
Submitted:
08 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
Anthracnose, caused by the fungus Colletotrichum lindemuthianum, poses a significant and widespread threat to the common bean crop. The use of plant genetic resistance has proven to be the most effective strategy for managing anthracnose disease. The Amendoim Cavalo (AC) Andean cultivar has resistance against multiple races of C. lindemuthianum, which is conferred by the Co-AC gene. Fine mapping of this resistance gene to common bean chromosome Pv01 enabled the identification of Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500 candidate genes for further validation. In this study, we assessed the relative expression of Co-AC candidate genes, as well as other putative genes in vicinity of this locus and known resistance genes, in the AC cultivar following inoculation with the race 73 of C. lindemuthianum. Gene expression analysis revealed significantly higher expression levels of Phvul.001G244500. Notably, Phvul.001G244500 encodes a putative Basic Helix-Loop-Helix transcription factor, suggesting its involvement in the regulation of defense responses. Furthermore, we observed a significant modulation of the expression of defense related genes PR1a, PR1b, and PR2, in a time-course experiment. These findings contribute to the development of improved strategies for breeding anthracnose-resistant common bean cultivars, thereby mitigating the impact of this pathogen on crop yields and ensuring sustainable bean production.
Keywords:
candidate gene expression
; common bean–anthracnose interaction
; plant defense genes
1. Introduction
The common bean (Phaseolus vulgaris L.) holds the distinction of being the most widely consumed legume in human diets [1]. Known for its affordability, the common bean seeds serve as a crucial source of protein, dietary fiber, complex carbohydrates, and other essential nutrients, particularly for low-income populations in Africa and Latin America [2,3]. However, the productivity and quality of common bean crops are significantly threatened by Colletotrichum lindemuthianum (Sacc. & Magnus) Briosi & Cavara, a hemibiotrophic ascomycete fungus that causes anthracnose (ANT) [4,5]. This pathogen represents one of the most severe, widespread, and recurring threats to the common bean cultivation. Under favorable environmental conditions, ANT can lead to reduced seed quality and significant yield losses [5,6].
Efforts to control C. lindemuthianum have primarily relied on genetic resistance, as the pathogen exhibits high genetic variability that challenges conventional breeding programs [4,5]. Unfortunately, the pathogen has shown a remarkable ability to overcome cultivated plant resistance through coevolution, rendering previously resistant cultivars to become susceptible over time [7,8]. The use of resistant cultivars remains the most effective and environmentally friendly approach for managing C. lindemuthianum in common bean cultivation [9]. These cultivars offer a cost-effective and user-friendly solution. However, developing cultivars that are resistant to the diverse range of physiological races of C. lindemuthianum poses significant challenges [7].
Anthracnose (ANT) resistance in common beans is conferred by independent loci known as ‘Co.’ These resistance loci have been mapped to various chromosomes of the common bean genome, often clustering in disease-resistance regions [10]. Among the identified resistance loci in the Andean genetic pool, many have been mapped to chromosome Pv01. Notable alleles include Co-1, Co-12, Co-3, Co-14, Co-15 and Co-1HY at the Co-1 locus [11,12,13,14], Co-x [15,16], Co-Pa [17], CoPv01CDRK [18], and Co-AC [19], all located at the end of Pv01.
Plants have successfully thrived for millions of years by developing sophisticated immune systems to combat pathogenic attacks [20,21]. Plants have evolved robust defense mechanisms to resist attacks by pathogens, relying on their innate immune responses. These responses are activated by specialized receptors known as pattern-recognition receptors (PRRs) located on the cell surface, as well as intracellular nucleotide-binding domain leucine-rich repeat containing receptors (NLRs). PRRs initiate pattern-triggered immunity (PTI), while NLRs trigger effector-triggered immunity (ETI). Together, PTI and ETI form a formidable defense system, safeguarding plants against various pathogenic threats [22,23]. Therefore, the identification of novel genes involved in the resistance response to C. lindemuthianum in common beans holds significant promise for the development of genetically resistant cultivars. Such discoveries can enhance our understanding of resistance mechanisms and contribute to the breeding of improved cultivars.
The identification and molecular characterization of novel resistance genes play a crucial role in breeding for disease resistance [24,25]. One effective approach to achieve this is by assessing the expression levels of candidate genes associated with resistance loci and known resistance genes in response to pathogens, utilizing the Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR) technique. Gene expression analysis offers valuable insights into the role and interaction of these genes in mounting an effective resistance response. Moreover, it provides additional knowledge necessary for the identification of promising genes for utilization in plant breeding programs. By employing RT-qPCR, researchers can gain a better understanding of the molecular mechanisms underlying disease resistance and ultimately enhance the selection and incorporation of effective genes into breeding strategies.
Gene expression analysis has proven to be a valuable tool in understanding the resistance response of common bean genotypes SEL 1308 and T9576R to the race 73 of C. lindemuthianum [26,27]. Several defense-related genes, including those encoding pathogenesis-related proteins (PR) such as PR1a, PR1b, and PR2, were found to be spatially and temporally induced by the pathogen [26,27,28]. Notably, a potential gene associated with the Co-12 locus was identified [27]. Similarly, changes in the expression levels of PR1a, PR1b, and PR2 genes were reported during incompatible interactions with race 2 of C. lindemuthianum [29].
In the evaluation of expression levels of candidate genes for the Co-12 allele of the Co-1 gene in response to race 73 of C. lindemuthianum, the gene Phvul.001G243800 exhibited high induction, suggesting its potential as a candidate gene for the Co-12 allele of the Co-1 gene [27]. Additionally, through fine mapping in the Hongyundou genotype, four candidate genes were identified for the Co-1HY allele. Among them, Phvul.001G243600 and Phvul.001G243700 showed higher induction in response to race 81 of C. lindemuthianum, indicating their potential as candidate genes for the Co-1HY locus [13]. Moreover, in a genetic study focusing on the anthracnose resistance Co-x gene, a gene called KTR2/3 was found within a CRINKLY4 kinase cluster located between the Phvul.001G243600 and Phvul.001G243700 genes. Gene expression analysis revealed that KTR2/3 was induced by strain 100 (race 3993) of C. lindemuthianum, and its transient expression in susceptible genotype BAT93 resulted in increased resistance to the pathogen [16]. The authors suggested that this gene may act as a decoy involved in indirectly recognizing fungal effectors [16]. These findings highlight the significance of gene expression analysis in uncovering potential resistance genes and their roles in the common bean’s defense response to C. lindemuthianum.
Through fine mapping, five candidate genes for the CoPv01CDRK/PhgPv01CDRK locus, which confer resistance to anthracnose and angular leaf spot in the CDRK common bean cultivar, were identified [18]. Among these candidate genes, gene expression analysis revealed that Phvul.001G246300, potentially encoding an Abscisic Acid Receptor (PYL5), exhibited the highest responsiveness to both pathogens, making it the primary candidate gene for the CoPv01CDRK/PhgPv01CDRK locus [30].
Another noteworthy common bean cultivar, Amendoim Cavalo (AC), is an Andean landrace collected in the state of Santa Catarina, Brazil [19]. AC possesses the Co-AC gene that confers resistance to races 2, 7, 9, 19, 23, 39, 55, 65, 73, 89, 1545, 2047, and 3481 of C. lindemuthianum [31]. Through fine mapping, the Co-AC gene was located at the end of the Pv01 chromosome, within a genomic region containing three candidate genes: Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500 [19].
We propose that candidate genes located within the Co-AC loci on Pv01 demonstrate unique expression patterns when inoculated with race 73 of C. lindemuthianum in the Amendoim Cavalo (AC) cultivar. The primary aim of this study was to investigate the dynamic expression profiles of Co-AC candidate genes Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500 within the AC cultivar upon exposure to C. lindemuthianum race 73. This investigation will be conducted through quantitative real-time PCR-based gene expression analysis. The present study hypothesizes that each of the candidate genes that overlap with the Co-AC loci on Pv01 exhibits distinct expression patterns in response to inoculations with race 73 of C. lindemuthianum in the Amendoim Cavalo cultivar. The objective of this study was to investigate the expression patterns of the Co-AC candidate genes (Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500) in the AC cultivar, in response to C. lindemuthianum race 73, using gene expression analysis employing quantitative real-time PCR. Specifically, we seek to gain insights into their potential roles in the plant’s defense mechanisms against these pathogens, contributing to a deeper understanding of disease resistance in common beans.
2. Results
2.1. Phenotypic Evaluation of the Cultivars
Inoculation of C. lindemuthianum race 73 on the resistant cultivar AC and the susceptible cultivar Cornell 49-242 resulted in disease development exclusively in the susceptible cultivar (Figure 1). Disease symptoms manifested as small water-soaked lesions on the underside of the leaves and small sunken lesions on the stems, ultimately leading to plant mortality. Notably, symptom expression occurred only after 72 hours post-inoculation (hpi), indicating the hemibiotrophic nature of the fungus. Conversely, no symptoms or hypersensitive response were observed in the resistant cultivar.
2.2. Differential Expression of Candidate in the Amendoim Cavalo Cultivar Inoculated with Race 73 of C. lindemuthianum
Aiming to identify the molecular mechanisms underlying Co-AC resistance, the relative expression of the following candidate genes was assessed: KTR2/3, Phvul.001G243800, Phvul.001G244300, Phvul.001G244400, Phvul.001G244500, Phvul.001G245300, and Phvul.001G246300). Additionally, the expression of defense genes PR1a, PR1b, and PR2, were evaluated as markers for resistance upon C. lindemuthianum race 73 inoculation. These evaluated genes with functional annotation are described in Table S1.
Phvul.001G244500 exhibited the most significant response to the pathogen, showing a 2.5-fold change at 72 hours post-inoculation (hpi). Additionally, an approximately 1.8-fold increase was observed at 24 hpi, while increases higher than 1.0-fold were observed at 96 and 120 hpi (Figure 2A and Figure 3 and Table 1). The gene Phvul.001G245300 displayed the second highest response to the pathogen, being induced at 24, 72, and 120 hpi with an average increase of 0.90-fold (Figure 2B and Figure 3 and Table 1). The gene Phvul.001G243800 was also induced at 24, 72, and 120 hpi, albeit with a relatively small average increase of 0.9-fold (Figure 2C and Figure 3 and Table 1).
