Submitted:
11 December 2023
Posted:
12 December 2023
You are already at the latest version
Abstract
A Staudinger reaction on solid phase between an electronodeficit organic azide such as sulfonyl azide, and the phosphite triester formed upon phosphoramidite coupling is a convenient method for chemical modification of oligonucleotides at the internucleotidic phosphate position. In this work, 4-carboxybenzenesulfonyl azide either with a free carboxy group or in the form of an activated ester such as pentafluorophenyl, 4-nitrophenyl, or pentafluorobenzyl ester were used to introduce a carboxylic acid function to the terminal or internal internucleotidic phosphate of an oligonucleotide via Staudinger reaction. A subsequent treatment with excess primary alkyl amine followed by usual work-up, after prior activation with a suitable peptide coupling agent such as HBTU/HOBt in the case of a free carboxyl, afforded amide-linked oligonucleotide conjugates in good yields including multiple conjugations of up to the exhaustive modification at each phosphate position for a weakly activated pentafluorobenzyl ester, whereas more strongly activated and, thus, more reactive aryl esters provided only single conjugations at the 5′-end. The conjugates synthesized include those with di- and polyamines that introduce a positively charged side-chain to potentially assist intracellular delivery of the oligonucleotide.
Keywords:
nucleic acid
; antisense oligonucleotide
; conjugation
; sulfonyl azide
; pentafluorophenyl and 4-nitrophenyl active esters
; carboxylic acid group modification
; pentafluorobenzyl
1. Introduction
The opening decades of this century witnessed an upsurge of interest in synthetic nucleic acids: single or double stranded DNA and RNA, and their chemically-modified analogues, which have firmly established their kind as promising therapeutics to engage various target genes and their products at the pre- or post-transcriptional level [1,2]. In comparison to small molecule drugs, oligonucleotides are capable of recognizing and directly binding the target, which is most often a biologically important RNA molecule, through Watson-Crick complementary interactions, which ensures high specificity of their action. The attention to oligonucleotide therapy sparkled in the late 1970s with the advent of antisense technology [3], subsequent discovery of RNA interference (RNAi) in the late 1990s adding another dimension to it [4]. To-date, many oligonucleotide analogues with chemical modifications in the various parts of the molecule: nucleobases, sugar or phosphate backbone have been developed. The first generation of antisense agents were the derivatives with modification in the phosphate group to ensure enzymatic resistance in the body, such as phosphorothioates [5], methyl phosphonates [6], and many others [7,8,9,10] up to the most recent mesyl phosphoramidates [11,12,13] and phosphoryl guanidines [14,15,16]. Next wave involved the compounds with modifications in the ribose ring, such as 2'-O-methyl [17,18] or 2'-O-methoxyethyl (MOE) RNAs [19,20], 2’-α- or β-fluoro-DNAs [21,22], bridged/locked nucleic acids (B/LNAs) [23,24,25,26], and tricyclo-DNAs [27]. A merger of the above two types are the gapmers with 2’-modified 3’ and 5’-terminal “wings”, and 6-10 nt DNA stretch in between, usually all-phosphate-modified [19,20]. A separate group encompasses DNA mimics in which the sugar and phosphate backbone is replaced by a unnatural substitute; examples are peptide nucleic acids (PNAs) [28] and phosphordiamidate morpholino oligomers (PMOs) [29,30]. Up until now, FDA has approved more than a dozen of nucleic acid-based drugs: notably, nusinersen (Spinraza) [31], eteplirsen (Exondys 51) [32] and its morpholino kin [33,34], all of them acting through the antisense mechanism on rare genetic disease targets; and the RNAi mediators small interfering RNAs (siRNAs) starting with patisiran (Onpattro) [35] that was quickly followed by others [36,37,38,39]. Furthermore, well over a hundred oligonucleotide drug candidates are currently going through various phases of clinical trials [40].
Thus, there is little doubt that oligonucleotides and their analogues have huge therapeutic potential, which is confirmed every year by FDA approval of yet another nucleic acid drug [41]. However, due to their intrinsically low cellular uptake and tissue distribution, and, in many cases, unfavorable pharmacokinetics and rapid clearance from the body, nucleic acid drugs did not yet unfold their full therapeutic potential [42,43]. Moreover, the inability more often than not to effectively overcome additional barriers inside the cell on the way to their biological target, such as endosomal entrapment and nuclear translocation [44,45,46,47], poses further obstacles on the path of oligonucleotides to the clinic.
To improve therapeutic activity of oligonucleotides, various delivery agents were proposed, such as lipids [48,49,50], polymers [51], in particular, polyethyleneimine (PEI) [52], dendrimers [53], inorganic nanoparticles [54], cell-penetrating peptides [55], and oligonucleotide conjugates with different transport molecules [56]. However, despite huge advances recently made in this field [57], there is still no generally applicable and, even more so, inexpensive, way for boosting in vivo efficacy of oligonucleotide therapeutics through the improved delivery. A potential step forward to the solution of this problem could be the reducing the total negative charge of the phosphate backbone, e.g. by introduction of cationic moieties, which has been previously shown to improve their intracellular penetration [58,59]. In particular, the attachment of multiple copies of spermine to neutralize a significant proportion of the net negative charge of the oligonucleotide has been shown to promote cellular uptake in the absence of transfection agents [60] as well as increase the affinity to the nucleic acid target through electrostatic interaction [61,62]. However, the attachment of spermine according to the above method required an expensive and bulky phosphoramidite synthon [63], which coupling efficacy, in particular, upon multiple incorporations, may be compromised by a generally lower stability of the primary alcohol-derived phosphoramidites compared to, e.g., nucleoside phosphoramidites (especially, after some storage in a freezer even at –20oC). Herein, a post-synthetic conjugation, e.g. via the amide bond formation between a carboxylic acid and a (poly)amine may offer a useful alternative.
We [64] and others [65] have described phosphoramidite synthons for modification of synthetic oligonucleotides with a carboxyl group, either in the form of a protected carboxylic ester, which can be cleaved selectively, or as the free carboxyl, which is then activated by a suitable peptide coupling agent either on solid phase [64] or post-synthetically in solution [66] to couple to a range of primary amines including polyamines, and short peptides. An alternative approach involved a phosphoramidite incorporating a pre-activated carboxyl group in the form of N-succinimidyl ester that can be converted to a range of amides upon amine treatment on solid phase [65]. However, this synthon (5'-Carboxy-Modifier C10) as well as the other commercially available reagent (5'-Carboxy-Modifier C5) can only work for the 5’-terminal conjugation, whereas the spermine phosphoramidite offers more flexibility as it can be incorporated on the 5’ or 3’-end, or on both ends at the same time [63]. Some extra flexibility can be achieved by switching to solid-phase conjugation at the 2’-position via 2’-O-(carboxymethyl)-uridine synthon but this approach is sequence-dependent [67]. In order to find a conjugation method that may be applicable to any position within oligonucleotide chain, we decided to focus on phosphate modification.
The Staudinger reaction has been explored as a way of converting the internucleotidic phosphate to the phosphoramidate moiety as early as in 1970s [68]. In particular, an early paper by Letsinger and Schott described a conjugation of Ethidium Bromide dye to the internucleotidic position of an oligonucleotide via Staudinger reaction [69]. Later, Heindl et al. introduced a convenient N-(sulfonyl)-phosphoramidate chemistry [70,71], which we recently extended to a range of novel internucleotidic groups [72,73] including, most notably, the mesyl phosphoramidate group [74] that proved to be useful as a backbone modification of antisense oligonucleotides [11,12,13].
Recently, an approach to oligonucleotide functionalization by various reporter groups via sulfonyl azides was published, which presented a further extension of the Staudinger chemistry at the internucleotidic position [75]. Very recently, it was successfully applied to siRNA to peptide conjugation [76]. In this Communication, we describe an orthogonal approach to oligonucleotide modification and conjugation at the internucleotidic position via sulfonyl azide-mediated Staudinger reaction in tandem with amide bond formation between a side-chain carboxyl group and a range of amines including di- and polyamines.
2. Results and Discussion
We have previously shown that the Staudinger reaction between an organic azide and the support-bound internucleotidic β-cyanoethyl phosphite triester (1, Figure 1) formed upon phosphoramidite coupling during solid-phase DNA synthesis, can serve as a versatile and convenient method for the chemical modification of oligonucleotides at the phosphate position [77]. In this work, we used commercially available 4-carboxybenzenesulfonyl azide A or its readily prepared activated esters (B-D) to modify one or more internucleotidic phosphate groups of an oligonucleotide at the ends or inside the sequence via Staudinger reaction (2a-d, Figure 1). Thus, a carboxylic acid function, either free or in its pre-activated form, can be introduced into various positions of an oligonucleotide chain at the phosphate position, and subsequently employed to react with a suitable primary amine (see Table 1) with the formation of a stable amide bond. The Staudinger reaction with azides A-D can be performed manually after the synthesis of the corresponding oligonucleotide at the last phosphate position adjacent the 5’-end, skipping usual iodine oxidation. More conveniently, it can be done automatically by substituting azide solution for standard oxidizer in the synthesizer bottle during solid-phase phosphoramidite DNA synthesis (see Section 4).
