
Submitted:
08 December 2023
Posted:
11 December 2023
You are already at the latest version
Abstract
Triphenylphosphonium (TPP) derivatives of a decapeptide related to sequences of bactenecin 7 (Bac7) and oncocin (Onc112) were synthesized and studied as potential antimicrobial compounds using various biochemical and microbiological assays. It was shown that while the reduction of the Bac7 length to 10 a.a. residues dramatically decreases the affinity to bacterial ribosomes, the modification of peptide with alkyl-TPP moieties leads to an increase in the affinity. New analogs which structure combines a decapeptide related to Bac7 and Onc112 — Bac(1-10, R/Y) — and TPP, attached to the C-terminal amino acid residue through alkylamide linkers, inhibit translation in vitro and are found to be more selective inhibitors of bacterial translation as compared with eukaryotic translation, than Onc112 and Bac7. Triphenylphosphonium analogs of a decapeptide related to Bac7 and Onc112 suppress the growth of both gram-negative bacteria, similar to Onc112 and Bac7, and gram-positive ones as alkyl-TPP derivatives, and also act against some resistant laboratory strains. Bac(1-10, R/Y)-C2-TPP, containing a short alkylamide linker between the decapeptide and TPP, is transferred into the E. coli cells via the SbmA transporter protein. TPP derivatives of the decapeptide Bac(1-10, R/Y) containing either decylamide or ethylamide linker cause B. subtilis membrane depolarization, similar to alkyl-TPP. The Bac(1-10, R/Y)-C2-TPP analog is proved to be non-toxic for mammalian cells by MTT-test.
Keywords:
alkyl(triphenyl)phosphonium
; proline arginine rich antimicrobial peptides
; bactenecin 7
; oncocin 112
; bacterial ribosome
; bacterial membrane potential
; antimicrobial activity
1. Introduction
Antimicrobial peptides (AMP) have recently attracted the attention of many researchers as promising antibacterial agents for medical uses [1]. This interest is caused by the ability of AMP to act on resistant bacteria, low bacterial resistance against them, a wide range of antimicrobial activity, a variety of mechanisms of action, and relatively simple approaches to modifying the structure [2].
Proline-arginine-rich antimicrobial peptides (PA-AMP) which include peptides with a high content of proline and a PR motif in the primary structure, have a non-lytic mechanism of action on bacteria and are present in a wide range of organisms: from insects to mammals, being the products of their innate immune system and providing protection against the penetration of bacteria [3,4]. The mechanism of antibacterial action of these peptides is associated with intracellular targets, and as has been shown, PA-AMP bind to ribosomes and inhibit protein synthesis [5,6,7].
Depending on the interaction with the ribosome and the mechanism of translation inhibition, PA-AMP are divided into two types. The first group includes oncocins, bactenecins and some other peptides, which bind inside the nascent peptide exit tunnel (NPET) in the opposite direction to the nascent peptide chain in such a way that the N-terminus is located at the peptidyl transferase center (PTC) and the C-terminus extends deep into the ribosomal tunnel. These PA-AMP in complex with ribosomes allow formation of the first peptide bond, but prevent the elongation of the nascent peptide [8,9,10,11,12]. The second group of studied PA-AMP includes natural peptides apidaecin and its synthetic analogs, as well as drosocin, which bind to the ribosome in the same orientation as the nascent peptide chain. These AMP cause ribosome stalling at the stop codon during translation, as a result of their interaction with class 1 release factors (RFs) in PTC, which leads to the depletion of the number of free RFs in the bacterial cell [13,14,15,16]. Mammalian PA-AMP bactenecin 7 (Bac7) identified in the cow (Bos taurus) and then in sheep (Ovis aries) and goats (Capra hircus) [4,10], as well as insect PA-AMP oncocin (Onc) from the milkweed bug Oncopeltus fasciatus [17], are among the most well-studied PA-AMP [5,18,19].
The main problem with the use of AMP as potential drugs is their short half-life in biological media as a result of proteolytic degradation [2]. A number of Onc analogs have been synthesized containing various substitutions of amino acid residues at all 19 positions of the peptide chain, as well as double and triple substitutions, to improve inhibitory activity and proteolytic stability; among them Onc112 turned out to be the most optimal (Figure 1A) [20]. Mammalian PA-AMP are generally longer than insect PA-AMP [5]. Shorter analogs of Bac7, whose sequence consists of 60 amino acid residues, have been obtained to identify a domain responsible for its antimicrobial activity; N-terminal peptides Bac7(1-35) and Bac7(1-16) (Figure 1A) were shown to retain antibacterial activity [21]. Crystal structures of the bacterial ribosome in complex with Onc112 and Bac7(1-16) revealed that despite the different origin of these peptides, they interact with similar regions of the ribosome, and both Bac7 and Onc112 compete effectively with other antibiotics targeting NPET, such as macrolides, chloramphenicol, lincosamides, and some others [8,9,10,12]. In these works, along with the proof of the mechanism of action of type 1 PA-AMP on the bacterial translation process, it was shown that N-terminal sequences of type 1 PA-AMP are important for binding to ribosomes, and these sequences are relatively short.
Despite the structural similarity of Onc112 and Bac7(1-16) in the region of the N-terminal sequence in complexes with bacterial ribosomes, the first six residues of Bac7 form a domain different from the structure of the N-terminus of Onc112. This compact domain in which Arg residues form an arranged positively charged structure makes a significant contribution to the interaction of Bac7 with the ribosome; moreover, N-terminal RRIR motif also plays an important role in the internalization of Bac7(1-35) into bacterial cells [10].
PA-AMP are synthesized in organisms in the form of inactive precursors that undergo proteolytic cleavage to form active AMP [3,5,10]. PA-AMP, including Bac7 and Onc, act mainly on gram-negative bacteria [5]. This specificity is determined by the fact that AMP penetrates into bacterial cells through the inner membrane transporter proteins, such as SbmA and YjiL-MdtM transporter system [8,10,22,23]. Positively charged residues distributed along the PA-AMP chain are believed to be necessary for their effective uptake by the cell via SbmA transporter [24].
PA-AMPs exhibit low toxicity in eukaryotes [25,26]. On the other hand, Bac(1-35) was shown to inhibit eukaryotic translation in vitro; the absence of its toxic effect on mammalian cells is explained by the ways of its activation and internalization into a bacterial cell [10].
The appearance of bacteria resistance to PA-AMP is not often observed, however the resistance can occur in the case of mutations or deletion of transporter protein genes required for the entry of PA-AMP into bacterial cells, in particular, sbmA [5,22,23,27]. Another type of bacterial resistance, due to ribosomal mutations, was observed for Onc112 [12]. Nucleotide substitutions A2503C and A2059C in the 23S RNA sequence and especially the double substitution A2503C/A2059G increased E. coli resistance to Onc112 but not to Bac7(1-35).
In recent years, the search has continued for derivatives of both oncocin and bactenecins that would be resistant to proteolytic degradation, have broad antibacterial activity, act against resistant strains of bacteria and possess other interesting and useful properties. This kind of derivatives have been created either by replacing various amino acid residues with others contributing to the stability to proteolysis [28,29,30], or by combining peptides related to Bac7 with other antibiotics [31], antiviral drugs [32] or PNA [33] within the same molecule, or by conjugating two different AMP [34], as well as on the basis of deep mutational scanning [35].
For some ribosome targeting antibiotics, it has been demonstrated that their modification by alkyl(triphenyl)phosphonium (alkyl-TPP) groups, leads to improved penetration into bacterial cells and cancer mitochondria, enhanced activity and increased affinity for ribosomes [36,37,38,39]. Moreover, it has also been shown that alkyl-TPP themselves exhibit antibacterial properties [40,41,42].
In the current study, we continued research on the synthesis and exploration of compounds with antimicrobial properties by combining two different biologically active agents within a single molecule [36,37,43]; a new series based on a decapeptide relative to sequences of bactenecin 7 and oncocin 112 and alkyl(triphenyl)phosphonium cations (Figure 1) was created in order to expand the spectrum of antimicrobial activity of AMP and give new properties to analogs as antimicrobial compounds. Using various biochemical and microbiological assays, we have shown which properties are preserved or changed in new derivatives compared to the original compounds, as well as what new properties appeared that might be valuable for the creation of new antibacterial agents.
2. Materials and Methods
2.1. Chemicals and Materials
The following reagents and solvents were used: amino acids, their derivatives, and 2-chlorotrityl chloride resin for solid phase peptide synthesis (2CTC Resin), HATU and TIS (Iris Biotech, Marktredwitz, Germany and GL Biochem Shanghai, Shanghai, China); TentaGel HL NH2 resin (Rapp Polymere, Tubingen, Germany); HBTU (hexafluorophosphate benzotriazole tetramethyl uranium), DIPEA (N,N-diisopropylethylamine), triphenylphosphine, 1,2-dibromoethane, 1,10-dibromodecane, 11-bromoundecanoic acid, and 7N NH3 in methanol (Sigma-Aldrich, Steinheim, Germany); 4-methylpiperidine (Mosinter Chemicals, Zhejiang, China); BODIPY-FL-C3-OSu (Lumiprobe, Moscow, Russia); CH2Cl2, dimethylformamide (DMF), and trifluoroacetic acid (TFA) (PanReac AppliChem, Darmstadt, Germany); TFA (Solvay S.A., Brussels, Belgium); acetonitrile (gradient grade) (Biosolve Chimie, Dieuze, France); hexafluoro-2-propanol (HFIP) (Acros Organics, Geel, Belgium); BODIPY-ERY, the fluorescent erythromycin derivative [44], were synthesized as described previously.
2.2. Chromatography
TLC was performed on Kieselgel 60 F254 plates (Merck, Germany); for column chromatography Silica gel 60 (0.063-0.200 and 0.04-0.063 mm, Macherey Nagel, Germany) was used. Compounds containing UV-absorbing groups were detected with a UV-cabinet Camag (England); substances containing amino groups were visualized by a ninhydrin reagent. TPP and its derivatives were detected using Dragendorff's reagent.
Preparative HPLC was performed using Knauer semi-preparative system Smatrline (Germany) equipped with HPLC pumps Smartline 1050, UV detector Smartline 2520, Smartline Manager 5050 and Beckman Coulter Ultrasphere ODS column (5 microns, 250×10 mm). ClarityChrom software were applied. Following conditions were used: flow rate 5 mL/min, ambient temperature, elution with appropriate gradient of CH3CN in 0.05% TFA. A preparative purification of Onc112 was carried out on a Gilson HPLC system (333/334 pump with a 215 liquid handler) equipped with an YMC Triart C18 (150 × 30 mm) column and a UV detector at 210 and 280 nm. The peptide was eluted in an aqueous gradient of CH3CN (from 10 to 55% for 30 min) with 0.1% TFA at a flow rate of 70 mL/min.
2.3. Liquid Chromatography-Mass Spectrometry
Liquid chromatography-mass spectrometry was performed using a UPLC/MS/MS system consisting of an Acquity UPLC chromatograph from Waters (USA) and a TQD quadrupole mass-spectrometer (Waters) with registration of positive ions using the ESI MS method with an Acquity BEH column C18 (1.7 microns, 50×2.1 mm, Waters), flow rate 0.5 mL/min, 35 °C, and elution with a gradient of 5-100% CH3CN in 20 mM of HCOOH for 4 min. UPLC-MS analysis of Onc112 was performed using a Thermo Finnigan LCQ Deca XP ion trap spectrometer with Thermo Accela UPLC system equipped with a Waters Atlantis T3 C18 (150×2 mm) column.
2.4. Mass-Spectrometry
MALDI-TOF mass spectra were recorded with a MALDI-TOF mass spectrometer UltrafleXtreme (Bruker Daltonics, Germany) equipped with an UV laser (Nd) in the reflectron positive ion mode.
High resolution mass spectra (HRMS) were recorded with an Orbitrap Elite Hybrid mass spectrometer (Thermo Fisher Scientific, USA) equipped with an electrospray ionization (ESI) source in positive ion mode. Detailed HRMS data for compounds 1–5 are presented in Supplementary Materials, chapter III.
