
Submitted:
06 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
Rapidly progressive glomerulonephritis, the term coined for a subtype of nephritic syndrome, is characterized by a state of acute loss of renal function, along with those components of renal damage on urinalysis, over a relatively short period (a few weeks or months). It is the aftermath of an acute, severe glomerular injury. RPGN, famously known as Crescentic glomerulonephritis (cGN) is not a single disease entity but a pattern that can occur in a variety of glomerular diseases caused by different pathologic lesions leading to the development of necrosis in the glomerular capillaries seen in both, systemic and kidney-restricted diseases. While still little is known about the exact etiologic cause behind the primary injury to the capillary wall leading to the development of the condition, many mechanisms are put forth to describe the pathogenesis leading to the progression. Immunopathologic studies have demonstrated that rapidly progressive glomerulonephritis can result from glomerular antibody deposition, can be immune complex-mediated, or from some as yet undefined mechanism that doesn’t involve glomerular antibody deposition. The condition is often severe with poor prognosis as it develops so rapidly and with extensive glomerular involvement, the damage is almost irreversible. Many elements can be associated with the occurrence and progression of the disease but it often involves a disruptive immune response one way or the other. RPGN’s morphological hallmark, the extensive extra-capillary cellular proliferation into the bowman’s space, commonly known as crescent formation which is often observed by electron microscopic view of the tissue biopsy, is considered one of the great diagnostic approaches for the detection of the disease. Also, the severity of the disease is related to the nature and type of immunologic process causing the disease and (in part) to the degree of crescent formation, that can only be observed by histopathological studies. This activity attempts to explain the etiology and pathogenesis involved in the development leading to the rapid progression of the disease.
Keywords:
RPGN
; Pathology
; Aetiology
Introduction
The term “Glomerulonephritis” (also, nephritic syndrome) refers to the group of diseases in which glomeruli contain inflammatory cells, namely, polymorphonuclear leukocytes (PMNs), tissue macrophages, or lymphocytes, beyond the number expected in normal glomeruli. (“resident” macrophages) RPGN is a subtype of nephritic syndrome that is characterized by the inflammation of the nephron, mainly the glomerulus that leads to disruption of the filtration barrier of the glomerulus. The term RPGN is a combined abbreviation describing the patient’s condition as well as the tissue pathology where the term “Rapid Progression” refers to the rapidly deteriorating renal function in the patient (over a few days to weeks) and the term “glomerulonephritis” refers to the inflammation of the glomeruli, here, rather severe.
The glomerulus is the interface between the blood and the nephron, from where the kidneys control the filtrate passing on into the urine using a filtration barrier. This filtration barrier is made up of three layers, the capillary endothelium, the glomerular basement membrane, and Bowman’s capsule epithelium. The specialty of this barrier is that it allows only some components of the blood to filter through it and prevents the majority of the cellular components and large proteins from filtering. Its specialized structure facilitates this, as the first layer, that is the capillary endothelium has lots of fenestrations, whose size is about 60-70nm [1], that allow small molecules of plasma to filter but don’t allow RBCs and other cells to filter into them thus preventing their filtration. The second layer i.e., the capillary basement membrane is heavily negatively charged due to the presence of a molecule known as heparan sulfate. Because almost all blood proteins are negatively charged, heparan sulfate acts as a charge barrier to prevent proteins from being filtered into Bowman’s space [2]. The third layer i.e., the epithelium of Bowman’s space has podocytes i.e., foot-processes which have small slits between them that are also coated in heparan sulfate to ensure further that no proteins are filtered into the ultrafiltrate. This is how the glomerulus regulates the quality and quantity of filtration when filtering the plasma.
Figure 1.
Cross section of the filtration barrier.
