
Submitted:
07 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
The catalytically inactive caspase-8-homologous protein, c-FLIP, is a potent antiapoptotic protein highly expressed in various types of cancers. c-FLIP competes with caspase-8 for binding to the adaptor protein FADD (Fas-Associated Death Domain) following Death Receptors (DRs) activation by the ligands of the TNF-R family. As a consequence, the extrinsic apoptotic signaling pathway involving DRs is inhibited. Inhibition of c-FLIP activity in tumor cells might enhance DR-mediated apoptosis and overcome immune and anticancer drug resistance. Based on an in silico approach, the aim of this work was to identify, new small inhibitory molecules able to bind selectively to c-FLIP and block its anti-apoptotic activity. Using a homology 3D model of c-FLIP, an in silico screening of 1880 compounds from the NCI database (National Cancer Institute) was performed. Nine molecules were selected for in vitro assays, based on their binding affinity to c-FLIP and their high selectivity compared to caspase-8. These compounds selectively bind to the Death Effector Domain 2 (DED2) of c-FLIP. We have tested in vitro the inhibitory effect of these 9 molecules using the human lung cancer cell line H1703 overexpressing c-FLIP. Our results showed that 6 of these newly identified compounds efficiently prevent FADD/c-FLIP interactions in a molecular pull-down assay, as well as in a DISC Immunoprecipitation assay. Overexpression of c-FLIP in H1703 prevents TRAIL-mediated apoptosis, however, combination of TRAIL with these selected molecules signifi-cantly restored TRAIL-induced cell death by rescuing caspase cleavage and activation. Altogether, our findings indicate that new inhibitory chemical compounds efficiently prevent c-FLIP recruit-ment into the DISC complex, thus restoring caspase-8 dependent apoptotic cascade. These results pave the way to design new c-FLIP inhibitory compounds that may serve as anticancer agents in tumors overexpressing c-FLIP.
Keywords:
c-FLIP
; TRAIL
; apoptosis
; protein-protein interaction
; drug resistance
; cancer treatment
Introduction
The development of drug resistance is one of several clinical challenges causing anti-cancer drugs limited efficacy or failure. The identification of resistance mechanisms to therapies helps to design novel therapeutics [1,2]. Over the past ten years, evidences have shown that abnormalities in apoptosis signaling pathways, such as the activation of anti-apoptotic proteins were highly associated with immune and drug resistance [3].
Among these proteins, c-FLIP (Cellular FLICE Inhibitory Protein) is a major anti-apoptotic and resistance protein, restraining apoptosis induced by the TNF (Tumor Necrosis Factor) superfamily members, including TRAIL (TNF-Related Apoptosis Inducing Ligand), Fas-L or TNFα, as well as apoptosis stimulated by chemotherapeutic drugs in cancer cells [4]. c-FLIP is overexpressed in many different types of human cancers such as ovarian carcinomas [5], colorectal carcinomas [6], gastric adenocarcinomas [7], prostate carcinomas [8]. An upregulated level of c-FLIP has also been detected in primary tissues from patients with lung adenocarcinomas [9], hepatocellular carcinomas [10], melanomas [11], B-cell chronic lymphocytic leukemia [12] and Hodgkin’s lymphomas [13].
c-FLIP has 13 distinct variants, but only three of them are expressed as proteins in human cells. These are known as c-FLIP (L), c-FLIP (s) and c-FLIP(R) [14]. The long form c-FLIP (L) with a 55 kDa molecular weight (MW) is similar to procaspase-8, containing two N-terminal tandem “Death effector domains” = DED1 and DED2, and a C-terminal caspase-like domain, lacking the catalytic cysteine residue responsible for the proteolytic activity of caspases. The short form c-FLIP (s) (26 kDa MW) is composed only of two DEDs (DED1 and DED2) without a caspase-Like domain and a short C-terminus [15]. Another short form of the protein called c-FLIP (R) (27 kDa) is specifically expressed in a number of T and B cells such as Raji cells as well as in human primary T cells. It also contains N-terminal DEDs ( DED1 and DED2), but a short C-terminus composed of a stretch of residues playing a key role in the ubiquitination of c-FLIP proteins (Chang et al., 2006; Golks et al., 2005)
c-FLIP is considered as being a key inhibitory molecule in the extrinsic apoptotic signaling pathway, preventing the homodimerization and autoactivation of procaspase-8/10, the initiator caspases of apoptosis. This extrinsic pathway, also called death receptor pathway, is induced by different ligands of the TNF superfamily (TRAIL, Fas-L, TNFα) binding to their respective death receptors DRs (TRAIL- R1/R2, Fas, TNF receptor). These ligands’ bindings induce the trimerization of the respective DRs which in turn recruit the adaptor protein FADD [17]. Once FADD is recruited to the DRs, procaspase-8/10 binds by its DED2 domain to the DED domain of FADD, leading to DISC (Death Inducing Signaling Complex) formation and the activation of downstream caspases and subsequent apoptosis [18]. However, this apoptotic signaling pathway can be attenuated or totally inhibited by c-FLIP. First, it was suggested that c-FLIP binds by its DED2 to DED of FADD and impedes the recruitment of procaspase-8 to the DISC, thereby precluding its activation [19]. Then, Scaffidi and coworkers have contradicted this hypothesis and demonstrated that caspase-8 is always recruited to the DISC at the same time as c-FLIP(s/L) proteins (Scaffidi et al., 1999). Procaspase-8 forms a heterodimeric complex with c-FLIP(L), resulting in an incomplete cleavage and limited activation of caspase-8, due to the lack of c-FLIP(L) enzymatic activity. This heterodimerization prevents further apoptotic signal transduction. By competition, c-FLIP(s) over-expression can also inhibit the processing of caspase-8 at the DISC, thus blocking the activation of the apoptotic cascade. These findings reflect different functional roles of c-FLIP(L) and c-FLIP(s) in inhibiting apoptosis [21].
TRAIL, also called APO-2L, is a member of TNF family and is mainly expressed by immune cells. It is a type II transmembrane protein with a C-terminal extracellular domain, which can be cleaved by a cysteine protease resulting in a soluble form [22]. TRAIL can bind to five distinct receptors. TRAIL-R1 (DR4) and TRAIL-R2 (DR5), are classical death receptors and can trigger apoptosis with their functional cytoplasmic death domain (DD). TRAIL-R3 (DcR1), TRAIL-R4 (DcR2) also known as decoy receptors, as well the circulating receptor Osteoprotegerin (OPG) are not able to propagate a death signal due to a lack of a functional cytoplasmic death domain [23]. TRAIL expressed on cytotoxic T cells and NK cells is a potent anti-tumor molecule since it has been proven to preferentially kill cancer cells in a wide variety of tumors and does not exhibit any toxicity in a majority of normal cells. However, a large number of cancers evade TRAIL-induced apoptosis and become TRAIL resistant through different mechanisms including the over expression of c-FLIP [24]. Selective knock-down of c-FLIP(L) sensitizes tumor cells to TRAIL-induced cell death in human lung cancer cell lines [25]. It has also been demonstrated that Withanolide E, a steroidal lactone derived from Physalis peruviana, can highly sensitize renal carcinoma cells and other human cancer cells to TRAIL-mediated apoptosis through rapid destabilization, aggregation and proteasomal degradation of c-FLIP proteins, confirming the key role of c-FLIP in protecting cells from death ligands mediated apoptosis [26].
So far, except siRNAs approaches, c-FLIP inhibitors that have been studied act indirectly on c-FLIP, such as Cisplatin which induces p53-dependent FLIP ubiquitination and degradation in ovarian cancer cells [27], or Actinomycin D which downregulates FLIP(L) and FLIP(s) expression in B chronic lymphocytic leukemia [28]. Thus, the identification of molecules directly targeting c-FLIP and which are more stable than siRNA represents a new promising strategy to overcome therapy resistance. c-FLIP is structurally similar to caspase-8. Each DED of c-FLIP shares ∼25% similarity with DEDs of caspase-8, and the C-terminus (270 amino acids) of c-FLIP is also ∼25% identical to the C-terminus of caspase-8 [29]. Thus, due to this homology, the identification of new compounds that selectively bind to c-FLIP versus caspase-8 and prevent its recruitment to the DISC is challenging.
Based on this rational, molecular modeling and docking experiments were set up to construct c-FLIP and caspase-8 homology models in order to find selective inhibitors targeting unique sequences of c-FLIP and not caspase-8. In vitro assays using recombinant c-FLIP(s) and FADD, coupled with in cellulo assays demonstrated the inhibitory role of these new compounds, restoring TRAIL-mediated apoptosis and selectively preventing c-FLIP/FADD interactions. Our findings suggest that blocking c-FLIP recruitment into the DISC by specific inhibitors decreases tumor resistance to death receptors mediated apoptosis and represent a new avenue for cancer treatment.
Materials and methods
Molecular modeling
- 1)
- Homology modeling of DED2 domains
Homology models were built using MOE software (Molecular Operating Environment). First, sequences of the DED2 domains of c-FLIP and of CASP8 were extracted from the Uniprot database and used to find homolog structures in the Protein Data Bank (PDB) using the BLAST software. Three sequences showed a significant percentage of identity with the target sequences (around 30%) to perform homology modeling. Three structures of the FADD DED domain (PDB.ID 1A1W, 1A1Z, 2GF5) and three structures of two v-FLIP DED2 domains (PDB.ID 2BBR, 2BBZ, 3CL3) were identified as suitable templates for modeling the DED2 domains of CASP8 and c-FLIP respectively.
- 2)
- Identification of the binding site
SiteFinder module of the MOE software was used to identify druggable pockets at the surface of our models and only the homology models which present such pockets at the vicinity of the F-L hydrophobic patch were retained. Two models of the CASP8 DED2 domain and one model of the c-FLIP DED2 were kept based on the PDB.ID 1A1W, 2GF5 and 2BBZ structure templates respectively.
- 3)
- Docking of chemical libraries
The 1880 molecules from the NCI DiversitySet3 and extracted from the ZINC database were virtually screened on the three models (2 for CASP8 DED2 and one for c-FLIP DED2) using two docking software, AutoDock and Glide. The ZINC database provides ready-to-dock sets of purchasable molecules. The results of these two virtual screenings were combined using a consensus scoring method and a root mean square deviation (RMSD) filter.
- 4)
- Consensus scoring function
In addition to the Glide and AutoDock scoring functions, the MOE GBVI/WS dG function was selected to rescore all poses of the docked ligands. Therefore, the binding modes of each docked ligand with Glide and AutoDock were assessed using four score values. The score values were normalized using the Z-score following formula:
where μ is the mean and σ is the standard deviation of the scores.