2.5. 1H and 13C NMR
1H and 13C NMR spectra were recorded with a Bruker Avance spectrometer (Bruker, Germany) with operating frequency 400 MHz for 1H, 101 MHz for 13C, and 162 MHz for 31P at 298 K in DMSO-d6 using tetramethylsilane as an internal reference. Spectra were processed and analyzed using Mnova software (Mestrelab Research, Spain).
2.6. Molecular Modelling
Molecular design was performed by means of static modeling using Avogadro [45]. and PyMOL 2.6 software (The PyMOL Molecular Graphics System, Version 2.6 Schrödinger, LLC).
2.7. Synthetic Procedures
The scheme for the synthesis of peptides 1 and 2 and their TPP derivatives 3–5 is represented in Figures S1 and S2. NH2-Cn-TPP (n = 2, 10) were obtained in two stages from TPP by the conjugation of TPP with dibromoalkane, according to [46] and amination of resulting bromoalkyl-TPP with 7M ammonia in methanol [47]. TPP-C10-COOH were synthesized from TPP and 11-bromoundecanoic acid as described in [48].
Peptides 1 and 2, and analog 5 were synthesized by standard Fmoc solid-phase peptide synthesis protocol using 2-chlorotrityl resin and HBTU/DIPEA activation. Synthesis of Onc112 was carried out using custom-made automated parallel peptide synthesizer. Fmoc strategy with HATU/DIPEA activation was applied. Compounds 3 and 4 were obtained by conjugation the corresponding protected peptide to NH2-Cn-TPP (n = 2, 10) using HBTU as activation agent. BODIPY-Bac(1-10) was synthesized from peptide 1 and BODIPY-FL-C3 succinimidyl ester. See also Supplementary Materials for more detailed information on synthetic procedures.
1, Bac(1-10). MALDI-TOF MS: m/z calculated for [C57H105N25O11+H]+: 1316.8; found 1316.8; HRMS: m/z calculated for [C57H105N25O11+2H]2+: 658.9286; found 658.9285; calculated for [C57H105N25O11+3H]3+: 439.6215; found 439.6215; calculated for [C57H105N25O11+4H]4+: 329.9679; found 329.9681; calculated for [C57H105N25O11+5H]5+: 264.1758; found 264.1758.
2, Bac(1-10, R/Y). MALDI-TOF MS: m/z calculated for [C60H102N22O12+H]+: 1323.8; found 1323.7; HRMS: m/z calculated for [C60H102N22O12+2H]2+: 662.4097; found 662.4100; calculated for [C60H102N22O12+3H]3+: 441.9422; found 441.9423; calculated for [C60H102N22O12+4H]4+: 331.7085; found 331.7084.
3, Bac(1-10, R/Y)-C2-TPP. MALDI-TOF MS: m/z calculated for [C80H121N23O11P]+: 1610.9; found 1610.9; HRMS: m/z calculated for [C80H121N23O11P+H]2+: 805.9710; found 805.9708; calculated for [C80H121N23O11P+2H]3+: 537.6498; found 537.6496; calculated for [C80H121N23O11P+3H]4+: 403.4892; found 403.4892; calculated for [C80H121N23O11P+4H]5+: 322.9928; found 322.9931.
4, Bac(1-10, R/Y )-C10-TPP. MALDI-TOF MS: m/z calculated for [C88H137N23O11P]+: 1723.1; found 1723.0; HRMS: m/z calculated for [C88H137N23O11P+2H]3+: 575.0249; found 575.0249; calculated for [C88H137N23O11P+3H]4+: 431.5205; found 431.5209; calculated for [C88H137N23O11P+4H]5+: 345.4178; found 345.4180.
5, TPP-C10-Bac(1-10, R/Y). MALDI-TOF MS: m/z calculated for [C89H136N22O13P]+: 1752.0; found 1751.9; HRMS: m/z calculated for [C89H136N22O13P+H]2+: 877.0248; found 877.0250; calculated for [C89H136N22O13P+2H]3+: 585.0189; found 585.0185; calculated for [C89H136N22O13P+3H]4+: 439.0160; found 439.0159; calculated for [C89H136N22O13P+4H]5+: 351.4143; found 351.4144.
BODIPY-Bac(1-10). Fluorescence (MeOH): λex = 505 nm, λem = 510 nm; LC-MS m/z calculated for [C71H118BF2N27O12+2H]2+: 795.98; found 794.82; τ (UPLC) = 0.89 min; MALDI TOF MS m/z calculated for [C71H118BF2N27O12+H]+: 1591.0; found 1591.0.
Onc112. LC-MS m/z calculated for [C109H177N37O24+H]+: 2390.4; found 2390.8.
2.8. In Vitro Binding Assay
70S ribosomes were isolated from E. coli MRE600 cells according to a published procedure [49]. Binding of BODIPY-Bac(1-10) to 70S E. coli ribosomes was performed as previously described [50]. BODIPY-Bac(1-10) (16 nM) was incubated with ribosomes (from 0.5 nM to 2000 nM) for 2 h at 26 °C in the buffer containing 20 mM HEPES-KOH (pH 7.5), 50 mM NH4Cl, 10 mM Mg(CH3COO)2, 4 mM β-mercaptoethanol, and 0.05% Tween-20. Binding affinities of peptides 1 and 2, their TPP analogs 3–5, as well as control compounds, for the E. coli ribosome were analyzed by a competition-binding assay using fluorescently labeled BODIPY-Bac(1-10) or BODIPY-ERY as described before [36,51]. Fluorescent compound (16 nM) was mixed with ribosomes (84 nM for BODIPY-Bac(1-10) and 46 nM for BODIPY-ERY) in the buffer. Solutions of tested compounds were added to obtained complexes to final concentrations ranging from 0.01 to 100 µM. The mixtures were incubated for 2 h at 26 °C, and then the values of fluorescence anisotropy were measured with VICTOR X5 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA) using a 384-well plate. The excitation wavelength was 485 nm, and the emission wavelength was 535 nm. From the data obtained, apparent dissociation constants were calculated [52].
2.9. In Vitro Translation Inhibition Assays
The inhibition of firefly luciferase synthesis by the tested compounds was assessed in vitro, as described previously [53] with minor modifications. Briefly, the in vitro transcribed firefly luciferase mRNA (fluc) was translated using the E. coli S30 Extract System for Linear Templates (Promega, Madison, WI, USA). Reaction mixtures (5 μL total volume) supplemented with 0.1 mM mixture of all canonical amino acids, 4 U of RiboLock RNase Inhibitor (Thermo Fisher Scientific, Waltham, MA, USA) and 0.1 mM of D-luciferin (Sigma-Aldrich, Burlington, MA, USA), were pre-incubated at RT for 5 min after the addition of the tested compounds at a final concentration of 30 µM. Then 50 ng of the mRNA was added to each reaction tube, and mixtures were immediately subjected to continuous chemiluminescence measurement using the VICTOR X5 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA) at 37 °C for 30 min. Maximal rates of the firefly luciferase (Fluc) accumulation were calculated using the OriginPro 7.5 software. The values were normalized to a positive control (0.3% DMSO, assigned a value of 100%).
The inhibition of eukaryotic translation was measured in a lysate of HEK293T cells, as described previously [43] with minor modifications. Briefly, the reaction was carried out in a total volume of 10 µL with the following reagents: 5 µL of HEK293T cell lysate, prepared as described here [54]; 1 µL of Translation Buffer 10x (200 mM HEPES-KOH pH 7.6, 10 mM DTT, 5 mM spermidine-HCl, 80 mM creatine phosphate, 10 mM ATP, 2 mM GTP, 0.25 mM of each amino acid); 0.5 µL of potassium acetate (2 M); 0.5 µL of magnesium acetate (20 mM); 0.5 µL of D-luciferin (10 mM) (Sigma-Aldrich, Burlington, MA, USA); 0.05 µL of RiboLock RNase Inhibitor (40 U/µL) (Thermo Fisher Scientific, Waltham, MA, USA); 0.5 µL of nuclease-free water; 1 µL of the tested compound or nuclease-free water; 1 µL of mRNA Fluc (100 ng), capped and polyadenylated. All compounds were tested at a final concentration of 30 μM. Reactions containing all components, except mRNA, were pre-incubated at 30 °C for 5 min. Then the mRNA was added, and the reactions were incubated in the CLARIOstar® Plus Microplate Reader (BMG Labtech, Ortenberg, Germany) at 30 °C for 1.5 h with continuous measurement of the luciferase activity. Maximal rates of the luciferase accumulation were calculated using the OriginPro 7.5 software. The values were normalized to a positive control (0.3% DMSO, assigned a value of 100%).
2.10. Bacteria Inhibition Assays
2.10.1. Detection of Translation Inhibitors Using pDualrep2 Reporter Strain
The “pDualrep2” system was used to evaluate the mechanism of antimicrobial action of the synthesised compounds. This system is based on hypersensitive strain E. coli JW5503 (∆tolC) (KanS) [55] transformed with “pDualrep2” plasmid, which allowed to sort out suppressors of protein synthesis or SOS-response induces [56,57]. Briefly, 1 µL of the solutions of Bac(1-10), Bac(1-10, R/Y), Bac(1-10, R/Y)-Cn-TPP, TPP-C10-Bac(1-10, R/Y), Onc112 (10 mM) in water were applied onto the agar plate that already contained a lawn of the reporter strain E. coli JW5503 (∆tolC) (KanS) and incubated overnight at 37 °C. The agar plate was scanned using the ChemiDoc Imaging System (Bio-Rad Laboratory, USA). This system consisted of two channels, “Cy3-blot” (553/574 nm, green pseudocolor) fluorescent for Turbo red fluorescent protein (TurboRFP) and “Cy5-blot” (588/633 nm, red pseudocolor) for Katushka2S fluorescence. Translation inhibitors triggered the induction of Katushka2S expression, while TurboRFP was upregulated by SOS response. Levofloxacin (LEV, 50 μg/mL, 1 μL) and erythromycin (ERY, 5 mg/mL, 1 μL) were utilized as positive controls for DNA and protein translation inhibitors, separately.
2.10.2. Testing the Antibacterial Activity of Substances on Plates with LB and Agar
Antibiotic activity was also evaluated against the following strains: E. coli JW5503 ΔtolC (KanS) ermC, modified by the plasmid pKH80 [58], providing resistance to erythromycin due to expression of ErmC methyltransferase; E. coli SQ171 ΔtolC transformed by a pAM552 plasmid [59]. E. coli SQ171 ΔtolC modified by a pAM552 plasmid with A2059G substitution in the 23S rRNA, E.coli JW1052 ΔmdtH, E. coli JW0912 ΔompF, E.coli JW0368 ΔsbmA [55]. The procedure was as described [31]. In short, Petri dishes filled with LB solid medium contained selective antibiotic and agar were covered with tested strain. After that 1 µL of the solutions of Bac(1-10), Bac(1-10, R/Y), Bac(1-10, R/Y)-Cn-TPP, TPP-C10-Bac(1-10, R/Y), Onc112 (10 mM), LEV (25 μg/mL) in water and ERY (5 mg/mL and 50 mg/mL) in DMSO were applied onto the agar plate. After overnight incubation at 37 °C, the plates were scanned by ChemiDoc (Bio-Rad, Benicia, CA, USA) in three channels (Cy2, Cy3, and Cy5). The obtained images were processed in the Image Lab software (Bio-Rad).
2.10.3. MIC Determination
The MICs for Bac(1-10), Bac(1-10, R/Y), Bac(1-10, R/Y)-Cn-TPP, TPP-C10-Bac(1-10, R/Y), and Onc112 were determined by LB broth microdilution, as recommended by CLSI in the Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard, 9th ed., CLSI document M07-A9, using in-house-prepared panels. The compounds were diluted in a 96-well microtiter plate to final concentrations ranging from 0.1 to 100 µM in a 200-µL aliquot of the bacterial suspension, followed by incubation at 37 °C for 18 h. Cell concentration was estimated according to the absorbance (A600). The measurements were performed on a Victor X5 2030 plate reader (Perkin Elmer, USA). The following strains of bacteria were used: E. coli BW25113, E. coli JW0368 ΔsbmA, B. subtilis 168, B. subtilis CFR. The lowest concentration of the test compound that completely inhibited bacterial growth was considered the minimum inhibitory concentration.
