
Submitted:
10 December 2023
Posted:
12 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Coronaviruses (CoVs) are enveloped positive-sense single-stranded RNA viruses with a genome 27-31kbases in length. Critical genes include the spike (S), envelope (E), membrane (M), nucleocapsid (N) and 9 accessory open reading frames encoding for non-structural proteins (NSPs) that have multiple roles in the replication cycle and immune evasion (1). There are 7 known human CoVs that most likely appeared after zoonotic transfer, the most recent being SARS-CoV-2 responsible for the Covid-19 pandemic. Antivirals that have been approved by the FDA for use against Covid-19 such as Paxlovid can potentially target and successfully inhibit the main protease (MPro) activity of multiple human CoVs, however alternative proteomes encoded by CoV genomes have a closer genetic similarity to each other, suggesting that antivirals could be developed now that target future CoVs. New zoonotic introductions of CoVs to humans are inevitable and unpredictable. Therefore, new antivirals are required to control not only the next human CoV outbreak, but also the 4 common human CoVs (229E, OC43, NL63, HKU1) that circulate frequently and to contain sporadic outbreaks of the severe human CoVs (SARS-CoV, MERS and SARS-CoV-2). The current study found that emerging antiviral drugs, such as Paxlovid, have only been proven to target SARS-CoV-2. Other drugs which have the potential to target other human CoVs, are still within clinical trial and are not yet available for public use. Monoclonal antibody (mAb) treatment and vaccines for SARS-CoV-2 can reduce mortality and hospitalisation rates, however they target the Spike protein whose sequence mutates frequently and drifts. Spike also is not applicable for targeting other HCoVs as these are not well conserved sequences among human CoVs. Thus, there is a need for readily available treatments globally that target all 7 human CoVs and improve the preparedness for inevitable future outbreaks. Here we discuss antiviral research contributing to the control of common and severe CoV replication and transmission, including the current SARS-CoV-2 outbreak. The aim was to identify common features of CoVs for antivirals, biologics and vaccines that could reduce the scientific, political, economic and public health strain caused by CoV outbreaks now and in the future.
Keywords:
coronavirus
; antiviral
; biologic
1. Introduction
1.1. History of Human Coronaviruses
The 2019 Human Coronavirus (Covid-19) outbreak, caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), has caused public health, economic and political devastation on a global scale [2,3]. Human coronaviruses (CoVs) are single-stranded, positive-sense RNA viruses, with genomes ranging between 27,000-31,000 bases (b) in length [4]. The CoVs are transmitted via aerosols from infected individuals and by direct contact with contaminated surfaces, which can be prevented by handwashing with soap, social distancing and utilising personal protective equipment (PPE) [5]. There are currently 7 CoVs which infect humans, with the first identification in the 1960s and the most recent in 2019, although new variants of SARS-CoV-2 are still appearing such as Omicron that has frequently emerging subvariants including Pirola (BA.2.86) [6].
CoV-229E and CoV-OC43 were the first CoVs identified in the 1960s [3], while the first severe CoV, known as SARS-CoV, was identified in 2003 in China [7]. CoV-NL63 and CoV-HKU1 were detected as a result of increased testing following the SARS-CoV outbreak [8]. CoV-229E, CoV-OC43, CoV-NL63 and CoV-HKU1 are self-limiting infections of the upper respiratory tract and present with mild common cold-like symptoms, whilst SARS-CoV, MERS-CoV and SARS-CoV-2 are responsible for increased clinical severity by infecting the lower respiratory tract, leading to complications such as pneumonia and other severe lung pathology [9].
In 2003 in Guangdong, China, SARS-CoV was the first severe HCoV to be identified. It caused acute respiratory distress syndrome (ARDS), pneumonia and even respiratory failure, as well as other complications such as liver or kidney impairment and diastolic cardiac dysfunction [10,11]. Nosocomial infections and transmission from very sick symptomatic individuals tended to follow within the immunocompromised and elderly populations [7]. However, whilst spreading across >30 different countries, ~83% of all 8096 laboratory-confirmed cases remained in China, with 774 deaths worldwide as of July 2003 and a case fatality rate of approximately 9.5% [7]. The first cases were documented from November 2002 and the SARS epidemic continued until June 2003, with a brief peak in April 2003, but a better understanding of control measures were implemented by then and the cases decreased with none having been documented since 2004 [12].
Middle Eastern Respiratory Syndrome CoV (MERS-CoV) is another severe CoV identified in Saudi Arabia in 2012, with camels being the known reservoir [13]. Transmission largely occurs directly from camels to humans, with occasional human to human transmission from very close contact with infected individuals. Between April 2012 and June 2023, there have been 2,605 laboratory-confirmed cases of MERS-CoV with 936 associated deaths and 36% case-fatality ratio [14]. Most of these cases were identified in Saudi Arabia that had 2,196 cases and 855 deaths in total [14]. Currently, ~40% of cases are nosocomial potentially due to overcrowded hospitals and co-morbidities increasing infection susceptibility [15]. In fact, MERS is only very rarely transmitted between people outside of hospital settings and is therefore considered low-risk for the general population [16].
SARS-CoV-2 is the most recent severe HCoV which emerged late 2019 in Wuhan, China, and subsequently spread globally with >700 million confirmed cases and >6.95 million deaths [17]. SARS-CoV-2 has been shown to have a higher transmission rate (reproductive number: 2.9, however in other studies it has been demonstrated that it can be anywhere between 2.6 and 4.71) than SARS-CoV (reproductive number: 1.77), but interestingly has a much lower fatality rate when measured against its infection rate, SARS-CoV had a case fatality rate of 9.5% and SARS-CoV-2 was 2.13% [18,19].
The severe human CoV’s are thought to have originated from bats [20]. Bats harbour the most zoonotic diseases compared to other mammalian taxonomic groups and are one of the most abundant mammalian orders known, comprising 20% of biodiversity [21]. Viruses such as filoviruses, paramyxoviruses and coronaviruses have evidence of emerging from various bat species, this may be because of the long lifespan of bats (20-40 years) worldwide distribution and high metabolic rates. Research shows that bats also have immunological characteristics that control virus propagation [21,22,23]. Viruses generally have very high evolution rates, especially RNA viruses, because of their rapid mutation rates and instable genetic heterogeneity. This is compounded by high host population density and short generation times [24]. SARS-CoV-2 has a much higher evolutionary rate compared to the other HCoVs which is reflected in numerous mutations within the Spike gene that have not mutated in others, therefore the unpredictability of the different CoV strains evolution creates difficulty in preparing for outbreaks and ultimately in providing control measures, however this may also be a result of predominantly sequencing SARS-CoV-2 compared to other CoVs because of the urgency at the time [21,23,25].
1.2. CoV Genes and Genetic Diversity
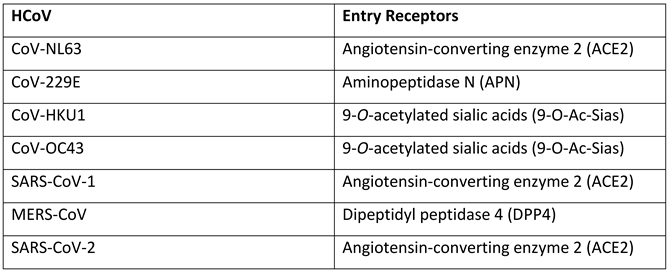
SARS-CoV and SARS-CoV-2 are ~80% genetically similar to each other, providing some evidence that the virus evolved and re-emerged from their original reservoirs, with epidemiological and genetic studies indicating SARS-CoV and SARS-CoV-2 originated from the BANAL coronaviruses found in horseshoe (Rhinolophus) bats in Laos [26,27]. Figure 1 presents the phylogenetic classes representing the genetic differences between SARS-CoV-2 and other HCoVs by tracking mutagenic shifts in their genomic sequences using FigTree [28,29,30] .
Figure 1 shows CoV-NL63 and CoV-229E having the closest similarity to each other, CoV-OC43 with CoV-HKU1, and MERS-CoV with SARS-CoV-1 and SARS-CoV-2 based on their genomic sequences. Bats and rodents are known to be the primary reservoir for all 7 human CoVs by using epidemiological data to track and trace the index, primary and secondary cases through human migration [34]. SARS-CoV-2 has ~96.8% genetic similarity to BANAL-52 CoV found in the R malayanus species of horseshoe bats in Laos [35], therefore providing evidence of bats generally being the original reservoir for severe CoVs. The CoVs are categorised in different genera based on their genetic similarity and evolutionary patterns, this is displayed in Figure 2. The α-CoV and ß-CoV include the 7 CoVs which infect humans, whilst γ-CoV and δ-CoV predominantly consist of rodent and avian CoVs [30].
The human CoV genomes have a typical structure containing multiple open reading frames (ORFs) that encode non-structural proteins (NSPs) 1-16 which are responsible for viral replication, virion formation and immune evasion [37]. This arrangement is shared by the seven human CoVs, with subtle differences found at the 3’ end of the genome where the viral structural protein genes and accessory ORFs are found. Further details of the SARS-CoV-2 schematics are shown in Figure 3 and Table 1 [37].
ORF1ab is the largest section of the genome which encodes for polyproteins 1a and 1b (pp1ab) [38], pp1a encodes for NSPs 1-11, and pp1b encodes for NSPs 12-16 which are made as long polypeptides before further processing [39]. A detailed summary of the different viral proteins is presented in Table 1.

Table 1.
A table summarising the different SARS-CoV-2 proteins including the S, E, M and N proteins, as well as the different ORFs and their functions where known.
Table 1.
A table summarising the different SARS-CoV-2 proteins including the S, E, M and N proteins, as well as the different ORFs and their functions where known.
