
Submitted:
11 December 2023
Posted:
11 December 2023
You are already at the latest version
Abstract
Alterations in microRNA (miRNA) expression have been reported in different cancers. We assessed the expression of 754 oncology-related miRNAs in esophageal adenocarcinoma (EAC) and evaluated their correlations with clinical parameters. We found that miR-221 and 483-3p were consistently upregulated in EAC cases vs. controls (Wilcoxon signed rank test: miR-221 P<0.0001; miR-483-3p P<0.0001). Kaplan‒Meier analysis showed worse cancer-related survival in patients expressing high miR-221 or miR-483-3p levels in all EACs (log-rank P=0.0025 and P=0.0235, respectively). Higher miR-221 or miR-483-3p levels also correlated with advanced tumor stages (Mann‒Whitney P=0.0195 and P=0.0085, respectively). Moreover, we found that overexpression of miR-221 was associated with worse survival in the low-risk EAC group. A significantly worse outcome was associated with the combined overexpression of miR-221 and miR-483-3p (log-rank P=0.0410).
To identify target genes modified by miRNAs, we evaluatedtheir expression in different EAC cell lines, we transfected the corresponding mimic RNA (miRVANA) for either miR-221 or miR-483-3p and performed RNA-seq analysis. We found converging dysregulated genes involved in cancer progression, apoptosis, ATP synthesis and angiogenesis in miRNA-overexpressing cell lines, including a long non-coding RNA associated with oncogenesis, i.e., MALAT1.
In conclusion, miR-221 and 483-3p overexpression in EAC correlated with low cancer-related survival, indicating that they might be considered new biomarkers for patient stratification.
Keywords:
Esophageal adenocarcinoma
; microRNA
; miR-221
; miR-483-3p
1. Introduction
Esophageal adenocarcinoma (EAC) is a severe malignancy with a low survival rate and increasing incidence in Western countries. The causes of its high lethality may be attributed to inadequate screenings and early diagnosis programs, as well as the relative inefficiency of treatments. Indeed, most patients are diagnosed at an advanced stage, and the overall 5-year survival rate is 10-15% [1] EAC may rise according to the widely accepted sequence gastro-esophageal reflux disease (GERD)/intestinal metaplasia/dysplasia/adenocarcinoma, but other oncogenic pathways cannot be ruled out [2]. Since the end of the past century, several screening and early diagnosis programmes promoted by scientific and professional medical societies have been implemented, particularly for patients with histologically proven Barrett's esophagus (BE), which are thought to have a 40–50-fold higher annual incidence of EAC than the general population. However, only 12% of EAC patients have a prior diagnosis of BE, implying either difficulty in detecting BE in the diagnostic phase/surgical specimen or BE-independent pathways of EAC development [3]. The American Joint Committee on Cancer TNM staging system and the international guidelines on therapy consider EAC as a single entity [4]. However, EAC is consistently heterogeneous; therefore, different biological behaviors may impair the efficacy of unmodulated therapy [5,6,7,8]. EAC is among the tumors with the highest incidence of copy number alterations (CNAs) and somatic structural rearrangements [9], exhibits a high mutation frequency, and recent omics studies suggest the existence of distinct EAC subtypes based on different mutational signatures [10,11] and epigenetic mechanisms [9,12,13]. These different subtypes showed a correlation with prognostic factors and potential response to therapy [13,14].
In this framework, we investigated the value of microRNA (miRNA) expression as a potential biomarker in EAC subclassification and correlation with survival. MicroRNAs (miRNAs) regulate many cell processes by binding to the 3′ untranslated region of target mRNAs and therefore modulating their expression through translational repression, mRNA degradation or cleavage [15]. MiRNA dysregulation is implicated in different stages of tumor progression, [16,17], and miRNA expression can be modulated for therapeutic purposes [18]. A few studies identified altered miRNA profiles in esophageal squamous cell carcinoma and in BE-derived cancer [19,20,21,22,23]. However, limited knowledge exists regarding miRNAs that could discriminate among different subtypes of EAC (i.e., according to different histological subgroups).
This study aimed to assess the expression of a large number of oncology-related miRNAs in EAC samples derived from patients who underwent surgery without preoperative chemo- and radiotherapy and to correlate miRNA dysregulation with clinical features and histological subtypes to improve the efficiency of diagnosis and therapy for this aggressive form of cancer.
2. Materials and Methods
2.1. Tumor Samples
Samples for which RNA was available from paraffin embedded surgical resections (FFPE) of EAC cases among the Esophageal Adenocarcinoma Study Group Europe (EACSGE) consortium were included (124 cases) in the study. Clinical and pathological data [8,24] and EACSGE morphological classification have been previously reported. [25] The EACGSE classification was based on morphological features of esophageal/esophagogastric junction adenocarcinoma, which divided the cases into two main categories with a different prognosis: lower risk, including glandular well differentiated, mucinous muco-nodular carcinoma and diffuse desmoplastic subgroups; and higher risk, including glandular poorly differentiated, diffuse anaplastic, invasive mucinous carcinomas and mixed subgroups.
Eight specimens of healthy gastric mucosa extracted from FFPE blocks were used as controls. The study was approved (# L3P1223) by the Ethical Committee “Comitato Etico IRST IRCCS AVR (CEIIAV)”- Italy (Reg. Sper. 109/2016 Protocol 7353/51/2016).
2.2. Cell Lines
The OE19 (RRID:CVCL_1622 / ECACC: 96071721) [26], OE33 (RRID:CVCL_0471) and FLO-1 (RRID:CVCL_2045) cell lines were used for functional studies. OE-19 and OE-33 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (EuroClone, Milan, Italy). FLO-1 cells were cultured in High Glucose DMEM (Dulbecco's Modified Eagle Medium). All cell lines were supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin (supplements from Sigma Aldrich, St. Louis, Missouri, USA) at 37°C in a 5% CO2 atmosphere. The experiments were performed within 8 passages of resuscitation and all experiments were performed with mycoplasma-free cells.
2.3. RNA Isolation from FFPE Surgical Specimens
Total RNA was isolated starting from two 10-μm thick FFPE sections enriched in the tumor area using the RecoverAllTM Total Nucleic Acid Isolation for FFPE Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and treated with DNase I under RNase-free conditions, according to the manufacturer’s protocol. The yield was assessed through a NanoDrop spectrophotometer reading (Thermo Fisher Scientific), and an aliquot was run on a 1% agarose gel in 1XTBE and visualized using Midori green staining (Nippon Genetics Europe, Düren, Germany) under UV light.
2.4. MicroRNA Expression Profiling
Array Card Expression Analysis for 754 miRNAs
The expression of 754 different human miRNAs was profiled in 8 FFPE EAC cases and a pool of 8 FFPE healthy gastric mucosa samples using the TaqMan MicroRNA Array card A2.1/B3.0 (Cat. Num. 4399966 – 4444303; Thermo Fisher Scientific). U6 snRNA, RNU44 and RNU48 were used as endogenous controls. Fifty nanograms of total RNA was reverse transcribed (RT) using the TaqMan microRNA Reverse Transcription Kit (Cat. Num. 00331121; Thermo Fisher Scientific) and Megaplex RT primer pools A or B (Thermo Fisher Scientific). A preamplification step was performed combining 2.5 μl of the RT reaction with the matching Megaplex PreAmp Primer Pool and TaqMan PreAmp Master Mix (Cat. Num. 4384266; Thermo Fisher Scientific) under the following conditions: 10 min at 95°C; 2 min at 55°C; 2 min at 72°C; 15 sec at 95°C, 4 min at 60°C for 12 cycles; 99°C for 10 min. A dilution of 1:4 in TE 0.1X was made, and 9 μl of each dilution was combined with the TaqMan Universal Master Mix, NoAmpErase UNG (2X) (Cat. Num.4440040; Thermo Fisher Scientific) and loaded on the matching TaqMan MicroRNA Array Card. The cards were run on a 7900 HT Real Time PCR system (Thermo Fisher Scientific) with the following cycling conditions: 10 min at 95°C; 15 sec at 95°C, 1 min at 60°C, for 40 cycles. We employed the comparative 2-ΔΔCT method to analyse raw data with Expression Suite software (Thermo Fisher Scientific).