The four normalized scores were then summed up and ranked by decreasing order, the best score being the lowest ones. To keep only ligands presenting similar poses with the two docking methods, we calculated the RMSD (Root Mean Square Deviation) between the poses obtained by the two methods. The ligands which present a RMSD value higher than 2 Å were removed from the ranking.
- 5)
- Hit selection
The goal of this docking study was to identify ligands that selectively bind to c-FLIP and not to CASP8. For this purpose, the best 20 screened molecules obtained on the c-FLIP target domain were compared with the best 20 molecules obtained on each CASP8 model. Molecules present in the top 20 for c-FLIP and absent in the top 20 for CASP8 models were considered selective ligands.
Cell culture
H1703 (Human non-small lung cancer cell line), mock transfected or overexpressing c-FLIP(L) cells were grown in RPMI 1640 (LONZA) culture media supplemented with 10 % fetal bovine serum and puromycin (2 µg/ml) from Sigma-Aldrich. The cells were kept in a humidified atmosphere in an incubator at 37°C and 5 % CO₂. H1703 were a kind gift from O. Micheau (INSERM, DIJON FRANCE).
Reagents and antibodies
KillerTRAIL (human recombinant) were purchased from Alexis Biochemicals. The nine most highly selective molecules targeting cFLIP were obtained from NCI–DS&CB (National Cancer Institute-Drug Synthesis and Chemistry Branch, USA). For western blotting (WB) experiments, we used the following antibodies: anti-FLIP antibodies DAVE2 and NF6 (ADIPOGEN), caspase-3 (8G10; OZYME), PARP (Asp214, 19F4; OZYME), caspase-8 (1C12; OZYME), anti-MBP (New England Biolabs), anti-His (C-ter) (INVITROGEN). Anti-His(ab81663) was used to immuno-precipitate the DISC complex, and the following antibodies: anti-Flip (DAVE II, Adipogen), anti-FADD (556402,BD), anti-casp8 (5F7,EnzoLife), anti-DR4 (1139, ProSci), anti-DR5 (3696, Cell Signaling were used for WB analysis. Anti-rabbit and anti-mouse HRP linked secondary antibodies (Santa Cruz Biotechnology), β-actin (Sigma Aldrich) were also used.
Flow cytometry analysis
Apoptotic cell death was confirmed by flow cytometry (cytoFLEX, Beckman Coulter) using Annexin V-PE Kit, according to the manufacturer’s instructions (BD Biosciences). In brief, H1703 cells were seeded at a density of 2 x 10⁵ cells/well in 24 well plates and incubated for 24 hours. Cells were then treated, as indicated in figure legends, for 18 hours. Thereafter, cells were collected, washed and re-suspended in 1X Annexin-V binding buffer. Annexin-V- PE was added to the cells and left 20 mins at room temperature in the dark. 7-AAD dye was added and Flow cytometric analysis was performed in the final step.
Recombinant protein production and purification
Full-length c-FLIP(s) and FADD were synthesized (Genscript, USA), and subcloned respectively into pET24b(+) (Novagen) and pMAL-C2X (New England Biolabs) expression vectors. The resulting constructs enabled the fusion of the corresponding protein with a C-terminal polyhistidine peptide, or a N-terminal Maltose Binding Protein (MBP).
All proteins were expressed in 1 mM IPTG-induced Rosetta transformed bacterial cells with the expression vectors. After 18 hours of induction at 37°C, cells were harvested and pellets were resuspended in a lysis buffer (50 mM Tris-HCl, 100 mM NaCl, pH 8, 0.1% Triton) in addition to 0,1 mg/ml lysozyme and 1mM PMSF (Phenylmethanesulfonyl fluoride) and incubated for 20 minutes at 4°C. Cells were then lyzed by freeze-thaw cycles, followed by sonification. Lysate was centrifuged at 34000 g for 20 minutes at 4°C, and supernatant was loaded on chromatographic media. HisPurTM Ni-NTA Chromatography Cartridge 1 mL (Thermo Scientific) or MBPTrapTM HP 1 mL (GE Healthcare) were respectively used for purification of c-FLIP(s) or FADD, following columns manufacturer procedures. Protein purity was assessed by SDS-PAGE analysis and concentrations were qualified using the Bio-Rad protein assay based on the Bradford dye-binding method.
Pull-Down binding assay
The purified proteins were mixed at a ratio of 1:7 for FADD:FLIP(s), equivalent to 0,7 mg/ml for FADD and 6,8 mg/ml for FLIP (26µM and 261 µM respectively) and incubated for 18 hours at 4°C with 0-3000 µM concentration range of each inhibitor. Then, the incubation mix was loaded on a MBPTrap chromatography cartridge and purified using 10 mM maltose as eluent. Negative controls were also performed without FADD. Unbound and eluted samples were used for Western Blot analysis.
DISC Immunoprecipitation
For DISC Immunoprecipitation, 50.10⁶ H1703-FLIP(L) cells seeded in F175 Flasks were incubated overnight. The next day, cells were collected in 10 ml of media, pre-treated with seletected Molecules 1,3,4 or 9 (500 µM) for 2 hours and then stimulated with 1 µg/mL His-TRAIL for 20 minutes. Reaction was blocked with cold PBS, and cells were lysed in 1 mL of IP-Lysis Buffer (1% NP40, 20 mM TRIS HCL Ph 7.4, 150 mM NaCl, 10 % Glycerol) containing COMPLETE Inhibitor of Proteases cocktail (Roche), for 30 mins on ice. Lysates were cleared by centrifugation at 15000 RPM, 20 minutes at 4°C. All supernatants were pre-cleared with 50 µL of Sepharose 6B (Sigma 6B100) for 1.5 hour at 4°C on a wheel, and then centrifugated at 1500 RPM at 4°C for 15 seconds. To analyze the DISC complex, supernatants were collected and incubated overnight at 4°C with 50 µL Protein G beads and coupled with 3 µg anti-His tag antibody (abcam ab81663) supplemented with 1 µM of caspase inhibitor Z-VAD. After immunoprecipitation (IP), beads were washed 4 times with IP-lysis buffer (without inhibitor of proteases) and eluted with 120 µL of LDS buffer with DTT. Samples were left 30 minutes at room temperature, then heated 5 minutes at 95°C before WB analysis.
Western blot analysis
Pull-down eluted samples were heated 5 minutes at 99°C with Laemmli Buffer and then passed on 4%-12% gradient SDS-PAGE and transferred to nitrocellulose membrane (GE Healthcare). To evaluate enzymatic activity, treated cells were lysed in RIPA buffer at 4°C and centrifuged at 15 000 rpm for 20 minutes. Protein concentration was then determined using Bradford assay. Equal amounts of proteins (50µg) were boiled for 5 minutes with 1X LDS sample buffer and then loaded on a 4%-12% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked by 5% non-fat milk in PBS-TWEEN 20 (0.1%) for one hour at room temperature and then incubated with different primary antibodies for one to two hours at room temperature. Membranes were then washed by PBS-TWEEN and incubated for 1 hour with HRP-conjugated secondary antibodies. Proteins bands were visualized by chemoluminescence protocol ECL (Thermo Scientific).
Statistical analysis
Statistical analyses were performed with the Student t test.
Results
Potential c-FLIP inhibitors selected by in silico screening
Three homology models were built for each target: DED2 domains of CASP8 and c-FLIP. Among the six homology models obtained, only those having at least one druggable pocket near the hydrophobic patch F-L were conserved (Figure 1A). We thus kept one model for c-FLIP DED2 domain, based on the PDB.ID 2BBZ structure, and two models for CASP8 DED2 domain, based on the PDB.ID 1A1W and 2GF5 structures.
The search for druggable pockets near the hydrophobic patch made it possible to select the models to be used in virtual screenings: the CASP8 models based on 1A1W and 2GF5 (which we will respectively call CASP8 (1A1W) and CASP8 (2GF5)) and the c-FLIP model 2BBZ c- FLIP (2BBZ). The MOE Site Finder module searches for cavities in a protein using a method based on filling with alpha centers. A deep cavity contains at least ten α centers. The pockets of the CASP8 (1A1W) model only contain seven, so they are rather in surface. One of the pockets of the CASP8 (2GF5) model contains 21 α centers and the other less than ten. This model therefore has a surface pocket and a buried one. The c-FLIP (2BBZ) model has the largest cavity deeper, tunnel-shaped (31 α centers). Pockets 1 and 2 of the CASP8 models are located on either side from the hydrophobic patch and always respectively present the triplets ARG19, LYS22 and PHE23 (pocket 1) and LEU24, VAL64 and GLN67 (pocket 2). The c-FLIP cavity is notably bordered by residues LEU24 and CYS64 and therefore corresponds to pocket 2 of the CASP8 models (Figure 2).
Finally, for each of the two models of CASP8 DED2 domain, two druggable pockets were found at the proximity of the hydrophobic patch F-L by the module SiteFinder of the MOE software. We only found one druggable pocket satisfying this condition for the homology model of c-FLIP DED2 domain. Five virtual screenings were performed using two docking software, AutoDock and Glide, four on the CASP8 target and one on c-FLIP Compounds were ranked using three scoring functions, AutoDock, Glide, and the consensus scoring function. We chose to select the three best selective compounds obtained by each of the ranking methods for biological testing. Nine compounds were selected as potentially selective inhibitors of c-FLIP (Figure 1B).
The newly identified c-FLIP inhibitors prevent DED-FADD/DED2-c-FLIP interaction.
Upon death receptors’ stimulation and DISC formation, the DD domain of the death receptors interacts with the DD of FADD, whereas the DED of FADD interacts with the DED2 of procaspases 8/10 [18]. When highly expressed, c-FLIP is incorporated and binds to FADD via its DED2 domain, particularly via the FL motif (F114/L115). Mutations in this FL motif abrogated FLIP recruitment to the DISC [30]. There are two contradictory evidence concerning c-FLIP binding to FADD. One study has shown that the DED of FADD is docked at the interface between DED1 and DED2 of c-FLIP, because mutations in this linker region exhibited a weak interaction with DED of FADD [31]. However, another study has demonstrated that the amino acid F114 of the hydrophobic patch of c-FLIP-DED2 domain is responsible for binding to the FADD α1-α4 groove, with a reciprocal interaction of H9 of FADD with α2-α5 hydrophobic patch of DED2-c-FLIP [32]. However, the direct interaction between c-FLIP and FADD DED is not very well determined. Here, to elucidate the molecular mechanism involved in the restoration of TRAIL-induced apoptosis by compounds 1 to 9, we investigated in cellulo the potentiality of these compounds to prevent c-FLIP(s)/FADD interaction. A mix containing recombinant proteins c-FLIP(s) and FADD was incubated in the presence of various concentration of each inhibitor, and FADD was purified using the MBP affinity tag. When a potent inhibitor prevented the interaction, then c-FLIP(s) was not co-eluted after FADD purification.
