
Submitted:
08 December 2023
Posted:
11 December 2023
You are already at the latest version
Abstract
Focal adhesions (FAs) play a crucial role in cell spreading and adhesion, and their autophagic degradation is an emerging area of interest. Our study investigates the role of Thrombospondin Type 1 Domain-Containing Protein 1 (THSD1) in regulating autophagy and FA stability in brain endothelial cells, shedding light on its potential implications for cerebrovascular diseases. Our research reveals a physical interaction between THSD1 and FAs. Depletion of THSD1 significantly reduces FA numbers, impairing cell spreading and adhesion, in an autophagy-dependent manner. Intriguingly, THSD1-mediated autophagy activation operates independently of changes in mTOR and AMPK activation, implying that THSD1 primarily governs FA dynamics rather than serving as a global regulator of nutrient and energy status. Interestingly, THSD1 negatively regulates Beclin 1, a central autophagy regulator, at FAs through interactions with focal adhesion kinase (FAK). The loss of THSD1 diminishes FAK activity, consequently relieving its inhibitory phosphorylation on Beclin 1. This, in turn, promotes the complex formation between Beclin 1 and ATG14, a critical event for the activation of the autophagy cascade. In summary, our findings identify THSD1 as a novel regulator of autophagy that degrades FAs in brain endothelial cells. This underscores the distinctive nature of THSD1-mediated, cargo-directed autophagy and its potential relevance to vascular diseases due to the loss of endothelial FAs. Investigating the underlying mechanisms of THSD1-mediated pathways holds promise for discovering novel therapeutic targets in vascular diseases.
Keywords:
THSD
; Autophagy
; Focal Adhesions
; Beclin 1
; Endothelial Cells
1. Introduction
Focal adhesions (FAs) represent subcellular organelles that tether cells to their extracellular matrix. Among their constituents, integrin proteins assume a prominent role. The extracellular domain of integrin engages various matrix proteins, such as collagens and laminins, while its intracellular segment interacts with a diverse array of adaptor proteins such as talin. Consequently, the integrin complex serves a conduit for sensing extracellular cues and transducing these signals into the cell, ultimately orchestrating cytoskeletal rearrangements for effective cell adhesion [1]. While originally observed in the context of two-dimensional cell culture systems, loss-of-function studies in animal models underscore the critical role of FAs in vascular integrity. For instance, the inactivation of αv or β8 integrin subunits in mice [2,3], or the knockdown of talin in zebrafish, leads to cerebral hemorrhage [4]. Furthermore, endothelial-specific depletion of FA kinase (FAK) is implicated in brain hemorrhage [5]. Despite the wealth of research on FA biogenesis, our understanding of the regulatory mechanisms governing FA turnover remains limited.
Emerging evidence indicates that autophagy, an intracellular self-digestion system, contributes to the degradation of FAs. Under various stress conditions, autophagy is induced, initiating the formation of autophagosomes—double-membrane organelles that engulf cargo and subsequently fuse with lysosomes for cargo degradation [6]. Intriguingly, in breast cancer cells, autophagy supports cell migration by facilitating the degradation of FA complexes [7,8,9]. Notably, distinct molecular mechanisms were uncovered in different cell lines, suggesting that autophagy-mediated FA turnover is context-dependent. However, our understanding of how autophagy degrades FAs in primary cells, particularly in primary human brain endothelial cells, where FAs are recognized as pivotal regulators of vascular integrity, remains largely unexplored. Bridging this knowledge gap is critical for identifying potential therapeutic targets in the context of cerebrovascular diseases due to the loss of endothelial integrity.
Multiple rare variants in the THSD1 gene have been identified in patients with intracranial aneurysms, a cerebrovascular disorder. Loss of THSD1 led to cerebral hemorrhage in both murine and zebrafish models [10], supporting its role in vascular integrity maintenance. Our group has further characterized THSD1 as a novel FA-associated protein, predominantly expressed in endothelial cells while being scarcely detectable in vascular smooth muscle cells [10,11]. However, the precise mechanisms by which THSD1 regulates endothelial FA stability have remained elusive. Our work reveals that THSD1 functions as a negative regulator of autophagy, with its loss inducing autophagy-mediated turnover of FAs in primary human brain endothelial cells. Consistently, blockade of autophagy restores FA stability, along with FA-associated cell spreading and attachment. Mechanistically, FAK, a master focal adhesion kinase, acts downstream of THSD1. Loss of THSD1 leads to reduced FAK activity, relieving its inhibitory effects on the complex formation between Beclin 1 and ATG14—a crucial event known to activate the autophagy cascade.
2. Results
Our previous investigations have established a physical interaction between THSD1 and Talin, a critical component of the integrin complex localized at FA sites [11]. To further confirm this observation, we conducted experiments in human brain microvascular endothelial cells (HBEMCs), an in vitro model with physiological relevance to cerebrovascular pathology. Endogenous THSD1 was detected in immunoprecipitates pulled down by anti-Talin antibody but was absent in Talin-deficient cells (as shown in the first and second lane of Figure 1A). Likewise, we performed a reciprocal co-immunoprecipitation, demonstrating that Talin could be pulled down by anti-FLAG antibody only when THSD1-FLAG was expressed (Figure 1B). These findings support our hypothesis that THSD1 modulates the stability of FAs in brain endothelial cells. To further test this hypothesis, we conducted loss-of-function analyses using small interfering RNAs (siRNAs). Depletion of THSD1 resulted in a significant reduction in the number of FAs, as indicated by immunofluorescent staining with paxillin antibodies (shown in green) in HBMECs (Figure 1C, comparing C3 to C4, and grey to black bars in Figure 1D). Similar results were observed when using zyxin, another marker predominantly identifying mature FAs (Supplemental Figure S1A-B). The diminished stability of FAs is often associated with impaired cell spreading, as this process relies on interactions between FAs and the extracellular matrix [12]. Indeed, our results demonstrated compromised cell spreading in THSD1-deficient endothelial cells, with a notable reduction in the percentage of cells exhibiting an area exceeding 1200 nm2 within 20 minutes after reseeding (Figure 1E, and comparison of grey to black bars in Figure 1F). Additionally, we observed a significant reduction in cell attachment ability in THSD1-deficient cells, as assessed by relative fluorescence units correlated with the total number of cells remaining attached to the pre-coated collagen IV matrix (Figure 1G). Collectively, these data suggest that THSD1 negatively modulates FA stability, cell spreading, and attachment.
To elucidate the mechanisms through which THSD1 downregulates FAs, we performed quantitative polymerase chain reaction (qPCR) analyses on the mRNA level of paxillin and zyxin. As shown in Supplemental Figure S2, there were no discernible changes in the mRNA levels of paxillin or zyxin following THSD1 depletion, indicating that THSD1 may not directly regulate the transcription of these FA genes in brain endothelial cells. Given that paxillin and zyxin mRNA levels remained unchanged after THSD1 knockdown, we investigated how THSD1 impacts their protein stability. It is well-established that both autophagy and the ubiquitin-proteasome system play crucial roles in regulating protein stability by influencing turnover rates [13,14,15]. To assess the involvement of THSD1 in these degradation pathways, we conducted autophagy flux analyses based on GFP-LC3 puncta formation, along with protein ubiquitination assays as we did previously [16]. LC3 serves as an autophagosomal marker, and GFP-LC3 can be used to monitor autophagic activity. Under conditions of starvation, we observed an increased number of GFP-LC3 puncta (comparison of A2 to A1 in Supplemental Figure S3A, and black bars in 3B). This effect was mitigated upon the knockdown of the essential autophagy gene, ATG5 (comparison of A4 to A2 in Supplemental Figure S3A, and grey to black bars in 3B). These data validated the sensitivity and specificity of brain endothelial cells in response to autophagy stimuli.