2.5. Single microRNA Expression Assays
The validation of miR-221 and miR-483-3p expression was performed through real-time quantitative PCR (RT‒qPCR) using single TaqMan probes (Thermo Fisher Scientific). Reverse transcription was performed starting from 150 ng of total RNA extracted from FFPE sections or OE19, OE33 and FLO-1 cell lines using the TaqMan MicroRNA Assay (Cat. Num. 4427975; Thermo Fisher Scientific) with primers for RNU44 (#001094), miR-221 (#000524) and miR-483-3p (#002339) and using the TaqMan MicroRNA Reverse Transcription Kit with a preamplification step as described above. RT‒qPCR was performed using 2 μl of the diluted preamplification product as outlined by the manufacturer. RNU44 was used as an endogenous control. For each case, the reaction was performed in triplicate. The relative miRNA expression levels for FFPE samples were calculated using the 2-ΔΔCT method, comparing FFPE tumor cases versus the healthy gastric mucosa. miRNA expression from cell line RNA was evaluated using a commercial RNA from human normal esophagus (Cat. Num B209050; BioChain, Newark, CA, USA).
2.6. miR-221 and miR-483-3p Mimic Transfection
A total of 3*105 cells were seeded in a 6-well plate to be 80% confluent at transfection. 25 nM of either mirVana™ miR-221 or miR-483-3p mimic (Cat. Num. 4464066; Thermo Fisher Scientific), and the corresponding negative controls were transfected using the TransIT-siQUEST transfection reagent (Cat. Num. MIR2114; Mirus Bio LCC; Madison USA) according to the manufacturer’s protocol. Cells were incubated at 37°C for 48 hours before RNA extraction. The validation of miR-221 and miR-483-3p overexpression was performed through RT‒qPCR using single TaqMan probes, as indicated above.
2.7. Bulk RNA-Sequencing (RNA-seq) in Transfected OE-19 Cells
RNA (250 ng) was extracted from transfected cells with a Recoverall kit (Thermo Fisher Scientific) and quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Library preparation and indexing for mRNA sequencing were performed with the Illumina TruSeq Stranded mRNA sample preparation kit (Illumina). Library sizes were verified using the Agilent High Sensitivity assay (Agilent Technologies) and quantified with the dsDNA High Sensitivity Assay for Qubit v.3.0 (Thermo Fisher). All samples were equally normalized, pooled and run onto the Illumina NexSeq550, with the Mid Output Kit v2.5 flow cell (150 cycles, paired-ends). Quality control of all the generated FASTQ files was performed with FastQC, [27] and results across all samples were summarized using MultiQC [28]. Reads were mapped on the reference human genome hg38 adopting STAR [29]; duplicate removal and sorting were carried out using SAMtools [30]. Gene expression was quantified and normalized as counts per million (CPM), starting from raw gene counts generated by the python package HTseq-count [31]. In both one-to-one comparisons (OE19-C1 vs miR-221 and OE19-C1 vs miR-483-3p), only genes with cpm > 0 in at least one sample were selected for further analysis. The log2 ratio of each gene was calculated as the difference between the log2cpm of samples derived from the control cells transfected with a scramble sequence and the log2cpm of samples from cells transfected with the corresponding miRVana from the two miRNAs. Subsequently, miR-221 and miR-483-3p duplicates were included to perform differential gene expression (DGE) analysis using the R-bioconductor limma package [32]. Differentially expressed genes with P ≤ 10−3 were selected for the evaluation of the functional classification of biological processes and pathway overrepresentation with the analytical tools PANTHER13.1 (Protein ANalysis THrough Evolutionary Relationships; http://pantherdb.org). PANTHER is strongly connected with a variety of other genomic resources, including the UniProt Reference Proteome datasets, the Quest for Orthologues Consortium, and the InterPro Consortium of protein classification resources. In addition to the phylogenetically derived annotations, the PANTHER Overrepresentation tool also provided functional annotations directly acquired from the Gene Ontology (GO) Consortium [33].
For gene validation from RNA-seq datasets, total RNA was isolated from transfected and control cells with TRIzol (Thermo Fisher Scientific) according to the manufacturer’s instructions. Five hundred nanograms of total RNA was retrotranscribed with SuperScript IV VILO MasterMix with ezDNase Enzyme (Thermo Fisher Scientific). Ten nanograms of cDNA was used as a template for the RT‒qPCRs with PowerTrack SYBR Green Master Mix 2X (Thermo Fisher Scientific) and 500 nM each of the forward- and reverse-specific primers, according to the protocol. The primers were as follows: MALAT1 forward 5΄- CGTAATGGAAAGTAAAGCCCT-3΄ and reverse TCTTGTGTTCTCTTGAGGGACA; ACTB forward 5´-CCTGGCACCCAGCACAAT-3´ and reverse 5´-GGGCCGGACTCGTCATACT-3´. ACTB, encoding β-actin, was used as an endogenous control. Each reaction was performed in triplicate. Data were analysed with the 2-ΔΔCT method using total RNA from nontransfected cell lines as a normal control.
2.8. Data Analysis
Quantitative analysis of the expression data derived from the microRNA Array Card experiments was performed with Expression Suite Software v.1.0 (Thermo Fisher Scientific). The ROC (Receiver Operating Characteristic) with Youden index method was used to optimize cut-off values for miRNA classification into “high expression” and “low expression” groups. Correlations between miRNA expression, tumor recurrence, cancer-related survival, tumor stage, and EACGSE classification were investigated using Mann‒Whitney and Kruskal‒Wallis tests. Survival analysis was performed using the Kaplan‒Meier method and the log-rank test. The multivariate analysis was performed according to the Cox regression analysis. The method of decision trees based on machine learning was adopted to develop a predictive algorithm of cancer-specific survival. Data analysis was performed using MedCalc 13.0.6.0 (MedCalc Software bvba, Østend, Belgium), SPSS15.0 software package (SPSS Inc., Chicago, IL, USA), Prism (GraphPad, San Diego, CA, USA) and the R software package (R Project for Statistical Computing, Vienna, Austria). The RT‒qPCR experiments were analysed with Student’s T test and one-way ANOVA. P values <0.05 were considered statistically significant.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
3. Results
3.1. Discovery Dataset: Identification of Deregulated miRNAs in EAC
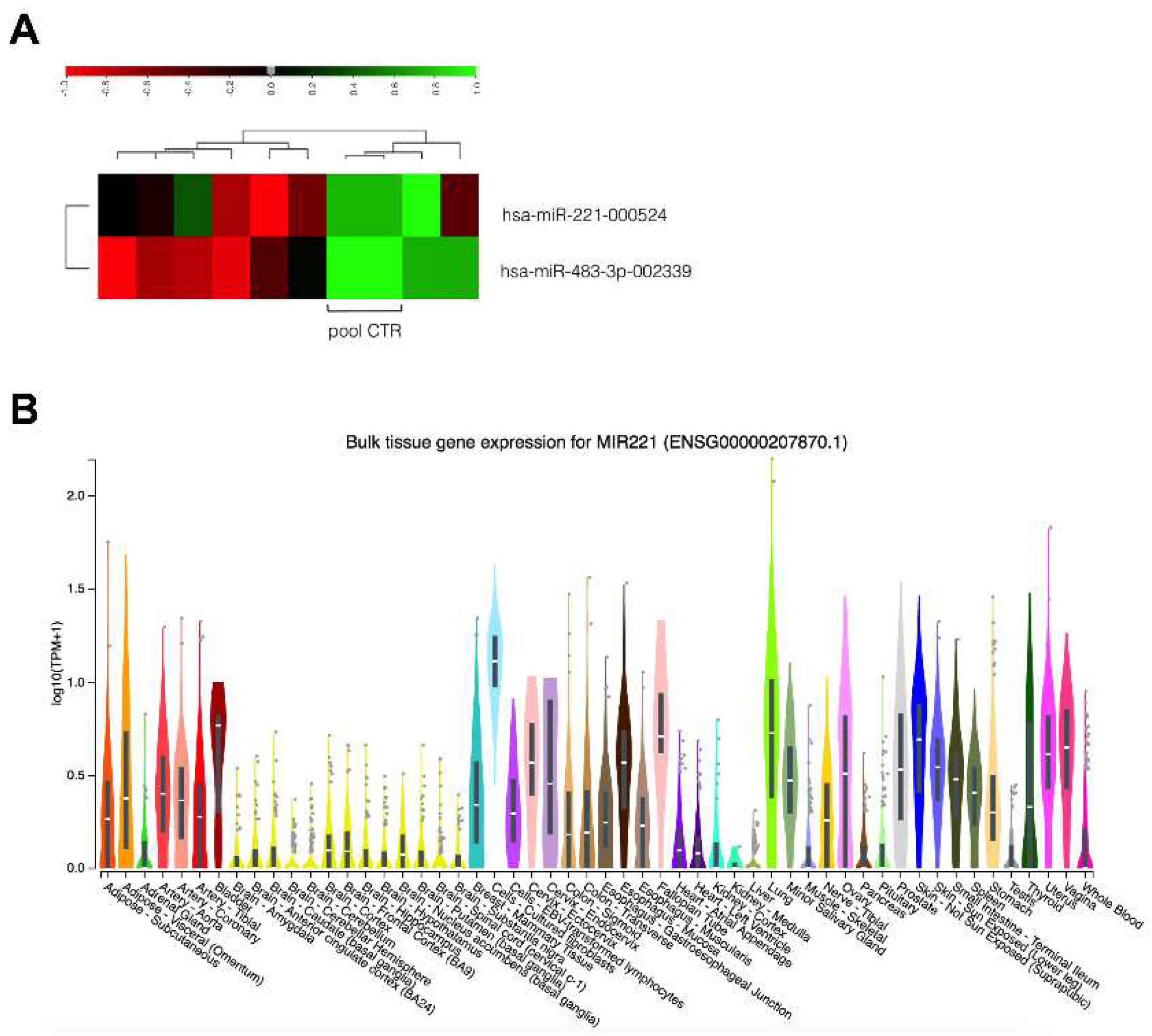
To study the differential expression of 754 tumor-related miRNAs, we first profiled 8 EAC cases vs. 8 healthy tissues. MiR-221 and miR-483-3p were consistently overexpressed, with mean fold increases of 2.746 and 11.33, respectively (Figure 1A). We verified the expression of miR-221 and miR-483-3p in human tissues in the Genotype-Tissue Expression (GTEx) portal: miR-221 was expressed in different tissues, including the esophagus, where it was more highly expressed in the esophageal mucosa compared to the esophageal junction (Figure 1B). Very low expression of miR-483-3p was identified in normal tissues (Supplementary Figure S1).