The expression of the Phvul.001G244300 gene was induced by the pathogen at 48 and 72 hpi, showing a small increase of 0.4-fold (Figure 2D and Figure 3 and Table 1). Conversely, the Phvul.001G244400 gene was downregulated only at 96 hpi, with a reduction of 0.9-fold (Figure 2E and Figure 3 and Table 1). The KTR2/3 and Phvul.001G246300 genes exhibited downregulation at 48, 96, and 120 hpi (Figure 2F, 2G, Figure 3 and Table 1).
The Phvul.001G244500 gene was the most responsive candidate gene to the pathogen, particularly at 72 hpi. Additionally, the Phvul.001G246300 candidate gene for CoPv01CDRK and the KTR2/3 candidate gene for Co-x were significantly downregulated in the Amendoim Cavalo cultivar upon inoculation with the race 73 of C. lindemuthianum. This suggests that different candidate genes are expressed in each anthracnose-resistant cultivar (Figure 2 and Figure 3 and Table 1).
2.3. Expression Profile of Defense Genes in the Amendoim Cavalo Cultivar Inoculated with Race 73 of C. lindemuthianum
The known disease resistance genes also exhibited responsiveness to race 73 of C. lindemuthianum in the AC cultivar, albeit with distinct expression patterns (Figure 4 and Figure 5 and Table 3). Specifically, PR1b, PR1a, and PR2 were induced only starting from 72 hpi, with PR1b showing prominent induction between 96 and 120 hpi. These findings indicate that the resistance response may be activated by the candidate gene Phvul.001G244500, and at 72 hpi, additional defense genes are triggering the resistance response.
The PR1b gene displayed repression at 24 and 48 hours post-inoculation (hpi), with a mean reduction of nearly 1.8-fold. However, at 96 and 120 hpi, a substantial mean induction of almost 4-fold was observed, making it the most prominently induced resistance gene in this study (Figure 4A and Figure 5 and Table 1). Similarly, the PR1a gene exhibited repression at 48 hpi, showing a reduction of over 1.6-fold. However, from 72 hpi onwards, we observed inductions higher than 1.4-fold, specifically 1.4-fold at 72 hpi, 2.3-fold at 96 hpi, and 1.8-fold at 120 hpi (Figure 4B and Table 1). The expression pattern of the PR2 gene mirrored that of PR1b. It experienced downregulation at 24 and 48 hpi, with a reduction of 1-fold, but demonstrated increased expression levels at 72, 96, and 120 hpi with fold changes of 0.5, 1.2, and 1.8, respectively (Figure 4C and Figure 5 and Table 1).
3. Discussion
Gene expression analysis plays a crucial role in understanding the genetic basis of disease resistance and can aid in the identification of effective resistance genes for plant breeding and molecular studies within specific pathosystems. In our study, we investigated the resistance response to C. lindemuthianum in the Amendoim Cavalo cultivar, focusing on the relative expression of candidate genes associated with resistance loci, namely Co-AC [19], CoPv01CDRK/PhgPv01CDRK [18], Co-x [15,16], and the Co-12 allele for the Co-1 locus [27] (Figure 2 and Figure 3). Additionally, we examined the relative expression of disease resistance genes PR1a, PR1b, and PR2 in the same pathosystem (Figure 4 and Figure 5).
Our study focused on a genomic region spanning 250 Kb at the end of Pv01, which encompasses the candidate genes for Co-AC and potential genes closely linked to this locus (Figure 6). Notably, we observed distinct expression patterns among the candidate genes of Co-AC resistance gene in the AC common bean cultivar. Among these candidate genes (Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500), Phvul.001G244500 displayed the most pronounced responsiveness to race 73 of C. lindemuthianum, particularly at 72 hpi, with a 2.5-fold change in gene expression (Figure 2 and Figure 3 and Table 1).
The identification and characterization of specific genes involved in the defense response against pathogens are crucial for understanding plant resistance mechanisms. Our findings highlight the potential role of the Phvul.001G244500 gene, which encodes a Basic Helix-Loop-Helix (bHLH) transcription factor, in regulating defense processes against C. lindemuthianum race 73. This gene exhibited the highest responsiveness in the Amendoim Cavalo cultivar, particularly at 72 hpi, suggesting its involvement in the defense against this specific pathogen. Unlike the robust induction of Phvul.001G244500 in the Amendoim Cavalo cultivar, a previous study showed a low expression pattern of this candidate gene in the California Dark Red Kidney cultivar inoculated with C. lindemuthianum race 73. Lovatto [30] et al. (2022) reported that Phvul.001G244500 displayed only a slight induction at 120 hpi, with less than a 1-fold change in gene expression. Phvul.001G244500 is a strong candidate for the Co-AC resistance gene in the Amendoim Cavalo cultivar, while it was not responsive in the California Dark Red Kidney cultivar.
Phvul.001G245300 expression in AC cultivar was the second most induced gene, although at lower levels, with approximately 1.0-fold change at 24, 72, and 120 hpi. This observation suggests that Phvul.001G245300 may function in a secondary layer of the resistance response.
In the AC cultivar Phvul.001G243800 showed only minor increases in expression levels at 24, 72, and 120 hpi (approximately 1-fold change). In contrast, the Phvul.001G244500 gene exhibited a substantial 2.5-fold change in gene expression (Figure 2 and Figure 3 and Table 1). These results suggest that Phvul.001G243800 has a limited effect on the resistance response in the AC cultivar. The Phvul.001G243800 gene, which is a candidate for the Co-12 allele of the Co-1 locus, was found to be highly induced at 72 hpi in the T9576R genotype inoculated with race 73 of C. lindemuthianum [27].
The present study demonstrated a different pattern for the KTR2/3 gene in the AC cultivar inoculated with race 73 of C. lindemuthianum. It was consistently downregulated at 48, 96, and 120 hpi, indicating that this gene may not trigger the resistance response in this specific pathosystem. Conversely, the KTR2/3, a candidate gene for the Co-x gene, was upregulated at 24 hpi in the JaloEEP558 cultivar [16].
In our study, the Phvul.001G246300 gene in AC cultivar was downregulated, suggesting that it may not be the responsive resistance gene in this specific pathosystem. On the other hand, in the CDRK cultivar, the Phvul.001G246300 gene showed the highest responsiveness to both C. lindemuthianum race 73 and P. griseola race 63-39, indicating that the CoPv01CDRK/PhgPv01CDRK resistance gene confers resistance to both diseases in common bean.
Taking together, it is possible to identify distinct roles of the genes Phvul.001G244500, Phvul.001G243800, KTR2/3, and Phvul.001G246300 involved in the resistance response to race 73 of C. lindemuthianum in different common bean cultivars. The contrasting expression patterns emphasize the complexity of resistance mechanisms and highlight the importance of the candidate gene Phvul.001G244500 for Co-AC in conferring robust resistance.
Plant resistance to pathogens involves the activation of genes encoding pathogenesis-related (PR) proteins, which are categorized into 17 families and are known to accumulate following pathogen infection in various plant species [32]. PR1 genes, a subset of the PR family, are commonly used as markers for systemic acquired resistance [33]. However, our understanding of PR1 genes remains limited, with only a small proportion having been studied thus far [34].
In the present study, we observed that among the tested known disease resistance genes, PR1b exhibited the highest responsiveness to race 73 of C. lindemuthianum in the AC cultivar, particularly at 96 and 120 hpi (Figure 4 and Figure 5). Interestingly, PR1b also demonstrated the most pronounced induction in the CDRK cultivar inoculated with race 73 of C. lindemuthianum, mainly at 120 hpi [29]. Hypothetically, PR1b encodes a PR1-like protein that is typically secreted into the extracellular spaces of plant leaves in response to pathogen infection [35]. In Arabidopsis thaliana, a homolog of PR1b is involved in defense responses against necrotrophic pathogens mediated by methyl jasmonate and ethylene, while being repressed by salicylic acid [36].
In our study, we also observed upregulation of PR1a in the AC cultivar inoculated with race 73 of C. lindemuthianum, particularly between 72 and 120 hpi, with a notable peak at 96 hpi (Figure 4, Figure 5 and Table 1). Similarly, in the CDRK cultivar inoculated with race 73 of C. lindemuthianum, PR1a showed the highest induction at 72 and 96 hpi [30]. Upregulation of PR1a was also observed in the SEL 1308 cultivar inoculated with race 73 of C. lindemuthianum (Borges et al., 2012 a; Oblessuc et al., 2012), as well as in T9576R common bean inoculated with race 73 of C. lindemuthianum [27] and in the ‘Naz’ cultivar when inoculated with race 2 of C. lindemuthianum [29]. PR1a hypothetically encodes a PR protein containing the Bet v I domain [37]. A recent transcriptome study investigating the incompatible interaction between strain C531 and the BAT93 cultivar emphasized the prominent role of PR10/Bet vI in common bean disease resistance [38].
In our experiment with the Amendoim Cavalo cultivar inoculated with race 73 of C. lindemuthianum, we observed a slight repression of PR2 at 24 hpi, followed by induction from 72 hpi, with a notable peak at 120 hpi. Similarly, PR2 was upregulated in response to race 73 of C. lindemuthianum in the CDRK cultivar, primarily between 72 and 96 hpi [30]. PR2 was also found to be upregulated in the SEL 1308 cultivar following inoculation with race 73 of C. lindemuthianum [26,28], as well as in the Naz cultivar, which is resistant to race 2 of C. lindemuthianum, particularly from 48 hpi [29]. In the T9576R genotype inoculated with race 73 of C. lindemuthianum, PR2 was upregulated by the pathogen at all evaluated time points except 96 hours post-inoculation [27]. Hypothetically, PR2 encodes a 1,3-beta-glucan endohydrolase [39]. Therefore, PR2 may play a role in the resistance response by degrading fungal cell walls, potentially triggering a plant’s pattern-triggered immunity (PTI) [40,41].