As depicted in Figure 3 A-D and I, compounds 1, 2, 3, 4, and 9 were able to prevent c-FLIP(s)/FADD direct interaction with a concentration below 3000 µM. This high concentration was required as c-FLIP(s) and FADD have to be mixed at high concentration to enable efficient pull-down in this assay. Compounds 5, 6, 7 and 8 (Figure 3 E, F, G, H) did not exhibit potent inhibition of the protein-protein interaction below the highest concentration tested. Interestingly, compounds 1 and 2 exhibit the highest potency as they prevent the interaction with the lowest concentration (500 µM), followed by compounds 3 and 9 (1000 µM), and finally compound 4 (2500 µM).
Newly identified molecules inhibit c-FLIP recruitment into the DISC by preventing its interaction with FADD
It was important to confirm that our new selected molecules inhibit c-FLIP interaction with FADD and its recruitment into the DISC, since it has been demonstrated that FLIP depletion from the cytosolic fraction prevents its binding to the DISC and allows caspase-8 cleavage and apoptosis [33]. To evaluate the inhibitory role of these molecules, in cellulo tests were proceeded using H1703 cells overexpressing c-FLIP protein. H1703 were treated with TRAIL alone or in combination with FLIP inhibitors, then the DISC complex was immunoprecipitated to investigate the presence of FLIP protein. Interestingly, when cells were treated with TRAIL combined with Molecule 1, 3, 4 or 9, FLIP recruitment into the DISC was significantly reduced (Molecule 2 was not tested because of a lack of its required concentration). This was demonstrated by a significantly reduced elution of FLIP with other DISC components (Fig 4. A and B). Remarkably, Molecule 3 and 9 seemed to be more efficient to prevent FLIP interaction with the DISC, according to the pull-down assay data (Fig 3). As a control, FLIP protein was always recruited to the DISC when H1703 cells were treated with TRAIL alone. Hence, the new molecules act as inhibitors to prevent FLIP incorporation into the DISC complex following TRAIL stimulation.
c-FLIP inhibitors rescue TRAIL-mediated apoptosis.
It was reported previously that up-regulated expression of c-FLIP(L) precludes the interaction of the initiator procaspase 8/10 with the adaptor protein FADD, thereby blocking cell death induced by members of the TNF superfamily (TNF-α, FasL, or TRAIL) [34]. Indeed, our previous study revealed that TRAIL is a promising cytotoxic molecule against Follicular lymphoma B cells while it does not affect normal B cells. We also showed that downregulation of c-FLIP(L/s) after inhibition of NF-κB signaling could restore TRAIL-mediated apoptosis in follicular lymphomas [35]. In this current study, we aimed to assess whether the newly identified FLIP inhibitors, could sensitize the extrinsic apoptosis pathway when combined with the death ligand TRAIL (Figure 5). The squamous non-small cell lung cancer (NSCLC) cell line, H1703 either Mock-transfected or H1703 carrying the long form c-FLIP-(L) were initially treated with or without TRAIL ligand in order to evaluate the influence of c-FLIP(L) on apoptosis. As expected, the high expression of c-FLIP(L) in the transfected cell line inhibits TRAIL-induced apoptosis compared to H1703-mock transfected cells (Figure 5A), confirming the anti-apoptotic effect of c-FLIP. Since c-FLIP(L) prevents death receptor-mediated apoptosis, we then examined whether its inhibition could increase the sensitivity of cancer cells to TRAIL, since it was reported that targeting c-FLIP directly or indirectly could overcome apoptosis resistance [36,37]. We first investigated the cytotoxic effect of our nine selected molecules administered alone at the “appropriate concentration” (chosen from a range of concentration for each molecule with no cytotoxicity on cells). As shown in Figure 5B., there is no enhancement of cell death when cells were treated with the inhibitors alone, compared to non-treated cells, either in mock or in FLIP(L) cells. This indicates that these FLIP-inhibitors at selected concentrations were not toxic against cancer cells. In contrast, a significant increase of apoptosis was observed in cells overexpressing c-FLIP(L), after a co-treatment of TRAIL with FLIP-inhibitors, compared to FLIP(L) cells treated with TRAIL alone or FLIP inhibitors alone. Molecules 1, 3, 4, 8 and 9 were the most efficient and showed a remarkable enhancement of cell death compared to other tested compounds ( 30%). These findings strongly suggest that blocking c-FLIP with this new Molecules overcomes c-FLIP resistance and sensitizes cancer cells to death receptor mediated apoptosis.
Combination treatment of TRAIL with c-FLIP targeting molecules restores caspases-dependent apoptosis.
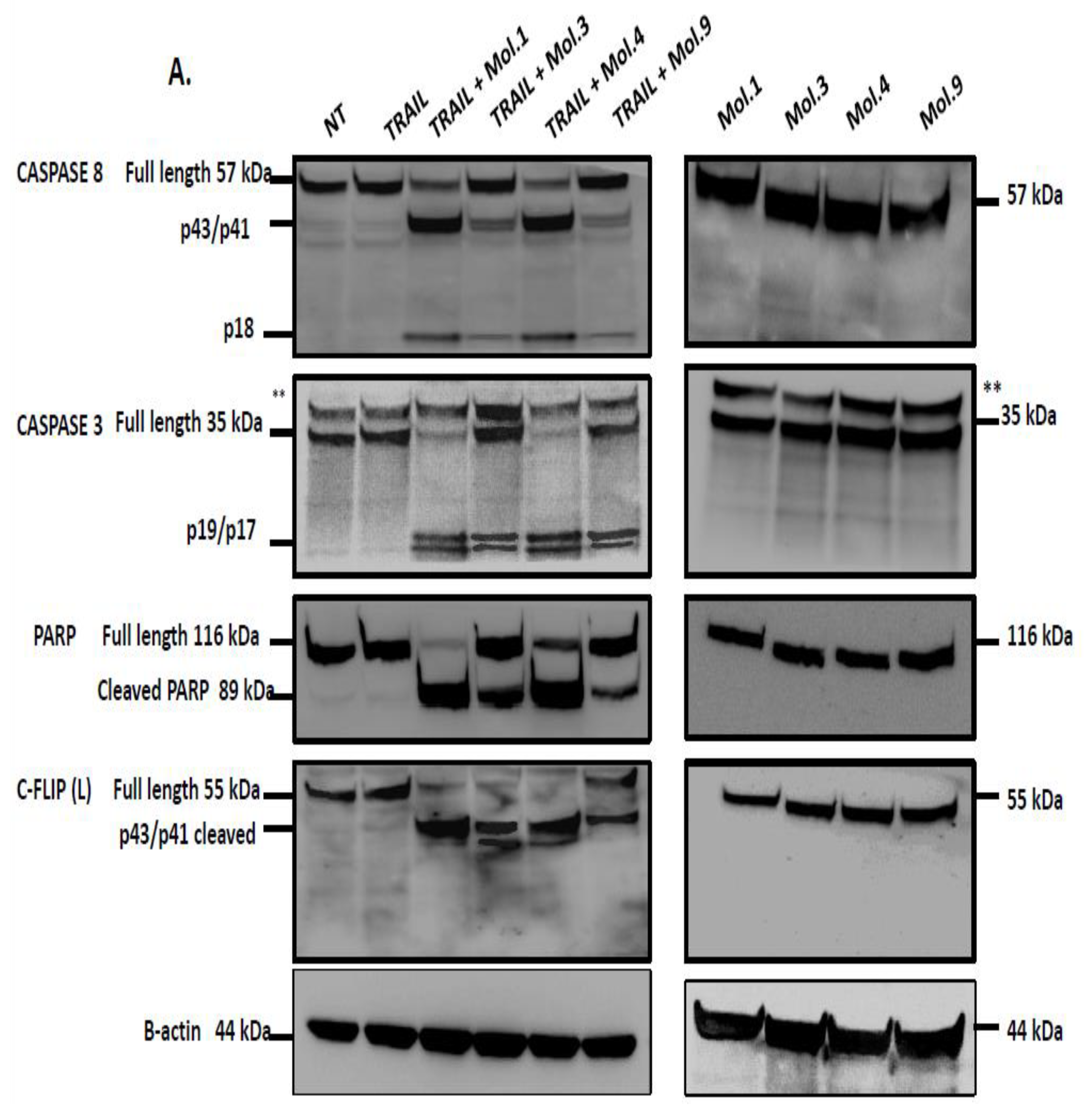
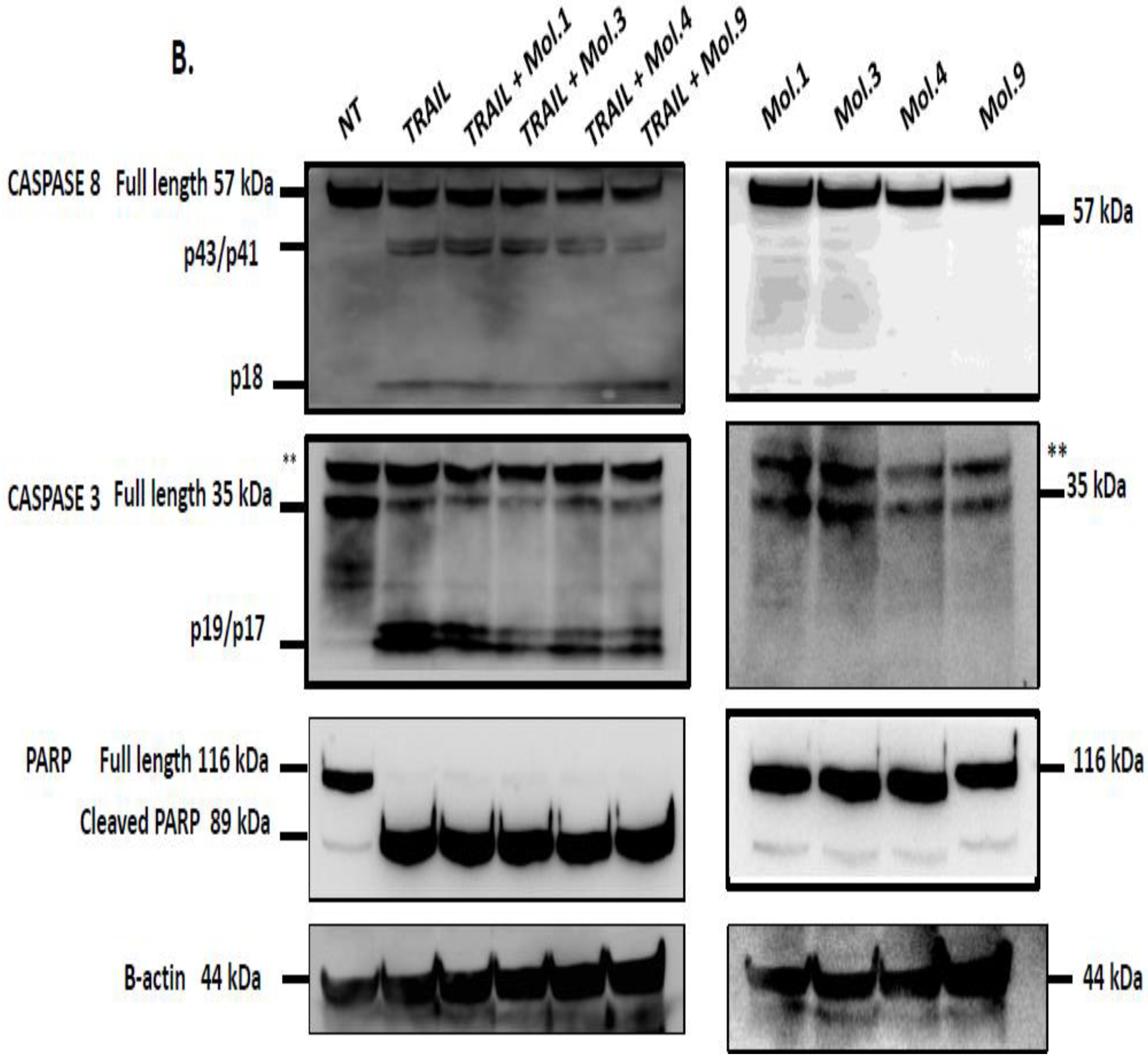
Caspases are the major effectors of apoptosis. Death signaling events induce dimerization and auto-processing of the initiator caspase-8, which activates downstream executioner caspases such as caspase-3 [38]. PARP cleavage is reported as an hallmark of caspase-3 activation and apoptosis [39]. At high concentrations, c-FLIP(L) forms an heterodimer with caspase-8, which prevent the formation of a fully active caspase-8, thereby blocking apoptosis [40]. To study the impact of the newly identified c-FLIP inhibitors on caspase activation and apoptosis induction, H1703 overexpressing c-FLIP(L) were treated with TRAIL combined with each c-FLIP inhibitor. Among the nine inhibitors, we have chosen only those that showed an inhibitory effect both in cellulo (Figure 4 and Figure 5) and in the pull-down assay (Figure 3), such as Molecules 1, 3, 4 and 9. H1703-c-FLIP(L) cells treated with 100 ng/ml of TRAIL alone, did not exhibit any caspase cleavage, confirming the role of c-FLIP in the inhibition of caspase-8/10 activation (Figure 6A, left panels). However, combination of TRAIL with the selected molecules targeting c-FLIP restored apoptosis, demonstrated by caspase-8, -3 and PARP cleavage. Interestingly, treated cells with c-FLIP inhibitors alone did not induce any caspase cleavage, confirming that these molecules do not exhibit a direct cytotoxic effect on cells (Figure 6A, right panels). Similarly, H1703 lacking c-FLIP(L) protein expression were treated in the same manner, either with TRAIL alone, inhibitors alone, or TRAIL combined with c-FLIP inhibitors. In the absence of c-FLIP (L), cells were sensitive to TRAIL and induced caspase-8 cleavage, as well as subsequently caspase-3 and PARP cleavage. Combination of TRAIL with c-FLIP inhibitors also induced caspase cleavage (Figure 6B, left panels), suggesting that only TRAIL is responsible for caspase activation as cells treated with the molecules alone did not exhibit any caspase cleavage (Figure 6B, right panels). Taken together, these experiments indicate that interacting with and blocking c-FLIP, using the newly characterized compounds, prevent its binding to FADD and its recruitment into the DISC. Consequently, apoptosis can be restored though efficient caspase-8/10 cleavage.
Discussion