Initially, we employed the commercially available 4-carboxybenzenesulfonyl azide A to introduce a free carboxy group at the internucleotidic phosphate position next to the 5’-end. A 0.25 M solution of A in acetonitrile was used that contained 5% pyridine to buffer the acidity of the carboxylic acid function to avoid any danger of the premature cleavage of the acid-labile 5’-DMTr group and the acid-mediated scission of the phosphite triester. After the Staudinger reaction, an activation of the carboxyl group was carried out on solid phase using a well-known peptide coupling agent HBTU in the presence of HOBt [78,79,80] similarly to previously published methods [64,67], which was followed by a treatment with a solution of a primary amine (Table 1) in acetonitrile at 25oC for 1 h. At the end of the reaction, cleavage from support and deprotection of the oligonucleotides under mild conditions was ensured by addition of conc. aq. ammonia solution and standby at 25◦C for 18 h. Following usual work-up, a series of novel oligonucleotide derivatives containing N-(4-carboxybenzenesulfonyl)-phosphoramidate (ξ) group with different amide-attached side-chains were obtained (Table 1). The crude oligonucleotides modified at the 5’-terminal internucleotidic position were usually cleaved retaining the 5’-DMTr group, which can be removed by acidic treatment (80% aq. acetic acid, 30 min) on or after the purification step. The structures of modified oligonucleotide and conjugates were confirmed by MALDI-TOF MS (Supplementary Material, Table S1), and the HPLC conversions were good to acceptable (Figure 2A, S1, S2). Thereby, after usual post-synthetic processing we obtained a number of amide-linked oligonucleotide conjugates with amines, including, in particular, polycyclic aromatic or polyamine residues, or zwitter-ionic groups in good yields (Table 1).
Reaction mixtures of oligonucleotide or conjugate syntheses were analyzed by reverse-phased (RP) HPLC. In some profiles such as for a diamine (5) or a polyamine (6) (Table 1), by-products associated with acrylonitrile addition to the free primary or secondary amino groups were obtained (Supplementary Material, Figure S2). To avoid this side-reaction, it was found to be advantageous to pre-treat the support-bound oligonucleotide with 50% triethylamine in acetonitrile for 30 min to remove the β-cyanoethyl groups from the internucleotidic phosphates and convert the carboxyl group to its triethylammonium salt to promote the subsequent activation with HBTU/HOBt.
Next, we set out to ascertain if the unprotected carboxyl group can stay unchanged during solid-phase DNA synthesis by the phosphoramidite chemistry to be introduced into the next to the 3’-end or internal position within oligonucleotide chain. Therefore, the activation and amine treatment steps were skipped and, after the mild aq. ammonia treatment at 25oC to avoid potential amide formation, the oligonucleotides were isolated as carboxylic acids (Table 1, A1-A5). Thus, we demonstrated that the carboxyl group does not need a special protection during DNA synthesis, and can be introduced into any internucleotidic position within oligonucleotide chain including 3’-terminal or internal position. This may open up further possibilities for conjugation at a modified phosphate group through amide formation upon activation in aqueous or aqueous – organic solution by, e.g. a water-soluble carbodiimide, as described previously [66]. It was possible to clearly distinguish the oligonucleotide with a free carboxyl group (A3) from the one with amide group (D2) by co-injection on RP-HPLC (Figure 3A). The paired peaks of the corresponding P-derived diastereomeric oligonucleotides were present.
To find out if one can skip the in situ activation of a free carboxyl group by a coupling agent on solid phase, which may potentially lead to unwanted side-reactions such as base modification [81], we synthesized the activated pentafluorophenyl ester of (4-azidosulfonyl)-benzoic acid B by a DCC-mediated reaction of A with pentafluorophenol as a stable crystalline solid (see Section 4 for details). It was successfully used to obtain amide-linked oligonucleotide conjugates at the next to 5’-end internucleotidic position (Table 1, the B series). However, the introduction the B modification at any internal position within oligonucleotide chain resulted in a multiple product formation indicating that the pentafluorophenyl ester is too reactive to withstand the conditions of solid-phase DNA synthesis (data not shown). Furthermore, the after-synthesis treatment with 50% triethylamine in acetonitrile to remove the β-cyanoethyl group from the phosphates to avoid potential acrylonitrile addition to the amino groups side-reaction in the case of B5 or B6 was found to be detrimental to the integrity of the active ester. Therefore, a less activated 4-nitrophenyl (4-azidosulfonyl)-benzoate C was obtained to see if the 4-nitrophenyl group can survive the conditions of DNA synthesis unchanged. However, neither the internally-linked conjugates nor multiple conjugates were available with 4-nitrophenyl ester C likewise, and the yields were lower than with B. Further attempts to lower the reactivity of aryl ester by making 4-chlorophenyl 4-(azidosulfonyl)-benzoate were likewise unsuccessful to obtain either 3’-terminal or internal modification in acceptable yields (data not shown). Thus, we tried a still less activated pentafluorobenzyl ester of 4-(azidosulfonyl)-benzoic acid D to introduce a more stable yet sufficiently reactive ester groups into oligonucleotides via Staudinger reaction. Contrary to both aryl esters B and C, a relatively weakly activated pentafluorobenzyl ester D proved to be stable under the conditions of solid-phase oligonucleotide synthesis. The application of the ester D for modification of internucleotidic phosphate groups allowed us to obtain amide-linked conjugates without pre-activation by post-synthetic treatment with conc. aq. ammonia (1) or 1,1-dimethylethylenediamine (7) (Table 1, Figure 5) including those with next to the 3’-terminal or internal modification such as D1, D3, D5/D7, or more than one conjugated position, e.g. D4, up to the exhaustive replacement of all the internucleotidic phosphates with modified groups, e.g. D6/D8 (Table 1).
Typical elution profiles of the amide-linked oligonucleotide conjugates obtained are presented in Figure 2, Figure 3, Figure 4 and Figure 5 (see also Figures S1-S7, Supplementary Material). As expected, the replacement of one phosphate group with N-(4-carboxybenzenesulfonyl)-phosphoramidate (ξ) in the model 6-mer hexathymidylate A1 led to a slight increase in the retention time compared to the unmodified one (Figure 2B). In the conjugate with benzylamine A9, the presence of a hydrophobic benzyl group, as expected, resulted in a significant increase in retention time compared to A1 (Figure 2B). A new chiral centre at the phosphorus atom to which the sulfonamide group is attached led to the separation of two diastereomers in both cases, more prominent in the case of the less polar benzylamide A9. For most 15- and 17-mer 5’-unprotected oligonucleotides with one modification in different positions of the chain, a single peak corresponding to the main product was observed in the profiles with no detectable separation of diastereomers (Figure 2A, S1-S4, S5A, S6, and S7A). Yet, for some of the longer sequences, two peaks of stereoisomers were clearly distinguishable (Figures 3B, S4B, S6B).
Longer 17-mer oligonucleotide conjugates A7, A8, A14, A15 obtained with azide A with HBTU/HOBt activation (Figure S3), and the same sequences B1, B2, B3, B4 derived from pentafluorophenyl ester B, respectively (Figure 3B) all show gradual increase in the retention time that correlates with the increase in hydrophobicity of the amines from 1 to 4. According to elution profiles, the conjugation reaction with aqueous ammonia, n-propylamine, benzylamine and 1-pyrenemethylamine proceeded with high yields in both series A and series B, the latter resulting in cleaner products and slightly higher yields. In the case of the azide C, only two derivatives were synthesized: amide C1 and benzylamide C2. As evident from their HPLC profiles (Figure S5), these two conjugates contained higher amounts of impurities than in the case of A and B. This prompts us to conclude that 4-nitrophenyl 4-(azidosulfonyl)-benzoate C may also be used for oligonucleotide modification and conjugation but with somewhat lesser efficiency.
The corresponding 15-mer oligonucleotide conjugates D1-D4 obtained from pentafluorobenzyl ester D and having one or two N-(4-carboxybenzenesulfonyl)-phosphoramidate (ξ) groups were prepared in high yield (Figure S6), and used later to study the thermal stability of complementary duplexes with DNA and RNA (see below). The fully modified oligonucleotide conjugate D6 with 1,1-dimethylethylenediamine residue at each internucleotidic position demonstrated a significant increase in retention time compared to the singly modified conjugate D5 (Figure 4A). In turn, the singly modified conjugate D5 showed a small decrease in the retention time compared to the unmodified control presumably due to the presence of a polar zwitter-ionic group [82], but this effect was not observed for the 5'-dimethoxytritylated D7 (Figure 4B). The separation of the main peak into the peaks of diastereomers was detected only for the fully modified D6 and D7, which are the mixtures of 32 stereoisomers due to the presence of a chiral centre at each of the five internucleotidic positions.
