
Submitted:
05 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
ERK1/2 phosphorylation is frequently downregulated in the early phase of colon tumorigenesis with subsequent activation of ERK5. In the current work, we studied the advantage of ERK1/2 downregulation for tumor growth by dissecting the individual functions of ERK1 and ERK2. The patient sample data demonstrated decreased ERK1/2 phosphorylation in the early phase of tumorigenesis followed by upregulation of the phosphorylation in late-stage colon adenocarcinomas with intratumoral invasion or metastasis. In vitro results indicated SOD3-mediated coordination of small GTPase RAS regulatory genes in the inhibition of RAS-ERK1/2 signaling. In vitro and in vivo studies suggested that ERK2 has a more prominent role in chemotactic invasion, collective migration, and cell proliferation than ERK1. Noteworthily, simultaneous ERK1 and ERK2 expression inhibited collective cell migration and proliferation but tended to promote invasion, therefore suggesting that ERK1 controls ERK2 function. According to the present data, phosphorylated ERK1/2 at the early phase of colon adenocarcinoma limits tumor mass expansion, whereas reactivation of the kinases at the later phase of colon carcinogenesis is associated with the initiation of metastasis. Additionally, our results suggest that ERK1 is a regulatory kinase, which coordinates ERK2-promoted chemotactic invasion, collective migration, and cell proliferation. Our findings indicate that ROS, especially H2O2, are associated with the regulation of ERK1/2 phosphorylation in colon cancer either increasing or decreasing the kinase activity. The data suggesting a growth-promoting role for ERK2 and a regulatory role for ERK1 could result in new avenues in the developmental strategies for cancer therapy.
Keywords:
Colon tumorigenesis
; ERK1/2
; SOD3
; migration
; invasion
; proliferation
1. Introduction
The four main MAPKs responding to stress signaling, p38 MAPK, extracellular-signal-regulated kinase (ERK1/2), Big MAP kinase (ERK5), and c-jun N-terminal kinase (JNK), have a pivotal function in cancer progression (Koul et al., 2013). The kinase p38 MAPK has been shown to have a profound effect in the early phase of inflammation-associated colon tumorigenesis stimulating inflammatory cytokine and chemokine production, thereby modulating the innate immune system and contributing to the initiation of tumorigenesis (Del Reino et al., 2014). Correspondingly, it has been shown that the inhibition of p38 MAPK activates epidermal growth factor receptor ErbB3 with subsequent activation of MEK1/2-ERK1/2 independently of RAS or RAF (Pattingre et al., 2003).
In metastatic colon cancer, deregulation of the RAS-MEK1/2-ERK1/2 signaling pathway downstream of epidermal growth factor receptor by RAS-associated mutations confers a selective growth and survival advantage to tumor cells giving them an acquired resistance to anti-EGFR therapy (Van Emburgh et al., 2014). ERK1/2 are intermediator molecules in signal transduction contributing to oncogene-induced senescence (OIS) (Leikam et al., 2008), the transformation of primary cells (Mitsushita et al., 2004), increased superoxide anion (O2.)- and hydrogen peroxide (H2O2) levels by stimulating nox1 mRNA synthesis, NOXO1 activation, and extracellular superoxide dismutase (SOD3) production (Parascandolo and Laukkanen, 2019). ERK1/2 kinases not only promote growth but also contribute to migration, cell cycle arrest, survival, and differentiation correlating on the cellular location of phosphorylated ERK1/2 and the type the activated downstream target molecules (Del Reino et al., 2014, Guo et al., 2011, Hacohen Lev-Ran and Seger, 2023, Lemieux et al., 2011, Wu et al., 2020).
Previous studies have demonstrated downregulation of ERK1/2 phosphorylation in colon tumorigenesis in adenomas and early phase adenocarcinomas (Eggstein et al., 1999, Park et al., 1999), with a subsequent increase in ERK5 phosphorylation to maintain the progression of tumorigenesis (de Jong et al., 2016) and the stem cell-like malignant phenotype of cancer cells (Pereira et al., 2019). The ERK1/2 activation is regulated by a complex network of tyrosine kinase receptors, cellular kinases, small GPTases and their regulatory proteins, protein scaffolds, phosphatases, mutations in the oncogenes, and ROS (Andreyev et al., 2001, Balmanno and Cook, 2009). RAS-ERK1/2 feedback regulatory mechanisms contribute to redox balance through fine-tuning of small GTPase activity, consequently affecting the activation status of the signaling pathway (Cammarota et al., 2015b, Laukkanen et al., 2015, Laurila et al., 2009).
In the current work, we studied the advantage of ERK1/2 inactivation for the progression of colon tumorigenesis. Our data corroborated previous observations (Eggstein et al., 1999) showing downregulation of the phosphorylation of ERK1/2 in adenomas and in early phase adenocarcinomas, which, based on our in vitro data, could be caused by ROS-mediated downregulation of RAS-BRAF-MEK1/2-ERK1/2 signaling cascade. Simultaneous ERK1 and ERK2 expression significantly inhibited cell proliferation but it enhanced in vitro cancer cell chemotactic invasion through extracellular matrix and metastasis observed in patients. In vitro and in vivo data suggested a more pronounced role for ERK2 tumorigenesis as compared to ERK1, which could function as a regulatory kinase promoting or inhibiting ERK2 function.
Therefore, the data may suggest that the downregulation of ERK1/2 phosphorylation is needed for the initial colon tumor expansion, whereas upregulation of ERK1/2 activation promotes the metastasis.
2. Methods
2.1. Tissue staining
Human adenocarcinoma sections were stained with ERK1/2 (Cell Signaling, Danvers, MA, USA) and hematoxylin-eosin (Sigma-Aldrich, St. Louis, MO, USA). The ethical permissions for the study were approved by Clinica Mediterranea ethical committee, Naples, Italy, Monaldi Hospital ethical committee (Deliberazione del Direttore Generale n:o 1239), Naples, Italy, and by the University of Federico II of Naples ethical committee (protocol number 394/19), Naples, Italy. Informed consent was asked from patients participating the study.
2.2. Cells
Normal colon epithelial CCD841cells (ATCC, Manassas, VA, USA) were cultured in EMEM/10% FBS/L-alanine-l-glutamine/ penicillin-streptomycin (ATCC). DLD1 cells (ATCC) were cultured in RPMI/10% FBS/L-alanine-l-glutamine (Life Technologies, Grand Island, NY, USA), penicillin-streptomycin (Sigma, St. Louis, MO, USA). HCT116 cells (ATCC) were cultured in DMEM/10% FBS/L-alanine-l-glutamine/ penicillin-streptomycin. Human colon cells were transduced with GFP, human ERK1, or ERK2 lentiviruses (MOI 1) or RNAi viruses for the kinases (MOI 1) (ABM, Vancouver, Canada). Mouse embryonic fibroblasts (MEF) clones (MEF GFP, MEF SOD3 cl6, MEF SOD3 cl8, and MEF SOD3 cl5) (Castellone et al., 2014) were cultured in αMEM (Mediatech, Manassas, VA, USA) supplemented with 10% FBS (Hyclone, Logan, UT, USA), non-essential amino acids (Mediatech) and L-alanine-l-glutamine (Life Technologies), and penicillin-streptomycin (100 mg/L) (Sigma).
Normal colon CCD841 epithelial cells is a cell line with limited life span of less than 100 population doublings. DLD1 cells, derived from adenocarcinoma, are tumorigenic in nude mice carrying several activated oncogenes. The culture contains 0.2-0.4% of Side Population (SP) stem cells and 1% of CD133 positive cells (Kai et al., 2009, Tavaluc et al., 2007) indicating their undifferentiation status. HCT116 cells lack the differentiation capacity almost completely, form primary tumors in nude mice, and have been demonstrated to metastasize. Analysis of HCT116 has suggested that the cultures are formed almost solely of CD166 positive cancer stem cells explaining their highly aggressive nature (Kai et al., 2009, Rajput et al., 2008) (Supplemental Figure S1). MEF clones cl5, cl6, and cl8 were derived from individual mouse embryos transduced with an ecotropic retrovirus containing GFP marker gene or rabbit sod3 cDNA (Castellone et al., 2014, Parascandolo and Laukkanen, 2021).
