
Submitted:
07 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
Antimicrobial resistance is one of the most important global health issues identified in recent decades. Different approaches have been used to establish the presence and abundance of antimicrobial resistance genes (ARGs) in the environment. In this study, we analyzed soil samples from Fildes Peninsula (King George Island, maritime Antarctica), exposed to different types of impact. The objective of the work was to identify ARGs in the samples, and evaluate if these genes were located in plasmids, using two different strategies. A metagenomic analysis was used to identify ARGs and if they were associated to plasmid sequences. The analysis showed that the site highly exposed to anthropogenic activity had a higher number of ARGs compared to other sites. A high percentage of those ARGs (19.4%), were located in plasmidic contigs. We also assessed replicon mobilization, using microbial communities from these soil samples as donors through an exogenous plasmid isolation method. In this case, we could recover plasmids with ARGs in a Tcr transconjugant clone. Although they could not be fully assembled, we could detect broad host range IncP1 and IncQ plasmid sequences. Our results indicate that sewage impacted soils could be a hotspot for ARGs spread into the Antarctic environment

Keywords:
Resistome
; plasmids
; Antimicrobial resistance genes
; Metagenomics.
1. Introduction
The Antimicrobial resistance (AMR) occurs when a bacterium is not affected by a specific compound to which other microorganisms are susceptible. This is an evolutionary phenomenon increased by misuse of antimicrobials, which has had an important impact on public health during the last decades [1]. ARGs usually identified in clinical settings, farms, sewage water and urban environments, arise as a consequence of a strong selective pressure associated with the use of antibiotics in human health and animal production. There is a consensus on the approach to tackle this public health issue, which considers the ability of many bacteria to thrive in different environments. The One Health concept takes into account the interconnection of animal, environmental and human microbiomes, to understand this complex adaptive bacterial process [2].
ARGs are prior to the use of antimicrobials, as well as to the identification of resistance in clinical settings. This has been evidenced by detection of clinical-type ARGs in ancient DNA purified from isolated permafrost samples [3]. In addition, a high diversity of ARGs has been detected in natural, low-impacted environments, suggesting that different compounds with potential antimicrobial activity could play a role in those communities [4]. Several human pathogens can survive in natural environments not associated with their hosts [5,6,7]. For example, sewage systems are settings where human-associated bacteria get in contact with environmental microorganisms. These sites have been described as hotspots for ARGs release and dispersion to the environment [8,9].
Plasmids are genetic elements that can regulate their own replication, independent of the host chromosome. Additionally, plasmids may contain genes that confer the ability to conjugate with other organisms. Also, they can integrate other intra-genomic mobile sequences, such as insertion sequences, transposons and integrons, which increase their ability to capture and spread genes that confer adaptive advantages such as ARGs or metal resistance genes (MRGs) [10,11]. It has been recognized that plasmids are common carriers of ARGs in clinical settings [12]. As a result, genomic surveillance of plasmids carrying ARGs is becoming essential to understand and monitor pathogen outbreaks.
The Antarctic continent has various regions with characteristic climates and associated biota. Classically, three biogeographical regions have been identified, which include the very cold and dry Continental region, the Maritime Antarctic region, with a less harsh climate, and the sub-Antarctic region, with a milder weather and a characteristic biota [13]. Antarctica is subject to a strict regulation in environmental management declared in the Environmental Protocol to the Antarctic Treaty (https://www.ats.aq/e/protocol.html). Thus, human activities that take place on the continent must be associated with an environmental impact assessment, defining even restricted access sites, identified as Antarctic Specially Protected Areas (ASPAs). These sites have been selected considering different aspects, like the presence of fossils, animal nests, history, etc.
South Shetland archipelago is located within the Maritime region, where King George Island stands out for hosting the highest number of scientific bases per area compared with the rest of the continent. Then, this island is considered as one of the most densely populated sites of the Antarctic continent, at least during the summer, and there are reports of AMR presence associated with human impact [14]. On the other hand, this island has nests and colonies of numerous marine mammals and birds. The fauna of this island, especially birds, have also been linked to spread of AMR bacteria, given their migration patterns and eventual contact with human [15].
Fildes Peninsula, the largest area of King George Island without a permanent ice cover, represents a small area of the Antarctic continent containing differentially impacted sites. Taking this into consideration, we estimated that it would be of interest to evaluate the microbial communities in soils of Fildes Peninsula, in sites exposed to different types of impact. In particular, the objective of this work was to analyze the presence of clinical ARGs and their potential association with plasmids. Using a metagenomic approach, we examined soils exposed to anthropogenic and animal influence as well as soils with a presumptive reduced exposition to this type of impact. Then, the analysis of these metagenomes was complemented with the identification of MRGs and their genomic location
2. Materials and Methods
2.1. Sample collection and metagenomic sequencing
Nine soil cores were collected in Fildes Peninsula during the summer campaign of 2017. Three sites were selected intending to collect soil exposed to different environmental conditions, specially considering influences of biotic origin. The first site, named IA, is located in Ardley Island (62º12´34´´ S; 58º55´44´´W). Some years ago, intending to protect the birds inhabiting this area, this island was classified as ASPA 150. This site hosts a large bird colony, and the soil could be considered as intensively impacted by bird activity (ornithogenic soil). The second sampled site, named HTP (ASPA 125d), is located in Half Three Point (62º13´38´´S; 58º57´12´´W). We selected this site, relatively far from scientific research stations and breeding sites for mammals and birds, assuming that it was less exposed to animal and human local activities. Finally, samples next to Artigas Research Scientific Station (BCAA) was also collected, specifically from soils impacted by the wastewater treatment system. This sample was designated BCAA (62º11´04´´S; 58º57´54´´W).
Three soil cores, separated by a distance of at least two meters between each other, were aseptically collected at each sample site and kept at 4ºC until further processing at the laboratory in Uruguay. Total DNA community was extracted using PowerSoil DNA kit from Qiagen (Hilden, Germany) following manufacturer instructions. Extracted DNA quantity and integrity were assessed by 0.9% (w/v) agarose gel electrophoresis in TAE buffer. The extractions that presented the best quality profile (both in quantity and integrity) from each of the three replicas were used for metagenomic sequencing at Macrogen Inc. (Seoul, Korea). Illumina technology was used through a HiSeq2500 sequencer, which yielded more than 2 Gbp of data for each metagenome with a read length of 101 bp. Read data is available at ENA web page under the project PRJEB67896.
