; Bailey, C.; van Hoek, M.L.; Kashanchi, F.; Hakami, R.M. The carrying pigeons of the cell: Exosomes and their role in infectious diseases caused by human pathogens. Pathog. Dis. 2014, 71, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Sidhom, K.; Obi, P.O.; Saleem, A. Review of exosomal isolation methods: Is size exclusion chromatography the best option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef] [PubMed]
- Szempruch, A.J.; Sykes, S.E.; Kieft Dennison, L.; Becker, A.C; Gartrell, A.; Martin, W.J; Nakayasu, E.S.; Almeida, I.C.; Hajduk, S.L.; Harrington, J.M. Extracellular vesicles from Trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell 2016, 14 164, 246–257. [Google Scholar] [CrossRef]
- Dong, G.; Filho, A.L.; Olivier, M. Modulation of Host-Pathogen Communication by Extracellular Vesicles (EVs) of the Protozoan Parasite Leishmania. Front. Cell Infect. Microbiol. 2019, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Wagner, V.; Minguez-Menendez, A.; Fernandez-Prada, C.; Olivier, M. Extracellular vesicles and leishmaniasis: Current knowledge and promising avenues for future development. Mol. Immunol. 2021, 135, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Clos, J.; de’Oliveira, C.C.; Shirvani, O.; Fang, Y.; Wang, C.; Foster, L.J.; Reiner, N.E. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J. Cell. Sci. 2010, 123, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, Á.M.; Galué-Parra, A.; Pereira, W.L.A.; Pedersen, K.W.; da Silva, E.O. Leishmania 360°: Guidelines for Exosomal Research. Microorganisms. 2021, 2, 2081. [Google Scholar] [CrossRef]
- Hassani, K.; Antoniak, E.; Jardim, A.; Olivier, M. Temperature-induced protein secretion by Leishmania mexicana modulates macrophage signaling and function. 2011, 3: e18724. PLoS ONE. [CrossRef]
- Cruz, A.B.; Carneiro, F.M.; Maia, M.M.; Pereira, I.S.; Taniwaki, N.N.; Namiyama, G.M.; Gava, R.; Hiramoto, R.M.; Pereira-Chioccola, VL. Dogs with canine visceral leishmaniasis have a boost of extracellular vesicles and miR-21-5p up-expression. Parasite Immunol. 2023, 45, e13004. [Google Scholar] [CrossRef]
- Colombo, F.A.; Odorizzi, R.M.; Laurenti, M.D.; Galati, E.A.; Canavez, F.; Pereira-Chioccola, VL. Detection of Leishmania (Leishmania) infantum RNA in fleas and ticks collected from naturally infected dogs. Parasitol Res. [CrossRef]
- Hippólito, D.D.C.; Gomes, A.H.S.; Maia, M.M.; Meira-Strejevitch, C.D.S.; Kanamura, C.T.; Lauletta Lindoso, J.A.; Pereira-Chioccola, V.L. Gene expression profile of cytokines produced in biopsies from patients with American cutaneous leishmaniasis. Acta Trop. 2019, 189, 69–75. [Google Scholar] [CrossRef]
- Cruz, A.B.; Maia, M.M.; Pereira, I.S.; Taniwaki, N.N.; Namiyama, G.M.; Telles, J.P.M.; Vidal, J.E.; Spegiorin, L.C.J.F.; Brandão de Mattos, C.C.; Mattos, L.C.; Meira-Strejevitch, C.D.S.; Pereira-Chioccola, V.L. Human extracellular vesicles and correlation with two clinical forms of toxoplasmosis. PLoS ONE 2020, 15, e0229602. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods. 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nogueira, P.M.; Ribeiro, K.; Silveira, A.C.O; Campos, J.H.; Martins-Filho, A.O.; Bela, S.R.; Campos, M.A.; Pessoa, N.L.; Colli, W.; Alves, M.J.M.; Soares, R.P.; Torrecilhas, A.C. Vesicles from differential innate and chronic imune responses. J. Extracell. Vesicles. 2015, 26, 28734. [Google Scholar] [CrossRef]
- Lambertz, U.; Silverman, J.M.; Nandan, D.; McMaster, W.R.; Clos, J.; Foster, L.J.; Reiner, N.E. Secreted virulence factors and immune evasion in visceral leishmaniasis. J. Leukoc. Biol. 2012, 91, 887–899. [Google Scholar] [CrossRef]
- Hassani, K.; Shio, M.T.; Martel, C.; Faubert, D.; Olivier, M. Absence of metalloprotease GP63 alters the protein content of Leishmania exosomes. PLoS ONE. 2014, 15, e95007. [Google Scholar] [CrossRef]