2.3. Gene expression analysis
RNA isolated using a RNeasy mini kit (Qiagen, Hilden, Germany) was reverse transcribed to cDNA by QuantiTect reverse transcription kit (Qiagen). The quantitative PCR was done using SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA). The primers are listed in the Supplemental Table S1.
2.4. Western blot analysis
The proteins were isolated from the tissues and cells were homogenized in lysis buffer (50 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1 mmol/L EGTA, 1.5 mmol/L MgCl2, 10 mmol/L NaF, 10 mmol/L sodium pyrophosphate, 1 mmol/L Na3VO4, 10 μg aprotinin/ml, and 10μg leupeptin/ml) (Sigma). The following antibodies were used: phospho-ERK1/2 (Thr202/Tyr204) (Cell Signaling), ERK1/2 (Cell Signaling), phospho-MEK1/2 (Ser217/221) (Cell Signaling), MEK1/2 (Cell Signaling), phopho-ERK5 (Thr218/Tyr220) (Merck KGaA, Darmstadt, Germania), ERK5 (Cell Signaling), RAS (Cell Signaling), RAC (Cell Signaling), RHO (Cell Signaling), CDC42 (Cell Signaling), and TUBULIN (Cell Signaling). The intensity of Western blot bands was analyzed using ImageJ software.
2.5. Pull-down assay for MEF SOD3 clones
Cells were grown to 60% confluence and were collected for small GTPase RAS and RAC pull-down analysis. The cells were lysed using an ice-cold buffer containing 20 mM HEPES (pH 7.4), 0.1 M NaCl, 1% Triton X-100, 10 mM EGTA, 40 mM glycerophosphate, 20 mM MgCl2, 1 mM Na3VO4, 1 mM dithiothreitol, a mixture of protease inhibitors, and 1 mM phenylmethylsulfonyl fluoride. The lysates were shaken gently for 15 min with a purified GST-fusion protein containing the CRIB domain of PAK1 (p21 activated kinase) bound to glutathione-Sepharose beads. The mix was washed three times using lysis buffer. The GTP-bound forms of RAS (Santa Cruz, Dallas, TX, USA) and RAC (Millipore, Burlington, MA, USA) were analyzed by Western blotting.
2.6. Growth analysis
To analyze cell proliferation, 10 000 cells were seeded on 24-well plates in triplicates and counted daily for four days.
2.7. Matrigel migration
To study the cellular invasion towards chemo-attractant using transwell migration assay, 100 μl of Matrigel (Corning, Corning, NY, USA) at 1 mg/ml was added to the migration chambers (8 microns, BD, San Jose, CA, USA) and allowed to stabilize at room temperature for 30 minutes. To analyze the invasion, 50000 cells were seeded on the Matrigel in 5% FBS medium and let to migrate toward 10% FBS medium for 48 to 72 hours depending on the cell line characteristics. To detect the migrated cells, Matrigel was removed from the chambers, cells were fixed with 7% paraformaldehyde (Sigma), washed with PBS, and stained with Cristal violet (Sigma). The migrated cells were counted from the high-power microscope fields (Leica DMI3000B microscope and Leica Application Suite camera software, Leica, Wetzlar, Germany). We used naïve counterpart fibroblasts isolated from the same patients as TAFs as controls.
2.8. Wound healing
For the collective cell migration assay, cells on 6-well plates (Corning) were grown to 60% confluency, the culture was broken with a scratch, and images (Leica) of the culture were taken at 24-hour intervals. The distance between the edges of the scratch was measured for the calculation of the closure percentage.
2.9. Mice
BALB/c Nude Mice (Plaisant, Rome, Italy) were xenografted with HCT116 cells transduced with GFP (n=6 xenografts), ERK1 (n=6 xenografts), ERK2 (n=6 xenografts), or ERK1/2 (n=6 xenografts). To study the specific effect of individual ERK1 or ERK2 kinases, we used 1x106 cells, which was the lowest number of cells resulting in tumorigenesis in GFP control animals. Tumor growth was measured with caliber twice a week for five weeks. Animal ethical permission was requested and approved by Plaisant.
2.10. Statistical analysis
The experiments were repeated three times. The p-values (*p<0.05, **p<0.01, ***p<0.001) were determined by two-tail independent samples t-tests. The results are expressed as the mean ± SD.
3. Results
3.1. Inverse correlation of ERK/2 phosphorylation and progression of colon tumorigenesis
A recent mouse model study emphasized the adaptability of colon cancer signaling suggesting increased ERK5 kinase phosphorylation to compensate for an abrogation of ERK1/2 activation in colon tumorigenesis in maintaining cell proliferation (de Jong et al., 2016). To dissect the characteristics of ERK1/2 kinase activation in colon tumorigenesis, were stained human normal colon, adenoma, and adenocarcinoma tissue sections with phosho-ERK1/2 antibody. We observed robust ERK1/2 phosphorylation in the normal mucosa, most prominently at the luminal regions, and markedly reduced phosphorylation of the kinase in adenomas and adenocarcinomas (Figure 1a,b).
The reduced activation of the kinases in adenomas and adenocarcinomas was confirmed by Western blot analysis, thereby corroborating the previous observations (Figure 1d,e) (Eggstein et al., 1999, Park et al., 1999). We further demonstrated recovery of the ERK1/2 phosphorylation in patients diagnosed with advanced pT3 and pT4 adenocarcinoma with intratumoral or peritumoral invasion to the vasculature, perineural invasion, or metastasis in lymph nodes (Figure 1c–e). Therefore, the patient data suggested that ERK1/2 activation contributes to the migration or invasion of colon cancer cells.
Figure 1.
ERK1/2 staining in histological tissue sections. a. Histological staining of normal colon, low- and high-grade adenoma, and stage 1-IV adenocarcinoma. b. Description of the staining data. Normal colon tissue close to the lumen of the colon was intensely positive suggesting a high endothelial cell proliferation rate. The positivity decreased in stages I-III. In stage III there was intensive staining in neoplastic glands. In stage IV the most intensive staining was observed in the infiltration areas near the fibroblast-rich region. c. Diagnosis of patients used for Western blot analysis in the panel d. Patient 1, patient 2, and patient 6 had intratumoral invasion or metastasis. Patient 3 had moderately differentiated adenocarcinoma without metastasis. Patient 4 had lipomatosis, and patient 5 had moderately differentiated adenocarcinoma without metastasis d. Western blot analysis of patients. We used normal tissue, adenoma, and adenocarcinoma tissues from each patient for the analysis. e. Intensity analysis of phosphoERK1/2 staining from the Western blot. All patients demonstrated reduced ERK1/2 activation in adenomas. The reduction was further augmented in patients 3, 4, and 5. Patients 1, patient 2, and patient 6 showed increased phosphorylation as compared to adenomas. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. To see this illustration in color, the reader is referred to the online version of this article.
Figure 1.