2.2. Metagenomic analysis
Metagenomic reads quality were assessed using Fastqc software [16]. Reads were filtered and trimmed using Trimmomatic software (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:80) [17]. Remaining and corrected reads were used as input for metagenomic assembly using IDBA-UD software with default parameters. Quast software [18] was used to assess quality of assemblies. Plasmid-derived sequences were detected using plaSquid pipeline [19]. Plasmids replication initiator proteins were retrieved using –ripextract option and compared against NCBI non-redundant protein database. Abricate software, using CARD database with 70% of identity and coverage thresholds, was used to detect antimicrobial resistance genes (ARGs) in plasmidic and metagenomic contigs.
In order to understand the dynamics of resistance with a genome-centered approach, we also ran metaWRAP software [20]. This wrapper software groups metagenomic contigs in bins composed by sequences derived from the same species genomes, called metagenome-assembled genomes (MAG). CheckM software was also run to filter out the assembled MAGs by completeness (lower than 50%) and contamination (higher than 10%) [21]. Additionally, Metaphlan3 software was run to detect phylogenetic composition of microbial communities from the three different samples [22]. Metal resistance genes were searched by using the experimentally confirmed resistance genes of BacMet database (v2.0) [23], blastp algorithm from BLAST+ software (v. 2.12.0) and filtered to 75% of identity and coverage using custom scripts [24].
In order to assess human impact in the sequenced microbial communities, the crAssphage genome was used as an indicator of human impact. The crAssphage genome was used as reference for mapping with bowtie2 using default parameters [25].
2.3. Construction of a strain for exogenous plasmid Isolation
A recombinant strain derived from Escherichia coli DH5α expressing kanamycin resistance (Kmr) and GFP protein (gfp), was constructed. For this, a mini-transposon mTn5-gusA-pgfp12 [26] was used to insert the aforementioned genes in the chromosome of E. coli DH5α . This was done by biparental conjugation using E. coli S17.1-λpir [27] with the plasmid pUT::mTn5-gusA-nptII-pgfp12 as donor. This plasmid can only replicate in λpir strains, able to express π protein. Resistant (Nalr, Kmr) transconjugants were selected, intending to work with recombinant DH5α clones in which just the mini-transposon mTn5-gusA-pgfp12 was inserted in the chromosome. Three clones were selected, further replicated in LB solid medium, and stability of fluorescent phenotype was checked.
2.4. Exogenous isolation assay
The recombinant fluorescent strain derived from E. coli DH5α carrying gfp gene in the chromosome was used as acceptor for Exogenous Isolation Assays. 5 mL of cell cultures were grown for 24 hs in LB broth at 37ºC and stirred at 200 rpm. The entire culture was centrifuged at 3900g for 5 minutes and resuspended in 1 mL LB broth. Microbial communities contained in nine soil samples were used as conjugation donors, three samples obtained from each site were evaluated. For this, 1 gr of soil was resuspended in 9 mL of 1/10 diluted sterile TSB with 5 autoclaved glass spheres of 5 mm diameter and stirred during 90 minutes at 200 rpm and 37ºC in order to disaggregate soil particles. A volume of 4 ml of soil suspension was thoroughly mixed with 1 mL of resuspended recipient cells and centrifuged at 3900g for 5 minutes. The mixture was resuspended in 100 µL of LB. These conjugation mixtures were thoroughly pipetted over a 45 µm pore-size filter, placed on LB agar, and incubated for 48 hs. at 25ºC [28]. This procedure was repeated for nine soil samples, three samples of each site were used as donor communities.
Then, filters were washed with 5 mL of 0.85% (w/v) NaCl, three serial dilutions were prepared from this suspension and volumes of 100 µl were plated on LB agar supplemented with kanamycin (50 μg/mL)(Km), nalidixic acid (25 μg/mL) (Nal), cycloheximide (10 μg/mL) (Chm) and one of the following antibiotics: tetracycline (10 μg/mL), ampicillin (50 μg/mL), trimethoprim (25 μg/mL) or gentamycin (10 μg/mL). Plates were incubated at 37ºC for 24 hs and fluorescent colonies were identified and selected under UV light.
2.5. Genomic sequencing analysis of receptor strain.
Fluorescent colonies grown on LB agar Nal Km Chm plus one of the antibiotics previously mentioned were transferred to fresh media in order to confirm their phenotypic stability. Selected colonies were grown in 5 mL LB broth with the corresponding antibiotics. Genomic DNA was extracted using Quick-DNA Fungal/Bacterial Miniprep kit (Zymo research, USA) following manufacturer instructions and DNA integrity and quantity was checked in a 0.95% agarose gel electrophoresis. Genomic DNA was sequenced at Genoma Mayor facilities, Universidad Mayor (Santiago, Chile) using Illumina technology. Raw reads were trimmed using the same software and parameters previously described for metagenomic data. Filtered reads were mapped against E. coli DH5α (GCA_000755445.1) reference assembly and miniTn5 assembly (HQ328084.1) and filtered out. Remaining reads were assembled using SPAdes software [29]. Additionally, plaSquid software was used to detect and classify plasmidic contigs [19], which were further analyzed looking for ARGs and MRGs as previously described.
In order to detect plasmidic contigs retrieved through exogenous isolation in the metagenome sequenced from the donor sample, metagenomic reads were mapped against plasmidic contigs using BWA software [30]. Additionally SAMtools toolkit was used to compute coverage of plasmidic contigs with quality-filtered metagenomic reads [31].
