
Submitted:
08 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
Methylation of cytosines at CpG sites in DNA, carried out by de novo by DNA methyltransferase Dnmt3a, is a basic epigenetic modification involved in gene regulation and genome stability. Aberrant CpG methylation in gene promoters leads to oncogenesis. In oncogene promoters, CpG sites often colocalize with guanine-rich sequences capable of folding into G-quadruplexes (G4s). Our in vitro study aimed to investigate how parallel G4s formed by a sequence derived from the c-MYC oncogene promoter region affect the activity of the Dnmt3a catalytic domain (Dnmt3a-CD). For this purpose, we designed synthetic oligonucleotide constructs: c-MYC G4-forming oligonucleotide and linear double-stranded DNA containing an embedded stable extrahelical c-MYC G4. The topology and thermal stability of G4 structures in these DNA models were analyzed by physicochemical techniques. We showed that Dnmt3a-CD specifically binds to an oligonucleotide containing c-MYC G4, resulting in inhibition of its methylation activity. c-MYC G4 formation in a double-stranded context significantly reduce Dnmt3a-CD-induced methylation of a CpG site located in close proximity to the quadruplex structure; this effect depends on the distance between the non-canonical structure and the specific CpG site. One would expect DNA hypomethylation near the G4 structure, while regions distant from this non-canonical form would maintain a regular pattern of high methylation levels. We hypothesize that the G4 structure sequesters the Dnmt3a-CD and impedes its proper binding to B-DNA, resulting in hypomethylation and activation of c-MYC transcription.
Keywords:
G-quadruplexes
; c-MYC promoter
; DNA methylation
; Dnmt3a
; DNA-protein interaction
1. Introduction
In addition to the double-stranded B-form, genomic DNA can fold into various sequence-dependent non-canonical structures. Among them, G-quadruplexes (G4) are the most studied and biologically significant form of nucleic acids.
Endogenous G4s are formed via intramolecular interactions of G-rich nucleic acids whose sequence contains at least four tandem G-tracts of consecutive guanosines separated by quasi-random nucleotide residues [1,2]. G4 structures are stabilized by two or three stacked G-tetrads, planar arrangements of four guanines from different G-tracts connected via Hoogsteen base pairing. Nucleotide sequences between G-tracts can form G4 loops that play an important role in determining quadruplex topology and stability. The G4 core is further stabilized by the coordination of guanine O6 with monovalent cations, predominantly K+, in the central cavity [3,4]. In vitro, it was shown that G4 structures can have a wide range of folds, differing in the orientation of G-tracts, adopting parallel, antiparallel, or mixed (3+1) topologies, type of loop and their length, the number of G-tetrads, as well as local structural parameters; some of the G4s are highly dynamic, while others adopt only one conformation. More than 700,000 G4 motifs have been identified in the human genome [5]. Bioinformatics analysis has shown that G4 motifs are frequently clustered in the promoter regions of many oncogenes and genes involved in growth control [6,7,8], replication origins, untranslated exon regions, telomeric DNA and micro(mini)satellite repeats [9,10,11]. G-rich promoter sequences are always present along with their C-rich complements, and G4 formation competes with the maintenance of the Watson–Crick duplex.
Currently, G4s are considered as novel regulatory elements that are involved in key genome functions such as transcription, telomere maintenance, DNA replication and repair [12,13]. In addition, numerous connections of G4 structures to cancer biology have been proposed. Thus, G4 in promoters of oncogenes performs regulatory functions, acting as a transcriptional silencer element that can be targeted with potential anticancer drugs [13,14,15,16].
Much less studied is the role of promoter G4s in another key biological process - methylation of cytosine residues at CpG sites. These basic epigenetic DNA modifications are involved in the regulation of gene expression, maintenance of genomic stability, aging, etc. In mammals, de novo DNA methylation, i.e., the implementation of the methylation pattern (specific alternation of methylated and unmethylated CpG sites) is performed by DNA methyltransferase (MTase) Dnmt3a [7,8]. The maintenance of the methylation pattern during DNA replication is carried out by the Dnmt1 MTase [17]. Despite the primary role of Dnmt1 in maintaining methylation, cooperation between Dnmt3a and Dnmt1 has been observed in some cases, particularly in embryonic stem cells. The promoter regions of actively transcribed genes containing CpG islands are usually unmethylated. In contrast, the promoters of oncogenes, unlike tumor suppressor genes, are hypermethylated. Methylated cytosines are potentially mutagenic due to their spontaneous deamination, leading to C>T point substitutions. Therefore, the presence of 5-methyl-2’-deoxycytosine residues in oncogene promoters can affect the DNA repair pathways and lead to somatic “driver mutations.” Thus, the level of methylation in regulatory regions of the mammalian genome is directly related to the progression of several types of cancers [18]. Evidence of the participation of the promoter G4s in transcriptional regulation, as well as the dependence of the transcription level on the methylation status of CpG islands in oncogene promoters, makes it relevant to analyze the G4 impact on the DNA methylation level. However, there are only a few studies on the involvement of G4 structures in the functioning of the DNA methylation machinery [12,19]. Nevertheless, studies based on whole-genome sequencing data identified a correlation between CpG methylation levels and G4 formation, namely, hypomethylation of the genome in regions enriched in G4 motifs [20,21]. The G4 formation in the promoters of imprinted genes was revealed [22]. Using surface plasmon resonance spectroscopy, the authors reported high binding affinity of recombinant human MTases to quadruplex structures formed in synthetic oligonucleotides containing G4 motifs of the corresponding promoter regions. The binding affinity of the enzyme to G4 has been shown to be comparable to that of other cellular proteins that specifically interact with G4s. Recently, the interplay between G4 formation and DNA methylation levels has been confirmed [23]. Using sequence analysis, it was shown that the majority of G4 motifs found in the human genome are localized in regions of unmethylated CpG islands. The authors also discovered co-localization of G4 motifs and binding sites of the maintenance MTase DNMT1 and proposed a mechanism for protecting CpG islands from methylation by G4 structures that effectively bind and inhibit DNMT1. However, the G4 effect was characterized indirectly by inhibiting methylation of a standard substrate with G4-forming oligonucleotides. G4 impact on de novo methylation by Dnmt3a has not been studied at all.
Given the crosstalk between DNA methylation status and G4 formation in oncogene promoters, the main goal of our work was to evaluate the effect of this non-canonical DNA structure on Dnmt3a function. In our study, we selected the G4 motif of the c-MYC promoter. This gene product is a major oncogenic driver in cancer involved in the regulation of cellular proliferation, differentiation, and apoptosis [24]. Aberrant expression of the c-MYC oncogene (usually c-MYC hyperexpression) leads to malignant transformation of cells. The 27-nt G-rich region of the c-MYC promoter (Pu27) located in the nuclease hypersensitive element (NHE) III1 has the ability to fold into intramolecular parallel G4 structures [6,16,25]. Five tandem G-tracts in its sequence: 5’-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3’, lead to the potential formation of multiple G4s [8]; the resulting G4 structures differ in the positions of the four G-tracts involved in their formation and, consequently, in their conformational features and thermodynamic stability. Nevertheless, a parallel arrangement of G-tracts characteristic of promoter G4 structures are realized in this mixture of G4s, which act as a transcriptional repressor.
In our work, we aimed to investigate how parallel G4s formed by a sequence derived from the c-MYC oncogene promoter region affect the activity of the murine Dnmt3a catalytic domain (Dnmt3a-CD) in a double-stranded context. Using this DNA model, we evaluated the role of the distance between the G4 structure and the analyzed CpG site on its methylation. Furthermore, we examined the influence of isolated c-MYC G4 on the Dnmt3a-CD function.
2. Results
2.1. Synthetic G4-Containing DNA and Control Single- and Double-Stranded Oligonucleotides
In this study, we developed a set of synthetic DNA models harboring G-rich sequence derived from the promoter region of the c-MYC oncogene (c-MYC G4). This set (the c-MYC system in Table 1, Figure 1) includes: (i) а 27-nt FAM-labeled oligonucleotide (27G4) and its mutant form (27G4-mut) containing two G>A substitutions to prevent quadruplex formation for control experiments [26]; (ii) a perfect 58-bp DNA duplex (c-MYC_58Uf/58Df) and a duplex carrying an extrahelical insert of 27-nt in one of the strands, capable of folding into a G4 structure (c-MYC_35Uf/62Df_G4) (Table 1, Figure 1). Oligonucleotides of arbitrary sequences (30X) and a known 30-bp MTase substrate (30Uf/30Df) were used as controls.
In addition, fluorescently labeled DNA duplexes were constructed with an extrahelical G4 motif (GGGT)4 located at varying distances from the CpG site to be methylated: proximal (32Uf/51Df_G4) and distal (76Uf/95Df_G4) (system (GGGG)4 in Table 1 and Figure 1). The (GGGT)4 motif, commonly found in eukaryotic genomes, folds into an exceptionally stable three-tetrad parallel G4 [23]; the duplex flank sequences in 32Uf/51Df_G4 and 76Uf/95Df_G4 are arbitrary. For comparison, we used perfect DNA duplexes 32Uf/32Df and 76Uf/76Df, which cannot form G4 structures (Table 1). Moreover, the same DNA molecules without fluorescent labels were generated for both systems.
To create DNA duplexes with stable G4 structures, we employed an approach that prevent the formation of a competing Watson-Crick double helix [27]. These DNA models were obtained via hybridizing partially complementary strands, one of which contained a G4 motif flanked by oligonucleotide fragments, while the opposite strand lacked a site complementary to the quadruplex-forming sequence.
A significant advantage of engineered DNA duplexes containing an extrahelical G4 structure is the ability to examine the methylation of specific CpG located within the restriction endonuclease site R.Hin6I (see below) in the duplex flanks, and to analyze the G4 impact on the methylation level directly within one duplex system.