- Barbosa, F.M.C.; Dupin, T.V.; Toledo, M.D.S.; Reis, N.F.D.C.; Ribeiro, K.; Cronemberger- Andrade, A.; Rugani, J.N.; De Lorenzo, B.H.P.; Novaes, E.; Brito, R.R.; Soares, R.P.; Torrecilhas, A.C.; Xander, P. Extracellular vesicles released by Leishmania (Leishmania) amazonensis promote disease progression and induce the production of different cytokines in macrophages and B-1 cells. Front. Microbiol. 2018, 9, 3056. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Reiner, N.E. Leishmania exosomes deliver preemptive strikes to create an environment permissive for early infection. Front. Cell Infect. Microbiol. 2012, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Tomiotto-Pellissier, F.; Bortoleti, B.T.S.; Assolini, J.P.; Gonçalves, M.D.; Carloto, A.C.M.; Miranda-Sapla, M.M.; Conchon-Costa, I.; Bordignon, J.; Pavanelli, W.R. Macrophage Polarization in Leishmaniasis: Broadening Horizons. Front Immunol. 2018, 9, 2529. [Google Scholar] [CrossRef]
- Barbiéri, C.L. Immunology of canine leishmaniasis. Parasite Immunol. 2006, 28, 329–337. [Google Scholar] [CrossRef]
- Dayakar, A.; Chandrasekaran, S.; Kuchipudi, S.V.; Kalangi, S.K. Cytokines: Key Determinants of Resistance or Disease Progression in Visceral Leishmaniasis: Opportunities for Novel Diagnostics and Immunotherapy. Front Immunol. 2019, 5, 10:670. [Google Scholar] [CrossRef] [PubMed]
- Stäger, S.; Maroof, A.; Zubairi, S.; Sanos, S.L.; Kopf, M.; Kaye, P.M. Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur. J. Immunol. [CrossRef]
- Bragato, J.P.; Rebech, G.T.; Freitas, J.H.; Santos, M.O.D; Costa, S.F.; Eugênio, F.R.; Santos, P.S.P.D; de Lima, V.M.F. miRNA-21 regulates CD69 and IL-10 expression in canine leishmaniasis. PLoS ONE. 2022, 24, e0265192. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004, 23, 281–297. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Shinagawa, K.; Castellino, F.J.; Schorey, J.S. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood. 2007, 110, 3234–3244. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Pandey, R.K.; Prajapati, P.; Prajapati, V.K. Circulating MicroRNAs: Potential and Emerging Biomarkers for Diagnosis of Human Infectious Diseases. Front. Microbiol. 2016, 15, 1274. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of α-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: Another mechanism for initiation and progression of Parkinson's disease? Acta Neuropathol. Commun. 2017, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Lin, X.; Zhou, F.; Li, C.; Wang, X.; Yu, H.; Pan, Y.; Fei, H.; Ma, L.; Zhang, S. A scaffold laden with mesenchymal stem cell-derived exosomes for promoting endometrium regeneration and fertility restoration through macrophage immunomodulation. Acta Biomater. 2020, 113, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Varikuti, S.; Verma, C.; Holcomb, E.; Jha, B.K.; Viana, A.; Maryala, R.; Lamenza, F.; McElwain, BK. , Doni, N.Y.; Papenfuss, T., Oghumu, S.; Gannavaram, S.; Nakhasi, H.L.; Satoskar, A.R. MicroRNA-21 Deficiency Promotes the Early Th1 Immune Response and Resistance toward Visceral Leishmaniasis. J. Immunol. 2021, 1, 1322–1332. [Google Scholar] [CrossRef]
- Ganguly, S.; Ghoshal, B.; Banerji, I.; Bhattacharjee, S.; Chakraborty, S.; Goswami, A.; Mukherjee, K.; Bhattacharyya, S.N. Leishmania survives by exporting miR-146a from infected to resident cells to subjugate inflammation. Life Sci. Alliance 2022, 24, e202101229. [Google Scholar] [CrossRef]
Figure 1.
L. (L.) infantum releasing Leish-EVs by membrane surface at 25°C. Panels A to E show the images captured by TEM and Panels F to J, by SEM of promastigotes shedding Leish-EVs by membranes in the beginning of incubation in culture medium at 25º C (Panels A and F); after 2h (Panels B and G); 4h (Panels C and H); 6h (Panels D and I) and 24 h (Panels E and J). The arrows indicate the Leish-EVs. Bars = 2 µm (Panels F–J).
Figure 1.
L. (L.) infantum releasing Leish-EVs by membrane surface at 25°C. Panels A to E show the images captured by TEM and Panels F to J, by SEM of promastigotes shedding Leish-EVs by membranes in the beginning of incubation in culture medium at 25º C (Panels A and F); after 2h (Panels B and G); 4h (Panels C and H); 6h (Panels D and I) and 24 h (Panels E and J). The arrows indicate the Leish-EVs. Bars = 2 µm (Panels F–J).