TRAIL’s ability to specifically kill tumor cells in vitro and in vivo makes this death ligand or its death receptors promising anticancer agent or target which exhibited potent anticancer activity in several preclinical studies [41]. However, a number of primary cancer cells are resistant to TRAIL monotherapy. This resistance can occur at the death receptor level via the upregulation of the anti-apoptotic protein c-FLIP [42]. c-FLIP is also a potent resistance factor against other TNFα superfamily members and chemotherapeutic drug-mediated apoptosis [43]. c-FLIP has been found to be overexpressed in many types of malignancies, and its overexpression is associated with poor prognosis and tumor progression due to its involvement in the inhibition of the apoptotic process [44]. The three known variants of c-FLIP are able to interfere with the FADD/caspase-8 interaction, thus inhibiting caspase-8 recruitment into the DISC and blocking its activation which ultimately lead to inhibition of apoptosis [14]. Because c-FLIP upregulation prevents the apoptotic machinery and leads to cancer promotion, its silencing has been shown to restore apoptosis. Therefore c-FLIP is considered as an important promising target in cancer therapies. In this context, we aimed to identify new inhibitory molecules specifically targeting c-FLIP and combine them with TRAIL in order to restore apoptosis in cancer cells. It was indeed previously reported in several studies that a combination of targeted anti-cancer therapies with TRAIL sensitizes resistant cells to TRAIL-induced apoptosis [45,46].
Because of the high structural homology to caspase-8 [14], c-FLIP is a difficult molecule to target since small molecules able to block its recruitment into the DISC may also inhibit caspase-8 recruitment, thereby preventing apoptosis. Therefore, small molecules selectively targeting c-FLIP without affecting caspase-8 functions are required. The challenge of our work was to identify new molecules that can selectively bind to c-FLIP and inhibit its interaction with FADD within the DISC, without affecting caspase-8 binding. We aimed to construct in silico DED2 domains of c-FLIP and caspase-8 based on homology models as their human crystallographic structures were not established. Searching for similar target sequences, Yang and collaborators [47] showed that v-FLIP DED2 and FADD DED were structurally similar to DED2 of c-FLIP and caspase-8 respectively. After having successfully generated a 3D-model homology domain of c-FLIP and caspase-8 DED2 and identify a potential unique druggable pocket in c-FLIP (Figure 1, 2), we initiated in silico screening and docking experiments of 1880 compounds from the NCI database targeting the homology model structures of c-FLIP and caspase-8 . This large set of chemical molecules led us to select compounds that only bound to c-FLIP and not caspase-8. These molecules selectively target and bind to DED2 of c-FLIP and not caspase-8.
Among the DED2-c-FLIP binding molecules, we selected nine compounds exhibiting the most potent binding affinity toward DED2 of c-FLIP versus DED2 of caspase-8. Further analyses were required to assess whether these new molecules were able to inhibit DED-FADD/DED2-cFLIP interaction. FADD is considered as the nucleus of the DISC assembly and responsible for initial caspase-8/-10 and c-FLIP recruitment through homotypic DEDs interactions [48]. FADD is composed of two distinct domains: DD (Death Domain) responsible for receptor engagement, and DED domain which contains a face-exposed hydrophobic patch, conserved in all other DEDs proteins, and thought to be crucial for DED-DED interactions with caspase-8 and c-FLIP [49]. Therefore, to elucidate the effect of the nine selected molecules, we investigated the interactions between the recombinant human FADD and c-FLIP(S) full length. The long form c-FLIP (L) was indeed impossible to produce and purify due to its rapid precipitation and low stability. Here we showed that Molecules 1, 2, 3, 4 and 9 were able to inhibit FADD-c-FLIP (S) interaction in a pull down assay (Figure 3 A-D and I). In addition, molecules 1, 3, 4 and 9 were able to prevent FLIP recruitment into the DISC using the Immunoprecipitation assay, confirming our previous results.
We further assessed whether these 9 chemicals could inhibit c-FLIP binding to FADD in a cellular model. It has been reported that c-FLIP is highly expressed in Non-small cell lung cancer (NSCLC) where its high cytoplasmic expression correlates with poor prognosis. Moreover, c-FLIP inhibits anticancer drug-induced apoptosis in preclinical models of NSCLC [50]. Thus, we overexpressed the long form of c-FLIP (L) protein in H1703 cancer cells (NSCLC) and we treated these cells first with TRAIL alone. As observed in Figure 5A, an ectopic expression of c-FLIP (L) inhibits TRAIL-mediated apoptosis. In contrast, when c-FLIP (L) is absent, cancer cells were sensitive to TRAIL, confirming the primary function of c-FLIP as an inhibitor of apoptosis and a possible biomarker of tumor resistance. As the activity of TRAIL is prevented by c-FLIP overexpression, targeting c-FLIP by our new molecules may restore TRAIL function, as it is broadly referenced in several studies showing that c-FLIP silencing sensitizes tumor cells to death ligands induced apoptosis [51]. To evaluate the efficiency of our new selected molecules in inhibiting c-FLIP function, a combination of them with TRAIL was assessed and showed a remarkable enhancement of the apoptotic level in c-FLIP-overexpressing cells; while the same molecules exhibit no significant cell death when administered alone compared to non-treated cells (Figure 5B). These findings revealed that the new identified molecules are distinctly efficient against c-FLIP and help to restore apoptosis in TRAIL-resistant cells, while they are not cytotoxic alone.
Molecules 1, 3, 4 and 9 are able to restore apoptosis when combined with TRAIL (Figure 5B), confirming their binding affinity to c-FLIP (Figure 3) and their role in preventing c-FLIP recruitment into the DISC (Figure 4).These molecules are poorly used in other studies but some results showed that they target and inhibit the following enzymes: Protein-tyrosine phosphatase (by molecule 1), MAP kinase ( by molecule 3 and 4), and matrix metalloproteinase (by molecule 9). In contrast, while molecules 2 and 6 showed an inhibitory role of FADD/c-FLIP interaction in the molecular assay (Figure 3B, F), they were poorly restoring apoptosis in resistant cancer cells overexpressing c-FLIP (Figure 5). Such results indicate that these two molecules may lose their binding potential to c-FLIP within the cell and they might require further chemical and structural modifications in order to enhance their binding affinity to c-FLIP. Surprisingly, we found that molecules 5, 7 and 8 could not prevent FADD/c-FLIP interaction in the pull-down assay (Figure 3E, G, H). In contrast they were able to sensitize resistant cells to TRAIL-induced apoptosis, especially molecule 8 which enhanced apoptosis by more than 30%, with no cytotoxicity by itself. This evidence led us to conclude that these three molecules are not selective to c-FLIP and they may target other signaling pathways contributing to cell death.
c-FLIP competes with caspase-8 recruitment into the DISC and prevents its activation, thereby blocking downstream caspases activation and apoptotic pathway [52]. Downregulation of c-FLIP using siRNAs sensitizes cells to caspases-dependent apoptosis [53]. In this study, we investigated caspases activity after combination of TRAIL with c-FLIP-inhibitory molecules to evaluate the restoration of extrinsic apoptosis. Our data indicates that caspase activity is blocked when c-FLIP is active and recruited into the DISC (Figure 6A). However, inhibiting c-FLIP function with these new molecules enhances caspases-8, -3 and PARP cleavages and promotes TRAIL-mediated apoptosis. Moreover, when cells overexpressing c-FLIP are treated with the molecules alone, no caspases activation and PARP cleavage have been observed indicating that the administration of these compounds is safe and does not induce any cytotoxicity. Similarly, when cells lacking c-FLIP (L) were treated with these compounds alone, we did not observe any caspase cleavage, suggesting that these molecules exhibit no side effect on cells with no expression of c-FLIP.