| Protein | Functions | 3D Structure Availability |
| Nucleocapsid (N) (ORF9a) | Nucleocapsid (~419 a.a. in SARS-CoV-2) binds viral genomic RNA and forms a helical ribonucleocapsid. Involved in genome protection, viral RNA replication, virion assembly, and immune evasion (including IFN-I suppression). Interacts with M and nsp3 proteins [40]. | ✓ |
| NSP1 | Non-structural protein 1 (nsp1; ~180 a.a. in SARS-CoV-2) promotes viral gene expression via interactions with the 40s ribosomal subunit [41]. It also inhibits immune functions by interfering with type 1 interferon expression and various cytokines [41]. | ✓ |
| NSP2 | Non-structural protein 2 (~638 a.a. in SARS-CoV-2) interacts with host factors prohibitin 1 and prohibitin 2, which are involved in many cellular processes including mitochondrial biogenesis. It appears that nsp2 may change the intracellular milieu and perturb host intracellular signaling, but many of its functions are unknown [42]. | ✕ |
| NSP3 | Non-structural protein 3 (~1945 a.a. in SARS-CoV-2) is a papain-like protease (PLpro) and multi-pass membrane protein that processes the viral polyprotein to cleave nsp1, nsp2, and nsp3. Interactions with NSP4 and NSP6 can induce double membrane vesicle (DMV) development for virion transport [43]. | ✓ |
| NSP4 | Non-structural protein 4 (~500 a.a. in SARS-CoV-2) is a transmembrane glycoprotein that forms DMVs in complex with NSP3, and has a high level of conservation across the HCoVs [44]. | ✕ |
| NSP5 | Non-structural protein 5 (3CLpro; ~306 a.a. in SARS-CoV-2) is the main protease of CoVs which cleave 11 sites in the polyprotein to release nsp4-nsp16. It is also responsible for viral polyprotein processing and NSP maturation [45]. | ✓ |
| NSP6 | Non-structural protein 6 (~290 a.a. in SARS-CoV-2) is a multi-pass membrane protein that forms complexes with NSP3 and NSP4 to induce DMVs in infected cells. It also interferes with autophagosome delivery of viral factors to lysosomes for destruction [46]. | ✕ |
| NSP7 | Non-structural protein 7 (~83 a.a. in SARS-CoV-2) forms a supercomplex with NSP8 and NSP12 (RNA-dependent RNA polymerase) in order to process and elongate viral RNA [47]. | ✓ |
| NSP8 | Non-structural protein 8 (~198 a.a. in SARS-CoV-2) forms a supercomplex with NSP7 and NSP12 (RNA-dependent RNA polymerase) in order to process and elongate viral RNA [47]. | ✓ |
| NSP9 | Non-structural protein 9 (~113 a.a. in SARS-CoV-2) is most likely associated with RNA synthesis because of its interactions with NSP12, but it has unclear specific functions [48]. | ✓ |
| NSP10 | Non-structural protein 10 (~139 a.a. in SARS-CoV-2) forms a dodecamer complex with both NSP14 and NSP16 to stimulate their respective 3’-5’ exoribonuclease and 2’-O-methyltransferase activities in the formation of the viral mRNA capping machinery [49]. | ✓ |
| NSP11 | Non-structural protein 11 (~13-23 a.a., depending on the CoV species) is a pp1a cleavage product at the nsp10/11 boundary. For pp1ab, it is a frameshift product that becomes the N-terminal of nsp12. Its function, if any, is unknown [50]. | ✕ |
| NSP12 | Non-structural protein 12 (~932 a.a. in SARS-CoV-2) is the RNA-dependent RNA polymerase (RdRp) performing both replication, transcription and elongation of the viral genome, therefore making it a crucial protein for viral replication [51]. | ✓ |
| NSP13 | Non-structural protein 13 (~601 a.a. in SARS-CoV-2) is the main helicase for the CoVs. It interacts with NSP12 for backtracking and to facilitate viral replication and mRNA capping [52]. | ✓ |
| NSP14 | Non-structural protein 14 (~527 a.a. in SARS-CoV-2) has a 3’-5’ exoribonuclease proofreading mechanism (ExoN) when in complex with NSP10 to prevent mismatches during RNA synthesis, and it has N7-guanine methyltransferase (viral mRNA capping) activities [53]. | ✓ |
| NSP15 | Non-structural protein 15 (~346 a.a. in SARS-CoV-2) is a uridine endoribonuclease that cleaves 3’ RNA. Its function is primarily important for immune evasion by preventing dsRNA sensor activation [54]. | ✓ |
| NSP16 | Non-structural protein 16 (~298 a.a. in SARS-CoV-2) has 2’-O-methyltransferase activity and is activated once in complex with NSP10. It is able to replicate CMTr1, a human homolog, in order to methylate mRNA and improve the efficiency of translation and viral mRNA capping [55]. | ✓ |
| ORF3a | ORF3a (~275 a.a. in SARS-CoV-2) is a viroporin iron channel in SARS-CoV which promotes viral movement and release. Importantly, it also activates inflammasomes such as NF-kB and NLRP3 to produce a cytokine storm [56]. | ✓ |
| ORF3b | ORF3b (~22 a.a. in SARS-CoV-2) varies in length amongst different CoV strains due to premature stop codon mutations. There is some evidence of interrupting interferon antagonistic functions, however it is not fully supported yet in CoV-infected cells [57]. | ✕ |
| ORF6 | ORF6 (~61 a.a. in SARS-CoV-2) is localised in the ER, lysosomes and autophagosomes of infected cells. It interferes with innate immune responses through suppressing various Janus kinases types I and II interferon pathways [58]. | ✓ |
| ORF7a | ORF7a (~121 a.a. in SARS-CoV-2) is a type I membrane protein that interacts with CD14+ monocytes resulting in drastic cytokine expression and increased glycosylation for immune evasion of presenting antigens [59]. | ✓ |
| ORF7b | ORF7b (~43 a.a. in SARS-CoV-2) is a transmembrane protein within the Golgi apparatus. It does not have a significant role in viral replication, but may have some interference with cellular processes regarding symptoms of infection, but there is not enough evidence to support this [59]. | ✕ |
| ORF8 | ORF8 (~121 a.a. in SARS-CoV-2) is not well conserved amongst CoVs, however it still has important roles in disease severity and symptoms across different strains. It is an interferon antagonist to promote signal transductions downstream to generate a cytokine storm [59]. | ✓ |
| ORF9b | ORF9b (~97 a.a. in SARS-CoV-2) is another accessory ORF within the N protein which is localised in mitochondrial membranes, suggesting hindered immune responses by interactions with TOM70, an outer membrane mitochondrial protein, which is associated with interferon responses [60]. | ✓ |
| ORF9c | ORF9c (~70 a.a. in SARS-CoV), also located in the N coding region, interacts with various host proteins including Sigma receptors, which have involvement in ER stress responses and lipid remodelling [59]. | ✕ |
| ORF10 | ORF10 (~38 a.a. in SARS-CoV-2) is not highly significant in viral replication, it is poorly conserved amongst CoVs and removal of this accessory protein as no effect on SARS-CoV-2 infection [59]. | ✕ |
| Spike (S) (ORF2) | Class I viral fusion protein cleaved into subunits 1 and 2 (~1273 a.a. in SARS-CoV-2). Assistance of host cell and viral membranes by binding of the S1 with the receptor binding domain (RBD) while S2 facilitates the fusion process [61]. | ✓ |
| Membrane (M) (ORF5) | Membrane protein (~222 a.a. in SARS-CoV-2) is the most abundant protein in SARS-CoV-2. It mediates assembly, packaging and budding of viral particles through recruitment of other structural proteins to “ER-Golgi-intermediate compartment (ERGIC)”. Once dimerised, it presents with a similar structure to accessory protein ORF3a, assuming interactions [62] | ✓ |
| Envelope (E) (ORF4) | Envelope protein (~75 a.a. in SARS-CoV-2) is a single-pass type III membrane protein involved in viral assembly, budding, and pathogenesis. It has roles in host immune responses and interacts with M, N, 3a, and 7a [63]. | ✓ |
| Nucleocapsid (N) (ORF9a) | Nucleocapsid (~419 a.a. in SARS-CoV-2) binds viral genomic RNA and forms a helical ribonucleocapsid. Involved in genome protection, viral RNA replication, virion assembly, and immune evasion (including IFN-I suppression). Interacts with M and nsp3 proteins [40]. | ✓ |
Each viral protein has specific functions which contribute to viral replication. For example, NSP3 is a protease which cleaves and divides NSP1, 2 and 3 into individual components, while NSP5 is the main protease (MPro) which cleaves the remaining NSPs (4-16) [64]. NSP12 is the RNA-dependent RNA polymerase (RdRp), and it is the central reservoir for RNA transcription and elongation, making it a critical NSP for viral replication [65]. RNA viruses normally have low RdRp fidelity resulting in high mutations, however HCoVs have an exonucleolytic proofreading mechanism regulated by NSP14 [66,67,68] . Favourable single nucleotide polymorphisms (SNPs) still occur to improve infectivity and immune evasion, negatively impacting the development of suitable treatments [68]. Most HCoVs possess almost all these genes/proteins in Table 1, however the betacoronaviruses (CoV-HKU1 and CoV-OC43) also have genes that encode the haemagglutinin-esterase (HE) protein. HE contains a receptor destroying sialate-O-acetylesterase domain and a receptor-binding lectin domain for O-Ac-Siac, this contributes to virion attachment and the breakdown of sialoglycotopes [69].
The Spike protein binds to the angiotensin converting enzyme 2 (ACE2) protein found on the surface of host cells which allows fusion between the cell membrane and viral envelope. The Nucleocapsid protein sheds viral RNA into the cytoplasm to form replication complexes [39,70,71]. The Envelope, Membrane, Spike and Nucleocapsid protein translation is supported by double membrane vesicles to form replication-transcription complexes [70]. After synthesis at the ER and Golgi apparatus, the proteins form into virions for budding and exocytosis to infect surrounding cells [57]. The final structure of the virus is presented in Figure 4.
The Spike protein is a common target for vaccine research due to its critical nature in virus entry and due to being the major target of the immune response to infection [74,75]. It is divided into sub-unit 1 (S1), which contains the receptor binding domain (RBD) that interacts with the entry receptors [39], and sub-unit 2 (S2) that facilitates cellular fusion for viral entry [76]. The Membrane protein is the most abundant in the viral particle, and it forms protein-RNA complexes for lipid bilayer stability [77]. The Nucleocapsid protein is critical for stabilising and shielding the genome, and facilitates exocytosis and has immunogenic properties, making it a vaccine and diagnostic target [78]. The Envelope protein is responsible for viral budding and envelope formation [79]. More details of each structural protein function can be found in Table 1. Most mutations amongst the HCoVs occur in the final third of their genomes, and mutations within the RBD of the Spike protein have resulted in the HCoVs evolving to bind different cell entry receptors, these are presented in Table 2 of section 1.3.
1.3. CoV Genetic Drift
Mutations are defined as a change in an organism’s genetic sequence, it can be an insertion, deletion or point mutation which does not always have a functional effect [80]. They can occur in both animal and human reservoirs. Multiple animals of different species could be carrying a progenitor SARS-CoV-2 and potentially infect humans in different locations, which can further impact how viruses may mutate considering population sizes [81].
Viral mutations often occur because of RNA polymerase instability and low fidelity [82]. CoVs possess an exoribonucleolytic proofreading mechanism (NSP14) that will maintain its long genome and protect from frequent mutations [24]. Viruses with a longer genome are less prone to sporadic mutations because of the evolutionary addition of this proofreading gene; simpler viruses with smaller genomes, such as influenza, tend to mutate at much higher rates [83]. Very high population rates combined with high levels of sporadic mutations will lead to a plethora of different variants which will be harder to control immunologically [84].
Even though CoVs have this proofreading mechanism, there has been evidence of multiple evolutionary mutations within its Spike protein, which is responsible for direct binding and entrance of viral material into host cells [85]. A very small portion of mutations are expected to be impactful on viral phenotype which will influence infectivity, pathogenicity and transmissibility positively [86]. Studies on 229E-CoV and NL63-CoV have shown that there are no correlation between phenotypic evolution and mutation rate, therefore mutations are rare with little evolutionary pressure [87,88]. The mutations that occur in the Spike protein can improve transmissibility and immune evasion, and this infers that mutations that happen here will have the same effect in all HCoVs [89].
Areas of a viral genome have different mutation rates that influence viral fitness for new hosts and are evidence of ongoing evolution. Studies have shown that the mutations that were most prominent in the Covid-19 pandemic were within the Spike (202 genomes had 34 Spike mutations, D614G being most frequent in 160 genomes) and Nucleocapsid (65 genomes had 25 variations, R203K being the most frequent in 21 genomes) [90]. The receptor-binding motif (RBM) is a ‘hotspot’ of unique mutations, in April 2020, the D614G mutation was highly prominent and contributed to improved infectivity/transmissibility and other mutations of spike resulted in the emergence of multiple variants of SARS-CoV-2, such as alpha, beta, delta and omicron [91,92]. Because of the fast rate of Spike mutations, it would not be considered an effective antiviral target for multiple CoVs. For example, there were ~>30 mutations in the Spike protein in the emergent Omicron SARS-CoV-2 strain which greatly increased its infectivity, and improved antiviral and immune evasion [93].
Genomic sequence changes or mutations can cause functional and structural changes to proteins , even as little as one amino acid placement can completely change a protein’s structure and function [94]. 229E-CoV, similarly to SARS-CoV-2, has significant variability in the Spike and Nucleocapsid proteins, most of which are found within the Spike RBD and affect its binding capability to host cells [87]. In January 2022, 2 novel OC43-CoV variants emerged and it was found that even though the critical sialoglycan receptor-interacting residues were conserved, there were significant sequence mutations in other areas of the receptor-binding motif across these OC43-CoV isolates [95,96]. Human CoV’s are clearly adaptable and open to selective pressure as the 7 HCoVs have several entry pathways and bind to different host receptors (Table 2).
SARS-CoV and SARS-CoV-2. NL63-CoV Spike protein has a very low sequence similarity compared to the severe beta CoVs (SARS-CoV: 23.7% and SARS-CoV-2: 25%) indicating significant divergence, however they all have a conserved glycine residue (NL63-CoV: G537, SARS-CoV: G488 and SARS-CoV-2: G502) which is essential for ACE2 binding. In studies conducted by Rawat, P., et al., 2020, it was revealed during bioinformatic analysis using FoldX and CUPSTAT, mutation of the conserved glycine reside destabilises the protein and interaction with ACE2 is hindered, proving that it is important for binding [99]. Once bound to ACE2, acid-dependent proteolytic cleavage occurs in the S1 unit by proteases such as the transmembrane protease serine proteases (TMPRSS), cathepsins and human airway trypsin-like proteases [100]. Spike cleavage exposes the fusion peptides of the subunit 2 (S2), subsequently the virus fuses with the host cell entry receptor of the cellular membrane [100].