Our findings contribute valuable knowledge regarding the genetic mechanisms underlying resistance to C. lindemuthianum in the AC cultivar and provide insights into potential genes involved in the resistance response to anthracnose in common beans. These results have implications for future research and can aid in the development of effective strategies for anthracnose resistance in common bean breeding programs. The characterization of a specific candidate gene, Phvul.001G244500, broadens our understanding of the defense networks activated in response to pathogen infection. Additionally, PR1a, PR1b and PR2 play significant roles in the defense responses of different common bean cultivars against C. lindemuthianum.
4. Conclusions
The observed upregulation of candidate genes in the compatible interaction may signify the host’s response to counterbalance the compromised resistance caused by pathogen infection. Our research has successfully pinpointed the candidate gene Phvul.001G244500 as an effective defense mechanism against the specific race 73 of C. lindemuthianum. Moreover, we have demonstrated the involvement of defense genes PR1a, PR1b, and PR2 in the resistance response, with a particular emphasis on PR1b. Our study has yielded invaluable insights into the genetic underpinnings of resistance to C. lindemuthianum race 73 within the Amendoim Cavalo (AC) cultivar. These discoveries significantly bolster our capacity to develop more effective strategies for breeding anthracnose-resistant common bean cultivars. By incorporating this resistance gene into breeding programs, we can enhance the resilience of common bean crops against this devastating pathogen, contributing to sustainable agriculture and food security.
5. Materials and Methods
5.1. Plant Material and Growth Conditions
The experiment was performed in a completely randomized design. Seedlings of the resistant Amendoim Cavalo and the susceptive Cornell 49-242 cultivars were inoculated with race 73 of C. lindemuthianum, and relative expression of ten genes only in Amendoim Cavalo cultivar was evaluated at 24, 48, 72, 96, and 120 hpi and in the mock. Three biological replicates (plants) were collected for each experimental condition evaluated, and for each biological replicate, three technical replicates (qPCR reactions) were performed in each experiment. The experiment was conducted at the Núcleo de Pesquisa Aplicada à Agricultura (Nupagri) at the Universidade Estadual de Maringá (UEM) in Maringá, Paraná, Brazil (latitude 23° 26′8” S, longitude 51° 53′42” W). Briefly, seeds were planted in plastic trays filled with a commercial substrate, MecPlant (MEC PREC—Ind. Com Ltda, Telemaco Borba, Brazil), that had been previously sterilized and fertilized. The seedlings were grown in greenhouses under natural light at a temperature of 25 °C until the first trifoliate leaf growth stage [8].
5.2. Pathogenesis Assay
Monosporic cultures of C. lindemuthianum were prepared following the methodologies described by Mathur et al. [42]. The inoculum was produced on a medium comprising green common bean pods incubated at 22 ± 2 °C without light for 14 days. Conidiospore quantification was conducted using a hemacytometer under an optical microscope. The plants were sprayed with a conidiospore suspension prepared in distilled water and Tween 20® (0.01%) at an approximate concentration of 1.2 x 106 mL-1. Inoculation was carried out by spraying the suspension onto the plants using a manual pressurized pump sprayer. For the negative control (mock), plants were sprayed only with distilled water and Tween 20® (0.01%). After inoculation, the plants were placed in a mist chamber at a temperature of 22 ± 2 °C, a photoperiod of 12 hours, and ≥95% relative humidity for 72 hours. Subsequently, the plants were transferred to a growth chamber with a temperature of 22 ± 2 °C and a photoperiod of 12 hours for the duration of the experiment. Anthracnose symptoms were evaluated using the 1-to-9 disease severity scales proposed by Pastor-Corrales et al. [8]. Plants with disease reaction scores between 1 and 3 were considered resistant, whereas plants with scores from 4 to 9 were considered susceptible.
5.3. RNA Extraction
For sample collection and total RNA extraction, leaf samples weighing approximately 100 ± 10 mg from the AC cultivar inoculated with race 73 of C. lindemuthianum were collected at 24, 48, 72, 96 and 120 hours post-inoculation (hpi), as well as from the mock control. The leaf samples were immediately submerged in liquid nitrogen (N2) and stored at -80 °C until further processing. Total RNA extraction was performed by macerating the tissue and adding 1000 µL of TRIzol® (Invitrogen™, Waltham, USA) to each microtube. The subsequent steps for RNA extraction and isolation followed the manufacturer’s recommendations. The precipitated total RNA pellets were washed with 70% ethanol (EtOH) and then suspended in RNase-free H2O.
The integrity of the total RNA was assessed by electrophoresis on a 1% m/v agarose gel, run for 80 minutes at 80 volts, at 5°C, and in the absence of light. To assess the quality and quantity of total RNA, a spectrophotometer (FEMTO 700STM) was used to measure absorbance at 230 nm, 240 nm, 260 nm, and 280 nm. The following absorbance ratios were used to determine RNA purity: A260/A230 between 1.9 and 2.4, A260/A240 ≥ 1.4, and A260/A280 between 1.8 and 2.2. The concentration of total RNA was calculated using the formula [RNA] (ng µL-1) = A260nm × 40 × 100 [43]. Total RNA samples that met the purity criteria and exhibited no visual signs of degradation were subjected to DNase I treatment using DNase ITM (Invitrogen™, Waltham, USA) to remove any residual DNA. The purification reaction was carried out using 1 µg of total RNA, following the manufacturer’s instructions.
5.4. Reverse Transcription (cDNA Synthesis)
The cDNA synthesis was carried out using the ‘Superscript® IV First-Strand Synthesis System’ kit (Invitrogen™, Waltham, USA) following the manufacturer’s protocol. The total volume of cDNA synthesis reaction was 20 µL with the following components: 1 µg of total RNA, primer-oligo d(T) (2.5 µM), dNTP mix (0.5 mM each), First-Strand Buffer (1X), DL-dithiothreitol (5 mM), ribonuclease inhibitor (2 U µL-1), MMLV-RT (10 U µL-1) and RNase-free water. Initially, total RNA, primer-oligo d(T), dNTP mix and RNase-free water (to 13 µL) were added to the reaction. The samples were incubated in a thermocycler (Applied Biosystems® Veriti® 96-Well Fast Thermal Cycler) at 65 °C for 5 minutes, followed by 4 °C for 1 minute. Then, the First-Strand Buffer, DL-dithiothreitol, ribonuclease inhibitor and 1 MMLV-RT were added to the reaction. The samples were incubated at 55 °C for 10 minutes for cDNA synthesis activation, followed by 80 °C for 10 minutes to inactivate the reaction. To remove residual RNA after cDNA synthesis, 1 µL of Escherichia coli RNase H was added, and the samples were incubated at 37 °C for 20 minutes. The cDNA synthesis product (20 µL) was diluted 1:100 for qPCR analysis. To assess the cDNA synthesis efficiency, positive control was included, in which HeLa-S3 RNA (10 ng) was used instead of total RNA.
For the control of cDNA synthesis, the PCR reaction was conducted using the following components: 5 µL of PCR buffer (10X), 2 µL of MgCl2 (50 mM), 1 µL of dNTP Mix (10 mM), 1 µL of sense primer (10 µM), 1 µL of antisense primer (10 µM), 2 µL of cDNA for the positive control, and 2 µL of ultrapure H2O for the negative control. Additionally, 0.2 µL of Taq PlatinumTM DNA polymerase (Invitrogen™, Waltham, USA) and 37.8 µL of ultrapure H2O were included in the reaction mixture. The PCR amplification was performed under the following thermocycling conditions: an initial denaturation step at 94 °C for 2 minutes, followed by 35 cycles of denaturation at 94 °C for 15 seconds, annealing at 55 °C for 30 seconds, and synthesis at 68 °C for 1 minute. After completion of the PCR reaction, both the positive and negative controls were subjected to electrophoretic analysis using a 1.5% (w/v) agarose gel. The expected fragment size of approximately 353 bp was observed in the positive control lane, confirming the successful amplification, while no fragment was detected in the negative control lane, validating the absence of non-specific amplification as indicated by the manufacturer’s instructions.
5.5. Target Genes and Primer Design
The candidate genes selected for expression analysis in the Amendoim Cavalo cultivar were associated with resistance to race 73 of C. lindemuthianum. Specifically, the genes Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500 that were identified within the Co-AC locus of the Amendoim Cavalo cultivar [19]. Additionally, the gene Phvul.001G246300, located within the CoPv01CDRK/PhgPv01 CDRK loci, that exhibited significant responsiveness in California Dark Red Kidney cultivar when inoculated with race 73 of C. lindemuthianum [30]. The gene Phvul.001G245300 is located in close proximity to the genomic region of the California Dark Red Kidney cultivar. The inclusion of the Phvul.001G243800 gene was based on its induction in the near-isogenic line T9576R, possessing the Co-12 resistance allele, when inoculated with race 73 of C. lindemuthianum [27]. The KTR2/3 candidate gene for Co-x in the Jalo EEP558 cultivar was also evaluated due to its induction in response to race 3993 of C. lindemuthianum [16]. Furthermore, the well-known plant defense genes Phvul.003G109100 (PR1a), Phvul.006G196900 (PR1b), and Phvul.009G256400 (PR2) were included in the analysis [26,27,28]. To standardize gene expression levels, we employed the reference genes Phvul.008G011000 (actin - ACT) and Phvul.001G133200 (insulin-degrading enzyme - IDE) [44]. ACT had previously been validated for quantifying the relative expression of candidate genes in studies [27,29], while IDE’s validation was previously established by Oblessuc et al. [28]. Both genes have been utilized as reference genes for quantifying the relative expression of resistance genes against ANT in studies [16,44]. For normalization purposes, the reference genes Phvul.008G011000 (ACT) and Phvul.001G133200 (IDE) were used [44].