In conclusion, we report that c-FLIP expression is a relevant biomarker of cancer resistance in general and for the anticancer agent TRAIL. In this current study, we have demonstrated that c-FLIP function can be inhibited by new small molecules while caspase-8 remains unaffected. Inhibition of c-FLIP interaction with FADD precludes c-FLIP from its recruitment into the DISC and allows caspase-8 binding to FADD, promoting TRAIL-induced apoptosis in resistant cancer cells. This combination therapy of TRAIL with these c-FLIP targeted new agents could be a promising approach to eradicate tumors with c-FLIP upregulation. However, maximizing the binding affinity of the most efficient molecules by structural modifications are needed in order to restore a higher level of apoptosis in tumor cells.
References
- ROBERTI, A; LA SALA, D; CINTI1, C. Multiple Genetic and Epigenetic Interacting Mechanisms Contribute to Clonally Selection of Drug-Resistant Tumors: Current Views and New Therapeutic Prospective. J Cell Physiol. 2006, 207, 12–22. [Google Scholar] [CrossRef]
- Safa, AR. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr Med Chem. 2004, 4, 1–17. [Google Scholar] [CrossRef]
- Stover, EH; Baco, MB; Cohen, O; et al. Pooled genomic screens identify anti-apoptotic genes as targetable mediators of chemotherapy resistance in ovarian cancer. Mol Cancer Res. 2019, 17, 2281–2293. [Google Scholar] [CrossRef]
- Seol, J-Y; Mihich, E; Berleth, ES. TNF Apoptosis Protection Fraction (TAPF) prevents apoptosis induced by TNF, but not by Fas or TRAIL, via NF-κB-induced increase in cFLIP. Cytokine. 2015, 75, 321–329. [Google Scholar] [CrossRef]
- Mezzanzanica, D; Balladore, E; Turatti, F; et al. CD95-mediated apoptosis is impaired at receptor level by cellular FLICE-inhibitory protein (long form) in wild-type p53 human ovarian carcinoma. Clin Cancer Res. 2004, 10, 5202–5214. [Google Scholar] [CrossRef] [PubMed]
- Longley, DB; Wilson, TR; McEwan, M; et al. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene. 2006, 25, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Nam, SY; Jung, GA; Hur, GC; et al. Upregulation of FLIPs by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci. 2003, 94, 1066–1073. [Google Scholar] [CrossRef]
- Zhang X, Jin T, Yang H, Dewolf WC, Khosravi-far R, Olumi AF. Persistent c-FLIP ( L ) Expression Is Necessary and Sufficient to Maintain Resistance to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand – Mediated Apoptosis in Prostate Cancer Sensitivity of Prostate Cancer Cells to Recombinant Human. 2004;(L):7086-7091. [CrossRef]
- Brambilla C, Brambilla E, Gazzeri S. E2F1 induces apoptosis and sensitizes human lung adenocarcinoma cells to death-receptor-mediated apoptosis through specific downregulation of. 2006:260-272. [CrossRef]
- Wilson, NS; Dixit, V; Ashkenazi, A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Bullani, RR; Huard, B; Viard-Leveugle, I; et al. Selective expression of FLIP in malignant melanocytic skin lesions. J Invest Dermatol. 2001, 117, 360–364. [Google Scholar] [CrossRef]
- MacFarlane, M; Harper, N; Snowden, RT; et al. Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia. Oncogene. 2002, 21, 6809–6818. [Google Scholar] [CrossRef]
- Mathas, S; Lietz, A; Anagnostopoulos, I; et al. c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor-induced apoptosis. J Exp Med. 2004, 199, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin Ther Targets. 2003, 7, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Chang, L; Kamata, H; Solinas, G; et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem. 2005, 280, 14507–14513. [CrossRef] [PubMed]
- Fu Q, Fu T-M, Cruz AC, et al. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol Cell. 2016, 61, 602–613. [CrossRef]
- Carrington PE, Sandu C, Wei Y, et al. The Structure of FADD and Its Mode of Interaction with Procaspase-8. Mol Cell. 2006, 22, 599–610. [CrossRef] [PubMed]
- Rasper DM, Vaillancourt JP, Hadano S, et al. Cell death attenuation by “Usurpin”, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998, 5, 271–288. [CrossRef] [PubMed]
- Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999, 274, 1541–1548. [CrossRef]
- Mahdizadeh SJ, Thomas M, Eriksson LA. Reconstruction of the Fas-Based Death-Inducing Signaling Complex (DISC) Using a Protein–Protein Docking Meta-Approach. J Chem Inf Model. 2021, 61, 3543–3558. [CrossRef]
- Kimberley FC, Screaton GR. Following a TRAIL : Update on a ligand and its five receptors. 2004, 14, 359–372. [CrossRef]
- Kundu M, Greer YE, Dine JL, Lipkowitz S. Targeting TRAIL Death Receptors in Triple-Negative Breast Cancers: Challenges and Strategies for Cancer Therapy. Cells 2022;11(23). [CrossRef]
- Roberts JZ, Holohan C, Sessler T, et al. The SCFSkp2 ubiquitin ligase complex modulates TRAIL-R2-induced apoptosis by regulating FLIP(L). Cell Death Differ. 2020, 27, 2726–2741. [CrossRef]
- Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005, 280, 19401–19409. [CrossRef]
- Henrich CJ, Brooks AD, Erickson KL, et al. Withanolide E sensitizes renal carcinoma cells to TRAIL-induced apoptosis by increasing cFLIP degradation. Cell Death Dis. 2015, 6, e1666. [CrossRef] [PubMed]
- Abedini MR, Muller EJ, Brun J, Bergeron R, Gray DA, Tsang BK. Cisplatin Induces p53-Dependent FLICE-Like Inhibitory Protein Ubiquitination in Ovarian Cancer Cells. Cancer Res. 2008;68(12).
- Olsson A, Diaz T, Aguilar-Santelises M, et al. Sensitization to TRAIL-induced apoptosis and modulation of FLICE-inhibitory protein in B chronic lymphocytic leukemia by actinomycin D. Leukemia. 2001, 15, 1868–1877. [CrossRef] [PubMed]
- Riley JS, Malik A, Holohan C, Longley DB. DED or alive: assembly and regulation of the death effector domain complexes. Cell Death Dis. 2015, 6, e1866. [CrossRef] [PubMed]
- Hughes MA, Powley IR, Jukes-Jones R, et al. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell. 2016, 61, 834–849. [CrossRef]
- Hwang EY, Jeong MS, Park SY, Jang SB. Evidence of complex formation between FADD and c-FLIP death effector domains for the death inducing signaling complex. BMB Rep. 2014, 47, 488–493. [CrossRef]
- Majkut J, Sgobba M, Holohan C, et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat Commun. 2014;5:3350. [CrossRef]
- Morlé A, Garrido C, Micheau O. Hyperthermia restores apoptosis induced by death receptors through aggregation-induced c-FLIP cytosolic depletion. Cell Death Dis. 2015, 6, e1633. [CrossRef]
- Piao X, Komazawa-Sakon S, Nishina T, et al. c-FLIP maintains tissue homeostasis by preventing apoptosis and programmed necrosis. Sci Signal. 2012, 5, ra93. [CrossRef]
- Travert M, Ame-Thomas P, Pangault C, et al. CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-kappaB activation and up-regulation of c-FLIP and Bcl-xL. J Immunol. 2008, 181, 1001–1011. [CrossRef]
- Day TW, Safa AR. RNA interference in cancer: targeting the anti-apoptotic protein c-FLIP for drug discovery. Mini Rev Med Chem. 2009, 9, 741–748. [CrossRef] [PubMed]
- El-Zawahry A, McKillop J, Voelkel-Johnson C. Doxorubicin increases the effectiveness of Apo2L/TRAIL for tumor growth inhibition of prostate cancer xenografts. BMC Cancer. 2005, 5, 2. [CrossRef] [PubMed]
- Logue SE, Martin SJ. Caspase activation cascades in apoptosis. Biochem Soc Trans. 2008;36(Pt 1):1-9. [CrossRef]
- Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000, 460, 1–15. [CrossRef] [PubMed]
- Pop C, Oberst A, Drag M, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011, 433, 447–457. [CrossRef] [PubMed]
- Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [CrossRef] [PubMed]
- Malhi H, Gores GJ. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene. 2006, 25, 7333–7335. [CrossRef] [PubMed]
- Safa AR, Pollok KE. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers (Basel) 2011, 3, 1639–1671. [CrossRef] [PubMed]
- Ullenhag GJ, Mukherjee A, Watson NFS, Al-Attar AH, Scholefield JH, Durrant LG. Overexpression of FLIPL Is an Independent Marker of Poor Prognosis in Colorectal Cancer Patients. Clin Cancer Res. 2007, 13, 5070–5075. [CrossRef] [PubMed]
- Galligan L, Longley DB, McEwan M, Wilson TR, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 2005, 4, 2026–2036. [CrossRef]
- Okano H, Shiraki K, Inoue H, et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest. 2003, 83, 1033–1043. [CrossRef]
- Yang JK, Wang L, Zheng L, et al. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell. 2005, 20, 939–949. [CrossRef] [PubMed]
- Scott FL, Stec B, Pop C, et al. The Fas–FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009, 457, 1019–1022. [CrossRef] [PubMed]
- Berglund H, Olerenshaw D, Sankar A, Federwisch M, McDonald NQ, Driscoll PC. The three-dimensional solution structure and dynamic properties of the human FADD death domain. J Mol Biol. 2000, 302, 171–188. [CrossRef] [PubMed]
- Riley JS, Hutchinson R, McArt DG, et al. Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer. Cell Death Dis. 2013;4:e951. [CrossRef]
- Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013, 332, 141–150. [CrossRef]
- Micheau, O; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Zou W, Chen S, Liu X, et al. c-FLIP downregulation contributes to apoptosis induction by the novel synthetic triterpenoid methyl-2-cyano-3, 12-dioxooleana-1, 9-dien-28-oate (CDDO-Me) in human lung cancer cells. Cancer Biol Ther. 2007, 6, 1614–1620. [CrossRef]
Figure 1.