ERK1/2 staining in histological tissue sections. a. Histological staining of normal colon, low- and high-grade adenoma, and stage 1-IV adenocarcinoma. b. Description of the staining data. Normal colon tissue close to the lumen of the colon was intensely positive suggesting a high endothelial cell proliferation rate. The positivity decreased in stages I-III. In stage III there was intensive staining in neoplastic glands. In stage IV the most intensive staining was observed in the infiltration areas near the fibroblast-rich region. c. Diagnosis of patients used for Western blot analysis in the panel d. Patient 1, patient 2, and patient 6 had intratumoral invasion or metastasis. Patient 3 had moderately differentiated adenocarcinoma without metastasis. Patient 4 had lipomatosis, and patient 5 had moderately differentiated adenocarcinoma without metastasis d. Western blot analysis of patients. We used normal tissue, adenoma, and adenocarcinoma tissues from each patient for the analysis. e. Intensity analysis of phosphoERK1/2 staining from the Western blot. All patients demonstrated reduced ERK1/2 activation in adenomas. The reduction was further augmented in patients 3, 4, and 5. Patients 1, patient 2, and patient 6 showed increased phosphorylation as compared to adenomas. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. To see this illustration in color, the reader is referred to the online version of this article.

3.2. ERK1 and ERK2 expression in vitro models
The mitogen signal transduction can be mediated by ROS, such as SOD3-produced H2O2, which modifies the activation of phosphotyrosine phosphatases (PTPs) (Oshikawa et al., 2010), small GTPases (Laukkanen et al., 2015), and their downstream kinases. To study the SOD3-related signaling involved in ERK1/2 phosphorylation we used a mouse embryonic fibroblast (MEF) model composed of MEF clones cl6, cl8, and cl5 and a GFP control (Castellone et al., 2014) demonstrating downregulation of ERK1/2 phosphorylation in cl5 with corresponding upregulation of ERK5 activation in MEF SOD3. The Western blot analysis further suggested moderately upregulated MEK1/2 and ERK1/2 activation in the cl8 (Figure 2a) confirming previous results demonstrating a regulatory role for the enzyme in mitogen signaling (Laukkanen et al., 2015). The analysis of the expression and activation of ERK1/2 in CCD841 normal colon cells, DLD1 colon adenocarcinoma cells, aggressive HCT116 colon carcinoma cells suggested downregulation of both the phosphorylation and the expression of total ERK1/2 proteins in DLD1 and HCT116 cells (Figure 2b).
To dissect the factors regulation ERK1/2 phosphorylation in SOD3 overexpressing cells, we first turned our focus on the expression of dual specific phosphatases (dusps) 5, 6, 7, and 9, which regulate the activation of mitogen-activated protein kinase (MAPK) family, especially ERK1/2 kinases.
Figure 2.
Characterization of ERK1/2 expression in vitro models. a. Western blot analysis of MEF GFP, MEF SOD3 cl6, MEF SOD3 cl8, and MEF SOD3 cl5. The analysis demonstrated moderately increased ERK1/2 phosphorylation in cl8 and markedly decreased phosphorylation in cl5. MEK1/2 staining supported the ERK1/2 activation results. The phosphorylation of ERK5 was moderately increased in MEF SOD3 cl5 cells. b. ERK1/2 phosphorylation in CCD841, DLD1, and HCT116 cells. Both the phosphorylation and total ERK1/2 levels are lower in DLD1 and HCT116 adenocarcinoma cells as compared to CCD841 normal colon epithelial cells. c. The mRNA expression analysis for dusp5, 6, 7, and 9 showed significantly decreased expression in all MEF SOD3 clones. d. Small GTPase regulatory gene expression analysis showed decreased rgl1, rgs4, adap2, and rasal mRNA levels in MEF SOD3 cl6 and cl8. In MEF SOD3 cl5 adap2 expression was at the same level as in MEF GFP controls, while rasal expression was significantly increased. e. Pull down assay for RAS, RAC, CDC42, and RHO in MEF GFP, MEF SOD3 cl6, MEF SOD3 cl8, and MEF SOD3 cl5. f–i. Intensity measurement of small GTPase pulldown assay. j. RAS, RAC, and ERK1/2 Western blot in MEF SOD3 cl5 cell overexpressing ras or rac. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using GFP expressing cells as a comparison control.
Figure 2.
Characterization of ERK1/2 expression in vitro models. a. Western blot analysis of MEF GFP, MEF SOD3 cl6, MEF SOD3 cl8, and MEF SOD3 cl5. The analysis demonstrated moderately increased ERK1/2 phosphorylation in cl8 and markedly decreased phosphorylation in cl5. MEK1/2 staining supported the ERK1/2 activation results. The phosphorylation of ERK5 was moderately increased in MEF SOD3 cl5 cells. b. ERK1/2 phosphorylation in CCD841, DLD1, and HCT116 cells. Both the phosphorylation and total ERK1/2 levels are lower in DLD1 and HCT116 adenocarcinoma cells as compared to CCD841 normal colon epithelial cells. c. The mRNA expression analysis for dusp5, 6, 7, and 9 showed significantly decreased expression in all MEF SOD3 clones. d. Small GTPase regulatory gene expression analysis showed decreased rgl1, rgs4, adap2, and rasal mRNA levels in MEF SOD3 cl6 and cl8. In MEF SOD3 cl5 adap2 expression was at the same level as in MEF GFP controls, while rasal expression was significantly increased. e. Pull down assay for RAS, RAC, CDC42, and RHO in MEF GFP, MEF SOD3 cl6, MEF SOD3 cl8, and MEF SOD3 cl5. f–i. Intensity measurement of small GTPase pulldown assay. j. RAS, RAC, and ERK1/2 Western blot in MEF SOD3 cl5 cell overexpressing ras or rac. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using GFP expressing cells as a comparison control.
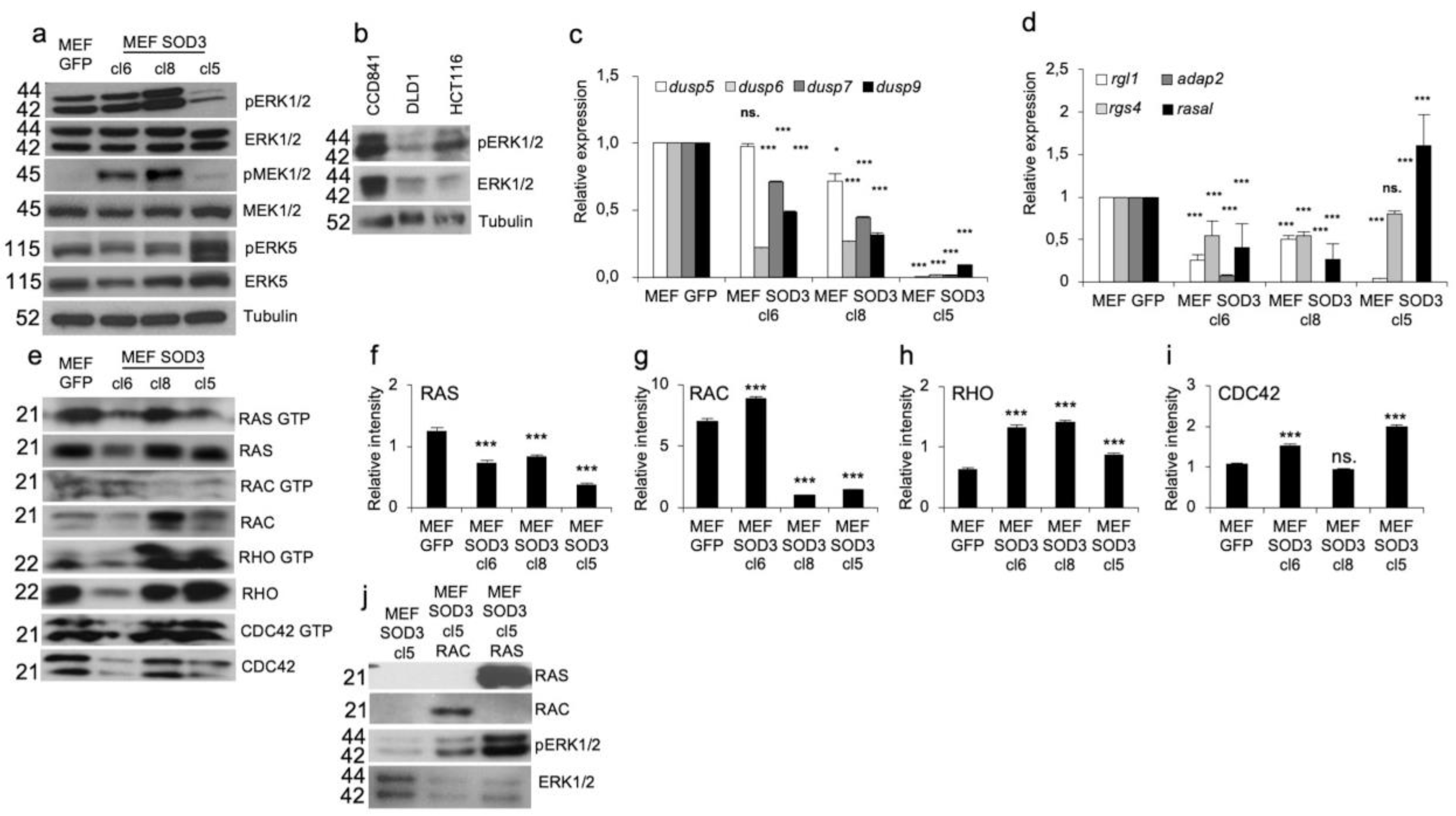
As shown in Figure 2c, the mRNA expression of the studied dusps was not increased in MEF SOD3 clones, thereby suggesting other factors causing the reduced ERK1/2 phosphorylation in MEF SOD3 cl5 (Figure 2c). The analysis of the expression of small GTPase and heterotrimeric G protein regulatory genes RGL1 (Ral guanine nucleotide dissociation stimulator-like 1), adap2 (ArfGAP with dual PH domains 2), rgs4 (regulator of G protein signaling 4), and rasal (RAS GAP activating protein) demonstrated significantly (p>0.001) upregulated rasal mRNA synthesis in MEF SOD3 cl5 (Fig, 2d). The data, therefore, suggested the contribution of small GTPase RAS regulatory protein GAP in the downregulation of ERK1/2 phosphorylation.
