
Submitted:
11 December 2023
Posted:
13 December 2023
You are already at the latest version
Abstract
The high prevalence of childhood obesity is known to increase the risk of non-communicable diseases typically found in adults, such as cardiovascular disease and type 2 diabetes mellitus. However, worldwide childhood obesity cases continue to rise, thus comprehending its multiple causes to build healthier approaches and revert this scenario is urgent. Obesity development is now strongly associated with high fructose intakes, so mitigating the idea that fructose is a ‘natural nutrient’ and therefore beneficial to health, may be required, since. Excessive consumption of this highly lipogenic sugar leads to white fat accumulation, stimulates adipogenesis and causes white adipose tissue (WAT) inflammation, oxidative stress and dysregulated adipokine release. Unfortunately, dietary consumption of fructose is not always evident to consumers, as it is commonly added as a sweetener in food and sugar-sweetened beverages (SSB). Global consumption of fructose has increased dramatically in recent years. Therefore, it is important that the population has access to reliable information about food ingredients, via food labels. Consumers also need scientific education to understand potential health risks to themselves and their children. Here we discuss the impact of excessive fructose intake on adipose tissue biology, its contribution to childhood obesity and make suggestions for reversing this trend.
Keywords:
fructose
; childhood obesity
; adipose tissue
1. Introduction
Childhood obesity, diagnosed by comparing body weight to age [1], is associated with non-communicable health complications such as cardiovascular disease, type 2 diabetes mellitus (T2DM), mental health problems and some types of cancer in later life [2,3]. These conditions are usually observed in adults, but the development of non-communicable disease (NCD) often begins in childhood. In children, excessive energy intake and/or a sedentary lifestyle can lead to positive energy balance with consequent expansion of adipose tissue, beyond what is expected in a healthy child [4]. Since treating this complex multifactorial condition [5,6] is so challenging, preventing childhood obesity is fundamental to improving public health worldwide. It is also important to successfully control obesity escalation, since a child living with obesity tends to become an adult living with obesity [2,3].
Globally, childhood obesity is predicted to rise, especially among male children and adolescents (aged 5-19 years) from 103 million in 2020 to 208 million in 2035, and from 72 to 175 million in female children and adolescents [7]. Although the causes of obesity are varied, the increasing consumption of fructose [8] has become a pivotal issue to understanding the pandemic status of obesity [5,9,10] and the exponential increase of childhood obesity [11]. Indeed, experimental protocols, epidemiological studies, and clinical trials have provided convincing evidence that sugar sweetened beverages (SSBs) increase the risk for obesity [12]. Here we discuss the impact of excessive fructose intake on the biology of adipose tissue and its contribution to development of childhood obesity.
2. Biology of adipose tissue
Adipose tissue can be classified into white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is distributed throughout the body in distinct depots, including visceral (vWAT) and subcutaneous WAT (sWAT) depots. vWAT is in omental, mesenteric, retroperitoneal, gonadal, and pericardial regions, while sWAT is subcutaneous and, in humans, also in the abdomen and gluteofemoral regions [13,14]. BAT is typically found in the interscapular and supraclavicular regions and represents approximately 4.3% of the fat mass in humans [15].
White adipocytes are responsible to the storage and release of lipids in response to systemic nutritional and metabolic needs, and brown adipocytes are specialized in thermogenesis. The brown adipocyte’s function is due to the expression of the uncoupling protein 1 (UCP1), which decouples the proton gradient from the mitochondrial ATP synthesis, dissipating energy in form of heat [15]. A third type of adipocytes have characteristics of both brown and white adipocytes (known as ‘beige’ or ‘brite’ adipocytes) and can be found in WAT depots. In addition, the morphological and functional characteristics of beige adipocytes are very similar to those of brown adipocytes, including thermogenic capacity due to the expression of UCP1 [16].
In addition to the function of acting as an energy reservoir for protecting vital organs and controlling body temperature, adipose tissue is also an endocrine organ that produces and secretes many signaling proteins called adipokines. These proteins modulate various functions in the body, including systemic metabolic state, inflammatory response, and cardiovascular function [17]. The role of adipokines such as leptin, adiponectin, interleukin-6(IL-6) and fibroblast growth factor 21 (FGF21) in health and disease were widely presented in a recent review by Clemente-Suárez et al. [17].
The development and expansion of adipose tissue can be driven by the formation of new adipocytes from preadipocyte differentiation in the process of adipogenesis (hyperplasia) and by increasing lipid storage through lipogenesis (hypertrophy). On the other hand, the process of lipolysis, which is the hydrolysis of triacylglycerol followed by fatty acids release to generate energy, is involved in the reduction of adipocyte size. Fat accumulation is determined by the balance between synthesis and breakdown of triacylglycerol [18].
Fetal adipogenesis begins early in the second trimester of pregnancy in a cranial to caudal and then medial to lateral manner [19]. At the 28th week of pregnancy, WAT is present in the principal fat depots throughout the body [19], while BAT can be identified earlier in development [20]. Considering that adipogenesis involves different precursors and distinct regulatory processes, factors such as maternal nutrition state and obesity can influence the developmental patterning of fetal adipose tissue with future consequences into adulthood [21].
After two weeks of birth, the expansion of adipose tissue rises rapidly in response to increased nutrient availability in the postnatal environment [22]. Adipocyte proliferation in human adipose tissue is markedly observed before two years and during puberty [23,24]. After puberty, adipocyte number and size become relatively static in lean individuals [25]. However, increases in adipose mass in individuals living with obesity can result from both adipocyte size and number elevation [26].
Regarding lipogenesis, fatty acids and monoacylglycerols taken up from the circulation are driven toward triacylglycerol synthesis and storage in the adipocyte. In this process, fatty acids are acylated with CoA forming acyl-CoA by acyl-CoA synthetase (ACS) [27]. Then, the enzymes glycerol-3 phosphate acyltransferase (GPAT) and diacylglycerol acyltransferase (DGAT) are responsible for the final part of the process esterifying acyl-CoA together with glycerol-3-phosphate in triacylglycerol [28].
Carbohydrates can be converted to fatty acids through a process known as de novo lipogenesis. This process occurs through the production of citrate followed by its conversion into acetyl-CoA by the enzyme ATP citrate lyase (ACL) [29]. Through the action of the enzyme fatty acid carboxylase (ACC) the acetyl-CoA formed is converted to malonyl-CoA, which will be used as a substrate for the synthesis of triacylglycerol by the enzyme fatty acid synthase (FAS) [28].
At the early stage of human pregnancy, the amount of fetal fat is provided by transplacental transfer of fatty acids. However, during the last 3 months, fetal fatty acids are derived predominantly from their own synthesis de novo [30]. In addition, the glucose used in lipogenesis is delivered by the mother via the placenta, because the fetus does not synthesize glucose. Thus, conditions such maternal obesity or gestational diabetes can impair glucose metabolism in fetal adipose tissue, and consequently increase adiposity and result in elevated birth weight [31].
The regulation of lipolysis has been extensively reviewed in another previous study [32]. Briefly, lipolysis starts from the binding of noradrenaline to beta-adrenergic receptors, which allows the increase of cyclic-AMP and the activation of protein kinase A (PKA). PKA phosphorylates hormone sensitive lipase (HSL) and perilipins, thus allowing triacylglycerol hydrolysis into fatty acids. Adipocyte triacylglycerol lipase (ATGL) and monoacylglycerol lipase (MAGL) also play important roles in the lipolysis, with ATGL participating in the initial phase of triacylglycerol hydrolysis by cleaving the first fatty acid and MAGL cleaving the last fatty acid of the triacylglycerol [32]. The 3 fatty acids liberated may be oxidized in muscle or BAT, and the glycerol may be used as a precursor for gluconeogenesis in the liver [33].
Lipolysis occurs during periods of energy demand such as fasting and physical exercise but seems to be suppressed during fetal life. In fact, adipose tissue favors lipid synthesis over lipolysis under normal conditions, actively contributing to progressive fat storage during the last weeks of gestation [34]. While in adults catecholamines are the main lipolytic hormones, thyrotropin hormone plays a key role in lipolysis in newborns. However, during childhood, catecholamines increase and replace its effect [35].
3. Biochemical aspects of fructose
Fructose is a monosaccharide made up of six carbon atoms bonded by single covalent bonds, presenting hydroxyl groups and a carbonyl group, formed by a double bond between carbon and oxygen. The position of this grouping will determine, after hydrolysis of the monosaccharide, if it will give rise to ketone or aldehyde. Fructose also called ketohexose, when hydrolysed gives ketone, since containing the carbonyl group at the end of the chain. Glucose, on the other hand, when hydrolysed it will give rise to aldehyde, being called aldohexose. In the same way that glucose 1g of fructose provides 16kJ of energy [36].