To obtain the coding sequences (CDS) and DNA sequences of the target genes, the common bean (P. vulgaris L.) genome available at v1.2 Phytozome [45] was accessed. Primer design for the qPCR assay was performed using the ‘Primer-Blast web tool’ [46] with the following specifications: primer size between 18 and 24 base pairs (bp), melting temperature between 59 and 61 °C, amplicon size between 80 and 160 bp, and, when possible, the primer pair should be separated by at least one intron in the corresponding genomic DNA sequence. Primer dimers and secondary structures were assessed using Gene Runner software (version 6.5.52), the ‘Multiple Prime Analyzer’ web tool (Thermo Fisher Scientific: https://bit.ly/34kZpnP), and ‘The Sequence Manipulation Suite’ web tool [47]. The secondary structure of the amplicons was verified using “The Mfold Web Server” platform [48] with coding sequences obtained from v1.2 Phytozome. All primer design and in silico validation procedures not explicitly mentioned followed established literature recommendations [49,50]. Table 2 provides the primer sequences for each candidate gene evaluated, with the primers for the KTR2/3 gene obtained from Richard et al. [16].
5.6. Quantitative PCR (qPCR) and Data Analysis
The determination of PCR efficiency for each primer involved establishing a standard curve through a fivefold serial dilution, utilizing the cDNA pool as the template. This process incorporated three replicates at every dilution point [51,52]. The amplification efficiency was computed employing the equation E = [10(-1/slope)] - 1 [52], using the slope values derived from linear regression analysis. This analysis encompassed the log10-transformed cDNA concentrations on the x-axis and corresponding Cq values on the y-axis. The calculated amplification efficiency for each primer pair ranged from 0.92 to 1.09, while maintaining a coefficient of determination (R2) for the linear regression of at least 0.98 (Table 2).
The cDNA quantification reactions were conducted in the StepOnePlus™ real-time PCR system (Applied Biosystems™; StepOnePlus™ Real-Time PCR Systems) using 96-well microplates [MicroAmp™ Fast 96 -well Reaction Plate (0.1 mL)] sealed with MicroAmp™ Optical Adhesive Film. The total reaction volume was 10 µL, consisting of 3.4 µL of cDNA, 1.6 µL of forward and reverse primer mix (800 nM), and 5 µL of PowerUp™ SYBR™ Green Master Mix (Applied Biosystems™). The thermocycling conditions included 50°C for 2 minutes, 95°C for 2 minutes, 40 cycles of 15 seconds at 95°C, and 30 seconds at 60°C.
After completing the cDNA quantitation reaction, a thorough assessment of target specificity was conducted through a dissociation curve analysis, employing the continuous melt curve setup as per the manufacturer’s specifications. Only samples demonstrating clear specificity in accordance with the dissociation curve were considered for subsequent analysis. Quantification cycle (Cq) values were extracted using StepOnePlus™ Software v2.3 (Applied Biosystems™). The baseline was automatically established, and the threshold was set manually during the exponential phase of amplification. For all cDNA quantification reactions, a consistent threshold value of 0.7707 was applied.”
The genes Phvul.008G011000 (IDE) and Phvul.001G133200 (ACT) served as reference genes [44]. The arithmetic mean of quantification cycle (Cq) values [53] was computed for each experimental condition under consideration. Relative expression levels were determined by normalizing Cq values with the reference genes, employing the 2-ΔΔCT method [54,55]. Mean Cq values were derived for each gene in every experimental condition through the calculation based on three biological repetitions and three replicates (n=3x3). Top of Form
The investigation into the relative expression of candidate genes at the Co-AC locus and known disease resistance genes was conducted in response to race 73 of C. lindemuthianum, spanning time points at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) in the Amendoim Cavalo cultivar. The calibrator condition for each gene was the relative expression observed in the mock (control, without pathogen). For data analysis and presentation of results, a logarithmic base 2 transformation was applied before statistical analysis. The Alexander-Govern test, with a significance level of 5%, was utilized to compare expression levels among experimental conditions. Pairwise comparisons of relative expression means at different time points for each gene were assessed, with significance levels adjusted using Bonferroni correction (p≤0.05). These statistical analyses employed the ‘oneway.test’ [56] and ‘companion’ R packages.
All data and statistical analyses were conducted using R software (version 4.0.3) (R Core Team), and plots were generated using the ggplot2 package [57] and R base. Error bars, representing the standard deviation of the means from three biological and three technical replicates (3x3), were incorporated into the visualizations. Heatmaps representing mean Cq values were generated using the ‘heatmaply’ R package, and the dendrogram was constructed based on the Euclidean distance measure and the average linkage function [58] among the relative expression values of the genes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org; Table S1: Gene model and predicted functional annotation based on Phytozome.
Author Contributions
Design and supervision of the research P.S.V.F., M.C.G-V., M.M.; Conduct of the experiments M.L., E.A.N., A.C.C.; Analyzed the data M.L., M.C.G-V., M.V-B.; Original draft of the manuscript M.L.; Revision and editing of the manuscript: M.C.G-V., T.A.S.G., M.V-B., P.S.V.F., M.M. All authors have read and agreed to the published version of the manuscript.
Funding
Federal Funding Institution National Council for Scientific and Technological Development (CNPq) Grant/Award Number: 408472/2018-9; and from Coordination for the Improvement of Higher Education Personnel (Capes), Award number 88887.470337/2019-00.
Data Availability Statement
All data are presented within the article or in the Supplementary Materials.
Acknowledgments
M. Lovatto, A.C. Calvi, E.A. Nascimento and M. Vaz Bisneta are grateful for grants or Scholarship from the National Council for Scientific and Technological Development (CNPq) and Capes. M.C. Gonçalves-Vidigal and P.S. Vidigal Filho are grateful for Grants from CNPq and Capes.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bitocchi, E.; Rau, D.; Bellucci, E.; Rodriguez, M.; Murgia, M.L.; Gioia, T.; Santo, D.; Nanni, L.; Attene, G.; Papa, R. Beans (Phaseolus ssp.) as a model for understanding crop evolution. Frontiers in Plant Science 2017, 8, 722. [Google Scholar] [CrossRef] [PubMed]
- Vaz Patto, M.C.; Amarowicz, R.; Aryee, A.N.A.; Boye, J.I.; Chung, H.-J.; Martín-Cabrejas, M.A.; Domoney, C. Achievements and Challenges in Improving the Nutritional Quality of Food Legumes. Critical Reviews in Plant Sciences 2015, 34, 105–143. [Google Scholar] [CrossRef]
- Vidigal Filho, P.S.; Gonçalves-Vidigal, M.C.; Vaz Bisneta, M.; Souza, V.B.; Gilio, T.A.S.; Calvi, A.C.; Lima, L.R.L.; Pastor-Corrales, M.A.; Melotto, M. Genome-wide association study of resistance to anthracnose and angular leaf spot in Brazilian Mesoamerican and Andean common bean cultivars. Crop Science 2020, 60, 2931–2950. [Google Scholar] [CrossRef]
- Padder, B.A.; Sharma, P.N.; Awale, H.E.; Kelly, J.D. Colletotrichum lindemuthianum, the causal agent of bean anthracnose. J. Plant Pathol. 2017, 99, 317–330. [Google Scholar] [CrossRef]
- Nunes, M.P.B.A.; Gonçalves-Vidigal, M.C.; Martins, V.S.R.; Xavier, L.F.S.; Valentini, G.; Vaz Bisneta, M.; Vidigal Filho, P.S. Relationship of Colletotrichum lindemuthianum races and resistance loci in the Phaseolus vulgaris L. genome. Crop Science 2021, 61, 3877–3893. [Google Scholar] [CrossRef]
- Singh, S.P.; Schwartz, H.F. Breeding common bean for resistance to diseases: A review. Crop Sci. 2010, 50, 2199–2223. [Google Scholar] [CrossRef]
- Kelly, J.D.; Afanador, L.; Cameron, L.S. New races of Colletotrichum lindemuthianum in Michigan and implications in dry bean resistance breeding. Plant Disease 1994, 78, 892–894. [Google Scholar] [CrossRef]
- Pastor-Corrales, M.A.; Otoya, M.M.; Molina, A.; Singh, S.P. Resistance to Colletotrichum lindemuthianum isolates from middle America and Andean South America in different common bean races. Plant Dis. 1995, 79, 63–67. [Google Scholar] [CrossRef]
- Pacheco, L.M.; Berrouet, K.V.; Yepes, M.S.; Sánchez, P.G.; Montoya, M.M. Detección por PCR de Colletotrichum lindemuthianum en cultivos y semillas de frijol en Antioquia, Colombia. Acta Agrómica 2014, 63, 377–387. [Google Scholar] [CrossRef]
- Vaz Bisneta, M.; Gonçalves-Vidigal, M.C. Integration of anthracnose resistance loci and RLK and NBS-LRR-encoding genes in the Phaseolus vulgaris L. genome. Crop Sci. 2020, 60, 2901–2918. [Google Scholar] [CrossRef]
- Gonçalves-Vidigal, M.C.; Cruz, A.S.; Garcia, A.; Kami, J.; Vidigal Filho, P.S.; Sousa, L.L.; McClean, P.; Gepts, P.; Pastor-Corrales, M.A. Linkage mapping of the Phg-1 and Co-14 genes for resistance to angular leaf spot and anthracnose in the common bean cultivar AND 277. Theor. Appl. Genet. 2011, 122, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Zuiderveen, G.H.; Padder, B.A.; Kamfwa, K.; Song, Q.; Kelly, J.D. Genome-wide association study of anthracnose resistance in Andean beans (Phaseolus vulgaris). PLoS One 2016, 11, e0156391. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wu, J.; Wang, L.; Mantri, N.; Zhang, X.; Zhu, Z.; Wang, S. Mapping and genetic structure analysis of the anthracnose resistance locus Co-1HY in the common bean (Phaseolus vulgaris L.). PLoS One, 2017; 12, e0169954. [Google Scholar] [CrossRef]