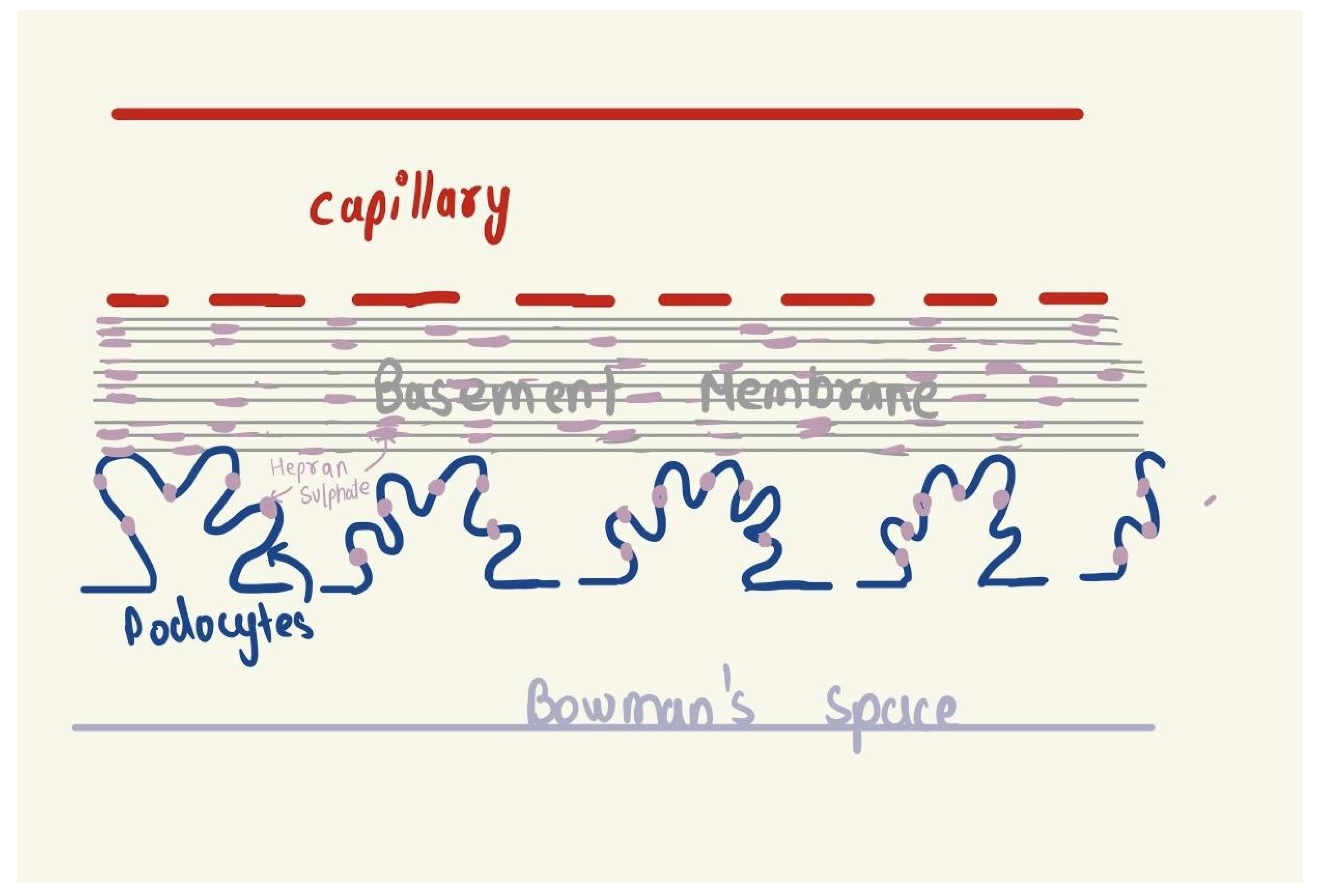
In GN, there is disruption of this barrier which commences with any kind of injury to the glomerular capillary wall, in the case of RPGN a severe injury or an unprecedented immune response to that injury [3].
RPGN is characterized by Rapid loss of renal function (Clinical manifestations include gross hematuria, proteinuria, oliguria, hypertension, and edema) over a very short period (days to weeks), Nephritic urine analysis: proteinuria, micro or macroscopic haematuria, dysmorphic red blood cells (RBC), RBC casts and Histopathological characteristic on renal biopsy finding; is an extreme form of glomerulonephritis that also reflects as a deteriorating patient condition over just few weeks or worse, a few days. The glomerular crescent formation, defined as two or more layers of proliferating cells in the Bowman’s space, is a hallmark of this inflammatory glomerulonephritis and a histologic marker of severe glomerular injury [4].