Figure 1.
Discovery of deregulated miRNAs in EAC. A. Heatmap showing the differential expression of miR-221 and miR-483-3p in EAC cases and controls. The color key indicates the expression levels from low (green) to high (red). B. Differential expression of miR-221 in human tissues, image from the Genotype-Tissue Expression (GTEx) portal.
Figure 1.
Discovery of deregulated miRNAs in EAC. A. Heatmap showing the differential expression of miR-221 and miR-483-3p in EAC cases and controls. The color key indicates the expression levels from low (green) to high (red). B. Differential expression of miR-221 in human tissues, image from the Genotype-Tissue Expression (GTEx) portal.
3.2. Replication Dataset (EACGSE Cohort): Single miRNA Analysis
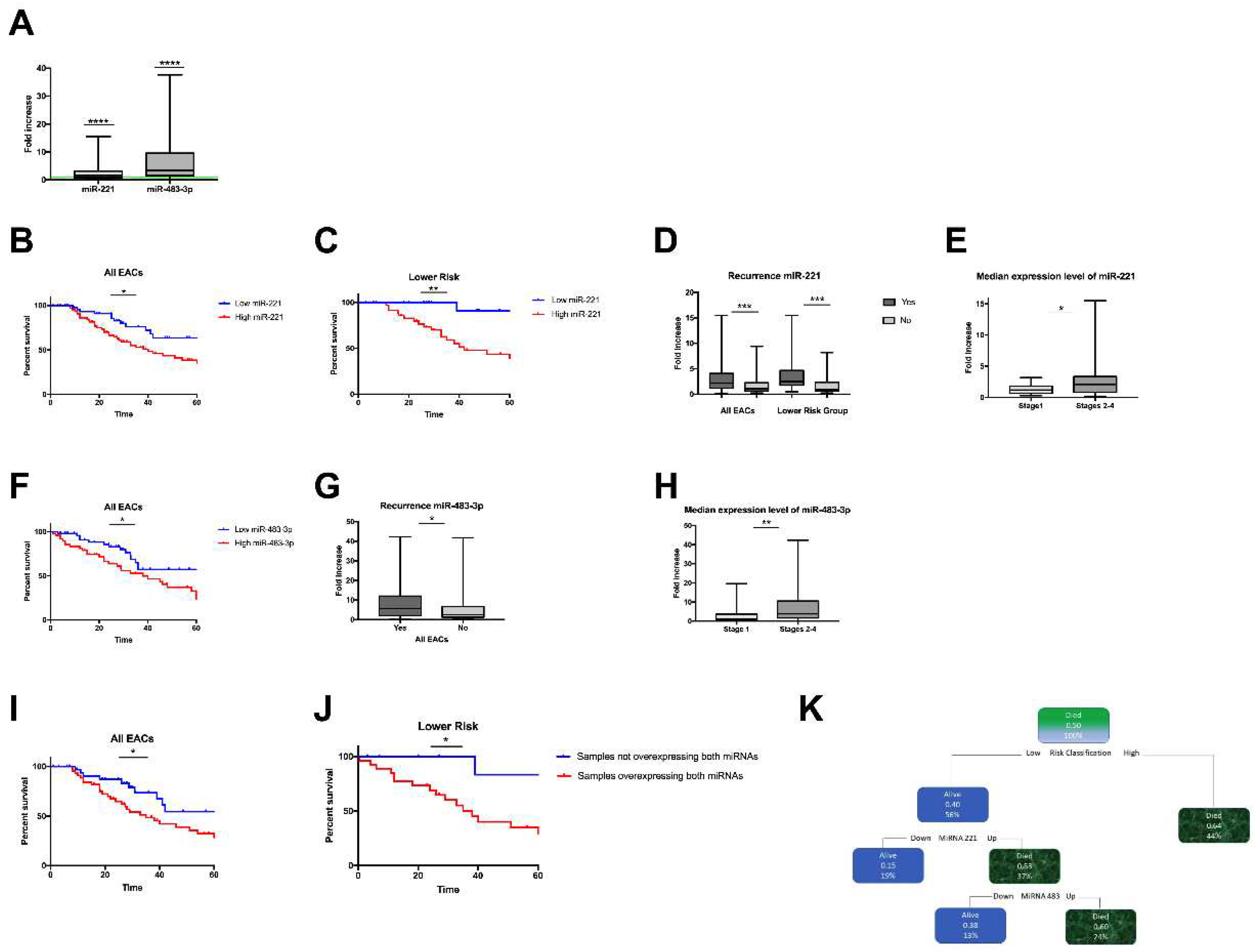
We investigated the expression of miR-221 and miR-483-3p in a separate cohort of 124 RNAs derived from FFPE surgical specimens of EAC patients (Supplementary Table S1). In accordance with our preliminary array data, these two miRNAs were significantly overexpressed in EAC tissues compared to normal tissues (miR-221 mean fold increase 2.276, Wilcoxon signed rank test: P<0.0001; miR-483-3p mean fold increase 5.964 P<0.0001; Figure 2A).
Figure 2.
miR-221 and miR-483-3p were significantly upregulated in the EAC replication group. A. miR-221 and miR-483-3p expression levels in a cohort of 124 EAC cases. The values are expressed as the fold increase compared to control FFPE-derived healthy gastric tissues (green baseline) (Wilcoxon signed rank test, P<0.0001). miR-221 expression levels and correlation with clinical outcomes. Kaplan‒Meier curves show the cancer-specific survival for EAC groups stratified by miR-221 expression levels. B. All EAC cases (log-rank P=0.0025). C. EACSGE lower risk subgroup (log-rank P=0.0065). Blue line: samples with low expression of miR-221, red line: samples with high expression of miR-221. D. Correlation between miR-221 expression and recurrence in all EACs (Mann‒Whitney P=0.0002) in the lower-risk EACSGE subgroup (Mann‒Whitney P=0.0005). E. Correlation between miR-221 expression and TNM stages (Mann‒Whitney P=0.0195 stage 1 versus stage 2-3-4). miR-483-3p expression levels and correlation with clinical outcomes. F. Kaplan‒Meier curves show cancer-specific survival for EAC groups stratified based on miR-483-3p expression levels in all EAC cases (log-rank P=0.0235). G. Recurrence in all EACs (Mann‒Whitney P=0.0173). H. Correlation between miR-483-3p and TNM stages (Mann‒Whitney P=0.0085 stage 1 versus stage 2-3-4). Combined overexpression of miR-221 and miR-483-3p and correlation with survival. Kaplan‒Meier curves for patients overexpressing both miRNAs versus patients not overexpressing both miRNAs showing cancer-specific survival in I. All EAC cases (log-rank P=0.0410), J. Lower risk EACSGE (log-rank P=0.0340). Predictive algorithm of cancer-specific survival. K. By using the decision tree method, a predictive algorithm of cancer-specific survival was developed. *=P ≤ 0.05; **=P ≤ 0.01; ***= P ≤ 0.001.