Usually, fructose is ingested as a monosaccharide or disaccharide (combined with glucose) via sucrose [37]. It can also be found in tri- and tetrasaccharides, such as raffinose and stachyose, present in legumes, soybeans, lentils, peas, and beans for example [38]. When sucrose is the source of fructose, it must be digested to liberate fructose and glucose for absorption. Dietary fructose is absorbed and transported through facilitative Glucose Transporters (GLUTs) located on both sides of the enterocyte membrane [39]. Absorption of fructose is performed by GLUT5, located on the enterocyte’s apical membrane, and transported to the portal bloodstream by GLUT2 present on the basolateral membrane of these cells [40]. GLUT5 exclusively transports fructose, while GLUT2 transports both glucose and fructose, also galactose. When co-ingested with glucose, intestinal fructose absorption is increased due to upregulation of GLUT5 [41]. Unlike glucose, the uptake of fructose into enterocytes is an insulin and sodium-independent process without the expenditure of ATP [42].
Most of the fructose absorbed by enterocytes is directed to the liver where it participates in carbohydrate metabolism and de novo lipogenesis. Hepatic fructose is rapidly phosphorylated by ketohexokinase (KHK) to fructose-1-phosphate (F1P), which is further metabolized to glyceraldehyde 3-phosphate and dihydroxyacetone phosphate by aldolase-B. Later, these products are used in glycolytic pathways, or even in lipogenic processes, especially during high fructose intake (Figure 1) [43]. The remaining fructose in the enterocyte is also metabolized by KHK to F1P and processed to further metabolites such as Short-Chain Fatty Acids (SCFAs), glucose, and organic acids by aldolase-B and triokinase sequentially [44].
A recent study by Jang et al. [45] showed that with low-dose fructose intake (<0.5 g/kg), the intestine can metabolize about 90% of this nutrient, mainly into glucose and organic acids, releasing only nonmetabolized small part into portal circulation. Conversely, with a high intake of fructose (≥1 g/kg), the intestinal capacity to metabolize this nutrient is exceeded, releasing the excess fructose to the liver [45]. Another problem with ingesting large amounts of fructose is that it is likely to exceed the absorptive capacity of the intestines, thought to be about 5–50 g per serving in a healthy adult [46]. Incomplete absorption of fructose is associated with diarrhea [47] and other gastrointestinal symptoms, such as gas accumulation, flatulence, and abdominal pain [48].
4. Sources and consumption of fructose
The main sources of fructose in the diet are fruit juice, fruit, yogurt, honey, ice cream, confectionary and soft drinks sweetened with either sucrose or high fructose corn syrup (HFCS) [49]. Naturally occurring fructose found in yogurt and fruit was shown to be protective against cardiometabolic disease, so there is no advice to limit these foods in the diet [49]. However, in recent decades, dietary consumption of sugar has increased, and this is often blamed for the increased prevalence of obesity and cardiometabolic disease [50]. The consumption of fructose in children and adolescents aged 2-18 years exceeds the desirable goal of less than 5% energy intake from free sugars [51]. Therefore, healthier approaches to beverage and dietary consumption should be adopted in infancy [51] to avoid the development of obesity and comorbidities [11].
Recommendations for reducing sugar intakes focus on ‘free sugar’ rather than the intrinsic sugars found in milk and plant food. Both the World Health Organization [52] and the UK’s Scientific Advisory Committee on Nutrition [53] recommend limiting free sugar intake to less than 5% of overall energy intake. Free sugar is defined as sugar added by the manufacturer, cook or consumer, plus sugar found in honey, syrup, or fruit juice [49,54]. It is often added as a sweetener to food and drink in the form of sucrose (table sugar) or HFCS (55% fructose) that are commonly used in sugar-sweetened beverages (SSB) [37]. It is estimated that Americans consume 25kg of HFCS per person, per year [55]. Most European countries consume far less HFCS, such as the United Kingdom (UK) consuming < 0.5kg per person, per year [55]. However, nutritional surveys in the UK show that most people consume far more free sugar compared to the recommendations, with a mean intake of 12.5% of energy coming from free sugar, including 9% and 3.5% of energy from sucrose and fructose [54]. There appears to be no figures for fructose consumption in Brazil, but consumption of SSB is reported to be high. In fact, young Brazilians consume an average of 281.5 ml or 1.6 SSB per day, which contribute to 5.9% of energy intake [56].
Fructose is often associated with obesity and cardiometabolic disorders. Countries with a high intake of fructose (from HFCS) are shown to have a significantly higher prevalence of type 2 diabetes (8%) compared with countries with low HFCS consumption (6.7%, p=0.03) [55]. Several countries, including the UK in 2018, have introduced financial penalties (a sugar levy) to dissuade the food industry from adding free sugar to food and beverages, aiming to reduce free sugar consumption by 20% [54]. Mexico also introduced a similar system in 2014, adding a 15% cost to SSBs, to discourage consumer purchase [57]. Both countries have observed a reduction in sugar intake since the introduction of the levy, by 4-12% in Mexico [57] and 10% in the UK [54].
A Canadian study with 1000 adolescents and young adults (aged 16-30) concluded that consumers appear to base healthiness perceptions on a sweetener’s level of “naturalness” rather than energy content, probably due to their perception about the level of product processing. Most of the respondents perceived HFCS (63.9%) and aspartame (52.4%) as less healthy than table sugar, and the perception of “naturalness” has important implications for understanding consumer preferences [58]. Therefore, an effective approach for limiting SSB consumption among young people might involve warning labels that includes calorie information [59,60]. A study conducted in 6 countries (Australia, Canada, Chile, Mexico, UK, and United States) with 10,762 children aged 10-17 showed that different types of front-of-package label induced the participants to perceive the product as unhealthy, especially with “high in” labels with intuitive symbols [59]. Additionally, another study performed with 2002 parents of children aged 11-16 in UK showed that SSB selection by parents for their children was lower when labels contained an image-based warning (35%) compared with no label (49%) or calorie information (43.5%) [60].
5. Excessive fructose intake and its metabolic implications
Excessive fructose intake has been associated with diseases [37,61] such as metabolic syndrome [62], blood hypertension [63], hypertriglyceridemia [64], non-alcoholic fatty liver disease (NAFLD) [65] and obesity in adults and infants [12] (Figure 2). Most of the fructose that exceeds the intestinal ability to absorb and metabolize this nutrient will bedriven to the liver, stimulating pathways that will simultaneously lead to hepatic fat accumulation and reduction of hepatic fat removal [65].
Fructose stimulates sterol regulatory element-binding protein 1 (SREBP-1c) and carbohydrate-responsive element–binding protein (ChREBP), both key transcriptional regulators of hepatic de novo lipogenesis [66]. In the other hand, fructose upregulates the synthesis of acetyl-CoA, which is then converted to malonyl-CoA leading to β-oxidation inhibition by limiting carnitine palmitoyl transferase action [67]. VLDL and triglyceride (TG) production are accentuated by the formation of glyceraldehyde-3-phosphate, a substrate for its synthesis from fructose-1-phosphate [67] (Figure 1).
In healthy subjects, a high-fructose diet consumed for 7 days resulted in an increase in ectopic fat accumulation in liver and skeletal muscle, increased fasting plasma TG and VLDL, and a decrease in hepatic insulin sensitivity, independently of family history of T2DM [68]. Glucose metabolism and uptake pathways may be impaired by dyslipidemia status generated from excessive fructose intake [69]. Mice fed a 66% fructose diet for 2 weeks decreased the number of insulin receptors in skeletal muscle and liver, as well as increased blood pressure and TG serum levels [70]. In addition, 28 days of high fructose feeding reduced insulin stimulated phosphorylation of insulin receptor substrate (IRS 1/2) in the skeletal muscle and liver of mice. Those mechanisms might be involved in fructose contributing to insulin resistance and T2DM pathogenesis [71].
Fructose metabolism in liver forms uric acid (UA) as a metabolite, which consequently induces endothelial dysfunction, inflammation and oxidative stress in hepatocytes [72]. By activating the proinflammatory NF-κB signaling cascade, hyperuricemia increases the expression of inflammatory biomarkers such as C-reactive protein, fibrinogen, ferritin, and complement C3 in HepG2 cells [73], which could contribute to the inflammation observed in metabolic and cardiovascular diseases. In accordance, fructose administration for 20 weeks in rats, induced hyperuricemia, increased serum proinflammatory cytokines IL-6, TNF-α, and MIP-2, and decreased anti-inflammatory cytokine IL-10 [74].