2.2. Formation, Topology and Thermal Stability of c-MYC G4 in the Designed DNA Models
We applied a combination of UV spectroscopy and circular dichroism (CD) to characterize the developed models. CD makes it possible to determine the G4 topology, whereas UV spectroscopy allows independent monitoring of the G4 and DNA duplex unfolding and estimation of their melting temperature (Tm). Temperature-dependent UV absorption at 295 nm is known to be a marker of the G4 structure [28]. Unlike the DNA duplex, whose melting is accompanied by a hyperchromic effect (usually at 260 nm), melting of G4 at 295 nm causes a decrease in optical density. Using CD spectroscopy, the Tm values of DNA secondary structures were determined independently. It is important to note that oligonucleotides without fluorescent labels were used for these physicochemical studies. The measurements were carried out in buffer solutions containing either 100 mM KCl, which corresponds to the conditions of Dnmt3a functioning (buffer A), or 2.5 mM KCl (to capture the entire conformational transition region within the available temperature range and compare the Tm values of G4 structures in different DNA models) (buffer B).
UV melting profiles of oligonucleotides 27G4 and 62G4 differing in the length of nucleotide sequences flanking the G4 motif, showed one-step conformational transitions corresponding to G4 unfolding, with an observed hypochromic effect and Tm values of 66 and 46±1°C, respectively (Figure 2A). The decrease in Tm value for 62G4 compared to 27G4 is explained by an unfavorable entropy effect due to the significant elongation of the G4 flanks. The same factor is responsible for reducing the hypochromic effect accompanying G4 unfolding in 62G4 compared to 27G4; an additional 35 nucleotide residues in 62G4 absorb UV light but do not contribute to the G4 unfolding.
These results were supported by CD data (Figure 3b). The CD spectrum of 27G4 at 35°C revealed a typical parallel G4 fold with positive peak at 264 nm and a negative one at 245 nm. CD spectra recorded at different temperatures made it possible to obtain melting profiles for the DNA models studied (Figure 2B, insets). As can be seen, the Tm of 65±1°C derived from CD data for 27G4 is in good agreement with the Tm value obtained from the temperature dependence of UV absorbance at 295 nm (66±1°C). Given that both methods used are highly correlated, we took advantages of each approach to characterize the topology and(or) the thermodynamic stability of G4 structures without duplicating the results of both methods. Thus, the CD data showed that 27G4-mut, containing two G>A substitutions, still retains the G4 structure with a Tm value of 43±1°C (Figure 2B); these measurements were performed in buffer A containing 100 mM KCl and used for the MTase-mediated methylation assay, but not in buffer B with 2.5 mM KCl, which is typically applied for our spectroscopic studies.
The parallel c-MYC G4 structure with Tm= 51±1°C is maintained when the quadruplex motif is flanked by DNA duplexes (Figure 3A, upper panel). According to CD data, the positive band of c-MYC_35U/62D_G4 is shifted to longer wavelengths due to the contribution of duplex flanks. While the G4 domain of c-MYC_35U/62D_G4 was monitored by CD spectroscopy, the thermal stability of duplex regions in the same DNA model was tested by UV melting (Tm=65±1°C) (Figure 3A, lower panel). The observed melting profile at 260 nm demonstrates a hyperchromic effect and reflects the superposition of the melting of two duplex regions flanking the G4 insert (Tm=65±1°C).
Addition of a fully complementary oligonucleotide to a DNA strand containing the G4 motif is known to shift the conformational equilibrium toward the B-form double helix [29]; in this case, the G4 formation becomes thermodynamically unfavorable. Using a combination of CD and UV spectroscopy, it was shown that hybridization of two fully complementary 58-nt DNA strands results in the formation of an extremely stable DNA duplex (c-MYC_58U/58D), and even minor G4 folding is not observed (Figure 3B).
2.3. Dnmt3a-CD Effectively Binds to Oligonucleotide 27fG4
The MTase Dnmt3a includes a C-terminal catalytic domain (Dnmt3a-CD) and an N-terminal regulatory region responsible for targeting chromatin and interactions with other proteins [7,8]. Dnmt3a-CD possesses catalytic activity in the absence of the regulatory region. The active form of Dnmt3a is a tetramer with two active sites, which, in complex with DNA, spans one turn of the double helix [30]. It is assumed that the tetrameric form of Dnmt3a-CD can further oligomerize on DNA [31]. The amino acid sequence of murine Dnmt3a-CD is 98% identical to that of human DNMT3A-CD [32]. The prokaryotic CpG-recognizing monomeric MTase M.SssI was also used as a control; the amino acid sequence of M.SssI contains all ten conserved motifs typical of C5-MTases, directly responsible for the catalytic reaction [33,34].
Here, we examined the binding of Dnmt3a-CD and M.SssI to the G4 structure formed by the purine-rich strand of the promoter region of cancer-associated c-MYC gene (27fG4). Fluorescence polarization was used to quantitatively characterize the parameters of MTases - G4 complexation in solution. We performed direct titration of the fluorescently labeled oligonucleotide 27fG4 and the control perfect DNA duplex 30Uf/30Df with Dnmt3a-CD or M.SssI in the presence of S-adenosyl-L-homocysteine (AdoHcy), an analog of the S-adenosyl-L-methionine (AdoMet) cofactor that facilitates the formation of a specific complex between MTase and DNA. The enzymes-DNA substrate binding curves presented in Figure 4 suggest that the G4-containing oligonucleotide 27fG4 forms a strong complex with Dnmt3a-CD with an equilibrium dissociation constant, Kd, (70 ± 20 nM), which is approximately four times less the Kd value (280 ± 70 nM) for the double-stranded substrate 30Uf/30Df lacking the G4 structure.
2.4. Inhibition of a 30-bp DNA Duplex Methylation in the Presence of 27G4
To determine whether G-quadruplexes could inhibit DNA methylation, the impact of 27G4 on the methylation of 30Uf/30Df was studied. The extent of methylation by Dnmt3a-CD was measured using a procedure similar to that described previously [36]. Specifically, 30Uf/30Df was methylated with Dnmt3a-CD followed by treatment with the endonuclease R.Hin6I, which cleaves only the unmethylated GCGC site overlapping the CpG site. Cleavage products were visualized in a denaturing polyacrylamide gel (Figure 5A), and the extent of methylation was calculated as the ratio of the fluorescence intensity of the 14-nt cleavage product to the total fluorescence intensity of the intact and cleaved DNA.
In the absence of 27G4, the 30Uf/30Df duplex was almost completely methylated (Figure 5B). With increasing 27G4 concentration, the extent of 30Uf/30Df methylation decreased, indicating inhibition of Dnmt3a-CD activity. The IC50 value determined from a plot of percent methylation, R, versus 27G4 concentration (Figure 5B) was 160 ± 10 nM. In contrast, the presence of the non-G4 control oligonucleotide 30X did not result in inhibition of the enzyme activity. The mutated control 27G4-mut turned out to be a significantly weaker MTase inhibitor (IC50 = 1100 ± 200 nM) compared to 27G4, which is consistent with CD data on the extremely low stability of the G4 structure formed by this oligonucleotide. Thus, 27G4 binds tightly to Dnmt3a-CD, resulting in inhibition of Dnmt3a-CD methylation activity by preventing MTase from binding to its regular DNA substrate.
2.5. Effect of c-MYC G4 on Methylation of the c-MYC Promoter Region
Further, we analyzed the impact of G4 formed and stabilized within the double-stranded c-MYC promoter region (c-MYC_35Uf/62Df_G4), on the methylation activity of Dnmt3a-CD. For comparison, G4-free c-MYC_58Uf/58Df, which mimics the native region of the c-MYC promotor, was used (Table 1, Figure 1). The methylation extent of c-MYC_58Uf/58Df and c-MYC_35Uf/62Df_G4 were determined as described above. FAM-labeled DNA cleavage products were analyzed by denaturing polyacrylamide gel (Figure 6A), followed by calculating the extent of methylation (Figure 7).
Figure 6 shows that after the treatment of the perfect c-MYC_58Uf/58Df duplex with Dnmt3a-CD (lane 3), the intensity of the bands corresponding the 11- and 45-nt cleavage products decreased compared to that of c-MYC_58Uf/58Df not treated with Dnmt3a-CD (lane 2). These data indicate that a significant portion of this duplex underwent methylation. In contrary, the quantity of 11-nt and 22-nt cleavage products of the c-MYC G4-containing c-MYC_35Uf/62Df_G4 substrate before and after methylation (lanes 5 and 6) remained almost identical, indicating the low efficiency of methylation of a DNA duplex with an embedded extrahelical G4. Overall, the formation of a G4 in the c-MYC_35Uf/62Df_G4 simulating a fragment of the c-MYC oncogene promoter leads to a 7-fold decrease in methylation efficiency compared to the c-MYC_58Uf/58Df DNA duplex enable to form a c-MYC G4 (Figure 7).
2.6. Influence of the Relative Positions of G4 and CpG Sites within a DNA Duplex on the Methylation Activity of Dnmt3a-CD
To evaluate the role of the distance between G4 and the analyzed CpG site on the functioning of Dnmt3a-CD, two DNA duplexes with an embedded stable parallel G4 structure formed by (GGGT)4 folding were compared. These included the new model duplex 32Uf/51Df_G4 and longer previously studied 76Uf/95Df_G4 [35], which differed in the distance between the CpG site and the G4 structure: 6 and 28 nt, respectively (Table 1 and Figure 1). In both cases, the efficiency of methylation of CpG located within the R.Hin6I site was examined and compared with the efficiency of corresponding control DNA duplexes lacking G4 structures. Figure 6B shows the cleavage product bands (8- and 41-nt) for 32Uf/51Df_G4, as well as for the control 32Uf/32Df before and after methylation. As can be seen, the G4 formation in close proximity (6 nt) to the CpG site reduces the methylation efficiency by approximately 5 times compared to that for a perfect DNA duplex (Figure 6B and Figure 7). In contrast, the hypomethylation effect induced by distal G4 (28 bp) is negligible (Figure 7). Thus, moving away the methylation site from G4 significantly reduces the impact of this non-B-DNA structure on Dnmt3a-CD activity.
2.7. M.SssI-Mediated Methylation of Model DNA with G4 Structure
To address the mechanism of G4 effect on CpG methylation, experiments were carried out using the prokaryotic CpG-recognizing MTase M.SssI and a DNA duplex (32Uf/51Df_G4) containing extrahelical stable G4 as a substrate. It was found that the quadruplex structure does not affect the efficiency of M.SssI-induced methylation (compare the methylation level in 32Uf/51Df_G4 and 32Uf/32Df lacking G4 in Figure 7 and Figure 8, and in a pair of substrates belonging to the c-MYC system: c-MYC_35Uf/62Df_G4 and c-MYC_58Uf/58Df (Figure 7)).