3. Results
3.1. Genome-centric resistome analysis
We could assemble a different number of contigs for each sequenced metagenome. N50 values ranged from 1280 bp in HTP metagenome to 3506 bp in BCAA. The total length of assembled metagenomes also varied between 33 and 103 Mbp. The presence of human fecal contamination was analyzed by mapping these metagenomes against the crAssphage genome as indicator of human impact (data not shown). This analysis showed that the BCAA sample was the only one containing reads that could be assigned to this phage. Additionally, by taxonomic profiling of the communities we could detect the presence of Faecalibacterium, Prevotella, Acinetobacter and other genera associated to human gut microbiome in BCAA(Supplementary Figure S1).
In order to analyze genomic resistance traits of the most abundant genomes in the samples, we assembled MAGs from shotgun metagenomic data (Figure 1). We could identify a total of 20 medium and high quality MAGs; nineteen of them (95%) included metal resistance genes (MRGs), while seven encoded ARGs (35%). Most of the MAGs detected were classified at family or genus level, corresponding to environmental bacteria. BCAA metagenome was the one with more MAGs identified. Five of them were classified within the Comamonadaceae family and another two within the Flavobacterium genus (Figure 1). Apart from that, we could also detect two genomes of Pseudomonadaceae and Pseudomonas in BCAA sample. Psychrobacter genus was also present in two of the three metagenomes analyzed. Additionally, an Aeromonadaceae MAG, found in IA sample, was the one with more resistance traits identified, with 30 different resistance genes, mainly MRGs. The only MAG detected without known resistance traits corresponded to Helicobacter genus and was found in the IA sample.
3.2. Gene-centric resistome analysis
Figure 2 shows all ARGs (38 genes encoding resistance against 9 different antibiotic classes) identified in the metagenomes of the three samples analyzed. BCAA contains a higher number of diverse ARGs compared to the other two samples. This was the only metagenome in which we could detect ARGs encoded in plasmidic contigs. In fact, nearly 20% of ARGs found, encoding resistance to 7 different antibiotic types, were assigned to plasmidic contigs. Within this metagenome, genes for resistance to chloramphenicol and tetracycline were found only in plasmidic contigs. Macrolide resistance genes were also evenly distributed in chromosomal and plasmidic contigs.
Diverse kinds of ARGs were found in the three metagenomes, encoding resistance to different antibiotics. Most of them are components of non-specific resistance mechanisms, which could confer resistance to multiple drugs. Various aminoglycoside resistance genes were found, some of them evenly distributed across samples, like aadA gene. However, the gene aph(3´)-IIa was found in metagenomes AI and HTP, but not in BCAA. In turn, in BCAA we could find a gene encoding a variant of the enzyme APH(3´)-Ia in a plasmidic contig, highly similar to Acinetobacter baumanii pAC30b plasmid (CP007579.1). Beta-lactam resistance was also an important trait found in these Antarctic metagenomes. TEM-4 was only found in metagenome IA, while OXA and VEB-1 variants were found in BCAA. OXA-15 gene was found in a highly fragmented plasmidic contig. Macrolide resistance was another trait found in metagenome BCAA. Genes mphD and msrE were located in the same plasmidic contig, with high sequence identity to a region of megaplasmid pXBB1-9 hosted in Acinetobacter johnsonii XB1. Additionally, sulfonamide resistance genes were detected in chromosomal contigs, while a tetracycline resistance gene was detected in a plasmidic contig, both in BCAA metagenome.
In order to compare the presence of resistance genes between samples, we normalized the count of ARGs and MRGs relative to 16S rRNA count. This analysis showed an important presence of MRGs in the metagenomes of the three samples analyzed, with a mean value of 1.5 genes per 16S rRNA gene (Supplementary Figure S2). When considering only plasmidic contigs, MRGs normalized count decayed more than 17 fold for BCAA metagenome. The main traits found confer resistance to multiple metals such as copper, zinc, cadmium, arsenic and lead. In the case of plasmidic ARGs, these were only detected in BCAA derived plasmid sequences (Figure 2).
3.3. Exogenous Plasmid Isolation Assay
Exogenous plasmid isolation assay was used to determine if the ARGs contained in these soil samples could be potentially transferred to human associated bacteria. Nine soil microbial communities were used as donors and E. coli DH5α::mTn5-gusA-nptII-pgfp12 as receptor strain. We could detect the acquisition of resistance to tetracycline antibiotic in one of the nine transconjugation assays; three Tcr colonies were detected, able to grow with a fluorescent phenotype (Supplementary Figure S3). In this case, one transconjugant strain was sequenced along with cargo plasmids. In order to facilitate plasmids detection and assembly, reads mapping against the E. coli chromosome were discarded. Plasmidic contigs were detected with plaSquid software and their classification and traits are shown in Table 1. Multiple plasmidic replication origins were found in the transconjugant strain. Three of them could be classified in replicon types IncP1, IncQ and ColE1. These three replicons have different mobility characteristics. MOBP1 and MOBQ relaxases were found in different contigs, which show the mobilization capacity of some of these replicons.
The largest contig encodes an IncP1 replicon which is part of a self-mobilizable plasmid, given the presence of the type 4 secretion system (TIVSS) genes (Figure 3A). This was the only self conjugative replicon found in the transconjugant strain. However, we could not find any ARG in this 43 kb contig. Notably, we could find a tetracycline resistance gene tetC in a 1345 bp contig (Contig-31, Table 1). This was the only selected resistance phenotype included in the transconjugation assay and it could not be linked to any particular replicon.
Another interesting contig identified was an IncQ-like plasmid, whose entire replicon could be assembled and had a coverage of more than 7X in the metagenome of BCAA site (Contig-4, Table 1). This mobilizable plasmid encodes for a relaxase, but lacks a type IV secretion system (TIVSS), in fact the only TIVSS detected was the one associated with the IncP1 replicon. In addition, two aminoglycoside resistance genes could be found in this replicon. One of them, rmtG, encodes a ribosomal methylase which gives high levels of aminoglycoside resistance (Figure 3B). This phenotype was verified by the growth of the transconjugant strain in LB medium supplemented with 250 μg/mL of kanamycin compared to the lack of growth of the receptor strain, which encodes nptll gene. Other aminoglycoside resistance gene, aadA6, and a multidrug efflux pump, qacH, were detected in a 2187 bp contig (Contig-12, Table 1). A significant fragmentation of these plasmidic contigs impeded to link most of the ARGs identified with their corresponding replicons.