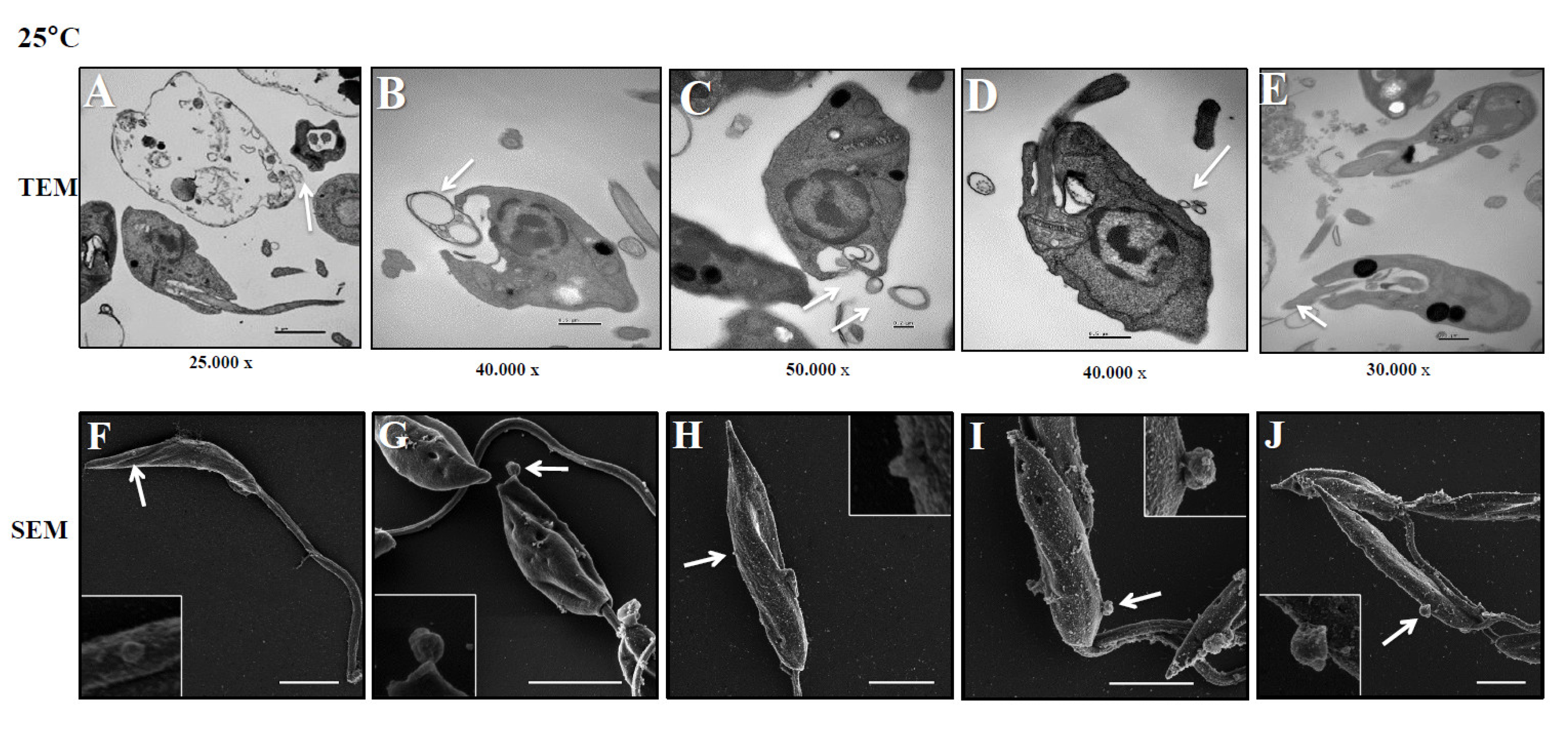
Figure 2.
L. (L.) infantum releasing Leish-EVs by membrane surface at 37°C. Panels A to E show the images captured by TEM and Panels F to J, by SEM of promastigotes shedding Leish-EVs by membranes in the beginning of incubation in culture medium at 37º C (Panels A and F); after 2h (Panels B and G); 4h (Panels C and H); 6h (Panels D and I) and 24 h (Panels E and J). The arrows indicate the Leish-EVs. Bars = 2 µm (Panels F–J).
Figure 2.
L. (L.) infantum releasing Leish-EVs by membrane surface at 37°C. Panels A to E show the images captured by TEM and Panels F to J, by SEM of promastigotes shedding Leish-EVs by membranes in the beginning of incubation in culture medium at 37º C (Panels A and F); after 2h (Panels B and G); 4h (Panels C and H); 6h (Panels D and I) and 24 h (Panels E and J). The arrows indicate the Leish-EVs. Bars = 2 µm (Panels F–J).