Figure 2.
miR-221 and miR-483-3p were significantly upregulated in the EAC replication group. A. miR-221 and miR-483-3p expression levels in a cohort of 124 EAC cases. The values are expressed as the fold increase compared to control FFPE-derived healthy gastric tissues (green baseline) (Wilcoxon signed rank test, P<0.0001). miR-221 expression levels and correlation with clinical outcomes. Kaplan‒Meier curves show the cancer-specific survival for EAC groups stratified by miR-221 expression levels. B. All EAC cases (log-rank P=0.0025). C. EACSGE lower risk subgroup (log-rank P=0.0065). Blue line: samples with low expression of miR-221, red line: samples with high expression of miR-221. D. Correlation between miR-221 expression and recurrence in all EACs (Mann‒Whitney P=0.0002) in the lower-risk EACSGE subgroup (Mann‒Whitney P=0.0005). E. Correlation between miR-221 expression and TNM stages (Mann‒Whitney P=0.0195 stage 1 versus stage 2-3-4). miR-483-3p expression levels and correlation with clinical outcomes. F. Kaplan‒Meier curves show cancer-specific survival for EAC groups stratified based on miR-483-3p expression levels in all EAC cases (log-rank P=0.0235). G. Recurrence in all EACs (Mann‒Whitney P=0.0173). H. Correlation between miR-483-3p and TNM stages (Mann‒Whitney P=0.0085 stage 1 versus stage 2-3-4). Combined overexpression of miR-221 and miR-483-3p and correlation with survival. Kaplan‒Meier curves for patients overexpressing both miRNAs versus patients not overexpressing both miRNAs showing cancer-specific survival in I. All EAC cases (log-rank P=0.0410), J. Lower risk EACSGE (log-rank P=0.0340). Predictive algorithm of cancer-specific survival. K. By using the decision tree method, a predictive algorithm of cancer-specific survival was developed. *=P ≤ 0.05; **=P ≤ 0.01; ***= P ≤ 0.001.
3.3. Correlation between miRNA-221 Expression and EAC Clinicopathological Features
We studied the relationship between miR-221 dysregulation and several clinical parameters in EAC. Using ROC curve analysis and the Youden index approach, the 124 EAC cases were categorized into "high" and "low" miR-221 expression groups (cut-off value of 1.32-fold change, P=0.003, Supplementary Figure S2A). Patients with high levels of miR-221 expression had a considerably worse prognosis, according to Kaplan‒Meier curves for cancer-specific survival (log-rank P= 0.0025, Figure 2B).
Next, we sought to correlate miRNA expression with EAC morpho-functional characteristics using the recently published EACSGE classification [25]. We found a significant correlation between miR-221 overexpression and worse outcome in the lower risk subgroup (log-rank P=0.0065; Figure 2C), whereas there was no significant correlation in the higher risk subgroup (Supplementary Figure S3A). Moreover, when considering all EAC cases, higher median expression levels of miR-221 were observed in relapsed patients than in nonrelapsed patients, but this association was more significant in the lower risk subgroup (Mann‒Whitney test P=0.0005 and P=0.0002, respectively, Figure 2D). In comparison to stage I patients, cases at advanced stages (stages I-IV) had significantly greater expression levels of miR-221 (Mann‒Whitney test P=0.0195, Figure 2E).
3.4. Correlation between miRNA-483-3p Expression and EAC Clinicopathological Features
We evaluated the correlation between miR-438-3p expression and clinical variables in our EAC cohort. All cases were divided into “high” or “low” miRNA-483-3p expression groups by evaluating the ROC curve with the Youden index method (cut-off value of 3.15-fold-change, P=0.0295, Supplementary Figure S2B).
In all EAC cases, patients with high miR-483-3p expression levels had a worse prognosis, according to Kaplan‒Meier analysis for cancer-specific survival (log-rank P=0.0235; Figure 2F), but it was not possible to observe specific differences in EACGSE lower vs higher risk groups (Supplementary Figures S3B and S3C, respectively).
Patients with relapses had significantly higher median expression levels of miR-483-3p (Mann‒Whitney test P=0.0173; Figure 2G). For patients overexpressing miR-483-3p, we also discovered a significant expression increase from stage I to later stages (Mann‒Whitney P=0.0085; Figure 2H).
3.5. Concurrent miRNA-221 and 483-3p Overexpression is Correlated with Poor Survival
To assess the combined effect of miR-221 and miR-483-3p on cancer-related survival, Kaplan‒Meier analysis was used to compare patients with both miRNAs overexpressed versus the rest of EAC patients (i.e., either only one miRNA overexpressed, or both not overexpressed). We found that the combined overexpression of miR-221 and miR-483-3p was linked to a significantly worse outcome in all EAC cases, particularly in the lower risk EACGSE subgroup (log-rank P=0.0410 and P=0.0340, respectively, Figures 2I and J).
Multivariate Cox regression analysis identified as statistically significant predictive variables for cancer-specific survival the histological classification in low-risk and high-risk groups of EAC (P<0.0001, HR 3.282, 95% CI 1.842-5.846) and the pathological stage (P=0.031, HR 9.279, 95% CI 1.229-70.056). The analysis to recognize a predictive prognostic value for miRNA dysregulation showed a trend toward significance (overexpression of miRNA-221, P=0.071).
Nonetheless, when we used a predictive algorithm of cancer-specific survival developed using the decision trees, the developed algorithm selected only the histological classification of EAC and the dysregulation of miR-221 and miR-483-3p in relation to cancer-specific survival (Figure 2K).
3.6. miRNA Overexpression and Transcriptome Analysis In Vitro
We evaluated miR-221 and miR-483-3p expression in three different EAC cell lines. Data were normalized using the RNA derived from a commercial pool of fresh normal human esophageal tissues [34]. Basal expression was present for both miRNAs, with no significant differences in the three cell lines (Supplementary Figures S4A and S4B). Therefore, we selected the OE-19 cells for further experiments to investigate the targets of miR-221 and miR-483-3p, since this cell line presents a TP53 inactivating mutation and ERBB2 amplifcation, conditions present in several of our EAC cases included in the analysis [11,24]. OE19 cells were transiently with either miR-221 or miR-483-3p mimic and a scramble negative control. Total RNA was extracted 48 hours post-transfection, and transfection efficiency was evaluated by RT‒qPCR for miR-221 and miR-483-3p, normalizing the expression with the endogenous control RNU44, using the scramble-transfected cells as controls (ANOVA test P<0.0001 and P<0.0001, respectively Supplementary Figures S4C). Next, we assessed the overall impact of miRNA 221 and 483-3p overexpression on gene expression via RNA-seq. RNA-seq was carried out on two independent transfected samples for each miRNA and scramble transfection. Data filtering, annotation and comparison were carried out according to our published pipeline [11]. Quantitative data analysis identified 220 altered genes with differential expression between miRNA-transfected and scramble-transfected cells when miR-221 was overexpressed (P≤ 10−3, Supplementary Table S2). A total of 868 altered genes were identified when miR483-3p was overexpressed (P≤10−3, Supplementary Table S3). Notably, the majority of the dysregulated genes were noncoding genes, such as long non-coding RNAs (lncRNAs), miRNAs and small nucleolar RNAs (snoRNAs), suggesting an important role of these miRNAs in the regulation of transcriptional complexes in EAC.
Analysis using the PANTHER Functional Classification test (PANTHER GO-Slim Process) for miR-221 led us to functionally map a total of 159 out of 220 differentially expressed genes to different biological processes, including ATP synthesis, the Wnt signaling pathway, p53 pathways, apoptosis, inflammation and neurodegenerative disorders (Figure 3A).
Figure 3.
Differentially expressed genes (DEGs) in transfected OE19 cell lines. After 48 hours of transient transfection with miRVANA for miR-221 or miR-483-3p, RNA-seq analysis of DEGs was performed vs cells transfected with negative control. Biological processes of genes (GO-Slim Biological processes) differentially expressed in EAC, as identified by PANTHER Functional Classification analysis, are reported for (A.) mir-221 and for (B.) mir-483-3p. C. Real-time qRT‒PCR data for MALAT1 expression. Data from transfected cells (overexpressing either miR-221 or miR-483-3p) were compared vs. cells transfected with a negative control (normalization was performed on a commercial pool of esophageal control tissues); human actin beta was used as an endogenous control gene (ANOVA test; P=0.007 and P=0.0469).
Figure 3.