As indicated in Figure 7, even when G4 is formed in close proximity to the CpG site, as in 32Uf/51Df_G4 and c-MYC_35Uf/62Df_G4, the activity of M.SssI is not affected, in contrast to Dnmt3a-CD, for which G4 formation becomes an obstacle to normal functioning.
3. Discussion
Both DNA methylation and G4 structures play multiple roles in cancer biology, DNA replication, and gene regulation. Therefore, the experimental evidences for crosstalk between c-MYC G4s and the functioning of the eukaryotic Dnmt3a presented in our in vitro study are of great importance. To address this issue, we used the catalytic domain of Dnmt3a and designed DNA models containing c-MYC G4: a single-stranded 27-nt oligonucleotide 27G4 mimicking the G4 motif (Pu27) in the c-MYC promoter, and a DNA duplex (c-MYC_35Uf/62Df_G4) bearing an extrahelical Pu27 insert in the center of one of its strands (Figure 1).
Using CD and UV spectroscopy, we revealed that 27G4 forms a stable c-MYC G4 structure with a parallel topology under Dnmt3a-CD operating conditions. The parallel topology and high thermal stability of the G4 structure were maintained when the 27G4 sequence was embedded into a double-stranded c-MYC promoter sequence lacking the fragment complementary to 27G4 (c-MYC_35Uf/62Df_G4) (Figure 3B). This approach allowing stabilization of G4 structure in a duplex context was elaborated to prevent its conformational transition to the more energetically favored B-DNA [27] (Figure 3A). The native c-MYC promoter fragment (c-MYC_58Uf/58Df) adopts a B-form DNA structure under Dnmt3a-CD operating conditions.
We further established that Dnmt3a-CD forms a stable specific complex with 27G4f. Comparison of the Dnmt3a-CD binding affinity to this oligonucleotide and to a known 30-bp DNA substrate 30Uf/30Df revealed a marked preference for the parallel G4 structure formed by 27G4f. This result is in line with the high binding affinity of full-length Dnmt3a and other proteins to G4 structures [22,23,37]. 27G4 was shown to promote significant inhibition of Dnmt3a-CD activity, in contrast to the control oligonucleotide of random sequence, 30X (Figure 5B); these data confirm the binding specificity of Dnmt3a-CD to the quadruplex structure formed in 27G4. The mutant oligonucleotide 27G4-mut was shown to be a weak inhibitor of Dnmt3a-CD activity due the low G4 stability (Figure 2B). Similar effects of specific binding and inhibition by c-MYC oligonucleotide were found for the human maintenance MTase DNMT1[23]. The IC50 value for 27G4 (160 ± 10 nM) is comparable to the IC50 values of known Dnmt3a-CD inhibitors, such as olivomycin A, which binds to the DNA minor groove [38], and the DNA-intercalating curaxin CBL0137 [36].
Inhibition of Dnmt3a-CD induced by 27G4 suggests hypomethylation of the c-MYC oncogene promoter due to c-MYC G4 binding to the enzyme. However, single-stranded oligonucleotides folded into G4 structures are not informative when studying the role of G4 in regions flanking CpG sites in the methylation process. Much more suitable are the combined duplex-quadruplex models, containing a c-MYC G4 structure stabilized in the DNA duplex context (e.g., c-MYC_35Uf/62Df_G4). In addition, the created model made it possible to monitor the methylation of specific CpG sites. It was previously shown that G4 formation in long dsDNA is promoted by conditions of molecular crowding created by PEG [39]. However, the G4 stabilization observed under these conditions occurs only during the process of in vitro transcription or heat denaturation/renaturation and is sequence-dependent.
In the DNA duplex c-MYC_35Uf/62Df_G4, the CpG site, which is the methylation target, is located at a distance of 6 bp from G4 (Table 1). It was shown that the degree of-MYC_35Uf/62Df_G4 methylation, decreased by approximately 7 times compared to the standard perfect DNA duplex c-MYC_58Uf/58Df (Figure 7). This is the first direct evidence that parallel G4 down-regulates Dnmt3a activity.
To answer the question of whether the G4 effect on the methylation level persists when the CpG is moved away the quadruplex structure, we compared the degrees of methylation of 32Uf/51Df_G4 and 76Uf/95Df_G4, containing an embedded extrahelical (GGGT)4 motif, which differed in the distance between G4 and the analyzed CpG site in flanking duplexes (Figure 7). Removal from the G4 structure on 28 bp from the analyzed CpG site (76Uf/95Df_G4) significantly reduces the hypomethylation effect of G4. DNA duplexes 32Uf/51Df_G4 and c-MYC_35Uf/62Df_G4 with parallel G4 insertions within the duplex structure located at approximately the same short distance from the CpG site showed equally high reduction in methylation compared to DNA substrates lacking G4.
Thus, one would expect DNA hypomethylation near c-MYC G4 structure and maintenance of elevated methylation levels in oncogene promoter regions distal to G4. Previous studies examined the impact of the relative position of quadruplex structures and CpG sites in gene promoters on the methylation status of CpG islands based on whole-genome bisulfite sequencing data [23,40]. It was noted that the hypomethylating effect of G4 was inversely proportional to the distance between G4 and CpG, but this analysis did not take into account the activity of DNA methyltransferases [40].
Understanding the molecular-level interactions between G4 structures in the c-MYC promoter region and Dnmt3a-CD remains unclear. High-resolution structural information on the nature of G4 interactions with various cell proteins and their selectivity in recognizing certain G4 structures is extremely limited [5]. Structural studies of Dnmt3a complexes with G4 DNA have not been performed. Two main mechanisms by which various proteins interact and function with intracellular G4s have been discussed [13,41]: (i) G4-unfolding proteins can unfold the G4 structure after binding to it, acting as helicases, and (ii) G4-recruited proteins can bind to G4 structures in specific functional regions of the DNA. We hypothesize that G4 structures may also serve as an obstacle to proper formation of the DNA-protein interface. Obviously, the hypomethylation effect that we discovered in vitro for Dnmt3a-CD is primarily due to the effective binding of MTase to the non-canonical structure in c-MYC_35Uf/62Df_G4. However, it is also necessary to take into account the ability of G4 to sterically interfere with Dnmt3a-mediated methylation of nearby CpG sites, preventing the formation of the correct DNA-protein complex. These assumptions are supported by the clear dependence of methylation efficiency on the distance between G4 and CpG (Figure 7) and by comparative experiments with the prokaryotic MTase M.SssI.
Despite the substantial similarity of the primary structures of M.SssI and Dnmt3a-CD, namely the presence of ten conserved amino acid motifs responsible for the catalytic reaction [7,34], profound differences were revealed in their interaction with G4. In the case of M.SssI, the G4 impact on the methylation of duplex-quadruplex models was negligible (Figure 7). Furthermore, M.SssI exhibits much weaker binding to 27G4f (Figure 4B) or TT(GGGT)4TT oligonucleotide [35] compared to Dnmt3a-CD. The key difference between these two enzymes may be the variable structure and composition of their complexes with DNA substrate [30,42]: firstly, the oligomerization of Dnmt3a-CD leading to the inability to properly bind to the double-stranded region of our model c-MYC_35Uf/62Df_G4, in contrast to the standard binding of monomeric M.SssI; secondly, the specific structure of the DNA-binding cavity in Dnmt3a formed by four monomers of the enzyme. Overall, the ability to tetramerize is a key characteristic that determines the Dnmt3a-CD’s affinity for parallel G4 structures, resulting in the development of a DNA-binding surface that appears to be more specific for G-quadruplex structures compared to monomeric M.SssI.
4. Materials and Methods
4.1. Reagents
All oligonucleotides (Table 1) were commercial products (Eurogene, Russia). Some of the oligonucleotides contained the fluorescent dye 6-carboxyfluorescein (FAM) or tetramethylrhodamine (TAMRA). The concentrations of oligonucleotides were determined spectrophotometrically, as described previously [27]. AdoMet and AdoHcy (Sigma, Germany) were used in the study The following buffer solutions were used: A - 20 mM HEPES-NaOH (pH 7.5), 100 mM KCl, 1 mM EDTA, 1 mM 1,4-dithiothreitol; B - 10 mM Tris-HCl (pH 7.3), 70 mM NaCl, 2.5 mM KCl. G4 structures in oligonucleotides and DNA duplexes were formed by annealing (by heating at 95 °C for 3 min and slowly cooling to 4 °C) in at appropriate buffer; DNA duplexes, including those containing G4 structures, were prepared by annealing complementary (or partly complementary) DNA strands under the same conditions.
4.2. Enzymes
To obtain Dnmt3a-CD), Escherichia coli BL21(DE3) cells were transformed with plasmid pET-28a(+) carrying the gene encoding Dnmt3a-CD with an N-terminal 6 × His tag. Subsequently, Dnmt3a-CD was isolated and purified using metal-affinity chromatography on Co2+-containing TALON® resin (GE Healthcare, USA) [43]. M.SssI was purified as previously described [33]. R.Hin6I is a commercial product (SibEnzyme, Russia). The purity of the protein samples was evaluated by electrophoresis in a 12% SDS-polyacrylamide gel. The protein concentrations were determined by the Bradford assay per protein monomer. Protein preparations were stored at -80°C.
4.3. CD Measurements
CD spectra of oligonucleotides and DNA duplexes that do not contain a fluorescent label were recorded in a quartz cuvette of 10-mm optical path length at room temperature or between 35 to 80 °C with a step of ~5 °C at average heating rate of 0.5 °C/min in buffer B or A on a Chirascan CD spectrometer (Applied Photophysics Ltd., Surrey, UK) equipped with a thermoelectric controller. The DNA concentration (~2 µM concentration per oligonucleotide strand) was chosen to attain an absorption of 0.6–0.8 at 260 nm, which gives an optimum signal-to-noise ratio. The measurements were performed in the 230–320 nm wavelength range at a scanning speed of 30 nm/min and a signal averaging time of 2 s with a constant flow of dry nitrogen. The CD spectra were baseline corrected for signal contributions caused by the buffer and processed with Graph Pad Prism 8.0.1 (Graph Pad Software, San Diego, CA). CD spectra were plotted as molar dichroism per oligonucleotide strand against wavelength. The CD melting profiles revealed the temperature dependence of a CD signal at a specific wavelength.