Notably, THSD1 knockdown resulted in an increased number of green puncta in this reporter cell line (comparison of A2 to A1 in Figure 2A and grey to black bars in 2B). This phenotype may result from either autophagy induction or compromised autophagic degradation that is otherwise indicative of reduced autophagic flux. To distinguish between these possibilities, we conducted standard flux analyses using Pepstatin/E-64D (P/E), two protease inhibitors commonly used to prevent cargo degradation within autolysosomes [17]. Our results indicated that autophagic degradation remained unimpaired, as evidenced by a further increase in the number of GFP-LC3 puncta with P/E treatment in both control and THSD1-deficient cells (comparison of A3 to A1, and A4 to A2 in Figure 2A, quantification depicted in Figure 2B,C). In line with these findings, a cargo-based endpoint autophagy assay revealed a significant reduction in the level of p62, a bona-fide autophagy substrate, in THSD1-deficient cells (Figure 2D-2E). Altogether, our data compellingly support the notion that THSD1 exerts a negative regulatory influence on autophagy in endothelial cells. Conversely, the overall level of protein ubiquitination remained unaltered following THSD1 knockdown, in comparison with the significant accumulation of ubiquitinated substrates in cells treated with MG132, a well-established proteasome inhibitor (Figure 2F).
To investigate whether THSD1-mediated autophagy contributes to FA stability, we subsequently knockdown ATG5 in both control and THSD1-deficient cells, followed by a FA assay. Our results revealed that ATG5 knockdown effectively rescued defective FAs in THSD1-deficient cells (comparison of the number of green puncta in A4 to A2, or A8 to A6 in enlarged pictures, as well as the grey bars in Figure 3B). Consistently, ATG5 knockdown also restored cellular spreading efficiency and attachment ability in THSD1-deficient cells (comparison of grey bars in Figure 3C-3D). The knockdown effectiveness of ATG5 and THSD1 was confirmed by Western blot (Supplemental Figure S4). In contrast, proteasome system inhibition via knockdown of the essential proteasome subunit 2 (PSMB2) failed to restore these phenotypes (Supplemental Figure S5), implying a lack of involvement of the ubiquitin proteasome system.
To examine the mechanisms underlying THSD1-mediated autophagy activation, we conducted a series of signaling pathway analyses. Prior literature suggests that downregulation of mTOR kinase, a central nutrient sensor, or upregulation of AMPK, a principal energy sensor, can potentiate autophagy under conditions of starvation [18,19]. To assess mTOR activity, we conducted immunoblotting to assess the phosphorylated levels of S6K and 4E-BP1, two canonical substrates of mTOR kinase. Intriguingly, THSD1 knockdown had no discernible effect on either of these readouts in terms of p-T389-S6K and p-T37/46-4E-BP1 (Figure 4A-4C). Similarly, we did not observe any alterations in AMPK activation, as evidenced by the levels of p-T172-AMPK and p-S79-ACC (Figure 4D-4F). ULK1, downstream of mTOR and AMPK, undergoes phosphorylation in response to autophagy induction [20,21]. We assessed the levels of phosphorylated ULK1 at Ser 757 (a target of mTOR) and at Ser 555 (a target of AMPK) but did not observe any discernible changes (Figure 4G-4I). These data collectively suggest that THSD1-mediated autophagy operates independently of both mTOR and AMPK.
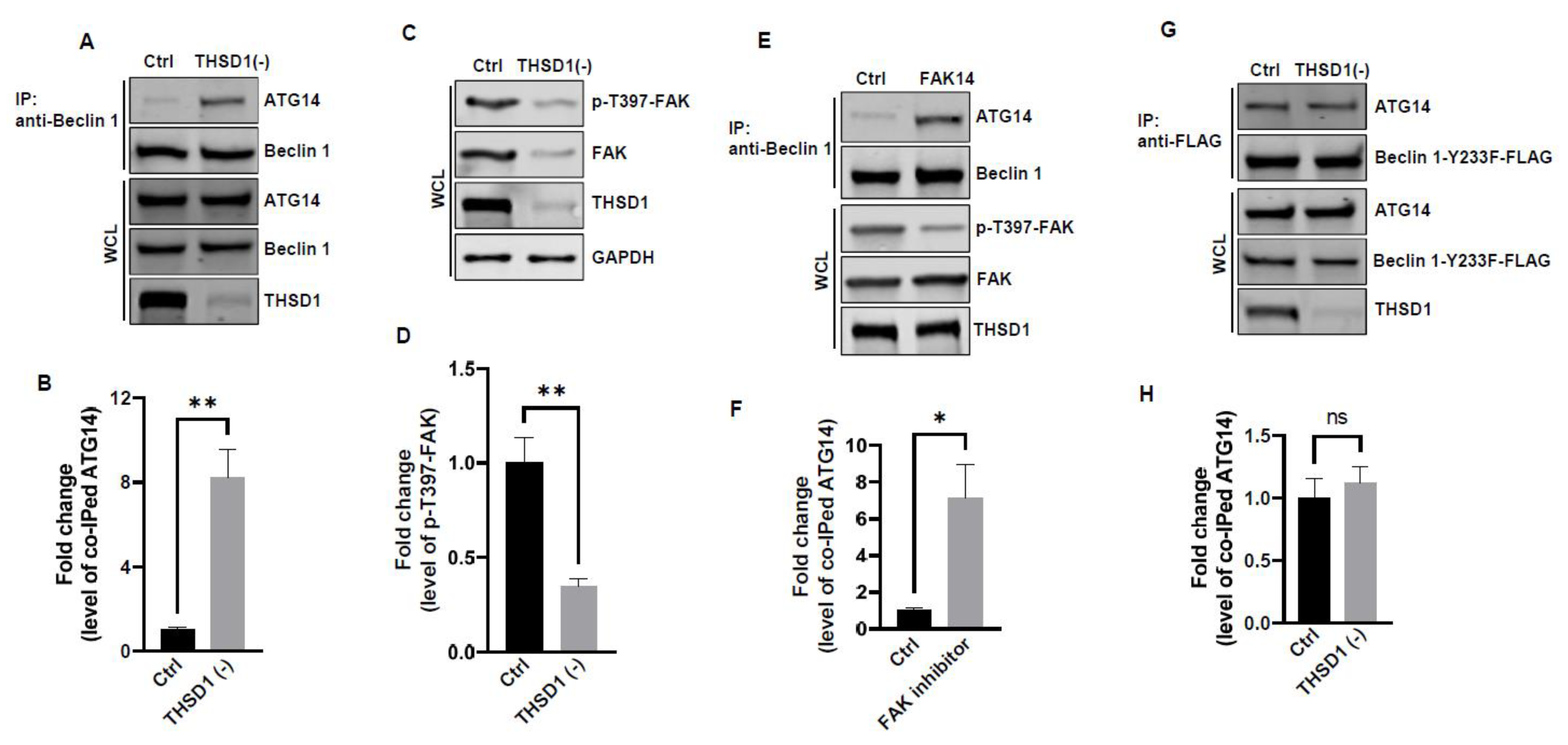
In light of these results, ULK1 was ruled out as a contributor to THSD1-mediated autophagy. We explored the possibility of other downstream molecules or signaling pathways being activated following THSD1 depletion. Beclin 1, an essential and sufficient component for autophagy induction, is positioned downstream of ULK1 within the autophagy pathway. In response to autophagic stimuli, Beclin 1 forms a complex with ATG14 to activate autophagy [22,23]. Interestingly, we observed a significant enhancement in the physical interaction between Beclin 1 and ATG14 in THSD1-deficient endothelial cells (Figure 5A-5B). This suggests that THSD1 negatively regulates the formation of the Beclin-ATG14 complex. Notably, FAK has been reported to phosphorylate Beclin 1 at Y233, a modification that impedes Beclin 1 binding to ATG14 [24]. Our data revealed that THSD1 knockdown resulted in reduced levels of both FAK and phosphorylated FAK at T397 (Figure 5C-5D). As such, we propose that THSD1 modulates the FAK/Beclin 1 pathway, which, in turn, regulates autophagy. To examine the role of FAK in Beclin 1-ATG14 protein complex formation, we treated HBMECs with FAK inhibitor 14 which inhibits FAK kinase activity (reduced phosphorylation of T397) without affecting the protein expression of FAK. Indeed, reduced FAK kinase activity augmented the interaction between Beclin 1 and ATG14 in brain endothelial cells (Figure 5E-5F). Importantly, a Beclin 1 mutant (Beclin 1-Y223F), resistant to FAK phosphorylation, consistently bound to ATG14 in both control and THSD1-deficient endothelial cells (Figure 5G-5H). These findings suggest that the loss of THSD1 reduces FAK activity, thereby relieving its inhibitory influence on Beclin 1-ATG14 complex formation, a pivotal step in the cascade of autophagy activation.