Additional characteristics of the modified oligonucleotides and conjugates were studied by 20% denaturing PAGE. Firstly, we compared the mobility of 15-mer oligodeoxyribonucleotides containing amide and benzylamide residues in different position of oligonucleotide backbone. The mobility of oligonucleotides A3, A4, and A5 containing amide residue (Figure S11, lanes 2, 4, and 7) was close to the unmodified control oligonucleotide (Figure S11, lane 1), and, at the same time, is higher than the mobility of the conjugates A11, A12, and A13 containing hydrophobic benzyl groups (Figure S11, lanes 3, 5, and 8). This data is in accord with previous observation that the amide-linked N-(4-carboxybenzenesulfonyl)-phosphoramidate group (ξ) in oligonucleotides likewise other sulfonyl phosphoramidate groups is negatively charged under physiological pH similarly to the natural phosphodiester group [70,72].
Next, we studied the difference in electrophoretic mobility between the oligonucleotide D5 with one zwitter-ionic group, and a fully modified sequence D6, as well as their 5’-DMTr versions D7 and D8. Presence of one zwitter-ionic group led to the decrease in the total negative charge of the oligonucleotide by one unit, which, in turn, resulted in a significant decrease in mobility (Figure 5, lanes 3 and 4) compared to the unmodified controls (Figure 5, lanes 1 and 2). Exhaustive substitution of all the phosphate groups with zwitter-ionic groups dramatically reduced the mobility (Figure 5, lanes 7 and 8), but nevertheless did not result in complete disappearance of the net negative charge of the oligonucleotide under the experimental conditions (pH 7.5). Interestingly, for the 5’-DMTr analogue of the fully modified oligonucleotide, we practically did not see any staining by the Stains-All dye (Figure 5B, lane 6), which may be explained by the screening of the remaining negative charge by the bulky DMTr group (compare Figure 5A and 5B). Other dyes need to be explored to ensure efficient visualization of extensively modified oligonucleotide sequences on PAGE such as described previously [83].
Figure 5.
Electrophoretic comparison of the mobility of oligonucleotide conjugates with zwitterionic group: lanes: (1) – unmodified 5’-d(TTTTTT), (2) – unmodified 5’-DMTr-d(TTTTTT), (3) – sample D5, (4) – sample D7, (5) – sample D6 and (6) – sample D8.
Figure 5.
Electrophoretic comparison of the mobility of oligonucleotide conjugates with zwitterionic group: lanes: (1) – unmodified 5’-d(TTTTTT), (2) – unmodified 5’-DMTr-d(TTTTTT), (3) – sample D5, (4) – sample D7, (5) – sample D6 and (6) – sample D8.

To ascertain the impact of the amide-linked N-(4-carboxybenzenesulfonyl)-phosphoramidate (ξ) group on thermal stability of complementary duplexes with DNA and RNA, we prepared four 15-mer oligodeoxynucleotides D1, D2, D3 and D4 containing a primary carboxamide modification (Figure 1, 3, R’ = H), which had either one group at the 3’ or 5’-end, or in the middle of the sequence, or two groups at both ends (Table 2). All the oligonucleotides were analyzed by HPLC and purified by PAGE. UV melting studies revealed that one group at either end, or two of the modifications at both ends did not have a significant adverse effect on the thermal stability of the duplexes of modified oligonucleotides with either DNA or RNA in comparison with those of the unmodified control. The most destabilizing effect had the modification in the middle of the sequence D3, which exceeded that of the doubly modified oligonucleotide D4 (Table 2). Thus, one may conclude that the N-(4-carboxamidobenzenesulfonyl)-phosphoramidate (ξ) group may be well suited for a single modification at any position of an oligonucleotide chain, or for multiple modifications at the ends of the sequence.
4. Materials and Methods
4.1. Synthesis of pentafluorophenyl, 4-nitrophenyl, and pentafluorobenzyl 4-(azidosulfonyl)-benzoates.
4-Carboxybenzenesulfonazide (A) was converted to either pentafluorophenyl (B) or 4-nitrophenyl (C) 4-(azidosulfonyl)-benzoate by reaction with the corresponding phenol promoted by dicyclohexylcarbodiimide (DCC) (see Supplementary Material for experimental details and characterization). Pentafluorobenzyl 4-(azidosulfonyl)-benzoate (D) was obtained by a two-step one-pot procedure from 4-(chlorosulfonyl)-benzoyl chloride by, firstly, reaction with pentafluorobenzyl alcohol in the presence of 4-dimethylaminopyridine (DMAP) as a base and catalyst, and, secondly, by treatment with NaN3 in acetonitrile, to give white crystalline D in good yield (see Supplementary Material for general information, experimental details and spectra).
4.2. Oligonucleotide Synthesis
Oligonucleotides were synthesized on an automated DNA/RNA ASM-800 synthesizer (Biosset, Russia) by the phosphoramidite method at 0.2–0.4 μM scale using the corresponding deoxyribonucleotide 2-cyanoethyl-N,N-diisopropyl phosphoramidites and Controlled Pore Glass (CPG) 500 Å supports with attached deoxyribonucleosides (Sigma-Aldrich, United States). If a ξ modification was introduced manually, the preceding synthesis cycle was interrupted immediately before the oxidation step, the column with the support-bound 5’-DMTr-oligonucleotide was removed from the synthesizer, and Staudinger reaction was carried out on solid-phase with 0.25 M solution of the corresponding sulfonyl azide B–D in acetonitrile for 30 min, or 1 h in the case of azide A in the presence of 5% v/v pyridine to buffer the acidity of the free carboxyl group. After the reaction, the support was washed 3×200 µL of CH3CN, and dried in vacuo. If necessary, the column with the support-bound oligonucleotide was placed back into the synthesizer, and the synthesis was continued until completion. In the case of automated insertion of the modification, a solution of the corresponding azide in acetonitrile (0.25 M) was placed into a synthesizer bottle, and pumped through column instead of iodine solution for the same time period. After the synthesis, the support-bound oligonucleotide was either treated with conc. aq. ammonia at 25◦C for 18 h to obtain the oligonucleotides with free carboxyl group A1-A5 (Table 1), or transferred to the conjugation step. Conversion of the pentafluorobenzyl ester to amide in D1-D4 was carried out after Staudinger reaction by treatment of the polymer-bound oligonucleotides with conc. aq. ammonia solution at 55◦C for 18 h.
4.3. Synthesis of Oligonucleotide Conjugates
For conjugation reaction, a 60 µl of a solution of the desired amine 2–6 was added to the support-bound oligonucleotides. In the case of the oligonucleotides with N-(4-carboxybenzenesulfonyl)-phosphoramidate group (ξ) obtained from sulfonyl azide A, the carboxyl group was activated prior to amine addition by treatment with HBTU/HOBt (1:1 mol. ratio) at 35˚C in dry DMF for 35 min as previously described [51]. The mixture was shaken for 1 h at 25oC for all amines except 4, when it was continued for 2 h at 37◦C. n-Propylamine (2) and benzylamine (3) were used as 10% v/v in DMF; 1-(pyrenemethylamine (4) was used as 0.25 M solution in DMF–THF (1:1 v/v); 4,7,10-trioxa-1,13-tridecanediamine (5) and tetraethylenepentamine (6) were used as 5% v/v solution in DMF; 1,1-dimethylethylenediamine (7) was used neat, thereby combining the conjugation reaction, final deprotection of the protecting groups, and cleavage from solid support. After completion of the conjugation reaction, the supernatant was removed, the support was washed by 3×200 µL of CH3CN, and dried in vacuo for 15 min. Finally, the protecting groups were removed and conjugate was cleaved from solid support by treatment with conc. aq. ammonia at 55◦C for 18 h for all conjugates except amine 7 (Table 1).
4.4. Reverse-phase HPLC analysis of oligonucleotides and conjugates
Analytical RP-HPLC was performed on a Milichrom A02 system (Econova Ltd., Chromatography Institute, Novosibirsk, Russia) with the use of ProntoSIL 120-5 C18 AQ (2×75 mm, 5 µ) column. The elution was carried out at a flow rate of 100 µL/min, and UV detection at 260 and 280 nm. Several elution gradients of acetonitrile in 20 mM TEAA, pH 7.0 were applied: from 0% to 50% in 30 min (i), from 0% to 60% in 30 min (ii), and from 0% to 50% in 30 min of 50% aq. acetonitrile in 20 mM TEAA buffer, pH 7.0 (iii).