CoV-OC43 and CoV-HKU1 use cellular glycocalyx sialylated compounds to enter host cells [101]. The Spike protein binds to sugar-based receptor-determinants such as 9-O-acetylated sialic acids, and the haemaglutin-esterase (HE) that these betacoronaviruses possess is an acetylesterase with a 9-O-acetylated sialic acid-specific lectin domain, which acts as a receptor and therefore breaks down the complex for viral entry [102]. Aminopeptidase N is the cell entry receptor for CoV-229E, also called CD13, which reside in fibroblastic epithelial cells of the lungs [103]. MERS-CoV utilises the dipeptidyl peptidase 4 (DPP4) cell entry receptor, once the Spike protein has undergone proteolytic activation via the TMPRSS2 or cathepsin L pathways, residues in S1 can directly bind to DPP4 [104].
2.0. Treatment of HCoV Infection
2.1. Antivirals
Antivirals are biopolymeric molecules that interfere with viral replication by targeting key stages in the viral life cycle, such as viral attachment, uncoating, genome replication/translation or budding [105,106,107] . The first antiviral was idoxuridine in 1963, a thymidine analogue that causes substitution mutations in herpes simplex virus (HSV), and since the 2000s, antiviral developments have improved exponentially [108]. Antivirals target many mechanisms that target viral enzymes such as proteases, polymerases or integrases, or they may be able to target viral surface membrane proteins, such as envelope or glycoproteins, the aim being to indirectly hinder the particular viral functions by allosterically altering enzymatic active sites and interrupt replication [109,110]. Some antivirals are designed without a 3’ hydroxyl group, which blocks the viral chain and prevents DNA/RNA synthesis and elongation [111]. Other inhibitor targets, for example HIV protease inhibitors, will commonly involve a reaction between a hydroxyl group from the inhibitor and a carboxyl group of the protease active site (Gly27, Gly48, Asp29 and Asp30 are conserved in many HIV strains, so these are common amino acid targets) [112]. Antiviral activity is achieved by either competitive binding to the substrate or by non-competitively binding to the enzyme to alter its 3D configurational shape [113,114]. The main antiviral groups that stop nucleic acid use and generation are nucleoside and non-nucleoside analogues. Nucleoside inhibitors are beneficial in mimicking naturally-occurring nucleosides and preventing protein synthesis and chain elongation, and non-nucleoside inhibitors with alter the 3D shape of the protein and it aims to overcome antiviral resistance that nucleoside analogues may be prone to [115,116]. These drug structures encompass many other sub-types of antivirals and are highly prevalent in drug research, more detail is explained below.
2.1.1. Nucleoside Analogues
Artificially derived nucleoside analogue antivirals resemble naturally produced nucleosides that bind to a target site on viral RNA [117]. By binding to a viral enzyme involved in replication, the nucleosides will be converted into an active form, nucleotide, and host cells will transcribe the nucleoside analogue as if it was part of the host’s natural make-up [117]. As a result, target enzymes cannot function normally and this interrupts its viral lifecycle [118].
Favipiravir is a guanosine nucleoside analogue that was approved by the FDA to target and inhibit the replication of novel and potentially re-emerging influenza viruses in 2014 in Japan [119]. Favipiravir targets the RdRp of RNA viruses, with specific activity against influenzas A, B and C, whilst also being able to inhibit rhinovirus replication in vitro [120]. Studies have shown that favipiravir activation takes places intracellularly due to the detection of favipiravir ribofuranosyl-5’-monophosphate, favipiravir ribofuranosyl-5’-triphosphate and favipiravir ribofuranose in MDCK cells and analysed by HPLC [121].
This antiviral is suggested to act as both a chain terminator and chain mutator, it is metabolically activated by phosphorylation and ribosylation and converted into favipiravir ribofuranosyl-5B-triphosphate [122]. Favipiravir binds to the RdRp’s active site and it is mistaken as a purine nucleotide, which causes a confirmational change in its binding pocket, impacting its original function and therefore the chain is terminated [122]. It also will induce C-to-U and G-to-A mutations in the RdRp active site which are lethal and will disrupt viral replication this way also [122]. The structure of the antiviral molecule can be found in Figure 5.
Many clinical trials have tested the use of favipiravir in Covid-19 patients. A multicentre, open-labelled, randomised control study found that favipiravir may have significant clinical symptomatic improvements compared to the control group in mild to moderate Covid-19 patients over 5-14 days, however it did not impact the viral load [124]. Those with a viral load at a lower baseline had greater viral reductions between 1-13 days during the favipiravir course [124]. However, other similar studies concluded that even though early infection may increase the likelihood of ventilation-free survival in patients younger than 60 whilst being treated with favipiravir, there were ultimately no improvements in clinical outcomes [125]. Overall, this study found that there was not enough evidence to support the hypothesis of favipiravir being used for Covid-19 patients. An antiviral drug that can bind to and inhibit the replication of multiple CoVs is crucial to reduce the severity of inevitable future CoV emergences, as well as improving the likelihood of being used for other RNA viruses too.
2.1.2. Non-nucleoside Analogues
Non-nucleoside analogues (NNAs) non-competitively bind to a viral protein involved in replication to interrupt its normal functions and indirectly prevent further replication [128]. They allosterically bind to a protein, away from the active site of an enzyme, to cause configurational changes within its 3D structure by forming or breaking hydrogen bonds/van der waals interactions, this inhibits surrounding viral substrates from binding to the active site and viral replication is therefore interrupted [128]. An example of an NNA is nevirapine which is a non-nucleoside reverse transcriptase inhibitor (NNRTI), and is commonly taken in combination with an NRTI such as ritonavir, a protease inhibitor that also inhibits the cytochrome P450 liver metabolism enzymes to prolong and increase plasma concentration levels of the drugs [129,130]. The chemical structures of both inhibitors are presented in Figure 6.
Nevirapine is an HIV RT inhibitor, whilst ritonavir is an HIV protease inhibitor with the capability of binding to the CYP liver enzymes for increased plasma concentrations. Studies have shown that patients who are HIV positive and have a SARS-CoV-2 co-infection have reduced symptoms in both illnesses when taking these medications, therefore showing that the drugs can bind to both HIV and SARS-CoV-2 to prevent replication [132]. Both drugs have many interacting nitrogen electrophilic and oxygen nucleophilic sites which can increase their binding reactivity [133].
2.2. Available Treatments
Repurposing antivirals is an effective method to control outbreaks more rapidly, reducing costs in research and reducing the risk of failure or adverse effects [134]. There are many drugs that have been approved to be used for the treatment of Covid-19, however most of them have been repurposed and were not designed for this use. The treatments that have been approved by the FDA include:
- Paxlovid (ritonavir and nirmatrelvir)
- Lagevrio (molnupiravir)
- Veklury (remdesivir) – this is only approved for the use in individuals who are at high risk of developing severe Covid-19 infection, including the vulnerable/immunocompromised population.
More detail on each of these drugs can be found in sections 1.5.4 and 1.5.5.
There are various monoclonal antibodies (mAbs) that block viral entry into human cells and neutralise the virus before being able to infect. These include:
- Sotrovimab
- Bebtelovimab
- Casirivimab/imdevimab
Although mAbs are an effective treatment, as they target the Spike protein to prevent viral entry into human cells, there is a significant risk of escape mutants developing as the Spike gene has a very high mutation rate. Therefore mAbs can become ineffective against new strains and variants [135,136]. One approach to avoid this problem is to use mAb cocktails to reduce the risk of escape mutants. Mutations such as N439K and Y453F within the S protein are proven to reduce the efficiency of mAb treatment [137]. These mutations have been found in SARS-CoV-2 spike sequences in >30 countries since January 2021, and provide improved affinity between RBD and the ACE2 receptor leading to increased viral loads compared to the ancestral Wuhan CoV [138].
There are many antiviral compounds being tested in clinical trials for Covid-19, such as Paxlovid, one of the first drugs designed specifically to target SARS-CoV-2, and each will bind to a particular protein that has crucial functions in viral replication. Examples are detailed below.
3.0. Novel Viral Druggable Targets
3.1. Non-Structural Protein 5 (NSP5)
All NSP5 proteases in CoVs identified are chymotrypsin cysteine proteases, and they are responsible for the cleavage of NSPs 4-16 (the majority of the polyprotein) and this function is highly conserved across other CoVs, making it an attractive drug target [45]. NSP5 proteases generally have a sequence identity of >80% within the same genera, other CoVs may have a similarity identity of ~50% across other genera, however the greatest conservation point is the enzymatic active site [139]. Because of its similarity, drug targets against NSP5 may have an increased chance of retaining activity in other SARS-CoV-2 strains and possibly other HCoVs, improving the chances for its widespread use [140].
Covid-19 can now be treated with Paxlovid, which was approved by the Food and Drug Administration (FDA) for emergency use in December 2021 for the vulnerable population. It consists of nirmatrelvir, the novel antiviral that targets MPro (NSP5) of SARS-CoV-2, and ritonavir, an HIV-1/2 protease inhibitor that targets cytochrome P450 3A (CYP3A) enzymes to increase the concentrations of nirmatrelvir [141]. CYP enzymes are drug-metabolising enzymes, and therefore when they are inhibited this prolongs a drug’s lifespan in the body by influencing drug-drug and drug-target interactions [142]. Nirmatrelvir is an analogue of GC373, a small molecule prodrug that was designed to target the MPro of feline and mink CoVs [143]. The adduct GC376, a bisulphate compound is converted readily to the peptide aldehyde GC373 when it reaches its target [143].
In a meta-analysis study identifying the efficiency of Paxlovid against Covid-19 in clinical trials, it was found that the drug has evidence of significantly decreasing mortality rates of individuals infected with the virus compared to a healthy control group [144]. Paxlovid was also compared against molnupiravir and fluvoxamine, 2 other drugs that have been shown to reduce disease severity in Covid-19 patients, and Paxlovid was the best [144]. In a phase 2-3 double-blind, randomised control trial to identify the efficacy and safety of Paxlovid in unvaccinated, symptomatic adults with a high risk of developing severe Covid-19 , the results showed that Paxlovid reduced the relative risk of disease severity by 89.1% in hospitalisation cases, after taking the medication every 12 hours for 5 days within 3 days of symptom onset [140].
3.2. Non-Structural Protein 12 (NSP12)
Remdesivir is an intravenous drug initially designed to target the NSP12 (RdRp) of Hepatitis C virus (HCV), and expanded to clinical trials for filoviruses such as ebolavirus and Marburg virus, and ultimately SARS-CoV-2 [145]. Remdesivir is an adenosine NA, and a phosphoramidate prodrug which is metabolised to form remdesivir triphosphate, its active form [146]. Polymerases across different viruses share very similar functions, and therefore have similar 3D structures and sequences, and because of this, pre-existing drugs targeting this enzyme are often repurposed to treat different viral infections [147].
Remdesivir generated results similar to molnupiravir in other studies, which is a prodrug of β-D-N4-hydroxycytidine (NHC) nucleoside analogue that promotes C-U and G-A point mutations in the RdRp of SARS-CoV-2. It transcribes viral RNA using NHC triphosphate to cause these point mutations, resulting in non-functioning proteins, reducing viral activity [148,149,150] . It can be produced on a large scale and does not require specific temperature-regulated or hospital conditions for storage, and it was proven in clinical trials that there are no significant adverse effects of administering the drug to infected individuals [151]. Molnupiravir was originally designed to target the RdRp (NSP12) of influenza in 2019, however NSP12 is a very versatile enzyme within RNA viruses and its structural features that are core in its function are highly conserved across different viruses, therefore the drug could be repurposed for Covid-19 [151,152].
In a retrospective study, hospitalised patients with Covid-19 were treated with remdesivir (they were predominantly 40-60 year old males who required oxygen therapy and were taking the treatment for 5 days) [153]. The infection improvement rate was reported in historical cohorts as 84% and only 13% of the individuals in the study experienced adverse side effects [153]. In a randomised control trial, remdesivir and casirivimab/imdevimab were tested in low-risk symptomatic adult individuals to identify the viral clearance rate with the intention to treat the wider population of Covid-19 by investigating the viral load by PCR across 7 days [154]. The results showed that both therapeutics increased viral clearance in individuals with early Covid-19 infection because the viral burden is at its highest, however there was still uncertainties of the use of remdesivir and casirivimab/imdevimab in hospitalised patients (once the infection is in the late-stage, anti-inflammatory treatments are more effective) [154]. Effects were also dependent on the SARS-CoV-2 strain, casirivimab was much less effective than imdevimab against the Omicron variant that was circulating at the time, due to the N440K and G446S mutations in the Spike protein that reduces the activity of the drug in vitro [154]. Viral clearance rates increased by 42% during remdesivir treatment, but only by 23% in the casirivimab/imdevimab group, indicating that remdesivir was more effective in reducing viral load in early Covid-19 infections [154]. A limitation of this approach is that intravenous monoclonal antibody and drug treatments are much more difficult to obtain, store and distribute across a large population, leading to increased costs and reductions in overall coverage. Therefore, it is likely more beneficial to opt for oral drug intake for non-hospitalised cases. Monoclonal antibody treatment such as casirivimab/imdevimab is also disadvantageous in targeting multiple different viral variants or genera and therefore are not ideal for being repurposed more widely for HCoVs [154].