2.11. Measurement of B. subtilis Membrane Potential
The membrane potential of B. subtilis was estimated by measuring the fluorescence of the potential-dependent probe, diS-C3-(5) [60]. B. subtilis from the overnight culture were seeded into a fresh LB medium, followed by growth for 24 h until reaching the optical density of 0.8 at 600 nm. Then the bacteria were diluted 20-fold in a buffer containing 100 mM of KCl and 10 mM of Tris, pH 7.4. The fluorescence was measured at 670 nm (excitation at 630 nm) using a Fluorat-02-Panorama fluorimeter (Lumex Instruments, St.Petersburg, Russia).
2.12. In Vitro Survival Assay (MTT Assay)
The cytotoxicity of the substances under study was tested using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [61] with some modifications. Two thousand five hundred cells per well for the MCF7, HEK293T, and A549 cell lines or 4000 cells per well for the VA13 cell line were plated in 135 µL of DMEM-F12 media with 10% FBS (both Gibco, Waltham, MA, USA) in a 96-well plate and incubated in a 5% CO2 incubator for the first 16 h, without treatment. Then 15 µL of media-DMSO solutions of the tested substances were added to the cells (the final DMSO concentrations in the media were 1% or less), and the cells were treated for 72 h with 25 nM–50 µM (eight dilutions) of our substances (triplicate each). Serial dilutions of DMSO and doxorubicin were used as controls. The MTT reagent (Paneco LLC, Moscow, Russia) was then added to the cells to a final concentration of 0.5 g/L (10× stock solution in PBS was used) and incubated for 2.5 h at 37 °C in the incubator under an atmosphere of 5% CO2. The MTT solution was then discarded, and 140 µL of DMSO (PharmaMed LLC, Krasnodar, Russia) was added. The plates were swayed on a shaker (60 rpm) to dissolve the formazan. The absorbance was measured using VICTOR X5 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA) at a wavelength of 565 nm (in order to measure formazan concentration). The results were used to construct a dose-response graph and to estimate IC50 values.
3. Results
3.1. Modelling of Triphenylphosphonium Analogs of Bac7
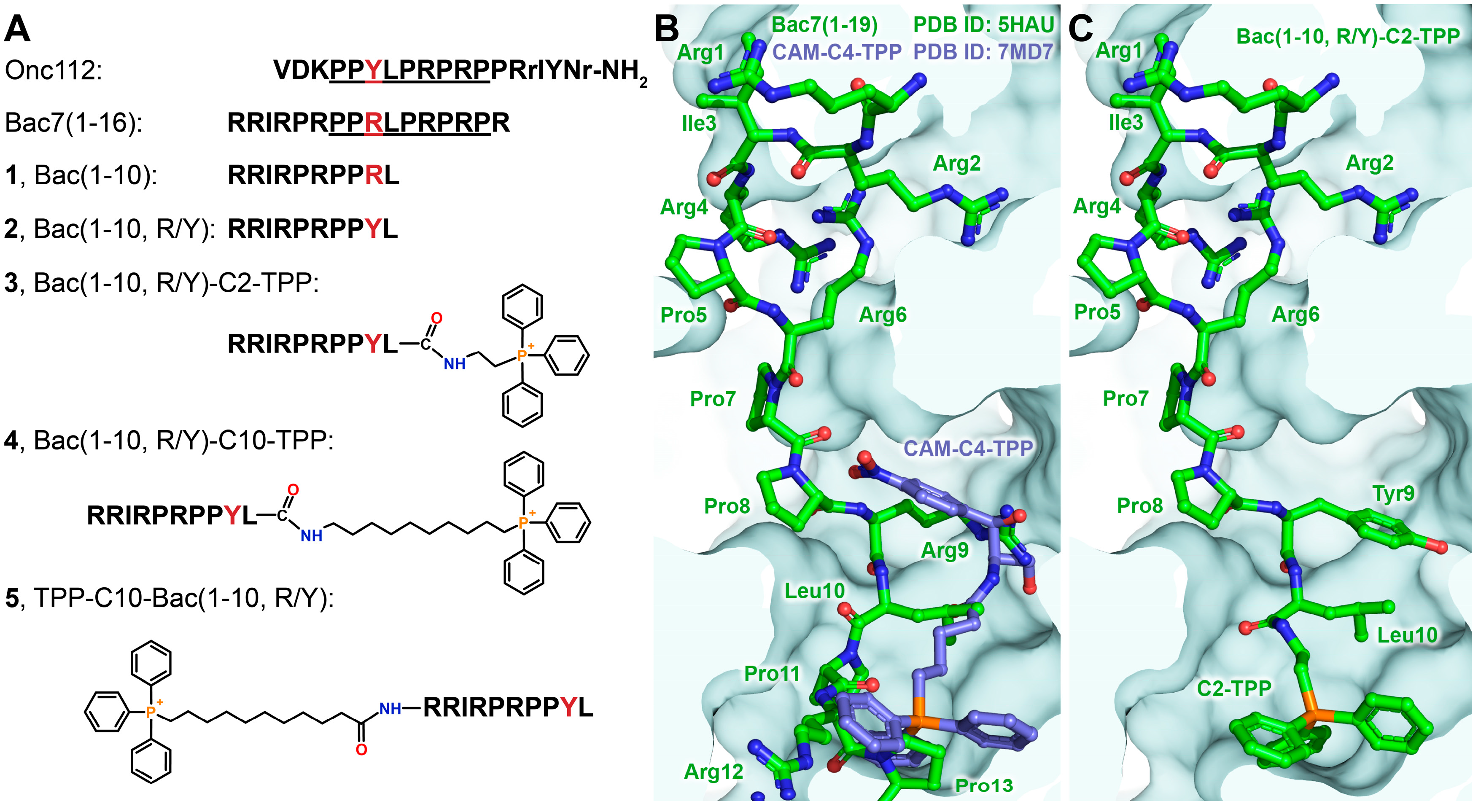
When designing analogs whose action is directed at bacterial ribosomes, we decided to use as short peptide as possible from the Bac7 N-terminal sequence and a triphenylphosphonium (TPP) cation attached to the peptide through linkers of various lengths (Figure 1A). It is known that the contacts formed by amino acid residues belonging to the N-terminal peptide sequence make the greatest contribution to the binding of Bac7, as well as Onc112, to bacterial ribosome, and almost identical conformations of the peptide backbone and side chains are observed for residues 7–13 of Bac7(1–16) and residues 4–10 of Onc112, with Arg9 of Bac7(1–16) substituting for Tyr6 of Onc112 [8,9,10]. Previous studies have shown that the introduction of a TPP group into the structure of chloramphenicol amine significantly increased its affinity for bacterial ribosomes [36,37]. In order to determine to which amino acid residue of short peptide from N-terminus Bac7 sequence, and through linkers of what length, alkyl(triphenyl)phosphonium cations could be attached, we performed superimposition the crystal structures of Bac7(1-16) (PDB ID: 5HAU) [12] and triphenylphosphonium analog of chloramphenicol (CAM-C4-TPP, PDB ID: 7MD7) [36] in complexes with the Thermus thermophilus ribosomes obtained by X-ray diffraction analysis (Figure 1B).
Therefore, a decapeptide 1-10 from the sequence of Bac7 (Figure 1A, Bac(1-10), 1) was chosen, moreover, the Arg9 residue was replaced by a Tyr one which is located in a similar position in the structure of Onc112 upon binding to the ribosome (compound 2 Bac(1-10, R/Y)). The alkyl(triphenyl)phosphonium cations should be attached to the C-terminal residue of peptide 2 (Leu10), so when the compounds bind to the ribosome, the TPP moieties would be directed deep into NPET. The length of the linker for compound 3 (Bac(1-10, R/Y)-C2-TPP) was chosen so that the TTP group fell into the same site on a ribosome as when binding the CAM-C4-TPP (Figure 1C). The choice of a linker for compound 4 (Bac(1-10, R/Y)-C10-TPP) was also based on data on the ribosome binding and the antibacterial activity of the TPP analog of chloramphenicol containing a longer linker [37]. Such alkyl-TPP-moiety can contribute to the interaction of the analog with the ribosome and facilitate the penetration inside bacterial cells by reducing the membrane potential. For comparison, a similar compound containing a decyl-TPP moiety at the N-terminus of the peptide 2 was also constructed (Figure 1A, TPP-C10-Bac(1-10, R/Y), 5).
Figure 1.
Structures of triphenylphosphonium analogs of Bac7. (A) Structures and abbreviations of the Bac7 analogs obtained in the work. The sequence that differs by only one amino acid, Tyr in the case of Onc112 and Arg in the case of Bac7(1-16), is underlined. (B) Superimposition of the crystal structures of Bac7(1-16) (green, PDB ID: 5HAU) [12] and triphenylphosphonium analog of chloramphenicol (blue, CAM-C4-TPP, PDB ID: 7MD7) [36] in complexes with the Thermus thermophilus ribosomes. (C) Model of the complex of compound 3 (Bac(1-10, R/Y)-C2-TPP) with the bacterial ribosome, derived from superimposition of X-ray diffraction data.
Figure 1.
Structures of triphenylphosphonium analogs of Bac7. (A) Structures and abbreviations of the Bac7 analogs obtained in the work. The sequence that differs by only one amino acid, Tyr in the case of Onc112 and Arg in the case of Bac7(1-16), is underlined. (B) Superimposition of the crystal structures of Bac7(1-16) (green, PDB ID: 5HAU) [12] and triphenylphosphonium analog of chloramphenicol (blue, CAM-C4-TPP, PDB ID: 7MD7) [36] in complexes with the Thermus thermophilus ribosomes. (C) Model of the complex of compound 3 (Bac(1-10, R/Y)-C2-TPP) with the bacterial ribosome, derived from superimposition of X-ray diffraction data.
3.2. Synthesis of Triphenylphosphonium Analogs of Bac7
Peptides 1 and 2 were synthesized by solid-phase synthesis on a 2-chlorotrityl resin using standard Fmoc/Pbf(tBu) procedure using HBTU as a condensation agent; Fmoc protective group was removed with piperidine solution, TFA with scavengers (reagent K) was used for cleavage of peptides from the resin and removing of side chain protective groups (Figure S1). For preparation of peptide 6, which served as started compound for synthesis of analogs 3 and 4, HFIP was used for cleavage from the resin while preserving Pbf and tBu protective groups (Figure S1). C-terminal carboxyl group of the peptide 6 was modified with amines, (2-aminoethyl)(triphenyl)phosphonium bromide or (10-aminodecyl)(triphenyl)phosphonium bromide (Figure S2), which were prepared in advance in two stages from dibromoalkanes and TPP with the formation of bromoalkyl derivatives of TPP [46] followed by their amination with 7M ammonia methanol solution [47]. Analog 5 was synthesized directly on the resin by condensation of carboxyl derivative of TPP [48] with peptidyl-polymer. The compounds were purified by silica gel column chromatography or HPLC and analyzed by LC-MS, NMR and HR-MS techniques (Supplementary Materials).
3.3. Triphenylphosphonium Derivatives of Decapeptide from N-terminal Sequence of Bac7 Bind to the Bacterial Ribosome
Since triphenylphosphonium analogs of Bac(1-10) were created as compounds acting on bacterial ribosomes, their ability to bind to 70S E. coli ribosomes was studied. For this purpose, starting from peptide 1, its fluorescent derivative BODIPY-Bac(1-10) was synthesized and its binding to E. coli ribosomes was investigated (Figure S3A). The apparent dissociation constant (KD) of BODIPY-Bac(1-10) was 77 ± 5 nM.