Next, to analyze the activation status of small GTPases we performed a pull-down assay for RAS, RAC, RHO, and CDC42, demonstrating a significant (p>0.001) downregulation of RAS GTP loading in all MEF SOD3 clones (Figure 2e–i). RAC activation was upregulated in MEF SOD3 clone 6 and significantly downregulated in clones 8 and 5. RHO was significantly (p<0.001) upregulated in all clones and CDC42 in clones 6 and 5, respectively (Figure 2e–i). To further characterize the contribution of SOD3 on ERK1/2 activation, we studied the effect of small GTPase RAC and RAS transient overexpression on the activation of ERK1/2 in the MEF SOD3 cl5 (Figure 2j). RAS had a pronounced role in the phosphorylation of ERK1/2 kinases although also RAC stimulated the activation, suggesting parallel RAS and RAC signaling in ERK1/2 activation.
3.3. ERK1 and ERK2 have different functions in cell migration and proliferation in vitro
Next, we studied the individual effect of ERK1 and ERK2 kinases in the colon in vitro models. The chemotactic invasion assay suggested different capacities for ERK1 and ERK2 to promote the cellular invasion towards chemo-attractant through extracellular matrix (Figure 3a–j). In normal colon CCD841 cells both ERK1 and ERK2 supported cell migration (p<0.001) that was further enhanced by the combined expression of ERK1/2 (p<0.001) (Figure 3a,b). RNA interference abolished the stimulatory effect of ERK1 and significantly (p<0.01) decreased the invasion in ERK2 and ERK1/2 cells as compared to GFP control cells (Figure 3c,d). In DLD1 cells ERK1 demonstrated a complete deficiency to promote invasion, whereas ERK2 significantly (p<0.01) stimulated invasion, which was further enhanced by simultaneous ERK1 and ERK2 expression similar to CCD841 cells (Figure 3e,f). In HCT116 cells, ERK2 demonstrated increased migration potential (p<0.001), whereas ERK1 and ERK1/2 invasion was at the level of control cells (Figure 3g,h). To study the effect of ERK1 and ERK2 in MEFs, we used MEF SOD3 cl5 cells that from now on are called MEF SOD3 GFP, MEF SOD3 ERK1, MEF SOD3 ERK2, or MEF SOD3 EKR1/2. The analysis of MEF clones further corroborated the results suggesting a more prominent role for ERK2 in the stimulation of invasion, which was further enhanced by simultaneous expression of ERK1 and ERK2 (Figure 3i,j).
The collective cell migration assay, which regulates the cellular movement as structural and functional unit (Friedl and Gilmour, 2009), corroborated the more prominent function of ERK2. The ERK2 overexpression demonstrated faster scratch recovery in all time points analyzed in CCD841, DLD1, HCT116, and MEF SOD3 as compared to ERK1 expressing cells, which had no effect on cell migration (Figure 4a,b,e–j). RNA interference in CCD841 cells decreased the migration capacity of ERK2 cells below the control cells, whereas it significantly (p<0.001) increased the collective migration of ERK1 and ERK1/2 expressing cells (Figure 4c,d). It is noteworthy that the combined expression of ERK1 and ERK2 in DLD1, HCT116, and in MEF SOD3 cells significantly (p<0.001) reduced cellular movement as compared to GFP transduced controls (Figure 4i,j).
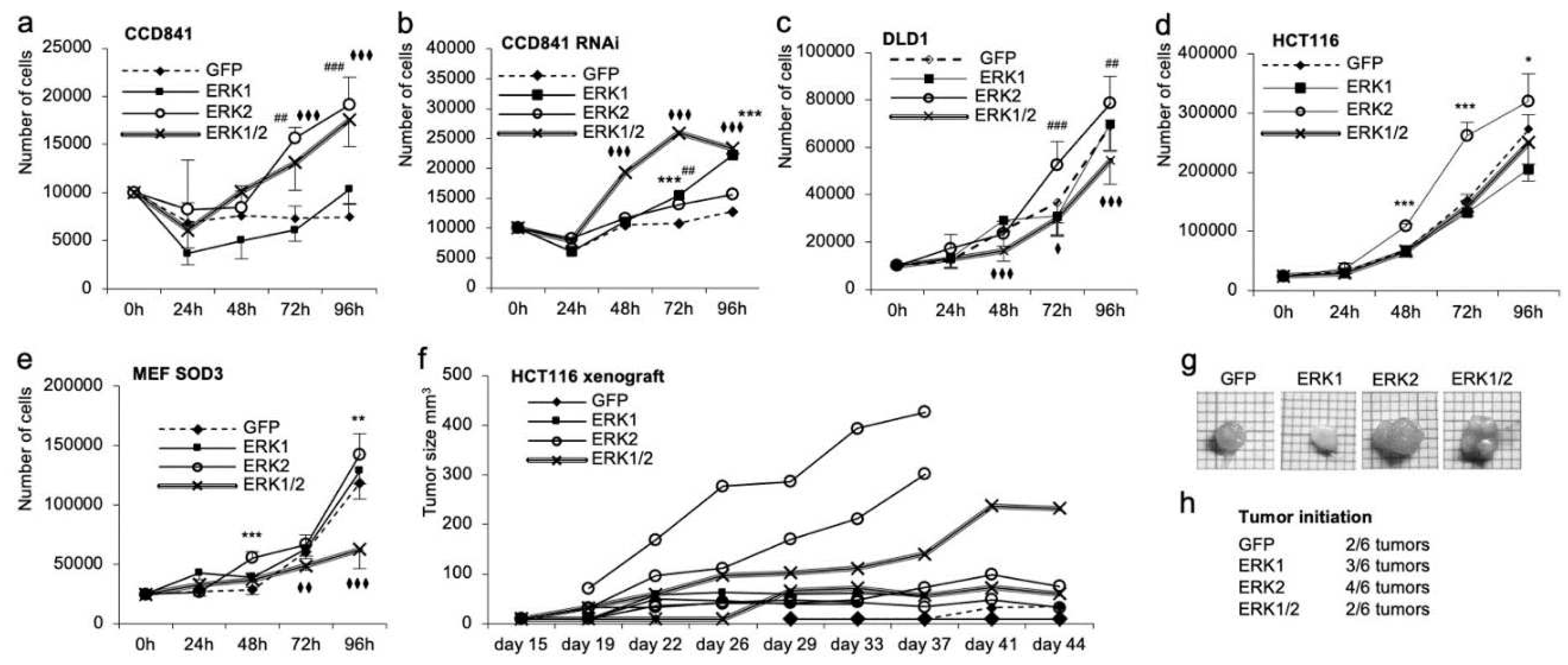
The analysis of the cell proliferation further strengthened the tumor promoter role of ERK2 in cancer cells. The ERK2 overexpression resulted in a higher proliferation rate as compared to the overexpression of ERK1 or GFP control gene in all cell lines studied (Figure 5a,c–e). The combined overexpression of ERK1 and ERK2 increased growth in CCD841 normal colon cells (Figure 5a) but decreased the growth in DLD1 and MEF SOD3 cultures as compared to GFP controls (Figure 5c,e), similar to collective cell migration (Figure 4e,f,i,j). RNAi in CCD841 cells eliminated the growth stimulatory role of ERK2 and increased cell proliferation in ERK1 and ERK1/2 cells as compared to control cells (Figure 5b).