Based on the cumulative findings described above, we propose a model delineating the role of THSD1 in the regulation of autophagy and FAs. In endothelial cells, FAs encompass the integrin complex, which engages with the extracellular matrix on one end and interacts with adaptor proteins, such as talin and paxillin, on the other. FAK, a critical focal adhesion kinase, is dynamically recruited to FAs through its interaction with paxillin. Under physiological conditions, THSD1 resides within FAs, stabilizing FAK activity via complex formation. FAK phosphorylates the autophagy essential scaffold protein Beclin 1 at Y233, preventing its binding to ATG14 and thereby inhibiting autophagy. However, under pathological conditions, the loss of THSD1 destabilizes FAK, leading to increased binding between Beclin 1 and ATG14. Consequently, Beclin 1-mediated autophagy degrades FAs, contributing to functional deficits in endothelial cells, such as impaired cell spreading and attachment, which may ultimately compromise endothelial integrity and contribute to vascular diseases.
3. Discussion
3.1. THSD1 and FA-Phagy
Our work supports THSD1 as a hitherto unrecognized player in the regulation of autophagy within endothelial cells. Notably, our investigation revealed that depleting THSD1 did not lead to changes in mTOR and AMPK activation, as depicted in Figure 4. These findings strongly suggest that the primary function of THSD1 may be closely tied to FA dynamics rather than serving as a global regulator of nutrient and energy status. This localized activation of autophagy aligns with the concept of "quality control autophagy," an emerging paradigm emphasizing the specificity of autophagic processes targeting distinct cellular organelles, such as FAs, to ensure cellular homeostasis. The phenomenon of targeted FA degradation through autophagy, often referred to as "FA-phagy," adds an important layer to our understanding of FA dynamics [13]. Our results emphasize that the activation of FA-phagy is a response to cellular stress arising from compromised cell adhesion, rather than being driven by nutrient or energy depletion.
FAs play a central role in various cellular functions, encompassing cell adhesion, migration, and tissue organization. The disruption of FA stability upon the loss of THSD1 serves as a trigger for localized autophagy at these sites, representing a mechanism of cellular adaptation. Intriguingly, previous studies have documented that Beclin 1 can be localized at the endoplasmic reticulum, and this compartmentalized Beclin 1 supports autophagosome biogenesis even in the absence of ULK1 and ULK2 kinases [25]. This observation is in line with our results, indicating that THSD1-mediated autophagy operates independently of ULK1 activation. Notably, it has been reported that FAK directly interacts with Beclin 1, suggesting that Beclin 1 may also be compartmentalized at FAs [24]. In the future, it would be highly valuable to investigate whether THSD1 facilitates the localization of Beclin 1 at FAs and potentially suppresses its activity by promoting the interaction between FAK and Beclin 1. Such protein complex formation may play a pivotal role in shaping the precise regulation of FA-phagy and its impact on cellular adhesion processes. This intriguing possibility highlights the multifaceted nature of THSD1 in autophagy and FA dynamics, warranting further exploration in subsequent studies.
3.2. Potential link between THSD1-mediated FA-phagy and vascular diseases
Our results extend beyond elucidating the fundamental mechanisms of THSD1-mediated autophagy and FA stability. Notably, multiple harmful rare variants in the THSD1 gene have been identified in patients with intracranial aneurysms—a cerebrovascular disorder characterized by the weakening of blood vessel walls. Previous research has underscored the essential role of THSD1 in maintaining endothelial integrity. Notably, these THSD1 rare variants exhibit varying degrees of impairment in their ability to interact with Talin [10,11]. By establishing THSD1 as a regulator of autophagy and FA stability (Figure 1, Figure 2 and Figure 3), our work opens a window of opportunity for investigating the contribution of THSD1-mediated pathways to the pathophysiology of vascular diseases, particularly those involving compromised endothelial integrity. It will be intriguing to determine whether THSD1 rare variants lack the capacity to suppress autophagy. Further investigations into the mechanisms by which THSD1 influences these processes may provide valuable insights into the development and progression of vascular diseases such as intracranial aneurysms, potentially unveiling novel therapeutic targets for intervention.
In conclusion, our study highlights the novel role of THSD1 in modulating autophagy that degrades FAs within endothelial cells. We elucidate a novel mechanism involving the regulation of the Beclin 1-ATG14 complex and emphasize that THSD1 as a FA-associated protein regulates cargo-initiated selective autophagy. These findings broaden our understanding of autophagy regulation and its implications in vascular integrity, paving the way for future investigations into the role of FA-phagy in vascular diseases and related pathologies.
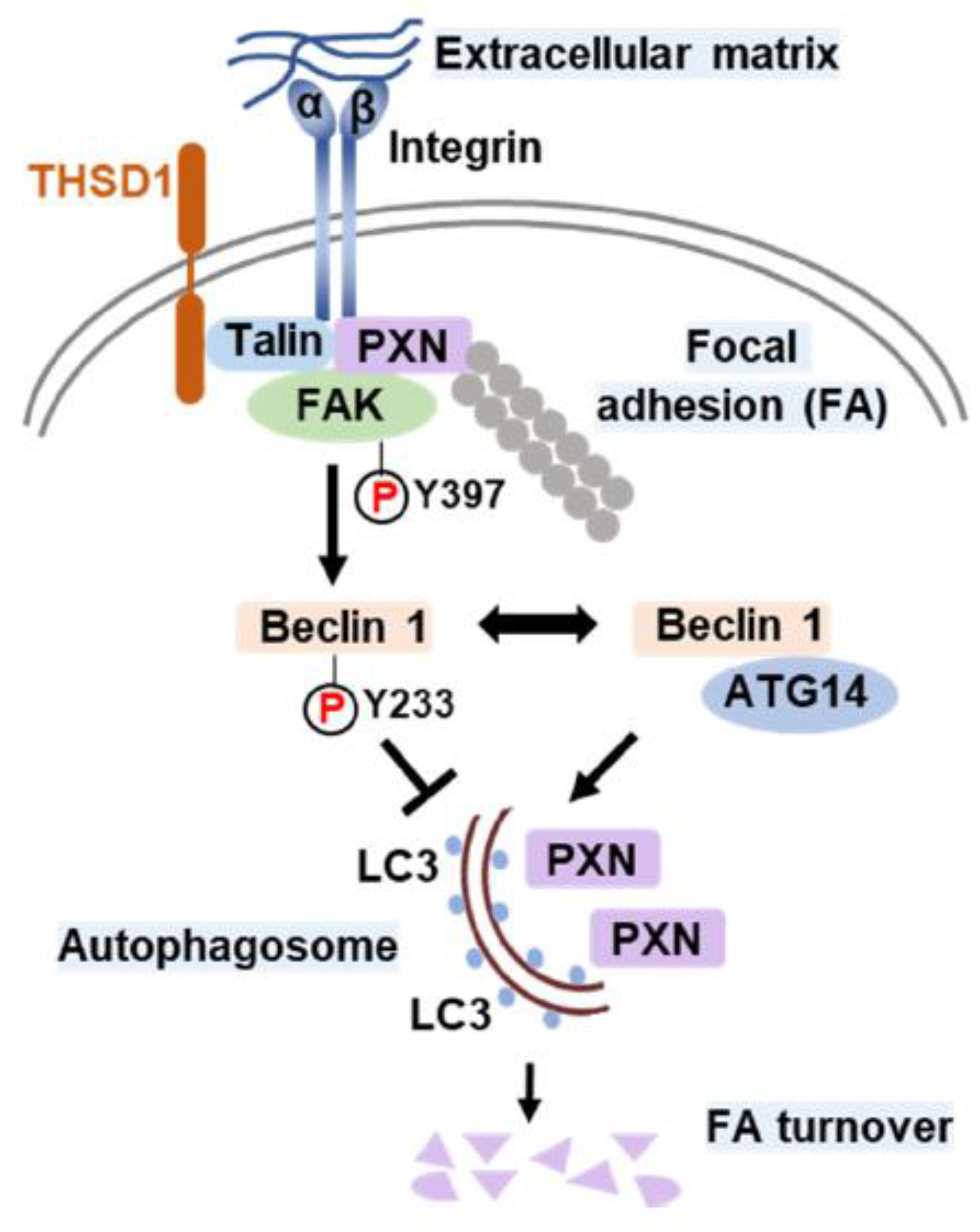
Figure 6.