The pathogenesis of this disease can be divided into three stages
- Etiologic factors
- Mechanisms of injury
- Response to that injury
Etiologic factors
While this represents the most compelling aspect of the disease pathology and its relevant therapeutic intervention, little is known about the exact etiologic factors responsible for disease occurrence. Nevertheless, common damaging/injuring agents present in nature have been suspected to play a role in the pathogenesis of RPGN. These include toxins, such as hydrocarbon solvents like fuels, and paints; various drugs including, acetaminophen, antibiotics like penicillins, propylthiouracil, hydralazine, etc; viral and bacterial infections, monoclonal gammopathy, autoimmune mechanisms, disruptive immunogenic factors, and malignancies [5].
This injury that leads to disruption of the filter action barrier to both cells and protein, leading to rapidly progressive glomerulonephritis, can manifest in many forms including both primary and secondary causes.
Primary causes: IgA nephropathy, MPGN, and anti-glomerular basement membrane (GBM) disease
Secondary causes: Antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis, lupus nephritis, post-streptococcal GN, IgAV (HSP) nephritis
The subsequent sections will discuss the categories of GN, the pathophysiology of glomerulonephritis leading to this rapid progression, and the diagnostic approaches best suitable for prompt detection and treatment.
Categories of GN
The term RPGN in the context of crescentic glomerulonephritis (GN), classified based on immunofluorescence staining studies of the immune deposits in the glomeruli, is usually due to one of the three broad mechanisms of glomerular injury. These include:
- Linear antibody deposition: Anti-GBM disease,
- Granular immune complex-mediated injury,
- pauci-immune narcotising and crescentic GN [https://www.ncbi.nlm.nih.gov/books/NBK557430/]
[A] Linear antibody deposition: Anti-GBM disease
The anti-glomerular basement membrane antibody disease is a subtype of RPGN where there is the presence of circulating IgG antibodies against an antigen normally present in the glomerular basement membrane and sometimes also the alveolar basement membrane (in that case, the disease is known as Goodpasture disease). The antibody is targeted against a noncollagenous domain of the alpha-3 chain of type IV collagen [6].
These antibodies will cause glomerular capillary wall injury by local complement activation and polymorphonuclear leukocytes. In addition, there’s a role played by T-cells which is independent of this mechanism. Some precipitating factors are environmental triggers for the formation of antibodies, like smoking, and hydrocarbons. Also, there is a genetic association in which HLA-DR15 increases the risk of anti-GBM disease [7].
[B] Granular immune ccomplex deposition
These patients form a heterogeneous group in which multiple stimuli lead to the development of crescentic GN. Immunohistologic studies show granular deposits of immunoglobulin and complement along capillary walls and in the mesangium. This may include either diffuse membranous nephropathy or post-infectious nephropathy.
The causes include infections, systemic diseases, and pre-existing primary GN [7].
- Post-infectious GN, especially after a Streptococcus infection (complex deposition mainly sub-epithelial)
- Collagen vascular disease
- Lupus nephritis (complex deposition mainly sub-endothelial)
- Henoch-Schönlein purpura - there are immunoglobulin A deposits and associated systemic vasculitis
- Immunoglobulin A nephropathy without vasculitis
- Mixed cryoglobulinemia
- Membranoproliferative glomerulonephritis
- Fibrillary glomerulonephritis
All these conditions can precipitate into crescentic GN or the development of granular immune complex deposition leading to crescentic GN can be entirely idiopathic
[C] Pauci-immune crescentic GN
These subtypes have no immune deposits in the glomerular space and so are immunofluorescence negative. Almost 90-95% of these cases are associated with the ANCA i.e., the Anti-neutrophil cytoplasm antibody and the rest 5-10% are idiopathic or even ANCA negative. ANCA is associated with neutrophil damage and vasculitis(necrotizing inflammation of small blood vessels) and can manifest into three different conditions, namely:
- Granulomatosis with polyangiitis {GPA} (previously known as Wegner’s disease)
- Microscopic polyangiitis
- Eosinophilic granulomatosis with polyangiitis {EPGA} (previously known as Chrug-Strauss syndrome)
All these conditions are grouped under the umbrella term ANCA-associated vasculitis because they’re associated with a key protein factor in the blood called ANCA and they all cause inflammation of the small blood vessels. The major organs affected are the Kidneys, lungs, joints, ears, and nose. ANCA is a type of IgG antibody that specifically binds to two proteins that are normally found in the fluid within the neutrophil (cytoplasm). The two proteins are named proteinase 3 (PR3) and myeloperoxidase (MPO). Patients with ANCA-associated vasculitis may have autoantibodies against PR3 (PR3-ANCA) or MPO (MPO-ANCA) but not both. The binding of ANCA to neutrophils in the blood, results in:
- The release of toxic substances from neutrophils leads to damage to the small blood vessel walls.