- Lima, L.R.L.; Gonçalves-Vidigal, M.C.; Vaz Bisneta, M.; Valentini, G.; Vidigal Filho, P.S.; Martins, V.S.R.; Souza, T.L.P.O. Genetic fine-mapping of anthracnose disease-resistance allele Co-14 present in the Andean common bean cultivar AND 277. Crop Sci. 2023; 1–14. [Google Scholar] [CrossRef]
- Richard, M.M.S.; Pflieger, S.; Sevignac, M.; Thareau, V.; Blanchet, S.; Li, Y.; Jackson, S.A.; Jackson, S.A.; Geffroy, V. Fine mapping of Co-x, an anthracnose resistance gene to a highly virulent strain of Colletotrichum lindemuthianum in common bean. Theor. Appl. Genet. 2014, 127, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.M.S.; Gratias, A.; Diaz, J.C.A.; Thareau, V.; Pflieger, S.; Meziadi, C.; Blanchet, S.; Marande, W.; Bitocchi, E.; Papa, R.; Miklas, P.N.; Geffroy, V. A common bean truncated CRINKLY4 kinase controls gene-for-gene resistance to the fungus Colletotrichum lindemuthianum. J. Exp. Bot. 2021, 72, 3569–3581. [Google Scholar] [CrossRef]
- Lima-Castro, S. A., Gonçalves-Vidigal, M. C., Gilio, T. A. S., Lacanallo, G. F., Valentini, G., Martins, V. S. R. et al. (2017). Genetics and mapping of a new anthracnose resistance locus in Andean common bean Paloma. BMC Genom. 18, 306. [CrossRef]
- Gonçalves-Vidigal, M.C.; Gilio, T.A.S.; Valentini, G.; Vaz Bisneta, M.; Vidigal Filho, P.S.; Song, Q.; Oblessuc, P.R.; Melotto, M. (2020). New Andean source of resistance to anthracnose and angular leaf spot: Fine-mapping of disease-resistance genes in California Dark Red Kidney common bean cultivar. PLoS One. 15, e0235215. [CrossRef]
- Gilio, T.A.S.; Hurtado-Gonzales, O.P.; Gonçalves-Vidigal, M.C.; Valentini, G.; Elias, J.C.F.; Song, Q.; Pastor-Corrales, M.A. Fine mapping of an anthracnose-resistance locus in Andean common bean cultivar Amendoim Cavalo. PLoS One 2020, 15, e0239763. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.L.; Puttick, M.N.; Clark, J.W.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Yang, Z.; Schneider, H.; Donoghue, P.C.J. The timescale of early land plant evolution. Proceedings of the National Academy of Sciences 2018, 115, E2274–E2283. [Google Scholar] [CrossRef]
- Gu, J.; Sun, J.; Liu, N.; Sun, X.; Liu, C.; Wu, L.; Liu, G.; Zeng, F.; Hou, C.; Han, S.; Zhen, W.; Wang, D. A novel cysteine-rich receptor-like kinase gene, TaCRK2, contributes to leaf rust resistance in wheat. Molecular Plant Pathology 2020, 21, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Ngou, B.P.M.; Ding, P.; Xin, X.-F. PTI-ETI crosstalk: an integrative view of plant immunity. Current Opinion in Plant Biology 2021, 62, 102030. [Google Scholar] [CrossRef] [PubMed]
- Ngou, B.P.M.; Ding, P.; Jones, J.D.G. Thirty years of resistance: Zig-zag through the plant immune system. The Plant Cell 2022, 34, 1447–1478. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, Y.; Ning, Y.; He, Z.; Wang, G.-L. Exploiting broad-spectrum disease resistance in crops: from molecular dissection to breeding. Annual Review of Plant Biology 2020, 71, 575–603. [Google Scholar] [CrossRef] [PubMed]
- Kankanala, P.; Nandety, R.S.; Mysore, K.S. Genomics of plant disease resistance in legumes. Frontiers in Plant Science 2019, 10, 1345. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.; Melotto, M.; Tsai, S.M.; Caldas, D.G.G. Changes in spatial and temporal gene expression during incompatible interaction between common bean and anthracnose pathogen. J. Plant Physiol. 2012, 169, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Mahiya-Farooq; Padder, B.A.; Bhat, N.N.; Shah, M.D.; Shikari, A.B.; Awale, H.E.; Kelly, J.D. Temporal expression of candidate genes at the Co-1 locus and their interaction with other defense related genes in common bean. Physiol. Mol. Plant Pathol. 2019, 108, 101424. [Google Scholar] [CrossRef]
- Oblessuc, P.R.; Baroni, R.M.; Garcia, A.A.F.; Chioratto, A.F.; Carbonell, S.A.M.; Camargo, L.E.A.; Benchimol, L.L. Mapping of angular leaf spot resistance QTL in common bean (Phaseolus vulgaris L.) under different environments. BMC Genet. 2012, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Shams, E.; Javan-Nikkhah, M.; Mirzadi Gohari, A. Dissecting molecular events and gene expression signatures involved in Colletotrichum lindemuthianum-Phaseolus vulgaris pathosystem in compatible and incompatible interactions. Eur. J. Plant Pathol. 2020, 156, 925–937. [Google Scholar] [CrossRef]
- Lovatto, M.; Gonçalves-Vidigal, M.C.; Calvi, A.C.; Vaz Bisneta, M.; Vidigal Filho, P.S. Gene expression analysis in California Dark Red Kidney common bean cultivar during incompatible interaction with Colletotrichum lindemuthianum. Annual Report of Bean Improvement Cooperative 2022, 65, 71–72. [Google Scholar] [CrossRef]
- Nanami, D.S.Y.; Gonçalves-Vidigal, M.C.; Castro, S.A.L.; Frias, A.A.T.; Vidigal Filho, P.S.; Elias, H.T. Characterization of genetic resistance in Andean common bean cultivar Amendoim Cavalo to Colletotrichum lindemuthianum. Agronomy Science and Biotechnology 2017, 3, 43–52. [Google Scholar] [CrossRef]
- Friesen, T.L.; Faris, J.D. Characterization of Effector–Target Interactions in Necrotrophic Pathosystems Reveals Trends and Variation in Host Manipulation. Annual Review of Phytopathology 2021, 59, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Riviere, M.-P.; Marais, A.; Ponchet, M.; Willats, W.; Galiana, E. Silencing of acidic pathogenesis-related PR-1 genes increases extracellular beta-(1->3)-glucanase activity at the onset of tobacco defence reactions. Journal of Experimental Botany 2008, 59, 1225–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guo, N.; Zhang, Y.; Yu, Y.; Liu, S. Genome-wide characterization and expression analysis of pathogenesis-related 1 (PR-1) gene family in tea plant (Camellia sinensis (L.) O. Kuntze) in response to blister-blight disease stress. International Journal of Molecular Sciences 2022, 23, 1292. [Google Scholar] [CrossRef]
- Dixon, D.C.; Cutt, J.R.; Klessig, D.F. Differential targeting of the tobacco PR-1 pathogenesis-related proteins to the extracellular space and vacuoles of crystal idioblasts. The EMBO Journal 1991, 10, 1317–1324. [Google Scholar] [CrossRef]
- Santamaria, M.; Thomson, C.J.; Read, N.D.; Loake, G.J. The promoter of a basic PR1-like gene, AtPRB1, from Arabidopsis establishes an organ-specific expression pattern and responsiveness to ethylene and methyl jasmonate. Plant Molecular Biology 2001, 47, 641–652. [Google Scholar] [CrossRef]
- Walter, M.H.; Liu, J.-W.; Grand, C.; Lamb, C.J.; Hess, D. Bean pathogenesis-related (PR) proteins deduced from elicitor-induced transcripts are members of a ubiquitous new class of conserved PR proteins including pollen allergens. Molecular and Genetics and Genomics 1990, 222, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Diaz, J.C.; Laugé, R.; Delannoy, E.; Huguet, S.; Roux, C.P.-L.; Gratias, A.; Geffroy, V. Genome-Wide Transcriptomic Analysis of the Effects of Infection with the Hemibiotrophic Fungus Colletotrichum lindemuthianum on Common Bean. Plants 2022, 11, 1995. [Google Scholar] [CrossRef] [PubMed]
- Edington, B.V.; Lamb, C.J.; Dixon, R.A. cDNA cloning and characterization of a putative 1,3-b-Dglucanase transcript induced by fungal elicitor in bean cell suspension cultures. Plant Molecular Biology 1991, 16, 81–94. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, R.; van Esse H.P.; Kombrink, A.; Shinya, T.; Desaki, Y.; Bours, R.; ... Thomma, B.P.H.J. Conserved fungal lysM effector ecp6 prevents chitin-triggered immunity in Plants. Science, 2010, 329, 953–955. [CrossRef] [PubMed]
- Barreto-Bergter, E.; Figueiredo, R.T. Fungal glycans and the innate immune recognition. Frontiers in Cellular and Infection Microbiology 2014, 4, 145. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.S.; Barnett, H.l.; Lilly, V.G. Sporulation of Colletotrichum lindemuthianum in culture. Phytopathol. 1950, 40, 104–114. [Google Scholar] [CrossRef]
- Farrell, R.E. RNA Methodologies: Laboratory guide for isolation and characterization, 5th ed.; Elsevier: Amsterdam, Netherlands, 2017; pp. 1–855. [Google Scholar]
- Borges, A.; Tsai, S.M.; Caldas, D.G.G. Validation of reference genes for RT-qPCR normalization in common bean during biotic and abiotic stresses. Plant Cell Rep. 2012, 31, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; Rokhsar, D.S. Phytozome: a comparative platform for green plant genomics. Nucleic. Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M. The MIQE Guidelines: minimum information for publication of quantitative real-time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.; Huggett, J. qPCR primer design revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R. Quantification on the LightCycler. In Rapid Cycle Real-Time PCR; Meuer, S., Wittwer, C., Nakagawara, K-I., Eds.; Springer: Heidelberg, Berlin, 2001; pp. 21–34. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Dag, O.; Dolgun, A.; Konar, N.M. onewaytests: An R Package for One-Way Tests in Independent Groups Designs. The R Journal 2018, 10, 175–199. [Google Scholar] [CrossRef]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistics Computing: Vienna, Austria. 2020. Available online: http://www.R-project.org/.