4.5. Polyacrylamide gel electrophoresis under denaturing conditions
To control the homogeneity of oligonucleotides, analytical electrophoresis was carried out in 0.4 mm thick 20% polyacrylamide gel (acrylamide – N,N′-methylene-bis-acrylamide (30:1) at 50 V/cm voltage in 90 mM Tris-borate buffer, pH 8.3 containing 8 M urea and 2 mM Na2EDTA. Oligonucleotides were loaded onto the gel as solutions containing 8 M urea, 0.05% Xylene Cyanol FF, and 0.05% Bromophenol Blue. Bands were visualized by staining the gel with a solution of Stains-All dye (500 mg/L) in formamide followed by rinsing with distilled water.
Oligonucleotides synthesized for UV melting studies were isolated by preparative polyacrylamide gel electrophoresis in 2–3 mm thick 20% gel under denaturing conditions (8 M urea) and desalting on a NAP25 column with Sephadex G-25 (GE Healthcare, Buckinghamshire, UK) in the form of sodium salts.
4.6. UV melting studies
RNA template 5'-r(ATTTGAGCCTGGGAG) was kindly provided by Dr. Maria I. Meshchaninova. Thermal denaturation analysis of the duplexes of modified oligonucleotides with complementary DNA or RNA sequences was carried out in a quartz micro multi-cell cuvette with optical path length of 1 mm using a UV-1800 UV-Vis spectrophotometer (Shimadzu, Japan) with a thermoelectric cooling. All the experiments were conducted in a buffer containing 10 mM Na-cacodylate, 5 mM MgCl2, and 100 mM NaCl, pH 7.4. The components were taken in stoichiometric ratios so that the total oligonucleotide duplex concentration in the buffer was 10 μM. The solutions were kept at 90°C for 5 min, followed by gradual decrease of temperature from 90oC to 15°C to anneal the duplexes. Thereafter, the samples were heated from 15oC to 90°C at a rate of 0.2°C min−1 to melt the duplex. Absorption spectra were recorded at 260 nm with a measurement step of 0.5°C.
5. Conclusions
In this Communication, we describe a convenient method for conjugation of oligonucleotides via the amide bond formation at a modified phosphate position suitable for a range of amines including di- and polyamines that introduce positive charges into the oligonucleotide chain, which could be a way to increasee cellular and tissue uptake, and improve bioavailability and activity of therapeutic oligonucleotides. Herein, we employed 4-carboxybenzenesulfonyl azide and its activated pentafluorophenyl, 4-nitrophenyl or pentafluorobenzyl esters to obtain oligonucleotides carrying one or more internucleotidic N-(4-carboxybenzenesulfonyl)-phosphoramidate groups (ξ) by a modified phosphoramidite synthesis protocol substuting Staudinger reaction for conventional aqueous iodine oxidation. The N-(4-carboxybenzenesulfonyl)-phosphoramidate group (ξ) is stable under the conditions of DNA synthesis, its free carboxyl function not requiring protection, and maintains a negative charge at physiological pH likewise the native phosphodiester group. Thus, the carboxyl group can be introduced into any internucleotidic position of the sequence, whether 5’ or 3’-terminal, or internal, and, after suitable pre-activation by a peptide coupling agent such as HBTU/HOBt, it can undergo amide bond formation with a range of amines including those with positively charged side-chain such as di- and polyamines. Although the activated aryl esters of the N-(4-carboxybenzenesulfonyl)-phosphoramidate proved to be too reactive to withstand the conditions of conventional DNA synthesis and, thus, allowed only the 5’-terminal conjugation to occur, the less activated pentafluorobenzyl ester was sufficiently robust to be introduced at every phosphate position within the sequence, yet reactive enough to allow for smooth conversion to fully amide-modified oligonucleotides. Melting temperatures of the duplexes formed by ξ-oligodeoxynucleotides modified by one or two carboxamide side-chains at the terminal or internal positions with complementary DNA or RNA were similar or only slightly lower than that of the unmodified DNA:DNA and DNA:RNA duplexes.
To conclude, the application of newly synthesized N-(4-carboxybenzenesulfonyl)-phosphoramidate-modified oligonucleotides either as activated esters or free carboxylic acids with subsequent HBTU/HOBt-mediated activation can be a useful way to prepare oligonucleotide conjugates with a range of suitable primary amine substrates. We are currently working on the extension of the approach described in this Communication to oligonucleotide conjugates with peptides including cell-penetrating peptides known to greatly improve cellular uptake of oligonucleotide therapeutics.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: An overlap of elution profiles of crude reaction mixtures from the syntheses of 5’-singly-modified 17-mer oligonucleotides 5’-AξGTCTCGACTTGCTACC obtained by A/HBTU/HOBt method; Figure S2: Elution profiles of the conjugates of 5’-AξGTCTCGACTTGCTACC obtained by A/HBTU/HOBt method; Figure S3: RP-HPLC profiles of crude oligonucleotide conjugates A7, A8, A14, A15; Figure S4: RP-HPLC profiles of crude modified oligonucleotides A3-A5 and their conjugates with benzylamine A10-A12; Figure S5: RP-HPLC profiles of crude oligonucleotide conjugates: (A) C1; and (B) C2; Figure S6: RP-HPLC profiles of crude oligonucleotide conjugates D1-D4; Figure S7: RP-HPLC profiles of crude oligonucleotide conjugates: (A) A6, A15, and A16; and (B) B5 and B6; Figure S8: RP-HPLC elution profiles of crude oligonucleotide A2 and its conjugate with benzylamine A9; Figure S9: MALDI-TOF MS analysis of oligonucleotide conjugates B1-B4; Figure S10: MALDI-TOF MS spectra of oligonucleotide conjugates B5-B6; Figure S11: Electrophoretic comparison of the mobility of modified oligonucleotides with ξ-group and their conjugates with benzylamine; Figure S12: Synthesis of pentafluorophenyl (B) and 4-nitrophenyl (C) esters of 4-(azidosulfonyl)-benzoic acid (A); Figure S13: 1H NMR spectrum of pentafluorophenyl 4-(azidosulfonyl)-benzoate (B); Figure S14: 13C spectrum of pentafluorophenyl 4-(azidosulfonyl)-benzoate (B); Figure S15: Expanded 162.0 to 127.000 ppm region of 13C NMR spectrum of pentafluorophenyl 4-(azidosulfonyl)-benzoate (B); Figure S16: 19F NMR spectrum of pentafluorophenyl 4-(azidosulfonyl)-benzoate (B); Figure S17: 1H NMR spectrum of 4-nitrophenyl 4-(azidosulfonyl)-benzoate (C). Inset: expanded peak region 8.30 – 7.10 ppm; Figure S18: 13C spectrum of 4-nitrophenyl 4-(azidosulfonyl)-benzoate (C); Figure S19: Synthesis of pentafluorobenzyl 4-(azidosulfonyl)-benzoate (D); Figure S20: 1H NMR spectrum of pentafluorobenzyl 4-(azidosulfonyl)-benzoate (D); Figure S21: 13C NMR spectrum of pentafluorobenzyl 4-(azidosulfonyl)-benzoate (D); Figure S22: 19F NMR spectrum of pentafluorobenzyl 4-(azidosulfonyl)-benzoate (D); Table S1: MALDI TOF MS of oligonucleotide conjugates.