An approach to decreasing efficacy could be to increase the dose and concentration of the mAb given, but this increases the risk of side effects and inflammation [137]. Developing an oral drug will improve the global coverage of treatment, accessibility and costs as well, therefore mAbs may not be a feasible long term treatment for CoVs [155,156]. NAs have vastly improved the HIV pandemic by reducing viral loads to undetectable levels in numerous individuals and therefore have the potential to reduce SARS-CoV-2 transmission, infections with the existing HCoVs, and improving preparedness for the next inevitable HCoV outbreak [157].
There are numerous potential drug compounds that target NSPs 5 and 12 in available drug libraries. Other viral proteins may not be strong contenders because of their conservation levels across strains/genera. For example, the Spike protein has one of the highest mutation levels as it is constantly and spontaneously adapting to improve its binding to host cells and we have seen mAb escape mutations., Others, such as NSP2, do not have significantly understood functions in viral activity and replication, and therefore it becomes more difficult to identify core amino acids that can be targeted with drugs to reduce protein function [158].
4.0. Alternative Treatment and Prophylaxis
Anti-malarials such as chloroquines have were tested in the Covid-19 emergency because of their interference with ACE2 glycosylation [159]. The drug is cheap and widely available, yet chloroquine resistance is already a significant public health problem [160]. Studies have shown that chloroquines may be able to inhibit SARS-CoV-2 replication by increasing the endosomal pH, and denature viral enzymes which require lower pH levels, however, Vero cells that were infected with SARS-CoV-2 and treated with chloroquines did not display effective inhibition of viral replication [161]. A clinical study showed that the odds ratio of mortality rates in SARS-CoV-2 infected individuals taking hydroxychloroquine and chloroquine were 1.11 and 1.77 respectively, indicating an association of increased mortality rates in SARS-CoV-2 cases with taking chloroquines, indicating that there were no molecular benefits [162]. Many drugs were trialled against SARS-CoV-2 during the pandemic due to the emergency nature, however the challenge remains to design antivirals which do not interfere with host cell functions and target viruses specifically. This is of utmost importance and an important research area to target for future epidemics, pandemics and novel infectious outbreaks [163].
5.0. Vaccine Approaches
Vaccines are a highly effective prevention method for numerous infectious diseases [164]. Examples of the key vaccine platforms include the AstraZeneca viral vector vaccine, the Pfizer/BionTech and Moderna mRNA vaccines and the Johnson & Johnson sub-unit vaccine [165]. The urgency of the pandemic meant that the first vaccine was prioritised and deployed in December 2020, however vaccine research was already in development for SARS-CoV and MERS-COV [166]. It was found the vaccines that targeted the Spike protein of the previous CoVs elicited strong immune responses, therefore it was continuously used in SARS-CoV-2 vaccine research as a target [166]. There is evidence of the mRNA vaccines to be more adaptable to CoV variants, and they have a longer period of immunity and induced protection against the Omicron variant compared to the viral vector vaccines [167].
During the Covid-19 pandemic, studies showed that between January 2021 and August 2022 the monthly rates of SARS-CoV-2 related hospitalisations were 5.2x greater in unvaccinated individuals aged >18 compared to those vaccinated, with general cases at the lowest rate within the vaccinated group who also received a booster [168,169]. However, As the strains mutate the spike sequences and generate dominant variants, the vaccines will become less effective because of decreased specificity and selection for viral escape. Therefore, designing an antiviral drug within a more conserved region of the CoVs will improve the chances of binding to multiple current and future CoVs, further improving for preparedness and control. Sufficient treatments are needed since the emergence of 3 severe CoVs over the last 20 years and no prior general antivirals being available. Targeting multiple HCoVs, including future emerging HCoVs, will thus be important in limiting infections, hospitalisations, deaths and disease severity. Reduced transmission rates will lead to fewer mutations and virus evolution resulting in better control [170].
6.0. The Design of a Novel Antiviral
An approach that is currently taking place involves the design and synthesis of novel small molecules with the capability of targeting multiple HCoVs. Proteins with functions critical for viral replication will often have a lower mutation rate, as mutations in active or enzymatic sites would likely negatively hinder that specific function. These regions will therefore be more conserved across different HCoVs making these an attractive drug target [171,172]. The challenge is finding the optimal target and designing a small molecule inhibitor that has broad CoV activity. The project began by researching all 7 HCoVs and identifying the roles of each protein in replication. Subsequently, a multiple sequence alignment (MSA) took place to visualise the most conserved proteomes and their active sites were identified using the available 3D PDB structures. It was identified that NSP3, a critical protease, will be the target focus of this research project. A lead compound was found by searching PDB for ligand-bound NSP3 3D structures and were remodelled into numerous analogues that will be developed in vitro. Cellular and molecular assays will take place using CoV-NL63, CoV-229E and CoV-OC43 to test whether these novel analogues will prevent cytopathic effect (CPE) in cell lines, along with other confirmatory assays such as MTT to measure cytotoxicity and qPCR to identify interruptions to replication. If proven successful, these newly designed drugs will have the capability of targeting more than one HCoV which will be hugely impactful for future inevitable CoVs and aim to help prevent detrimental impacts to public health.
7.0. Conclusions
Future coronavirus outbreaks are inevitable, as zoonotic diseases cannot simply be eradicated, therefore preparation, prevention and prophylaxis are key for their control [173]. CoVs have existed for at least ~70 years, most likely much longer, yet the first successful antiviral was approved by the FDA, Paxlovid, for emergency use in 2021. There have been many chemical compounds in clinical trials to reduce mortality and hospitalisation rates of Covid-19 patients such as various mAb treatment, vaccines and other small molecule compounds, however all of which seem to focus solely on SARS-CoV-2. Targets such as NSP5 and NSP12 have highly conserved binding sites which make them attractive drug targets, and there has been minimal research into how trialled drugs will affect both mild and severe CoVs but Covid-19 always remained the prime focus. Vaccines and mAbs typically target the Spike protein, an area with a very high mutation rate and significant differences between the existing CoVs, and therefore they would only be most successful in the particular target strain. They are unlikely to be successful for mutant strains or future CoVs. The novel approach proposed here uses a bioinformatic approach to identify conserved viral proteins and their functions, then using the PDB to screen for potential lead compounds. Analogues are designed based on the identified lead compound and synthesised in vitro to be tested in cellular and molecular assays in vitro to first evaluate antiviral effects. Other confirmatory assays will generate solid evidence of a novel compound that can interrupt viral replication of mild CoVs in vitro, ultimately the data collected should be extrapolated for use in current (SARS, MERS, SARS-CoV-2) and future severe CoVs such as SARS-X.
Author Contributions
All authors contributed to study conceptualization, methodology, formal analysis, and investigation; MO and GRM writing—original draft preparation; All authors writing—review and editing; CN, KD, BP, GRM supervision; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. MO is a VC scholar of London Metropolitan University.
Data Availability Statement
Data contained within are from source reference material and the authors can provide additional data following reasonable written request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Michel CJ, Mayer C, Poch O, Thompson JD. Characterization of accessory genes in coronavirus genomes. Virol J. 2020 Aug 27;17(1):131. [CrossRef]
- Agarwal, R. The aftermath of coronavirus disease 2019: devastation or a new dawn for nephrology? Nephrol Dial Transplant. 2020 Jun;35(6):904–7. [CrossRef]
- Liu DX, Liang JQ, Fung TS. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encycl Virol. 2021;428–40. [CrossRef]
- Ye ZW, Yuan S, Yuen KS, Fung SY, Chan CP, Jin DY. Zoonotic origins of human coronaviruses. Int J Biol Sci. 2020 Mar 15;16(10):1686–97. [CrossRef]
- Lotfi M, Hamblin MR, Rezaei N. COVID-19: Transmission, prevention, and potential therapeutic opportunities. Clin Chim Acta Int J Clin Chem. 2020 Sep;508:254–66. [CrossRef]
- Mahase, E. Covid-19: New “Pirola” variant BA.2.86 continues to spread in UK and US. BMJ. 2023 Sep 13;382:p2097. [CrossRef]
- Hodgens A, Gupta V. Severe Acute Respiratory Syndrome. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 [cited 2022 May 3]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK558977/.
- Abdul-Rasool S, Fielding BC. Understanding Human Coronavirus HCoV-NL63. Open Virol J. 2010 ;4:76–84. 25 May. [CrossRef]
- Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 May 9]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK554776/.
- Zhu Z, Lian X, Su X, Wu W, Marraro GA, Zeng Y. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res. 2020 Aug 27;21(1):224. [CrossRef]
- Abdelrahman Z, Li M, Wang X. Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses. Front Immunol [Internet]. 2020 [cited 2023 Jul 26];11. Available online. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2020.552909.
- Yang Y, Peng F, Wang R, Guan K, Jiang T, Xu G, et al. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J Autoimmun. 2020 May;109:102434. [CrossRef]
- Azhar EI, Hui DSC, Memish ZA, Drosten C, Zumla A. The Middle East Respiratory Syndrome (MERS). Infect Dis Clin North Am. 2019 Dec;33(4):891–905. [CrossRef]
- CSR. World Health Organization - Regional Office for the Eastern Mediterranean. [cited 2023 Jul 26]. MERS outbreaks. Available online: http://www.emro.who.int/health-topics/mers-cov/mers-outbreaks.html.
- Barry M, Phan MVT, Akkielah L, Al-Majed F, Alhetheel A, Somily A, et al. Nosocomial outbreak of the Middle East Respiratory Syndrome coronavirus: A phylogenetic, epidemiological, clinical and infection control analysis. Travel Med Infect Dis. 2020;37:101807. [CrossRef]
- Middle East respiratory syndrome coronavirus (MERS-CoV) [Internet]. [cited 2023 Nov 22]. Available online: https://www.who.int/news-room/fact-sheets/detail/middle-east-respiratory-syndrome-coronavirus-(mers-cov).
- WHO Coronavirus (COVID-19) Dashboard [Internet]. [cited 2023 May 8]. Available online: https://covid19.who.int.
- Zhou H, Yang J, Zhou C, Chen B, Fang H, Chen S, et al. A Review of SARS-CoV2: Compared With SARS-CoV and MERS-CoV. Front Med. 2021 Dec 7;8:628370. [CrossRef]
- Pustake M, Tambolkar I, Giri P, Gandhi C. SARS, MERS and CoVID-19: An overview and comparison of clinical, laboratory and radiological features. J Fam Med Prim Care. 2022 Jan;11(1):10–7. [CrossRef]
- Lau SKP, Luk HKH, Wong ACP, Li KSM, Zhu L, He Z, et al. Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg Infect Dis. 2020 Jul;26(7):1542–7. [CrossRef]
- Ghai RR, Carpenter A, Liew AY, Martin KB, Herring MK, Gerber SI, et al. Animal Reservoirs and Hosts for Emerging Alphacoronaviruses and Betacoronaviruses - Volume 27, Number 4—April 2021 - Emerging Infectious Diseases journal - CDC. [cited 2023 Jul 26]; Available online: https://wwwnc.cdc.gov/eid/article/27/4/20-3945_article.
- Gorbunova V, Seluanov A, Kennedy BK. The World Goes Bats: Living Longer and Tolerating Viruses. Cell Metab. 2020 Jul 7;32(1):31–43. [CrossRef]
- Banerjee A, Baker ML, Kulcsar K, Misra V, Plowright R, Mossman K. Novel Insights Into Immune Systems of Bats. Front Immunol. 2020 Jan 24;11:26. [CrossRef]
- Peck KM, Lauring AS. Complexities of Viral Mutation Rates. J Virol. 2018 Jun 29;92(14):10.1128/jvi.01031-17. [CrossRef]
- Kaur N, Singh R, Dar Z, Bijarnia RK, Dhingra N, Kaur T. Genetic comparison among various coronavirus strains for the identification of potential vaccine targets of SARS-CoV2. Infect Genet Evol. 2021 Apr;89:104490. [CrossRef]
- Mallapaty, S. Closest known relatives of virus behind COVID-19 found in Laos. Nature. 2021 Sep 24;597(7878):603–603. [CrossRef]
- Cyranoski, D. Bat cave solves mystery of deadly SARS virus — and suggests new outbreak could occur. Nature. 2017 Dec 1;552(7683):15–6. [CrossRef]
- Rossi GA, Sacco O, Mancino E, Cristiani L, Midulla F. Differences and similarities between SARS-CoV and SARS-CoV-2: spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection. 2020;48(5):665–9. [CrossRef]
- FigTree [Internet]. [cited 2022 Feb 25]. Available online: http://tree.bio.ed.ac.uk/software/figtree/.