- Wickham, H. ggplot2: elegant graphics for data analysis. 2nd ed. Cham: Springer International Publishing: Imprint: Springer. 2016. [CrossRef]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Wren, J., editor. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Disease reaction of the resistant cultivar Amendoim Cavalo and susceptible cultivar Cornell 49-242 after 72 hours post-inoculation with race 73 of Colletotrichum lindemuthianum.
Figure 1.
Disease reaction of the resistant cultivar Amendoim Cavalo and susceptible cultivar Cornell 49-242 after 72 hours post-inoculation with race 73 of Colletotrichum lindemuthianum.

Figure 2.
Relative expression of candidate genes (A) Phvul.001G244500, (B) Phvul.001G245300, (C) Phvul.001G243800, (D) Phvul.001G244300, (E) Phvul.001G2444400, (F) KTR2/3, (G) Phvul.001G246300 in the Amendoim Cavalo at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) with the race 73 of C. lindemuthianum and mock. The results are presented as logarithmic base 2 of the fold change of gene expression. Means with the same letter, for each gene, are not significantly different at the 5% significance level, using the Alexander-Govern test.
Figure 2.
Relative expression of candidate genes (A) Phvul.001G244500, (B) Phvul.001G245300, (C) Phvul.001G243800, (D) Phvul.001G244300, (E) Phvul.001G2444400, (F) KTR2/3, (G) Phvul.001G246300 in the Amendoim Cavalo at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) with the race 73 of C. lindemuthianum and mock. The results are presented as logarithmic base 2 of the fold change of gene expression. Means with the same letter, for each gene, are not significantly different at the 5% significance level, using the Alexander-Govern test.
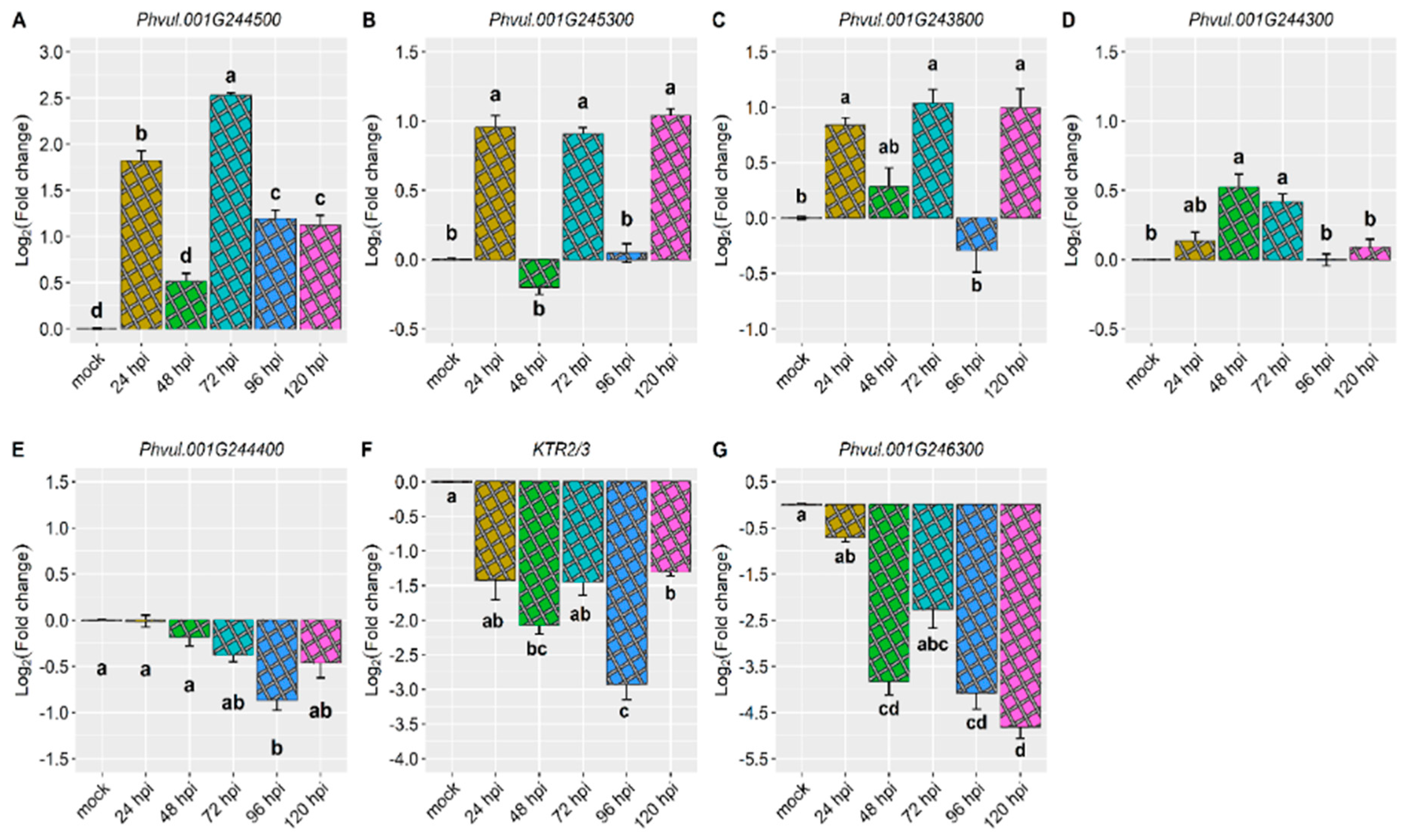
Figure 3.
Heatmap of the relative expression of candidate genes for the Co-AC and genes proximal to this locus in the Amendoim Cavalo cultivar at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) of race 73 of C. lindemuthianum and mock. The genes evaluated were Phvul.001G244500, Phvul.001G245300, Phvul.001G243800, Phvul.001G244300, Phvul.001G2444400, KTR2/3, Phvul.001G246300. Yellow shading indicates higher expression and dark blue shading has lower expression than reference genes.
Figure 3.
Heatmap of the relative expression of candidate genes for the Co-AC and genes proximal to this locus in the Amendoim Cavalo cultivar at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) of race 73 of C. lindemuthianum and mock. The genes evaluated were Phvul.001G244500, Phvul.001G245300, Phvul.001G243800, Phvul.001G244300, Phvul.001G2444400, KTR2/3, Phvul.001G246300. Yellow shading indicates higher expression and dark blue shading has lower expression than reference genes.
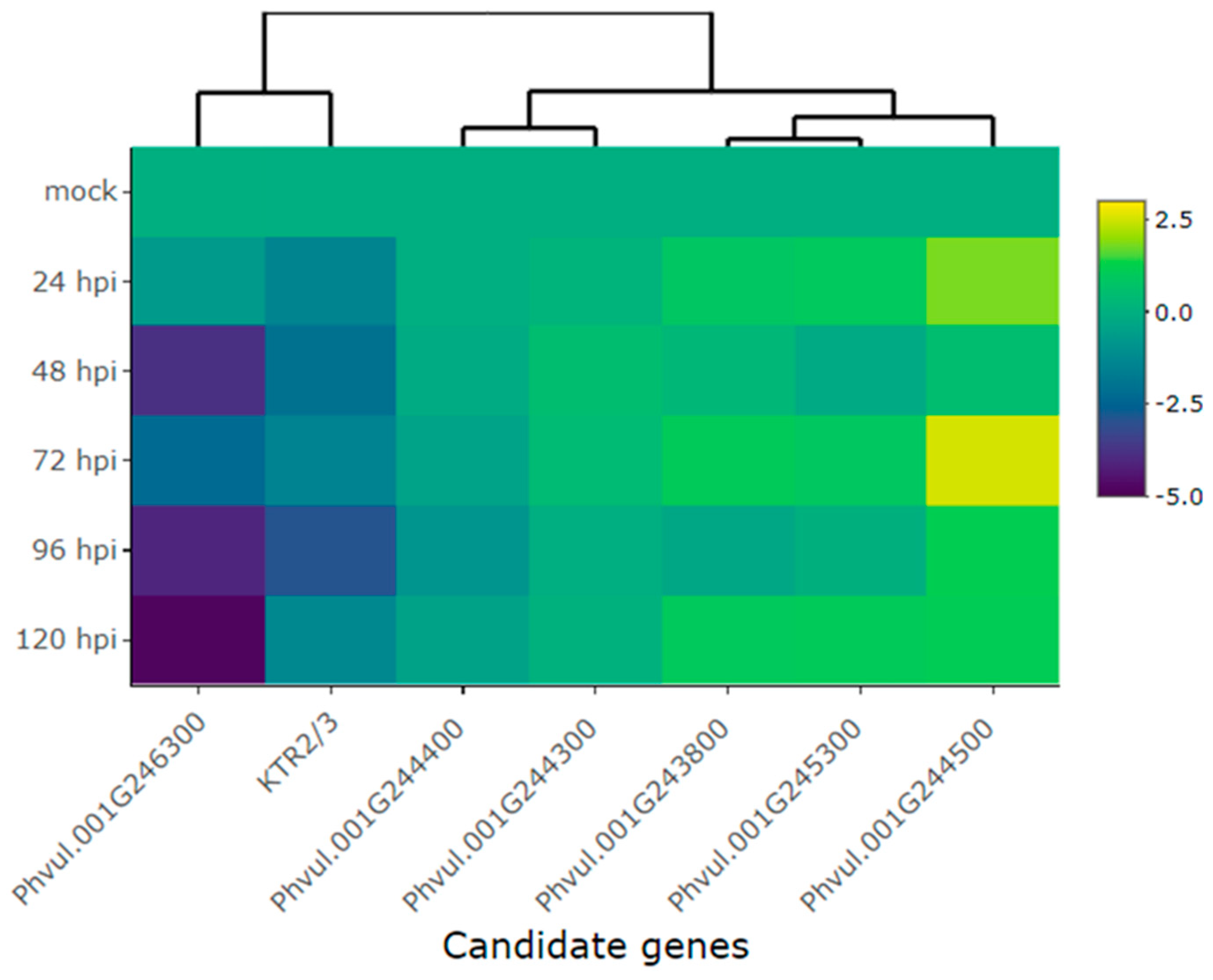
Figure 4.