Differentially expressed genes (DEGs) in transfected OE19 cell lines. After 48 hours of transient transfection with miRVANA for miR-221 or miR-483-3p, RNA-seq analysis of DEGs was performed vs cells transfected with negative control. Biological processes of genes (GO-Slim Biological processes) differentially expressed in EAC, as identified by PANTHER Functional Classification analysis, are reported for (A.) mir-221 and for (B.) mir-483-3p. C. Real-time qRT‒PCR data for MALAT1 expression. Data from transfected cells (overexpressing either miR-221 or miR-483-3p) were compared vs. cells transfected with a negative control (normalization was performed on a commercial pool of esophageal control tissues); human actin beta was used as an endogenous control gene (ANOVA test; P=0.007 and P=0.0469).
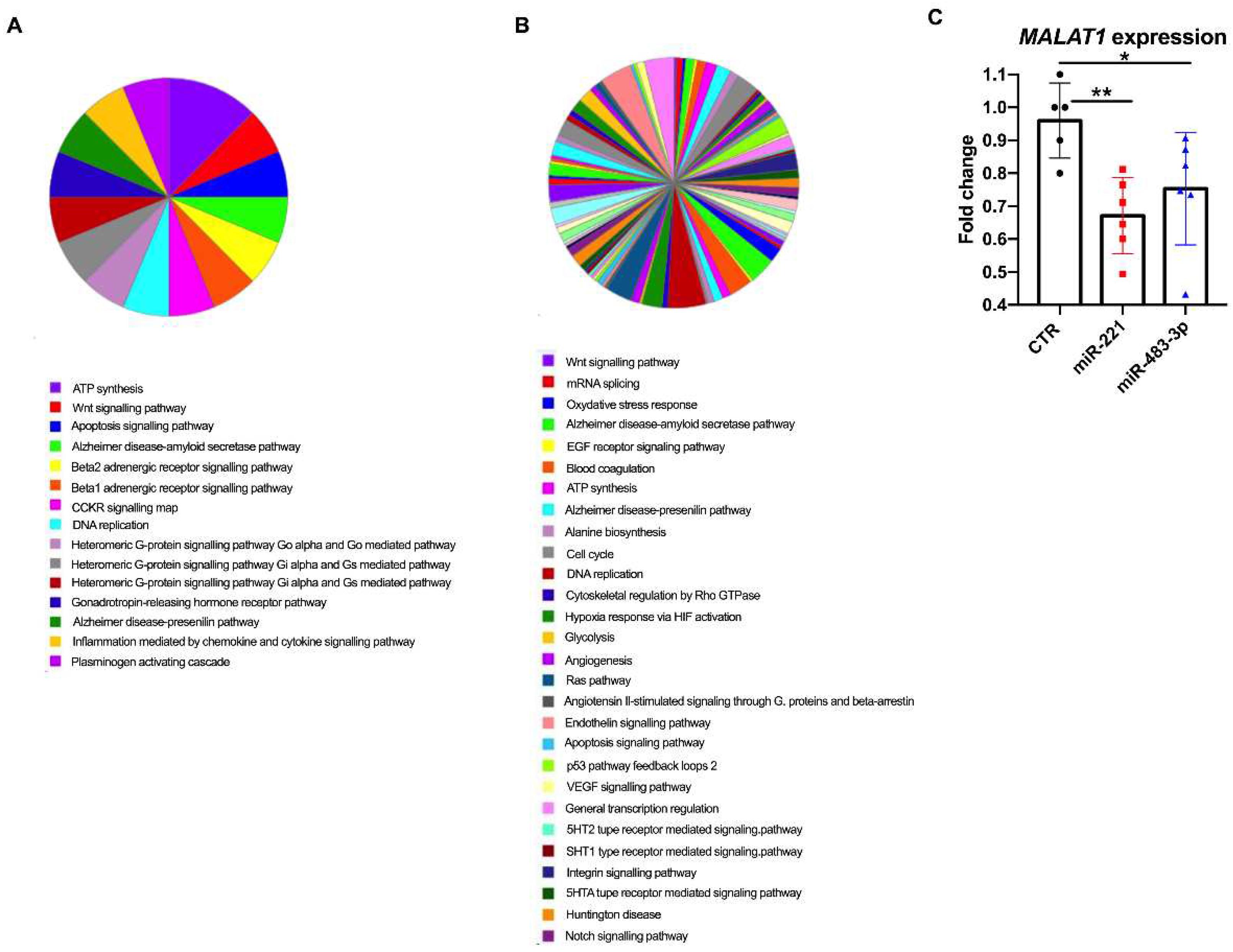
For miR-483-3p, we were able to map 172 of the 868 differentially expressed genes. Pathway analysis revealed that angiogenesis, Notch and Ras signaling, and cell cycle regulation were the main enriched pathways (Figure 3B). Several pathways were shared between the two miRNAs, suggesting a possible convergence in regulating oncogenic pathways.
Among the shared dysregulated genes, MALAT1 (metastasis-associated lung adenocarcinoma transcript 1) [35] was downregulated in both miR221 and miR483-3p overexpression analysis (P=3,32*10-55 and P=3.52*10-21, respectively). Thus, we further investigated MALAT1 via RT‒qPCR in transfected OE-19 cells vs. those transfected with the scramble sequence. RNA from nontransfected cell lines was used for data normalization using the ΔΔCt method. As reported in Figure 3C, we confirmed the dysregulated expression of MALAT1 in cells overexpressing either miR221 or miR483-3p (ANOVA test, P=0.007 and P=0.0469, respectively).
4. Discussion
EAC is characterized by high aggressiveness and poor prognosis [36,37]. The possibility to apply highly powered prognostic algorithms based on pathology and biomolecular patterns would have a capital role in tailoring the therapy according to biological characteristics of EAC and in driving an appropriate choice and timing of therapeutic and surgical options to improve their efficacy. EAC biological heterogeneity might be a barrier to achieving this fundamental goal. Several cancer-related characteristics are accelerated by EAC genomic instability, and some acquired mutations can benefit altered cells in specific ways [38]. However, when a cancer genome is very heterogeneous, as in EAC, it is difficult to fully characterize all the mutations, chromosomal rearrangements and epigenetic changes that give rise to tumor development and progression [38,39]. In our recent study, we showed that a combination of high-throughput sorting technology and massive sequencing could lead to a better definition of the EAC mutational status and inter- and intratumor heterogeneity than analysis of whole-tumor samples [11]. The identification of more/better prognostic markers, however, would help to subclassify the different forms of EACs, since the number of drivers per sample is frequently insufficient to fully explain the disease. Our most recent research developed a diagnostic algorithm that classified specific histotypes from adenocarcinomas with glandular architecture, further grading the former and subclassifying the latter. When combined with stage, this morphologic differentiation was shown to have a statistically significant prognostic influence either on its own or when dichotomized into lower- and higher-risk carcinomas. Indeed, the stage plus combination showed a high discriminating power for five-year cancer-specific survival [25].
It is a well-known concept that miRNAs can play an active role in tumor development and progression [16,40]. Specific miRNA signatures have been identified and translated into clinically relevant diagnostic and prognostic markers in thyroid cancer and hematological diseases [41,42]. In our study, we found two dysregulated miRNAs, miR-221 and miR-483-3p, that were reproducibly overexpressed in EAC. Their overexpression had previously been reported in many human cancers, and in vitro and in vivo studies supported a causal role for tumor progression in their dysregulated expression. In particular, in hepatocellular carcinoma, miR-221 overexpression correlated with tumor aggressiveness in terms of the number of metastases and multifocal lesions [43]. A possible role of miR-221 in EAC progression was provided by Matsuzaki and colleagues, since they reported an increased level of miR-221/222 in EAC compared to the surrounding BE [44]. Furthermore, in EAC, miR-221 is involved in 5-fluorouracile (5-FU) chemoresistance, leading to alteration of the Wnt/β-catenin pathway [45]. In our EAC cohort, we discovered an inverse relationship between increased miR-221 expression and cancer-specific survival and a significant correlation between increased miR-221 expression and tumor recurrence. Moreover, when we evaluated miRNA expression within the framework of the EACGSE categorization that distinguishes different histotypes [25], we observed that in lower risk carcinomas, patients with high levels of miR-221 expression had inferior cancer-specific survival and a significant correlation with recurrence. The correlation with EACGSE lower and higher risk, miR-221 overexpression and prognosis was further corroborated by the analysis using a predictive algorithm of cancer-specific survival.
The hsa-mir-483 gene (encoding both miR-483-5p and miR-483-3p) is a mammalian-conserved microRNA located within intron 2 of the human insulin growth factor 2 (IGF2) locus [46], an imprinted gene. Defects in the imprinting of the IGF2 locus are observed in Beckwith-Wiedemann syndrome, characterized, among other features, by an increased incidence of pediatric malignancies (nephroblastoma or Wilms’ tumor, hepatoblastoma, and rhabdomyosarcoma) [47]. miR-483-3p is overexpressed in Wilms’ tumors [48] but also in adult cancers such as colon, breast, and hepatocellular carcinoma [49,50,51]. In our EAC cohort, a lower expression of miR-483-3p was found in EAC tumors at stage I compared to other more advanced stages, suggesting that the miR-483-3p signature might also be useful for patient stratification.