4. Discussion
The Antarctic continent is home to very different regions, considering the climate and associated biota. Classically, some islands that surround the continent and the West coast of the Antarctic Peninsula have been grouped as a biogeographic region named as Maritime Antarctica. This region, unlike the largest area of the continent, has a climate that is not as cold and dry as the Continental region. This determines the establishment of a characteristic associated biota, which has been impacted in recent years as a consequence of global climate change [13] and by the local activity in the scientific bases established in the region. According to previous studies [32], the presence of numerous bases in some sites is associated with a high environmental impact. Global warming is a process that also impacts microbial communities in the Antarctic soil, with defined trends. All these factors affect the evolution and mixture of microbial communities within this region.
Sewage treatment and disposal policies in the Antarctic continent are under discussion nowadays. It has been demonstrated that non-treated sewage deposition into sea water may have an important impact on marine wildlife [33]. Additionally, there have been previous reports of human microbiome-associated bacteria delivered into the environment from sewage water of several Antarctic [34,35]. In this work we found in the BCAA sample a higher number of different ARGs of clinical-type than in the other two sites. This sample was collected from soil near the septic chamber, where there had previously been reported leaking events during high occupancy periods, which directly dispersed human microbiome components into the environment [36]. Additionally, samples were taken during the austral summer when scientific bases have high-occupancy with scientific and maintenance staff. Environmental monitoring protocols should be established in order to quantify this kind of impact.
Some of the MAGs recovered belong to taxonomic groups that have been reported and studied in the Antarctic environment. For instance, Psychrobacter and Flavobacterium are commonly found in the Antarctic environment. Several bacteria belonging to these genera are adapted to extreme conditions and on occasions, some isolates were studied for biotechnological applications [37,38,39]. The impact of penguins on Ardley island soil samples could be confirmed by the presence of a Fusobacterium MAG in the assembled metagenome. This genus has been reported as dominant in the gut microbiome of Adèlie and Gentoo penguins established in this island [40]. In addition, metal resistance traits seem to be widely distributed in bacteria that are dominant in the three samples analyzed. For example, we could detect several traits of metal resistance in environmental bacteria such as Burkholderia. Interestingly, the highest load of MRGs was detected in a MAG belonging to Aeromonadaceae family, present in the sample collected from the ornithogenic soil of Ardley Island. Penguin colonies have been demonstrated to accumulate high concentrations of metals in feces and feathers, thus metal resistance genes could be an adaptive trait to thrive in ornithogenic soils [41,42].
This work constitutes the first approach aimed at determining the mobilization capacity of clinical ARGs in Antarctic soils exposed to different kinds of environmental impact. We could establish the presence of 36 classes of ARGs in a soil sample collected near the septic chamber of BCAA, a higher number than those ARGs identified in the other two metagenomes. We could also determine that some of these resistance genes are associated with mobile genetic elements and are potentially transferable to an exogenous bacterial acceptor.
We could obtain a transconjugant clone of E. coli, using the microbial community contained in a soil sample from BCAA as donor. Although optimal conditions of temperature and incubation time were not adjusted to the Antarctic environmental conditions, we could find that some plasmids could be mobilized to another recipient bacteria. In these community-wide conjugation experiments, we could mobilize together different types of plasmids, ranging from totally self-conjugative, like the IncP1 replicon, to other plasmids containing just an OriT as mobilization element, like ColEI replicon. These multiple-replicon transfer events catalyzed by the presence of IncP1 conjugative plasmids have already been reported in other environments [43,44]. Even more interesting is the fact that IncP1 backbones with high similarity to pKJK5 plasmid, as the one reported in this work, seem to be highly effective thriving in soil microbiomes and mobilizing ARGs [45].
Another remarkable feature of IncP1- and IncQ-like replicons is their broad host range, which enables ARG transfer between phylogenetically distant bacterial species. This may be a key step for ARG dispersal among different environmental compartments [46,47]. Soil microbial communities impacted with sewage or manure, are important hotspots for ARG transfer events, given the ecological connectivity of microbial communities that have evolved and adapted to different environments [48,49]. Additionally, there is evidence for the presence and release of antibiotics in Antarctic research stations sewage, which could further act as a selective pressure for newly resistant clones [35]. If we also consider that the Antarctic environment could be a source of novel beta-lactamase genes [50], genetic exchange, coupled with selection pressures represent a microbiological risk for the selection of these genes within the Antarctic microbiosphere [51].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, M.G., G.A. and S.B.; methodology, M.G.; software, M.G..; validation, G.A., S.B.; formal analysis, M.G., G.A. and S.B.; investigation, M.G., G.A.; resources, S.B.; data curation, M.G.; writing—original draft preparation, M.G.; writing—review and editing, G.A. and S.B.; visualization, M.G.; supervision, G.A., S.B.; project administration, S.B.; funding acquisition, S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Agencia Nacional de Investigación e Innovación, grant number POS_NAC_2016_1_130134.
Data Availability Statement
Raw reads generated for this publication are available at SRA in the Bioproject PRJEB67896, all softwares used are freely available.
Acknowledgments
The authors acknowledge Instituto Antártico Uruguayo (IAU) for the technical and logistical contribution during sample collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- O’Neill, J. Tackling drug-resistant infections globally: final report and recommendations. 2016.