Schematic Model of THSD1-Mediated Autophagy in Focal Adhesion Stability. Focal adhesions (FAs) are composed of integrin α/β transmembrane proteins that link extracellular matrix components like collagens to intracellular adaptors such as talin and paxillin (PXN). The actin cytoskeleton, represented by aligned round grey dots, is tethered to integrins through PXN. Focal adhesion kinase (FAK) plays a central role in focal adhesion regulation, binding to both PXN and THSD1. Under normal conditions, THSD1 interacts with FAK, enhancing its kinase activity, resulting in the phosphorylation of Beclin 1 at tyrosine 233. This phosphorylation event inhibits the binding of ATG14 to Beclin 1, a critical step in autophagy activation. When THSD1 is inactivated, FAK kinase activity is reduced, alleviating its negative control over the formation of the Beclin-ATG14 complex. This, in turn, promotes Beclin 1-dependent autophagosome formation, where LC3 proteins are incorporated on both sides of the phagophore. These autophagosomes can sequester paxillin and other focal adhesion proteins, leading to autophagy-mediated focal adhesion turnover.
Figure 6.
Schematic Model of THSD1-Mediated Autophagy in Focal Adhesion Stability. Focal adhesions (FAs) are composed of integrin α/β transmembrane proteins that link extracellular matrix components like collagens to intracellular adaptors such as talin and paxillin (PXN). The actin cytoskeleton, represented by aligned round grey dots, is tethered to integrins through PXN. Focal adhesion kinase (FAK) plays a central role in focal adhesion regulation, binding to both PXN and THSD1. Under normal conditions, THSD1 interacts with FAK, enhancing its kinase activity, resulting in the phosphorylation of Beclin 1 at tyrosine 233. This phosphorylation event inhibits the binding of ATG14 to Beclin 1, a critical step in autophagy activation. When THSD1 is inactivated, FAK kinase activity is reduced, alleviating its negative control over the formation of the Beclin-ATG14 complex. This, in turn, promotes Beclin 1-dependent autophagosome formation, where LC3 proteins are incorporated on both sides of the phagophore. These autophagosomes can sequester paxillin and other focal adhesion proteins, leading to autophagy-mediated focal adhesion turnover.
4. Materials and Methods
4.1. Plasmids
The pLVX-TetOne-Puro vector was obtained from Takara Inc and subsequently modified to incorporate a new polylinker with several 8-cutter sites, including PacI and NotI, to facilitate cloning. This customized vector was referred to as pLTO-PANBR. Full-length THSD1-FLAG and Beclin 1-Y223F-FLAG were cloned into the pLTO-PANBR vector using the PacI and NotI restriction sites through standard PCR protocols. The constructs were verified by Sanger sequencing as previously reported[16]. The pBabepuro-GFP-LC3 plasmid was sourced from Addgene (#22405).
4.2. siRNAs
All stealth small interfering RNAs (siRNAs) were purchased from ThermoFisher Scientific and included siRNAs targeting talin (HSS110804 and HSS186350), thsd1 (HSS148179 and HSS148180), atg5 (HSS114103 and HSS114104), and psmb2 (HSS108676 and HSS108677). The effectiveness of each siRNA in knockdown experiments was validated through Western blotting.
4.3. Antibodies
S6K (9202), p-S6K-T389 (9205), 4E-BP1 (9452), p-4E-BP1-T37/46 (9459), AMPK-α (2532), p-AMPKα-T172 (2535), Acetyl-CoA Carboxylase/ACC (3662), p-ACC-S79 (3661), ULK1 (8054), p-ULK1-S757 (14202), p-ULK1-S555 (5869), ATG14 (5504), Beclin 1 (3495), FAK (3285), p-FAK-T397 (3283), p62 (8025) from Cell Signaling Technology; THSD1 (Novus Biological, NBP1-86930); Paxillin (BD Biosciences, 612405); Zyxin (Millipore, MAB2610); Ubiquitin (Millipore, 04-262); GAPDH ( Santa Cruz Technology, sc-32233); Anti-FLAG antibody (Sigma, F1804).
4.4. Cell Culture
Human brain microvascular endothelial cells (HBMECs), obtained from Cell Systems (ACBRI 376), were cultured in endothelial cell medium (#1001, ScienCell Research Laboratories). Inducible gene expression in HBMECs was achieved through lentiviral infection. Lentiviruses were generated in HEK293T cells with the assistance of two other plasmids, pMD2.G (#12259, Addgene) and psPAX2 (#12260, Addgene). For siRNA-mediated knockdown experiments, siRNAs were introduced into HBMECs using standard electroporation techniques (Neon Transfection System, Invitrogen). Autophagy was induced by treating HBMECs with Earle’s Balanced Salt Solution (EBSS) for 2 hours, followed by LC3 puncta or p62 turnover assays. All chemicals, including pepstatin/E-64D, MG132, and FAK inhibitor 14, were purchased from Cayman Chemical.
4.5. Quantitative RT-PCR
Total mRNA was isolated from HBMECs using TRIzol (15596026, ThermoFisher Scientific) and quantified following the Fast SYBR green protocol. The primer sequences are provided below.
paxillin: 5’- CTGATGGCTTCGCTGTCGGATT /5’- GCTTGTTCAGGTCAGACTGCAG
zyxin: 5’- TTCCACATCGCCTGCTTCACCT /5’- CGCAGGTGTTACACTTCTCCAG
4.6. Western Blot
HBMECs were subjected to siRNA or compound treatments in 60-mm dishes and lysed in Triton X-100 lysis buffer, as previously described [26]. The lysates were sonicated briefly and centrifuged at 13,000 rpm for 30 minutes at 4°C. Total cell lysates were supplemented with 2X SDS sample buffer and then subjected to SDS-PAGE analysis. The proteins were subsequently transferred onto nitrocellulose membranes using a Bio-Rad mini transfer apparatus. Following this, the membranes were blocked with 5% non-fat milk, and primary and secondary antibodies were applied at dilutions of 1:1000 and 1:10000, respectively. The Odyssey Imager system (LI-COR Biosciences) was used to detect fluorescence signals.
4.7. Immunoprecipitation (IP)
HBMECs treated with siRNAs or compounds in 60-mm dishes were lysed in an IP-lysis buffer, as previously described [26]. The cell lysate was briefly sonicated and then centrifuged at 13,000 rpm for 30 minutes at 4°C. Endogenous or exogenous proteins were immunoprecipitated from the cell lysate using either anti-THSD1 or anti-FLAG antibody in combination with protein A/G Plus agarose beads (Santa Cruz Biotechnology, sc-2003). The immunoprecipitates and whole cell lysates (WCL) were subjected to standard Western blotting.
4.8. Immunostaining
HBMECs treated with siRNAs were reseeded onto 8-well chamber slides at appropriate cell concentrations. After fixation with 4% paraformaldehyde and permeabilization with Triton X-100, the cells were blocked with 10% normal goat serum for 1 hour at room temperature. Next, the cells were incubated with primary antibodies at 4°C overnight and subsequently stained with Alexa-594 or Alexa-488 conjugated secondary antibodies (Invitrogen). The stained samples were mounted in Prolong Gold solution (Invitrogen), and images were captured using a Leica TCS SP5 confocal microscope.
4.9. Cell Spreading Assay
HBMECs treated with various siRNAs were trypsinized and then inactivated by endothelial cell medium before resuspension in 1X PBS. The cells were immediately reseeded onto 8-well chamber slides pre-coated with Collagen-IV. After 20 minutes, the cells were fixed with 4% paraformaldehyde in PBS, and the overall morphology was highlighted by immunostaining against the cytoskeleton protein actin using Alexa 594-phalloidin, as previously reported [11]. Widefield images were acquired using a Leica DM4 microscope with a cMOS camera, and at least 9-12 different fields were chosen from each chamber. Cell areas were measured using ImageJ, and data were analyzed by Student t-test.