To identify target genes modulated by miRNA upregulation, we enhanced the expression of miR-221 and miR-483-3p in OE-19 cells, a cell model of EAC that carries both a loss-of-function TP53 mutation and ERBB2 amplification. We performed a transcriptome analysis to identify the differentially expressed genes in cells overexpressing the two different miRNAs vs. OE19 cells transfected with a control negative sequence. Among the transcripts that exhibited significant dysregulation, many were noncoding genes, suggesting a complex regulatory system influenced by these miRNAs and targeting the transcription regulatory machinery. Among the protein-coding genes that we found dysregulated, several genes of interest have already been reported in the literature to be associated with cancer. For instance, in cells overexpressing miR-221, we observed an upregulation of FRAT2, AMD1 and MTHFD1L, genes linked to tumor progression, severity, invasiveness and worse prognosis [52,53,54,55,56,57,58,59,60]. ENTP6 was found to be downregulated, as previously reported in testicular cancer associated with cisplatin resistance [61].
It is interesting to identify MALAT1, a long non-coding RNA involved in cancer metastasis, as a target of both miR221 and miR483-3p overexpression. MALAT1 is aberrantly expressed in pancreatic cancer, lung cancer, breast cancer, colorectal cancer, gastric cancer, nasopharyngeal carcinoma, hepatocellular carcinoma and osteosarcoma [62]. MALAT1 is a nuclear-enriched and highly conserved lncRNA abundantly expressed in cells and tissues and is involved in mitochondrial homeostasis, cell proliferation and apoptosis. It has been shown that in lung epithelia, MALAT1 downregulation led to reduced apoptosis and promoted cell viability [63], suggesting a context-dependent regulation of different cell processes for this lncRNA. It will be interesting to further evaluate the role and expression of MALAT1 in EAC samples [26,34].
In conclusion, the study of miR-221 and miR-483-3p expression in our EAC cohort revealed that they are significantly overexpressed, and this dysregulation is correlated with worse clinical parameters. RNA sequencing analysis has demonstrated that this dysregulation leads to differential expression of genes previously reported to have a role in cancer development and progression.
Moreover, miR-221 profiling seems to be a promising strategy to identify patients with worse survival, especially in the EACGSE lower risk group, providing a valuable molecular parameter to stratify EAC cases.
It will be of great importance to characterize the expression of these miRNAs among circulating fluids in EAC patients (liquid biopsy). Indeed, liquid biopsy has emerged as a promising tool for diagnosis and prognosis and patient stratification for personalized therapy in various solid tumors. Therefore, combining this molecular approach with clinical parameters could help stratify EAC patients to improve their management and address specific therapeutic options and targets for tailored therapies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The present study was developed and performed in the framework of the EACSGE group (Esophageal Adenocarcinoma Study Group Europe) research program. We thank Dr. C. Diquigiovanni (DIMEC, University of Bologna) and Dr. A. Tomezzoli (University of Verona) for critical reading and discussion. Scientific english writing was edited using Curie (https://www.aje.com/curie).
References
- Dubecz, A.; Gall, I.; Solymosi, N.; Schweigert, M.; Peters, J. H.; Feith, M.; Stein, H. J. Temporal Trends in Long-Term Survival and Cure Rates in Esophageal Cancer: A SEER Database Analysis. J Thorac Oncol 2012, 7 (2), 443–447. [CrossRef]
- Velanovich, V.; Hollingsworth, J.; Suresh, P.; Ben-Menachem, T. Relationship of Gastroesophageal Reflux Disease with Adenocarcinoma of the Distal Esophagus and Cardia. Dig Surg 2002, 19 (5), 349–353. [CrossRef]
- Curtius, K.; Rubenstein, J. H.; Chak, A.; Inadomi, J. M. Computational Modelling Suggests That Barrett’s Oesophagus May Be the Precursor of All Oesophageal Adenocarcinomas. Gut 2020, 70 (8), 1435–1440. [CrossRef]
- Rice, T. W.; Ishwaran, H.; Ferguson, M. K.; Blackstone, E. H.; Goldstraw, P. Cancer of the Esophagus and Esophagogastric Junction: An Eighth Edition Staging Primer. J Thorac Oncol 2017, 12 (1), 36–42. [CrossRef]
- Mattioli, S.; Ruffato, A.; Di Simone, M. P.; Corti, B.; D’Errico, A.; Lugaresi, M. L.; Mattioli, B.; D’Ovidio, F. Immunopathological Patterns of the Stomach in Adenocarcinoma of the Esophagus, Cardia, and Gastric Antrum: Gastric Profiles in Siewert Type I and II Tumors. Ann Thorac Surg 2007, 83 (5), 1814–1819. [CrossRef]
- Ruffato, A.; Mattioli, S.; Perrone, O.; Lugaresi, M.; Di Simone, M. P.; D’Errico, A.; Malvi, D.; Aprile, M. R.; Raulli, G.; Frassineti, L. Esophagogastric Metaplasia Relates to Nodal Metastases in Adenocarcinoma of Esophagus and Cardia. Ann Thorac Surg 2013, 95 (4), 1147–1153. [CrossRef]
- Al-Batran, S.-E.; Hofheinz, R. D.; Pauligk, C.; Kopp, H.-G.; Haag, G. M.; Luley, K. B.; Meiler, J.; Homann, N.; Lorenzen, S.; Schmalenberg, H.; Probst, S.; Koenigsmann, M.; Egger, M.; Prasnikar, N.; Caca, K.; Trojan, J.; Martens, U. M.; Block, A.; Fischbach, W.; Mahlberg, R.; Clemens, M.; Illerhaus, G.; Zirlik, K.; Behringer, D. M.; Schmiegel, W.; Pohl, M.; Heike, M.; Ronellenfitsch, U.; Schuler, M.; Bechstein, W. O.; Königsrainer, A.; Gaiser, T.; Schirmacher, P.; Hozaeel, W.; Reichart, A.; Goetze, T. O.; Sievert, M.; Jäger, E.; Mönig, S.; Tannapfel, A. Histopathological Regression after Neoadjuvant Docetaxel, Oxaliplatin, Fluorouracil, and Leucovorin versus Epirubicin, Cisplatin, and Fluorouracil or Capecitabine in Patients with Resectable Gastric or Gastro-Oesophageal Junction Adenocarcinoma (FLOT4-AIO): Results from the Phase 2 Part of a Multicentre, Open-Label, Randomized Phase 2/3 Trial. Lancet Oncol 2016, 17 (12), 1697–1708. [CrossRef]
- van der Kaaij, R. T.; Snaebjornsson, P.; Voncken, F. E. M.; van Dieren, J. M.; Jansen, E. P. M.; Sikorska, K.; Cats, A.; van Sandick, J. W. The Prognostic and Potentially Predictive Value of the Laurén Classification in Oesophageal Adenocarcinoma. Eur J Cancer 2017, 76, 27–35. [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature 2017, 541 (7636), 169–175. [CrossRef]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M. D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; Yang, T.-P.; Bower, L.; Chettouh, H.; Crawte, J.; Galeano-Dalmau, N.; Grabowska, A.; Saunders, J.; Underwood, T.; Waddell, N.; Barbour, A. P.; Nutzinger, B.; Achilleos, A.; Edwards, P. A. W.; Lynch, A. G.; Tavaré, S.; Fitzgerald, R. C.; Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Consortium. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat Genet 2016, 48 (10), 1131–1141. [CrossRef]
- Isidori, F.; Bozzarelli, I.; Mastracci, L.; Malvi, D.; Lugaresi, M.; Molinari, C.; Söderström, H.; Räsänen, J.; D’Errico, A.; Fiocca, R.; Seri, M.; Krishnadath, K. K.; Bonora, E.; Mattioli, S. Targeted Sequencing of Sorted Esophageal Adenocarcinoma Cells Unveils Known and Novel Mutations in the Separated Subpopulations. Clin Transl Gastroenterol 2020, 11 (9), e00202. [CrossRef]