- Aslam, B.; Khurshid, M.; Arshad, M.I.; Muzammil, S.; Rasool, M.; Yasmeen, N.; Baloch, Z. Antibiotic resistance: one health one world outlook. Frontiers in Cellular and Infection Microbiology 2021, 1153. [Google Scholar] [CrossRef] [PubMed]
- D'Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; Golding, G.B.; Poinar, H.N.; Wright, G.D. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.C.; Lee, N.; Aw, T.G. Antibiotic Resistance in Minimally Human-Impacted Environments. Int J Environ Res Public Health 2020, 17, 3939. [Google Scholar] [CrossRef] [PubMed]
- Struve, C.; Krogfelt, K.A. Pathogenic potential of environmental Klebsiella pneumoniae isolates. Environmental microbiology 2004, 6, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H.; Whiteley, M.; Kolter, R.; Bjarnsholt, T. The environmental occurrence of Pseudomonas aeruginosa. APMIS 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Van Elsas, J.D.; Semenov, A.V.; Costa, R.; Trevors, J.T. Survival of Escherichia coli in the environment: fundamental and public health aspects. The ISME journal 2011, 5, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, A.; Kett, S.; Purchase, D.; Marvasi, M. Antibiotic-resistant genes and bacteria as evolving contaminants of emerging concerns (e-cec): is it time to include evolution in risk assessment? Antibiotics, 2021 10, 1066. [CrossRef]
- Delgado-Blas, J.F.; Ovejero, C.M.; David, S.; et al. Population genomics and antimicrobial resistance dynamics of Escherichia coli in wastewater and river environments. Commun Biol, 2021 4, 457. [CrossRef]
- Che, Y.; Yang, Y.; Xu, X.; Břinda, K.; Polz, M.F.; Hanage, W.P.; Zhang, T. Conjugative plasmids interact with insertion sequences to shape the horizontal transfer of antimicrobial resistance genes. Proceedings of the National Academy of Sciences, 2021, 118, e2008731118. [CrossRef]
- Di Cesare, A.; Eckert, E.M.; D'Urso, S.; Bertoni, R.; Gillan, D.C.; Wattiez, R.; Corno, G. Co-occurrence of integrase 1, antibiotic and heavy metal resistance genes in municipal wastewater treatment plants. Water research, 2016, 94, 208-214. [CrossRef]
- Rozwandowicz, M.; Brouwer, S.M.; Fischer, J.; Wagenaar, J.M.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae, Journal of Antimicrobial Chemotherapy, 2018, 73, 1121–1137. [CrossRef]
- Convey, P. Antarctic terrestrial biodiversity in a changing world. Polar Biology, 2011, 34, 1629-1641. [CrossRef]
- Hwengwere, K.; Paramel Nair, H.; Hughes, K.A.; Peck, L.S.; Clark, M.S.; Walker, C.A. Antimicrobial resistance in Antarctica: is it still a pristine environment? Microbiome, 2022, 10, 1-13. [CrossRef]
- Cerdà-Cuéllar, M.; Moré, E.; Ayats, T.; Aguilera, M.; Muñoz-González, S.; Antilles, N.; Ryan, P.G.; González-Solís, J. Do humans spread zoonotic enteric bacteria in Antarctica? Sci Total Environ. 2019, 654, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Bioinformatics. 2010. Available online: http://www.bioinformatics.babraham.ac.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014, 30, 2114-2120. [CrossRef]
- Gurevich, A.; Saveliev, A.; Vyahhi, N.; Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Giménez, M.; Ferrés, I.; Iraola, G. Improved detection and classification of plasmids from circularized and fragmented assemblies. 2022. Biorxiv, 2022-08. [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome, 2018, 6, 1-13. [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome research, 2015, 25, 1043-1055. [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Segata, N. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. elife, 2021, 10, e65088. [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: antibacterial biocide and metal resistance genes database, Nucleic Acids Res., 2014, 42, D737-D743. [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. Journal of molecular biology, 2022, 215, 403-410. [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nature methods, 2012, 9, 357–359. [CrossRef]
- Xi, C.; Lambrecht, M.; Vanderleyden, J.; Michiels, J. Bi-functional gfp-and gusA-containing mini-Tn5 transposon derivatives for combined gene expression and bacterial localization studies. Journal of Microbiological Methods, 2024, 35, 85-92. [CrossRef]
- Simon RU PA, P.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology, 1983, 1, 784-791. [CrossRef]
- Kopmann, C.; Jechalke, S.; Rosendahl, I.; Groeneweg, J.; Krögerrecklenfort, E.; Zimmerling, U.; Weichelt, V.; Siemens, J.; Amelung, W.; Heuer, H.; Smalla, K. Abundance and transferability of antibiotic resistance as related to the fate of sulfadiazine in maize rhizosphere and bulk soil. FEMS Microbiology Ecology, 2013, Volume 83, Issue 1, Pages 125–134. [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Pevzner, P.A. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology, 2012, 19, 455-477. [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics, 2010, 26, 589–595. [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. ; 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 2009, 25, 2078-9. [CrossRef]
- Tin, T.; Fleming, Z.; Hughes, K.; Ainley, D.; Convey, P.; Moreno, C.; Snape, I. Impacts of local human activities on the Antarctic environment. Antarctic Science, 2009, 21, 3-33. [CrossRef]
- Stark, J.S.; Smith, J.; King, C.K.; Lindsay, M.; Stark, S.; Palmer, A.S.; Snape, I.; Bridgen, P.; Riddle, M. Physical, chemical, biological and ecotoxicological properties of wastewater discharged from Davis Station, Antarctica. Cold Regions Science and Technology, 2015, 113, Pages 52-62. [CrossRef]