The previous reports have suggested that ERK1/2 could influence differentiation of the cells, such as PKC-Ca2+-stimulated differentiation of epidermal keratinocytes and stem cell factor-erythropoietin-mediated maturation and expansion of erythroid progenitor cells (Lemieux et al., 2011, Seo et al., 2004, Sui et al., 1998). To dissect the function of ERK1 in colon tumorigenesis, we analyzed OCT4, VIMENTIN, CHI3, and CDX2 differentiation marker mRNA expression. According to the analysis, ERK1 overexpression does not promote differentiation DLD1 cells (Supplemental Figure S2).
To enforce the data showing variable growth potential of ERK1 and ERK2 expressing cells, we injected 1x106 HCT116 cells subcutaneously in BALB c/A nude mice. HCT116 cells expressing ERK2 cells resulted in larger tumors as compared to ERK1 or ERK1/2 cells, thereby validating the in vitro data (Figure 5f,g). Besides their increased tumorigenic potential, ERK2 cells had a higher incidence of tumor initiation: 66% (four out of 6 injections) of HCT116 ERK2 transplantations resulted in detectable tumors, whereas 50% (three out of 6 injections) of HCT116 ERK1 cells, 33% (two out of 6 injections) of HCT116 ERK1/2 cells transplantations, and 33% (two out of 6 injections) of HCT116 GFP cells yielded tumors (Figure 5h).
4. Discussion
The ability of RAS-ERK1/2 signaling to coordinate diverse cellular processes is essential for different biological functions including ontogenesis (Cammarota et al., 2015a). In cancer, ROS, predominantly H2O2, along with O2._, coordinate cell signaling, tumor initiation, oncogene-induced senescence, and benign to malignant transformation (Irani et al., 1997, Kamiya et al., 2016, Mitsushita et al., 2004, Parascandolo and Laukkanen, 2019, Sarkisian et al., 2007). ROS and redox enzymes, such as SOD3, are involved in a variety of cellular phenomena ranging from DNA damage and point mutations to tissue-level metabolic disorders (Chini et al., 2021, Laukkanen et al., 2001, Lee et al., 2019, Parascandolo and Laukkanen, 2021). SOD3, by balancing the local concentrations of O2._ and H2O2, forms a positive feedback loop to regulate the RAS-MEK1/2-ERK1/2 signaling pathway (Laurila et al., 2009) through the coordination of guanine nucleotide exchange factor (GEF), GTPase activating protein (GAP), and guanosine nucleotide dissociation inhibitor (GDI) expression (Laukkanen et al., 2015). The current data demonstrating SOD3-derived downregulation of ERK1/2 phosphorylation (Figure 2) suggest the involvement of H2O2 in the coordinated interaction of RAS, ERK1/2, and ERK5 in colon tumorigenesis.
ERK1 and ERK2 kinases are generally co-phosphorylated by the same extracellular stimuli, although the kinases are suggested to have different biological functions ranging from a severe abnormality of the placenta, with subsequent embryonic lethality (Hatano et al., 2003), to a distinct expression pattern in the adult murine brain (Di Benedetto et al., 2007). After the activation, ERK1 and ERK2 are generally translocated to the nucleus, although they can be also detected at the cytoplasmic compartment, e.g., at Golgi and the cell membrane (Casar et al., 2008, Kondoh et al., 2005). The inactivation of ERK1 and ERK2 isoform phosphorylation in the early phase of colon tumorigenesis (Eggstein et al., 1999, Park et al., 1999) with subsequent increased phosphorylation of ERK5 (de Jong et al., 2016) is an intriguing phenomenon proposing an inhibitory function for ERK1/2 in the initial phases of adenocarcinoma development. Another ambiguous phenomenon is the activation of ERK1/2 kinases in metastatic patients (Figure 1).
In the current work, we demonstrated a more pronounced role for ERK2 than for ERK1 in promoting chemotactic invasion, collective migration, and proliferation (Figure 3, Figure 4 and Figure 5). Notably, ERK1 alone had only a minor, if any, stimulatory role on cellular functions or differentiation marker expression (Supplemental Figure S2), whereas the simultaneous overexpression of ERK1 and ERK2 reduced collective migration and cell proliferation. The reduced cellular growth caused by ERK1/2 transduction could be a growth limiting factor in the early phase of tumor expansion and therefore a reason for the downregulation of ERK1/2 phosphorylation in colon adenomas. The loss of ERK1/2 phosphorylation at the early benign phase of colon tumorigenesis (Eggstein et al., 1999, Park et al., 1999) with subsequent ERK5 activation (de Jong et al., 2016) could provide newly transformed cancer cells with a growth advantage.
ERK1/2 have at least 49 direct nuclear and cytoplasmic downstream substrates, mostly transcriptional factors Ets family members, including Ets-1 and Elk1, Smad proteins, c-Fos, c-Myc, and ATF2, and a markedly higher number of indirect targets, which, as a network, coordinate cellular functions (Whitehurst et al., 2004, Yang et al., 2019). Therefore, the decreased cell proliferation of simultaneous ERK1/2 expression in our data could result from the inhibition of nuclear translocation of the kinases. The entry into the cell cycle, G1 to S phase progression, occurs only after nuclear translocation of phosphorylated ERK1/2 that stimulates ELK1 transcription and reinitiation of DNA replication (Brunet et al., 1999, Mebratu and Tesfaigzi, 2009), which then activates the DNA replication machinery.
Another plausible explanation for reduced growth and collective cell migration caused by simultaneous ERK1 and ERK2 expression could be the inhibition of dimerization of ERKs, which has been reported to halt cell proliferation. Dimerization enhances ligand binding in the nucleus with subsequent entry to cell cycling. Correspondingly, inhibition of dimerization inhibits loss of cellular differentiation, growth, and tumorigenesis (Casar et al., 2008, Khokhlatchev et al., 1998).