4.4. UV Spectroscopy Melting of Oligonucleotides Containing G4 Motifs and DNA Duplexes
UV absorbance versus temperature profiles of DNA samples (at ~3 µM concentration per oligonucleotide strand) were recorded in a 600-μL quartz microcuvette with an optical path length of 10 mm on a double-beam Hitachi U-2900 UV/visible spectrophotometer (Japan) equipped with a Hitachi thermoelectric controller. UV absorbance changes were monitored between 20 and 85 °C at 295 or 260 nm at a heating rate of 1.0 °C/min. Tm value defined as the temperature of the mid-point, was estimated from a maximum/minimum value of the first derivative of the fitted curve for data smoothed with the Savitzky–Golay filter.
4.5. Complex Formation of Dnmt3a-CD and M.SssI with DNA
Complex formation was investigated by a fluorescence polarization assay via direct titration of FAM-labeled oligonucleotides or DNA duplexes with Dnmt3a-CD or M.SssI in the presence of AdoHcy as described before [36]. Various amounts of the protein were added to a mixture containing 10 nM DNA substrate in buffer A with 100 µM AdoHcy, and fluorescence polarization (P) was measured using a Cary Eclipse spectrofluorometer (Varian). The parameter P was calculated according to the equation P = (Iv - G·Ih)/(Iv + Ih), where Iv and Ih are vertical and horizontal components of emitted light, respectively, and G is a correction factor. The experimental data are presented as a dependence of P on the total concentration of the MTase. The titration curves of each DNA duplex with the MTase were obtained in at least two technical replicates. Dissociation constants (Kd) of the complexes were obtained by the fitting of the observed P dependences on the protein concentration to a logistic equation in the Graph Pad Prism 8.0.1 (Graph Pad Software, San Diego, CA).
4.6. Methylation of DNA Duplexes
Methylation activity of Dnmt3a-CD and M.SssI was analyzed by protection of methylated DNA from cleavage by R.Hin6I (G↓CGC) [43]. FAM- or TAMRA-labeled DNA duplexes containing or not containing G4 structures (75 nM) were incubated in buffer A for 2 hour at 37 °C in the presence of Dnmt3a-CD (2.5 µM) or M.SssI (1 µM) and 25 µM AdoMet. After methylation, the samples were treated with proteinase K (100 µg/mL) for 30 min at 55 °C to disrupt the Dnmt3a-CD•DNA complex and facilitate the entry of the DNA strands into a polyacrylamide gel. Proteinase K was subsequently inactivated via heating of the samples to 95 ◦C followed by slow cooling to room temperature. Next, the mixtures were incubated with 1 U of R.Hin6I for 1 hour at 37 °C in the presence of 3 mM Mg2+. In control mixtures, cleavage was performed without prior methylation. The mixtures were analyzed by electrophoresis in a 10% polyacrylamide gel with 7 M urea. The resulting gels were visualized using a Typhoon FLA 9500 scanner (GE Healthcare Life Sciences, UK), and the fluorescence intensities of intact DNA and cleavage products were determined. The extent of DNA cleavage (w) was calculated using the GelQuantNET software. The extent of methylation (R) was calculated using the equation [43]:
where w0 is the extent of DNA cleavage before methylation, and wDnmt3a is the extent of DNA cleavage after methylation by Dnmt3a-CD.
When calculating the extent of cleavage, we considered total fluorescence intensities of the initial DNA duplexes, because under denaturing conditions, the strands do not separate completely due to high stability of the extended DNA double helix. The fluorescence intensities of the cleavage products were also summed in the calculations.
4.7. Inhibition of Methylation of a Standard DNA Substrate by a G4 Oligonucleotide
A FAM-labeled DNA duplex (30Uf/30Df) containing a CpG site within the R.Hin6I site (300 nM) was incubated in buffer A for 1 hour at room temperature at various concentrations of the 27G4 oligonucleotide. 27G4-mut and 30X oligonucleotides were used as controls. Subsequently, the 30Uf/30Df was methylated by 2 µM Dnmt3a-CD in the presence of 25 µM AdoMet for 1.5 hours at 37 °C [35]. Then, R.Hin6I cleavage was performed, and the mixtures were analyzed on 20% polyacrylamide gels containing 7 M urea, with the determination of the extent of methylation as described above.
IC50 values were calculated via fitting the dependence of the extent of methylation on the concentration of 27G4 using the Graph Pad Prism 8.0.1 (Graph Pad Software, San Diego, CA).
5. Conclusions
The c-MYC oncogene, overexpressed in the majority of solid tumors, is a well-known as a gene with a G4-forming sequence in its promoter region.
In this study, we demonstrated strong specific binding of Dnmt3a-CD to the oligonucleotide 27G4 folded into a parallel c-MYC G4 structure. The c-MYC G4-enzyme complex inhibits Dnmt3a-CD activity by preventing standard DNA substrate binding to the enzyme. Using a specially designed DNA construct that mimics the G/C-rich promoter region of the c-MYC oncogene and stabilizes c-MYC G4 in a duplex context, we found that the presence of c-MYC G4 reduces the methylation activity of Dnmt3a-CD. The hypomethylation effect depends on the distance between G4 and the methylated CpG site. It can be assumed that the G4 effect on the Dnmt3a-CD function is determined by the sequestration of the enzyme on this non-canonical structure and the disruption of enzyme’s proper binding to the double-stranded region of the c-MYC promoter. In vivo, G4 formation in the c-MYC oncogene promoter may be one of the reasons of DNA hypomethylation resulting in overexpression of c-MYC protein and cancer progression. Overall, the G4 formation in gene promoters partially explains their hypomethylation compared to the rest of the genome [23,40].
Our findings and hypotheses contribute to the understanding of the relationships between the promoter G4′s ability to bind MTases, DNA methylation activity, and functional consequences. Future research should provide clear evidences of in vivo effect of G4 structures on the methylation machinery. It is known that genome G4 structures may interfere with the fidelity of DNA replication and repair. Their involvement in regulation of the DNA methylation revealed in this study suggests a complex network of G4-protein interactions that govern the main biological processes associated with genome stability and oncogenesis.
Author Contributions
Conceptualization, E.S.G. and A.V.S.; methodology, N.G.D., E.A.K. and A.V.S.; investigation, A.V.S., A.I.G, A.G.L., A.S.P. and N.G.D.; data curation, E.S.G. and E.A.K.; writing—original manuscript draft preparation, A.V.S., A.I.G., N.G.D. and E.S.G.; writing—review and editing, E.S.G. and N.G.D.; funding acquisition, E.S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 22-24-00368.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank A. Jeltsch for providing the plasmid pET-28a(+) containing the genes encoding Dnmt3a-CD, and A.M. Arutyunyan for CD measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Todd, A.K.; Johnston, M.; Neidle, S. Highly Prevalent Putative Quadruplex Sequence Motifs in Human DNA. Nucleic Acids Res 2005, 33, 2901–2907. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res 2005, 33, 2908–2916. [Google Scholar] [CrossRef]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and Kinetics of G-Quadruplex Structures. Nucleic Acids Res 2008, 36, 5482–5515. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, V. G4-Quadruplex-Binding Proteins: Review and Insights into Selectivity. Biophysical Reviews 2022 14:3 2022, 14, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Mammalian DNA Methyltransferases: New Discoveries and Open Questions. Biochem Soc Trans 2018, 46, 1191–1202. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. New Concepts in DNA Methylation. Trends Biochem Sci 2014, 39, 310–318. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Hurley, L.H. Structures, Folding Patterns, and Functions of Intramolecular DNA G-Quadruplexes Found in Eukaryotic Promoter Regions. Biochimie 2008, 90, 1149–1171. [Google Scholar] [CrossRef]
- Chen, L.; Dickerhoff, J.; Sakai, S.; Yang, D. DNA G-Quadruplex in Human Telomeres and Oncogene Promoters: Structures, Functions, and Small Molecule Targeting. Acc Chem Res 2022, 55, 2628–2646. [Google Scholar] [CrossRef]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The Regulation and Functions of DNA and RNA G-Quadruplexes. Nature Reviews Molecular Cell Biology 2020 21:8 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Dolinnaya, N.G.; Ogloblina, A.M.; Yakubovskaya, M.G. Structure, Properties, and Biological Relevance of the DNA and RNA G-Quadruplexes: Overview 50 Years after Their Discovery. Biochemistry (Moscow) 2016, 81, 1602–1649. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Chen, H.; Zhang, Q.; Xu, M.; Yuan, G.; Zhou, J. Exploration of the Structure and Recognition of a G-Quadruplex in the Her2 Proto-Oncogene Promoter and Its Transcriptional Regulation. Sci Rep 2019, 9, 3966. [Google Scholar] [CrossRef] [PubMed]