Author Contributions
Conceptualization, D.S.; methodology, K.K., S.B., E.B. and D.S.; software, K.K. and D.S.; validation, K.K., E.B. and S.B.; formal analysis, K.K. and D.S.; investigation, K.K. and P.Z.; resources, A.F.; data curation, K.K.; writing—original draft preparation, K.K.; writing—review and editing, D.S.; visualization, K.K.; supervision, D.S.; project administration, A.F.; funding acquisition, D.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Russian Science Foundation (grant no. 22-13-00212) and, in part, by the Ministry of Science and Higher Education of the Russian Federation (project of Novosibirsk State University no. FSUS-2020-0035, oligonucleotide purification and thermal melting analysis).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Acknowledgments
The authors thank Dr. M.I. Meshchaninova for the generous gift of the RNA template.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Corey, D.R.; Damha, M.J.; Manoharan, M. Challenges and Opportunities for Nucleic Acid Therapeutics. Nucleic Acid Ther. 2022, 32, 8–13. [CrossRef]
- Barresi, V.; Musmeci, C.; Rinaldi, A.; Condorelli, D.F. Transcript-Targeted Therapy Based on RNA Interference and Antisense Oligonucleotides: Current Applications and Novel Molecular Targets. Int. J. Mol. Sci. 2022, 23, 8875. [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 280–284. [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther 2014, 24, 374–387. [CrossRef]
- Miller, P.S. Oligonucleoside methylphosphonates as antisense reagents. Biotechnology (N.Y.) 1991, 9, 358–362. [CrossRef]
- Marshall, W.S.; Caruthers, M.H. Phosphorodithioate DNA as a potential therapeutic drug. Science. 1993, 259, 1564–1570. [CrossRef]
- Summers, J.S.; Shaw, B.R. Boranophosphates as mimics of natural phosphodiesters in DNA. Curr Med Chem. 2001, 8, 1147–1155. [CrossRef]
- Dellinger, D.J.; Sheehan, D.M.; Christensen, N.K.; Lindberg, J.G.; Caruthers, M.H. Solid-phase chemical synthesis of phosphonoacetate and thiophosphonoacetate oligodeoxynucleotides. J Am Chem Soc 2003, 125, 940–950. [CrossRef]
- Yamada, C.M.; Dellinger, D.J.; Caruthers, M.H. Synthesis and biochemical evaluation of phosphonoformate oligodeoxyribonucleotides. Journal of the American Chemical Society 2006, 128, 5251–5261. [CrossRef]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 1229–1234. [CrossRef]
- Patutina, O.A.; Gaponova Miroshnichenko, S.K.; Sen'kova, A.V.; Savin, I.A.; Gladkikh, D.V.; Burakova, E.A.; Fokina, A.A.; Maslov, M.A.; Shmendel, E.V.; Wood, M.J.A.; et al. Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting miR-21 with enhanced in vivo therapeutic potency. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 32370–32379. [CrossRef]
- Anderson, B.A.; Freestone, G.C.; Low, A.; De-Hoyos, C.L.; Drury III, W.J.; Østergaard, M.E.; Migawa, M.T.; Fazio, M.; Wan, W.B.; Berdeja, A.; Scandalis, E.; Burel, S.A.; Vickers, T.A.; Crooke, S.T.; Swayze, E.E.; Liang, X.; Seth, P.P. Towards next generation antisense oligonucleotides: mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021, 49, 9026–9041. [CrossRef]
- Kupryushkin, M.S.; Pyshnyi, D.V.; Stetsenko, D.A. Phosphoryl guanidines: a new type of nucleic acid analogues. Acta Naturae. 2014, 6, 116–118. PMID: 25558402; PMCID: PMC4273099.
- Kupryushkin, M.S.; Filatov, A.V.; Mironova, N.L.; Patutina, O.A.; Chernikov, I.V.; Chernolovskaya, E.L.; Zenkova, M.A.; Pyshnyi, D.V.; Stetsenko, D.A.; Altman, S.; Vlassov, V.V. Antisense oligonucleotide gapmers containing phosphoryl guanidine groups reverse MDR1-mediated multiple drug resistance of tumor cells. Mol. Ther. Nucleic Acids. 2022, 27, 211–226. [CrossRef]
- Kandasamy, P.; Liu, Y.; Aduda, V.; Akare, S.; Alam, R.; Andreucci, A.; Boulay, D.; Bowman, K.; Byrne, M.; Cannon, M.; Chivatakarn, O.; Shelke, J.D.; Iwamoto, N.; Kawamoto, T.; Kumarasamy, J.; Lamore, S.; Lemaitre, M.; Lin, X.; Longo, K.; Looby, R.; Marappan, S.; Metterville, J.; Mohapatra, S.; Newman, B.; Paik, I.H.; Patil, S.; Purcell-Estabrook, E.; Shimizu, M.; Shum, P.; Standley, S.; Taborn, K.; Tripathi, S.; Yang, H.; Yin, Y.; Zhao, X.; Dale, E.; Vargeese, C. Impact of guanidine-containing backbone linkages on stereopure antisense oligonucleotides in the CNS. Nucleic Acids Res. 2022, 50, 5401–5423. [CrossRef]
- Lamond, A.I.; Sproat, B.S. Antisense oligonucleotides made of 2'-O-alkyl RNA: their properties and applications in RNA biochemistry. FEBS Lett 1993, 325, 123–127. [CrossRef]
- Majlessi, M.; Nelson, N.C.; Becker, M.M. Advantages of 2'-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998, 26, 2224–2229. [CrossRef]
- Altmann, K.H.; Fabbro, D.; Dean, N.M.; Geiger, T.; Monia, B.P.; Müller, M.; Nicklin, P. Second-generation antisense oligonucleotides: structure-activity relationships and the design of improved signal-transduction inhibitors. Biochem. Soc. Trans. 1996, 24, 630-637. [CrossRef]
- Altmann, K.-H.; Dean, N. M.; Fabbro, D.; Freier, S. M.; Geiger, T.; Häner, R.; Hüsken, D.; Martin, P.; Monia, B. P.; Müller, M.; Natt, F.; Nicklin, P.; Phillips, J.; Pieles, U.; Sasmor, H.; Moser, H.E. Second Generation of Antisense Oligonucleotides: From Nuclease Resistance to Biological Efficacy in Animals. Chimia. 1996, 50, 168. [CrossRef]
- Kawasaki, A.M.; Casper, M.D.; Freier, S.M.; Lesnik, E.A.; Zounes, M.C.; Cummins, L.L.; Gonzalez, C.; Cook, P.D. Uniformly modified 2'-deoxy-2'-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J. Med. Chem. 1993, 36, 831–841. [CrossRef]
- El-Khoury, R.; Damha, M.J. 2'-Fluoro-arabinonucleic Acid (FANA): A Versatile Tool for Probing Biomolecular Interactions. Acc. Chem. Res. 2021, 54, 2287–2297. [CrossRef]
- Imanishi, T.; Obika, S. BNAs: novel nucleic acid analogs with a bridged sugar moiety. Chem Commun (Camb) 2002, 1653-1659. [CrossRef]
- Kaur, H.; Babu, B.R.; Maiti, S. Perspectives on chemistry and therapeutic applications of Locked Nucleic Acid (LNA). Chem Rev 2007, 107, 4672-4697. [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem Biodivers 2010, 7, 536-542. [CrossRef]
- Soler-Bistue, A.; Zorreguieta, A.; Tolmasky, M.E. Bridged Nucleic Acids Reloaded. Molecules 2019, 24. [CrossRef]
- Renneberg, D.; Bouliong, E.; Reber, U.; Schumperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acids Res 2002, 30, 2751-2757. [CrossRef]
- Nielsen, P.E. Peptide nucleic acids (PNA) in chemical biology and drug discovery. Chemistry Biodiversity 2010, 7, 786-804. [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Develop. 1997, 7, 187-195. [CrossRef]
- Summerton, J. History and Properties of Morpholino Antisense Oligos. J. Drug Discov. Develop. Deliv. 2016, 3, 1019.