The present data showing increased chemotactic invasion in cells transduced with both ERK1 and ERK2 kinases (Figure 3a–j) may suggest that the kinases are needed for the initiation and progression of metastasis in colon carcinogenesis. This is supported by a previous article suggesting that KRAS and BRAF mutations with consequent activation of MEK1/2-ERK1/2 signaling, increase the risk of lung metastasis of colorectal cancer patients (Tie et al., 2011). A recent work consolidates the current results by proposing increased risk of colon cancer liver metastasis caused by the ERK1/2-stimulated upregulation of ANGPT2 and CXCR4 (Urosevic et al., 2020), therefore proving a mechanism of how ERK1/2 signaling may promote colon cancer cell invasion.
According to our results, ERK1 expressed alone had a minor or no role in colon cancer behavior but together with ERK2 expression, the kinase pair had an impact on in vitro invasion, migration, proliferation and in vivo tumorigenesis. Correspondingly, RNA interference of both ERK1 and ERK2 in CCD481 cells abolished the ability of cells to invade through extracellular matrix, and significantly increased the collective migration and cell proliferation as compared to CCD841 cells overexpressing ERK1 and ERK2. Therefore, ERK1 could be a regulatory kinase coordinating the activity of ERK2 either by enhancing or inhibiting the ERK2 response in the cells.
In conclusion, we demonstrated that in colon adenocarcinoma in vitro and in vivo models ERK2 stimulates migration and proliferation of cells, while ERK1 alone has minor or no effect on cellular functions. Simultaneous ERK1/2 expression in cancer cells markedly reduced colon cancer cell proliferation and tumor formation, therefore suggesting that ERK1 regulates ERK2. The growth disadvantage of ERK1/2 expressing cells could explain the inhibition of the kinase phosphorylation in the early stages of colon tumorigenesis. Lastly, the variable function for ERK1 and ERK2 kinases in colon cancer models could result in novel innovations as part of future anti-cancer therapies for colon cancer.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Mikko Laukkanen; Formal analysis, Alessia Parascandolo, Giulio Benincasa, Francesco Corcione and Mikko Laukkanen; Funding acquisition, Mikko Laukkanen; Investigation, Alessia Parascandolo, Giulio Benincasa and Mikko Laukkanen; Methodology, Alessia Parascandolo, Giulio Benincasa and Mikko Laukkanen; Project administration, Francesco Corcione and Mikko Laukkanen; Resources, Giulio Benincasa, Francesco Corcione and Mikko Laukkanen; Supervision, Mikko Laukkanen; Validation, Alessia Parascandolo, Giulio Benincasa and Mikko Laukkanen; Visualization, Giulio Benincasa; Writing—original draft, Mikko Laukkanen; Writing—review & editing, Alessia Parascandolo and Francesco Corcione.
Institutional Review Board Statement
The study with patient samples was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Clinica Mediterranea ethical committee, Naples, Italy, Monaldi Hospital ethical committee, Naples, Italy, and by the University of Federico II of Naples ethical committee, Naples, Italy. Informed consent was asked from patients participating the study. All animal experimental procedures were approved by the Italian Ministry of Health, authorization number 289/2021-PR del 27/04/2021 (A69A0.86) and were performed following the guidelines of the European Union Directive on the Protection of Animals.
Acknowledgments
The study was funded by POR Campania CUP B63D18000210007.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer 2001;85(5):692-696. [CrossRef]
- Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell death and differentiation 2009;16(3):368-377. [CrossRef]
- Brunet A, Roux D, Lenormand P, et al. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J 1999;18(3):664-674. [CrossRef]
- Cammarota F, de Vita G, Salvatore M, et al. Ras oncogene-mediated progressive silencing of extracellular superoxide dismutase in tumorigenesis. Biomed Res Int 2015a;2015:780409. [CrossRef]
- Cammarota F, Fiscardi F, Esposito T, et al. Clinical relevance of thyroid cell models in redox research. Cancer Cell Int 2015b;15:113. [CrossRef]
- Casar B, Pinto A, Crespo P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaffold complexes. Mol Cell 2008;31(5):708-721. [CrossRef]
- Castellone MD, Langella A, Cantara S, et al. Extracellular superoxide dismutase induces mouse embryonic fibroblast proliferative burst, growth arrest, immortalization, and consequent in vivo tumorigenesis. Antioxid Redox Signal 2014;21(10):1460-1474. [CrossRef]
- Chini CCS, Zeidler JD, Kashyap S, et al. Evolving concepts in NAD(+) metabolism. Cell Metab 2021;33(6):1076-1087. [CrossRef]
- de Jong PR, Taniguchi K, Harris AR, et al. ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nat Commun 2016;7:11551. [CrossRef]
- Del Reino P, Alsina-Beauchamp D, Escos A, et al. Pro-oncogenic role of alternative p38 mitogen-activated protein kinases p38gamma and p38delta, linking inflammation and cancer in colitis-associated colon cancer. Cancer Res 2014;74(21):6150-6160. [CrossRef]
- Di Benedetto B, Hitz C, Holter SM, et al. Differential mRNA distribution of components of the ERK/MAPK signalling cascade in the adult mouse brain. J Comp Neurol 2007;500(3):542-556. [CrossRef]
- Eggstein S, Franke M, Kutschka I, et al. Expression and activity of mitogen activated protein kinases in human colorectal carcinoma. Gut 1999;44(6):834-838. [CrossRef]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 2009;10(7):445-457. [CrossRef]
- Guo D, Zhou H, Wu Y, et al. Involvement of ERK1/2/NF-kappaB signal transduction pathway in TF/FVIIa/PAR2-induced proliferation and migration of colon cancer cell SW620. Tumour Biol 2011;32(5):921-930. [CrossRef]
- Hacohen Lev-Ran A, Seger R. Retention of ERK in the cytoplasm mediates the pluripotency of embryonic stem cells. Stem Cell Reports 2023;18(1):305-318. [CrossRef]
- Hatano N, Mori Y, Oh-hora M, et al. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 2003;8(11):847-856. [CrossRef]