- Power, M.L.; Samuel, A.; Smith, J.J.; Stark, J.S.; Gillings, M.R.; Gordon, D.M. Escherichia coli out in the cold: Dissemination of human-derived bacteria into the Antarctic microbiome. Environ Pollut. 2016, 215:58-65. [CrossRef]
- Hernandez, F.; Calısto-Ulloa, N.; Gómez-Fuentes, C.; Gómez, M.; Ferrer, J.; González-Rocha, G.; Montory, M. Occurrence of antibiotics and bacterial resistance in wastewater and sea water from the Antarctic. Journal of hazardous materials, 2019, 363, 447-456. [CrossRef]
- Tort, L.F.L.; Iglesias, K.; Bueno, C.; Lizasoain, A.; Salvo, M.; Cristina, J.; Kandratavicius, N.; Pérez, R.; Figueira, M.C.; Bícego, S.; Taniguchi; Venturini, N.; Brugnoli, E.; Colina, R.; Victoria, M. Wastewater contamination in Antarctic melt-water streams evidenced by virological and organic molecular markers. Science of The Total Environment, 2017, 609, 225-231. [CrossRef]
- Paun, V.I.; Banciu, R.M.; Lavin, P.; et al. Antarctic aldehyde dehydrogenase from Flavobacterium PL002 as a potent catalyst for acetaldehyde determination in wine. Sci Rep, 2022, 12, 17301. [CrossRef]
- Herrera, L.M.; Braña, V.; Fraguas, L.F.; Castro-Sowinski, S. Characterization of the cellulase-secretome produced by the Antarctic bacterium Flavobacterium sp. AUG42. Microbiological research, 2019, 223, 13-21. [CrossRef]
- Acevedo-Barrios, R.; Rubiano-Labrador, C.; Navarro-Narvaez, D.; Escobar-Galarza, J.; González, D.; Mira, S.; Miranda-Castro, W. Perchlorate-reducing bacteria from Antarctic marine sediments. Environmental Monitoring and Assessment, 2022, 194, 1-13. [CrossRef]
- Zeng, Y.-X.; Li, H.-R.; Han, W.; Luo, W. Comparison of Gut Microbiota between Gentoo and Adélie Penguins Breeding Sympatrically on Antarctic Ardley Island as Revealed by Fecal DNA Sequencing. Diversity, 2021, 13, 500. [Google Scholar] [CrossRef]
- Romaniuk, K.; Ciok, A.; Decewicz, P.; et al. Insight into heavy metal resistome of soil psychrotolerant bacteria originating from King George Island (Antarctica). Polar Biol, 2018, 41, 1319–1333. [CrossRef]
- Castro, M.F.; Neves, J.C.; Francelino, M.R.; Schaefer CE, G.; Oliveira, T.S. Seabirds enrich Antarctic soil with trace metals in organic fractions. Science of the Total Environment, 2021, 785, 147271. [CrossRef]
- Brown, C.J.; Sen, D.; Yano, H.; Bauer, M.L.; Rogers, L.M.; Van der Auwera, G.A.; Top, E.M. Diverse broad-host-range plasmids from freshwater carry few accessory genes. Applied and environmental microbiology, 2013, 79, 7684-7695. [CrossRef]
- Schlüter, A.; Szczepanowski, R.; Pühler, A.; Top, E.M. Genomics of IncP-1 antibiotic resistance plasmids isolated from wastewater treatment plants provides evidence for a widely accessible drug resistance gene pool. FEMS microbiology reviews, 2007, 31, 449-477. [CrossRef]
- Heuer, H.; Binh, C.T.; Jechalke, S.; Kopmann, C.; Zimmerling, U.; Krögerrecklenfort, E.; Ledger, T.; González, B.; Top, E.; Smalla, K. IncP-1ε Plasmids are Important Vectors of Antibiotic Resistance Genes in Agricultural Systems: Diversification Driven by Class 1 Integron Gene Cassettes. Front Microbiol. 2012, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Klümper, U.; Riber, L.; Dechesne, A. Broad host range plasmids can invade an unexpectedly diverse fraction of a soil bacterial community. ISME J, 2015, 9, 934–945. [CrossRef]
- Smalla, K.; Heuer, H.; Götz, A.; Niemeyer, D.; Krögerrecklenfort, E.; Tietze, E. Exogenous isolation of antibiotic resistance plasmids from piggery manure slurries reveals a high prevalence and diversity of IncQ-like plasmids. Applied and Environmental Microbiology, 2000, 66, 4854-4862. [CrossRef]
- Cohen, O.; Gophna, U.; Pupko, T. The complexity hypothesis revisited: connectivity rather than function constitutes a barrier to horizontal gene transfer. Mol. Biol. Evol. 2011, 28, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Coque, T.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat Rev Microbiol, 2015, 13, 116–123. [CrossRef]
- Azziz, G.; Giménez, M.; Romero, H.; Valdespino-Castillo, P.M.; Falcón, L.I.; Ruberto, L.A.; Batista, S. Detection of presumed genes encoding beta-lactamases by sequence based screening of metagenomes derived from Antarctic microbial mats. Frontiers of Environmental Science & Engineering, 2019, 13, 1-12. [CrossRef]
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L. Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nature microbiology, 2019, 4, 1432-1442. [CrossRef]
Figure 1.
Phylogenetic tree of MAGs detected by MetaWRAP software using 43 marker genes detected with CheckM software. Concatenated genes at protein level were used for phylogenetic tree construction with the Neighbour-Joining algorithm in ape R package. Tip colors represent the taxonomic classification assigned by MetaWRAP software and tip shapes indicate the sample from which the MAG was assembled. MRGs and ARGs for each MAG were represented as light-blue and red bars respectively.
Figure 1.
Phylogenetic tree of MAGs detected by MetaWRAP software using 43 marker genes detected with CheckM software. Concatenated genes at protein level were used for phylogenetic tree construction with the Neighbour-Joining algorithm in ape R package. Tip colors represent the taxonomic classification assigned by MetaWRAP software and tip shapes indicate the sample from which the MAG was assembled. MRGs and ARGs for each MAG were represented as light-blue and red bars respectively.