- Neutrophil migration through blood vessel walls causing inflammation in surrounding tissues.
- Release of signaling factors which in turn attract more neutrophils, perpetuating the inflammation and destruction of small blood vessels.
They can cross the placental barrier (especially the MPO-ANCA)and damage the fetal lungs and kidneys if they’re present in the mother.
Furthermore, many medicines are associated with the development of ANCA-positive vasculitis leading to RPGN. These include:
- Hydralazine
- Levamisole-contaminated cocaine
- Propylthiouracil and methimazole
- Allopurinol
- Sulfasalazine
- Minocycline
- Penicillamine
- Rifampicin
- Aminoguanidine
- Sofosbuvir
- Anti-TNF therapy for rheumatoid arthritis and ankylosing spondylitis (Naik and Shawar 2023)
The appearance of ANCA antibodies in the body is poorly understood but certain speculations are that they may be triggered by infections mainly Staph aureus, or by molecular mimicry between the bacterial and self-antigens or by defective neurotrophic cell death resulting in the exposure of immune cells to the internal neutrophil contents.
Whatever may be the pathway the ultimate result is damage to the glomerular capillary wall which leads to fibrin deposition and parietal epithelial infiltration into the bowman’s space culminating in crescent formation.
Mechanisms of glomerular capillary wall injury:
The second stage in the pathogenesis after the discussion on etiologic factors is the discussion on the mechanism of injury to the glomerular capillary wall.
Figure 2.
Schematic diagram of the relationship between the mechanisms of injury and the pathogenesis of Crescents formation (dotted lines may suggest a relation but it isn’t established yet).
Figure 2.
Schematic diagram of the relationship between the mechanisms of injury and the pathogenesis of Crescents formation (dotted lines may suggest a relation but it isn’t established yet).
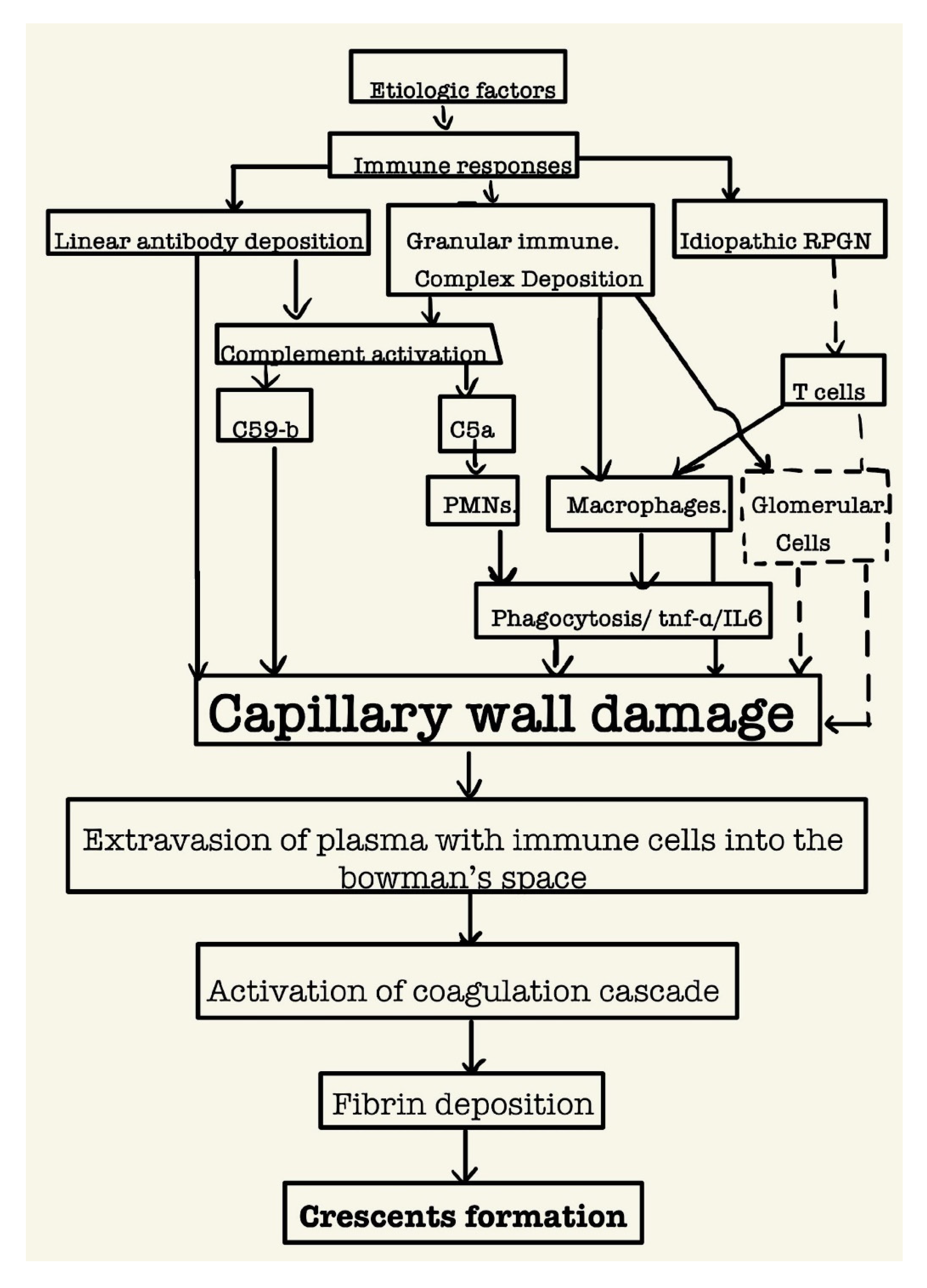
Studies show that the deposition of anti-GBM antibodies directly leads to capillary wall damage as they’re targeted against the collagen in the basement membrane [8]. Furthermore, both linear antibody deposition as well as granular immune complex deposition lead to the activation of the complement system leading to the activation of membrane attack complex C59-b that can produce severe capillary wall damage independent of inflammatory cells [9]. Neutrophils too play a direct role in damaging the capillary wall as established through ANCA associated vasculitis as well their activation via the complement system. Similarly, macrophages damage the capillary wall through their activation via the complement system [10]. Lastly, cell-mediated immunity also plays a significant role in damaging the glomerular capillary wall, particularly in those idiopathic cases.