- Irani K, Xia Y, Zweier JL, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997;275(5306):1649-1652. [CrossRef]
- Kai K, Nagano O, Sugihara E, et al. Maintenance of HCT116 colon cancer cell line conforms to a stochastic model but not a cancer stem cell model. Cancer Sci 2009;100(12):2275-2282. [CrossRef]
- Kamiya T, Courtney M, Laukkanen MO. Redox-Activated Signal Transduction Pathways Mediating Cellular Functions in Inflammation, Differentiation, Degeneration, Transformation, and Death. Oxid Med Cell Longev 2016;2016:8479718. [CrossRef]
- Khokhlatchev AV, Canagarajah B, Wilsbacher J, et al. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 1998;93(4):605-615. [CrossRef]
- Kondoh K, Torii S, Nishida E. Control of MAP kinase signaling to the nucleus. Chromosoma 2005;114(2):86-91. [CrossRef]
- Koul HK, Pal M, Koul S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013;4(9-10):342-359. [CrossRef]
- Laukkanen MO, Cammarota F, Esposito T, et al. Extracellular superoxide dismutase regulates the expression of small gtpase regulatory proteins GEFs, GAPs, and GDI. PloS one 2015;10(3):e0121441. [CrossRef]
- Laukkanen MO, Leppanen P, Turunen P, et al. Gene transfer of extracellular superoxide dismutase to atherosclerotic mice. Antioxid Redox Signal 2001;3(3):397-402. [CrossRef]
- Laurila JP, Castellone MD, Curcio A, et al. Extracellular superoxide dismutase is a growth regulatory mediator of tissue injury recovery. Mol Ther 2009;17(3):448-454. [CrossRef]
- Lee BWL, Ghode P, Ong DST. Redox regulation of cell state and fate. Redox Biol 2019;25:101056. [CrossRef]
- Leikam C, Hufnagel A, Schartl M, et al. Oncogene activation in melanocytes links reactive oxygen to multinucleated phenotype and senescence. Oncogene 2008;27(56):7070-7082. [CrossRef]
- Lemieux E, Boucher MJ, Mongrain S, et al. Constitutive activation of the MEK/ERK pathway inhibits intestinal epithelial cell differentiation. Am J Physiol Gastrointest Liver Physiol 2011;301(4):G719-730. [CrossRef]
- Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009;8(8):1168-1175. [CrossRef]
- Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res 2004;64(10):3580-3585. [CrossRef]
- Oshikawa J, Urao N, Kim HW, et al. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PloS one 2010;5(4):e10189. [CrossRef]
- Parascandolo A, Laukkanen MO. Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1-3 Signal Transduction Pathways. Antioxid Redox Signal 2019;30(3):443-486. [CrossRef]
- Parascandolo A, Laukkanen MO. SOD3 Is a Non-Mutagenic Growth Regulator Affecting Cell Migration and Proliferation Signal Transduction. Antioxidants (Basel) 2021;10(5). [CrossRef]
- Park KS, Kim NG, Kim JJ, et al. Differential regulation of MAP kinase cascade in human colorectal tumorigenesis. Br J Cancer 1999;81(7):1116-1121. [CrossRef]
- Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. The Journal of biological chemistry 2003;278(19):16667-16674. [CrossRef]
- Pereira DM, Gomes SE, Borralho PM, et al. MEK5/ERK5 activation regulates colon cancer stem-like cell properties. Cell Death Discov 2019;5:68. [CrossRef]
- Rajput A, Dominguez San Martin I, Rose R, et al. Characterization of HCT116 human colon cancer cells in an orthotopic model. J Surg Res 2008;147(2):276-281. [CrossRef]
- Sarkisian CJ, Keister BA, Stairs DB, et al. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 2007;9(5):493-505. [CrossRef]
- Seo HR, Kwan YW, Cho CK, et al. PKCalpha induces differentiation through ERK1/2 phosphorylation in mouse keratinocytes. Exp Mol Med 2004;36(4):292-299. [CrossRef]
- Sui X, Krantz SB, You M, et al. Synergistic activation of MAP kinase (ERK1/2) by erythropoietin and stem cell factor is essential for expanded erythropoiesis. Blood 1998;92(4):1142-1149.
- Tavaluc RT, Hart LS, Dicker DT, et al. Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle 2007;6(20):2554-2562. [CrossRef]
- Tie J, Lipton L, Desai J, et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res 2011;17(5):1122-1130. [CrossRef]
- Urosevic J, Blasco MT, Llorente A, et al. ERK1/2 Signaling Induces Upregulation of ANGPT2 and CXCR4 to Mediate Liver Metastasis in Colon Cancer. Cancer Res 2020;80(21):4668-4680. [CrossRef]
- Van Emburgh BO, Sartore-Bianchi A, Di Nicolantonio F, et al. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Molecular oncology 2014;8(6):1084-1094. [CrossRef]
- Whitehurst AW, Robinson FL, Moore MS, et al. The death effector domain protein PEA-15 prevents nuclear entry of ERK2 by inhibiting required interactions. The Journal of biological chemistry 2004;279(13):12840-12847. [CrossRef]
- Wu PK, Becker A, Park JI. Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. International journal of molecular sciences 2020;21(15). [CrossRef]
- Yang L, Zheng L, Chng WJ, et al. Comprehensive Analysis of ERK1/2 Substrates for Potential Combination Immunotherapies. Trends Pharmacol Sci 2019;40(11):897-910. [CrossRef]
Figure 3.
Characterization of the effect of ERK1/2 overexpression on migration in vitro in CCD841, CCD84 RNAi, DLD1, HCT116, and MEF SOD3 cells. a–e. Chemotactic invasion through Matrigel. According to the results, ERK2 overexpression resulted in a higher invasion rate as compared to ERK1 overexpression. RNAi significantly reduced the invasion potential of ERK2 and ERK1/2 cells below GFP control cells. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using GFP expressing cells as a comparison control. (* refers to ERK1, # refers to ERK2, ♦ refers to ERK1/2).
Figure 3.
Characterization of the effect of ERK1/2 overexpression on migration in vitro in CCD841, CCD84 RNAi, DLD1, HCT116, and MEF SOD3 cells. a–e. Chemotactic invasion through Matrigel. According to the results, ERK2 overexpression resulted in a higher invasion rate as compared to ERK1 overexpression. RNAi significantly reduced the invasion potential of ERK2 and ERK1/2 cells below GFP control cells. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using GFP expressing cells as a comparison control. (* refers to ERK1, # refers to ERK2, ♦ refers to ERK1/2).
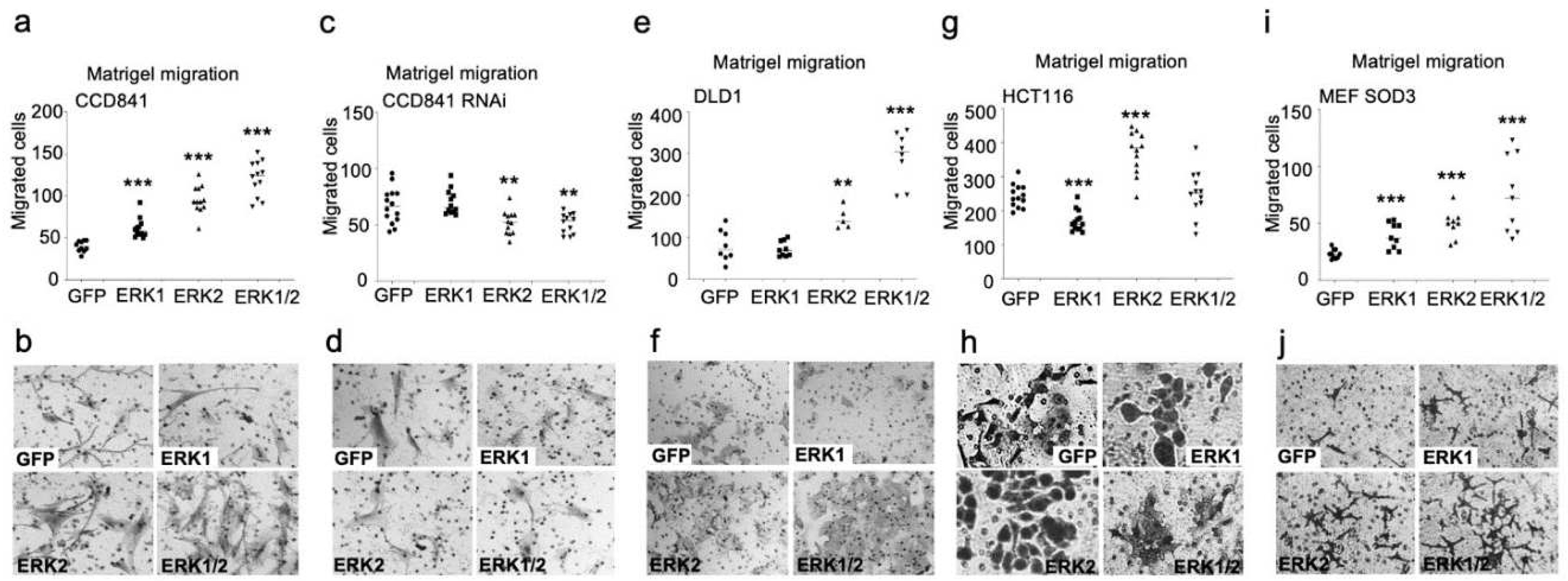
Figure 4.