6. Fructose and obesity: what happens with adipose tissue
High fructose consumption and obesity development in modern societies has become indissociable [75]. This highly lipogenic sugar stimulates adipogenesis and results in WAT accumulation [76], and may also cause WAT inflammation [77], oxidative stress [75], dysregulated adipokine release [78] and BAT whitening [79], involving different tissues and metabolic pathways (Figure 2).
In mice, a 15% increase in fructose intake via water consumption, increased adiposity [76], while a high-fat (31% g/kg) and high-fructose (24% g/kg) diet increased WAT accumulation and adipocyte hypertrophy, glucose intolerance and reduced insulin secretion by isolated pancreatic islets [80]. Moreover, rats fed a fructose-rich diet for 8 weeks showed increased epididymal and mesenteric WAT, with more large adipocytes and less small adipocytes in visceral WAT, compared to controls [81]. Increased adipocytes size following exposure to a fructose-rich diet was also observed in rat retroperitoneal WAT, with increased number of adipocyte precursor cells (APCs) and adipogenic potential, inferred by the higher expression level of two adipogenic competency markers, PPARγ2 and Zfp423 [82].
Fructose-stimulated adipogenesis is also mediated by the increasing of active glucocorticoids (GC) levels through 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) activity in WAT [83]. Fructose metabolism in 3T3-L1 adipocytes [85] and epidydimal WAT [85] generates NADPH that is required for the activation of 11β-HSD1, an enzyme that converts inert GC metabolite into active hormone, resulting in high levels of plasma GC [83]. GCs are required to fully differentiate adipocytes by induction of key adipogenic transcription factors such as C/EBPα, C/EBPβ, C/EBPδ, KLF5, KLF9, and PPARγ in the early phase of differentiation [86,87]. In vitro, 3T3-L1 preadipocytes cultured with only fructose as the carbohydrate source differentiated into adipocytes [88].
High-fat and high-fructose diets have a negative impact on the metabolome of liver, muscle, BAT and WAT [89], and long-term fructose consumption increased mRNA levels of genes involved in fatty acids synthesis such as FAS and SCD1, and decreased expression of genes involved in lipid mobilization like ATGL and HSL, contributing to adipocytes enlargement [90]. A high-fructose diet for 3 weeks increased adipocytes area, FAS activity and GLUT5 expression in rats [91]. Large adipocytes and increased plasma TG persisted even after fructose withdrawal from diet, which might indicate a longstanding negative effect of a fructose-rich diet in the young that may persist into adulthood [91].
Excessive fructose intake also might contribute to low-grade systemic inflammation. Male and female rats fed a high-fructose diet during 24 weeks presented a visceral WAT accumulation with high concentrations of inflammatory markers such as iNOS, TNFα, IL-1β, IL-18, MDA, and ALT [77]. Kovacevi et. al [92] found that visceral WAT inflammation precedes obesity in female rats maintained with 10% added fructose in water. In this study inflammatory markers like IL-1β, IL-6, TNFαand nuclear accumulation of NFκB were higher even before increases in visceral adiposity. The authors also observed insulin resistance in visceral WAT, detected by IRS1 inhibitory phosphorylation and decreased Akt activity, and hyperuricemia [92], which in turn activates NFκB inflammatory pathway [73], and may be a key factor responsible for the proinflammatory endocrine imbalance in WAT [93]. Local inflammation was found in WAT from high-fructose fed rats, with increased TNF-α levels, MPO activity, and decreased adiponectin, an anti-inflammatory molecule, that persisted after fructose diet withdrawal [91].
Singh et. al [94] found that a 60% fructose diet, fed over 10 weeks, induced the already mentioned fructose effects (visceral adiposity, insulin resistance and increased TG levels) accompanied by the activation of NLRP3 in rat epidydimal adipose tissue. In turn, NLRP3 inhibition mitigated inflammatory signaling and lipogenesis in WAT, as well as improved insulin sensitivity [93]. NLRP3 is an intracellular sensor that, when activated, forms an inflammasome complex and results in caspase-1 expression and secretion of IL-1β and IL-18 in macrophages [93]. Therefore, NLRP3 represents another mediator of high-fructose deleterious effects, including the increased lipogenesis rates that contributes to development of obesity. However, the relation between high fructose ingestion and inflammation in human is less clear. The excessive consumption of fructose over a period of 8 days from SSBs did not worsen the low-grade chronic systemic inflammation or increase body weight in adults, probably due to the short period of nutrient ingestion [95].
In obesity, systemic oxidative stress is accentuated by several mechanisms, including the activation of NADPH oxidase (NOX) with superoxide production as consequence of elevated FFA, inflammatory cytokines or hyperglycemia [96]. Fructose-induced obesity caused oxidative stress in hypertrophic visceral adipose tissue in male Wistar rats [97], and ROS production in cultured macrophages was higher in fructose-treated cells compared with untreated or glucose-treated cells [98]. Hyperuricemia might mediate this fructose induction of oxidative stress [75] since UA is involved with ROS production by activating NOX and reducing endothelial level of the NO [99].
Additionally, fructose increases the production of GC [83], that such as dexamethasone and cortisol also play a role in inducing the over production of ROS [100]. Such oxidative stress status could be implicated in adipogenesis and lipid accumulation in adipocytes, representing another link between high fructose intake and obesity development [75]. In addition, oxidative stress in 3T3-L1 cells mediated the expression of pro-adipogenic genes PPARγ, RXRα and C/EBPα and resulted in adipogenesis and lipid accumulation [101], and the impairment of ROS production decreased the expression of adipogenic markers and lipid deposition in this cell line [102].
The profile of adipokines produced by different adipose tissue depots reflects its health status [103]. WAT from lean subjects releases an anti-inflammatory adipokine profile with high levels of adiponectin, IL-10 and IL-4, and low levels of leptin, TNF-α, IL-6 and MCP-1, while the WAT from obese releases the same adipokines but in inverse proportion [104]. Interestingly, high-fructose intake has also been associated with damaged adipokine profile secretion [78]. Fructose may lead to reduced adiponectin levels [91] as consequence of hyperuricemia [93], impairing its anti-inflammatory and anti-diabetic effects [105]. Adiponectin is very sensitive to fructose intake, since a single high-fructose meal in rats was enough to reduce its serum concentration after 2 or 4 hours, as well as increasing TNF-α content and neutrophil recruitment in liver [106]. These effects limit the potential of adiponectin to decrease hepatic gluconeogenesis and WAT inflammation, or increase fatty acid oxidation in skeletal muscle and liver, contributing to the maintenance of obesity phenotype [107].
WAT from individuals with obesity increases leptin secretion, leading to leptin resistance by desensitizing its receptors in the hypothalamus, prejudicing its regulation of food intake and contributing to obesity [108]. High-fructose intake seems also to induce hyperleptinemia in rats [78,109,110] by leptin gene overexpression in WAT, which was further decreased by treatment with hypouricemic agents, suggesting that fructose-induced hyperuricemia is directly associated with hyperleptinemia [109]. However, further investigations are required, since this effect was not observed in other studies [111,112]. Despite that, high-fructose ingestion induced peripheral leptin resistance in rats, in combination or not with high-fat diets [110,111,112,113]. This effect was reversed after fructose withdrawal from diet [110]. Intraperitoneal leptin injections successfully reduced food intake in adult rats fed a fructose-free diet but had no effect in animals fed a 60% fructose diet for 6 months. This leptin resistance was associated with decreased phosphorylation in hypothalamic signal transducer and activator of transcription 3 (STAT3) [111]. When fructose was added in the diet of post-weaning rats, it resulted in decreased leptin receptors and SOCS3 in WAT, suggesting that long-term fructose diet alters paracrine signaling of leptin [112], alerting for the risks of a high-fructose consumption already early in life.
Recent studies indicate an effect of fructose also in BAT metabolism [79,114,115,116]. C57BL/6 mice fed a high-fructose diet (50% of energy as fructose) during 12 weeks showed an interscapular BAT (iBAT) whitening process, with a reduction in UCP1 immunodensity [79]. A similar study from the same group did not find a pronounced whitening process in high-fructose fed mice, only in high-fat fed animals. However, high-fructose diet caused lipid accumulation in iBAT and had a negative immunostaining for vascular endothelial growth factor A (VEGF-A), which could indicate a hypoxic state that precedes mitochondrial loss and suggests that whitening might occur in prolonged high-fructose intake. Those effects were reduced after PPAR-α activation, known to induce mitochondrial biogenesis, β-oxidation, fatty acid uptake, and UCP1 expression [114].