In silico identification of selective c-FLIP DED-binding compounds. A) Best docking poses of the nine compounds in the binding pocket of the c-FLIP structural model. The binding pocket is a “hydrophobic patch” Phenylalanine-Leucine (F-L) highly conserved in DED domains. The SiteFinder module (MOE) allows to identify the druggable pockets. The compounds are in stick representation. c-FLIP structure model is represented as blue surface. The F-L patch is highlighted in green. B) 2D structures of the nine compounds selected by in silico methods.
Figure 1.
In silico identification of selective c-FLIP DED-binding compounds. A) Best docking poses of the nine compounds in the binding pocket of the c-FLIP structural model. The binding pocket is a “hydrophobic patch” Phenylalanine-Leucine (F-L) highly conserved in DED domains. The SiteFinder module (MOE) allows to identify the druggable pockets. The compounds are in stick representation. c-FLIP structure model is represented as blue surface. The F-L patch is highlighted in green. B) 2D structures of the nine compounds selected by in silico methods.
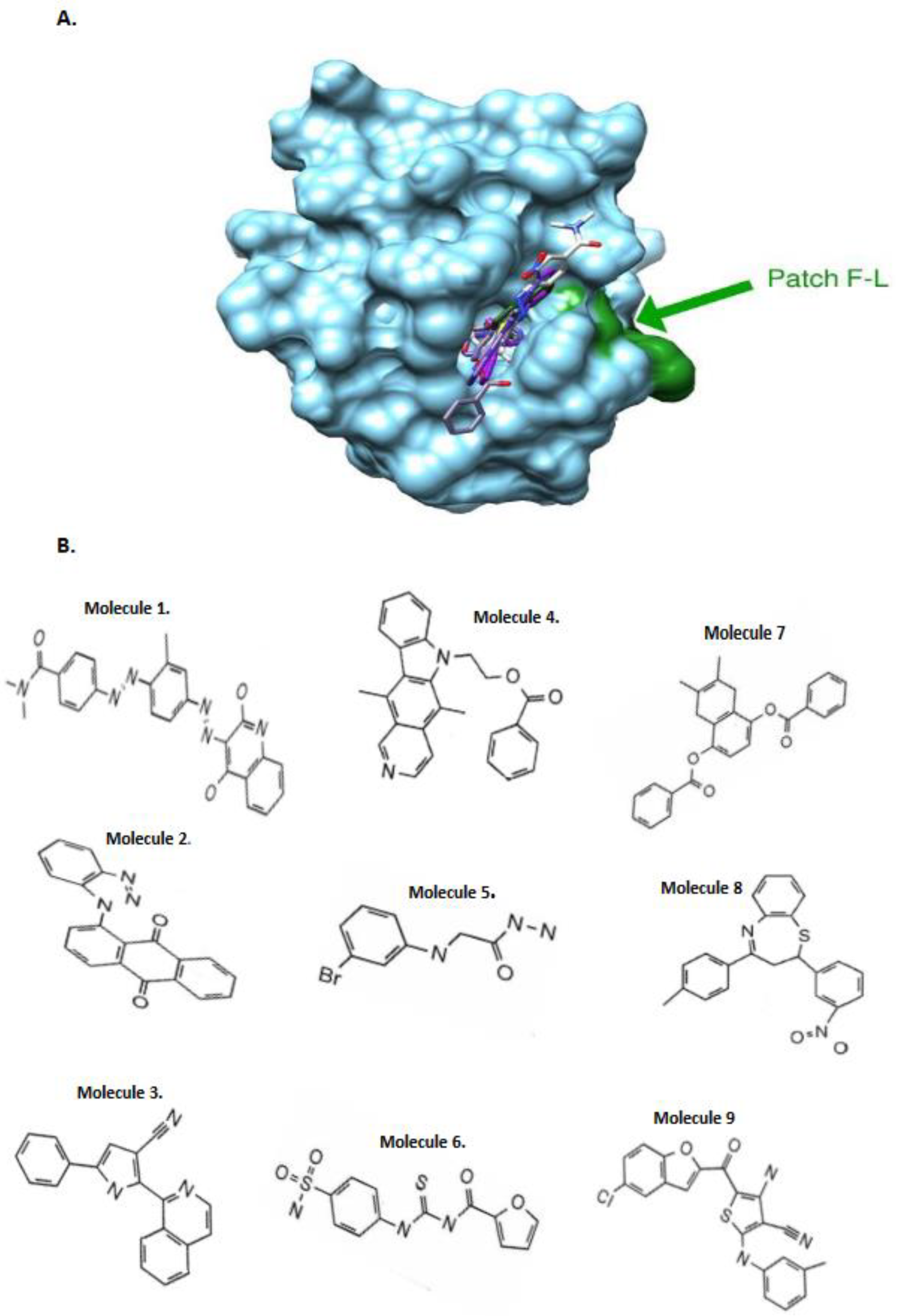
Figure 2.
Representation of the CASP8 and c-FLIP models chosen to carry out molecular docking with location of the pockets near the hydrophobic patch (yellow).
Figure 2.
Representation of the CASP8 and c-FLIP models chosen to carry out molecular docking with location of the pockets near the hydrophobic patch (yellow).
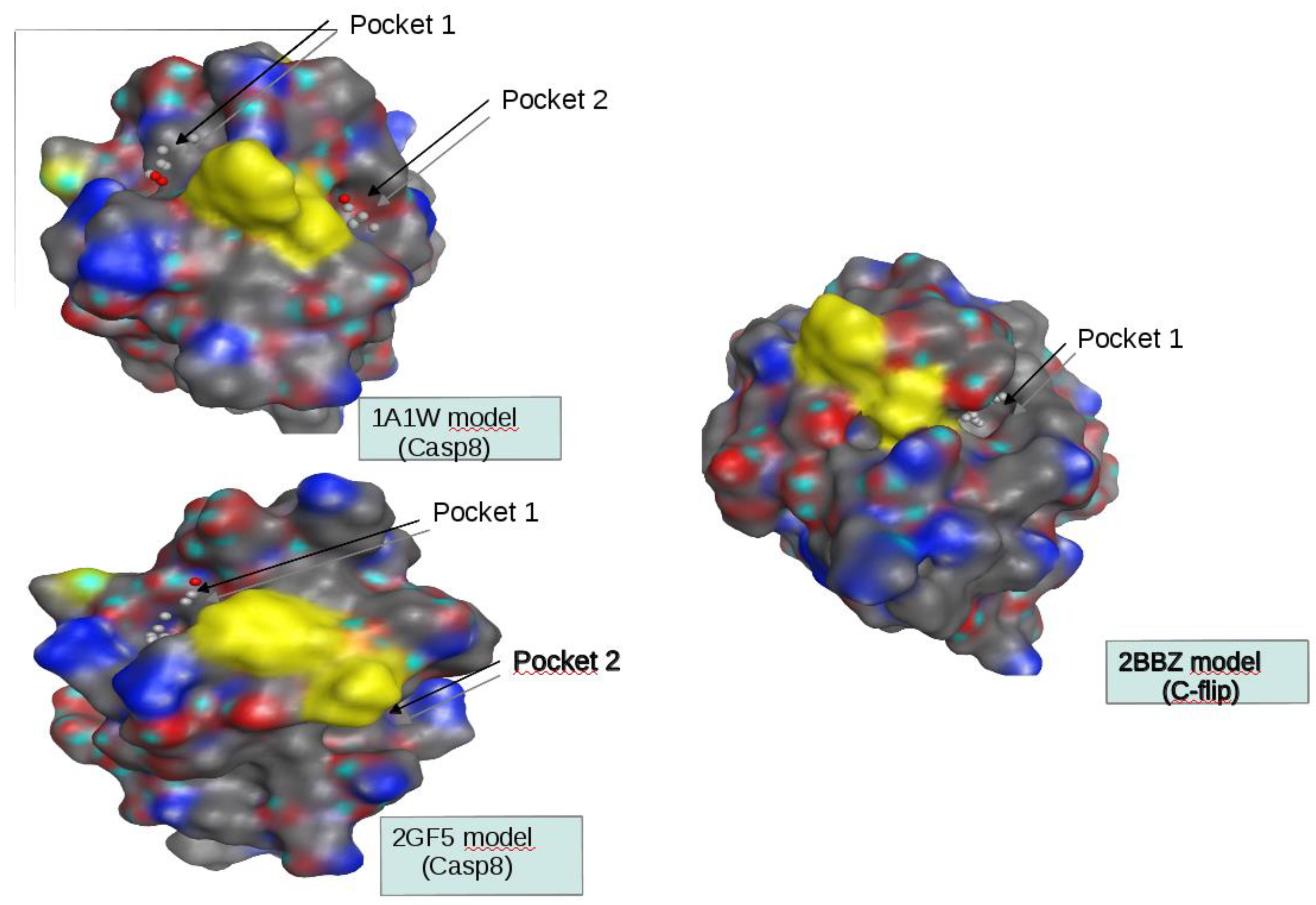
Figure 3.
The newly identified inhibitors prevent DED-FADD/DED2-c-FLIP interaction. MBP-tagged FADD and His-tagged FLIP(s) were produced and purified using chromatography columns, and incubated with each inhibitor using the indicated range of concentrations, for 18 hours. The different mixtures were passed on MBP-trap chromatography columns and eluted samples were analyzed by Western Blot using antibodies directed against Histidine and MBP; C -: Negative control without FADD, C+: Positive control FADD and FLIP without any inhibitor.
Figure 3.
The newly identified inhibitors prevent DED-FADD/DED2-c-FLIP interaction. MBP-tagged FADD and His-tagged FLIP(s) were produced and purified using chromatography columns, and incubated with each inhibitor using the indicated range of concentrations, for 18 hours. The different mixtures were passed on MBP-trap chromatography columns and eluted samples were analyzed by Western Blot using antibodies directed against Histidine and MBP; C -: Negative control without FADD, C+: Positive control FADD and FLIP without any inhibitor.