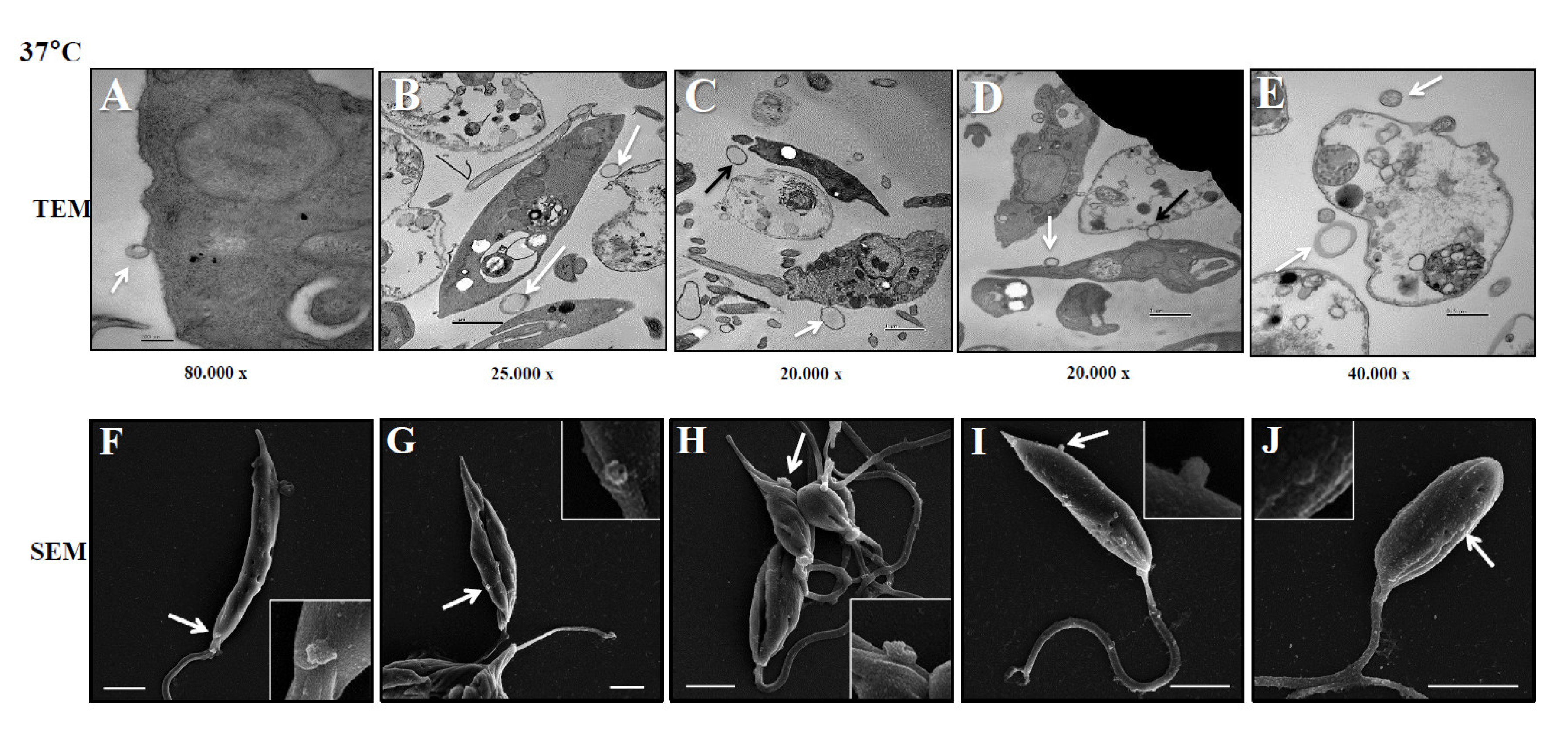
Figure 3.
Biological characteristics of Leish-EVs. Panel A shows a TEM image of Leish-EVs released by promastigotes with size compatible with microvesicles and exosomes. Distribution of concentration/mL (Panel B) and size (nm) (Panel C) of Leish-EVs released by 1 x 108 promastigotes incubated in culture medium at 25°C or 37°C for 2, 4, 6 and 24 h. The data represent three reads per aliquot and each experiment was performed at least 3 times.
Figure 3.
Biological characteristics of Leish-EVs. Panel A shows a TEM image of Leish-EVs released by promastigotes with size compatible with microvesicles and exosomes. Distribution of concentration/mL (Panel B) and size (nm) (Panel C) of Leish-EVs released by 1 x 108 promastigotes incubated in culture medium at 25°C or 37°C for 2, 4, 6 and 24 h. The data represent three reads per aliquot and each experiment was performed at least 3 times.