- Bornschein, J.; Wernisch, L.; Secrier, M.; Miremadi, A.; Perner, J.; MacRae, S.; O’Donovan, M.; Newton, R.; Menon, S.; Bower, L.; Eldridge, M. D.; Devonshire, G.; Cheah, C.; Turkington, R.; Hardwick, R. H.; Selgrad, M.; Venerito, M.; Malfertheiner, P.; OCCAMS Consortium; Fitzgerald, R. C. Transcriptomic Profiling Reveals Three Molecular Phenotypes of Adenocarcinoma at the Gastroesophageal Junction. Int J Cancer 2019, 145 (12), 3389–3401. [CrossRef]
- Jammula, S.; Katz-Summercorn, A. C.; Li, X.; Linossi, C.; Smyth, E.; Killcoyne, S.; Biasci, D.; Subash, V. V.; Abbas, S.; Blasko, A.; Devonshire, G.; Grantham, A.; Wronowski, F.; O’Donovan, M.; Grehan, N.; Eldridge, M. D.; Tavaré, S.; Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) consortium; Fitzgerald, R. C. Identification of Subtypes of Barrett’s Esophagus and Esophageal Adenocarcinoma Based on DNA Methylation Profiles and Integration of Transcriptome and Genome Data. Gastroenterology 2020, 158 (6), 1682-1697.e1. [CrossRef]
- Antonowicz, S.; Bodai, Z.; Wiggins, T.; Markar, S. R.; Boshier, P. R.; Goh, Y. M.; Adam, M. E.; Lu, H.; Kudo, H.; Rosini, F.; Goldin, R.; Moralli, D.; Green, C. M.; Peters, C. J.; Habib, N.; Gabra, H.; Fitzgerald, R. C.; Takats, Z.; Hanna, G. B. Endogenous Aldehyde Accumulation Generates Genotoxicity and Exhaled Biomarkers in Esophageal Adenocarcinoma. Nat Commun 2021, 12 (1), 1454. [CrossRef]
- Bartel, D. P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116 (2), 281–297. [CrossRef]
- Macfarlane, L.-A.; Murphy, P. R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics 2010, 11 (7), 537–561. [CrossRef]
- Di Leva, G.; Croce, C. M. Roles of Small RNAs in Tumor Formation. Trends Mol Med 2010, 16 (6), 257–267. [CrossRef]
- Shah, M. Y.; Calin, G. A. MicroRNAs as Therapeutic Targets in Human Cancers. Wiley Interdiscip Rev RNA 2014, 5 (4), 537–548. [CrossRef]
- Feber, A.; Xi, L.; Luketich, J. D.; Pennathur, A.; Landreneau, R. J.; Wu, M.; Swanson, S. J.; Godfrey, T. E.; Litle, V. R. MicroRNA Expression Profiles of Esophageal Cancer. J Thorac Cardiovasc Surg 2008, 135 (2), 255–260; discussion 260. [CrossRef]
- Gu, J.; Wang, Y.; Wu, X. MicroRNA in the Pathogenesis and Prognosis of Esophageal Cancer. Curr Pharm Des 2013, 19 (7), 1292–1300. [CrossRef]
- Gao, S.; Zhao, Z.-Y.; Zhang, Z.-Y.; Zhang, Y.; Wu, R. Prognostic Value of MicroRNAs in Esophageal Carcinoma: A Meta-Analysis. Clin Transl Gastroenterol 2018, 9 (11), 203. [CrossRef]
- Smith, C.-M.; Watson, D. I.; Michael, M. Z.; Hussey, D. J. MicroRNAs, Development of Barrett’s Esophagus, and Progression to Esophageal Adenocarcinoma. World J Gastroenterol 2010, 16 (5), 531–537. [CrossRef]
- Revilla-Nuin, B.; Parrilla, P.; Lozano, J. J.; de Haro, L. F. M.; Ortiz, A.; Martínez, C.; Munitiz, V.; de Angulo, D. R.; Bermejo, J.; Molina, J.; Cayuela, M. L.; Yélamos, J. Predictive Value of MicroRNAs in the Progression of Barrett Esophagus to Adenocarcinoma in a Long-Term Follow-up Study. Ann Surg 2013, 257 (5), 886–893. [CrossRef]
- 24. Orsini A, Mastracci L, Bozzarelli I, Ferrari A, Isidori F, Fiocca R, Lugaresi M, D'Errico A, Malvi D, Cataldi-Stagetti E, Spaggiari P, Tomezzoli A, Albarello L, Ristimäki A, Bottiglieri L, Krishnadath KK, Rosati R, Fumagalli Romario U, De Manzoni G, Räsänen J, Martinelli G, Mattioli S, Bonora E, On Behalf Of The Eacsge Consortium. Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma. Cancers (Basel). 2023, 15(5):1408. doi: 10.3390/cancers15051408.
- Fiocca, R.; Mastracci, L.; Lugaresi, M.; Grillo, F.; D’Errico, A.; Malvi, D.; Spaggiari, P.; Tomezzoli, A.; Albarello, L.; Ristimäki, A.; Bottiglieri, L.; Bonora, E.; Krishnadath, K. K.; Raulli, G. D.; Rosati, R.; Fumagalli Romario, U.; De Manzoni, G.; Räsänen, J.; Mattioli, S. The Prognostic Impact of Histology in Esophageal and Esophago-Gastric Junction Adenocarcinoma. Cancers (Basel) 2021, 13 (20), 5211. [CrossRef]
- Rockett, J. C.; Larkin, K.; Darnton, S. J.; Morris, A. G.; Matthews, H. R. Five Newly Established Oesophageal Carcinoma Cell Lines: Phenotypic and Immunological Characterization. Br J Cancer 1997, 75 (2), 258–263. [CrossRef]
- Babraham, B. FastQC A Quality Control Tool for High Throughput Sequence Data. Accessed July 4, 2023. Https://Www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/.
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32 (19), 3047–3048. [CrossRef]
- Dobin, A.; Davis, C. A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29 (1), 15–21. [CrossRef]
- Danecek, P.; Bonfield, J. K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M. O.; Whitwham, A.; Keane, T.; McCarthy, S. A.; Davies, R. M.; Li, H. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10 (2), giab008. [CrossRef]
- Putri, G. H.; Anders, S.; Pyl, P. T.; Pimanda, J. E.; Zanini, F. Analysing High-Throughput Sequencing Data in Python with HTSeq 2.0. Bioinformatics 2022, 38 (10), 2943–2945. [CrossRef]
- Ritchie, M. E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C. W.; Shi, W.; Smyth, G. K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res 2015, 43 (7), e47. [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P. D. PANTHER Version 11: Expanded Annotation Data from Gene Ontology and Reactome Pathways, and Data Analysis Tool Enhancements. Nucleic Acids Res 2017, 45 (D1), D183–D189. [CrossRef]
- Boonstra, J. J.; van Marion, R.; Beer, D. G.; Lin, L.; Chaves, P.; Ribeiro, C.; Pereira, A. D.; Roque, L.; Darnton, S. J.; Altorki, N. K.; Schrump, D. S.; Klimstra, D. S.; Tang, L. H.; Eshleman, J. R.; Alvarez, H.; Shimada, Y.; van Dekken, H.; Tilanus, H. W.; Dinjens, W. N. M. Verification and Unmasking of Widely Used Human Esophageal Adenocarcinoma Cell Lines. J Natl Cancer Inst 2010, 102 (4), 271–274. [CrossRef]
- Hutchinson, J. N.; Ensminger, A. W.; Clemson, C. M.; Lynch, C. R.; Lawrence, J. B.; Chess, A. A Screen for Nuclear Transcripts Identifies Two Linked Noncoding RNAs Associated with SC35 Splicing Domains. BMC Genomics 2007, 8, 39. [CrossRef]
- Njei, B.; McCarty, T. R.; Birk, J. W. Trends in Esophageal Cancer Survival in United States Adults from 1973 to 2009: A SEER Database Analysis. J Gastroenterol Hepatol 2016, 31 (6), 1141–1146. [CrossRef]
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal Cancer. Lancet 2017, 390 (10110), 2383–2396. [CrossRef]
- Dulak, A. M.; Stojanov, P.; Peng, S.; Lawrence, M. S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S. E.; Shefler, E.; McKenna, A.; Carter, S. L.; Cibulskis, K.; Sivachenko, A.; Saksena, G.; Voet, D.; Ramos, A. H.; Auclair, D.; Thompson, K.; Sougnez, C.; Onofrio, R. C.; Guiducci, C.; Beroukhim, R.; Zhou, Z.; Lin, L.; Lin, J.; Reddy, R.; Chang, A.; Landrenau, R.; Pennathur, A.; Ogino, S.; Luketich, J. D.; Golub, T. R.; Gabriel, S. B.; Lander, E. S.; Beer, D. G.; Godfrey, T. E.; Getz, G.; Bass, A. J. Exome and Whole-Genome Sequencing of Esophageal Adenocarcinoma Identifies Recurrent Driver Events and Mutational Complexity. Nat Genet 2013, 45 (5), 478–486. [CrossRef]
- Malumbres, M.; Carnero, A. Cell Cycle Deregulation: A Common Motif in Cancer. Prog Cell Cycle Res 2003, 5, 5–18.