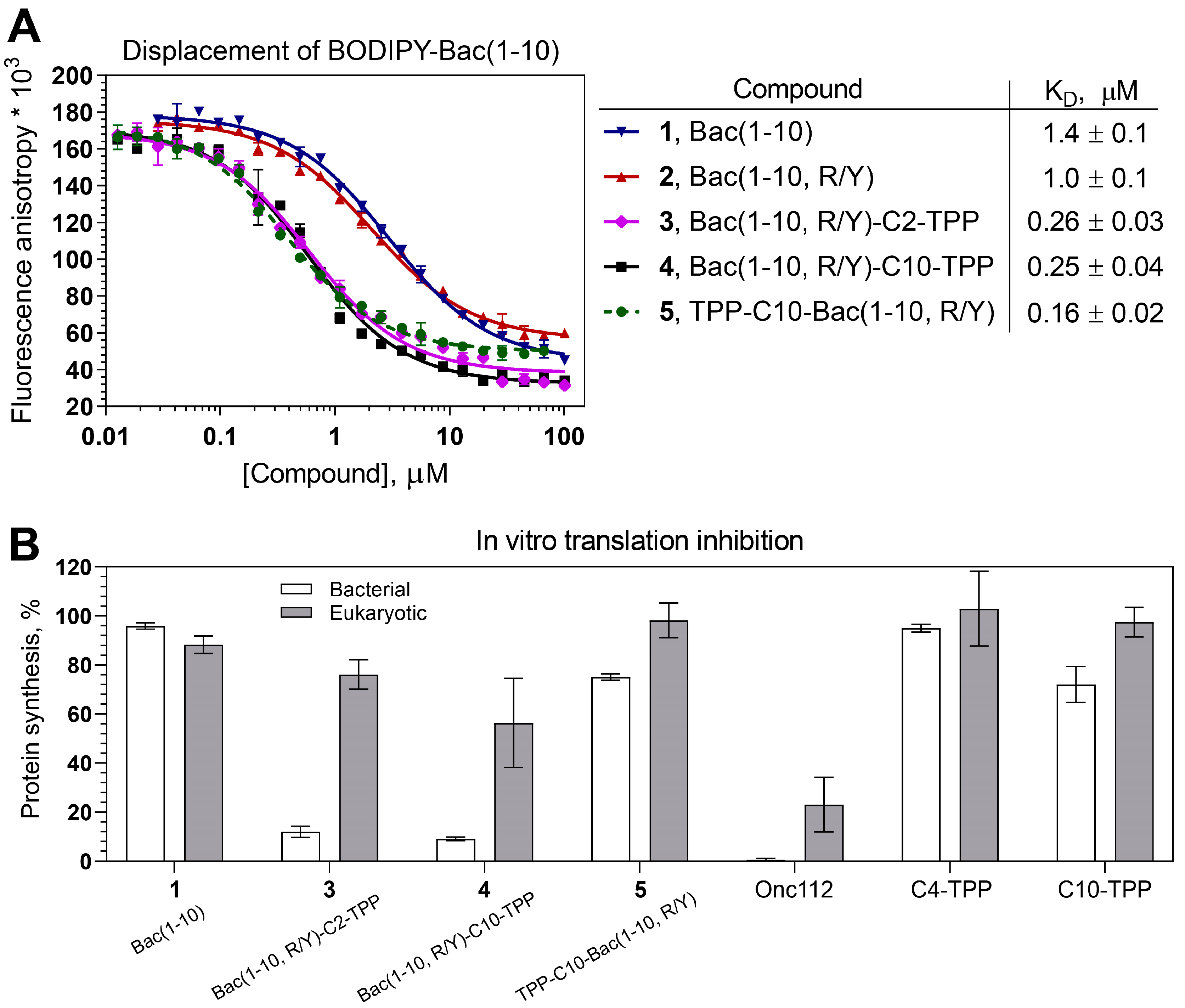
To assess the affinity of peptides 1 and 2 and analogs 3–5 for the 70S E. coli ribosome, a competition-binding assay with BODIPY-labeled decapeptide BODIPY-Bac(1-10) was used (Figure 2A). According to the results, the decapeptide (1-10) from the Bac sequence (1) retained the ability to bind to bacterial ribosomes (KD = 1.4 ± 0.1 µM), although its affinity was significantly lower than that of Onc112 (KD = 3 ± 1 nM, Figure S3B) and Bac7(1-16) [10]. Replacing of the Arg residue at position 9 with Tyr, which occupies the corresponding position in the Onc112 structure upon binding to bacterial ribosome, slightly increased the affinity of the peptide 2 (KD = 1.0 ± 0.1 µM) compared to that of the peptide 1. The introduction of TPP moieties significantly improved the affinity of analogs 3–5 to bacterial ribosomes in comparison with unmodified peptides 1 and 2. Compounds 3 and 4 exhibited approximately the same affinity (KD = 0.26 ± 0.03 µM and 0.25 ± 0.04 µM, respectively), while analog 5 showed the strongest binding (KD = 0.16 ± 0.02 µM). Despite having lower affinity for ribosomes compared to Onc112, the TPP derivatives still exhibit dissociation constants that are typical of numerous PA-AMPs [7,62,63].
Compounds 1–5 also displace fluorescently labeled erythromycin BODIPY-ERY [51] from its binding site (Figure S3C). This observation suggests that the compounds interact with the NPET, which contains overlapping binding sites for both ERY and Bac7 [12].
3.4. Bac(1-10, R/Y)-C2-TPP and Bac(1-10, R/Y)-C10-TPP Selectively Inhibit Prokaryotic Translation
The next step in testing antimicrobial compounds targeting the bacterial ribosome involves testing their ability to inhibit protein biosynthesis which was tested using the in vitro translation reaction in a cell-free transcription-translation system based on the E. coli S30 extract. Analogs 3 and 4 inhibited bacterial translation of firefly luciferase (Fluc) mRNA almost completely like Onc112, while peptide 2 practically did not inhibit this process, as well as the analog 5 which weakly inhibited bacterial translation (Figure 2B, white bars). These data confirm that analogs 3 and 4, designed so that they bind to the ribosome in such a way that the TPP group is directed deep into the NPET, are able to inhibit bacterial translation as it was observed earlier for TPP and berberine analogs of chloramphenicol [36,37,43]. It is noteworthy that even though compound 5 exhibits strong binding to ribosome, it shows very low inhibitory activity. This phenomenon has been observed among various derivatives of chloramphenicol, which bind to the upper part of the NPET and PTC [64,65,66].
It has been shown that Bac7(1-35) and Bac7(1-16), like some other Pro-Arg-rich AMP, can affect not only the process of bacterial translation, but also eukaryotic translation [10]. This property can be considered as a disadvantage for antibacterial compounds, since it can lead to various side effects when using these agents as medicals. To tested how TPP analogs of Bac(1-10) influence eukaryotic translation in vitro, we applied a cell-free translation system based on HEK293T lysates (Figure 2B, grey bars). At a concentration of 30 µM, compounds 2–5 inhibited translation much weaker than Onc112. Thus, the triphenylphosphonium analogs 3 and 4 were found to be more selective inhibitors of bacterial translation than Onc112 and Bac7 [10].
Figure 2.
Binding affinity to bacterial ribosomes and the inhibition of protein synthesis by triphenylphosphonium analogs of short peptide from Bac7. (A) A competitive binding assay to test the affinity of peptides 1 and 2 and their TPP analogs 3–5 to E. coli 70S ribosomes measured by fluorescence anisotropy of fluorescently labeled peptide, BODIPY-Bac(1-10). All experiments were performed at least twice, error bars are SD. The table displays the average apparent dissociation constants (KD) along with their CI (α = 0.05). (B) The inhibition of protein synthesis in vitro in the presence of Bac(1-10) (1) and triphenylphosphonium analogs – Bac(1-10, R/Y)-Cn-TPP (3: n = 2, 4: n = 10) and TPP-C10-Bac(1-10, R/Y) (5) – in a cell-free bacterial (white bars) and eukaryotic (grey bars) systems. Onc112 and alkyl-TPPs (C4-TPP and C10-TPP) were used as controls. All compounds were tested at a final concentration of 30 μM. Relative maximal rates of the firefly luciferase (Fluc) accumulation in vitro are shown. Experiments were performed at least two times, the error-bars represent the SD.
Figure 2.
Binding affinity to bacterial ribosomes and the inhibition of protein synthesis by triphenylphosphonium analogs of short peptide from Bac7. (A) A competitive binding assay to test the affinity of peptides 1 and 2 and their TPP analogs 3–5 to E. coli 70S ribosomes measured by fluorescence anisotropy of fluorescently labeled peptide, BODIPY-Bac(1-10). All experiments were performed at least twice, error bars are SD. The table displays the average apparent dissociation constants (KD) along with their CI (α = 0.05). (B) The inhibition of protein synthesis in vitro in the presence of Bac(1-10) (1) and triphenylphosphonium analogs – Bac(1-10, R/Y)-Cn-TPP (3: n = 2, 4: n = 10) and TPP-C10-Bac(1-10, R/Y) (5) – in a cell-free bacterial (white bars) and eukaryotic (grey bars) systems. Onc112 and alkyl-TPPs (C4-TPP and C10-TPP) were used as controls. All compounds were tested at a final concentration of 30 μM. Relative maximal rates of the firefly luciferase (Fluc) accumulation in vitro are shown. Experiments were performed at least two times, the error-bars represent the SD.
3.5. Triphenylphosphonium Derivatives of Decapeptide Related to Bac7 and Onc112 Exhibit Antibacterial Activity on Various Strains
3.5.1. Using the Double Reporter System pDualrep2 for the Preliminary Assessment of the Mechanism of Action of Triphenylphosphonium Derivatives of Decapeptide Related to Bac7 and Onc112
The JW5503 (∆tolC) (KanS) pDualrep2 strain was used for a preliminary assessment of the antibacterial action of the obtained compounds on bacterial cells and possible mechanisms of their antibacterial activity [56]. This E. coli ∆tolC reporter system is based on two proteins, RFP and Katushka2S, whose fluorescence can be independently detected, and can be used for screening inhibitors targeting either protein synthesis or DNA replication. The expression of far-red fluorescent protein, Katushka2S, occurs in pDualrep2 reporter system in the presence of ribosome-stalling compounds and is detected as red pseudocolor (Figure S4, Ery). The expression of red fluorescent protein, RFP, induced by compounds that trigger SOS response can be also detected as green pseudocolor (Figure S4, LEV).
Bac(1-10, R/Y)-C2-TPP (3), Bac(1-10, R/Y)-C10-TPP (4), and TPP-C10-(Bac(1-10, R/Y) (5) had an inhibitory effect on the growth of bacterial cells in this test. On the contrary, the peptides 1 and 2 did not show activity (Figure S4, data for peptide 2 are not shown), which is consistent with the data available in the literature [21]. In the future, experiments we used either peptide 1 or peptide 2 as control compounds when checking the antibacterial activity of the synthesized analogs. For Bac(1-10, R/Y)-C2-TPP (3) and Bac(1-10, R/Y)-C10-TPP (4), as well as for the Onc112 and positive controls, ERY, red pseudocolor rings were observed, indicating that these compounds specifically inhibit protein synthesis. Peptide 2 did not cause zone of inhibition, TPP-C10-Bac(1-10, R/Y) (5) slightly inhibited bacterial cell growth (dark area in the middle). C10-TPP caused a large zone of inhibition, but does not induce the reporters, which suggests that it has antibacterial properties, but the mechanism is different from those that can be detected by pDualrep2 reporter system (Figure S4).
Thus, testing of the compounds using a double reporter system showed that the mechanism of action of the triphenylphosphonium analogs of Bac(1-10, R/Y) 3 and 4 on bacterial cells is similar to the parent PA-AMP Bac7 and Onc112.