4.10. Cell Adhesion Assay
To evaluate cell adhesion, HBMECs subjected to various siRNA treatments were assessed using the CytoSelect Cell Adhesion Assay Kit (MBS168514, Cell Biolabs). Cells were trypsinized and resuspended in serum-free endothelial cell medium before adding 150 µl of the cell suspension to each well of a 96-well plate pre-coated with collagen IV. Following a 1-hour incubation in a cell culture incubator, the culture medium was removed, and the adherent cells were washed three times with 1X PBS. The remaining adherent cells on the well bottoms were lysed using the 1X Lysis Buffer/CyQuant Dye provided in the kit. Subsequently, a 150 µl aliquot of the lysate was transferred to a 96-well plate, and fluorescence signals were measured using a plate reader at an excitation wavelength of 480 nm and an emission wavelength of 520 nm.
4.11. LC3 Puncta Formation and p62 Turnover Assays
The LC3 puncta formation and p62 turnover assays were conducted following established protocols [16]. To assess LC3 puncta formation, cells were initially fixed using 4% paraformaldehyde, followed by permeabilization with 50 µg/ml digitonin. The primary antibody utilized was anti-GFP (GFP-1020, Aves Labs), and the secondary antibody was Alexa-488 anti-chicken IgG (Invitrogen). For each assay condition, puncta profile data were calculated by averaging the total number of GFP-LC3-positive puncta per cell, drawing from approximately 30–50 representative cells. In the p62 turnover assay, HBMECs treated with various siRNAs for 48 hours were harvested in Triton lysis buffer. Western blot analysis was then carried out to determine the protein levels of p62.
4.12. Statistics Analysis
Statistical analysis was conducted using GraphPad Prism 6. p-values were calculated using the Student’s t-test or one- or two-way ANOVA, with Bonferroni correction applied for multiple comparison tests between selected pairs. Data are presented as mean ± standard error of the mean (s.e.m.). A significance level of p < 0.05 was considered statistically significant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
The project was supported in part by R21NS108310 and R21NS130570 (to Y.R.).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors declare no competing or financial interests.
References
- N. Yamaguchi, H. Knaut, Focal adhesion-mediated cell anchoring and migration: from in vitro to in vivo. Development 2022, 149, dev200647. [Google Scholar] [CrossRef] [PubMed]
- J. H. McCarty, R.A. Monahan-Earley, L.F. Brown, M. Keller, H. Gerhardt, K. Rubin, M. Shani, H.F. Dvorak, H. Wolburg, B.L. Bader, A.M. Dvorak, R.O. Hynes, Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol 2002, 22, 7667–7677. [Google Scholar] [CrossRef]
- J. Zhu, K. Motejlek, D. Wang, K. Zang, A. Schmidt, L.F. Reichardt, beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 2002, 129, 2891–2903. [Google Scholar] [CrossRef] [PubMed]
- Q. Wu, J. Zhang, W. Koh, Q. Yu, X. Zhu, A. Amsterdam, G.E. Davis, M.A. Arnaout, J.W. Xiong, Talin1 is required for cardiac Z-disk stabilization and endothelial integrity in zebrafish. FASEB J 2015, 29, 4989–5005. [Google Scholar] [CrossRef]
- T. L. Shen, A.Y. Park, A. Alcaraz, X. Peng, I. Jang, P. Koni, R.A. Flavell, H. Gu, J.L. Guan, Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol 2005, 169, 941–952. [Google Scholar] [CrossRef] [PubMed]
- T. Ichimiya, T. Yamakawa, T. Hirano, Y. Yokoyama, Y. Hayashi, D. Hirayama, K. Wagatsuma, T. Itoi, H. Nakase, Autophagy and Autophagy-Related Diseases: A Review. Int J Mol Sci 2020, 21, 8974. [Google Scholar] [CrossRef]
- M. N. Sharifi, E.E. Mowers, L.E. Drake, C. Collier, H. Chen, M. Zamora, S. Mui, K.F. Macleod, Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep 2016, 15, 1660–1672. [Google Scholar] [CrossRef]
- C. M. Kenific, S.J. Stehbens, J. Goldsmith, A.M. Leidal, N. Faure, J. Ye, T. Wittmann, J. Debnath, NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 2016, 212, 577–590. [Google Scholar] [CrossRef]
- E. A. Assar, D.A. Tumbarello, Loss of the Essential Autophagy Regulators FIP200 or Atg5 Leads to Distinct Effects on Focal Adhesion Composition and Organization. Front Cell Dev Biol 2020, 8, 733. [Google Scholar] [CrossRef]
- T. Santiago-Sim, X. Fang, M.L. Hennessy, S.V. Nalbach, S.R. DePalma, M.S. Lee, S.C. Greenway, B. McDonough, G.W. Hergenroeder, K.J. Patek, S.M. Colosimo, K.J. Qualmann, J.P. Hagan, D.M. Milewicz, C.A. MacRae, S.M. Dymecki, C.E. Seidman, J.G. Seidman, D.H. Kim, THSD1 (Thrombospondin Type 1 Domain Containing Protein 1) Mutation in the Pathogenesis of Intracranial Aneurysm and Subarachnoid Hemorrhage. Stroke 2016, 47, 3005–3013. [Google Scholar] [CrossRef]
- Y. N. Rui, Z. Xu, X. Fang, M.R. Menezes, J. Balzeau, A. Niu, J.P. Hagan, D.H. Kim, The Intracranial Aneurysm Gene THSD1 Connects Endosome Dynamics to Nascent Focal Adhesion Assembly. Cell Physiol Biochem 2017, 43, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- P. A. Janmey, B. Hinz, C.A. McCulloch, Physics and Physiology of Cell Spreading in Two and Three Dimensions. Physiology (Bethesda) 2021, 36, 382–391. [Google Scholar] [CrossRef]
- J. Lu, B. Linares, Z. Xu, Y.N. Rui, Mechanisms of FA-Phagy, a New Form of Selective Autophagy/Organellophagy. Front Cell Dev Biol 2021, 9, 799123. [Google Scholar] [CrossRef]
- C. M. Kenific, T. Wittmann, J. Debnath, Autophagy in adhesion and migration. J Cell Sci 2016, 129, 3685–3693. [Google Scholar] [CrossRef]
- A. Teckchandani, J.A. Cooper, The ubiquitin-proteasome system regulates focal adhesions at the leading edge of migrating cells. Elife 2016, 5, e17440. [Google Scholar] [CrossRef]
- Y. N. Rui, Z. Xu, B. Patel, Z. Chen, D. Chen, A. Tito, G. David, Y. Sun, E.F. Stimming, H.J. Bellen, A.M. Cuervo, S. Zhang, Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol 2015, 17, 262–275. [Google Scholar] [CrossRef]
- T. Ueno, M. Komatsu, Monitoring Autophagy Flux and Activity: Principles and Applications. Bioessays 2020, 42, e2000122. [Google Scholar] [CrossRef]
- A. J. Meijer, S. Lorin, E.F. Blommaart, P. Codogno, Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef]
- S. Wang, H. Li, M. Yuan, H. Fan, Z. Cai, Role of AMPK in autophagy. Front Physiol 2022, 13, 1015500. [Google Scholar] [CrossRef]
- J. Kim, M. Kundu, B. Viollet, K.L. Guan, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011, 13, 132–141. [Google Scholar] [CrossRef]
- D. F. Egan, D.B. Shackelford, M.M. Mihaylova, S. Gelino, R.A. Kohnz, W. Mair, D.S. Vasquez, A. Joshi, D.M. Gwinn, R. Taylor, J.M. Asara, J. Fitzpatrick, A. Dillin, B. Viollet, M. Kundu, M. Hansen, R.J. Shaw, Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- J. H. Hurley, L.N. Young, Mechanisms of Autophagy Initiation. Annu Rev Biochem 2017, 86, 225–244. [Google Scholar] [CrossRef] [PubMed]
- K. Obara, Y. Ohsumi, Atg14: a key player in orchestrating autophagy. Int J Cell Biol 2011, 2011, 713435. [Google Scholar] [CrossRef]
- Z. Cheng, Q. Zhu, R. Dee, Z. Opheim, C.P. Mack, D.M. Cyr, J.M. Taylor, Focal Adhesion Kinase-mediated Phosphorylation of Beclin1 Protein Suppresses Cardiomyocyte Autophagy and Initiates Hypertrophic Growth. J Biol Chem 2017, 292, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- T. Anwar, X. Liu, T. Suntio, A. Marjamaki, J. Biazik, E.Y.W. Chan, M. Varjosalo, E.L. Eskelinen, ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases. Cells 2019, 8, 475. [Google Scholar] [CrossRef]
- Y. N. Rui, Y. Chen, Y. Guo, C.E. Bock, J.P. Hagan, D.H. Kim, Z. Xu, Podosome formation impairs endothelial barrier function by sequestering zonula occludens proteins. J Cell Physiol 2020, 235, 4655–4666. [Google Scholar] [CrossRef]
Figure 1.