- Ducani, C.; Bernardinelli, G.; Högberg, B.; Keppler, B.K.; Terenzi, A. Interplay of Three G-Quadruplex Units in the KIT Promoter. J Am Chem Soc 2019, 141, 10205–10213. [Google Scholar] [CrossRef]
- Monsen, R.C.; DeLeeuw, L.W.; Dean, W.L.; Gray, R.D.; Chakravarthy, S.; Hopkins, J.B.; Chaires, J.B.; Trent, J.O. Long Promoter Sequences Form Higher-Order G-Quadruplexes: An Integrative Structural Biology Study of c-Myc, k-Ras and c-Kit Promoter Sequences. Nucleic Acids Res 2022, 50, 4127–4147. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and Function of Mammalian DNA Methyltransferases. ChemBioChem 2011, 12, 206–222. [Google Scholar] [CrossRef]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark Insights 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Varizhuk, A.; Isaakova, E.; Pozmogova, G. DNA G-Quadruplexes (G4s) Modulate Epigenetic (Re)Programming and Chromatin Remodeling. BioEssays 2019, 41, 1900091. [Google Scholar] [CrossRef]
- Halder, R.; Halder, K.; Sharma, P.; Garg, G.; Sengupta, S.; Chowdhury, S. Guanine Quadruplex DNA Structure Restricts Methylation of CpG Dinucleotides Genome-Wide. Mol Biosyst 2010, 6, 2439–2447. [Google Scholar] [CrossRef]
- Rauchhaus, J.; Robinson, J.; Monti, L.; Di Antonio, M. G-Quadruplexes Mark Sites of Methylation Instability Associated with Ageing and Cancer. Genes (Basel) 2022, 13, 1665. [Google Scholar] [CrossRef] [PubMed]
- Cree, S.L.; Fredericks, R.; Miller, A.; Pearce, F.G.; Filichev, V.; Fee, C.; Kennedy, M.A. DNA G-Quadruplexes Show Strong Interaction with DNA Methyltransferases in Vitro. FEBS Lett 2016, 590, 2870–2883. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.-Q.; Ghanbarian, A.T.; Spiegel, J.; Martínez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hänsel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-Quadruplex Structures Mold the DNA Methylome. Nat Struct Mol Biol 2018, 25, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I.; Alves, J. The C-myc Promoter: Still MysterY and Challenge. Adv Cancer Res 2008, 99, 113–333. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.; Bhattacharya, S.; Dash, J.; Bhattacharya, S. Recent Update on Targeting c-MYC G-Quadruplexes by Small Molecules for Anticancer Therapeutics. J Med Chem 2021, 64, 42–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Hurley, L. Structure of the Biologically Relevant G-Quadruplex in The c-MYC Promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A. V.; Monakhova, M. V.; Ogloblina, A.M.; Andreeva, N.A.; Laptev, G.Y.; Polshakov, V.I.; Gromova, E.S.; Zvereva, M.I.; Yakubovskaya, M.G.; Oretskaya, T.S.; et al. Responses of DNA Mismatch Repair Proteins to a Stable G-Quadruplex Embedded into a DNA Duplex Structure. Int J Mol Sci 2020, 21, 8773. [Google Scholar] [CrossRef] [PubMed]
- Mergny, J.L.; Lacroix, L. UV Melting of G-Quadruplexes. Curr Protoc Nucleic Acid Chem 2009, 37, 17.1.1–17.1.15. [Google Scholar] [CrossRef] [PubMed]
- Sekibo, D.A.T.; Fox, K.R. The Effects of DNA Supercoiling on G-Quadruplex Formation. Nucleic Acids Res 2017, 45, 12069–12079. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural Basis for DNMT3A-Mediated de Novo DNA Methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Rajavelu, A.; Anspach, N.; Urbanke, C.; Jankevicius, G.; Ragozin, S.; Nellen, W.; Jeltsch, A. Oligomerization and Binding of the Dnmt3a DNA Methyltransferase to Parallel DNA Molecules: Heterochromatic Localization and Role of Dnmt3L. Journal of Biological Chemistry 2011, 286, 24200–24207. [Google Scholar] [CrossRef] [PubMed]
- Chédin, F. The DNMT3 Family of Mammalian De Novo DNA Methyltransferases. Prog Mol Biol Transl Sci 2011, 101, 255–285. [Google Scholar] [CrossRef] [PubMed]
- Darii, M. V; Cherepanova, N.A.; Subach, O.M.; Kirsanova, O. V; Raskó, T.; Ślaska-Kiss, K.; Kiss, A.; Deville-Bonne, D.; Reboud-Ravaux, M.; Gromova, E.S. Mutational Analysis of the CG Recognizing DNA Methyltransferase SssI: Insight into Enzyme–DNA Interactions. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2009, 1794, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Pósfai, J.; Bhagwat, A.S.; Pósfai, G.; Roberts, R.J. Predictive Motifs Derived from Cytosine Methyltransferases. Nucleic Acids Res 1989, 17, 2421–2435. [Google Scholar] [CrossRef] [PubMed]
- Loiko, A.G.; Sergeev, A. V.; Genatullina, A.I.; Monakhova, M. V.; Kubareva, E.A.; Dolinnaya, N.G.; Gromova, E.S. Impact of G-Quadruplex Structures on Methylation of Model Substrates by DNA Methyltransferase Dnmt3a. Int J Mol Sci 2022, 23, 10226. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, A.; Vorobyov, A.; Yakubovskaya, M.; Kirsanova, O.; Gromova, E.S. Novel Anticancer Drug Curaxin CBL0137 Impairs DNA Methylation by Eukaryotic DNA Methyltransferase Dnmt3a. Bioorg Med Chem Lett 2020, 30, 127296. [Google Scholar] [CrossRef]
- Brázda, V.; Hároníková, L.; Liao, J.C.C.; Fojta, M. DNA and RNA Quadruplex-Binding Proteins. Int J Mol Sci 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, A. V.; Tevyashova, A.N.; Vorobyov, A.P.; Gromova, E.S. The Effect of Antitumor Antibiotic Olivomycin A and Its New Semi-Synthetic Derivative Olivamide on the Activity of Murine DNA Methyltransferase Dnmt3a. Biochemistry (Moscow) 2019, 84, 62–70. [Google Scholar] [CrossRef]
- Zheng, K. wei; Chen, Z.; Hao, Y. hua; Tan, Z. Molecular Crowding Creates an Essential Environment for the Formation of Stable G-Quadruplexes in Long Double-Stranded DNA. Nucleic Acids Res 2010, 38, 327–338. [Google Scholar] [CrossRef]
- Jara-Espejo, M.; Peres Line, S.R. DNA G-Quadruplex Stability, Position and Chromatin Accessibility Are Associated with CpG Island Methylation. FEBS J 2020, 287, 483–495. [Google Scholar] [CrossRef]
- Dai, Y.; Teng, X.; Zhang, Q.; Hou, H.; Li, J. Advances and Challenges in Identifying and Characterizing G-Quadruplex–Protein Interactions. Trends Biochem Sci 2023, 48, 894–909. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, M.; Czapinska, H.; Bochtler, M. CpG Underrepresentation and the Bacterial CpG-Specific DNA Methyltransferase M.Mpel. Proceedings of the National Academy of Sciences 2013, 110, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, A. V.; Kirsanova, O. V.; Loiko, A.G.; Nomerotskaya, E.I.; Gromova, E.S. Detection of DNA Methylation by Dnmt3a Methyltransferase Using Methyl-Dependent Restriction Endonucleases. Mol Biol 2018, 52, 272–278. [Google Scholar] [CrossRef]
Figure 1.
DNA models: c-MYC system (containing the c-MYC promoter sequence) and (GGGT)4 system. CpG sites (in red) are underlined; TAMRA and FAM (in yellow) indicate fluorophores; the R.Hin6I site is in red and arrows indicate cleavage site; G4 motifs are highlighted in purple.
Figure 1.
DNA models: c-MYC system (containing the c-MYC promoter sequence) and (GGGT)4 system. CpG sites (in red) are underlined; TAMRA and FAM (in yellow) indicate fluorophores; the R.Hin6I site is in red and arrows indicate cleavage site; G4 motifs are highlighted in purple.

Figure 2.
Thermal stability and topology of G4 structures formed by oligonucleotides 27G4, 27G4-mut and 62G4. Temperature-dependent UV absorption measured at 295 nm (~3 µM oligonucleotide strand concentration) in buffer B (A). CD spectra recorded at increasing temperature, starting from 35 °C (upper curve) with a step of 5 °C, are shown by multi-colored lines. (Insets) CD-monitored melting profiles at the wavelength corresponding to the positive maximum of the CD spectrum (~2 µM oligonucleotide strand concentration); 27G4 was measured in buffer B, 27G4-mut – in buffer A (B).
Figure 2.
Thermal stability and topology of G4 structures formed by oligonucleotides 27G4, 27G4-mut and 62G4. Temperature-dependent UV absorption measured at 295 nm (~3 µM oligonucleotide strand concentration) in buffer B (A). CD spectra recorded at increasing temperature, starting from 35 °C (upper curve) with a step of 5 °C, are shown by multi-colored lines. (Insets) CD-monitored melting profiles at the wavelength corresponding to the positive maximum of the CD spectrum (~2 µM oligonucleotide strand concentration); 27G4 was measured in buffer B, 27G4-mut – in buffer A (B).
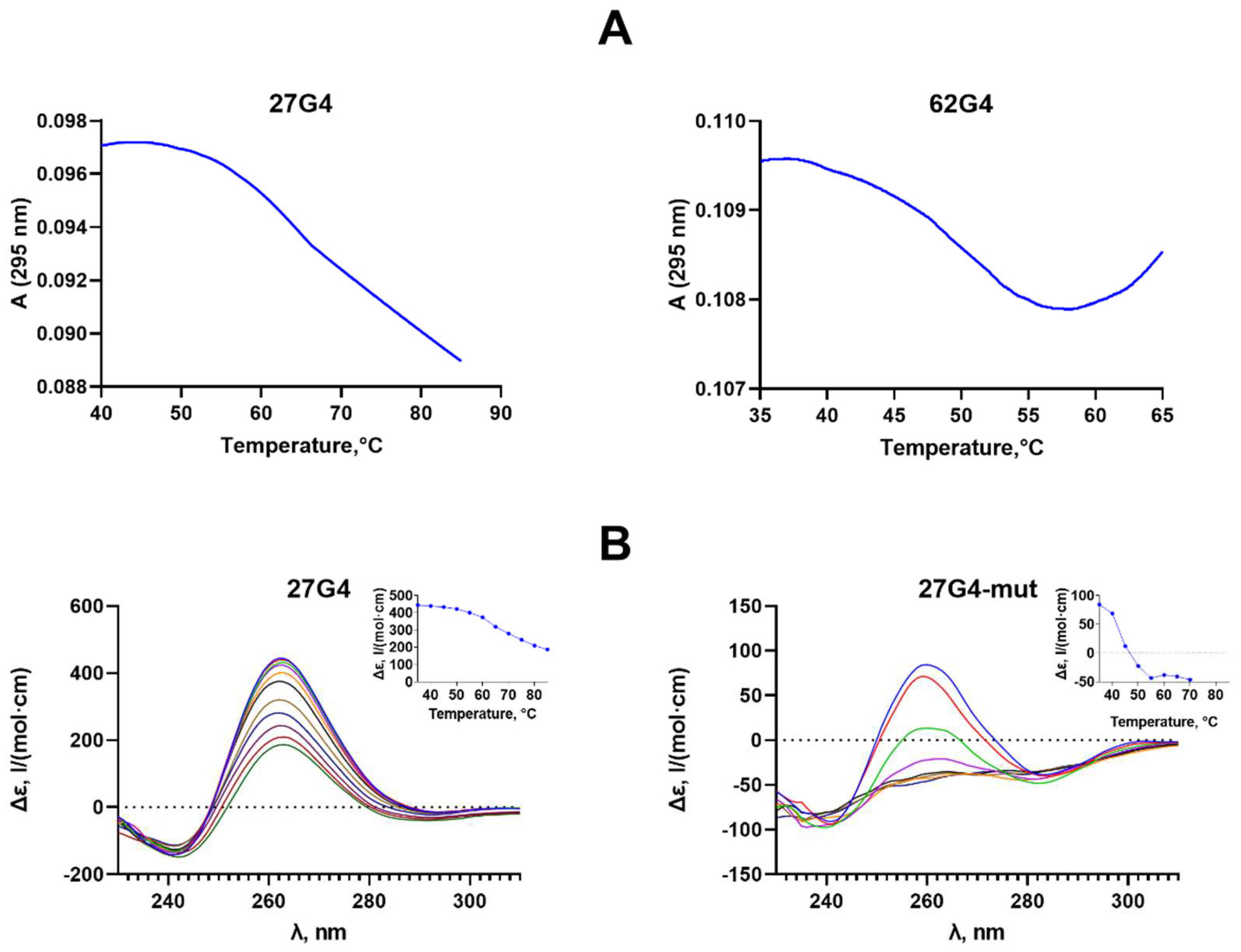
Figure 3.