3.5.2. Triphenylphosphonium Derivatives of Decapeptide from N-terminal Sequence of Bac7 Exhibit Antibacterial Activity on Various Strains Including Some Resistant Laboratory Strains
Like most PA-AMP, Onc112 and Bac7 inhibit the growth of gram-negative bacteria [5,25], while alkyl-TPP and their derivatives act on gram-positive bacteria [40]. Since compounds 3–5 combine in their structure a decapeptide, which is related to both Bac7 and Onc112, as well as the alkyl-TPP fragment, we tested the effect of these analogs on various gram-negative and gram-positive strains (Figure 3, Table 1). According to the results obtained, decapeptides 1 and 2 did not inhibit bacterial growth, this is in accordance with the known data that peptides containing less than 16 a.a. from the sequence of Bac7 do not exhibit antibacterial activity [21].
Unlike peptides 1 and 2, triphenylphosphonium derivatives 3–5 containing modifications at both the N- or C-terminus of the peptide 2 were active against E. coli strains, and also inhibited the growth of B. subtilis, unlike Onc112 (Table 1). It is also worth noting that if analogs 4 and 5 contain a decyl-TPP fragment (C10-TPP) that was shown to have an antibacterial effect itself, then analog 3 contains ethyl-TPP, which does not have such activity [40].

Table 1.
Suppression of the growth of E. coli strain (BW25113), E. coli strain with the deletion of the sbmA gene (∆sbmA) or B. subtilis (168) and the harboring CFR resistant (B. subtilis-CFR) strains by TPP analogs of decapeptide related to Bac7 and Onc112. The values of a minimal inhibitory concentration (MIC, µM) are shown1.
Table 1.
Suppression of the growth of E. coli strain (BW25113), E. coli strain with the deletion of the sbmA gene (∆sbmA) or B. subtilis (168) and the harboring CFR resistant (B. subtilis-CFR) strains by TPP analogs of decapeptide related to Bac7 and Onc112. The values of a minimal inhibitory concentration (MIC, µM) are shown1.
| E. coli BW25113 | E. coli ∆sbmA | B. subtilis 168 | B. subtilis-CFR | ||
|---|---|---|---|---|---|
| 1 | - | - | >50 | >50 | |
| 2 | >100 | >100 | - | - | |
| 3 | 26.3 | >100 | 16.7 | 41.7 | |
| 4 | 100 | 100 | 1.3 | 0.8 | |
| 5 | 5.3 | 5.3 | 10.4 | 12.5 | |
| Onc112 | 11.6 | >90 | >50 | >50 | |
| C4-TPP | >100 | >100 | >100 | >100 | |
| C10-TPP | 50 | 50 | 1.3 | 0.8 | |
| Ery | 170 | 170 | <0.1 | <0.1 | |
1The MIC values were determined using the double-dilution method. The MIC for each compound was determined in triplicate in two independent sets.
It have been shown, that mutations of 23S rRNA nucleotides in the peptide exit ribosome tunnel, such as A2503C, A2059C, and especially the double mutation (A2503C/A2059G) lead to increased resistance to Onc112, but not to Bac7(1-35) [5,12]. The authors [12] have explain this effect by the structural features of the binding of Onc112 in the ribosomal tunnel when the enhanced stacking between a backbone chain of the Arg11 residue from the Onc112 and the key for ribosome stalling nucleotide A2062, which interacts with both nucleotides A2503 and A2059. As a result, any mutation in these nucleotides can affect the position of A2062 and to have a greater effect on the binding of Onc112 than Bac7(1–35). In the structures of analogs 3 and 4, there is no amino acid residue corresponding to the position of Arg11 of the Onc112, but they contain bulky alkyl-TPP substituent directed towards a region of nucleotides A2062, A2503 and A2059 when binding to the ribosome. Therefore, the triphenylphosphonium analogs of Bac7 were tested on CHL-resistant B. subtilis strain (B. subtilis-CFR) harboring plasmid encoding a methyltransferase, that modifies A2503 of 23S rRNA (cfr) (Table 1), on macrolide-resistant E. coli strains SQ171 ΔtolC (A2059G) in which the A2059 nucleotide is replaced by G2059 in the 23S rRNA gene (Figure 3B), and E. coli JW5503 (∆tolC) (KanS) ermC harboring plasmid encoding a methyltransferase, that catalyzes methylation of A2058 of 23S rRNA (Figure 3C).
As it was previously shown for B. subtilis, analogs 3 and 4, containing alkyl-TPP moieties at C-terminus of peptide, also act on a resistant B. subtilis strain (Table 1). On E. coli strains resistant to macrolides (Figure 3), the same analogs 3 and 4 also showed bacterial growth inhibiting activity, unlike erythromycin. However, it should be noted that Onc112 in these experiments also showed activity against resistant E. coli bacteria comparable to that demonstrated on the wild type.
3.5.3. Bac(1-10, R/Y)-C2-TPP Penetrates into the E. coli Cell with the Participation of the Transporter Protein SbmA
PA-AMP have been shown to act on gram-negative bacteria by penetrating through the inner membrane without destroying it, using special protein transporters such as SbmA protein [22,24,67]. E. coli strains with deletions of a SbmA gene have been less sensitive to Bac7(1-35) and Bac7(1-16) as well as to oncocins, including Onc112, compared with wild type strains [21,23]. Onc112 also suppressed to a lesser extent the growth of strains with deletions of a gene of another transporter protein, MdtH, or of both SbmA and MdtH genes simultaneously compared with a wild type E. coli strain [23].
The TPP analogs of decapeptide Bac(1-10, R/Y) 3–5, as well as peptides 1 and Onc112 were tested on three laboratory E. coli strains with deletions of genes of transporter proteins of the inner membrane sbmA and mdtH or the outer membrane protein F gene (ompF, Figure 4, Table 1). The latter protein is a porin wich is responsible for the passive transport of small molecules, such as nutrients and antibiotics into cells [68].
As follows from the results obtained on the plates there were not any difference in the inhibition of E. coli ΔompF, E. coli ΔmdtH and a wild-type strain under the action of both the synthetic analogs 3–5 and Onc112 (Figure 4). Only in the case of the strain E. coli ΔsbmA for the analog 3 and Onc112, the difference was observed in activity compared to that against the wild-type strain (Figure 4B). Values of MICs were determined for all compounds on these strains (Table 1). It turned out that the corresponding values differed by about 8 times for Onc112 and 4 times for analog 3, while for analog 4, as well as for C10-TPP, the MIC values were the same for strains with deletion of the transporter protein and the wild-type strain. Based on these results, we conclude that analog 3 is transported through the E. coli membrane mainly with the participation of a transporter protein SbmA, like Onc112. Unlike analog 3, analog 4, containing a decyl-TPP fragment in its structure, is transferred to a bacterial cell as C10-TPP using a different mechanism peculiar to alkyl derivatives of TPP [40,69].
Thus, as a result of testing of antibacterial activity on various strains, it was shown that TPP analogs of a decapeptide related to Bac7 and Onc112 have antimicrobial action, and are able to act both against gram-negative bacteria, like oncocin and bactenecin, and against gram-positive ones, like alkyl derivatives of TPP, and also inhibit the growth of some resistant laboratory strains. It has also shown that decapeptide as part of its triphenylphosphonium derivative containing a short alkyl linker is transferred into the E. coli cell with the participation of the transporter protein SbmA.
3.6. Bac(1-10, R/Y)-Cn-TPP Cause a Decrease in the Membrane Potential of B. subtilis
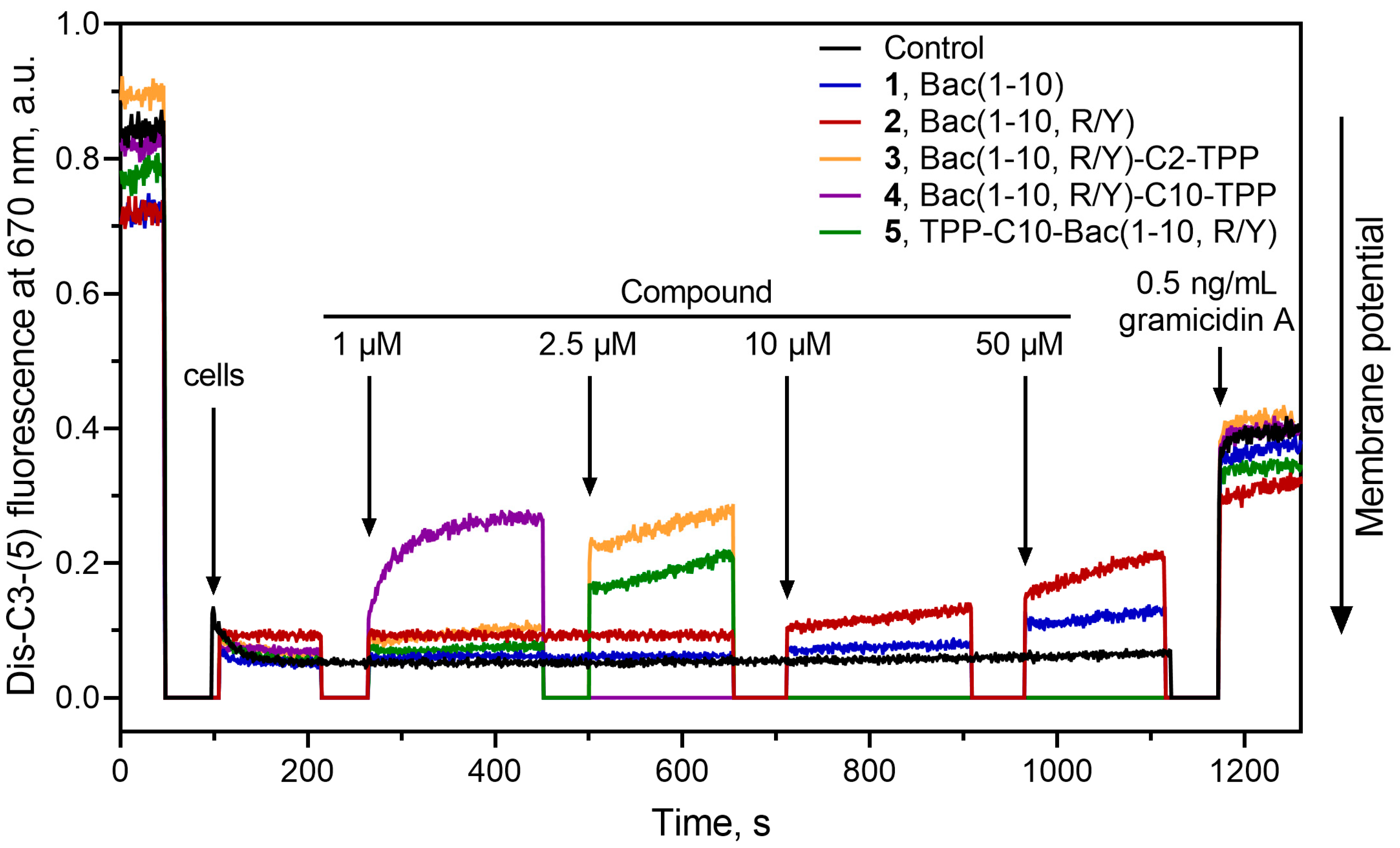
Previously it has been shown that alkyl(triphenyl)phosphonium molecules, containing eight or more methylene groups, as well as, their derivatives showed the ability to reduce the membrane potential of bacteria [37,40,41]. Thus, we further examined the effect of triphenylphosphonium analogs of decapeptide related to Bac7 and Onc112 3–5, as well as parent peptides 1 and 2 on the bacterial membrane potential of B. subtilis. Gramicidin A, a channel-forming antibiotic, was used to cause the disappearance of the bacterial membrane potential. The fluorescent potential-sensitive dye diS-C3-(5) was applied for measuring the change in the membrane potential of B. subtilis [60].