Additionally, in high-fructose fed mice a proportional reduction in BAT mass was observed in comparison with visceral WAT and morphological remodeling, with increased lipid deposition in enlarged intracellular lipid droplets [115]. In humans, a 14-day high-fructose diet impaired glucose uptake in BAT, without changes in cold-stimulated thermogenesis [116].
7. The multiple causes of childhood obesity
Obesity is a multifactorial condition, therefore some factors such as maternal lifestyle during the pre-gestational and gestational phases, birth weight, nutrition and physical activity, socioeconomic and genetic factors are associated with childhood obesity pathogenesis [117,118]
Prenatal and early postnatal factors increase the risk of developing obesity and several additional diseases throughout life [117,119]. Children born from mothers with obesity or from women that developed gestational diabetes are more likely to develop obesity and metabolic problems compared to children born from healthy mothers [119]. Therefore, factors such as pre-pregnancy body mass index (BMI), maternal weight gain and glucose metabolism during pregnancy and lactation correlates with the occurrence of childhood obesity [120,121,122]. Additionally, there is a greater propensity for obesity in adulthood in children with high birthweight, and an increase in the central distribution of fat in those with low birthweight, reaffirming that the gestational phase is an important moment for an increase in later body adiposity [119].
Birthweight is an indicator of fetal health that reflects both intrauterine growth and gestational age. Abnormal fetal growth is correlated with increased risk for cardiometabolic disease [118,119] and there is a linear association between birth weight and BMI in adulthood [123].
Since a lack of physical activity and an inadequate diet are closely related to energy balance, these are often identified as the main risk factors for obesity in childhood [118]. Diets that lack a good fruit and vegetable intake, alongside increased consumption of ultra-processed foods and SSBs are thought to be among the various nutritional patterns that contribute to childhood obesity [124]. Since the family is primarily responsible for the child's development, the provision of food, and that children look to their parents for examples, parental involvement in lifestyle changes and obesity prevention are essential. Promoting an environment that prioritizes a balanced diet and encourages the practice of regular physical activity are essential ingredients for obesity prevention [125].
Low socioeconomic status is also identified as one of the risk factors for childhood obesity, since individuals from lower socioeconomic groups tend to have a different lifestyle than individuals with higher purchasing power. Individuals in higher socioeconomic groups can have improved access to more nutritional diets that contribute to better food choices, and greater opportunities for regular physical activity, thus reducing the risk of overweight and obesity [118,126].
In addition to environmental factors, genetic factors are involved with excessive weight gain, such as the mutations in the genes for leptin (LEP) and leptin receptors (LEPR), proopiomelanocortin (POMC), melanocortin-4 receptor (MC4R) and prohormone convertase, that all alter appetite regulation [121,127]. However, genetic defects directly leading to obesity are rare, representing less than 1% of cases. So, genetic factors play a key secondary role in the development of childhood obesity, by increasing an individual’s predisposition to body weight gain, in the presence of other factors such as the environmental and behavior [121].
8. Fructose and childhood obesity
High fructose ingestion and childhood obesity are linked through several paths taken at different stages of a child's life (Figure 3), even before the birth, since fructose crosses the placental barrier [128] and fetal membranes exhibit nutrient transporter expression profiles like the placenta [129]. Supplementation of maternal diet with carbohydrate (glucose, fructose, or both) provoked a significant increase in amniotic fluid glucose and a significant decrease in amniotic fluid uric acid, as the level of carbohydrate increased in the maternal diet [130]. Furthermore, in this study, the glucose content of amniotic fluid was predictive of fetal body weight [130]. Thus, maternal excessive intake of fructose may affect fetal development very early, impacting the developmental patterning of fetal adipose tissue, with its highly adipogenic effect [76]. Whether this intrauterine exposure to excess fructose modulates the expression of adipogenic factors such as C/EBPα, C/EBPβ, C/EBPδ, KLF5, KLF9, PPARγ and Zfp423 in fetal adipose tissue remains a gap in knowledge.
The impact of fructose consumption during pregnancy and the impact on fetal metabolic programming is of particular concern. Animal studies have shown that the offspring of mice fed fructose (via fructose syrup), during pregnancy and lactation, consumed more food and gained more weight than controls [131,132]. Furthermore, offspring from the mice fed a high fructose diet, showed evidence of DNA damage in blood, liver, kidney, and brain [131], or induced dyslipidemia and hyperglycemia [132]; all suggestive of increased long-term susceptibility to cardiometabolic disease. High fructose consumption during pregnancy and lactation was also associated with hypertension in offspring during adulthood; thought to be due to fetal renal programming [133]. Additionally, in pregnant mice consumption of water with 20% of added fructose, resulted in elevated circulating levels of fructose in dams and their fetuses, which led to impaired fetal BAT development, attenuated diet-induced thermogenesis, and metabolic disorders in adult offspring [134].
The evidence in humans is less clear as it is an under researched area. However, a Norwegian study [135] demonstrated an increased risk of preterm birth, when pregnant women regularly consumed SSB (high in sucrose/fructose). It is unclear whether the risks were associated directly with the beverages or indirectly, due to other dietary or socioeconomic factors, as regular intakes of SSBs are associated with inferior quality diets and poorer socio-economic status [134]. In China, a significant increased risk of gestational diabetes was found in pregnant women with higher fasting serum fructose; however, the authors believe that the higher serum fructose was due to endogenous production of fructose, rather than an association with diet [136].
An American study [137] assessed the intake of SSBs and fructose in 1,068 pregnant women using a food frequency questionnaire. Higher consumption of SSBs were associated with non-white ethnicity, younger maternal age, lower education and income, plus higher pre-pregnancy BMI. Furthermore, they found that higher intake of SSBs (odds ratio, 1.70; 95% confidence interval, 1.08-2.67) and total fructose (odds ratio, 1.58; 95% confidence interval, 0.98-2.53) during pregnancy was associated with an increased prevalence of asthma in offspring during mid-childhood [137]. Cohen et al. [138] found that sugar consumption during pregnancy, especially from SSBs, adversely affected childhood cognition scores in offspring. In terms of food, breast milk can be the child's first contact with fructose, if the mother consumes foods excessively rich in this nutrient, with negative consequences for weight control in progeny. Although the presence of fructose in breast milk does not eliminate the many other benefits of exclusive breastfeeding, the possibility of this nutrient interfering with child's taste formation cannot be ruled out, since the foods eaten by the mother during pregnancy and lactation form the basis of the child’s weaning patterns [139].
Fructose consumption during lactation may also be problematic, as research [140] has found that consumption of SSBs during lactation increases the fructose content of breastmilk, and this increase remained for up to five hours post consumption. So, infants who are breastfed by mothers consuming SSBs, will be consuming significantly higher levels of fructose during early life. In guinea pigs fed a high fructose diet, an increased FFA content in milk was found, which resulted in offspring altered serum FFA, as well as increased levels of UA and TG [141]. Goran et al. [142] found higher growth in infants at 6 months of age (an increase in both lean and fat mass), when fructose was identified in breastmilk. Moreover, high consumption of SSBs in women during early lactation was associated with lower neurological development scores in infants at 24 months of age [143].
After birth, the development of infant's gut microbiota becomes a very important issue. When compared with human milk or a traditional lactose-based infant formula, consumption of lactose-reduced infant formula with added corn syrup, for a period of six months, was found to shape an infant’s microbiome prematurely and was directly associated with the consumption of a high fat and high carbohydrate diet during childhood [144]. Alternatively, the numerous human milk oligosaccharides serve as substrate for the proliferation of beneficial bacteria, contributing to an intestinal microbiota composition with health benefits for the breast-fed neonate [145]. Considering that fructose is not the most abundant sugar found in human milk (lactose is made of glucose and galactose), the early introduction of fructose into infant's diet certainly figures as a risk factor for childhood obesity.
Moreover, consumers erroneously tend to perceive fructose as a “natural” nutrient [146] originating from fruits and therefore considering it neutral or even beneficial to health [147]. Conversely, SSB intake was correlated with healthy diet and physical activity among adolescents [148], while a study of 548 children over 19 months showed an association between SSB consumption and obesity, increasing BMI by 0.24kg/m2 (p=0.03) with a 60% rise in obesity (p=0.02) [149]. These findings point to the importance of guidance from parents and tutors, regarding healthy consumption of SSB, but also the requirement that the population in general, have access to adequate information about food, its components, and the consequences of its ingestion for people's health.