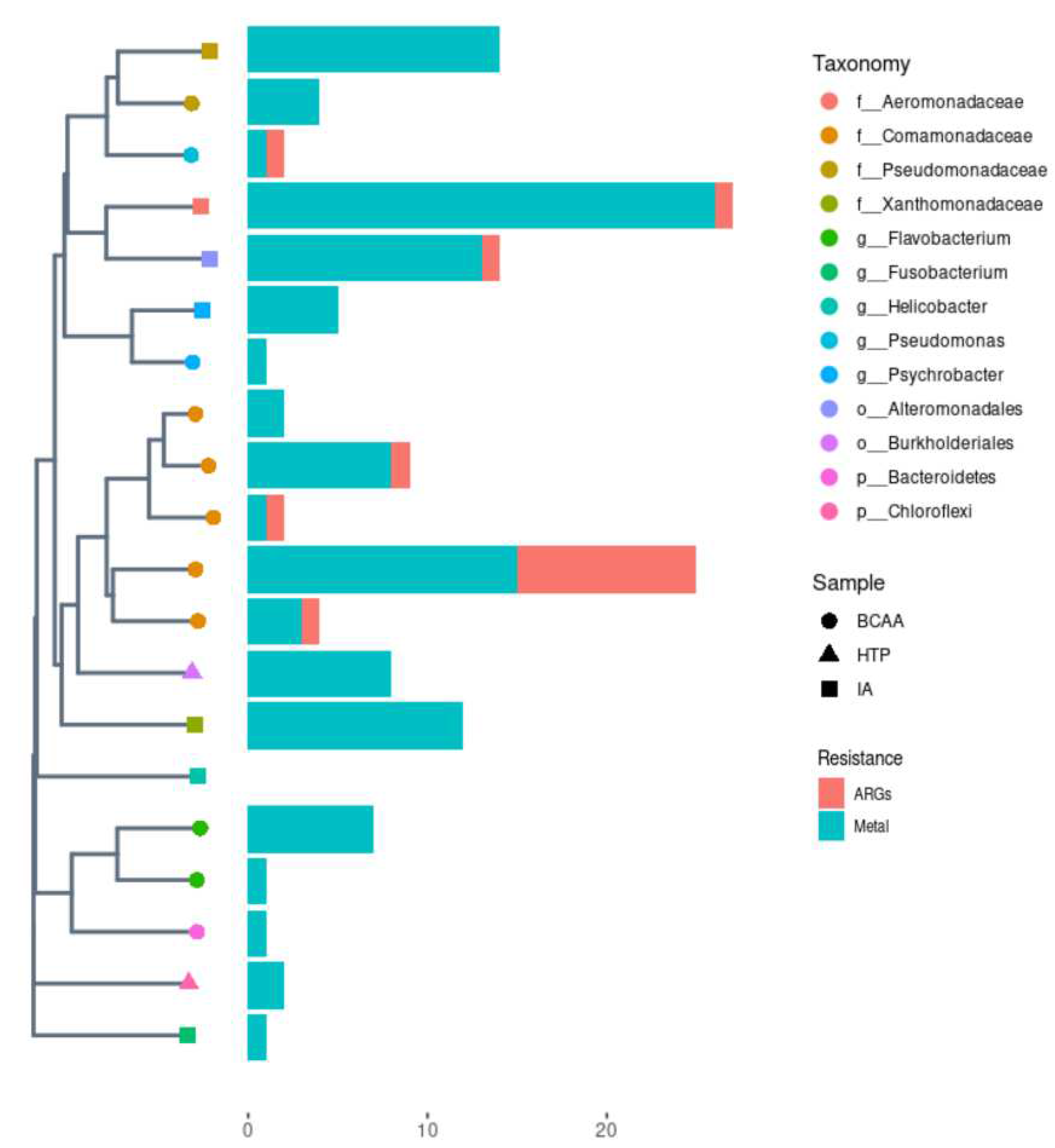
Figure 2.
Tile plot of ARGs genes in the three metagenomes analyzed, ARGs genes encoded in plasmidic contigs, detected by plaSquid analysis, are indicated in navy blue. Colors at the top bar indicate the antibiotic class for each detected ARGs gene.
Figure 2.
Tile plot of ARGs genes in the three metagenomes analyzed, ARGs genes encoded in plasmidic contigs, detected by plaSquid analysis, are indicated in navy blue. Colors at the top bar indicate the antibiotic class for each detected ARGs gene.
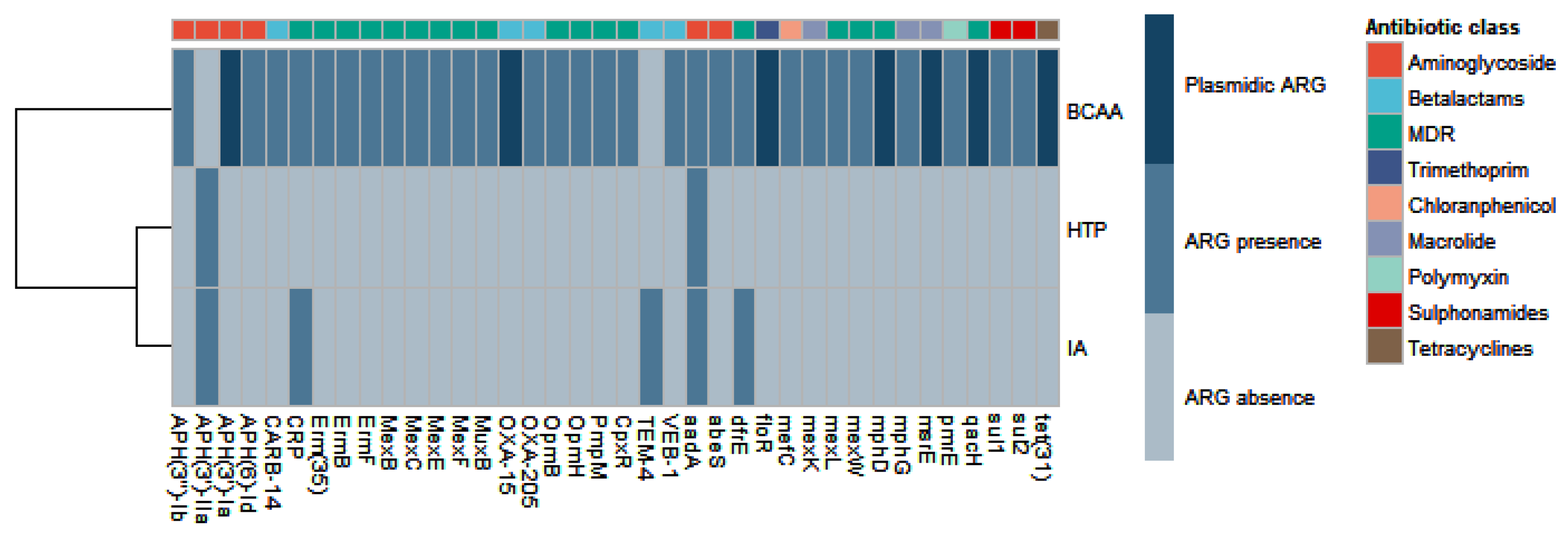
Figure 3.