Collective cell migration in wound healing assay. ERK2 overexpression promoted significantly higher migration in CCD841, DLD1, and MEF SOD3 cells as compared to gfp expressing controls. ERK1 overexpression had an obsolete effect on cell movement, whereas simultaneous ERK1 and ERK2 overexpression reduced collective cell migration in DLD1, HCT116, and MEF SOD3 cells. RNAi for ERK1 and ERK2 significantly reduced the collective migration of RNAi ERK2 cells below the GFP control cells, whereas it significantly increased migration of RNAi ERK1/2 cells. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using gfp expressing cells as a comparison control. (* refers to ERK1, # refers to ERK2, ♦ refers to ERK1/2).
Figure 4.
Collective cell migration in wound healing assay. ERK2 overexpression promoted significantly higher migration in CCD841, DLD1, and MEF SOD3 cells as compared to gfp expressing controls. ERK1 overexpression had an obsolete effect on cell movement, whereas simultaneous ERK1 and ERK2 overexpression reduced collective cell migration in DLD1, HCT116, and MEF SOD3 cells. RNAi for ERK1 and ERK2 significantly reduced the collective migration of RNAi ERK2 cells below the GFP control cells, whereas it significantly increased migration of RNAi ERK1/2 cells. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using gfp expressing cells as a comparison control. (* refers to ERK1, # refers to ERK2, ♦ refers to ERK1/2).
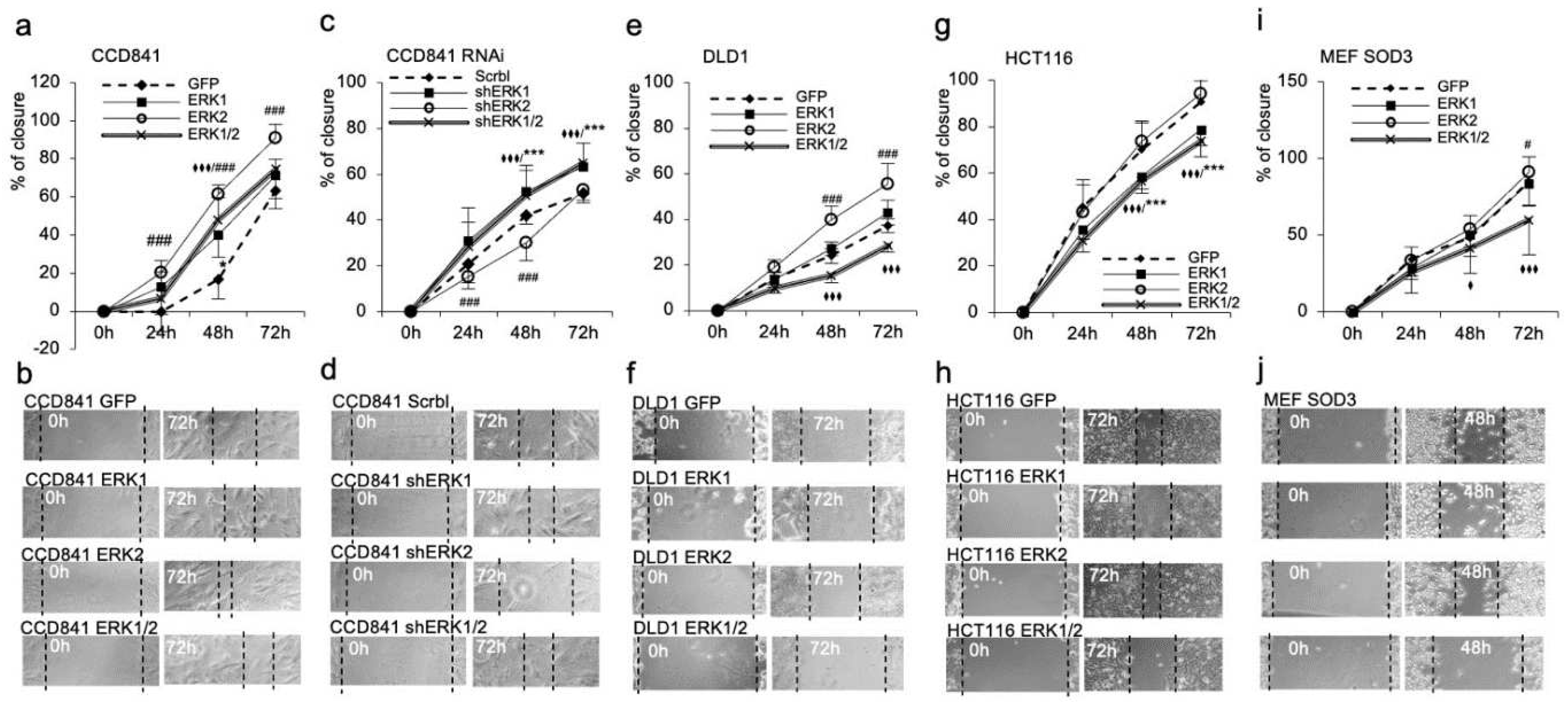
Figure 5.
Characterization of the effect of ERK1/2 overexpression on cell proliferation in vitro and in vivo in CCD841, CCD84 RNAi, DLD1, HCT116, and MEF SOD3 cells. a–e. ERK2 overexpression significantly increased cell proliferation in all cell models studied, whereas ERK1 overexpression had an obsolete effect. Simultaneous ERK1/2 expression promoted cell proliferation in CCD841 cells, whereas it inhibited growth in DLD1 and MEF SOD3 cells. Similarly to collective migration, RNAi significantly reduced cell proliferation of RNAi ERK2 cells and increased RNAi ERK1/2 cells. f. In vivo tumorigenesis of HCT116 cells overexpressing GFP, ERK1, ERK2 or ERK1/2 in nude mice. ERK2 overexpressing cells demonstrated the highest tumor formation capacity corroborating in vitro data. g–h. Tumor size and tumor initiation capacity further strengthened the obtained results suggesting moderately reduced tumor sizes in ERK1 overexpressing cells and highest tumor sizes in ERK2 expressing cells. The tumor formation capacity data showed two tumors out of six transplantations for GFP control cells, three tumors out of six transplantations for ERK1 cells, four tumors out of six transplantations for ERK2 cells, and two tumors out of six transplantations for ERK1/2 cells. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using GFP expressing cells as a comparison control. (* refers to ERK1, # refers to ERK2, ♦ refers to ERK1/2).
Figure 5.
Characterization of the effect of ERK1/2 overexpression on cell proliferation in vitro and in vivo in CCD841, CCD84 RNAi, DLD1, HCT116, and MEF SOD3 cells. a–e. ERK2 overexpression significantly increased cell proliferation in all cell models studied, whereas ERK1 overexpression had an obsolete effect. Simultaneous ERK1/2 expression promoted cell proliferation in CCD841 cells, whereas it inhibited growth in DLD1 and MEF SOD3 cells. Similarly to collective migration, RNAi significantly reduced cell proliferation of RNAi ERK2 cells and increased RNAi ERK1/2 cells. f. In vivo tumorigenesis of HCT116 cells overexpressing GFP, ERK1, ERK2 or ERK1/2 in nude mice. ERK2 overexpressing cells demonstrated the highest tumor formation capacity corroborating in vitro data. g–h. Tumor size and tumor initiation capacity further strengthened the obtained results suggesting moderately reduced tumor sizes in ERK1 overexpressing cells and highest tumor sizes in ERK2 expressing cells. The tumor formation capacity data showed two tumors out of six transplantations for GFP control cells, three tumors out of six transplantations for ERK1 cells, four tumors out of six transplantations for ERK2 cells, and two tumors out of six transplantations for ERK1/2 cells. The p-values are *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. The p-value was determined using GFP expressing cells as a comparison control. (* refers to ERK1, # refers to ERK2, ♦ refers to ERK1/2).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