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther. 2017, 2, 67–69. [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [CrossRef]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [CrossRef]
- Scott, L.J.; Keam, S.J., Lumasiran: First Approval. Drugs 2021, 81, 277–282. [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; Kastelein, J.J.P.; ORION-9 Investigators. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. New Engl. J. Med. 2020, 382, 1520–1530. [CrossRef]
- Keam, S.J. Vutrisiran: First Approval. Drugs 2022, 82, 1419–1425. [CrossRef]
- Moumne, L.; Marie, A.C.; Crouvezier, N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics 2022, 14. [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrod, B.; Pietri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [CrossRef]
- Juliano, R.L.; Ming, X.; Nakagawa, O. The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res. 2012, 45, 1067–1076. [CrossRef]
- Juliano, R.L.; Carver, K.; Cao, C.; Ming, X. Receptors, endocytosis, and trafficking: the biological basis of targeted delivery of antisense and siRNA oligonucleotides. J. Drug Target. 2013, 21, 27–43. [CrossRef]
- Juliano, R.L.; Ming, X.; Carver, K.; Laing, B. Cellular uptake and intracellular trafficking of oligonucleotides: implications for oligonucleotide pharmacology. Nucleic Acid Ther. 2014, 24, 101–113. [CrossRef]
- Patwa, A.; Gissot, A.; Bestel, I.; Barthélémy, P. Hybrid lipid oligonucleotide conjugates: synthesis, self-assemblies and biomedical applications. Chem. Soc. Rev. 2011, 40, 5844–5854. [CrossRef]
- Zhao, B.; Tian, Q.; Bagheri, Y.; You, M. Lipid-Oligonucleotide Conjugates for Simple and Efficient Cell Membrane Engineering and Bioanalysis. Curr. Opin. Biomed. Eng. 2020, 13, 76–83. [CrossRef]
- Li, X.; Feng, K.; Li, L.; Yang, L.; Pan, X.; Yazd, H.S.; Cui, C.; Li, J.; Moroz, L.; Sun, Y.; et al. Lipid–oligonucleotide conjugates for bioapplications. Nat. Sci. Rev. 2020, 7, 1933–1953. [CrossRef]
- Khan, A.; Benboubetra, M.; Sayyed, P.Z.; Ng, K.W.; Fox, S.; Beck, G.; Benter, I.F.; Akhtar, S. Sustained polymeric delivery of gene silencing antisense ODNs, siRNA, DNAzymes and ribozymes: in vitro and in vivo studies. J. Drug Target. 2004, 12, 393–404. [CrossRef]
- Sabin, J.; Alatorre-Meda, M.; Miñones, J., Jr.; Domínguez-Arca, V.; Prieto, G. New insights on the mechanism of polyethylenimine transfection and their implications on gene therapy and DNA vaccines. Colloids Surf. B Biointerfaces. 2022, 210, 112219. [CrossRef]
- Ravina, M.; Paolicelli, P.; Seijo, B.; Sanchez, A. Knocking down gene expression with dendritic vectors. Mini Rev. Med. Chem. 2010, 10, 73–86. [CrossRef]
- Parveen, S.; Misra, R.; Sahoo, S.K. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine 2012, 8, 147–166. [CrossRef]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; El Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [CrossRef]
- Craig, K.; Abrams, M.; Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Exp. Opin. Drug Deliv. 2018, 15, 629–640. [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [CrossRef]
- Michel, T.; Martinand-Mari, C.; Debart, F.; Lebleu, B.; Robbins, I.; Vasseur, J.J. Cationic phosphoramidate alpha-oligonucleotides efficiently target single-stranded DNA and RNA and inhibit hepatitis C virus IRES-mediated translation. Nucleic Acids Res. 2003, 31, 5282–5290. [CrossRef]
- Fraley, A.W.; Pons, B.; Dalkara, D.; Nullans, G.; Behr, J.P.; Zuber, G. Cationic oligonucleotide-peptide conjugates with aggregating properties enter efficiently into cells while maintaining hybridization properties and enzymatic recognition. J. Am. Chem. Soc. 2006, 128, 10763–10771. [CrossRef]
- Gagnon, K.T.; Watts, J.K.; Pendergraff, H.M.; Montaillier, C.; Thai, D.; Potier, P.; Corey, D.R. Antisense and antigene inhibition of gene expression by cell-permeable oligonucleotide-oligospermine conjugates. J. Am. Chem. Soc. 2011, 133, 8404–8407. [CrossRef]
- Noir, R.; Kotera, M.; Pons, B.; Remy, J.S.; Behr, J.P. Oligonucleotide-oligospermine conjugates (zip nucleic acids): a convenient means of finely tuning hybridization temperatures. J. Am. Chem. Soc. 2008, 130, 13500–13505. [CrossRef]
- Nothisen, M.; Bagilet, J.; Behr, J.P.; Remy, J.S.; Kotera, M. Structure Tuning of Cationic Oligospermine-siRNA Conjugates for Carrier-Free Gene Silencing. Mol. Pharm. 2016, 13, 2718–2728. [CrossRef]
- Pons, B.; Kotera, M.; Zuber, G.; Behr, J.P. Online synthesis of diblock cationic oligonucleotides for enhanced hybridization to their complementary sequence. ChemBioChem. 2006, 7, 1173–1176. [CrossRef]
- Kachalova, A.V.; Stetsenko, D.A.; Romanova, E.A.; Tashlitsky, V.N.; Gait, M.J.; Oretskaya, T.S. A New and Efficient Method for Synthesis of 5’-Conjugates of Oligonucleotides through Amide-Bond Formation on Solid Phase. Helv. Chim. Acta. 2002, 85, 2409–2416. doi: 0.1002/1522-2675(200208)85:8<2409::AID-HLCA2409>3.0.CO;2-P.
- Hogrefe, R.I.; Vaghefi, M.M. US Patent US6,320,041B1, Nov 20, 2001.
- Kachalova, A.V.; Stetsenko, D.A.; Gait, M.J.; Oretskaya, T.S. Synthesis of oligonucleotide 2'-conjugates via amide bond formation in solution. Bioorg. Med. Chem. Lett. 2004, 14, 801–804. [CrossRef]
- Kachalova, A.; Zubin, E.; Stetsenko, D.; Gait, M.; Oretskaya, T. Oligonucleotides with 2'-O-carboxymethyl group: synthesis and 2'-conjugation via amide bond formation on solid phase. Org. Biomol. Chem. 2004, 2, 2793–2797. [CrossRef]
- Letsinger, R.L.; Heavner, G.A. Synthesis of phosphoromonoamidate diester nucleotides via the phosphite-azide coupling method. Tetrahedron Lett. 1975, 16, 147–150. [CrossRef]
- Letsinger, R.L.; Schott, M.E. Selectivity in binding a phenanthridinium-dinucleotide derivative to homopolynucleotides. J. Am. Chem. Soc. 1981, 103, 7394–7396. [CrossRef]
- Heindl, D.; Kessler, D.; Schube, A.; Thuer, W.; Giraut, A. Easy method for the synthesis of labeled oligonucleotides. Nucleic Acids Symp. Ser. 2008, 405–406. [CrossRef]
- Heindl, D. Patent WO2008/128686A1, Apr 18, 2007.
- Prokhorova, D.V.; Chelobanov, B.P.; Burakova, E.A.; Fokina, A.A.; Stetsenko, D.A. New Oligodeoxyribonucleotide Derivatives Bearing Internucleotide N-Tosyl Phosphoramidate Groups: Synthesis and Complementary Binding to DNA and RNA. Russ. J. Bioorg. Chem. 2017, 43, 38–42. [CrossRef]
- Burakova, E.A.; Derzhalova, A.S.; Chelobanov, B.P.; Fokina, A.A.; Stetsenko, D.A. New Oligodeoxynucleotide Derivatives Containing N-(Sulfonyl)-Phosphoramide Groups. Russ. J. Bioorg. Chem. 2019, 45, 662–668. [CrossRef]
- Chelobanov, B.P.; Burakova, E.A.; Prokhorova, D.V.; Fokina, A.A.; Stetsenko, D.A. New oligodeoxynucleotide derivatives containing N-(methanesulfonyl)-phosphoramidate (mesyl phosphoramidate) internucleotide group. Russ. J. Bioorg. Chem. 2017, 43, 664–668. [CrossRef]
- Santorelli, A.; Gothelf, K.V. Conjugation of chemical handles and functional moieties to DNA during solid phase synthesis with sulfonyl azides. Nucleic Acids Res. 2022, 50, 7235–7246. [CrossRef]
- Smidt, J.M.; Lykke, L.; Stidsen, C.E.; Pristovšek, N.; Gothelf, K.V. Synthesis of peptide-siRNA conjugates via internal sulfonylphosphoramidate modifications and evaluation of their in vitro activity. Nucleic Acids Res. 2023 Nov 16:gkad1015. Epub ahead of print. PMID: 37971296. [CrossRef]
- Kupryushkin, M.S.; Apukhtina, V.S.; Vasilyeva, S.V.; Pyshnyi, D.V.; Stetsenko, D.A. A new simple and convenient method for preparation of oligonucleotides containing a pyrene or a cholesterol moiety. Russian Chemical Bulletin 2015, 64, 1678–1681. [CrossRef]
- Dourtoglou, V.; Ziegler, J.-C.; Gross, B. L’hexafluorophosphate de O-benzotriazolyl-N,N-tetramethyluronium: Un reactif de couplage peptidique nouveau et efficace. Tetrahedron Lett. 1978, 19, 1269–1272. [CrossRef]
- Fields, C.G.; Lloyd, D.H.; Macdonald, R.L.; Otteson, K.M.; Noble, R.L. HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept Res. 1991, 4, 95–101. PMID: 1815783.
- Reid, G.E.; Simpson, R.J. Automated solid-phase peptide synthesis: use of 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate for coupling of tert-butyloxycarbonyl amino acids. Anal Biochem. 1992; 200, 301–309. [CrossRef]
- Pritz, S.; Wolf, Y.; Klemm, C.; Bienert, M. Modification of guanine residues in PNA-synthesis by PyBOP. Tetrahedron Lett. 2006, 47, 5893–5896. [CrossRef]
- Patrushev, D.E.; Burakova, E.A.; Bizyaev, S.N.; Fokina, A.A.; Stetsenko, D.A. New Zwitter-Ionic Oligonucleotides: Preparation and Complementary Binding. Mol. Biol. 2023, 57, 320–328. [CrossRef]
- Fokina, A.; Wang, M.; Ilyina, A.; Klabenkova, K.; Burakova, E.; Chelobanov, B.; Stetsenko, D. Analysis of new charge-neutral DNA/RNA analogues phosphoryl guanidine oligonucleotides (PGO) by gel electrophoresis. Anal. Biochem. 2018, 555, 9–11. [CrossRef]
Figure 1.
A general scheme for the preparation of a carboxyl group containing phosphate-modified oligonucleotides and their amide-linked conjugates. Key: Bt – 1H-benzotriazol-1-yl, DMTr – 4,4’-dimethoxytrityl, HBTU – O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate, HOBt – 1-hydroxy-1H-benzotriazole; for R’ see Table 1.
Figure 1.