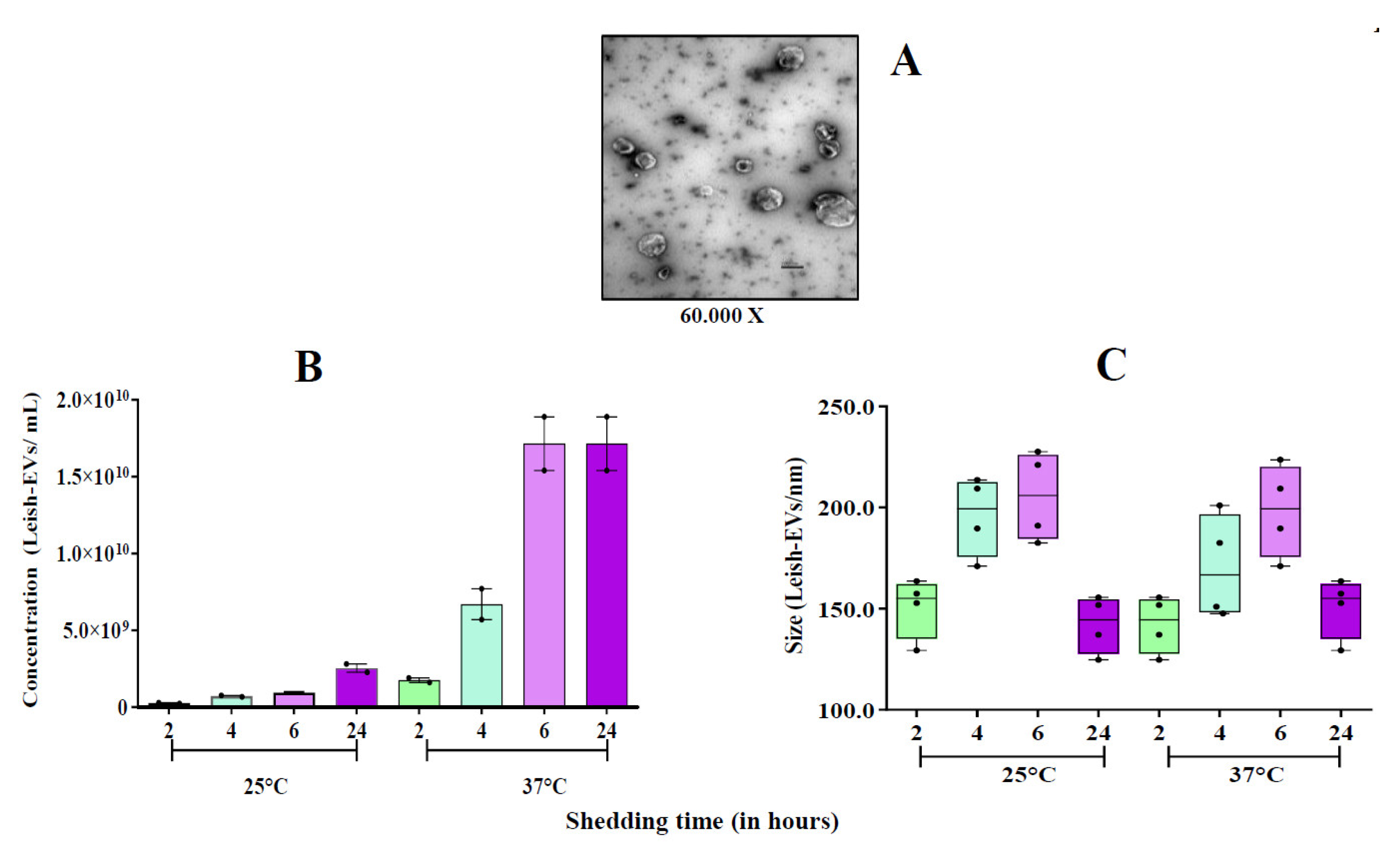
Figure 4.
Stimulation of THP-1-EVs releasing. Mean concentration of THP-1-EVs in supernatants analyzed by NTA after incubations during 6 h and 24 h (red bars) with promastigotes (MOI=5:1); after CLA treatment (20µg/well) (green bars); and Leish-EVs (20 µg/well) (yellow bars). THP-1 cells received only culture medium in controls (blue bars). The data represent three reads per sample.
Figure 4.
Stimulation of THP-1-EVs releasing. Mean concentration of THP-1-EVs in supernatants analyzed by NTA after incubations during 6 h and 24 h (red bars) with promastigotes (MOI=5:1); after CLA treatment (20µg/well) (green bars); and Leish-EVs (20 µg/well) (yellow bars). THP-1 cells received only culture medium in controls (blue bars). The data represent three reads per sample.