Thermodynamic stability and topology of c-MYC_35U/62D_G4 containing the c-MYC G4 structure stabilized in a duplex surrounding (A) and a control perfect duplex c-MYC_58U/58D derived from the c-MYC promoter region (B). Upper panel: CD spectra recorded at different temperatures. (Inserts) CD-monitored melting profiles at the wavelength corresponding to the positive maximum of the CD spectrum (~2 µM oligonucleotide strand concentration). Lower panel: temperature dependence of UV absorption at 260 nm. All measurements were carried out in buffer B.
Figure 3.
Thermodynamic stability and topology of c-MYC_35U/62D_G4 containing the c-MYC G4 structure stabilized in a duplex surrounding (A) and a control perfect duplex c-MYC_58U/58D derived from the c-MYC promoter region (B). Upper panel: CD spectra recorded at different temperatures. (Inserts) CD-monitored melting profiles at the wavelength corresponding to the positive maximum of the CD spectrum (~2 µM oligonucleotide strand concentration). Lower panel: temperature dependence of UV absorption at 260 nm. All measurements were carried out in buffer B.
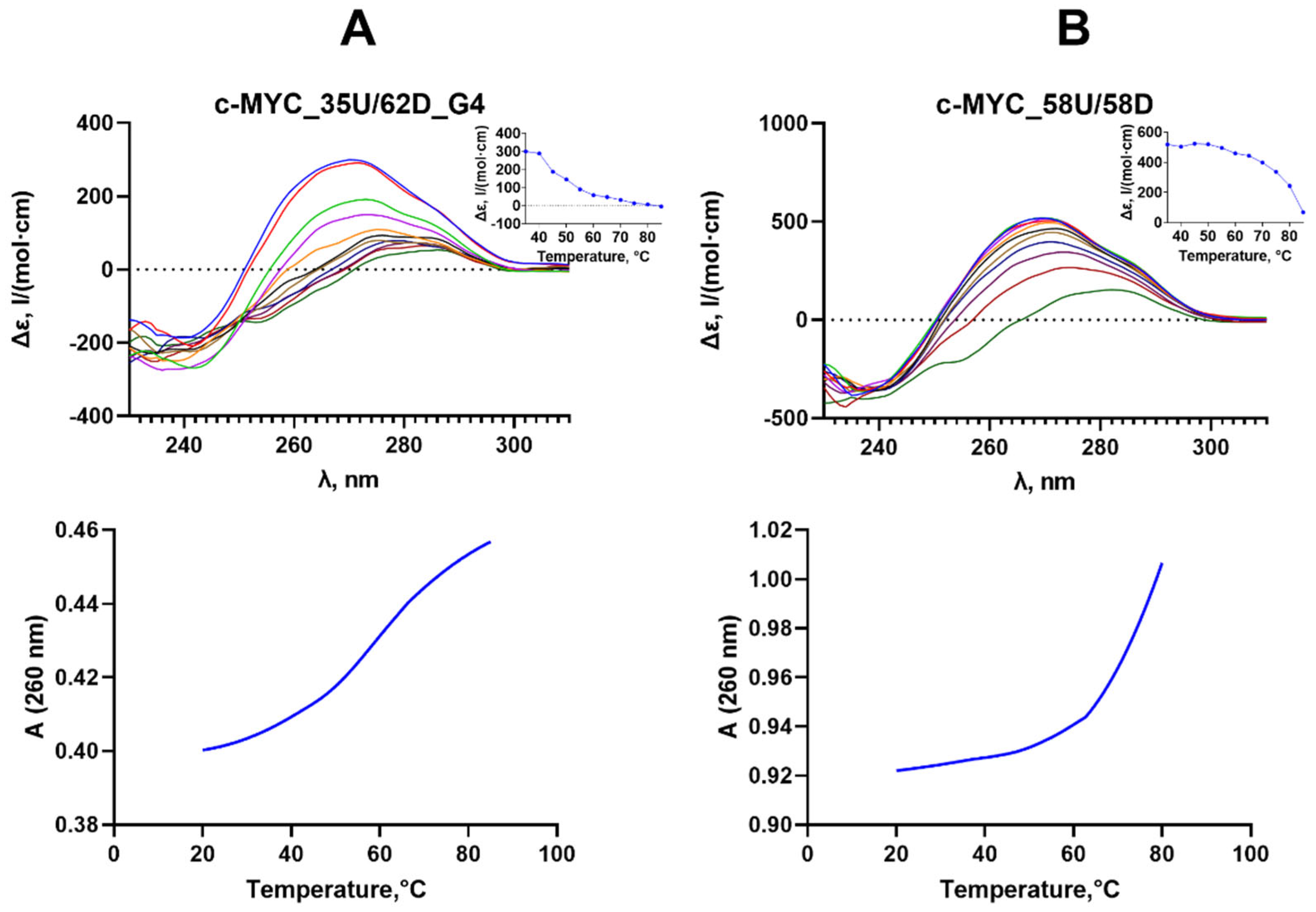
Figure 4.
Binding curves of FAM-tagged 27fG4 and 30Uf/30Df to Dnmt3a-CD (A) and M.SssI (B) derived from fluorescence polarization data.
Figure 4.
Binding curves of FAM-tagged 27fG4 and 30Uf/30Df to Dnmt3a-CD (A) and M.SssI (B) derived from fluorescence polarization data.
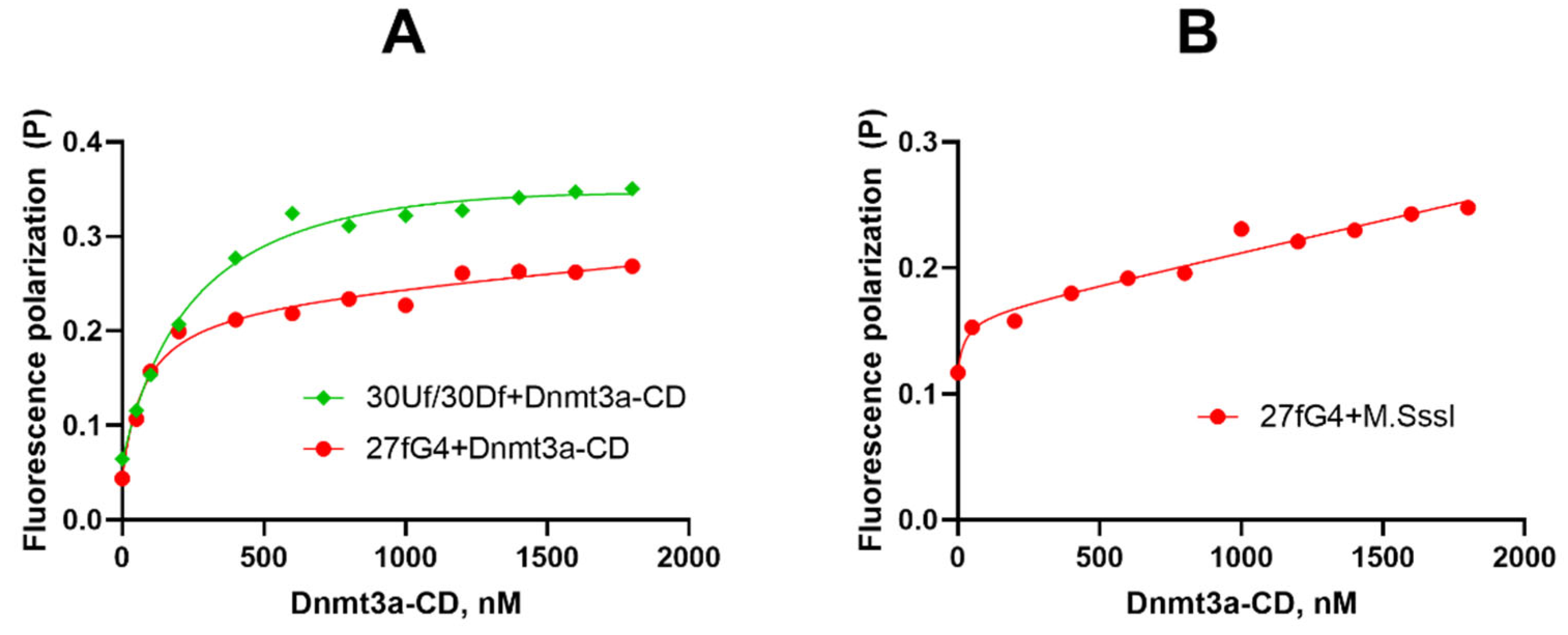
Figure 5.