THSD1 Interacts with Talin and Modulates Focal Adhesion Stability. (A) Western blot detection of co-immunoprecipitated THSD1 in the Talin protein complex from HBMECs treated with siRNAs against control (Ctrl, 5 nM) and talin (Talin (-), 5 nM) for 48 hours. n=3 independent experiments (B) Induction of FLAG-THSD1 in HBMECs with doxycycline (100 ng/ml) for 48 hours followed by pull-down using anti-FLAG antibody and Western blot analysis against Talin or THSD1. n=3 independent experiments. (C-D) Representative images of focal adhesions (FAs) immunostained with paxillin (green) and phalloidin (red) in HBMECs treated with siRNAs against control (Ctrl, 10 nM) and thsd1 (THSD1 (-), 10 nM) for 48 hours. The number of FAs was quantified from at least 12 different fields (20X objective) for each experiment (n=3) and presented in (D). Enlarged images were shown in white dashed boxes. (E-F) Representative images of cell morphologies revealed by phalloidin staining for control or THSD1-deficient HBMECs at 20 minutes after re-seeding onto chamber slides. Quantitative analysis was performed by counting the number of cells with surface areas exceeding 1200 nm2 in each well. At least 9 different fields (10X objective) were randomly recorded for each experiment (n=3). (G) Measurement of relative fluorescence units of CyQuant Dye at 480/520 nm from control or THSD1-deficient HBMECs after reseeding onto collagen IV pre-coated 48-well plates. n=3 independent experiments. *p<0.05; **p<0.01. Scale bar: 10 µm.
Figure 1.
THSD1 Interacts with Talin and Modulates Focal Adhesion Stability. (A) Western blot detection of co-immunoprecipitated THSD1 in the Talin protein complex from HBMECs treated with siRNAs against control (Ctrl, 5 nM) and talin (Talin (-), 5 nM) for 48 hours. n=3 independent experiments (B) Induction of FLAG-THSD1 in HBMECs with doxycycline (100 ng/ml) for 48 hours followed by pull-down using anti-FLAG antibody and Western blot analysis against Talin or THSD1. n=3 independent experiments. (C-D) Representative images of focal adhesions (FAs) immunostained with paxillin (green) and phalloidin (red) in HBMECs treated with siRNAs against control (Ctrl, 10 nM) and thsd1 (THSD1 (-), 10 nM) for 48 hours. The number of FAs was quantified from at least 12 different fields (20X objective) for each experiment (n=3) and presented in (D). Enlarged images were shown in white dashed boxes. (E-F) Representative images of cell morphologies revealed by phalloidin staining for control or THSD1-deficient HBMECs at 20 minutes after re-seeding onto chamber slides. Quantitative analysis was performed by counting the number of cells with surface areas exceeding 1200 nm2 in each well. At least 9 different fields (10X objective) were randomly recorded for each experiment (n=3). (G) Measurement of relative fluorescence units of CyQuant Dye at 480/520 nm from control or THSD1-deficient HBMECs after reseeding onto collagen IV pre-coated 48-well plates. n=3 independent experiments. *p<0.05; **p<0.01. Scale bar: 10 µm.
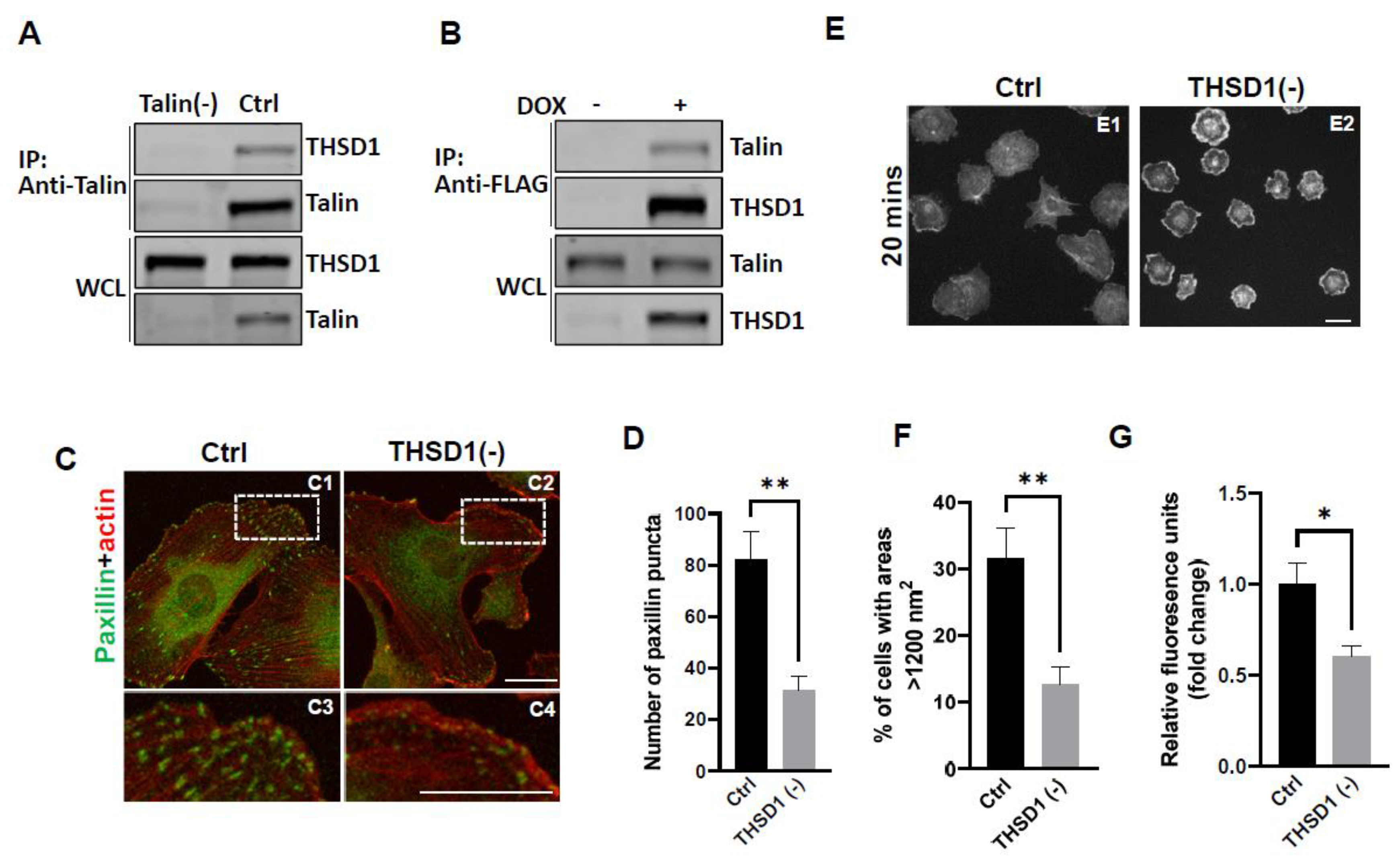
Figure 2.
THSD1 Inactivation Promotes Autophagy in Endothelial Cells. (A) Representative images of GFP-LC3 puncta formation in control or THSD1-deficient HBMECs in the absence and presence of pepstatin (50 µg/ml)/E-64D (50 µM) treatment (abbreviated as P/E) for 6 hours. (B-C) Analysis of the effects of THSD1 inactivation on LC3 puncta formation (B) and autophagic flux (C) in control or THSD1-deficient cells. The number of LC3 puncta per cellular profile was quantified. (D) Western blot analysis of p62 protein levels in control or THSD1-deficient HBMECs, with quantification in (E). (F) Representative images of Western blots against ubiquitinated proteins using the FK1 antibody. GAPDH served as a loading control. Fold changes were calculated after normalization to the total ubiquitinated protein level in the control sample (n=3 independent experiments). *p<0.05; **p<0.01. n.s: not significant. Scale bar: 10 µm.
Figure 2.