9. Conclusions
High fructose intake has several different harmful effects on metabolism and adipose tissue function, including stimulation of adipogenesis and lipogenesis that ultimately can lead to WAT accumulation and obesity. Early exposition to high fructose levels, from the intrauterine environment and breast-feeding (both associated with maternal fructose consumption), and in childhood, all represent a significant concern for childhood obesity development and its current prevalence globally. In this review we explored the multiple relationships between excessive fructose ingestion and childhood obesity with particular interest in WAT pathophysiology. Finally, we highlight the importance of ensuring that the population has sufficient access to reliable information about the food they are consuming, including nutrient content, indicated on food labels. Consumers also need to have reliable knowledge about diet, through scientific education to be able to make good choices about what foods will or will not enter their homes. A population able to choose high quality food will be a population capable of preventing and managing childhood obesity.
Author Contributions
Conception of the manuscript idea, A.K.A.M. and F.S.E; Abstract and introduction writing, M.P.S. and A.K.A.M; Biology of adipose tissue writing, F.S.E; Biochemical aspects of fructose writing, A.K.A.M; Sources and consumption of fructose writing, J.A. and M.P.S; Excessive fructose intake and its metabolic implications, and Fructose and obesity: what happens with adipose tissue writing, M.P.S; The multiple causes of childhood obesity writing, N.J.R.F; Fructose and childhood obesity writing, A.K.A.M and J.A.; Conclusion writing, A.K.A.M, M.P.S and F.S.E.; Conception and development of the figures, M.P.S., A.K.A.M and F.S.E; Article review, A.K.A.M, F.S.E. and J.A.; English review, J.A.
Funding
São Paulo Research Foundation (FAPESP processes 2018/22361-9 and 2020/15748-4)
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this manuscript. Data sharing is not applicable to this review article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Apperley LJ, Blackburn J, Erlandson-Parry K, Gait L, Laing P, Senniappan S. Childhood obesity: A review of current and future management options. Clin Endocrinol. 2022, 96, 288–301. [CrossRef]
- Thomas-Eapen N. Childhood Obesity. Prim Care - Clin Off Pract. 2021, 48, 505–15. [CrossRef]
- Pandita A, Sharma D, Pandita D, Pawar S, Tariq M, Kaul A. Childhood obesity: Prevention is better than cure. Diabetes, Metab Syndr Obes. 2016, 9, 83–9. [CrossRef]
- Sahoo K, Sahoo B, Choudhury A, Sofi N, Kumar R, Bhadoria A. Childhood obesity: causes and consequences. J Fam Med Prim Care. 2015, 4, 187-192. [CrossRef]
- Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019,15, 288–98. [CrossRef]
- Lin X, Li H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front Endocrinol. 2021, 12, 1–9. [CrossRef]
- World Obesity Federation. World Obesity Atlas 2023. Available online: https://data.worldobesity.org/publications/?cat=19 (accessed on 27 May 2023).
- Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease1-3. Am J Clin Nutr. 2007, 86, 899–906. [CrossRef]
- Pereira RM, Botezelli JD, da Cruz Rodrigues KC, Mekary RA, Cintra DE, Pauli JR, et al. Fructose consumption in the development of obesity and the effects of different protocols of physical exercise on the hepatic metabolism. Nutrients. 2017, 9, 1–21. [CrossRef]
- Helsley RN, Moreau F, Gupta MK, Radulescu A, Debosch B, Softic S. Tissue-Specific Fructose Metabolism in Obesity and Diabetes. Curr Diab Rep. 2020, 20, 1–16. [CrossRef]
- Czerwonogrodzka-Senczyna A, Rumińska M, Majcher A, Credo D, Jeznach-Steinhagen A, Pyrżak B. Fructose Consumption and Lipid Metabolism in Obese Children and Adolescents. Adv Exp Med Biol. 2019, 1153, 91–100. [CrossRef]
- Johnson RJ, Sánchez-lozada LG, Andrews P, Lanaspa MA. Perspective : A Historical and Scienti fi c Perspective of Sugar and Its Relation with Obesity and Diabetes. Adv Nutr. 2017, 8, 412–22. [CrossRef]
- Kwok KH, Lam KS, XU A. Heterogeneity of white adipose tim ssue: molecular basis and clinical implications. Exp Mol Med. 2016, 48, e215. [CrossRef]
- Schoettl T, Fischer IP, Ussar S. Heterogeneity of adipose tissue in development and metabolic function. J Exp Biol. 2018, 221, jeb162958. [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004, 84, 277-359. [CrossRef]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends in Endocrinol Metabol. 2018, 29, 191-200. [CrossRef]
- Clemente-Suárez, V. J. et al. The role of adipokines in health and disease. Biomedicines, 2023, 11, 1290. [CrossRef]
- Song Z, Xiaoli AM, Yang F. Regulation and metabolic significance of De Novo lipogenesis in adipose tissues. Nutrients. 2018, 10,1383. [CrossRef]
- Poissonnet, C. M., Burdi, A. R. & Garn, S. M. The chronology of adipose tissue appearance and distribution in the human fetus. Early Hum. Dev. 1984, 10, 1–11,1. [CrossRef]
- Billon N, Dani C. Developmental origins of the adipocyte lineage: new insights from genetics and genomics studies. Stem Cell Rev Rep. 2012, 8, 55–66. [CrossRef]
- Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019, 20, 242–58. [CrossRef]
- Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011, 12, 722–34. [CrossRef]
- Knittle JL, Timmers K, Ginsberg-Fellner F, Brown RE, Katz DP. The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell number and size. J Clin Invest 1979, 63, 239-46. [CrossRef]
- Cameron M, Demerath EW. Critical Periods in Human Growth and Their Relationship to Diseases of Aging. Am J Phys Anthropol. 2002, 45, 159–184. [CrossRef]
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, N€aslund E, Britton T, et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783-7. [CrossRef]
- Jeffery E, Church CD, Holtrup B, Colman L & Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat. Cell Biol. 2015, 17, 376–385. [CrossRef]
- Schwenk RW, Holloway GP, Luiken JJFP, Bonen A, Glatz JFC. Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins leukot essent fatty acids, 2010, 82, 4-6, 49-54. [CrossRef]
- Bódis, K.; Roden, M. Energy metabolism of white adipose tissue and insulin resistance in humans. Europ J Clin Invest. 2018, 48, 13017. [CrossRef]
- Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [CrossRef]
- Hirsch J, Farquhar JW, Ahrens Jr EH, Peterson ML, Stoffel W. Studies of adipose tissue in man. A microtechnic for sampling and analysis. Am J Clin Nutr. 1960, 8, 499-511. [CrossRef]
- Schaefer-Graf UM, Meitzner K, Ortega-Senovilla H, Graf K, Vetter K, Abou-Dakn M, Herrera E. Differences in the implications of maternal lipids on fetal metabolism and growth between gestacional diabetes mellitus and control pregnancies. Diabet Med. 2011, 28, 1053-9. [CrossRef]
- Fruhbeck G, Mendez-Gimenez L, Fernandez-Formoso JA, Fernandez S, Rodriguez A. Regulation of adipocyte lipolysis. Nutr Res Rev. 2014, 27, 63–93. [CrossRef]
- Bolsoni-Lopes A., Alonso-Vale MI. Lipolysis and lipases in white adipose tissue—An update. Arch. Endocrinol. Metab. 2015, 59, 335–342. [CrossRef]
- Desoye G, Herrera E. Adipose tissue development and lipid metabolism in the human fetus: The 2020 perspective focusing on maternal diabetes and obesity. Prog Lipid Res. 2021, 81, 101082. [CrossRef]
- Marcus C, Ehren H, Bolme P, Arner P. Regulation of lipolysis during the neonatal period: importance of thyrotropin. J. Clin. Invest., 1988, 82. 1793-7. [CrossRef]
- Barreiros RC, Bossolan G, Trindade CEP. Fructose in humans: Metabolic effects, clinical utilization, and associated inherent errors. Rev Nutr. 2005, 18, 377–89. [CrossRef]
- Herman MA, Birnbaum MJ. Molecular aspects of fructose metabolism and metabolic disease. Cell metabolism. 2021, 7, 33, 2329-54. [CrossRef]
- Hallfrisch J. Metabolic effects of dietary fructose.FASEB J. 1990, 4, 2652-60. [CrossRef]
- Mueckler M. Facilitative glucose transporters. Eur J Biochem. 1994, 725, 713–25. [CrossRef]
- Mueckler M, Thorens B. Molecular Aspects of Medicine The SLC2 ( GLUT ) family of membrane transporters. Mol Aspects Med. 2013, 34, 121–38. [CrossRef]
- Truswell S, Thorburn AW. Incomplete absorption of pure fructose in healthy subjects. AM J Clin Nutr. 1988, 48, 1424-1430. [CrossRef]
- Ferraris RP, Choe J, Patel CR. Intestinal Absorption of Fructose. Annu Rev Nutr. 2018, 38, 41–67. [CrossRef]
- Hannou SA, Mckeown NM, Herman MA, Hannou SA, Haslam DE, Mckeown NM, et al. Fructose metabolism and metabolic disease Fructose metabolism and metabolic disease. J Clin Invest. 2018, 128, 545–55. [CrossRef]
- Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, Hayward BE, Asipu A, Bonthron DT. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme, J. Histochem. Cytochem, 2009, 57, 763–774. [CrossRef]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, Rabinowitz JD. The small intestine converts dietary fructose intoglucose and organic acids, Cell Metabol. 2019, 27, 351–361. [CrossRef]
- Bidwell AJ, Chronic fructose ingestion as a major health concern: is a sedentary lifestyle making it worse? A Review, Nutrients, 2017, 9. [CrossRef]
- Duro D, Rising R, Cedillo M, Lifshitz F. Association between infantile colic and carbohydrate malabsorption from fruit juices in infancy, Pediatrics, 2002, 109, 797–805. [CrossRef]
- Gaby AR. Adverse effects of dietary fructose, Alternative Med. Rev., 2005, 10, 294–306.