A general scheme for the preparation of a carboxyl group containing phosphate-modified oligonucleotides and their amide-linked conjugates. Key: Bt – 1H-benzotriazol-1-yl, DMTr – 4,4’-dimethoxytrityl, HBTU – O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate, HOBt – 1-hydroxy-1H-benzotriazole; for R’ see Table 1.
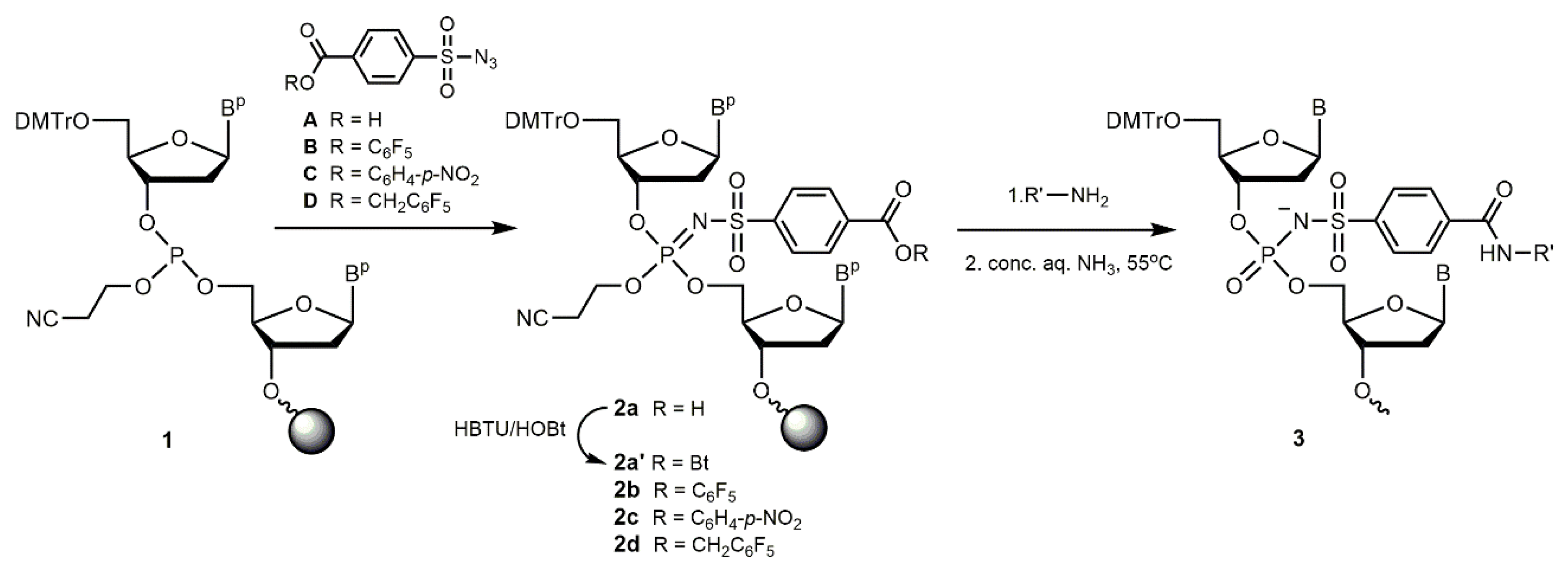
Figure 2.
RP-HPLC elution profiles of crude oligonucleotides and conjugates: (A) unmodified oligonucleotide 5’-d(AGTCTCGACTTGCTACC) (1), A6 (2), and conjugate A14 (3); (B) unmodified oligonucleotide 5’-d(TTTTTT) (1), A1 (2), and conjugate A9 (3). Elution gradient (i) (Section 4).
Figure 2.
RP-HPLC elution profiles of crude oligonucleotides and conjugates: (A) unmodified oligonucleotide 5’-d(AGTCTCGACTTGCTACC) (1), A6 (2), and conjugate A14 (3); (B) unmodified oligonucleotide 5’-d(TTTTTT) (1), A1 (2), and conjugate A9 (3). Elution gradient (i) (Section 4).
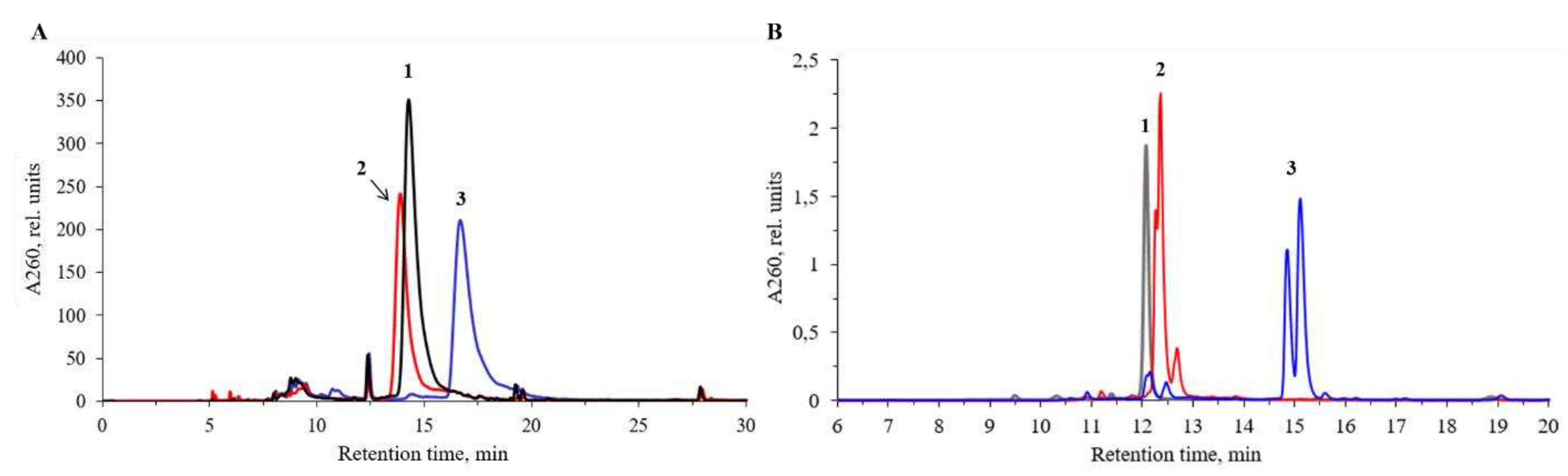
Figure 3.
RP-HPLC elution profiles of crude reaction mixtures of: (A) a co-injection of the carboxylic oligonucleotide A3 (1) and the amide D2 (2), elution gradient (iii); (B) conjugates B1, B2, B3, and B4, elution gradient (ii) (Section 4).
Figure 3.
RP-HPLC elution profiles of crude reaction mixtures of: (A) a co-injection of the carboxylic oligonucleotide A3 (1) and the amide D2 (2), elution gradient (iii); (B) conjugates B1, B2, B3, and B4, elution gradient (ii) (Section 4).
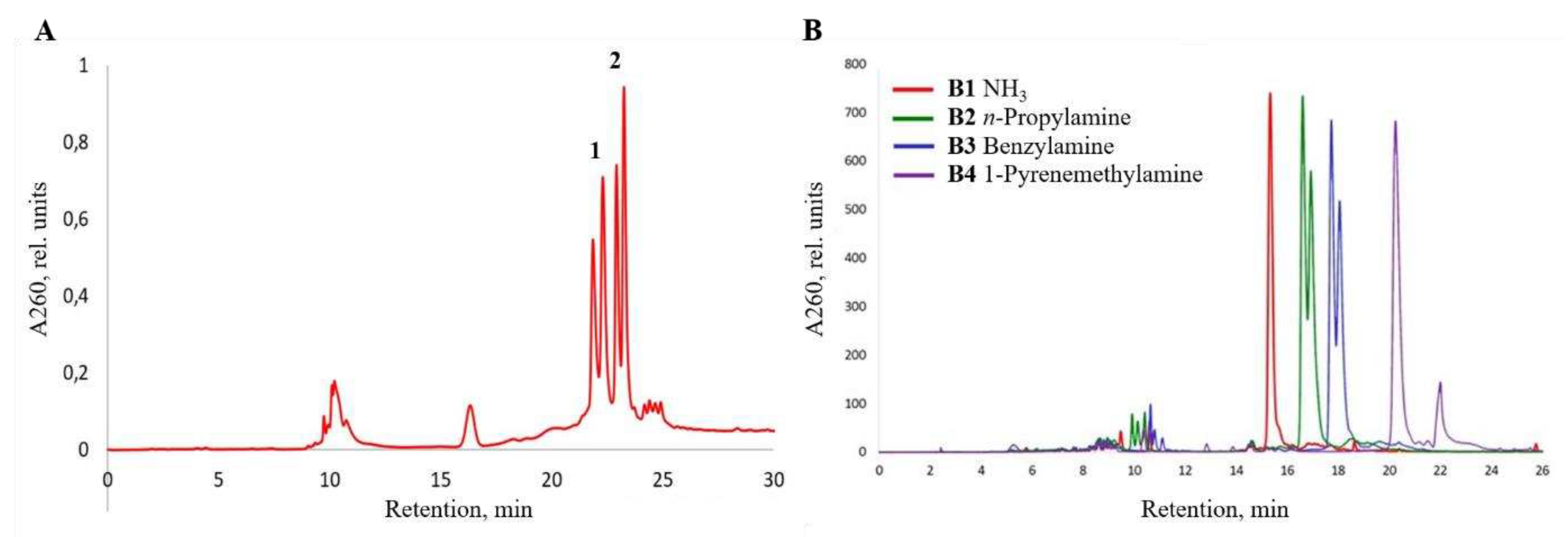
Figure 4.