Diagram of annotated plasmids that could be assembled from sequencing of transconjugant clones obtained by exogenous plasmids isolation assays. A) IncP plasmid contains the modules needed for self-conjugation, while B) IncQ-like plasmid contains a relaxase gene that makes it mobilizable.
Figure 3.
Diagram of annotated plasmids that could be assembled from sequencing of transconjugant clones obtained by exogenous plasmids isolation assays. A) IncP plasmid contains the modules needed for self-conjugation, while B) IncQ-like plasmid contains a relaxase gene that makes it mobilizable.
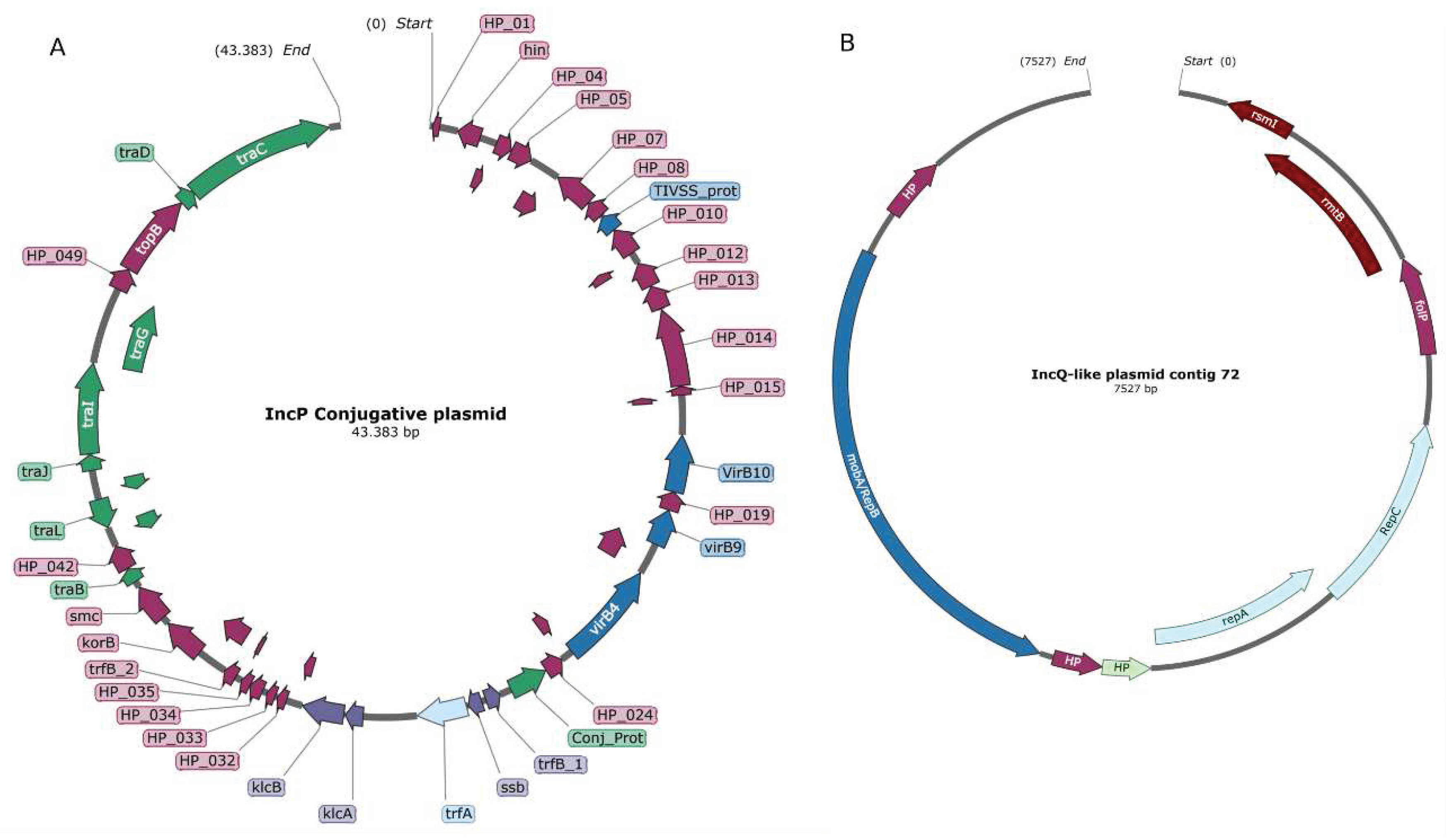

Table 1.
Plasmidic contigs sequenced from receptor strain E. coli DH5ɑGfp+.
| Contig | RIP domain | MOB group | Rep type | Length (bp) | Metagenomic coverage (X) | Resistance genes | Contig | RIP domain | MOB group | Rep type |
| contig-1 | TrfA | MOBP1 | IncP | 43383 | 2.3752 | NA | contig-1 | TrfA | MOBP1 | IncP |
| contig-15 | Rop | NA | NA | 1909 | 0 | NA | contig-15 | Rop | NA | NA |
| contig-4 | RepC | MOBQ | IncQ | 7527 | 7.5709 | rmtB, rsmI | contig-4 | RepC | MOBQ | IncQ |
| contig-13 | NA | NA | ColRNAI | 2121 | 3.7807 | NA | contig-13 | NA | NA | ColRNAI |
| contig-3 | NA | MOBP1 | NA | 11349 | 1.8938 | NA | contig-3 | NA | MOBP1 | NA |
| Contig-12 | NA | NA | NA | 2187 | 0 | qacH, aadA6 | Contig-12 | NA | NA | NA |
| Contig-11 | NA | NA | NA | 2210 | 1.85 | dfrB1, cmlA1 | Contig-11 | NA | NA | NA |
| Contig-31 | NA | NA | NA | 1345 | 1.27 | tetC | Contig-31 | NA | NA | NA |
| Contig-6 | NA | NA | NA | 4703 | 4.64 | sul1 | Contig-6 | NA | NA | NA |
NA Not available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