- Wang N, Shang J, Jiang S, Du L. Subunit Vaccines Against Emerging Pathogenic Human Coronaviruses. Front Microbiol [Internet]. 2020 [cited 2022 Mar 24];11. Available online: https://www.frontiersin.org/article/10.3389/fmicb.2020. Available online: https://www.frontiersin.org/article/10.3389/fmicb.2020.00298.
- Binet M, Gascuel O, Scornavacca C, P. Douzery EJ, Pardi F. Fast and accurate branch lengths estimation for phylogenomic trees. BMC Bioinformatics. 2016 Jan 7;17:23.
- Information NC for B, Pike USNL of M 8600 R, MD B, Usa 20894. National Center for Biotechnology Information [Internet]. [cited 2022 Jan 13]. Available online: https://www.ncbi.nlm.nih.gov/.
- Artic Network [Internet]. [cited 2023 Jul 26]. Available online: https://artic.network/how-to-read-a-tree.html#.
- El-Sayed A, Kamel M. Coronaviruses in humans and animals: the role of bats in viral evolution. Environ Sci Pollut Res Int. 2021;28(16):19589–600. [CrossRef]
- Temmam S, Vongphayloth K, Baquero E, Munier S, Bonomi M, Regnault B, et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature. 2022 Apr;604(7905):330–6. [CrossRef]
- BioRender [Internet]. [cited 2022 May 11]. Available online: https://app.biorender.com/illustrations/627b7981d323fa4528ad7c00.
- Ellis P, Somogyvári F, Virok DP, Noseda M, McLean GR. Decoding Covid-19 with the SARS-CoV-2 Genome. Curr Genet Med Rep. 2021;9(1):1–12. [CrossRef]
- Krichel B, Falke S, Hilgenfeld R, Redecke L, Uetrecht C. Processing of the SARS-CoV pp1a/ab nsp7–10 region. Biochem J. 2020 Mar 13;477(5):1009–19. [CrossRef]
- Fehr AR, Perlman S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Coronaviruses. 2015 Feb 12;1282:1–23. [CrossRef]
- Mu J, Xu J, Zhang L, Shu T, Wu D, Huang M, et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci China Life Sci. 2020 Sep;63(9):1413–6. [CrossRef]
- Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler LA, et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. 2020 Oct;27(10):959–66. [CrossRef]
- Cornillez-Ty CT, Liao L, Yates JR, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. 2009 Oct;83(19):10314–8. [CrossRef]
- Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 2018 Jan;149:58–74. [CrossRef]
- Davies JP, Almasy KM, McDonald EF, Plate L. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus non-structural proteins identifies unique and shared host-cell dependencies. bioRxiv. 2020 Jul 14;2020.07.13.201517. [CrossRef]
- Scott BM, Lacasse V, Blom DG, Tonner PD, Blom NS. Predicted coronavirus Nsp5 protease cleavage sites in the human proteome. BMC Genomic Data. 2022 Apr 4;23(1):25. [CrossRef]
- Sun X, Liu Y, Huang Z, Xu W, Hu W, Yi L, et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022 Jun;29(6):1240–54. [CrossRef]
- Reshamwala SMS, Likhite V, Degani MS, Deb SS, Noronha SB. Mutations in SARS-CoV-2 nsp7 and nsp8 proteins and their predicted impact on replication/transcription complex structure. J Med Virol. 2021 Jul;93(7):4616–9. [CrossRef]
- El-Kamand S, Du Plessis MD, Breen N, Johnson L, Beard S, Kwan AH, et al. A distinct ssDNA/RNA binding interface in the Nsp9 protein from SARS-CoV-2. Proteins Struct Funct Bioinforma. 2022;90(1):176–85. [CrossRef]
- Lin S, Chen H, Chen Z, Yang F, Ye F, Zheng Y, et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 2021 ;49(9):5382–92. 21 May. [CrossRef]
- Gadhave K, Kumar P, Kumar A, Bhardwaj T, Garg N, Giri R. Conformational dynamics of 13 amino acids long NSP11 of SARS-CoV-2 under membrane mimetics and different solvent conditions. Microb Pathog. 2021 Sep;158:105041. [CrossRef]
- Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol. 2021 Apr;18(4):945–53. [CrossRef]
- Fung SY, Siu KL, Lin H, Chan CP, Yeung ML, Jin DY. SARS-CoV-2 NSP13 helicase suppresses interferon signaling by perturbing JAK1 phosphorylation of STAT1. Cell Biosci. 2022 Mar 22;12(1):36. [CrossRef]
- Ma Y, Wu L, Shaw N, Gao Y, Wang J, Sun Y, et al. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc Natl Acad Sci. 2015 Jul 28;112(30):9436–41. [CrossRef]
- Frazier MN, Dillard LB, Krahn JM, Perera L, Williams JG, Wilson IM, et al. Characterization of SARS2 Nsp15 nuclease activity reveals it’s mad about U. Nucleic Acids Res. 2021 Sep 27;49(17):10136–49. [CrossRef]
- Vithani N, Ward MD, Zimmerman MI, Novak B, Borowsky JH, Singh S, et al. SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys J. 2021 Jul 20;120(14):2880–9. [CrossRef]
- Azad GK, Khan PK. Variations in Orf3a protein of SARS-CoV-2 alter its structure and function. Biochem Biophys Rep. 2021 Jul 1;26:100933. [CrossRef]
- V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021 Mar;19(3):155–70. [CrossRef]
- Miyamoto Y, Itoh Y, Suzuki T, Tanaka T, Sakai Y, Koido M, et al. SARS-CoV-2 ORF6 disrupts nucleocytoplasmic trafficking to advance viral replication. Commun Biol. 2022 ;5(1):1–15. 19 May. [CrossRef]
- Redondo N, Zaldívar-López S, Garrido JJ, Montoya M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front Immunol [Internet]. 2021 [cited 2022 Sep 18];12. [CrossRef]
- Gao X, Zhu K, Qin B, Olieric V, Wang M, Cui S. Crystal structure of SARS-CoV-2 Orf9b in complex with human TOM70 suggests unusual virus-host interactions. Nat Commun. 2021 ;12(1):2843. 14 May. [CrossRef]
- Suzuki YJ, Gychka SG. SARS-CoV-2 Spike Protein Elicits Cell Signaling in Human Host Cells: Implications for Possible Consequences of COVID-19 Vaccines. Vaccines. 2021 Jan 11;9(1):36. [CrossRef]
- Zhang Z, Nomura N, Muramoto Y, Ekimoto T, Uemura T, Liu K, et al. Structure of SARS-CoV-2 membrane protein essential for virus assembly. Nat Commun. 2022 Aug 5;13(1):4399. [CrossRef]
- Chai J, Cai Y, Pang C, Wang L, McSweeney S, Shanklin J, et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat Commun. 2021 Jun 8;12(1):3433. [CrossRef]
- Narayanan A, Narwal M, Majowicz SA, Varricchio C, Toner SA, Ballatore C, et al. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun Biol. 2022 Feb 25;5(1):1–17. [CrossRef]
- RdRp inhibitors and COVID-19: Is molnupiravir a good option? - ScienceDirect [Internet]. [cited 2022 Aug 19]. Available online: https://www.sciencedirect.com/science/article/pii/S0753332221013044.
- Robson F, Khan KS, Le TK, Paris C, Demirbag S, Barfuss P, et al. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol Cell. 2020 Sep 3;79(5):710–27. [CrossRef]
- McLean G, Kamil J, Lee B, Moore P, Schulz TF, Muik A, et al. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio. 13(2):e02979-21. [CrossRef]
- Akkiz, H. Implications of the Novel Mutations in the SARS-CoV-2 Genome for Transmission, Disease Severity, and the Vaccine Development. Front Med. 2021 ;8:636532. 7 May. [CrossRef]
- Lang Y, Li W, Li Z, Koerhuis D, van den Burg ACS, Rozemuller E, et al. Coronavirus hemagglutinin-esterase and spike proteins coevolve for functional balance and optimal virion avidity. Proc Natl Acad Sci. 2020 Oct 13;117(41):25759–70. [CrossRef]
- Wong NA, Saier MH. The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. Int J Mol Sci. 2021 Jan 28;22(3):1308. [CrossRef]
- Liu D, Tedbury PR, Lan S, Huber AD, Puray-Chavez MN, Ji J, et al. Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus. Viruses. 2019 Nov 8;11(11):1039. [CrossRef]
- Afzal, A. Molecular diagnostic technologies for COVID-19: Limitations and challenges. J Adv Res. 2020 Aug 6;26:149–59. [CrossRef]
- Boopathi S, Poma AB, Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J Biomol Struct Dyn. 2020 Apr 30;1–10. [CrossRef]
- Suzuki YJ, Gychka SG. SARS-CoV-2 Spike Protein Elicits Cell Signaling in Human Host Cells: Implications for Possible Consequences of COVID-19 Vaccines. Vaccines. 2021 Jan 11;9(1):36. [CrossRef]
- Huang Y, Yang C, Xu X feng, Xu W, Liu S wen. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020 Sep;41(9):1141–9. [CrossRef]
- S N N, B N R, C P, K S S, Ramakrishnappa T, B T K, et al. SARS-CoV 2 spike protein S1 subunit as an ideal target for stable vaccines: A bioinformatic study. Mater Today Proc. 2022;49:904–12. [CrossRef]
- Thomas, S. The Structure of the Membrane Protein of SARS-CoV-2 Resembles the Sugar Transporter SemiSWEET. Pathog Immun. 2020 Oct 19;5(1):342–63. [CrossRef]
- Zeng W, Liu G, Ma H, Zhao D, Yang Y, Liu M, et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem Biophys Res Commun. 2020 Jun 30;527(3):618–23. [CrossRef]
- Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J. 2019 ;16:69. 27 May. [CrossRef]
- Fitzgerald DM, Rosenberg SM. What is mutation? A chapter in the series: How microbes “jeopardize” the modern synthesis. PLoS Genet. 2019 Apr 1;15(4):e1007995. [CrossRef]
- Mallapaty, S. Did the coronavirus jump from animals to people twice? Nature. 2021 Sep 16;597(7877):458–9. [CrossRef]
- Halley JM, Vokou D, Pappas G, Sainis I. SARS-CoV-2 mutational cascades and the risk of hyper-exponential growth. Microb Pathog. 2021 Dec;161:105237. [CrossRef]
- Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73(23):4433–48. [CrossRef]
- Barr JN, Fearns R. Genetic Instability of RNA Viruses. Genome Stab. 2016;21–35. [CrossRef]
- Islam MdA, Shahi S, Marzan AA, Amin MR, Hasan MN, Hoque MN, et al. Variant-specific deleterious mutations in the SARS-CoV-2 genome reveal immune responses and potentials for prophylactic vaccine development. Front Pharmacol. 2023 Feb 7;14:1090717. [CrossRef]
- Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021 Jul;19(7):409–24. [CrossRef]
- Eguia RT, Crawford KHD, Stevens-Ayers T, Kelnhofer-Millevolte L, Greninger AL, Englund JA, et al. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021 Apr 8;17(4):e1009453. [CrossRef]
- Forni D, Cagliani R, Clerici M, Sironi M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017 Jan;25(1):35–48. [CrossRef]
- Lazarevic I, Pravica V, Miljanovic D, Cupic M. Immune Evasion of SARS-CoV-2 Emerging Variants: What Have We Learnt So Far? Viruses. 2021 Jun 22;13(7):1192. [CrossRef]
- Mohammadi E, Shafiee F, Shahzamani K, Ranjbar MM, Alibakhshi A, Ahangarzadeh S, et al. Novel and emerging mutations of SARS-CoV-2: Biomedical implications. Biomed Pharmacother. 2021 Jul;139:111599. [CrossRef]
- Manathunga SS, Abeyagunawardena IA, Dharmaratne SD. A comparison of transmissibility of SARS-CoV-2 variants of concern. Virol J. 2023 Apr 2;20(1):59. [CrossRef]
- Padhan K, Parvez MK, Al-Dosari MS. Comparative sequence analysis of SARS-CoV-2 suggests its high transmissibility and pathogenicity. Future Virol. [CrossRef]
- Ou J, Lan W, Wu X, Zhao T, Duan B, Yang P, et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct Target Ther. 2022 Apr 26;7:138. [CrossRef]
- LaPelusa A, Kaushik R. Physiology, Proteins. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 [cited 2022 Oct 26]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK555990/.