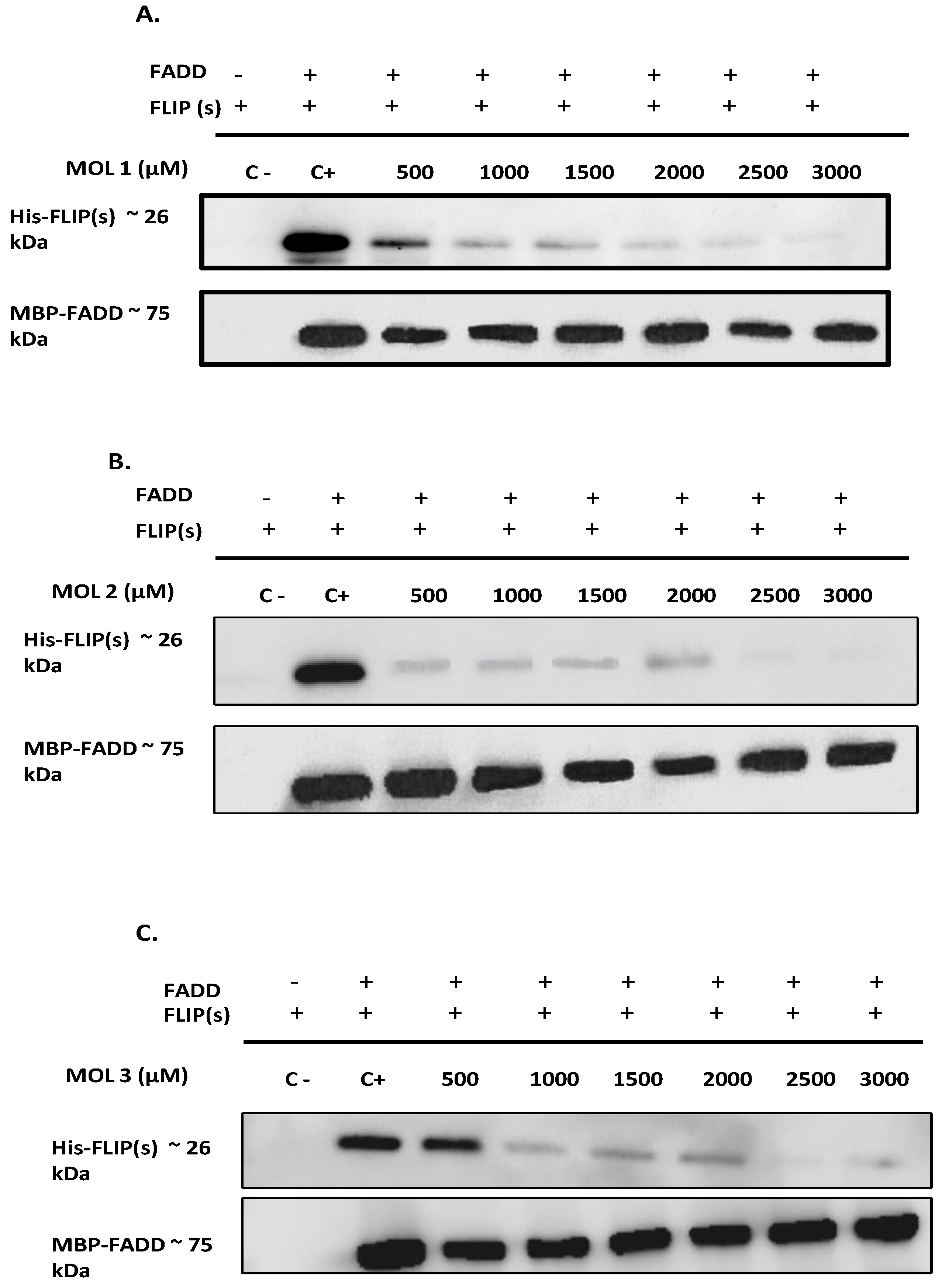
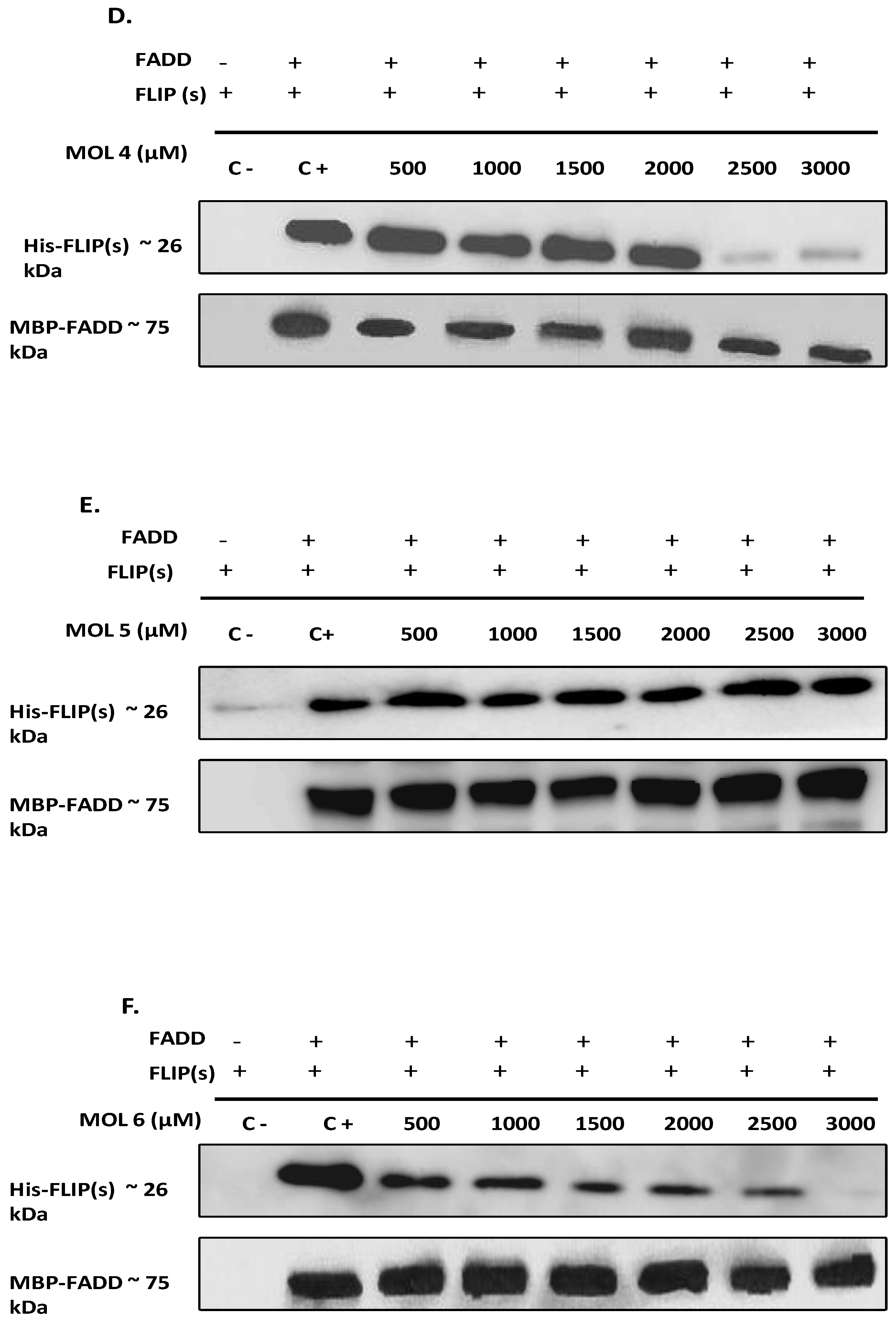
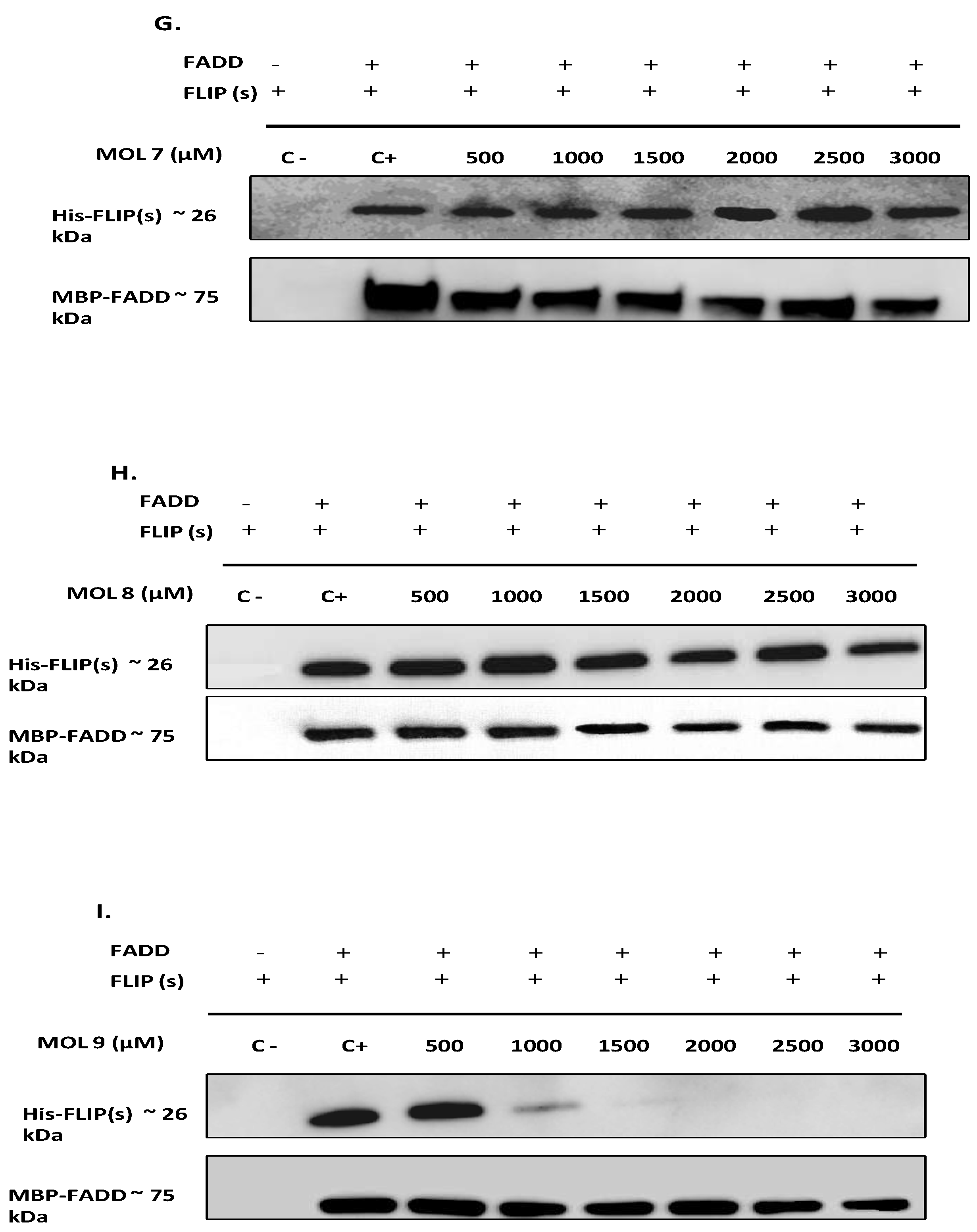
Figure 4.
New identified molecules inhibit c-FLIP recruitment to the DISC complex and promote apoptosis. 50.10e6 of H1703-FLIP(L) cells were seeded and exposed the next day to a “Pre-treatment” with 500 µM Molecule 1 and 3 (A), or Molecule 4 and 9 (B), for 2 hours. Cells were treated later with 1 µg/mL of His-TRAIL for 20 minutes. DISC complexes were Immunoprecipitated using anti-HIS antibody and analyzed by Western Blot.