Bac(1-10, R/Y)-C10-TPP (4) at a concentration of 1 µM caused a rapid decrease in membrane potential of B. subtilis to a level closed to that observed in the presence of the gramicidin A (Figure 5). A decrease of the membrane potential to a somewhat lesser extent was observed in the presence of TPP-C10-Bac(1-10, R/Y) (5) at a concentration of 2.5 µM. These effects were expected, because both these compounds contain decyl-TPP moiety in their structures. Moreover, in the case of Bac(1-10, R/Y)-C2-TPP (3) containing a short ethyl linker between the TPP and the C-terminus of the peptide, there was also a release of a fluorescent dye from cells, which indicates a decrease of the membrane potential of B. subtilis. It is possible that the combination of several positive charges and a hydrophobic TPP group in this amphiphilic compound with linear flexible structure might contribute to its interaction with negatively charged lipids (in particular, cardiolipin) of the B. subtilis membrane and promote peptide accumulation on the bacterial surface [70,71]. This also does not exclude the possible interaction of the compound 3 with endogenous fatty acids, as it happens in the case of penetration of alkyl-TPP derivatives through the mitochondrial membrane [69].
Peptides 1 and 2 did not cause some significant decrease in membrane potential even at concentration of 50 µM.
Thus, the action of TPP analogs of decapeptide related to Bac7 and Onc112 on bacterial cells can be associated with the inhibition of protein biosynthesis and depolarization of the bacterial membrane, similar to that it was previously shown for the TPP and berberine analogs of chloramphenicol [37,43]. Moreover, unlike compounds from our previously studied series of conjugates of penetrating hydrophobic cations with chloramphenicol, in this series of TPP derivatives of decapeptide related to Bac7 and Onc112 all compounds, both containing a long alkyl linker between TPP and the peptide and a short one, had an effect on the bacterial membrane. An arginine-rich peptide in the structure of the Bac(1-10, R/Y)-C2-TPP, apparently contributes to the effect of the analog on the membrane potential, however, the mechanism of this action needs to be clarified and further investigated.
Figure 5.
Effect of triphenylphosphonium analogs Bac(1-10, R/Y)-Cn-TPP (3: n = 2, 4: n = 10), TPP-C10-Bac(1-10, R/Y) (5), and the parent peptides 1 and 2 on the kinetics of the membrane potential of B. subtilis cells measured using the fluorescent probe diS-C3-(5). Arrows mark moments at which appropriate amounts of compounds were added. The Gramicidin A at a concentration of 0.5 ng/mL was used as a control.
Figure 5.
Effect of triphenylphosphonium analogs Bac(1-10, R/Y)-Cn-TPP (3: n = 2, 4: n = 10), TPP-C10-Bac(1-10, R/Y) (5), and the parent peptides 1 and 2 on the kinetics of the membrane potential of B. subtilis cells measured using the fluorescent probe diS-C3-(5). Arrows mark moments at which appropriate amounts of compounds were added. The Gramicidin A at a concentration of 0.5 ng/mL was used as a control.
3.7. Bac(1-10, R/Y)-C2-TPP is Non-toxic for Mammalian Cells
It is known that alkyl derivatives of TPP can be toxic to mammalian cells, herewith, both antibacterial and toxic effects are observed for TPP derivatives containing a relatively long alkyl radical [37]. For test the cytotoxicity of triphenylphosphonium analogs of Bac7 for mammalian cells we used the Mosmann (“MTT”) assay [61].
As the results showed, the analog 3 containing a short linker between the C-terminal carbonyl group of the peptide and TPP turned out to be non-toxic to cell lines used: HEK293T, MCF7, VA13, and A549 (Table 2). The toxicity of analogs 4 and 5 containing decyl-TPP fragments, both at the C-end or at the N-end of the peptide, was reduced compared to C10-TPP.
The absence of a toxic effect in combination with an inhibitory effect on the bacterial translation process and the ability of the Bac(1-10, R/Y)-C2-TPP to suppress the growth of both gram-negative and gram-positive bacteria, including some resistant strains, is an important property from the point of view of using the found structural and functional patterns to create antibacterial drugs.
4. Conclusions
Triphenylphosphonium derivatives of a decapeptide related to sequences of Bac7 and Onc112 were synthesized in this study as potential antimicrobial compounds. It was shown that while reducing the length of the antimicrobial peptide to 10 a.a. residues, the affinity to bacterial ribosomes dramatically decreases, the modification of peptide with alkyl-TPP moieties leads to an increase in affinity. Triphenylphosphonium analogs, designed so that when they bind to the ribosome the TPP group could be directed deep into the NPET, were able to inhibit bacterial translation more selectively than Onc112 and Bac7. Triphenylphosphonium analogs of a decapeptide related to Bac7 and Onc112 due to its antimicrobial activity were able to act against gram-negative bacteria similar to oncocin and bactenecin and against gram-positive bacteria as alkyl derivatives of TPP. Also they inhibited the growth of some resistant laboratory strains. It has also shown that Bac(1-10, R/Y)-C2-TPP, containing a short alkylamide linker between the decapeptide and TPP, is transferred into the E. coli cell by the transporter protein SbmA. Triphenylphosphonium derivatives of the decapeptide Bac(1-10, R/Y) containing decylamide as well as ethylamide linker caused B. subtilis membrane depolarization similar to alkyl-TPP. The analog Bac(1-10, R/Y)-C2-TPP which has been shown to combine the antibacterial properties of alkyl-TPP and Pro-Arg-rich AMP and to differ in reduced side effects inherent in the original molecules, such as toxicity and effect on eukaryotic translation, stands out in the series. These properties follow from the structure of the compound, which combines a short peptide related to the sequences of Bac7 and Onc112 and TPP, attached to the C-terminal amino acid residue of the peptide through a short alkylamide linker. The structural features of triphenylphosphonium analogs of short peptide related to Bac7 and Onc112 found in this study can be applied in the future for the creation of therapeutic antibacterial agents based on AMP.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Detailed procedures of the synthesis and HRMS data; Figure S1. Scheme of the chemical synthesis of short peptide from Bac 7 (Bac(1-10), 1), its R9/Y-analog (Bac(1-10, R/Y), 2), their triphenylphosphonium analog TPP-C10-Bac(1-10, R/Y), 5, and protected peptide 6: i – 20% Pip/DMF, ii – HBTU/DIPEA/DMF, iii – reagent K, iv – HFIP/CH2Cl2, v – TPP-(CH2)10-COOH/HBTU/DIPEA/DMF; Figure S2. Scheme of the chemical synthesis of triphenylphosphonium analogs of short peptide from Bac7: (Bac(1-10, R/Y)-C2-TPP, 3) and (Bac(1-10, R/Y)-C10-TPP, 4); Figure S3. Binding affinity to bacterial ribosomes of triphenylphosphonium analogs of short peptide from Bac7; Figure S4. Testing of antibacterial activity of triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112 using E. coli (ΔtolC) pDualrep2 strain. Figure S5. HRMS data for Bac(1-10) (1); Figure S6. HRMS data for Bac(1-10, R/Y) (2); Figure S7. HRMS data for Bac(1-10, R/Y)-C2-TPP (3); Figure S8. HRMS data for Bac(1-10, R/Y)-C10-TPP (4); Figure S9. HRMS data for TPP-C10-Bac(1-10, R/Y) (5).
Author Contributions
Synthesis, N.V.S., A.G.T., Z.Z.K., D.A.S.; purification, N.V.S., A.G.T., Z.Z.K.; LC-MS analysis, V.N.T.; binding assay, A.G.T.; translation inhibition assay, I.A.V., J.A.P.; molecular modelling, A.G.T.; membrane potential assays; P.A.N.; 70S ribosomes preparation, A.L.K., A.P.; MTT-tests, D.A.I.; bacteria inhibition assays, D.A.L., E.A.R., O.V.E.; HRMS analysis, Y.V.T., I.A.R.; supervision, N.V.S., I.A.O., P.V.S., I.A.R., A.A.B.; writing—original draft preparation, N.V.S., Z.Z.K., A.G.T.; writing—review and editing, N.V.S., A.G.T., O.A.D., P.V.S., I.A.O., A.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation [project No. 23-24-00247 to N.V.S.].
Acknowledgments
We thank M.V. Serebriakova for MALDI mass spectrometry analyses. This study was carried out using equipment purchased with funds from the Lomonosov Moscow State University Development Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-Rich Antimicrobial Peptides Targeting Protein Synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-Rich Antimicrobial Peptides: Converging to a Non-Lytic Mechanism of Action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, M.; Grzela, R.; Giglione, C.; Meinnel, T.; Gennaro, R.; Mergaert, P.; Scocchi, M. The Host Antimicrobial Peptide Bac71-35 Binds to Bacterial Ribosomal Proteins and Inhibits Protein Synthesis. Chem. Biol. 2014, 21, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Volke, D.; Weinert, S.; Sträter, N.; Knappe, D.; Hoffmann, R. Insect-Derived Proline-Rich Antimicrobial Peptides Kill Bacteria by Inhibiting Bacterial Protein Translation at the 70 S Ribosome. Angew. Chem. Int. Ed. 2014, 53, 12236–12239. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, A.C.; Nguyen, F.; Antunes, S.; Pérébaskine, N.; Graf, M.; Arenz, S.; Inampudi, K.K.; Douat, C.; Guichard, G.; Wilson, D.N.; et al. The Proline-Rich Antimicrobial Peptide Onc112 Inhibits Translation by Blocking and Destabilizing the Initiation Complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.N.; Lomakin, I.B.; Gagnon, M.G.; Steitz, T.A. The Mechanism of Inhibition of Protein Synthesis by the Proline-Rich Peptide Oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469. [Google Scholar] [CrossRef]
- Seefeldt, A.C.; Graf, M.; Pérébaskine, N.; Nguyen, F.; Arenz, S.; Mardirossian, M.; Scocchi, M.; Wilson, D.N.; Innis, C.A. Structure of the Mammalian Antimicrobial Peptide Bac7(1–16) Bound within the Exit Tunnel of a Bacterial Ribosome. Nucleic Acids Res. 2016, 44, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Yang, M.; Sun, R.N.; Liu, Y.; Wang, W.; Xi, Q.; Gong, H.; Chen, C. Mechanism of Actions of Oncocin, a Proline-Rich Antimicrobial Peptide, in Early Elongation Revealed by Single-Molecule FRET. Protein Cell 2018, 9, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of Proline-Rich Peptides Bound to the Ribosome Reveal a Common Mechanism of Protein Synthesis Inhibition. Nucleic Acids Res. 2016, 44, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An Antimicrobial Peptide That Inhibits Translation by Trapping Release Factors on the Ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Huter, P.; Maracci, C.; Peterek, M.; Rodnina, M.V.; Wilson, D.N. Visualization of Translation Termination Intermediates Trapped by the Apidaecin 137 Peptide during RF3-Mediated Recycling of RF1. Nat. Commun. 2018, 9, 3053. [Google Scholar] [CrossRef] [PubMed]
- Skowron, K.J.; Baliga, C.; Johnson, T.; Kremiller, K.M.; Castroverde, A.; Dean, T.T.; Allen, A.C.; Lopez-Hernandez, A.M.; Aleksandrova, E.V.; Klepacki, D.; et al. Structure–Activity Relationships of the Antimicrobial Peptide Natural Product Apidaecin. J. Med. Chem. 2023, 66, 11831–11842. [Google Scholar] [CrossRef] [PubMed]
- Mangano, K.; Klepacki, D.; Ohanmu, I.; Baliga, C.; Huang, W.; Brakel, A.; Krizsan, A.; Polikanov, Y.S.; Hoffmann, R.; Vázquez-Laslop, N.; et al. Inhibition of Translation Termination by the Antimicrobial Peptide Drosocin. Nat. Chem. Biol. 2023, 19, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Dorn, A. Differential Infectivity of Two Pseudomonas Species and the Immune Response in the Milkweed Bug, Oncopeltus Fasciatus (Insecta: Hemiptera). J. Invertebr. Pathol. 2001, 78, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Knappe, D.; Piantavigna, S.; Hansen, A.; Mechler, A.; Binas, A.; Nolte, O.; Martin, L.L.; Hoffmann, R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH 2 ): A Novel Antibacterial Peptide Optimized against Gram-Negative Human Pathogens. J. Med. Chem. 2010, 53, 5240–5247. [Google Scholar] [CrossRef] [PubMed]