THSD1 Inactivation Promotes Autophagy in Endothelial Cells. (A) Representative images of GFP-LC3 puncta formation in control or THSD1-deficient HBMECs in the absence and presence of pepstatin (50 µg/ml)/E-64D (50 µM) treatment (abbreviated as P/E) for 6 hours. (B-C) Analysis of the effects of THSD1 inactivation on LC3 puncta formation (B) and autophagic flux (C) in control or THSD1-deficient cells. The number of LC3 puncta per cellular profile was quantified. (D) Western blot analysis of p62 protein levels in control or THSD1-deficient HBMECs, with quantification in (E). (F) Representative images of Western blots against ubiquitinated proteins using the FK1 antibody. GAPDH served as a loading control. Fold changes were calculated after normalization to the total ubiquitinated protein level in the control sample (n=3 independent experiments). *p<0.05; **p<0.01. n.s: not significant. Scale bar: 10 µm.
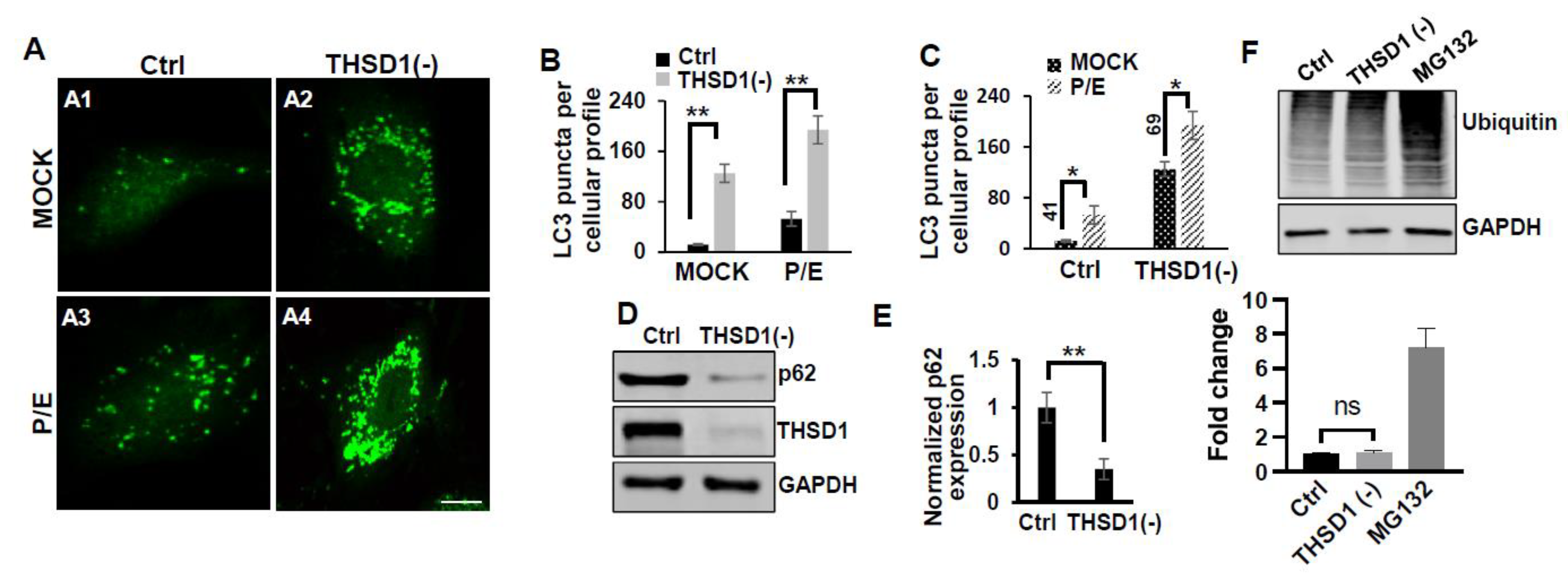
Figure 3.
Autophagy Inhibition Rescues Focal Adhesion Stability. (A) Representative images of FAs immunostained with paxillin (green) and actin (red) in control or THSD1-deficient HBMECs treated with siRNAs against control (5 nM) or atg5 (5 nM). Enlarged images are shown in white dashed boxes. (B) Quantification of the number of paxillin puncta per cellular profile from at least 12 different fields (20X objective) for each experiment (n=3). (C-D) Evaluation of cell spreading (C) and attachment (D) in control or THSD1-deficient HBMECs in the absence and presence of atg5 siRNA (5 nM) treatment for 48 hours. Data were analyzed by two-way ANOVA followed by Bonferroni correction. *p<0.05; **p<0.01. Scale bar: 10 µm.
Figure 3.
Autophagy Inhibition Rescues Focal Adhesion Stability. (A) Representative images of FAs immunostained with paxillin (green) and actin (red) in control or THSD1-deficient HBMECs treated with siRNAs against control (5 nM) or atg5 (5 nM). Enlarged images are shown in white dashed boxes. (B) Quantification of the number of paxillin puncta per cellular profile from at least 12 different fields (20X objective) for each experiment (n=3). (C-D) Evaluation of cell spreading (C) and attachment (D) in control or THSD1-deficient HBMECs in the absence and presence of atg5 siRNA (5 nM) treatment for 48 hours. Data were analyzed by two-way ANOVA followed by Bonferroni correction. *p<0.05; **p<0.01. Scale bar: 10 µm.
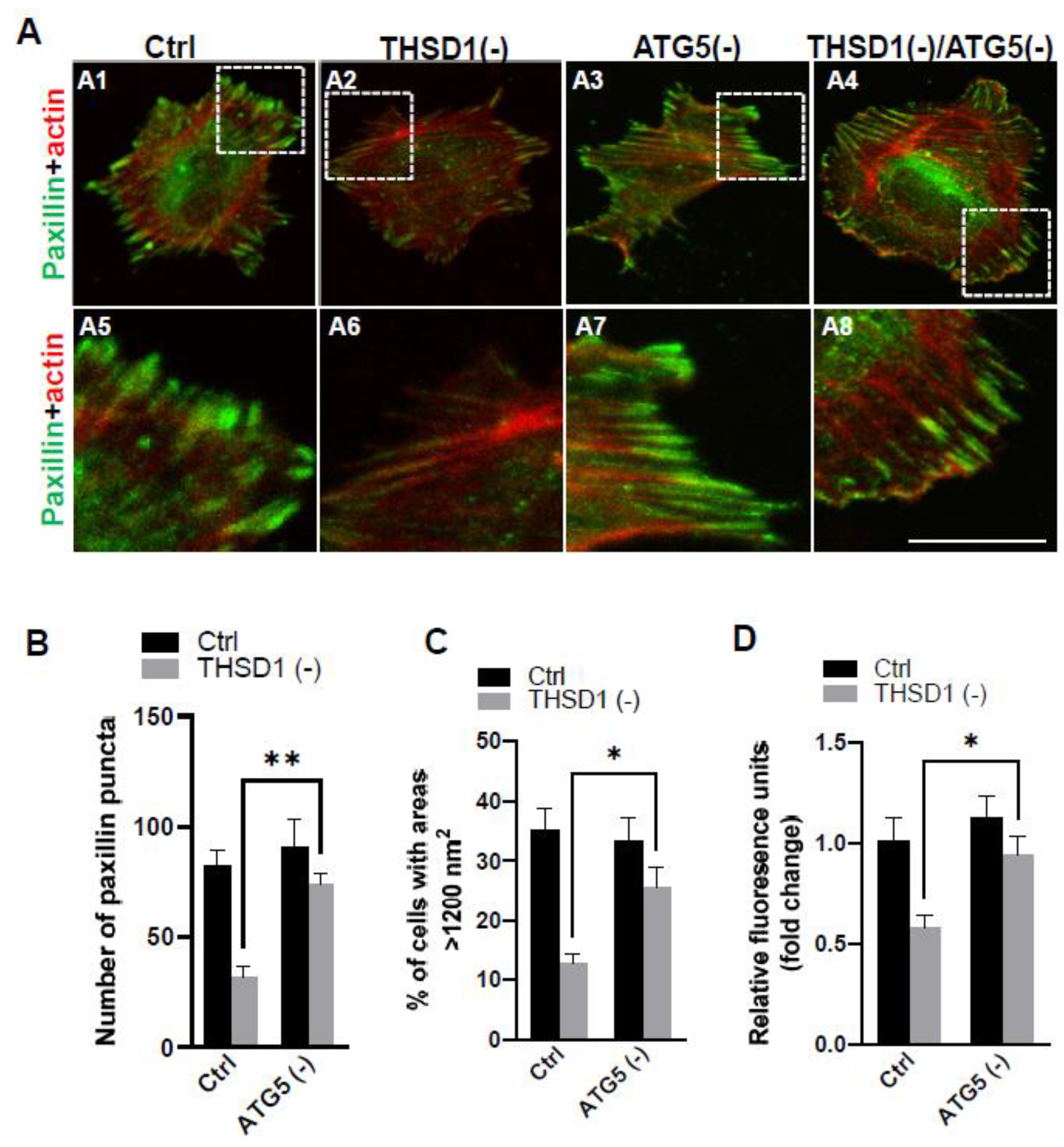
Figure 4.