- Zhang Z, Liu W, Zhang S, Wei P, Zhang L, Chen D, et al. Two novel human coronavirus OC43 genotypes circulating in hospitalized children with pneumonia in China. Emerg Microbes Infect. 11(1):168–71. [CrossRef]
- Wang C, Hesketh EL, Shamorkina TM, Li W, Franken PJ, Drabek D, et al. Antigenic structure of the human coronavirus OC43 spike reveals exposed and occluded neutralizing epitopes. Nat Commun. 2022 ;13(1):2921. 25 May. [CrossRef]
- Millet JK, Jaimes JA, Whittaker GR. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol Rev. 2020 Oct 28;45(3):fuaa057. [CrossRef]
- Nassar A, Ibrahim IM, Amin FG, Magdy M, Elgharib AM, Azzam EB, et al. A Review of Human Coronaviruses’ Receptors: The Host-Cell Targets for the Crown Bearing Viruses. Molecules. 2021 Oct 26;26(21):6455. [CrossRef]
- Rawat P, Jemimah S, Ponnuswamy PK, Gromiha MM. Why are ACE2 binding coronavirus strains SARS-CoV/SARS-CoV-2 wild and NL63 mild? Proteins Struct Funct Bioinforma. 2021;89(4):389–98. [CrossRef]
- Samavati L, Uhal BD. ACE2, Much More Than Just a Receptor for SARS-COV-2. Front Cell Infect Microbiol [Internet]. 2020 [cited 2022 Aug 18];10. 0031. [CrossRef]
- González-Morelo KJ, Vega-Sagardía M, Garrido D. Molecular Insights Into O-Linked Glycan Utilization by Gut Microbes. Front Microbiol [Internet]. 2020 [cited 2022 Aug 23];11. [CrossRef]
- Hulswit RJG, Lang Y, Bakkers MJG, Li W, Li Z, Schouten A, et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc Natl Acad Sci. 2019 Feb 12;116(7):2681–90. [CrossRef]
- Lachance C, Arbour N, Cashman NR, Talbot PJ. Involvement of Aminopeptidase N (CD13) in Infection of Human Neural Cells by Human Coronavirus 229E. J Virol. 1998 Aug;72(8):6511–9. [CrossRef]
- Kleine-Weber H, Schroeder S, Krüger N, Prokscha A, Naim HY, Müller MA, et al. Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg Microbes Infect. 2020 Jan 21;9(1):155–68. [CrossRef]
- de Castro KC, Costa JM. Polymeric surfaces with biocidal action: challenges imposed by the SARS-CoV-2, technologies employed, and future perspectives. J Polym Res. 2021;28(6):230. [CrossRef]
- Paintsil E, Cheng YC. Antiviral Agents. Encycl Microbiol. 2009;223–57. [CrossRef]
- Pruijssers AJ, Denison MR. Nucleoside analogues for the treatment of coronavirus infections. Curr Opin Virol. 2019 Apr;35:57–62. [CrossRef]
- De Clercq E, Li G. Approved Antiviral Drugs over the Past 50 Years. Clin Microbiol Rev. 2016 Jul;29(3):695–747. [CrossRef]
- Li G, Jing X, Zhang P, De Clercq E. Antiviral Classification. Encycl Virol. 2021;121–30. [CrossRef]
- Lenard, J. Viral Membranes. Encycl Virol. 2008;308–14. [CrossRef]
- Nucleoside Analogues. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012 [cited 2022 Sep 30]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK548938/.
- Weber IT, Kneller DW, Wong-Sam A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med Chem. 2015 Jun;7(8):1023–38. [CrossRef]
- Midde NM, Patters BJ, Rao P, Cory TJ, Kumar S. Investigational protease inhibitors as antiretroviral therapies. Expert Opin Investig Drugs. 2016 Oct;25(10):1189–200. [CrossRef]
- Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIVAIDS Auckl NZ. 2015 Apr 8;7:95–104. [CrossRef]
- Eyer L, Nencka R, de Clercq E, Seley-Radtke K, Růžek D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir Chem Chemother. 2018 Mar 13;26:2040206618761299. [CrossRef]
- Denel-Bobrowska M, Olejniczak AB. Non-nucleoside structured compounds with antiviral activity—past 10 years (2010–2020). Eur J Med Chem. 2022 Mar 5;231:114136. [CrossRef]
- Seley-Radtke KL, Yates MK. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 2018 Jun;154:66–86. [CrossRef]
- Zenchenko AA, Drenichev MS, Il’icheva IA, Mikhailov SN. Antiviral and Antimicrobial Nucleoside Derivatives: Structural Features and Mechanisms of Action. Mol Biol. 2021 Nov 1;55(6):786–812. [CrossRef]
- Shiraki K, Daikoku T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Ther. 2020 May;209:107512. [CrossRef]
- Nirwan S, Kakkar R. Rhinovirus RNA Polymerase. Viral Polym. 2019;301–31. [CrossRef]
- FURUTA Y, KOMENO T, NAKAMURA T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017 Aug 2;93(7):449–63. [CrossRef]
- Padhi AK, Dandapat J, Saudagar P, Uversky VN, Tripathi T. Interface-based design of the favipiravir-binding site in SARS-CoV-2 RNA-dependent RNA polymerase reveals mutations conferring resistance to chain termination. Febs Lett. 2021 Sep;595(18):2366–82. [CrossRef]
- PubChem. Favipiravir [Internet]. [cited 2023 Dec 2]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/492405.
- Sirijatuphat R, Manosuthi W, Niyomnaitham S, Owen A, Copeland KK, Charoenpong L, et al. Early treatment of Favipiravir in COVID-19 patients without pneumonia: a multicentre, open-labelled, randomized control study. Emerg Microbes Infect. 2022 Dec 31;11(1):2197–206. [CrossRef]
- Shah PL, Orton CM, Grinsztejn B, Donaldson GC, Ramírez BC, Tonkin J, et al. Favipiravir in patients hospitalised with COVID-19 (PIONEER trial): a multicentre, open-label, phase 3, randomised controlled trial of early intervention versus standard care. Lancet Respir Med. 2023 ;11(5):415–24. [CrossRef]
- Ray AS, Fordyce MW, Hitchcock MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Res. 2016 Jan 1;125:63–70. [CrossRef]
- Boyer PL, Sarafianos SG, Arnold E, Hughes SH. The M184V Mutation Reduces the Selective Excision of Zidovudine 5′-Monophosphate (AZTMP) by the Reverse Transcriptase of Human Immunodeficiency Virus Type 1. J Virol. 2002 Apr;76(7):3248. [CrossRef]
- Sluis-Cremer, N. Future of nonnucleoside reverse transcriptase inhibitors. Proc Natl Acad Sci U S A. 2018 Jan 23;115(4):637–8. [CrossRef]
- Rehman N, Nguyen H. Nevirapine. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 [cited 2022 Oct 11]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK554477/.
- Rock BM, Hengel SM, Rock DA, Wienkers LC, Kunze KL. Characterization of ritonavir-mediated inactivation of cytochrome P450 3A4. Mol Pharmacol. 2014 Dec;86(6):665–74. [CrossRef]
- PubChem. PubChem [Internet]. [cited 2022 Oct 11]. Available online: https://pubchem.ncbi.nlm.nih.gov/.
- Heidary M, Asadi A, Noorbakhsh N, Dashtbin S, Asadollahi P, Dranbandi A, et al. COVID-19 in HIV-positive patients: A systematic review of case reports and case series. J Clin Lab Anal. 2022 Apr;36(4):e24308. [CrossRef]
- Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease | Nature Communications [Internet]. [cited 2022 Oct 27]. Available online: https://www.nature. 4146. Available online: https://www.nature.com/articles/s41467-021-23751-3.
- Punekar M, Kshirsagar M, Tellapragada C, Patil K. Repurposing of antiviral drugs for COVID-19 and impact of repurposed drugs on the nervous system. Microb Pathog. 2022 Jul;168:105608. [CrossRef]
- Heo, YA. Sotrovimab: First Approval. Drugs. 2022;82(4):477–84. [CrossRef]
- Research C for DE and. FDA. FDA; 2023 [cited 2023 Aug 30]. Coronavirus (COVID-19) | Drugs. Available online: https://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs.
- Challenges and opportunities for antiviral monoclonal antibodies as COVID-19 therapy - ScienceDirect [Internet]. [cited 2022 Oct 27]. Available online: https://www.sciencedirect.com/science/article/pii/S0169409X20302751.
- Thomson EC, Rosen LE, Shepherd JG, Spreafico R, da Silva Filipe A, Wojcechowskyj JA, et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021 Mar 4;184(5):1171-1187.e20. [CrossRef]
- Roe MK, Junod NA, Young AR, Beachboard DC, Stobart CC. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J Gen Virol. 2021 Jan 28;102(3):001558. [CrossRef]
- Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N Engl J Med. 2022 Feb 16;NEJMoa2118542. [CrossRef]
- Liu J, Pan X, Zhang S, Li M, Ma K, Fan C, et al. Efficacy and safety of Paxlovid in severe adult patients with SARS-Cov-2 infection: a multicenter randomized controlled study. Lancet Reg Health – West Pac [Internet]. 2023 Apr 1 [cited 2023 Aug 1];33. Available online: https://www.thelancet.com/journals/lanwpc/article/PIIS2666-6065(23)00012-3/fulltext.
- Zhao M, Ma J, Li M, Zhang Y, Jiang B, Zhao X, et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int J Mol Sci. 2021 Nov 1;22(23):12808. [CrossRef]
- Vuong W, Khan MB, Fischer C, Arutyunova E, Lamer T, Shields J, et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2020 Aug 27;11(1):4282. [CrossRef]
- Amani B, Amani B. Efficacy and safety of nirmatrelvir/ritonavir (Paxlovid) for COVID-19: A rapid review and meta-analysis. J Med Virol. 2023 Feb;95(2):e28441. [CrossRef]
- Eastman RT, Roth JS, Brimacombe KR, Simeonov A, Shen M, Patnaik S, et al. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent Sci. 2020 ;6(5):672–83. [CrossRef]
- Kokic G, Hillen HS, Tegunov D, Dienemann C, Seitz F, Schmitzova J, et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat Commun. 2021 Jan 12;12(1):1–7. [CrossRef]
- Choi, KH. Viral Polymerases. Adv Exp Med Biol. 2012;726:267–304. [CrossRef]
- Gordon CJ, Tchesnokov EP, Schinazi RF, Götte M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J Biol Chem. 2021 ;297(1):100770. [CrossRef]
- Kabinger F, Stiller C, Schmitzová J, Dienemann C, Kokic G, Hillen HS, et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat Struct Mol Biol. 2021 Sep;28(9):740–6. [CrossRef]
- Wen W, Chen C, Tang J, Wang C, Zhou M, Cheng Y, et al. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19:a meta-analysis. Ann Med. 54(1):516–23. [CrossRef]
- Zarenezhad E, Marzi M. Review on molnupiravir as a promising oral drug for the treatment of COVID-19. Med Chem Res. 2022;31(2):232–43. [CrossRef]
- Venkataraman S, Prasad BVLS, Selvarajan R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses. 2018 Feb 10;10(2):76. [CrossRef]
- Gupte V, Hegde R, Sawant S, Kalathingal K, Jadhav S, Malabade R, et al. Safety and clinical outcomes of remdesivir in hospitalised COVID-19 patients: a retrospective analysis of active surveillance database. BMC Infect Dis. 2022 Jan 4;22(1):1. [CrossRef]
- Clinical antiviral efficacy of remdesivir and casirivimab/imdevimab against the SARS-CoV-2 Delta and Omicron variants | medRxiv [Internet]. [cited 2023 Sep 1]. Available online: https://www.medrxiv.org/content/10.1101/2022.10.17.22281161v1.full-text.