- Knappe, D.; Kabankov, N.; Hoffmann, R. Bactericidal Oncocin Derivatives with Superior Serum Stabilities. Int. J. Antimicrob. Agents 2011, 37, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Knappe, D.; Ruden, S.; Langanke, S.; Tikkoo, T.; Ritzer, J.; Mikut, R.; Martin, L.L.; Hoffmann, R.; Hilpert, K. Optimization of Oncocin for Antibacterial Activity Using a SPOT Synthesis Approach: Extending the Pathogen Spectrum to Staphylococcus Aureus. Amino Acids 2016, 48, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, M.; Scocchi, M.; Podda, E.; Skerlavaj, B.; Dolzani, L.; Gennaro, R. Antimicrobial Activity of Bac7 Fragments against Drug-Resistant Clinical Isolates. Peptides 2004, 25, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia Coli SbmA in the Antimicrobial Activity of Proline-rich Peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Knappe, D.; Hoffmann, R. Influence of the yjiL-mdtM Gene Cluster on the Antibacterial Activity of Proline-Rich Antimicrobial Peptides Overcoming Escherichia Coli Resistance Induced by the Missing SbmA Transporter System. Antimicrob. Agents Chemother. 2015, 59, 5992–5998. [Google Scholar] [CrossRef] [PubMed]
- Ghilarov, D.; Inaba-Inoue, S.; Stepien, P.; Qu, F.; Michalczyk, E.; Pakosz, Z.; Nomura, N.; Ogasawara, S.; Walker, G.C.; Rebuffat, S.; et al. Molecular Mechanism of SbmA, a Promiscuous Transporter Exploited by Antimicrobial Peptides. Sci. Adv. 2021, 7, eabj5363. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar] [PubMed]
- Stalmans, S.; Wynendaele, E.; Bracke, N.; Knappe, D.; Hoffmann, R.; Peremans, K.; Polis, I.; Burvenich, C.; Spiegeleer, B. Blood-Brain Barrier Transport of Short Proline-Rich Antimicrobial Peptides. PPL 2014, 21, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Safronova, V.N.; Kruglikov, R.N.; Bolosov, I.A.; Ovchinnikova, T.V. Genomic Insights into Bacterial Resistance to Proline-Rich Antimicrobial Peptide Bac7. Membranes 2023, 13, 438. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Arasteh, S.; Haney, E.F.; Hancock, R.E.W. Tryptic Stability of Synthetic Bactenecin Derivatives Is Determined by the Side Chain Length of Cationic Residues and the Peptide Conformation. J. Med. Chem. 2016, 59, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.Y.; Björkling, F.; Zabicka, D.; Tomczak, M.; Urbas, M.; Domraceva, I.; Kreicberga, A.; Franzyk, H. Structure-Activity Study of Oncocin: On-Resin Guanidinylation and Incorporation of Homoarginine, 4-Hydroxyproline or 4,4-Difluoroproline Residues. Bioorg. Chem. 2023, 141, 106876. [Google Scholar] [CrossRef] [PubMed]
- Kolano, L.; Knappe, D.; Berg, A.; Berg, T.; Hoffmann, R. Effect of Amino Acid Substitutions on 70S Ribosomal Binding, Cellular Uptake, and Antimicrobial Activity of Oncocin Onc112. ChemBioChem 2022, 23, e202100609. [Google Scholar] [CrossRef] [PubMed]
- Khairullina, Z.Z.; Makarov, G.I.; Tereshchenkov, A.G.; Buev, V.S.; Lukianov, D.A.; Polshakov, V.I.; Tashlitsky, V.N.; Osterman, I.A.; Sumbatyan, N.V. Conjugates of Desmycosin with Fragments of Antimicrobial Peptide Oncocin: Synthesis, Antibacterial Activity, Interaction with Ribosome. Biochem. (Mosc.). 2022, 87, 871–889. [Google Scholar] [CrossRef] [PubMed]
- Samizadeh, M.; Zhang, X.; Gunaseelan, S.; Nelson, A.G.; Palombo, M.S.; Myers, D.R.; Singh, Y.; Ganapathi, U.; Szekely, Z.; Sinko, P.J. Colorectal Delivery and Retention of PEG-Amprenavir-Bac7 Nanoconjugates—Proof of Concept for HIV Mucosal Pre-Exposure Prophylaxis. Drug Deliv. and Transl. Res. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Bonke, G.; Larsen, C.J.; Yavari, N.; Nielsen, P.E.; Franzyk, H. Antibacterial Peptide Nucleic Acid–Antimicrobial Peptide (PNA–AMP) Conjugates: Antisense Targeting of Fatty Acid Biosynthesis. Bioconjugate Chem. 2016, 27, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Goldbach, T.; Knappe, D.; Reinsdorf, C.; Berg, T.; Hoffmann, R. Ribosomal Binding and Antibacterial Activity of Ethylene Glycol-bridged Apidaecin Api137 and Oncocin Onc112 Conjugates. J. Pept. Sci. 2016, 22, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Schmitt, S.; Heynisch, A.; Gumpinger, A.; Wüthrich, I.; Gysin, M.; Shcherbakov, D.; Hobbie, S.N.; Panke, S.; Held, M. Optimization of the Antimicrobial Peptide Bac7 by Deep Mutational Scanning. BMC Biol. 2022, 20, 114. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Pavlova, J.A.; Lukianov, D.A.; Tereshchenkov, A.G.; Makarov, G.I.; Khairullina, Z.Z.; Tashlitsky, V.N.; Paleskava, A.; Konevega, A.L.; Bogdanov, A.A.; et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics 2021, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, J.A.; Khairullina, Z.Z.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Volynkina, I.A.; Skvortsov, D.A.; Makarov, G.I.; Abad, E.; Murayama, S.Y.; et al. Triphenilphosphonium Analogs of Chloramphenicol as Dual-Acting Antimicrobial and Antiproliferating Agents. Antibiotics 2021, 10, 489. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, E.J.; Hulit, J.; Lagasse, F.P.; Lechertier, T.; Stevenson, B.; Tudor, C.; Trebicka, D.; Sparey, T.; Ratcliffe, A.J. Impact of Mitochondrial Targeting Antibiotics on Mitochondrial Function and Proliferation of Cancer Cells. ACS Med. Chem. Lett. 2021, 12, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Retamal, S.; Sandoval-Acuña, C.; Peredo-Silva, L.; Guzmán-Rivera, D.; Pavani, M.; Torrealba, N.; Truksa, J.; Castro-Castillo, V.; Catalán, M.; Kemmerling, U.; et al. Complex Mitochondrial Dysfunction Induced by TPP+-Gentisic Acid and Mitochondrial Translation Inhibition by Doxycycline Evokes Synergistic Lethality in Breast Cancer Cells. Cells 2020, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Khailova, L.S.; Nazarov, P.A.; Sumbatyan, N.V.; Korshunova, G.A.; Rokitskaya, T.I.; Dedukhova, V.I.; Antonenko, Yu.N.; Skulachev, V.P. Uncoupling and Toxic Action of Alkyltriphenylphosphonium Cations on Mitochondria and the Bacterium Bacillus Subtilis as a Function of Alkyl Chain Length. Biochem. (Mosc.). 2015, 80, 1589–1597. [Google Scholar] [CrossRef]
- Nazarov, P.A.; Osterman, I.A.; Tokarchuk, A.V.; Karakozova, M.V.; Korshunova, G.A.; Lyamzaev, K.G.; Skulachev, M.V.; Kotova, E.A.; Skulachev, V.P.; Antonenko, Y.N. Mitochondria-Targeted Antioxidants as Highly Effective Antibiotics. Sci. Rep. 2017, 7, 1394. [Google Scholar] [CrossRef] [PubMed]
- Pinto, T.C.A.; Banerjee, A.; Nazarov, P.A. Triphenyl phosphonium-based substances are alternatives to common antibiotics. Bulletin of RSMU 2018, 7, 16–21. [Google Scholar] [CrossRef]
- Pavlova, J.A.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Skvortsov, D.A.; Polshakov, V.I.; Vasilieva, B.F.; Efremenkova, O.V.; Kaiumov, M.Y.; Paleskava, A.; et al. Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics 2022, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, I.H.; Roche, E.D.; Beeman, D.; Lynch, A.S.; Ding, C.Z.; Ma, Z. Design, Synthesis, and Biological Evaluation of BODIPY®–Erythromycin Probes for Bacterial Ribosomes. Bioorganic Med. Chem. Lett. 2006, 16, 794–797. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Dicker, D.W.; Whiting, M.C. 407. Synthetical Studies on Terpenoids. Part I. The Synthesis of Squalene. J. Chem. Soc. 1958, 1994. [Google Scholar] [CrossRef]
- S. G. Romero. Modified Creatine Compounds 2015000 5258, 2015.
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Yu.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 1. Cationic Plastoquinone Derivatives: Synthesis and in Vitro Studies. Biochem. (Mosc.). 2008, 73, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V.; Wintermeyer, W. GTP Consumption of Elongation Factor Tu during Translation of Heteropolymeric mRNAs. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 1945–1949. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Shishkina, A.V.; Karpenko, V.V.; Chertkov, V.A.; Konevega, A.L.; Kasatsky, P.S.; Bogdanov, A.A.; Sumbatyan, N.V. New Fluorescent Macrolide Derivatives for Studying Interactions of Antibiotics and Their Analogs with the Ribosomal Exit Tunnel. Biochem. (Mosc.). 2016, 81, 1163–1172. [Google Scholar] [CrossRef]
- Yan, K.; Hunt, E.; Berge, J.; May, E.; Copeland, R.A.; Gontarek, R.R. Fluorescence Polarization Method To Characterize Macrolide-Ribosome Interactions. Antimicrob. Agents. Chemother. 2005, 49, 3367–3372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X. An Exact Mathematical Expression for Describing Competitive Binding of Two Different Ligands to a Protein Molecule. FEBS Letters 1995, 360, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Lukianov, D.A.; Buev, V.S.; Ivanenkov, Y.A.; Kartsev, V.G.; Skvortsov, D.A.; Osterman, I.A.; Sergiev, P.V. Imidazole Derivative As a Novel Translation Inhibitor. Acta Naturae 2022, 14, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Terenin, I.M.; Andreev, D.E.; Dmitriev, S.E.; Shatsky, I.N. A Novel Mechanism of Eukaryotic Translation Initiation That Is Neither m7G-Cap-, nor IRES-Dependent. Nucleic Acids Res. 2013, 41, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia Coli K-12 In-frame, Single-gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting Out Antibiotics’ Mechanisms of Action: A Double Fluorescent Protein Reporter for High-Throughput Screening of Ribosome and DNA Biosynthesis Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef]
- Zakalyukina, Y.V.; Birykov, M.V.; Lukianov, D.A.; Shiriaev, D.I.; Komarova, E.S.; Skvortsov, D.A.; Kostyukevich, Y.; Tashlitsky, V.N.; Polshakov, V.I.; Nikolaev, E.; et al. Nybomycin-Producing Streptomyces Isolated from Carpenter Ant Camponotus Vagus. Biochimie 2019, 160, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.; Haefeli, C. Expression in Escherichia Coli of a Staphylococcal Gene for Resistance to Macrolide, Lincosamide, and Streptogramin Type B Antibiotics. J. Bacteriol. 1982, 152, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents. Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Denisov, S.S.; Khailova, L.S.; Nazarov, P.A.; Rokitskaya, T.; Tashlitsky, V.N.; Firsov, A.M.; Korshunova, G.A.; Kotova, E.A. Alkyl-Substituted Phenylamino Derivatives of 7-Nitrobenz-2-Oxa-1,3-Diazole as Uncouplers of Oxidative Phosphorylation and Antibacterial Agents: Involvement of Membrane Proteins in the Uncoupling Action. Biochim. Biophys. Acta - Biomembr. 2017, 1859, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Krizsan, A.; Mohammed, G.K.; Hoffmann, R. Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate. Antibiotics 2022, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short Proline-Rich Antimicrobial Peptides Inhibit Either the Bacterial 70S Ribosome or the Assembly of Its Large 50S Subunit. ChemBioChem 2015, 16, 2304–2308. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenkov, A.G.; Shishkina, A.V.; Tashlitsky, V.N.; Korshunova, G.A.; Bogdanov, A.A.; Sumbatyan, N.V. Interaction of Chloramphenicol Tripeptide Analogs with Ribosomes. Biochem. (Mosc.). 2016, 81, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Khairullina, Z.Z.; Tereshchenkov, A.G.; Zavyalova, S.A.; Komarova, E.S.; Lukianov, D.A.; Tashlitsky, V.N.; Osterman, I.A.; Sumbatyan, N.V. Interaction of Chloramphenicol Cationic Peptide Analogues with the Ribosome. Biochem. (Mosc.). 2020, 85, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Runti, G.; Lopez Ruiz, M.D.C.; Stoilova, T.; Hussain, R.; Jennions, M.; Choudhury, H.G.; Benincasa, M.; Gennaro, R.; Beis, K.; Scocchi, M. Functional Characterization of SbmA, a Bacterial Inner Membrane Transporter Required for Importing the Antimicrobial Peptide Bac7(1-35). J. Bacteriol. 2013, 195, 5343–5351. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Yu.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating Cation/Fatty Acid Anion Pair as a Mitochondria-Targeted Protonophore. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Scheinpflug, K.; Krylova, O.; Nikolenko, H.; Thurm, C.; Dathe, M. Evidence for a Novel Mechanism of Antimicrobial Action of a Cyclic R-,W-Rich Hexapeptide. PLoS ONE 2015, 10, e0125056. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular Mechanisms of Membrane Targeting Antibiotics. Biochim. Biophys. Acta - Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef]
Figure 3.