Inhibition of Dnmt3a-CD-mediated methylation of DNA duplex 30Uf/30Df (300 nM) by the G4-forming oligonucleotide 27G4. The products of 30Uf/30Df cleavage induced by the R.Hin6I after its methylation with 2 µM Dnmt3a-CD in buffer A in the presence of 25 µM AdoMet and 27G4 (27G4 concentrations indicated above the gel lanes) were analyzed on 20% polyacrylamide gel containing 7 M urea (A). Methylation efficiency is plotted against the concentration of 27G4 or controls: 30X lacking G4 and 27G4-mut. Error bars represent SEM from at least three independent experiments (B).
Figure 5.
Inhibition of Dnmt3a-CD-mediated methylation of DNA duplex 30Uf/30Df (300 nM) by the G4-forming oligonucleotide 27G4. The products of 30Uf/30Df cleavage induced by the R.Hin6I after its methylation with 2 µM Dnmt3a-CD in buffer A in the presence of 25 µM AdoMet and 27G4 (27G4 concentrations indicated above the gel lanes) were analyzed on 20% polyacrylamide gel containing 7 M urea (A). Methylation efficiency is plotted against the concentration of 27G4 or controls: 30X lacking G4 and 27G4-mut. Error bars represent SEM from at least three independent experiments (B).
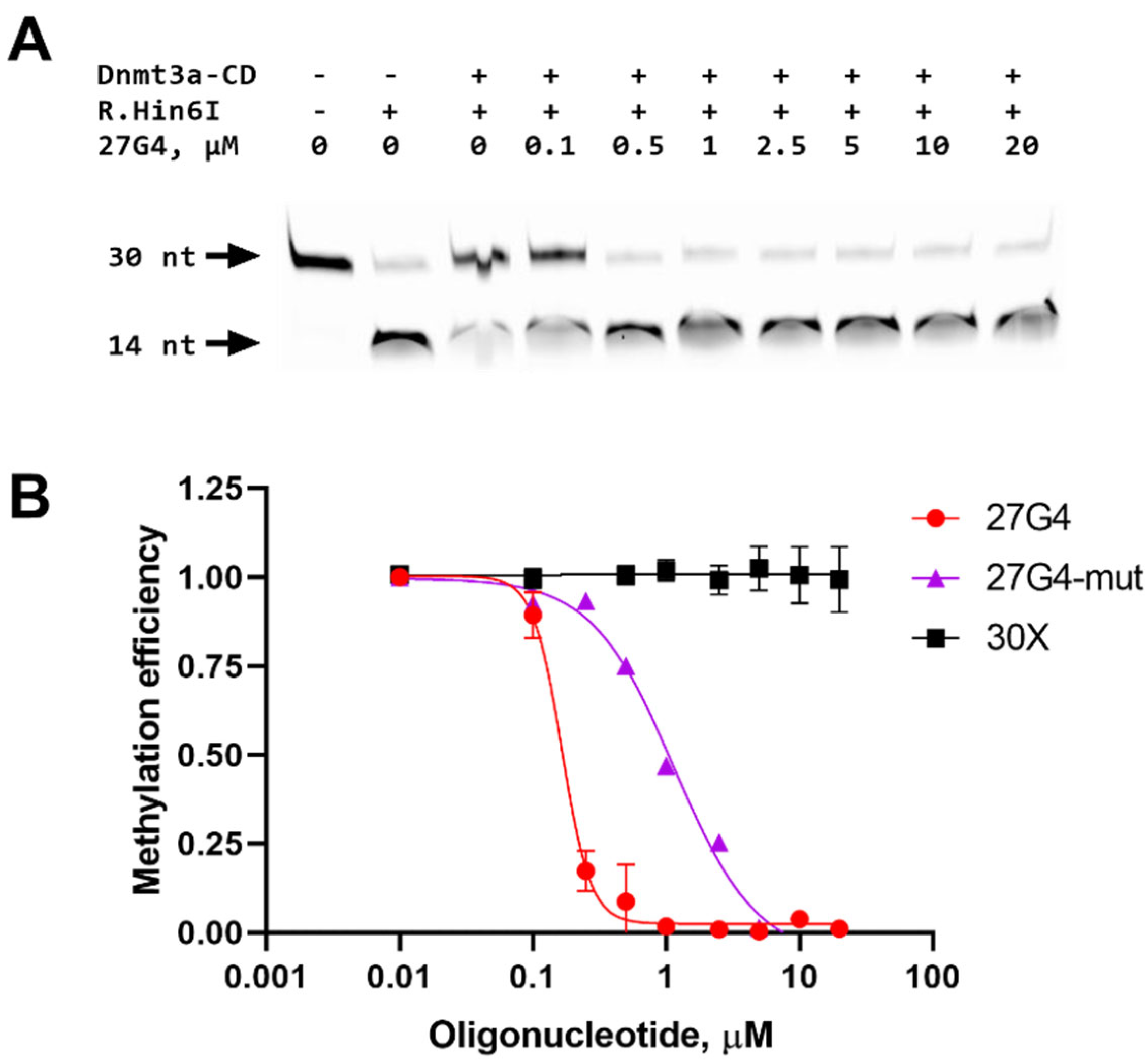
Figure 6.
Effect of the G4 structure in c-MYC_35Uf/62Df_G4 (A) and in (GGGT)4-containing 32Uf/51Df_G4 (B) on their methylation with Dnmt3a-CD in comparison with the control c-MYC_58Uf/58Df (A) and 32Uf/32Df (B). The products of DNA duplex (75 nM) cleavage by R.Hin6I after methylation with Dnmt3a-CD (2.5 µM) were separated on a 10% polyacrylamide gel containing 7 M urea. DNA duplexes that do not dissociate during gel-electrophoresis are marked with an asterisk.
Figure 6.
Effect of the G4 structure in c-MYC_35Uf/62Df_G4 (A) and in (GGGT)4-containing 32Uf/51Df_G4 (B) on their methylation with Dnmt3a-CD in comparison with the control c-MYC_58Uf/58Df (A) and 32Uf/32Df (B). The products of DNA duplex (75 nM) cleavage by R.Hin6I after methylation with Dnmt3a-CD (2.5 µM) were separated on a 10% polyacrylamide gel containing 7 M urea. DNA duplexes that do not dissociate during gel-electrophoresis are marked with an asterisk.
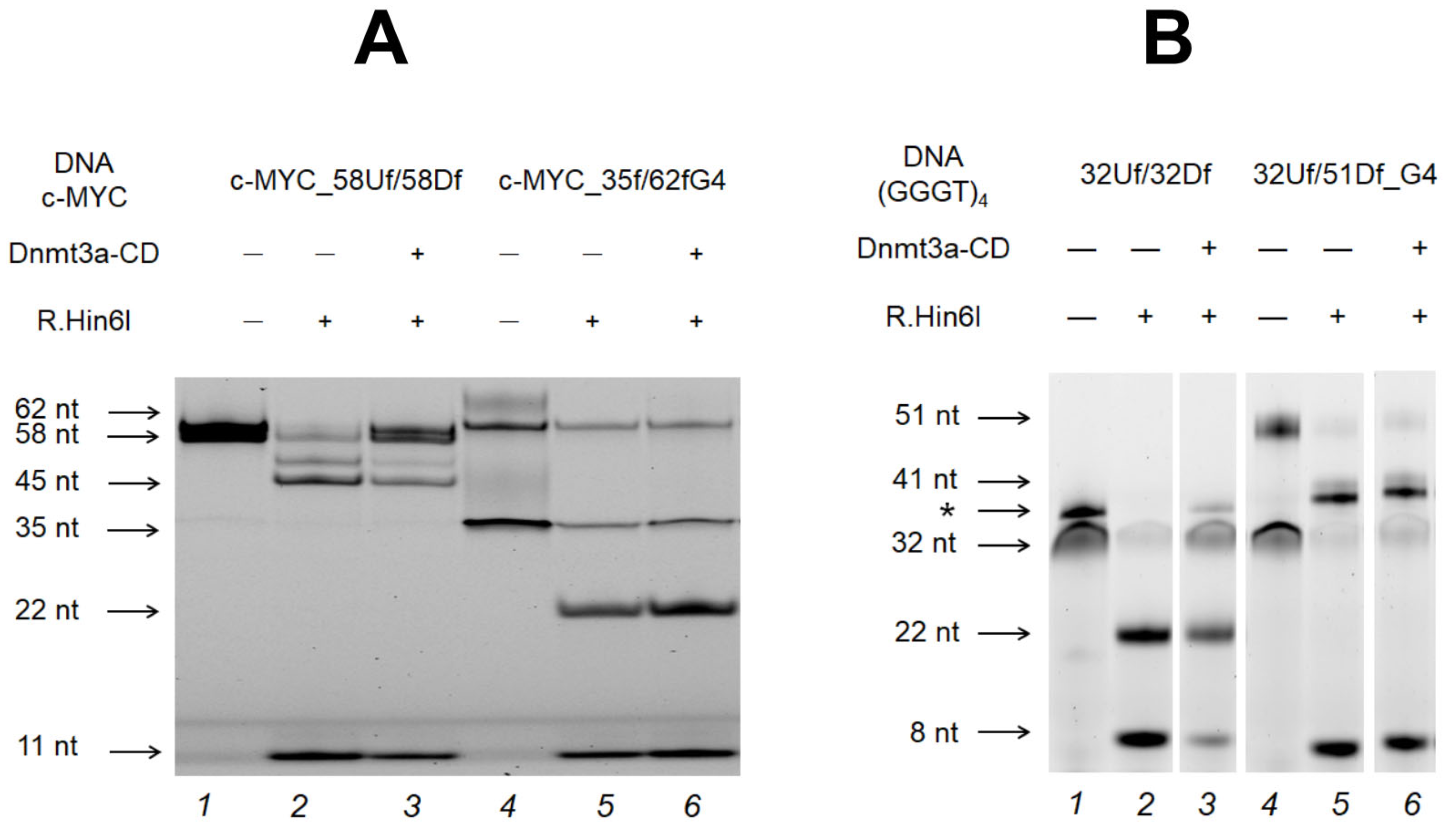
Figure 7.
Extent of methylation of model DNA duplexes (75 nM) with Dnmt3a-CD (2.5 µM) and M.SssI (1 µM). Error bars represent the SDs derived from at least three independent experiments. * Data from [35].
Figure 7.
Extent of methylation of model DNA duplexes (75 nM) with Dnmt3a-CD (2.5 µM) and M.SssI (1 µM). Error bars represent the SDs derived from at least three independent experiments. * Data from [35].