- Aleem A, Slenker AK. Monoclonal Antibody Therapy For High-Risk Coronavirus (COVID 19) Patients With Mild To Moderate Disease Presentations. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 [cited 2022 Mar 30]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK570603/.
- Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020 Jun 15;eabd0831. [CrossRef]
- Advances in Nucleoside and Nucleotide Analogues in Tackling Human Immunodeficiency Virus and Hepatitis Virus Infections - Ramesh - 2021 - ChemMedChem - Wiley Online Library [Internet]. [cited 2022 Mar 31]. 2020. [CrossRef]
- Melo-Filho CC, Bobrowski T, Martin HJ, Sessions Z, Popov KI, Moorman NJ, et al. Conserved coronavirus proteins as targets of broad-spectrum antivirals. Antiviral Res. 2022 Aug;204:105360. [CrossRef]
- Lasheras I, Santabárbara J. Use of antimalarial drugs in the treatment of COVID-19: A window of opportunity? Med Clin Engl Ed. 2020 Jul 10;155(1):23–5. [CrossRef]
- Shibeshi MA, Kifle ZD, Atnafie SA. Antimalarial Drug Resistance and Novel Targets for Antimalarial Drug Discovery. Infect Drug Resist. 2020 Nov 10;13:4047–60. [CrossRef]
- Ho TC, Wang YH, Chen YL, Tsai WC, Lee CH, Chuang KP, et al. Chloroquine and Hydroxychloroquine: Efficacy in the Treatment of the COVID-19. Pathogens. 2021 Feb 17;10(2):217. [CrossRef]
- Axfors C, Schmitt AM, Janiaud P, van’t Hooft J, Abd-Elsalam S, Abdo EF, et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat Commun. 2021 Apr 15;12(1):2349. [CrossRef]
- Kausar S, Said Khan F, Ishaq Mujeeb Ur Rehman M, Akram M, Riaz M, Rasool G, et al. A review: Mechanism of action of antiviral drugs. Int J Immunopathol Pharmacol. 2021 Mar 16;35:20587384211002621. [CrossRef]
- Pollard AJ, Bijker EM. A guide to vaccinology: from basic principles to new developments. Nat Rev Immunol. 2021 Feb;21(2):83–100. [CrossRef]
- Kudlay D, Svistunov A. COVID-19 Vaccines: An Overview of Different Platforms. Bioengineering [Internet]. 2022 Feb [cited 2023 Nov 27];9(2). Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8869214/.
- Li YD, Chi WY, Su JH, Ferrall L, Hung CF, Wu TC. Coronavirus vaccine development: from SARS and MERS to COVID-19. J Biomed Sci. 2020 Dec 20;27(1):104. [CrossRef]
- Mirtaleb MS, Falak R, Heshmatnia J, Bakhshandeh B, Taheri RA, Soleimanjahi H, et al. An insight overview on COVID-19 mRNA vaccines: Advantageous, pharmacology, mechanism of action, and prospective considerations. Int Immunopharmacol. 2023 Apr;117:109934. [CrossRef]
- CDC. Centers for Disease Control and Prevention. 2020 [cited 2022 Oct 24]. COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker.
- Johnson, AG. COVID-19 Incidence and Death Rates Among Unvaccinated and Fully Vaccinated Adults with and Without Booster Doses During Periods of Delta and Omicron Variant Emergence — 25 U.S. Jurisdictions, April 4–December 25, 2021. MMWR Morb Mortal Wkly Rep [Internet]. 2022 [cited 2022 Oct 24];71. Available online: https://www.cdc.gov/mmwr/volumes/71/wr/mm7104e2.htm.
- Malik JA, Ahmed S, Mir A, Shinde M, Bender O, Alshammari F, et al. The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J Infect Public Health. 2022 Feb;15(2):228–40. [CrossRef]
- Jaroszewski L, Iyer M, Alisoltani A, Sedova M, Godzik A. The interplay of SARS-CoV-2 evolution and constraints imposed by the structure and functionality of its proteins. PLoS Comput Biol. 2021 Jul 8;17(7):e1009147.
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020 ;10(5):766–88. [CrossRef]
- Bhatia, R. Addressing challenge of zoonotic diseases through One Health approach. Indian J Med Res. 2021 Mar;153(3):249–52. [CrossRef]
Figure 1.
A phylogenetic tree of all 7 HCoVs including a calculated distance between branches and nodes in nucleotides substitution per site [29,30,31]. The genetic sequences and reference genetic codes were obtained from the National Centre for Biotechnology Information (NCBI) [32]. The nodes represent the most recent common ancestor of the lineages, and the values are the measure of support for each node, the values are between 0-1 and the higher the value the stronger the evidence that the sequences cluster together this way [33].
Figure 1.
A phylogenetic tree of all 7 HCoVs including a calculated distance between branches and nodes in nucleotides substitution per site [29,30,31]. The genetic sequences and reference genetic codes were obtained from the National Centre for Biotechnology Information (NCBI) [32]. The nodes represent the most recent common ancestor of the lineages, and the values are the measure of support for each node, the values are between 0-1 and the higher the value the stronger the evidence that the sequences cluster together this way [33].
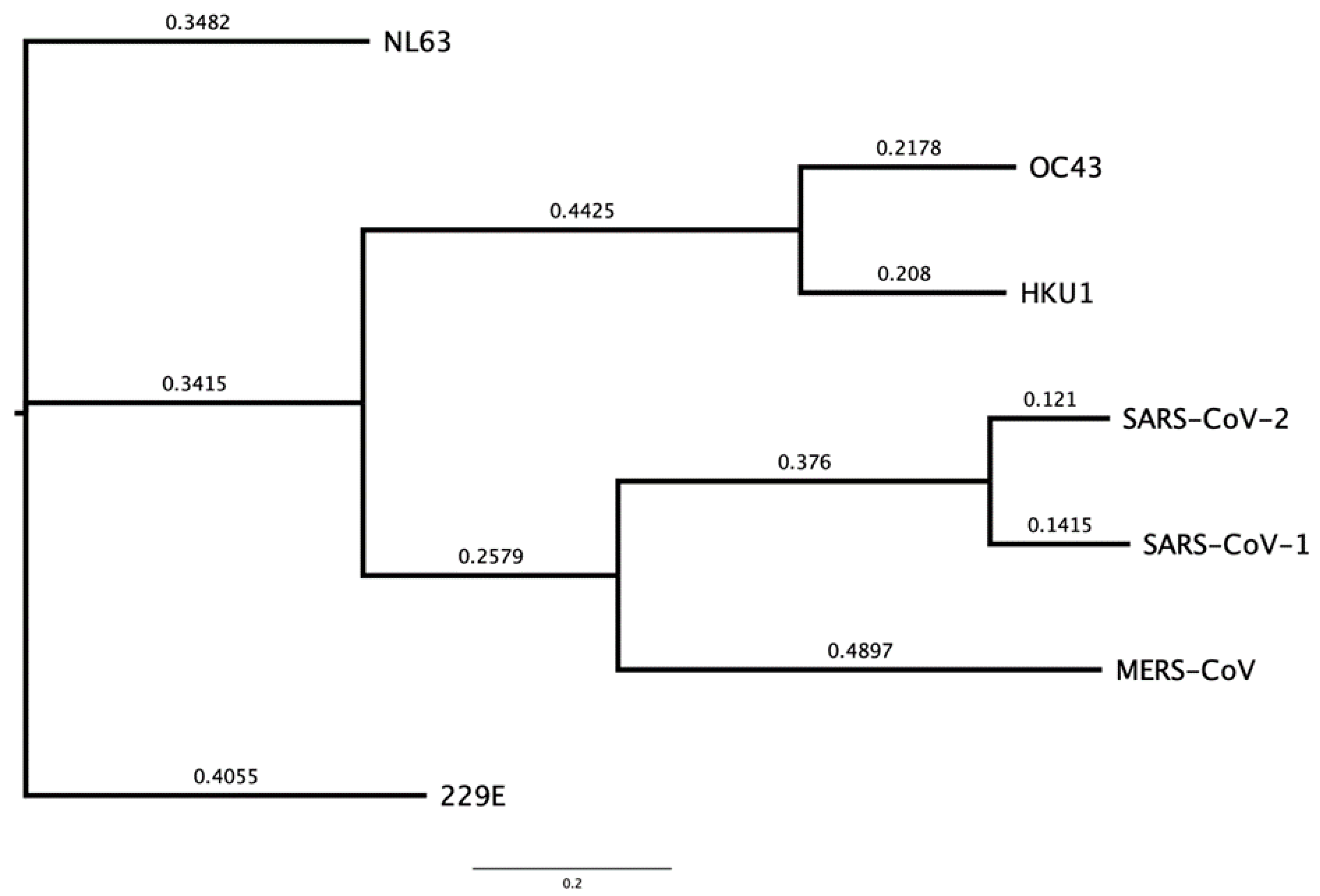
Figure 2.
The taxonomic groups of human and zoonotic CoVs which include alpha, beta, gamma and delta groups created using FigTree and BioRender [30,36]. Those with branches highlighted in red are the HCoVs also presented in Figure 1. The accession codes for the CoV genomes were found using NCBI (MN996532.2 (Bat CoV RaTG13), MZ081381.1 (RpYN06 betacoronavirus) , MZ802777.1 (porcine deltacoronavirus), MH532440.1 (quail deltacoronavirus), MK841495.1 (porcine endemic diarrhea virus), MN535737.1 (mink coronavirus 1), KM347965.1 (FRCoV-NL-2010), NC_002306.3 (feline infectious peritonitis virus), NC_026011.1 (HKU24 betacoronavirus), MZ368698.1 (avian coronavirus), NC_010646.1 (beluga whale coronavirus SW1), and the HCoVs accession codes are in Table 2 [32].
Figure 2.
The taxonomic groups of human and zoonotic CoVs which include alpha, beta, gamma and delta groups created using FigTree and BioRender [30,36]. Those with branches highlighted in red are the HCoVs also presented in Figure 1. The accession codes for the CoV genomes were found using NCBI (MN996532.2 (Bat CoV RaTG13), MZ081381.1 (RpYN06 betacoronavirus) , MZ802777.1 (porcine deltacoronavirus), MH532440.1 (quail deltacoronavirus), MK841495.1 (porcine endemic diarrhea virus), MN535737.1 (mink coronavirus 1), KM347965.1 (FRCoV-NL-2010), NC_002306.3 (feline infectious peritonitis virus), NC_026011.1 (HKU24 betacoronavirus), MZ368698.1 (avian coronavirus), NC_010646.1 (beluga whale coronavirus SW1), and the HCoVs accession codes are in Table 2 [32].