Antibacterial activity of triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112. Onc112, erythromycin (ERY), C10-TPP and C4-TPP are used as the controls. Testing of the activity of Bac(1-10, R/Y)-C2-TPP (3) and Bac(1-10, R/Y)-C10-TPP (4) using E. coli ΔtolC strain SQ171 (A), macrolide-resistant E. coli strains SQ171 ΔtolC (A2059G) in which the A2059 nucleotide is replaced by G2059 in the 23S rRNA gene (B) and JW5503 (∆tolC) (KanS) ermC harboring plasmid encoding a methyltransferase, that catalyzes methylation of A2058 of 23S rRNA (C).
Figure 3.
Antibacterial activity of triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112. Onc112, erythromycin (ERY), C10-TPP and C4-TPP are used as the controls. Testing of the activity of Bac(1-10, R/Y)-C2-TPP (3) and Bac(1-10, R/Y)-C10-TPP (4) using E. coli ΔtolC strain SQ171 (A), macrolide-resistant E. coli strains SQ171 ΔtolC (A2059G) in which the A2059 nucleotide is replaced by G2059 in the 23S rRNA gene (B) and JW5503 (∆tolC) (KanS) ermC harboring plasmid encoding a methyltransferase, that catalyzes methylation of A2058 of 23S rRNA (C).
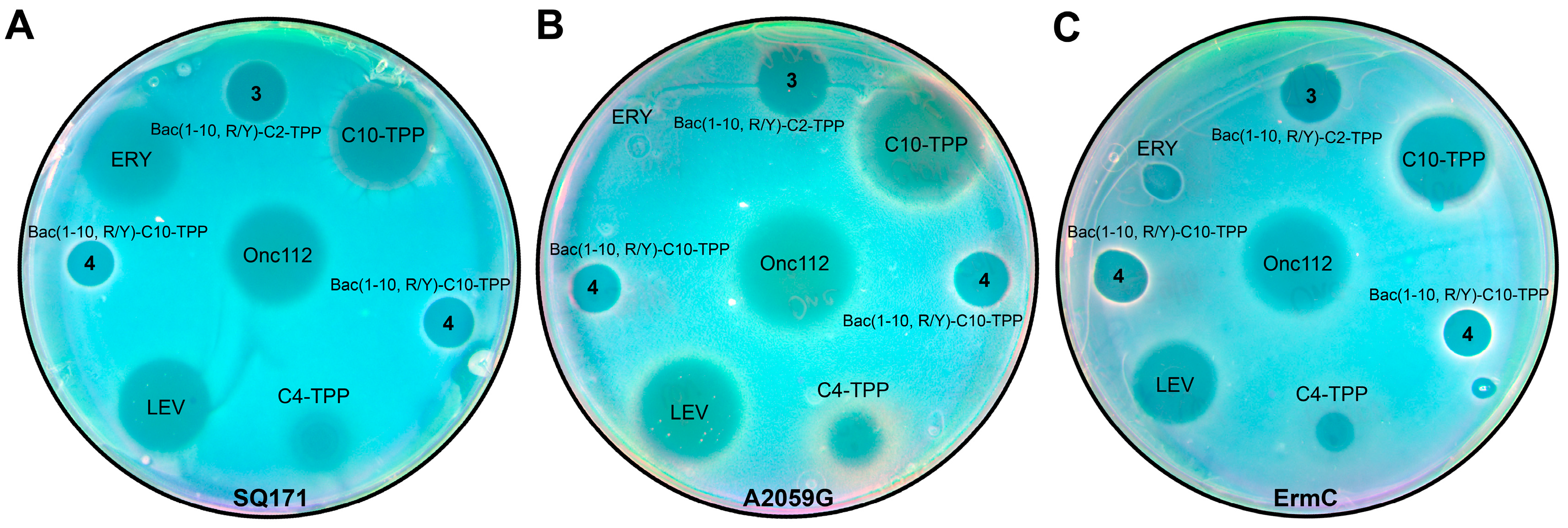
Figure 4.
Antibacterial activity of triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112. Onc112, Levofloxacin (LEV), erythromycin (ERY), C10-TPP and C4-TPP are used as the controls. Testing of the activity of Bac(1-10) (1), Bac(1-10, R/Y)-C2-TPP (3), Bac(1-10, R/Y)-C10-TPP (4), and TPP-C10-Bac(1-10, R/Y) (5) using E. coli strain 25113 (A) and E. coli strains 25113 with deletion of genes of transporter protein SbmA (B), OmpF (C), and MdtH (D).
Figure 4.
Antibacterial activity of triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112. Onc112, Levofloxacin (LEV), erythromycin (ERY), C10-TPP and C4-TPP are used as the controls. Testing of the activity of Bac(1-10) (1), Bac(1-10, R/Y)-C2-TPP (3), Bac(1-10, R/Y)-C10-TPP (4), and TPP-C10-Bac(1-10, R/Y) (5) using E. coli strain 25113 (A) and E. coli strains 25113 with deletion of genes of transporter protein SbmA (B), OmpF (C), and MdtH (D).
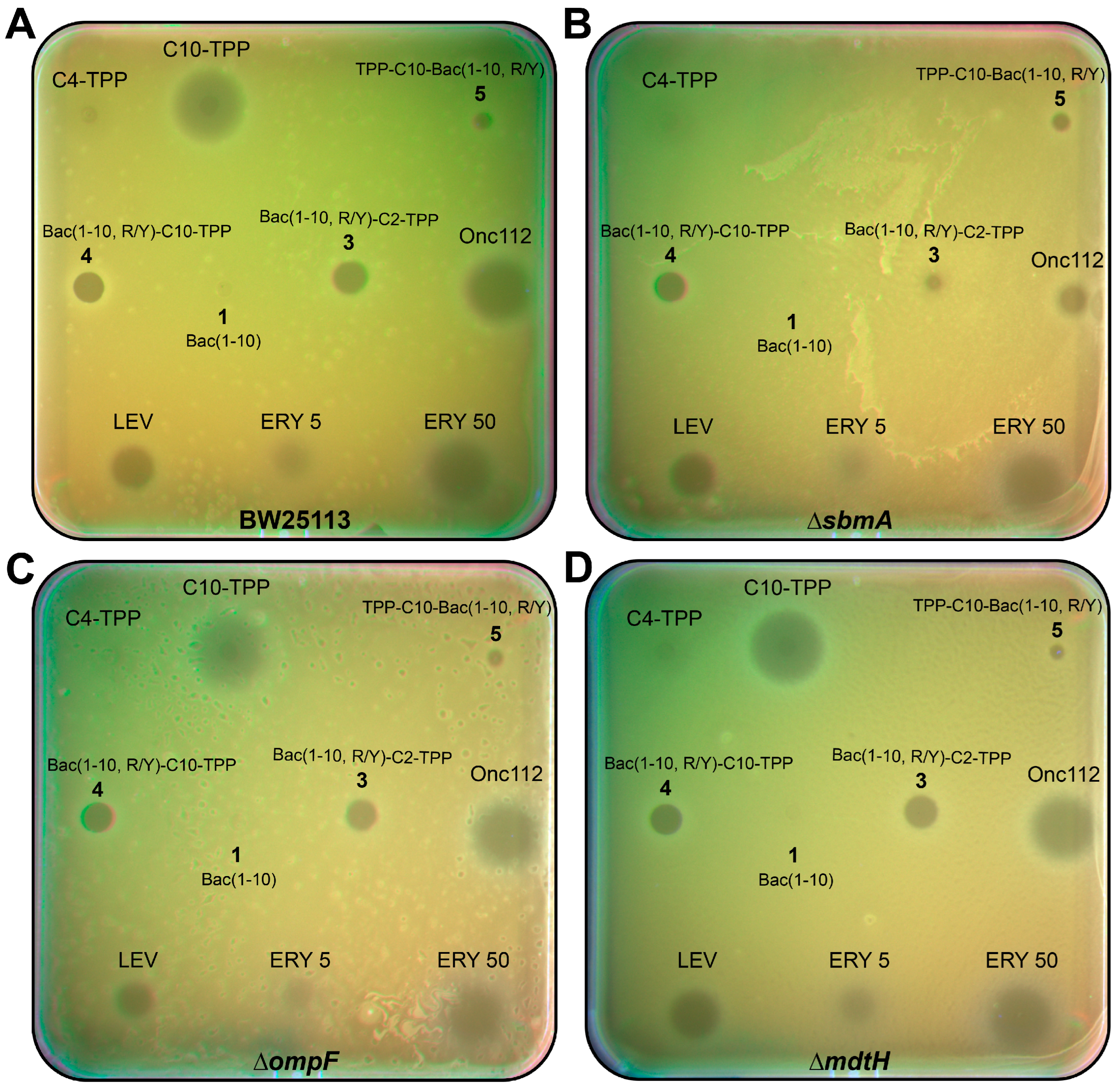
Table 2.
Growth inhibition by triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112 (3–5) and the parent peptides Bac(1-10) (1) and Bac(1-10, R/Y) (2) in relation to a number of cell lines according to the MTT assay. Values of a 50% growth inhibition concentration (GI50, µM) are shown.
Table 2.
Growth inhibition by triphenylphosphonium derivatives of decapeptide related to Bac7 and Onc112 (3–5) and the parent peptides Bac(1-10) (1) and Bac(1-10, R/Y) (2) in relation to a number of cell lines according to the MTT assay. Values of a 50% growth inhibition concentration (GI50, µM) are shown.
| HEK293T | MCF7 | VA13 | A549 | |
|---|---|---|---|---|
| 2 | >50 | >20 | >50 | >50 |
| 3 | >50 | >20 | >50 | >50 |
| 4 | 3.1 ± 0.5 | 10 ± 1 | 12 ± 1 | 9.2 ± 0.9 |
| 5 | 36 ± 4 | >20 | >50 | >50 |
| Onc112 | >50 | >20 | >50 | >50 |
| C4-TPP | 2.9 ± 0.5 | 8 ± 2 | 11 ± 2 | 6.2 ± 0.9 |
| C10-TPP | <0.16 | 0.25 ± 0.07 | <0.16 | <0.16 |
| Doxorubicin | <1.6 | 1.7 ± 0.2 | 1.2 ± 0.3 | 1.2 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