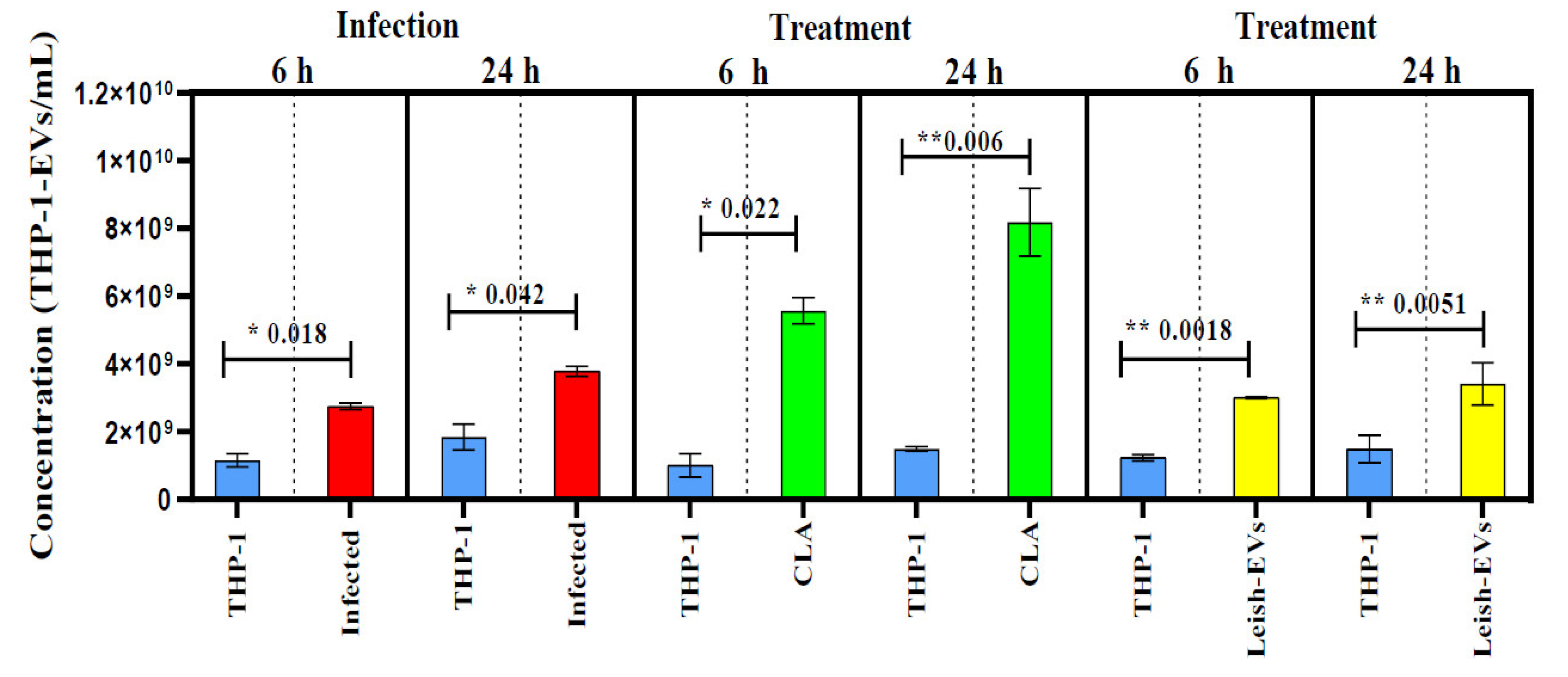
Figure 5.
Reactivity of Leish-EVs against canine sera. Panel A: Electrophoretic analysis of Leish-EVs on 12% SDS-PAGE stained with silver nitrate (strip 1). Immunoblotting using Leish-EVs as antigen against a positive (strip 2) and negative (strip 3) canine sera for Can-LV. CLA, as positive antigen control was used against the same sera used for Leish-EVs. Positive (strip 3) and negative (strip 5) sera. Panel B. Reactivity in Dot-Blot using Leish-EVs as antigen against five positive and five negative sera for Can-LV. As positive control CLA was used as antigen against 4 sera (two positive and two negative for Can-LV).
Figure 5.
Reactivity of Leish-EVs against canine sera. Panel A: Electrophoretic analysis of Leish-EVs on 12% SDS-PAGE stained with silver nitrate (strip 1). Immunoblotting using Leish-EVs as antigen against a positive (strip 2) and negative (strip 3) canine sera for Can-LV. CLA, as positive antigen control was used against the same sera used for Leish-EVs. Positive (strip 3) and negative (strip 5) sera. Panel B. Reactivity in Dot-Blot using Leish-EVs as antigen against five positive and five negative sera for Can-LV. As positive control CLA was used as antigen against 4 sera (two positive and two negative for Can-LV).