- Semnani-Azad Z, Khan TA, Mejia SB, de Souza RJ, Leiter LA, Kendall CW, Hanley AJ, Sievenpiper JL. Association of major food sources of fructose-containing sugars with incident metabolic syndrome: a systematic review and meta-analysis. JAMA Netw open, 2020, 3, e209993. [CrossRef]
- Merino B, Fernández-Díaz CM, Cózar-Castellano I, Perdomo G. Intestinal fructose and glucose metabolism in health and disease. Nutrients. 2019, 12, 94. [CrossRef]
- Fidler Mis, N.; Braegger, C.; Bronsky, J.; Campoy, C.; Domellöf, M.; Embleton, N. D.; Hojsak, I.; Hulst, J.; Indrio, F.; Lapillonne, A.; Mihatsch, W.; Molgaard, C.; Vora, R.; Fewtrell, M. Sugar in Infants, Children and Adolescents: A Position Paper of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition Committee on Nutrition. J. Pediatr. Gastroenterol. Nutr. 2017, 65 , 681–696. [CrossRef]
- World Health Organization. Sugars intake for adults and children. Available online: https://www.who.int/publications/i/item/9789241549028 (accessed on 02 May 2023).
- UK’s Scientific Advisory Committee on Nutrition. Carbohydrates and health. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/445503/SACN_Carbohydrates_and_Health.pdf (accessed on 02 May 2023).
- Young J, Scott S, Clark L, Lodge JK. Associations between free sugar intake and markers of health in the UK population: an analysis of the National Diet and Nutrition Survey rolling programme. Br J Nutr, 2022, 128, 225-36. [CrossRef]
- Kmietowicz Z. Countries that use large amounts of high fructose corn syrup have higher rates of type 2 diabetes BMJ 2012, 345, 7994. [CrossRef]
- Bragança, M.L.B.M.; Bogea, E.G.; de Almeida Fonseca Viola, P.C.; dos Santos Vaz, J.; Confortin, S.C.; Menezes, A.M.B.; Gonçalves, H.; Bettiol, H.; Barbieri, M.A.; Cardoso, V.C.; da Silva, A.A.M. High Consumption of Sugar-Sweetened Beverages Is Associated with Low Bone Mineral Density in Young People: The Brazilian Birth Cohort Consortium. Nutrients 2023, 15, 324. [CrossRef]
- Colchero MA, Guerrero-Lopez CM, Molina M, et al. Beverages sales in Mexico before and after Implementation of a sugar sweetened beverage tax. PLoS One. 2016, 11, e0163463. [CrossRef]
- Goodman S, Vanderlee L, Jones A, White C, Hammond D. Perceived healthiness of sweeteners among young adults in Canada. Can J Diet Pract Res. 2021, 82, 90–4. [CrossRef]
- Hock K, Acton RB, Jáuregui A, Vanderlee L, White CM, Hammond D. Experimental study of front-of-package nutrition labels’ efficacy on perceived healthfulness of sugar-sweetened beverages among youth in six countries. Prev Med Reports. 2021, 24, 101577. [CrossRef]
- Mantzari E, Vasiljevic M, Turney I, Pilling M, Marteau T. Impact of warning labels on sugar-sweetened beverages on parental selection: An online experimental study. Prev Med Reports.2018, 259–67. [CrossRef]
- Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease1-3. Am J Clin Nutr. 2007, 86, 899–906. [CrossRef]
- Taskinen MR, Packard CJ, Borén J. Dietary fructose and the metabolic syndrome. Nutrients. 2019, 11, 1–16. [CrossRef]
- Klein AV, Kiat H. The mechanisms underlying fructose-induced hypertension: A review. J Hypertens. 2015, 33, 912–20. [CrossRef]
- Ichigo Y, Takeshita A, Hibino M, Nakagawa T, Hayakawa T, Patel D, et al. High-fructose diet-induced hypertriglyceridemia is associated with enhanced hepatic expression of ACAT2 in Rats. Physiol Res. 2019, 68, 1021–6. [CrossRef]
- DiStefano JK. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. cell mol life sci. 2020, 77, 2079–90. [CrossRef]
- Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. 2018, 128, 545–55. [CrossRef]
- Muriel P, López-sánchez P, Ramos-tovar E. Fructose and the liver. Int J Mol Sci. 2021, 22, 6969. [CrossRef]
- Lê KA, Ith M, Kreis R, Faeh D, Bortolotti M, Tran C, et al. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr. 2009, 89, 1760–5. [CrossRef]
- Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab. 2005, 2, 1–14. [CrossRef]
- Catena C, Giacchetti G, Novello M, Colussi G, Cavarape A, Sechi LA. Cellular Mechanisms of Insulin Resistance in Rats with Fructose-Induced Hypertension. Am J Hypertens. 2003, 16, 973–8. [CrossRef]
- Ueno M, Bezerra RMN, Silva MS, Tavares DQ, Carvalho CR, Saad MJA. A high-fructose diet induces changes in pp185 phosphorylation in muscle and liver of rats. Brazilian J Med Biol Res. 2000, 33, 1421–7. [CrossRef]
- Russo E, Leoncini G, Esposito P, Garibotto G, Pontremoli R, Viazzi F. Fructose and uric acid: Major mediators of cardiovascular disease risk starting at pediatric age. Int J Mol Sci. 2020, 21, 1–13. [CrossRef]
- Spiga R, Marini MA, Mancuso E, Di Fatta C, Fuoco A, Perticone F, et al. Uric Acid Is Associated with Inflammatory Biomarkers and Induces Inflammation Via Activating the NF-κB Signaling Pathway in HepG2 Cells. Arterioscler Thromb Vasc Biol. 2017, 37, 1241–9. [CrossRef]
- Wang Y, Qi W, Song G, Pang S, Peng Z, Li Y, et al. High-fructose diet increases inflammatory cytokines and alters gut microbiota composition in rats. Mediators Inflamm. 2020, 2020, 6672636. [CrossRef]
- Hernández-Díazcouder A, Romero-Nava R, Carbó R, Sánchez-Lozada LG, Sánchez-Muñoz F. High fructose intake and adipogenesis. Int J Mol Sci. 2019, 20, 2787. [CrossRef]
- Jürgens H, Haass W, Castañeda TR, Schürmann A, Koebnick C, Dombrowski F, et al. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes Res. 2005, 13, 1145–56. [CrossRef]
- Pektas MB, Koca HB, Sadi G, Akar F. Dietary Fructose Activates Insulin Signaling and Inflammation in Adipose Tissue: Modulatory Role of Resveratrol. Biomed Res Int. 2016, 2016, 8014252. [CrossRef]
- Yahia H, Hassan A, El-Ansary MR, Al-Shorbagy MY, El-Yamany MF. IL-6/STAT3 and adipokine modulation using tocilizumab in rats with fructose-induced metabolic syndrome. Naunyn Schmiedebergs Arch Pharmacol. 2020, 393, 2279–2292. [CrossRef]
- Miranda CS, Silva-Veiga FM, Santana-Oliveira DA, Fernandes-da-Silva A, Brito GC, Martins FF, Souza-Mello V. Chronic Excessive Fructose Intake Maximizes Brown Adipocyte Whitening but Causes Similar White Adipocyte Hypertrophy Than a High-Fat Diet in C57BL/6 Mice. J Am Nutr Assoc. 2023, 42, 435-444. [CrossRef]
- Santos MP, Cauduro LFR, Ferreira MM, Martucci LF, Vecchiatto B, Vilas-boas EA, et al. Effect of Low-Dose Progesterone on Glycemic Metabolism , Morphology and Function of Adipose Tissue and Pancreatic Islets in Diet-Induced Obese Female Mice. 2023, 28, 312. [CrossRef]
- Crescenzo R, Bianco F, Coppola P, Mazzoli A, Valiante S, Liverini G, et al. Adipose tissue remodeling in rats exhibiting fructose-induced obesity. Eur J Nutr. 2014, 53, 413–9. [CrossRef]
- Zubiría MG, Alzamendi A, Moreno G, Rey MA, Spinedi E, Giovambattista A. Long-term fructose intake increases adipogenic potential: Evidence of direct effects of fructose on adipocyte precursor cells. Nutrients. 2016, 8, 198. [CrossRef]