Relative expression of plant defense genes (A) Phvul.006G196900 (PR1b), (B) Phvul.003G109100 (PR1a), and (C) Phvul.009G256400 (PR2) in the common bean cultivar Amendoim Cavalo at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) with the race 73 of C. lindemuthianum and mock. The results are presented as logarithmic base 2 of the fold change of gene expression. Means with the same letter, for each gene, are not significantly different at the 5% significance level, using the Alexander-Govern test.
Figure 4.
Relative expression of plant defense genes (A) Phvul.006G196900 (PR1b), (B) Phvul.003G109100 (PR1a), and (C) Phvul.009G256400 (PR2) in the common bean cultivar Amendoim Cavalo at 24, 48, 72, 96, and 120 hours post-inoculation (hpi) with the race 73 of C. lindemuthianum and mock. The results are presented as logarithmic base 2 of the fold change of gene expression. Means with the same letter, for each gene, are not significantly different at the 5% significance level, using the Alexander-Govern test.
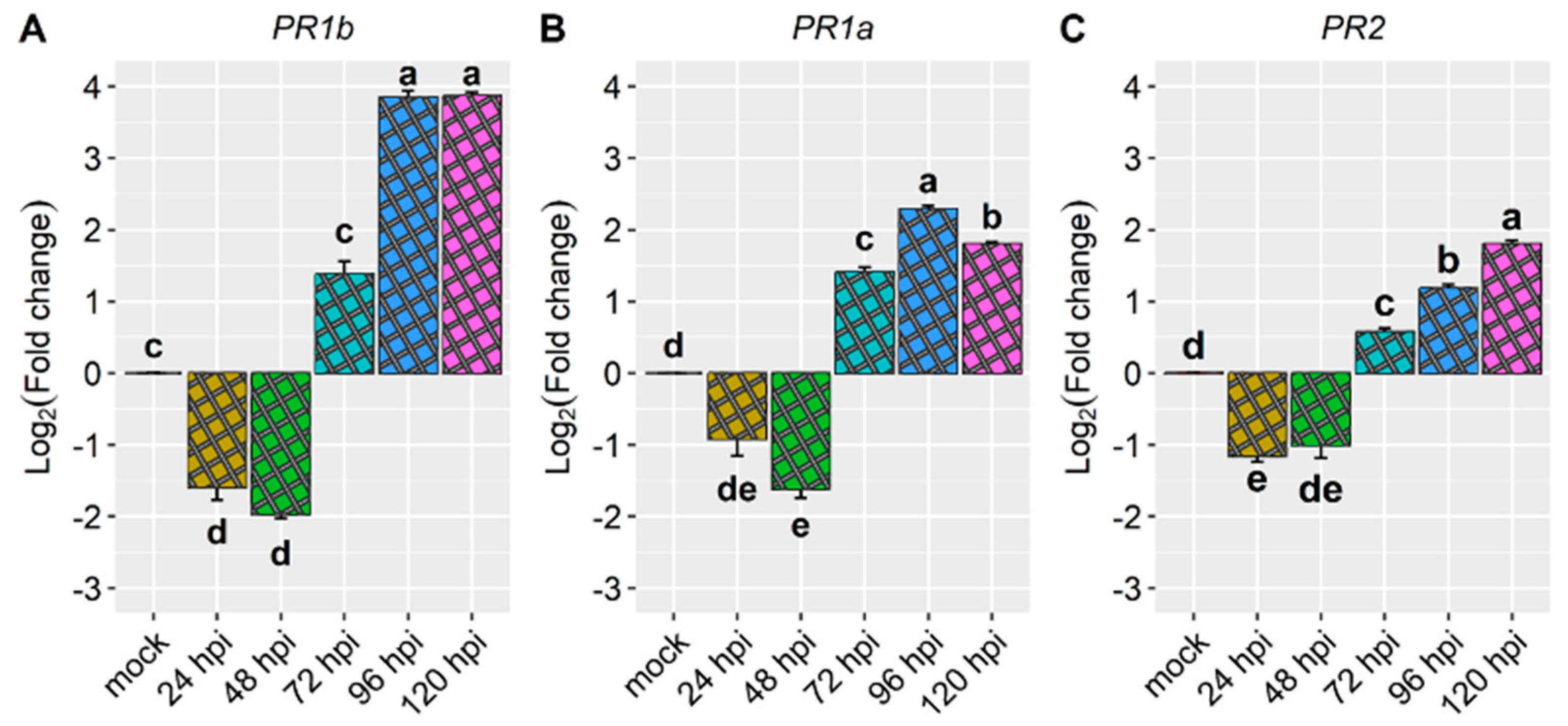
Figure 5.
Heatmap of the relative expression of plant defense genes Phvul.006G196900 (PR1b), Phvul.003G109100 (PR1a), and Phvul.009G256400 (PR2) in the common bean cultivar Amendoim Cavalo at 24, 48, 72, 96 and 120 hours post-inoculation (hpi) of race 73 of C. lindemuthianum and mock. Yellow shading indicates higher expression and dark blue shading lower expression than that of reference genes.
Figure 5.
Heatmap of the relative expression of plant defense genes Phvul.006G196900 (PR1b), Phvul.003G109100 (PR1a), and Phvul.009G256400 (PR2) in the common bean cultivar Amendoim Cavalo at 24, 48, 72, 96 and 120 hours post-inoculation (hpi) of race 73 of C. lindemuthianum and mock. Yellow shading indicates higher expression and dark blue shading lower expression than that of reference genes.
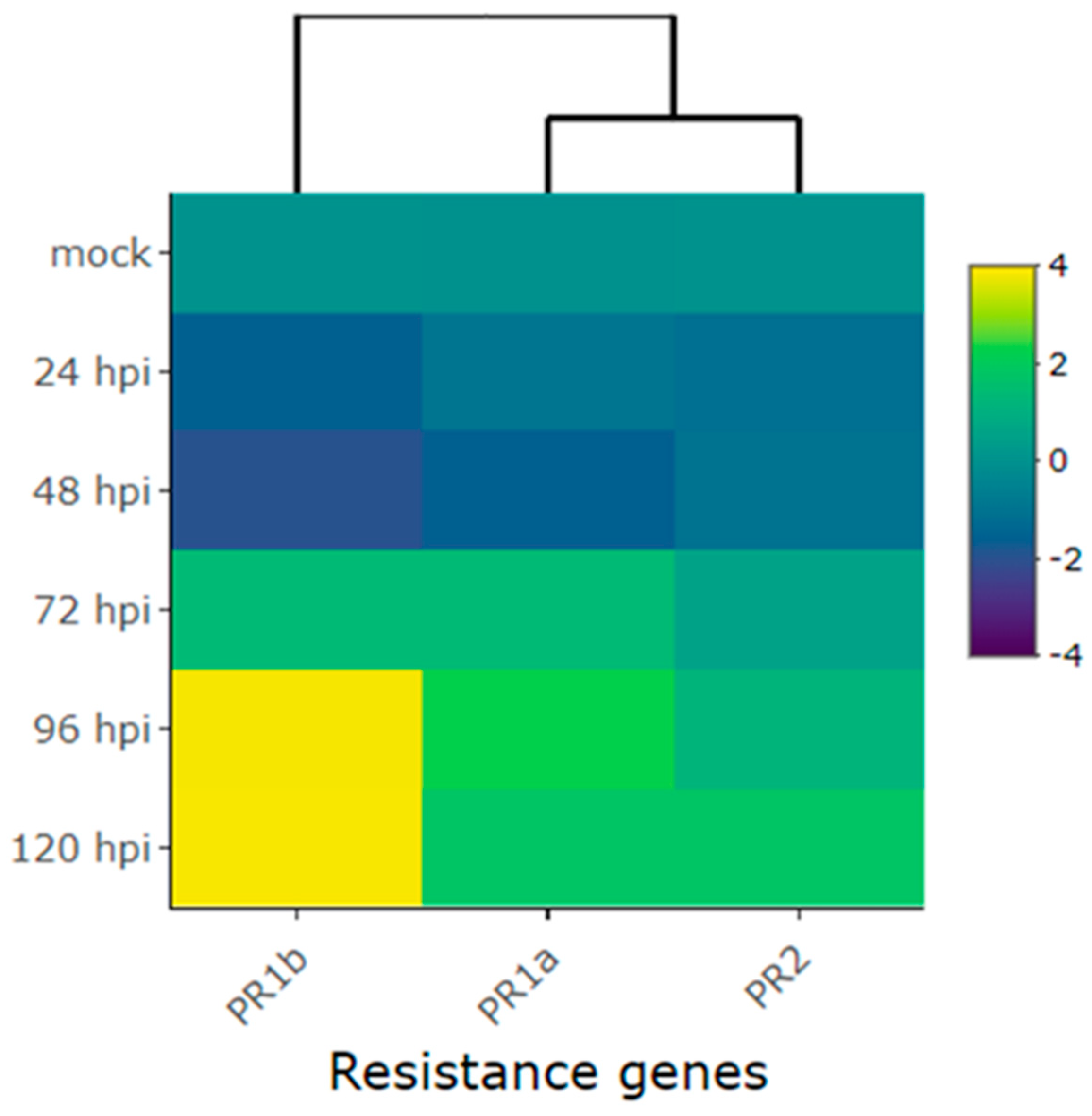
Figure 6.