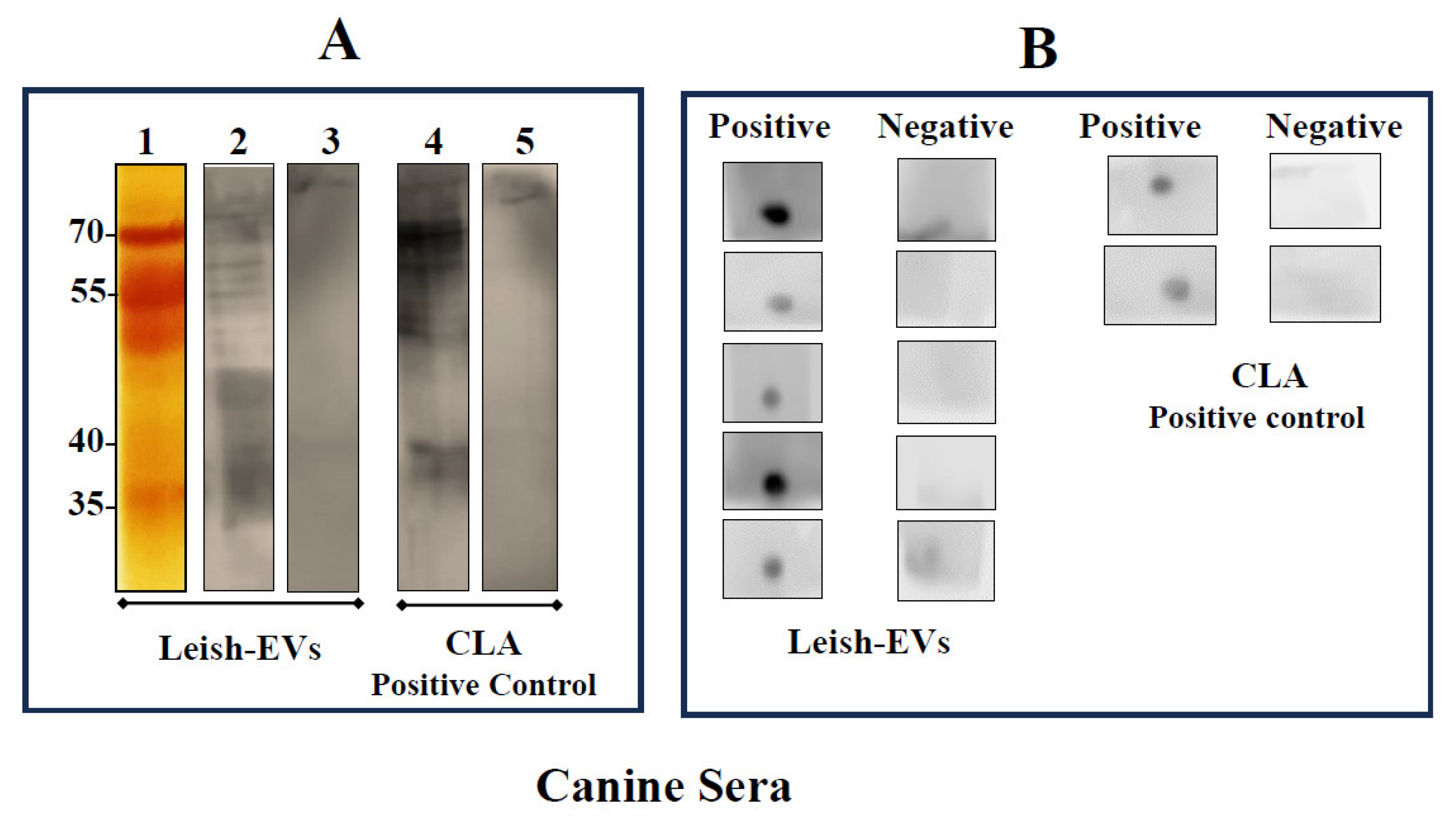
Figure 6.
Interaction of THP-1 cells and Leish-EVs. Relative quantifications of the cytokines IL-10, IL-12, TGF-β, TNF, IL-6 (Panel A) and the miRNAs species miR-21-5p, miR-146a-5p, miR-125b-5p, miR-144-3p (Panel B) in THP-1 cells treated with Leish-EVs. Values are expressed as mean ± SEM of Relative Quantification after calculation by comparative CT method (2-∆∆CT) as described in the Material and Methods section.
Figure 6.
Interaction of THP-1 cells and Leish-EVs. Relative quantifications of the cytokines IL-10, IL-12, TGF-β, TNF, IL-6 (Panel A) and the miRNAs species miR-21-5p, miR-146a-5p, miR-125b-5p, miR-144-3p (Panel B) in THP-1 cells treated with Leish-EVs. Values are expressed as mean ± SEM of Relative Quantification after calculation by comparative CT method (2-∆∆CT) as described in the Material and Methods section.