- London E, Castonguay TW. High fructose diets increase 11β-hydroxysteroid dehydrogenase type 1 in liver and visceral adipose in rats within 24-h exposure. Obesity. 2011, 19, 925–32. [CrossRef]
- Legeza B, Balázs Z, Odermatt A. Fructose promotes the differentiation of 3T3-L1 adipocytes and accelerates lipid metabolism. FEBS Lett. 2014, 588, 490–6. [CrossRef]
- Prince PD, Santander YA, Gerez EM, Höcht C, Polizio AH, Mayer MA, et al. Fructose increases corticosterone production in association with NADPH metabolism alterations in rat epididymal white adipose tissue. J Nutr Biochem. 2017, 46, 109–16. [CrossRef]
- Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta - Mol Basis Dis. 2014, 1842, 473–81. [CrossRef]
- Park Y-K, Ge K. Glucocorticoid Receptor Accelerates, but Is Dispensable for, Adipogenesis. Mol Cell Biol. 2017, 37, e00260-16. [CrossRef]
- Du L, Heaney AP. Regulation of adipose differentiation by fructose and GluT5. Mol Endocrinol. 2012, 26, 1773–82. [CrossRef]
- Meneses MJ, Sousa-Lima I, Jarak I, Raposo JF, Alves MG, Macedo MP. Distinct impacts of fat and fructose on the liver, muscle, and adipose tissue metabolome: An integrated view. Front Endocrinol (Lausanne). 2022, 13, 898471. [CrossRef]
- Li J xiu, Ke D zhi, Yao L, Wang S, Ma P, Liu L, et al. Response of genes involved in lipid metabolism in rat epididymal white adipose tissue to different fasting conditions after long-term fructose consumption. Biochem Biophys Res Commun. 2017, 484, 336–41. [CrossRef]
- Mazzoli A, Porzio A Di, Gatto C, Crescenzo R, Nazzaro M, Spagnuolo MS, et al. Skeletal muscle insulin resistance and adipose tissue hypertrophy persist beyond the reshaping of gut microbiota in young rats fed a fructose-rich diet. J Nutr Biochem . 2023, 113, 109247. [CrossRef]
- Kovačević S, Brkljačić J, Vojnović Milutinović D, Gligorovska L, Bursać B, Elaković I, et al. Fructose Induces Visceral Adipose Tissue Inflammation and Insulin Resistance Even Without Development of Obesity in Adult Female but Not in Male Rats. Front Nutr. 2021, 8, 1–18. [CrossRef]
- Baldwin W, McRae S, Marek G, Wymer D, Pannu V, Baylis C, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes. 2011, 60, 1258–69. [CrossRef]
- Singh S, Sharma A, Guru B, Ahmad S, Gulzar F, Kumar P, et al. Fructose-mediated NLRP3 activation induces inflammation and lipogenesis in adipose tissue. J Nutr Biochem. 2022, 107, 109080. [CrossRef]
- Kuzma JN, Cromer G, Hagman DK, Breymeyer KL, Roth CL, Foster-Schubert KE, et al. No differential effect of beverages sweetened with fructose, high-fructose corn syrup, or glucose on systemic or adipose tissue inflammation in normal-weight to obese adults: A randomized controlled trial. Am J Clin Nutr. 2016, 104, 306–14. [CrossRef]
- Manna P, Jain SK. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab Syndr Relat Disord. 2015, 13, 423–44. [CrossRef]
- Bratoeva K, Radanova M, Merdzhanova A, Donev I. Protective role of S-Adenosylmethionine against fructose-induced oxidative damage in obesity. J Mind Med Sci. 2017, 4, 163–71. [CrossRef]
- Araoye E, Ckless K. Effects of High Fructose/Glucose on Nlrp3/Il1β Inflammatory Pathway. J Young Investig. 2016, 31, 25–30. [CrossRef]
- Gherghina ME, Peride I, Tiglis M, Neagu TP, Niculae A, Checherita IA. Uric Acid and Oxidative Stress—Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int J Mol Sci. 2022, 23, 3188. [CrossRef]
- Baiċc J, Bjelakoviċ G, Pavloviċ D, Kocić G, Jevtoviċ T, Stojanoviċ I, et al. Glucocorticoids and Oxidative Stress. J Basic Clin Physiol Pharmacol. 2007, 18, 115–28. [CrossRef]
- Imhoff BR, Hansen JM. Extracellular redox environments regulate adipocyte differentiation. Differentiation. 2010, 80, 31–9. [CrossRef]
- Han J, Choi HY, Dayem AA, Kim K, Yang G, Won J, et al. Regulation of Adipogenesis Through Differential Modulation of ROS and Kinase Signaling Pathways by 3,4′-Dihydroxyflavone Treatment. J Cell Biochem. 2017, 118, 1065–77. [CrossRef]
- Zorena K, Jachimowicz-Duda O, Ślęzak D, Robakowska M, Mrugacz M. Adipokines and obesity. Potential link to metabolic disorders and chronic complications. Int J Mol Sci. 2020, 21, 3570. [CrossRef]
- Taylor EB. The complex role of adipokines in obesity, inflammation, and autoimmunity. Clin Sci. 2021, 135, 731–52. [CrossRef]
- Maslov LN, Naryzhnaya N V., Boshchenko AA, Popov S V., Ivanov V V., Oeltgen PR. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J Clin Transl Endocrinol. 2019, 15, 1–5. [CrossRef]
- Rodrigues DF, Henriques MC do C, Oliveira MC, Menezes-Garcia Z, Marques PE, Souza D da G, et al. Acute intake of a high-fructose diet alters the balance of adipokine concentrations and induces neutrophil influx in the liver. J Nutr Biochem . 2014, 25, 388–94. [CrossRef]
- Chait A, den Hartigh LJ. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front Cardiovasc Med. 2020, 7, 22. [CrossRef]
- Mendoza-herrera K, Florio AA, Moore M, Marrero A, Tamez M, Bhupathiraju SN, et al. The Leptin System and Diet : A Mini Review of the Current Evidence. 2021, 12, 1–12. [CrossRef]
- Jian-mei LI, Chuang W, Qing-hua HU, Ling-dong K. Fructose Induced Leptin Dysfunction and Improvement by Quercetin and Rutin in Rats. Chinese J Nat Med. 2008, 6, 466-473. [CrossRef]
- Shapiro A, Tu N, Gao Y, Cheng K, Scarpace PJ. Prevention and reversal of diet-induced leptin resistance with a sugar-free diet despite high fat content. Br J Nutr. 2011, 106, 390–7. [CrossRef]
- Shapiro A, Mu W, Roncal C, Cheng K, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. 2008, 32610, 1370–5. [CrossRef]
- Äijälä M,Malo E, Ukkola O, Bloigu R, Lehenkari P, Autio-harmainen H, Santaniemi M, et al. Long-term fructose feeding changes the expression of leptin receptors and autophagy genes in the adipose tissue and liver of male rats : a possible link to elevated triglycerides. 2013, 8, 623-35. [CrossRef]
- Haring SJ, Harris RBS. The relation between dietary fructose , dietary fat and leptin responsiveness in rats. Physiol Behav. 2011, 104, 914–22. [CrossRef]
- Miranda CS, Silva-Veiga F, Martins FF, Rachid TL, Mandarim-De-Lacerda CA, Souza-Mello V. PPAR-α activation counters brown adipose tissue whitening: a comparative study between high-fat– and high-fructose–fed mice. Nutrition. 2020, 78, 110791. [CrossRef]
- Machado TQ, Pereira-Silva DC, Goncalves LF, Fernandes-Santos C. Brown Adipose Tissue Remodeling Precedes Cardiometabolic Abnormalities Independent of Overweight in Fructose-Fed Mice. Integr Diabetes Cardiovasc Dis. 2019, 3, 72–82. [CrossRef]
- Richard G, Blondin DP, Syed SA, Rossi L, Fontes ME, Fortin M, et al. High-fructose feeding suppresses cold-stimulated brown adipose tissue glucose uptake independently of changes in thermogenesis and the gut microbiome. Cell Reports Med. 2022, 3, 100742. [CrossRef]