Whatever may be the mechanism of the injury to the glomerular capillary wall, the ultimate consequence is the rents/gaps in the capillary wall (wider than the already present fenestrations) along with rupture in the glomerular capillary basement membrane that lead to fibrin exudation as well as cellular and humoral components of inflammation into the Bowman’s capsule, generating a response from the parietal epithelial cells and causing them to proliferate. This leads to extra capillary proliferations that narrow the remaining space in the capsule and appear as crescents on renal biopsy.
Response to the glomerular capillary wall injury: formation of the glomerular crescents
The initiating event or the very first stage in the formation of glomerular crescents is the induction of rents in the glomerular capillary wall, resulting in the movement of plasma products, including fibrinogen, into Bowman’s space and with subsequent fibrin formation, the influx of macrophages and T cells (Th1 and Th17 CD4+ T cells), and the release of proinflammatory cytokines, such as interleukin-1 and tumor necrosis factor-alpha and procoagulant and fibrinolytic inhibitory factors [11]. As crescent formation is the final common pathway of severe glomerular inflammatory disease, many immune factors cross often generating an elaborated response.
Crescents contain inflammatory cells including macrophages, the proliferating parietal epithelial cells of the glomerulus, and also fibroblasts. Fibrin deposits are central in the role played by the infiltration of glomerular macrophages near the site of injury [12] as thrombin in chemotactic for monocytes it is believed that macrophages in glomeruli generate procoagulant activity, presumably in the form of tissue factor that can activate the extrinsic coagulation pathway. subsequently, the glomerular procoagulant activity may also be derived from the endothelial damage. This results in the second stage of crescents formation i.e., active proliferative inflammation resulting in the development of cellular, microcellular, and then fibrous crescents with time. Fibroblast growth factors promote fibroblast proliferation and collagen deposition mainly type III. This final fibrous stage is unlikely to respond to immunosuppressive therapy and has a high risk of ESKD [13] Apart from macrophages and T-cells, parietal and visceral epithelial cells also contribute to active crescent formation. The stimuli for the proliferation of Epithelial cells and fibroblasts are unknown, but macrophages, platelets, or intrinsic glomerular cell-derived cytokines are considered likely candidates. While the presence of scarring will not decrease renal function per se, the age of presence does serve as a marker of disease duration and hence the probability of successful therapeutic intervention.