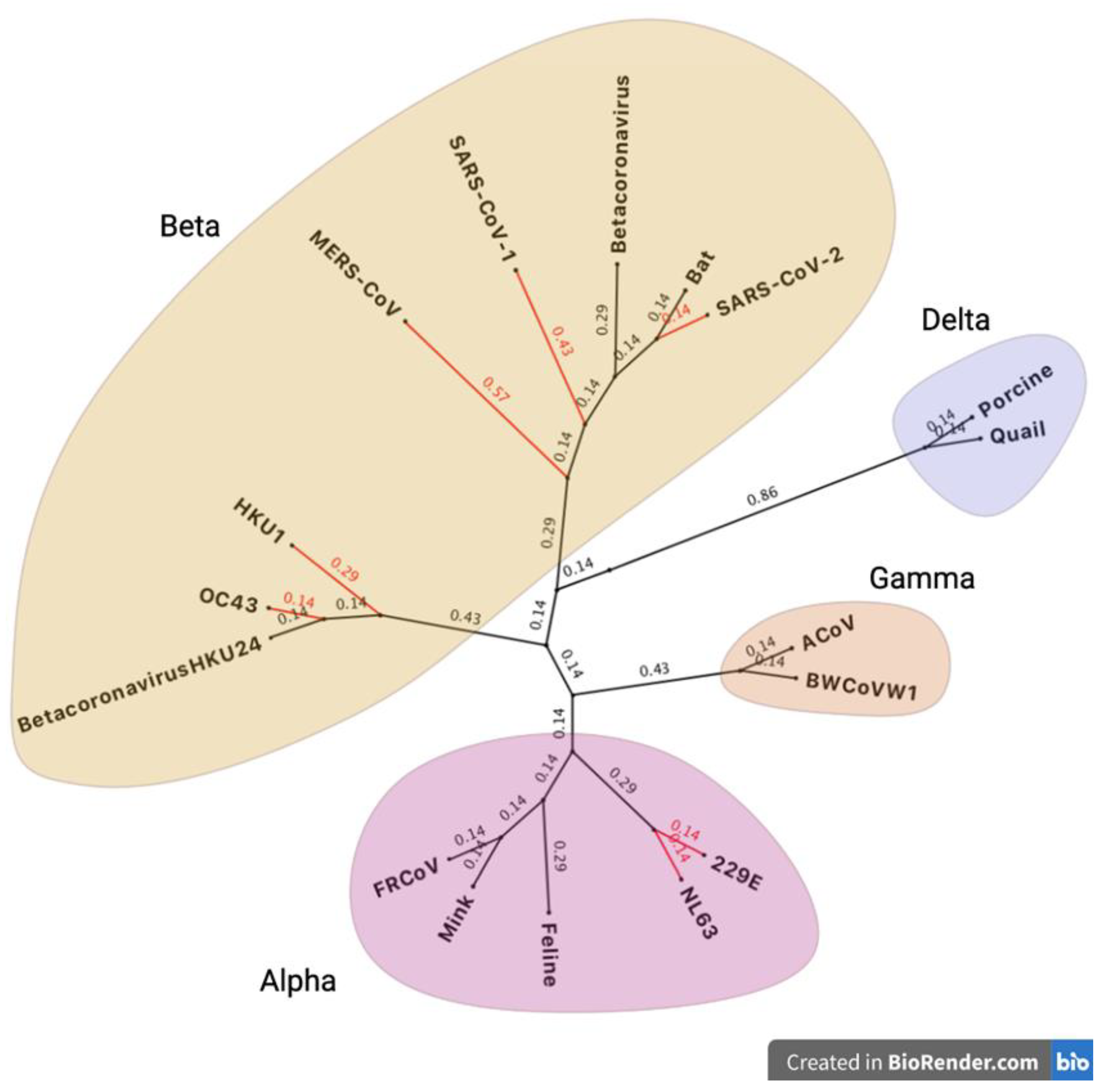
Figure 3.
The SARS-CoV-2 genome divided into its NSPs, ORFs, and notable structural proteins such as the spike (S), envelope (E), membrane (M) and nucleocapsid (N) [37]. Other important genes include ORFs 3 and 6-8 at the 3’ UTR. Diagram inspired by Ellis, et al, 2021 and created using BioRender [36,37].
Figure 3.
The SARS-CoV-2 genome divided into its NSPs, ORFs, and notable structural proteins such as the spike (S), envelope (E), membrane (M) and nucleocapsid (N) [37]. Other important genes include ORFs 3 and 6-8 at the 3’ UTR. Diagram inspired by Ellis, et al, 2021 and created using BioRender [36,37].
Figure 4.
A visual presentation of a generic human CoV virion, with image inspiration from Boopathi, S and Afzal, A and created in BioRender [36,72,73].
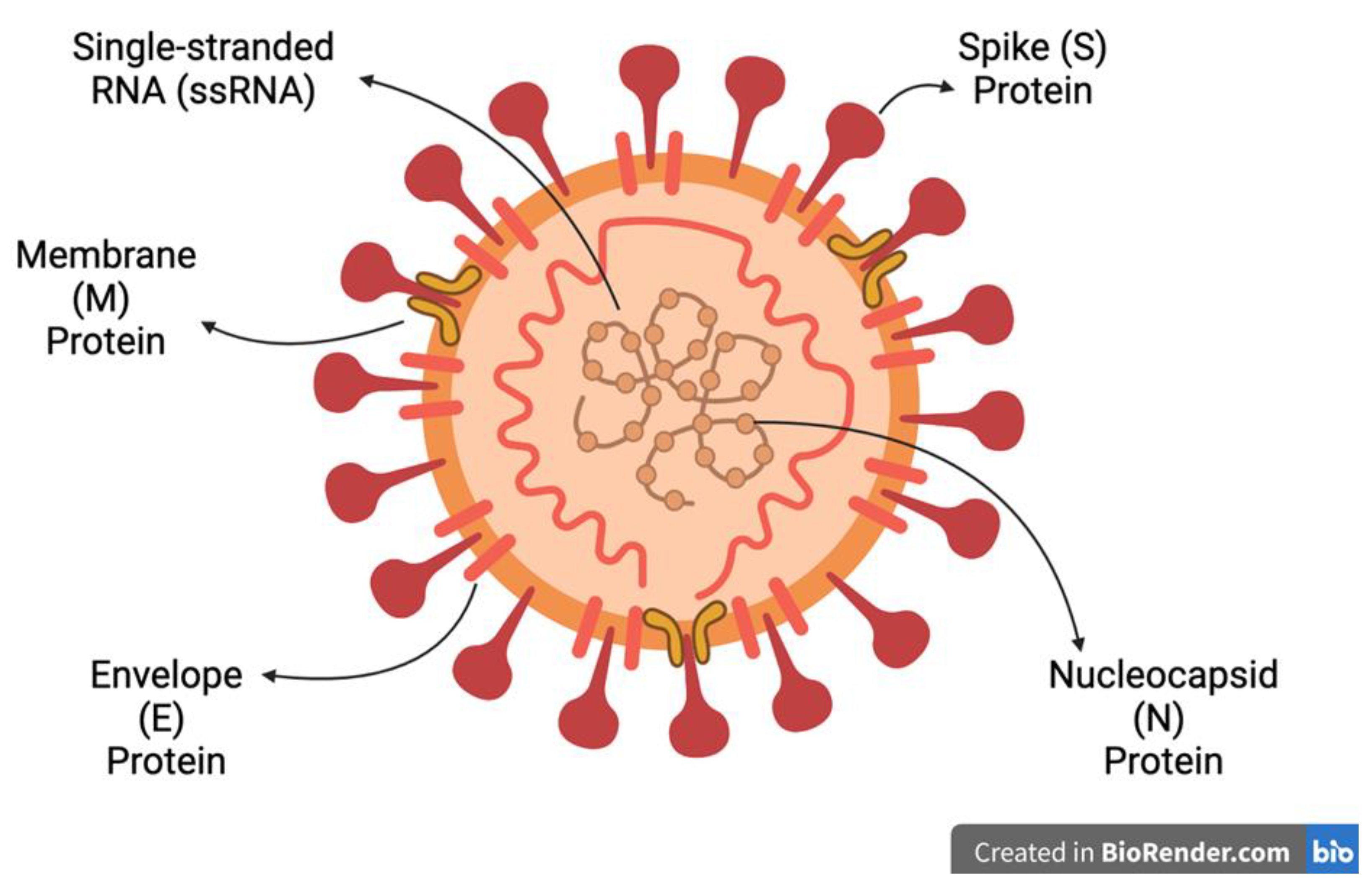
Figure 5.
The chemical structure of favipiravir [123].
Figure 5.
The chemical structure of favipiravir [123].
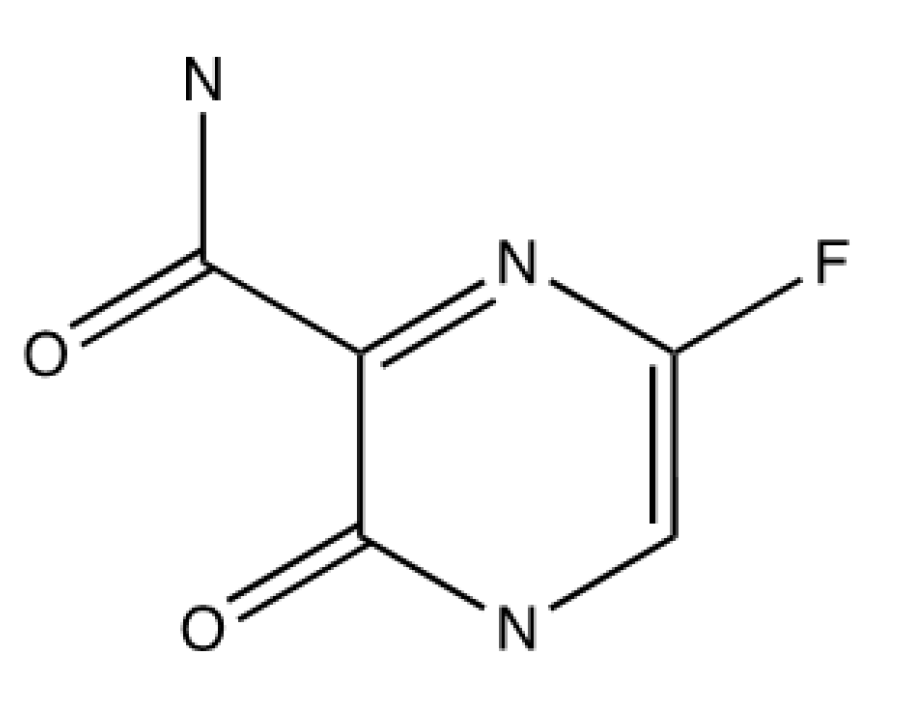
Figure 6.
The chemical structures of nevirapine and ritonavir [131].
Figure 6.
The chemical structures of nevirapine and ritonavir [131].
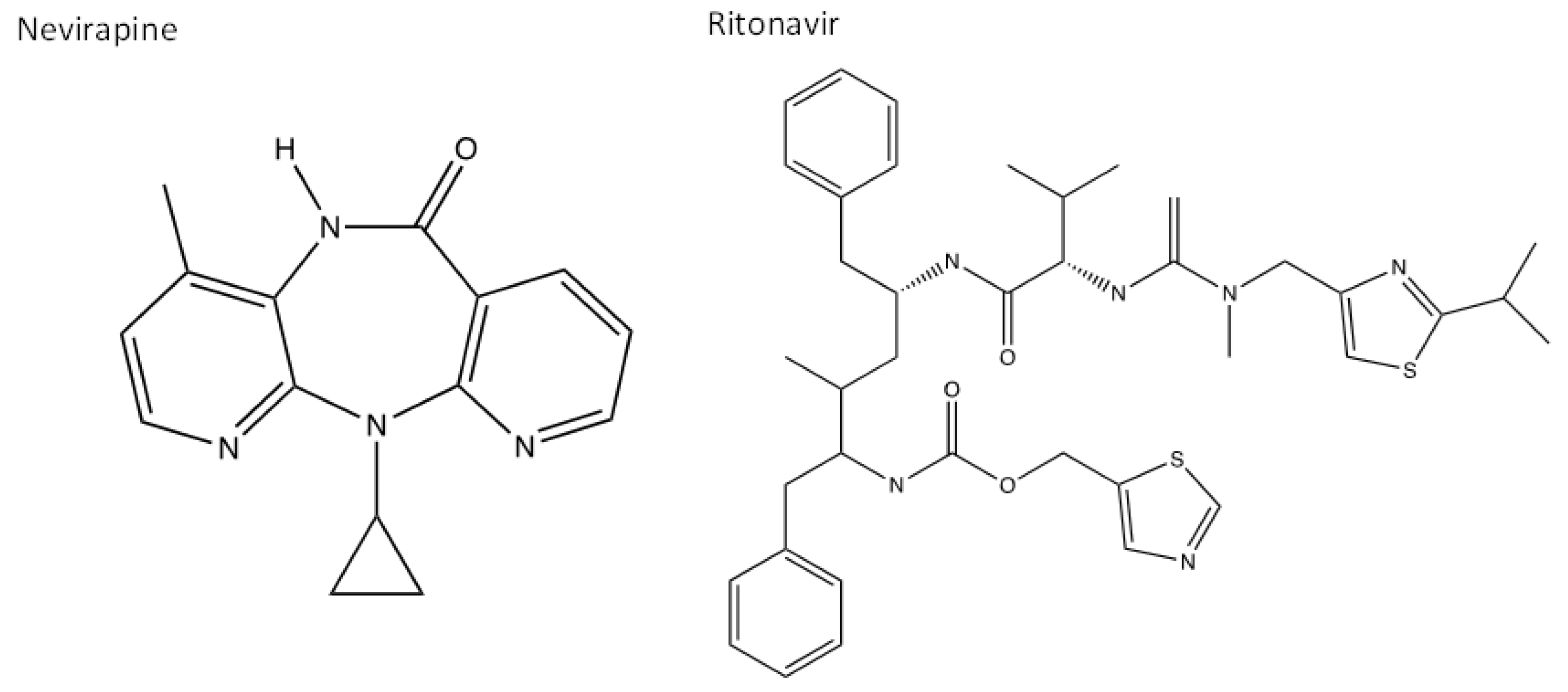
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