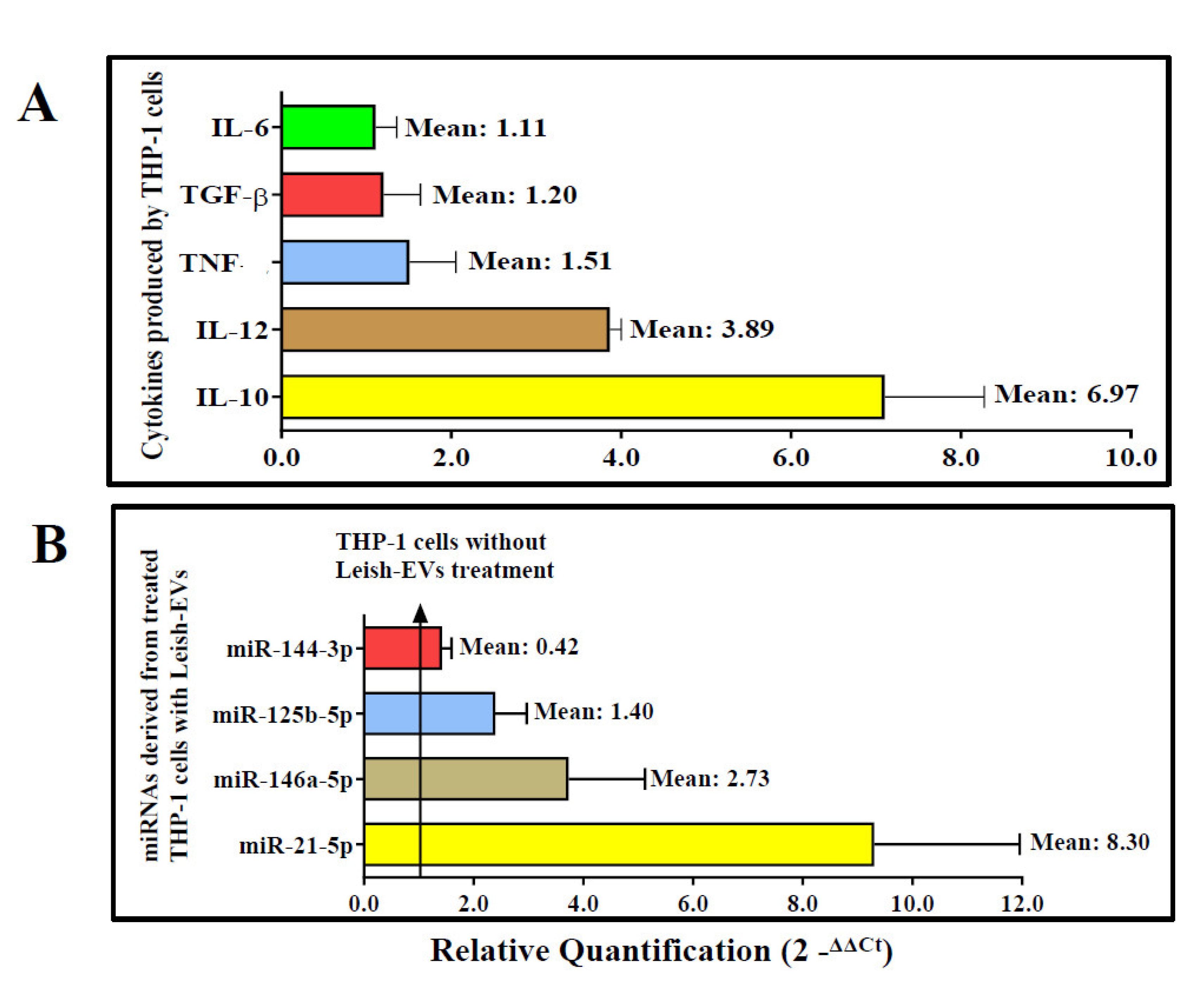

Table 1.
Cytokines assayed in this study.
| Gene symbol | Gene name | Biological function | Assay ID1 | AmpliconLength | Chromosome location |
|---|---|---|---|---|---|
| IFN-γ | Interferon gamma | Protein coding | Hs00989291_m1 | 73 | 12 |
| IL10 | Interleukin 10 | Protein coding | Hs00961622_m1 | 74 | 1 |
| IL12 | Interleukin 12 | Protein coding | Hs01011518_m1 | 72 | 5 |
| IL6 | Interleukin 6 | Protein coding | Hs00985639_m1 | 66 | 7 |
| TNF | Tumor necrosis factor | Protein coding | Hs01113624_g1 | 143 | 6 |
| TGF-β | Transforming growth factor beta 1 | Protein coding | Hs00998133_m1 | 57 | 19 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | Protein coding | Hs02758991_g1 | 93 | 12 |
TaqMan real-time PCR assays were chosen to span at least one exon-exon boundary and were purchased as assay IDs from Thermo Fisher Scientific 1.
Table 2.
miRNAs species assayed in this study.
| Assay name 1 | Gene family | Assay ID | Chromosome location | Mature miRNA sequence |
|---|---|---|---|---|
| 1: miR-21-5p | MI0000077 | 477975_miR | 17 | UAGCUUAUCAGACUGAUGUUGA |
| 2: miR-146a-5p | MI0000477 | 478399_miR | 5 | UGAGAACUGAAUUCCAUGGGUU |
| 3: miR-125b-5p | MIMAT0000423 | 477885_miR | 11 | UCC CUG AGA CCC UAA CUU GUGA |
| 4: miR-144-3p | MI0000460 | MC11051 | 17 | UACAGUAUAGAUGAUGUACU |
| 5: cel-miR-39-3p 2 | MIMAT0000010 | 478293_miR | ND | UCACCGGGUGUAAAUCAGCUUG |
ND, not determined; 1 purchased from Applied Biosystems; 2 external control. Source: http://www.thermofisher.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