THSD1-Mediated Autophagy is Independent of mTOR or AMPK Pathway. (A-C) Western blot analysis of whole cell lysates (WCL) from control or THSD1-deficient HBMECs using antibodies against p-S6K-T389, S6K, p-4E-BP1-37/46, and 4E-BP1. GAPDH served as a loading control. Quantification of p-S6K-T389 (B) and p-4E-BP1-T37/46 (C) levels were analyzed by Student t-test (n=3 independent experiments). (D-I) Representative Western blots of WCL from control or THSD1-deficient HBMECs for analyzing AMPK signaling (D-F) or ULK1 activation (G-I). Quantification of p-AMPKα-T172 (E), p-ACC-S79 (F), p-ULK1-S757, and p-ULK1-S555 were analyzed by Student t-test (n=3 independent experiments). n.s: not significant.
Figure 4.
THSD1-Mediated Autophagy is Independent of mTOR or AMPK Pathway. (A-C) Western blot analysis of whole cell lysates (WCL) from control or THSD1-deficient HBMECs using antibodies against p-S6K-T389, S6K, p-4E-BP1-37/46, and 4E-BP1. GAPDH served as a loading control. Quantification of p-S6K-T389 (B) and p-4E-BP1-T37/46 (C) levels were analyzed by Student t-test (n=3 independent experiments). (D-I) Representative Western blots of WCL from control or THSD1-deficient HBMECs for analyzing AMPK signaling (D-F) or ULK1 activation (G-I). Quantification of p-AMPKα-T172 (E), p-ACC-S79 (F), p-ULK1-S757, and p-ULK1-S555 were analyzed by Student t-test (n=3 independent experiments). n.s: not significant.
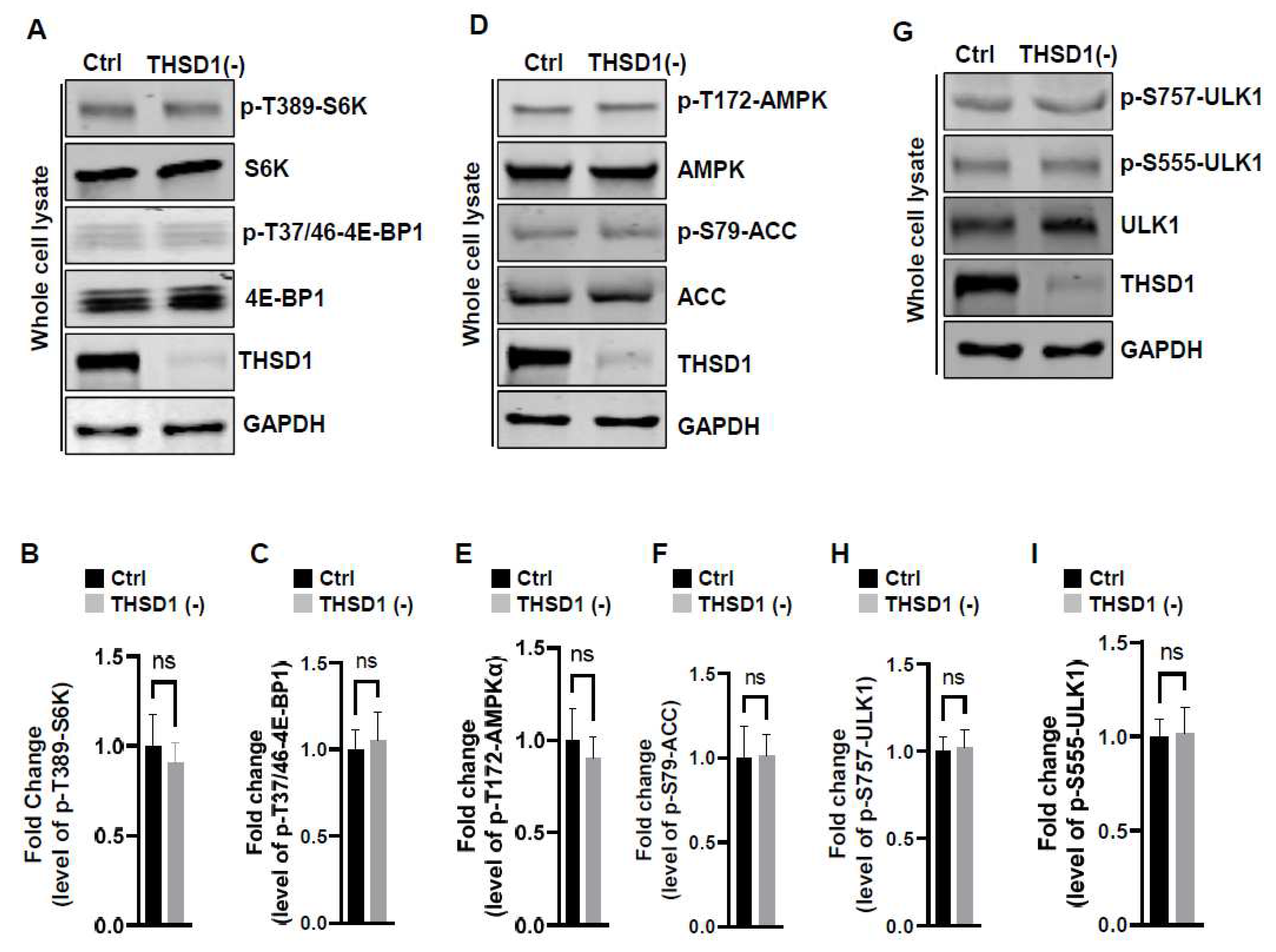
Figure 5.
FAK and Beclin 1 are Required for THSD1-Mediated Autophagy. (A-B) Western blot analysis of immunoprecipitated Beclin 1 protein complexes from control or THSD1-deficient HBMECs using the ATG14 antibody, with quantification in (B) (n=3). (C-D) Western blot analysis of p-FAK-T397 levels in control and THSD1-deficient HBMECs, with quantification in (D) (n=3 independent experiments). (E-F) Immunoprecipitation of Beclin 1 protein complex from HBMECs treated with water or FAK inhibitor 14 (FAK14, 2 µM) for 1 hour. Western blot detection of co-immunoprecipitated ATG14 (E) and quantification in (F) (n=3 independent experiments). (G-H) Pull-down of Beclin 1-Y233F-FLAG from WCL in control or THSD1-deficient HBMECs, with Western blot detection of co-immunoprecipitated ATG4 (G) and quantification in (H) (n=3 independent experiments). *p<0.05; **p<0.01. n.s: not significant.
Figure 5.
FAK and Beclin 1 are Required for THSD1-Mediated Autophagy. (A-B) Western blot analysis of immunoprecipitated Beclin 1 protein complexes from control or THSD1-deficient HBMECs using the ATG14 antibody, with quantification in (B) (n=3). (C-D) Western blot analysis of p-FAK-T397 levels in control and THSD1-deficient HBMECs, with quantification in (D) (n=3 independent experiments). (E-F) Immunoprecipitation of Beclin 1 protein complex from HBMECs treated with water or FAK inhibitor 14 (FAK14, 2 µM) for 1 hour. Western blot detection of co-immunoprecipitated ATG14 (E) and quantification in (F) (n=3 independent experiments). (G-H) Pull-down of Beclin 1-Y233F-FLAG from WCL in control or THSD1-deficient HBMECs, with Western blot detection of co-immunoprecipitated ATG4 (G) and quantification in (H) (n=3 independent experiments). *p<0.05; **p<0.01. n.s: not significant.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