RP-HPLC profiles of crude oligonucleotide conjugate D5, D6 (A) and D7, D8 (B) compared to unmodified control 5’-d(TTTTTT) and 5’-DMTr-d(TTTTTT), respectively.
Figure 4.
RP-HPLC profiles of crude oligonucleotide conjugate D5, D6 (A) and D7, D8 (B) compared to unmodified control 5’-d(TTTTTT) and 5’-DMTr-d(TTTTTT), respectively.
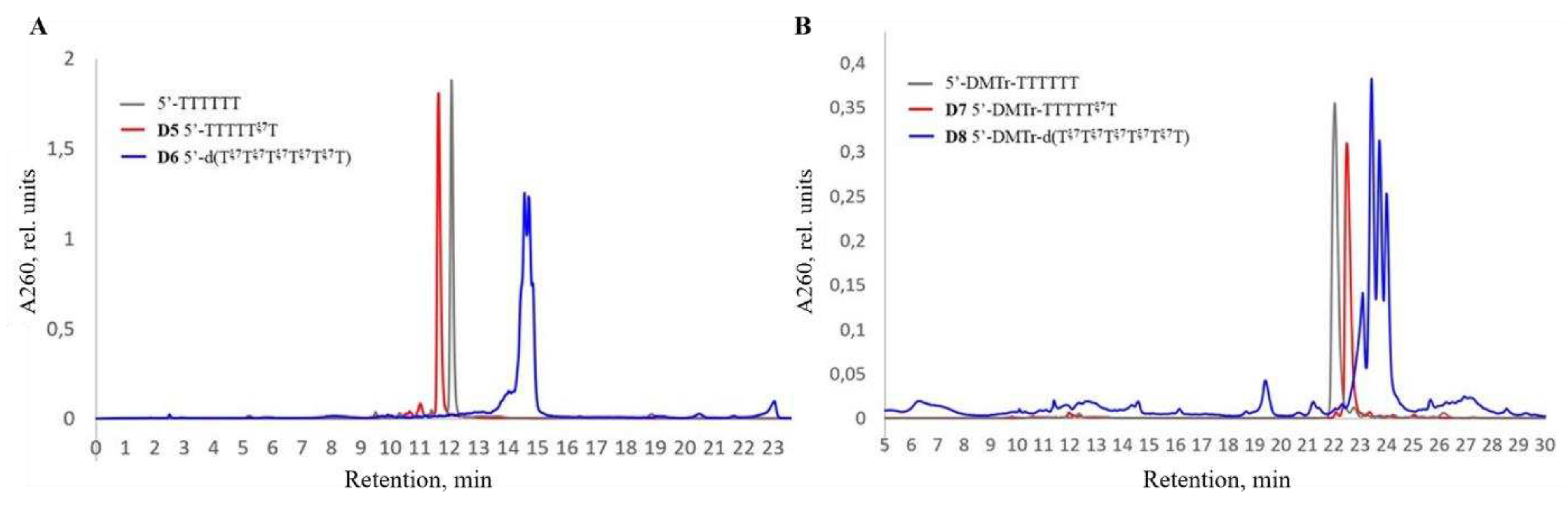

Table 1.
Amide-linked oligonucleotide conjugates with various primary amines obtained in this work.
| No. | Primary amine R'NH2 | Oligonucleotide sequence, 5’-3’a | Codeb |
|---|---|---|---|
| 0 | Aqueous NH3 (25°C) Carboxylic acids |
TcTTTTξT | A1 |
| DMTr-TTTTTξT | A2 | ||
| CξTCCCAGGCTCAAAT | A3 | ||
| CTCCCAGGCTCAAAξT | A4 | ||
| CTCCCAGGξCTCAAAT | A5 | ||
| AξGTCTCGACTTGCTACC | A6 | ||
| 1 | Aqueous NH3 (55°C) R’ = –HAmides |
Aξ1GTCTCGACTTGCTACC | A7 |
| Aξ1GTCTCGACTTGCTACC | B1 | ||
| Aξ1GTCTCGACTTGCTACC | C1 | ||
| CTCCCAGGCTCAAAξ1T | D1 | ||
| Cξ1TCCCAGGCTCAAAT | D2 | ||
| CTCCCAGGξ1CTCAAAT | D3 | ||
| Cξ1TCCCAGGCTCAAAξ1T | D4 | ||
| 2 |
n-Propylamine R’ = –(CH2)2Me |
Aξ2GTCTCGACTTGCTACC | A8 |
| Aξ2GTCTCGACTTGCTACC | B2 | ||
| 3 | Benzylamine R’ = –CH2Ph |
TTTTTξ3T | A9 |
| DMTr-TTTTTξ3T | A10 | ||
| Cξ3TCCCAGGCTCAAAT | A11 | ||
| CTCCCAGGCTCAAAξ3T | A12 | ||
| CTCCCAGGξ3CTCAAAT | A13 | ||
| Aξ3GTCTCGACTTGCTACC | A14 | ||
| Aξ3GTCTCGACTTGCTACC | B3 | ||
| Aξ3GTCTCGACTTGCTACC | C2 | ||
| 4 | 1-Pyrenemethylamine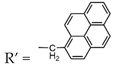
|
Aξ4GTCTCGACTTGCTACC | A15 |
| Aξ4GTCTCGACTTGCTACC | B4 | ||
| 5 | 4,7,10-Trioxa-1,13-tridecanediamine R’ = –(CH2)3[O(CH2)2]2O(CH2)3NH2 |
Aξ5GTCTCGACTTGCTACC | A16 |
| Aξ5GTCTCGACTTGCTACC | B5 | ||
| 6 | TetraethylenepentamineR’ = –(CH2)2NH[(CH2)2NH]2(CH2)2NH2 | Aξ4GTCTCGACTTGCTACC | A17 |
| Aξ6GTCTCGACTTGCTACC | B6 | ||
| 7 | 1,1-Dimethylethylenediamine R’ = –(CH2)2NMe2 |
TTTTTξ7T | D5 |
| Tξ7Tξ7Tξ7Tξ7Tξ7T | D6 | ||
| DMTr-TTTTTξ7T | D7 | ||
| DMTr-Tξ7Tξ7Tξ7Tξ7Tξ7T | D8 |
a The symbol (ξ) marks the positions of N-(4-carboxybenzenesulfonyl)-phosphoramidate groups, the superscript numbers (1-7) next to the symbol refer to the corresponding conjugated amine. b In the codes, the letters indicate the corresponding sulfonyl azides A-D from which oligonucleotides or conjugates were obtained. Note: the phosphate modification is the same but obtained using different azides A–D (Figure 1). c All the oligonucleotides were oligodeoxynucleotides, prefix ‘d’ was omitted throughout. Color code: white – A only; grey – A/HBTU/HOBt; green – B; blue – C; beige – D.
Table 2.
Thermal stability of the complementary duplexes of modified oligonucleotides with DNA and RNA.
Table 2.
Thermal stability of the complementary duplexes of modified oligonucleotides with DNA and RNA.
| Code | Oligonucleotide sequence, 5’-3’ |
DNA template 5'-d(ATTTGAGCCTGGGAG) |
RNA template 5'-r(ATTTGAGCCTGGGAG) |
||
| Tm, °C | ΔTm, °C | Tm, °C | ΔTm, °C | ||
| Control | d(CTCCCAGGCTCAAAT) | 61.20 | – | 65.68 | – |
| D1 | d(CTCCCAGGCTCAAAξ1T) | 61.23 | –0.03 | 65.59 | –0.09 |
| D2 | d(Cξ1TCCCAGGCTCAAAT) | 61.29 | –0.22 | 64.95 | –0.73 |
| D3 | d(CTCCCAGGξ1CTCAAAT) | 59.61 | –1.59 | 64.33 | –1.35 |
| D4 | d(Cξ1TCCCAGGCTCAAAξ1T) | 60.54 | –0.66 | 65.13 | –0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