Conclusion
- RPGN, characterized by Rapid loss of renal function (Clinical manifestations include gross hematuria, proteinuria, oliguria, hypertension, and edema.) over a very short period (days to weeks)
- Glomerular crescents are the histologic hallmark of the disease
- They are best diagnosed by renal biopsy electron microscopic studies.
- Mainly three types of crescentic GN: Anti GBM antibody disease; immune complex deposition, pauci immune GN
- Diagnostic approaches should include immunofluorescence studies on the biopsy as well as the detection of plasma ANCA.
- Prompt diagnosis is a must to reverse the damage. As final fibrous stages are unlikely to respond to immunosuppressive therapy.
Institutional Review Board Statement
Being a Short note, there were no ethical issues and IRB permission is not required.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Satchell, S.C.; Braet, F.; Qureshi, S.H.; Patel, N.N.; Murphy, G.J.; Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C.; et al. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. Am. J. Physiol. Physiol. 2009, 296, F947–F956. [Google Scholar] [CrossRef]
- Immunotactoid Glomerulopathy. Diagnostic Pathology: Kidney Diseases [Internet]. 2016 [cited 2023 Dec 5];266–9. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780323377072500549.
- discussant P, Charles Jennette J. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164–77. https://doi.org/10.1046/j.1523-1755.2003.00843.x. [CrossRef]
- Moorani KN, Aziz M, Amanullah F. Rapidly progressive glomerulonephritis in children. Pak J Med Sci [Internet]. 2022 [cited 2023 Dec 2];38(2):417. https://doi.org/10.12669/pjms.38.icon-2022.5774. [CrossRef]
- Lionaki S, Liapis G, Boletis JN. Pathogenesis and Management of Acute Kidney Injury in Patients with Nephrotic Syndrome Due to Primary Glomerulopathies. Medicina (B Aires) [Internet]. 2019 [cited 2023 Dec 2];55(7). https://doi.org/10.3390/medicina55070365. [CrossRef]
- Naik RH, Shawar SH. Rapidly Progressive Glomerulonephritis. StatPearls [Internet]. 2023 May 1 [cited 2023 Dec 2].. https://doi.org/10.20944/preprints202312.0614.v1. [CrossRef]
- Naik RH, Shawar SH. Rapidly Progressive Glomerulonephritis. StatPearls [Internet]. 2023 May 1 [cited 2023 Dec 2]. https://doi.org/10.20944/preprints202312.0614.v1. [CrossRef]
- Naik RH, Shawar SH. Rapidly Progressive Glomerulonephritis. StatPearls [Internet]. 2023 May 1 [cited 2023 Dec 2]. https://doi.org/10.20944/preprints202312.0614.v1. [CrossRef]
- Kaartinen K, Safa A, Kotha S, Ratti G, Meri S. Complement dysregulation in glomerulonephritis. 2019 [cited 2023 Dec 2]. https://doi.org/10.1016/j.smim.2019.101331. [CrossRef]
- Kaartinen K, Safa A, Kotha S, Ratti G, Meri S. Complement dysregulation in glomerulonephritis. 2019 [cited 2023 Dec 2]. https://doi.org/10.1016/j.smim.2019.101331. [CrossRef]
- Overview of the classification and treatment of rapidly progressive (crescentic) glomerulonephritis - UpToDate [Internet]. [cited 2023 Dec 2]. Available from: https://www.uptodate.com/contents/overview-of-the-classification-and-treatment-of-rapidly-progressive-crescentic-glomerulonephritis.
- Overview of the classification and treatment of rapidly progressive (crescentic) glomerulonephritis - UpToDate [Internet]. [cited 2023 Dec 2]. Available from: https://www.uptodate.com/contents/overview-of-the-classification-and-treatment-of-rapidly-progressive-crescentic-glomerulonephritis.
- Moorani KN, Aziz M, Amanullah F. Rapidly progressive glomerulonephritis in children. Pak J Med Sci [Internet]. 2022 [cited 2023 Dec 2];38(2):417–25. https://doi.org/10.12669/pjms.38.ICON-2022.5774. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