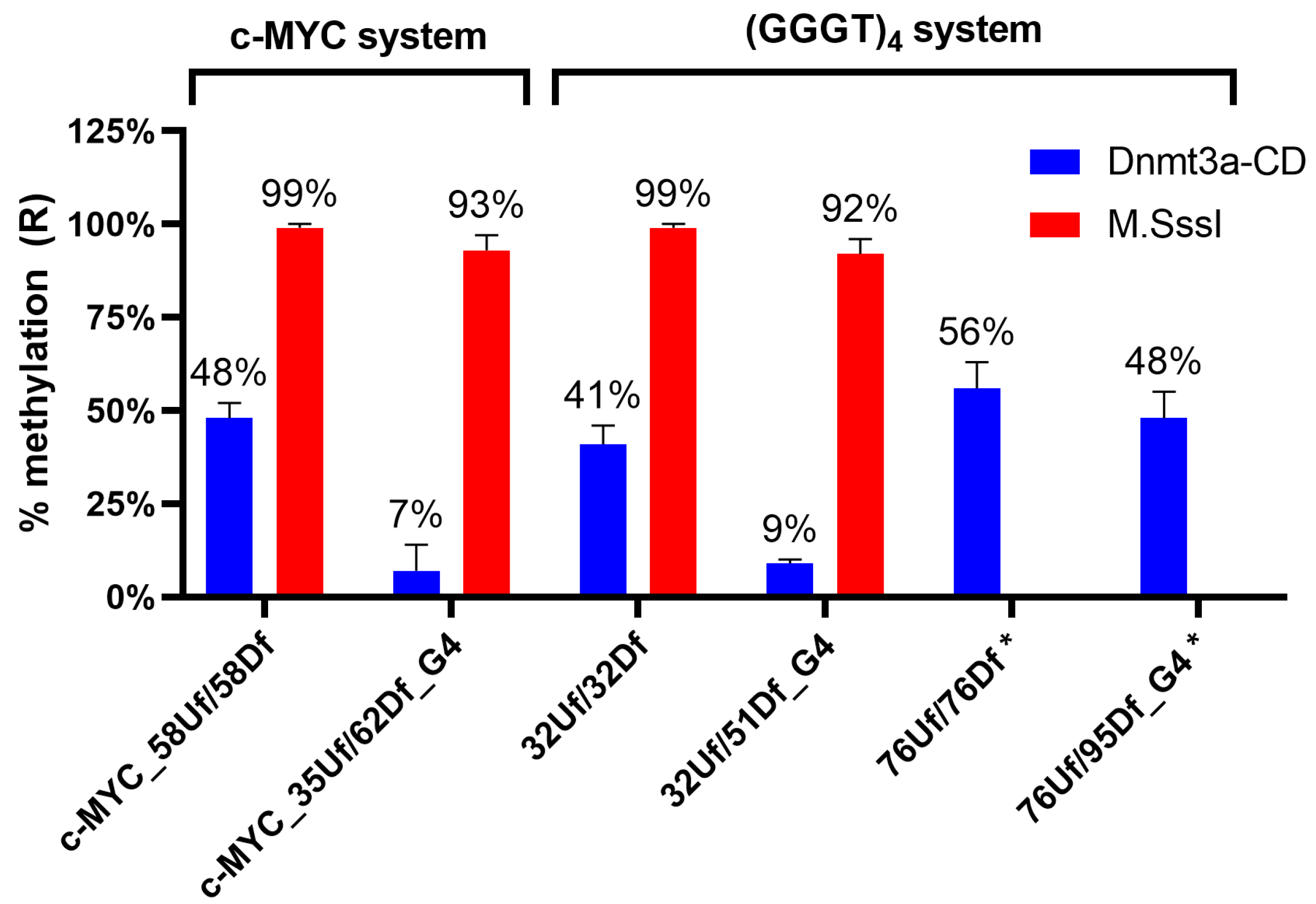
Figure 8.
Effect of G4 formation in 32Uf/51Df_G4 on M.SssI-mediated methylation compared to the control 32Uf/32Df. The cleavage products of the DNA duplex (75 nM) by R.Hin6I after methylation with Dnmt3a-CD (1.0 µM) were separated on a 10% polyacrylamide gel containing 7 M urea. DNA duplexes that do not dissociate during gel-electrophoresis are marked with an asterisk.
Figure 8.
Effect of G4 formation in 32Uf/51Df_G4 on M.SssI-mediated methylation compared to the control 32Uf/32Df. The cleavage products of the DNA duplex (75 nM) by R.Hin6I after methylation with Dnmt3a-CD (1.0 µM) were separated on a 10% polyacrylamide gel containing 7 M urea. DNA duplexes that do not dissociate during gel-electrophoresis are marked with an asterisk.
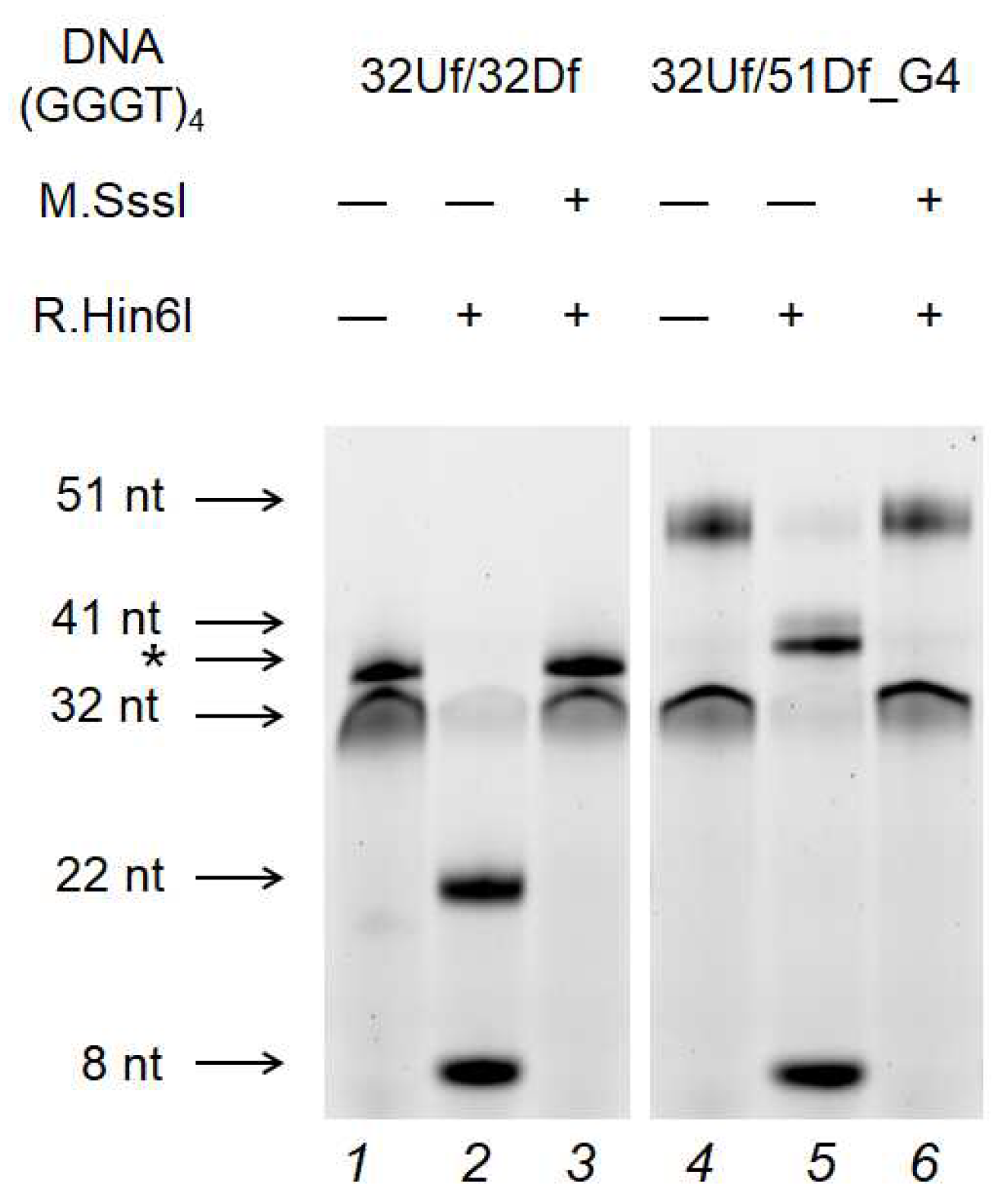

Table 1.
Designation and sequence of oligonucleotides and DNA duplexes used in this work.
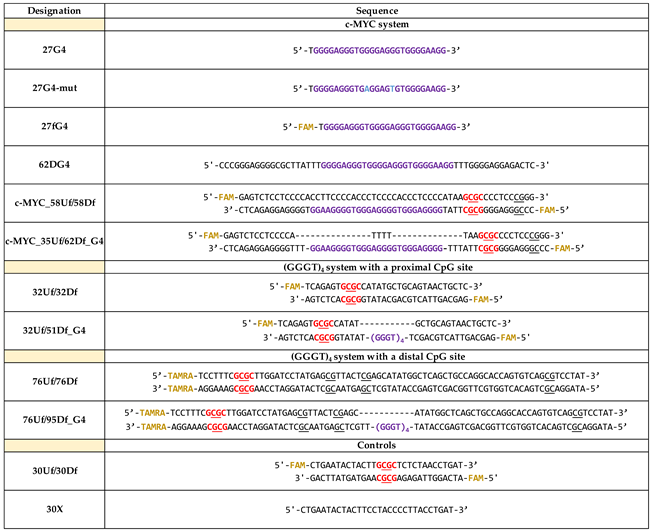
* DNA model designations included: oligonucleotide length, strand positions (indicated as “upper” (U) or “downstream” (D)), the presence of a fluorescent label (f), 5-carboxytetramethylrhodamine (TAMRA) or 6-carboxyfluorescein (FAM), and embedded G4 motif (G4). The CpG sites are underlined; GCGC Hin6I restriction sites are highlighted in red. FAM and TAMRA fluorophores are highlighted in yellow, G4 motifs — in purple, and substituted nucleotide residues — in blue.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