Figure 4.
New identified molecules inhibit c-FLIP recruitment to the DISC complex and promote apoptosis. 50.10e6 of H1703-FLIP(L) cells were seeded and exposed the next day to a “Pre-treatment” with 500 µM Molecule 1 and 3 (A), or Molecule 4 and 9 (B), for 2 hours. Cells were treated later with 1 µg/mL of His-TRAIL for 20 minutes. DISC complexes were Immunoprecipitated using anti-HIS antibody and analyzed by Western Blot.
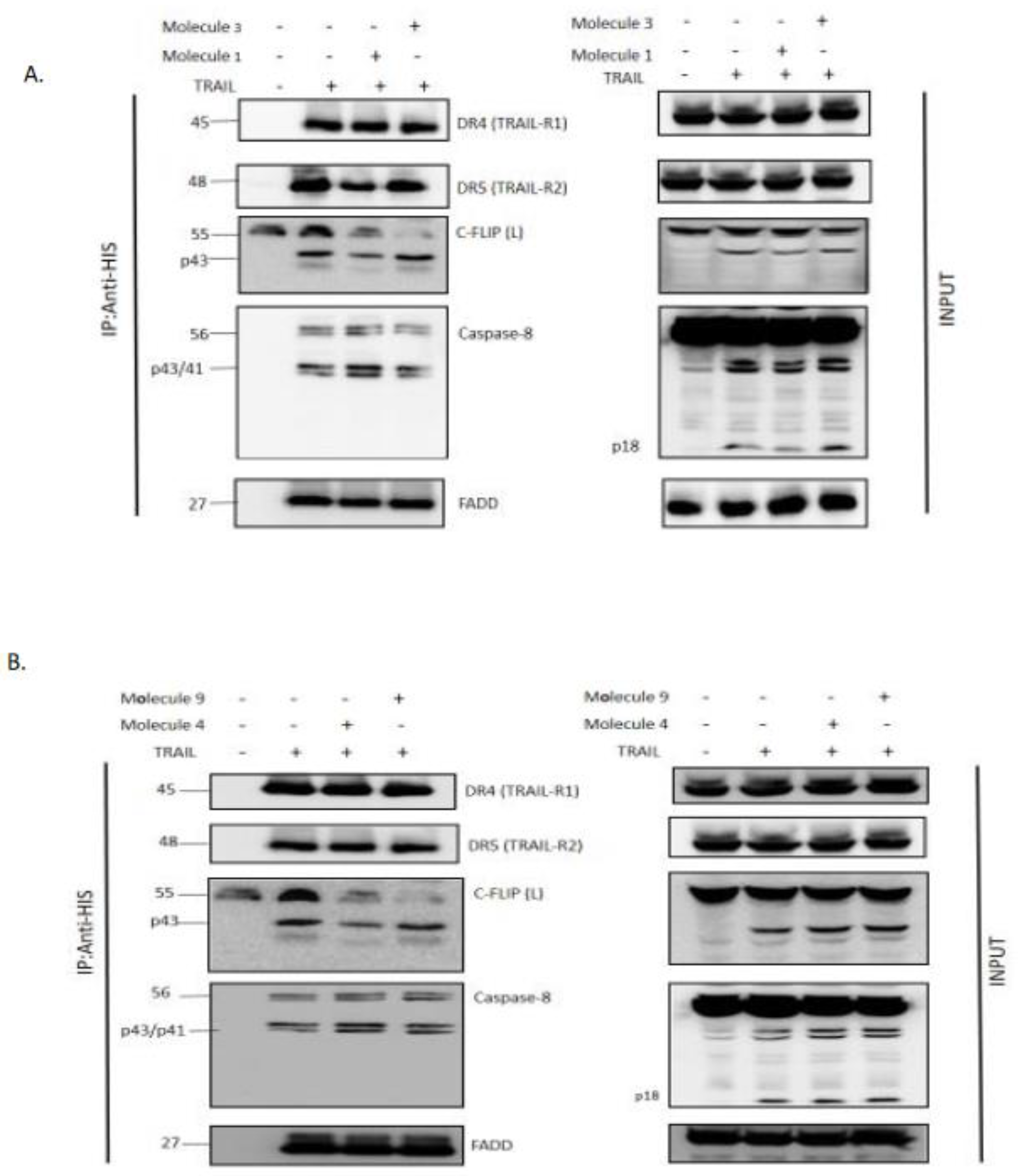
Figure 5.
Inhibition of c-FLIP rescues TRAIL-mediated apoptosis. (A), 2x10⁵ of H1703-Mock transfected and H1703-FLIP (L) cells were seeded and treated with or without 100 ng/ml TRAIL for 18 hours. Apoptosis was evaluated with Annexin V-PE staining by flow cytometry (n=3). (B), 2x10⁵ of H1703-Mock transfected and H1703-FLIP (L) cells were seeded and treated with or without 100 ng/ml TRAIL alone, inhibitors of FLIP(L) alone or with co-treatment of TRAIL/FLIP inhibitors for 18 hours. Apoptosis was evaluated with Annexin V-PE staining by flow cytometry (n=3). .
Figure 5.
Inhibition of c-FLIP rescues TRAIL-mediated apoptosis. (A), 2x10⁵ of H1703-Mock transfected and H1703-FLIP (L) cells were seeded and treated with or without 100 ng/ml TRAIL for 18 hours. Apoptosis was evaluated with Annexin V-PE staining by flow cytometry (n=3). (B), 2x10⁵ of H1703-Mock transfected and H1703-FLIP (L) cells were seeded and treated with or without 100 ng/ml TRAIL alone, inhibitors of FLIP(L) alone or with co-treatment of TRAIL/FLIP inhibitors for 18 hours. Apoptosis was evaluated with Annexin V-PE staining by flow cytometry (n=3). .
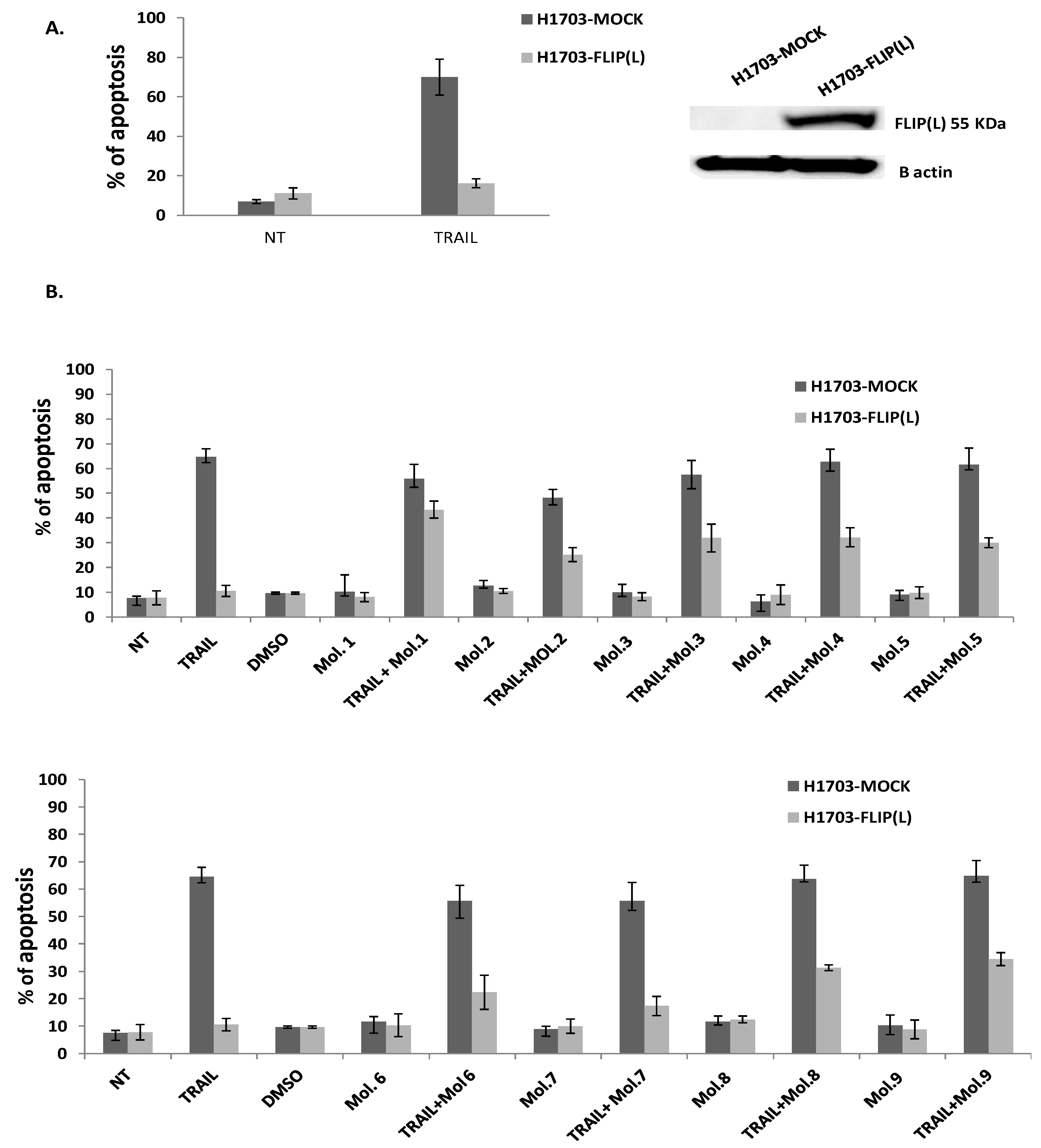
Figure 6.
Inhibition of c-FLIP restores apoptosis mediated by caspases activation. A) H1703 overexpressing c-FLIP (L), and B) H1703-lacking c-FLIP (L) were treated with 100 ng/ml of TRAIL alone, 500 µM of c-FLIP-inhibitors alone, or TRAIL combined with inhibitor 1, 3, 4 or 9 at their appropriate concentrations for 8 hours. Lysates were separated by SDS-PAGE and analyzed by western Blot using specific antibodies. The bands marked with an asterisk are unspecific bands for the anti-caspase-3 (8G10).
Figure 6.
Inhibition of c-FLIP restores apoptosis mediated by caspases activation. A) H1703 overexpressing c-FLIP (L), and B) H1703-lacking c-FLIP (L) were treated with 100 ng/ml of TRAIL alone, 500 µM of c-FLIP-inhibitors alone, or TRAIL combined with inhibitor 1, 3, 4 or 9 at their appropriate concentrations for 8 hours. Lysates were separated by SDS-PAGE and analyzed by western Blot using specific antibodies. The bands marked with an asterisk are unspecific bands for the anti-caspase-3 (8G10).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