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C. M. MicroRNA and Cancer--a Brief Overview. Adv Biol Regul 2015, 57, 1–9. [CrossRef]
- Santiago, K.; Chen Wongworawat, Y.; Khan, S. Differential MicroRNA-Signatures in Thyroid Cancer Subtypes. J Oncol 2020, 2020, 2052396. [CrossRef]
- Garzon, R.; Croce, C. M. MicroRNAs in Normal and Malignant Hematopoiesis. Curr Opin Hematol 2008, 15 (4), 352–358. [CrossRef]
- Gramantieri, L.; Fornari, F.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G. A.; Grazi, G. L.; Croce, C. M.; Bolondi, L.; Negrini, M. MicroRNA-221 Targets Bmf in Hepatocellular Carcinoma and Correlates with Tumor Multifocality. Clin Cancer Res 2009, 15 (16), 5073–5081. [CrossRef]
- Matsuzaki, J.; Suzuki, H.; Tsugawa, H.; Watanabe, M.; Hossain, S.; Arai, E.; Saito, Y.; Sekine, S.; Akaike, T.; Kanai, Y.; Mukaisho, K.; Auwerx, J.; Hibi, T. Bile Acids Increase Levels of microRNAs 221 and 222, Leading to Degradation of CDX2 during Esophageal Carcinogenesis. Gastroenterology 2013, 145 (6), 1300–1311. [CrossRef]
- Wang, Y.; Zhao, Y.; Herbst, A.; Kalinski, T.; Qin, J.; Wang, X.; Jiang, Z.; Benedix, F.; Franke, S.; Wartman, T.; Camaj, P.; Halangk, W.; Kolligs, F. T.; Jauch, K. W.; Nelson, P. J.; Bruns, C. J. miR-221 Mediates Chemoresistance of Esophageal Adenocarcinoma by Direct Targeting of DKK2 Expression. Ann Surg 2016, 264 (5), 804–814. [CrossRef]
- Fu, H.; Tie, Y.; Xu, C.; Zhang, Z.; Zhu, J.; Shi, Y.; Jiang, H.; Sun, Z.; Zheng, X. Identification of Human Fetal Liver miRNAs by a Novel Method. FEBS Lett 2005, 579 (17), 3849–3854. [CrossRef]
- Lapunzina, P. Risk of Tumorigenesis in Overgrowth Syndromes: A Comprehensive Review. Am J Med Genet C Semin Med Genet 2005, 137C (1), 53–71. [CrossRef]
- Pepe, F.; Visone, R.; Veronese, A. The Glucose-Regulated MiR-483-3p Influences Key Signaling Pathways in Cancer. Cancers (Basel) 2018, 10 (6), 181. [CrossRef]
- Livingstone, C. IGF2 and Cancer. Endocr Relat Cancer 2013, 20 (6), R321-339. [CrossRef]
- Rainier, S.; Johnson, L. A.; Dobry, C. J.; Ping, A. J.; Grundy, P. E.; Feinberg, A. P. Relaxation of Imprinted Genes in Human Cancer. Nature 1993, 362 (6422), 747–749. [CrossRef]
- Veronese, A.; Lupini, L.; Consiglio, J.; Visone, R.; Ferracin, M.; Fornari, F.; Zanesi, N.; Alder, H.; D’Elia, G.; Gramantieri, L.; Bolondi, L.; Lanza, G.; Querzoli, P.; Angioni, A.; Croce, C. M.; Negrini, M. Oncogenic Role of miR-483-3p at the IGF2/483 Locus. Cancer Res 2010, 70 (8), 3140–3149. [CrossRef]
- Tang, W.; Pei, M.; Li, J.; Xu, N.; Xiao, W.; Yu, Z.; Zhang, J.; Hong, L.; Guo, Z.; Lin, J.; Dai, W.; Xiao, Y.; Wu, X.; Liu, G.; Zhi, F.; Li, G.; Xiong, J.; Chen, Y.; Zhang, H.; Xiang, L.; Li, A.; Liu, S.; Wang, J. The miR-3648/FRAT1-FRAT2/c-Myc Negative Feedback Loop Modulates the Metastasis and Invasion of Gastric Cancer Cells. Oncogene 2022, 41 (43), 4823–4838. [CrossRef]
- Saitoh, T.; Katoh, M. FRAT1 and FRAT2, Clustered in Human Chromosome 10q24.1 Region, Are up-Regulated in Gastric Cancer. Int J Oncol 2001, 19 (2), 311–315. [CrossRef]
- Sari, I. N.; Yang, Y.-G.; Wijaya, Y. T.; Jun, N.; Lee, S.; Kim, K. S.; Bajaj, J.; Oehler, V. G.; Kim, S.-H.; Choi, S.-Y.; Park, S.-H.; Kim, D.-W.; Reya, T.; Han, J.; Kwon, H. Y. AMD1 Is Required for the Maintenance of Leukemic Stem Cells and Promotes Chronic Myeloid Leukemic Growth. Oncogene 2021, 40 (3), 603–617. [CrossRef]
- Gao, H.; Li, H.; Wang, J.; Xu, C.; Zhu, Y.; Tuluhong, D.; Li, X.; Wang, S.; Li, J. Polyamine Synthesis Enzyme AMD1 Is Closely Related to the Tumorigenesis and Prognosis of Human Breast Cancer. Exp Cell Res 2022, 417 (2), 113235. [CrossRef]
- Xu, L.; You, X.; Cao, Q.; Huang, M.; Hong, L.-L.; Chen, X.-L.; Lei, L.; Ling, Z.-Q.; Chen, Y. Polyamine Synthesis Enzyme AMD1 Is Closely Associated with Tumorigenesis and Prognosis of Human Gastric Cancers. Carcinogenesis 2020, 41 (2), 214–222. [CrossRef]
- Agarwal, S.; Behring, M.; Hale, K.; Al Diffalha, S.; Wang, K.; Manne, U.; Varambally, S. MTHFD1L, A Folate Cycle Enzyme, Is Involved in Progression of Colorectal Cancer. Transl Oncol 2019, 12 (11), 1461–1467. [CrossRef]
- Lee, D.; Xu, I. M.-J.; Chiu, D. K.-C.; Lai, R. K.-H.; Tse, A. P.-W.; Lan Li, L.; Law, C.-T.; Tsang, F. H.-C.; Wei, L. L.; Chan, C. Y.-K.; Wong, C.-M.; Ng, I. O.-L.; Wong, C. C.-L. Folate Cycle Enzyme MTHFD1L Confers Metabolic Advantages in Hepatocellular Carcinoma. J Clin Invest 2017, 127 (5), 1856–1872. [CrossRef]
- He, Z.; Wang, X.; Zhang, H.; Liang, B.; Zhang, J.; Zhang, Z.; Yang, Y. High Expression of Folate Cycle Enzyme MTHFD1L Correlates with Poor Prognosis and Increased Proliferation and Migration in Colorectal Cancer. J Cancer 2020, 11 (14), 4213–4221. [CrossRef]
- Yang, Y.-S.; Yuan, Y.; Hu, W.-P.; Shang, Q.-X.; Chen, L.-Q. The Role of Mitochondrial Folate Enzyme MTHFD1L in Esophageal Squamous Cell Carcinoma. Scand J Gastroenterol 2018, 53 (5), 533–540. [CrossRef]
- Tada, Y.; Yokomizo, A.; Shiota, M.; Song, Y.; Kashiwagi, E.; Kuroiwa, K.; Oda, Y.; Naito, S. Ectonucleoside Triphosphate Diphosphohydrolase 6 Expression in Testis and Testicular Cancer and Its Implication in Cisplatin Resistance. Oncol Rep 2011, 26 (1), 161–167. [CrossRef]
- Sun, Y.; Ma, L. New Insights into Long Non-Coding RNA MALAT1 in Cancer and Metastasis. Cancers (Basel) 2019, 11 (2), 216. [CrossRef]
- Li, Z.; Zhang, Q.; Wu, Y.; Hu, F.; Gu, L.; Chen, T.; Wang, W. lncRNA Malat1 Modulates the Maturation Process, Cytokine Secretion and Apoptosis in Airway Epithelial Cell-Conditioned Dendritic Cells. Exp Ther Med 2018, 16 (5), 3951–3958. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