Common bean chromosome Pv01 containing candidate genes for anthracnose resistance genes Co-x (KTR2/3), Co-1 (Phvul.001G243800), Co-AC (Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500), and CoPv01CDRK/PhgPv01CDRK (Phvul.001G245300, Phvul.001G246000, Phvul.001G246100, Phvul.001G246200, Phvul.001G246300, Phvul.001G246400 and Phvul.001G246800).
Figure 6.
Common bean chromosome Pv01 containing candidate genes for anthracnose resistance genes Co-x (KTR2/3), Co-1 (Phvul.001G243800), Co-AC (Phvul.001G244300, Phvul.001G244400, and Phvul.001G244500), and CoPv01CDRK/PhgPv01CDRK (Phvul.001G245300, Phvul.001G246000, Phvul.001G246100, Phvul.001G246200, Phvul.001G246300, Phvul.001G246400 and Phvul.001G246800).
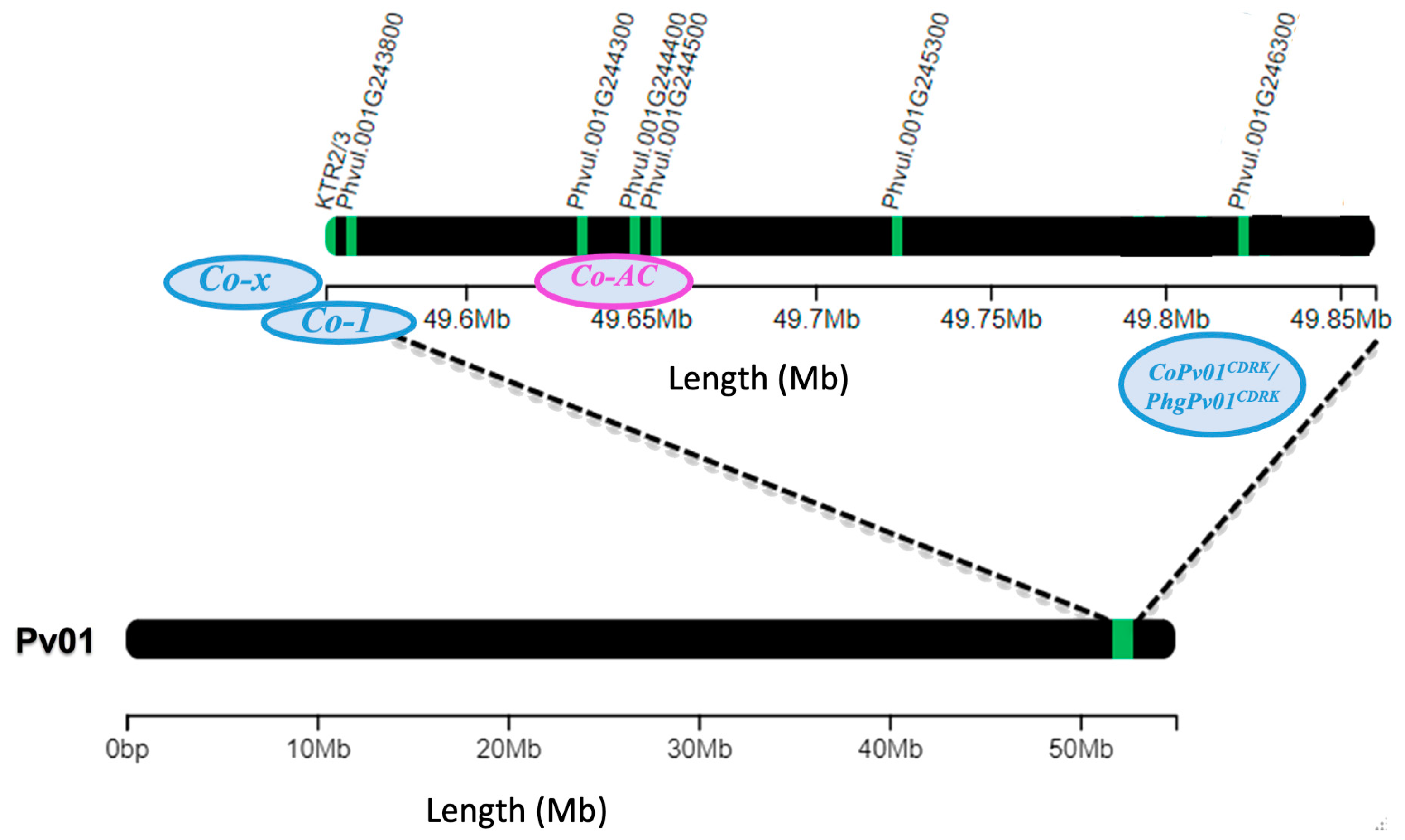

Table 1.
Gene model, previously mapped genes and functional annotation using Phytozome.
| Gene | Gene model | C. lindemuthianum race 73 | ||||
|---|---|---|---|---|---|---|
| 24 hpi | 48 hpi | 72 hpi | 96 hpi | 120 hpi | ||
| Co-x | KTR2/3 | -1.4 | -2.1 | -1.4 | -2.9 | -1.3 |
| Co-1 | Phvul.001G243800 | 0.8 | 0.3 | 1.0 | -0.3 | 1.0 |
| Co-AC | Phvul.001G244300 | 0.1 | 0.5 | 0.4 | 0.0 | 0.1 |
| Phvul.001G244400 | 0.0 | -0.2 | -0.4 | -0.9 | -0.5 | |
| Phvul.001G244500 | 1.8 | 0.5 | 2.5 | 1.2 | 1.1 | |
| CoPv01CDRK /PhgPv01CDRK | Phvul.001G245300 | 1.0 | -0.2 | 0.9 | 0.0 | 1.0 |
| Phvul.001G246300 | -0.7 | -3.8 | -2.3 | -4.1 | -4.8 | |
| Pathogenesis-related genes | Phvul.003G109100 (PR1a) | -0.9 | -1.6 | 1.4 | 2.3 | 1.8 |
| Phvul.006G196900 (PR1b) | -1.6 | -2.0 | 1.4 | 3.8 | 3.9 | |
| Phvul.009G256400 (PR2) | -1.2 | -1.0 | 0.6 | 1.2 | 1.8 | |
Table 2.
Target genes, primers used, qPCR product size (amplicon), primer melting temperature (Tm), amplification efficiency (E) and coefficient of determination of linear regression (R²).
Table 2.
Target genes, primers used, qPCR product size (amplicon), primer melting temperature (Tm), amplification efficiency (E) and coefficient of determination of linear regression (R²).
| Genea | Reference | Primers Forward (F) and | Tm | Amplicon | E | R² |
|---|---|---|---|---|---|---|
| Reverse (R) (5`-3`) | (°C) | (bp) | ||||
| Phvul.001G133200* | IDE | F: AAGCAGGTATCTTGGCCATCTC | F: 60.16 | 126 | 0.92 | 0.99 |
| R: AAAGCAAACTCCAAGCTCCAATC | R: 59.99 | |||||
| Phvul.008G011000* | ACT | F: ACAGCCAGGACCAGTTCATC | F: 59.67 | 154 | 0.93 | 0.98 |
| R: TGTATGTGGTCTCGTGAATGC | R: 58.38 | |||||
| Phvul.001G243800 | Co-1 | F: CCTCAAGGTGGGGCTTTTGAG | F: 61.16 | 118 | 1.01 | 0.99 |
| R: TCACCGAGAAACTCCCATTGC | R: 60.61 | |||||
| KTR2/3 | Co-x | F: ATGCACAGGGGAATGGGATG | F: 60.11 | 279 | 1.06 | 0.98 |
| R: GCCATAGCGAGTGAGAGTGCG | R: 63.42 | |||||
| Phvul.001G244300 | Co-AC | F: GAAACGTCTCCGCAGAATAGTG | F: 59.40 | 150 | 0.99 | 0.99 |
| R: GTCTTGTGTTGTTCCTTGGAGTTG | R: 60.44 | |||||
| Phvul.001G244400 | Co-AC | F: TACAGCAAGAGAGCGGTTAAAGG | F: 60.62 | 121 | 1.07 | 0.99 |
| R: CCCTTTGTCACTTTGTTTTGAAGC | R: 59.67 | |||||
| Phvul.001G244500 | Co-AC | F: CAATGCACAGCTCGCAACTC | F: 60.45 | 141 | 1.09 | 0.98 |
| R: GGAACTGTGAAAGCTCTGCTAAC | R: 59.81 | |||||
| Phvul.001G245300 | CoPv01CDRK | F: TCTGCTGGAAGGGTGGTAGTC | F: 61.17 | 93 | 1.04 | 0.99 |
| R: GGACGTTATGTGAACAAGGTTTGC | R: 61.08 | |||||
| Phvul.001G246300 | CoPv01CDRK | F: CTTCTTCCCTTCACTTCGATACC | F: 58.57 | 87 | 0.95 | 0.99 |
| R: GTTGAGAGTGTTTGTGGCAGT | R: 58.98 | |||||
| Phvul.003G109100 | PR1a | F: GTCCTAACGGAGGATCACTCA | F: 58.62 | 148 | 1.01 | 0.98 |
| R: CAGGGATTGGCCAGAAGGTAT | R: 59.50 | |||||
| Phvul.006G196900 | PR1b | F: GGTTTGCCTATGATCCCAATGC | F: 59.96 | 115 | 0.99 | 0.99 |
| R: TGTTGTGAGCGTTGAGGAAGTC | R: 61.06 | |||||
| Phvul.009G256400 | PR2 | F: CAGAGGTTCTCATTTGCTGCTTTC | F: 60.62 | 98 | 1.09 | 0.99 |
| R: ATGCCATAACACACCCCGATTTG | R: 61.75 |
a Based on the Phaseolus vulgaris genome available on the Phytozome v1.2 platform: https://phytozome.jgi.doe.gov/pz/portal.html#; * Reference genes; b Amplification efficiency obtained from the Equation E=[10(-1/slope)]- 1 (Rasmussen, 2001); c Coefficient of determination of linear regression.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