- Berger PK, Plows JF, Jones RB, et al. Associations of maternal fructose and sugar-sweetened beverage and juice intake during lactation with infant neurodevelopmental outcomes at 24 months. Am J Clin Nutr. 2020, 112, 1516-1522. [CrossRef]
- Larqué E, Labayen I, Flodmark CE, Lissau I, Czernin S, Moreno LA, Pietrobelli A, Widhalm K. From conception to infancy — early risk factors for childhood obesity. In Nature Rev Endocrinol. 2019, 15, 456-478. [CrossRef]
- Shaban Mohamed, M. A., AbouKhatwa, M. M., Saifullah, A. A., Hareez Syahmi, M., Mosaad, M., Elrggal, M. E., Dehele, I. S., Elnaem, M. H. Risk Factors, Clinical Consequences, Prevention, and Treatment of Childhood Obesity. In Children, 2022, 9. [CrossRef]
- Drozdz D., Alvarez-Pitti J., Wójcik M., Borghi C., Gabbianelli R., Mazur A., Herceg-čavrak V., Lopez-Valcarcel BG., Brzeziński M., Lurbe E., Wühl E. Obesity and cardiometabolic risk factors: From childhood to adulthood. In Nutrients. 2021, 13. [CrossRef]
- Avelar Rodriguez D., Toro Monjaraz EM., Ignorosa Arellano KR., Ramirez Mayans J. Childhood obesity in Mexico: Social determinants of health and other risk factors. BMJ Case Rep. 2018, bcr2017223862. [CrossRef]
- Lee EY., Yoon KH. Epidemic obesity in children and adolescents: risk factors and prevention. Front Med. 2018, 12, 658–666. [CrossRef]
- Williams CB., MacKenzie KC., Gahagan,S. The effect of maternal obesity on the offspring. Clin Obstet Gynecol. 2014, 57, 508–515. [CrossRef]
- Lakshman R., Elks CE., Ong KK. Childhood obesity. Circulation, 2012, 126, 1770–1779. [CrossRef]
- Mahumud RA., Sahle BW., Owusu-Addo E. et al. Association of dietary intake, physical activity, and sedentary behaviours with overweight and obesity among 282,213 adolescents in 89 low and middle income to high-income countries. Int J Obes. 2021, 45, 2404–2418. [CrossRef]
- Mittal, M., Jain, V. Management of Obesity and Its Complications in Children and Adolescents. In Indian Journal of Pediatrics, 2021, 88, 1222–1234. [CrossRef]
- Hemmingsson E. Early Childhood Obesity Risk Factors: Socioeconomic Adversity, Family Dysfunction, Offspring Distress, and Junk Food Self-Medication. In Curr Obes Reps, 2018, 7, 204–209. [CrossRef]
- Kostovski M., Tasic V., Laban N., Polenakovic M., Danilovski D., Gucev, Z. Obesity in childhood and adolescence, genetic factors. Pril (Makedonska Akademija Na Naukite i Umetnostite. Oddelenie Za Medicinski Nauki). 2017, 34, 85–89. [CrossRef]
- Holmberg NG, Kaplan B, Karvonen MJ, Lind J, Malm. M. Permeability of Human Placenta to Glucose, Fructose, and Xylose. Acta Physiol Scand. 1956, 36, 291–9. [CrossRef]
- Lintao RCV, Kammala AK, Vora N, Yaklic JL, Menon R. Fetal membranes exhibit similar nutrient transporter expression profiles to the placenta. Placenta. 2023, 135, 33–42. [CrossRef]
- Magenis ML, Damiani AP, de Bem Silveira G, Dagostin LS, de Marcos PS, de Souza E, de Roch Casagrande L, Longaretti LM, Silveira PC, de Andrade VM. Metabolic programming in offspring of mice fed fructose during pregnancy and lactation. Journal of Developmental Origins of Health and Disease. 2022, 13, 441-54. [CrossRef]
- Koo S, Kim M, Cho HM, Kim I. Maternal high-fructose intake during pregnancy and lactation induces metabolic syndrome in adult offspring. Nutrition Research and Practice. 2021, 15, 160-72. [CrossRef]
- Jia G, Hill MA, Sowers JR. Maternal exposure to high fructose and offspring health. Hypertension. 2019, 74, 499-501. [CrossRef]
- Wang P, Wu T, Fu Q, Liao Q, Li Y, Huang T, et al. Maternal High-Fructose Intake Activates Myogenic Program in Fetal Brown Fat and Predisposes Offspring to Diet-Induced Metabolic Dysfunctions in Adulthood. 2022, 9, 848983. [CrossRef]
- Englund-Ögge L, Brantsæter AL, Haugen M, Sengpiel V, Khatibi A, Myhre R, Myking S, Meltzer HM, Kacerovsky M, Nilsen RM, Jacobsson B. Association between intake of artificially sweetened and sugar-sweetened beverages and preterm delivery: a large prospective cohort study. The American journal of clinical nutrition. 2012, 96, 552-9. [CrossRef]
- Zhang H, Li X, Niu Y, et al. Fasting serum fructose is associated with risk of gestational diabetes mellitus. Bmc pregnancy and childbirth. 2022, 22, 446. [CrossRef]
- Wright LS, Rifas-Shiman SL, Oken E, Litonjua AA, Gold DR. Prenatal and Early Life Fructose, Fructose-Containing Beverages, and Mid childhood Asthma. Ann Am Thorac Soc. 2018, 15, 217-224. [CrossRef]
- Cohen JFW, Rifas-Shiman SL, Young J, Oken E. Associations of prenatal and child sugar intake with child cognition. Am J Prev Med. 2018, 54, 727-735. [CrossRef]
- Koski KG, Fergusson MA. Amniotic fluid composition responds to changes in maternal dietary carbohydrate and is related to metabolic status in term fetal rats. J Nutr. 1992, 122, 385–92. [CrossRef]
- Berger PK, Fields DA, Demerath EW, Fujiwara H, Goran MI. High-fructose corn syrup-sweetened beverage intake increases 5-hour breast milk fructose concentrations in lactating women. Nutrients. 2018, 10, 669. [CrossRef]
- Smith EVL, Dyson RM, Berry MJ, Gray C. Fructose Consumption During Pregnancy Influences Milk Lipid Composition and Offspring Lipid Profiles in Guinea Pigs. Front Endocrinol (Lausanne). 2020, 11, 550. [CrossRef]
- Goran MI, Martin AA, Alderete TL, Fujiwara H, Fields DA. Fructose in Breast Milk Is Positively Associated with Infant Body Composition at 6 Months of Age. Nutrients. 2017, 9, 146. [CrossRef]
- Berger PK, Plows JF, Jones RB, et al. Associations of maternal fructose and sugar-sweetened beverage and juice intake during lactation with infant neurodevelopmental outcomes at 24 months. Am J Clin Nutr. 2020, 112, 1516-1522. [CrossRef]
- Jones RB, Berger PK, Plows JF, Alderete TL, Millstein J, Fogel J, et al. Lactose-reduced infant formula with added corn syrup solids is associated with a distinct gut microbiota in Hispanic infants. Gut Microbes. 2020, 12. [CrossRef]
- Bode L. Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology. 2012, 22, 1147–62. [CrossRef]
- Giussani M, Lieti G, Orlando A, Parati G, Genovesi S. Fructose Intake, Hypertension and Cardiometabolic Risk Factors in Children and Adolescents: From Pathophysiology to Clinical Aspects. A Narrative Review. Front Med. 2022, 9, 1–19. [CrossRef]
- Febbraio MA, Karin M. “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 2021, 33, 2316–28. [CrossRef]
- Ranjit N, Evans MH, Byrd-Williams C, Evans AE, Hoelscher DM. Dietary and activity correlates of sugar-sweetened beverage consumption among adolescents. Pediatrics. 2010, 126, e754-e761. [CrossRef]
- Berkey CS, Rockett HRH, Field AE, Gillman MW, Colditz GA. Sugar-added beverages and adolescent weight change. Obes Res. 2004, 12, 778–88. [CrossRef]
Figure 1.
Consequences of high fructose intake for hepatic metabolism. After meal, the fructose absorbed and not metabolized in the intestine is directed to liver through portal vein and enter the hepatocyte via GLUT2, to then be phosphorylated to fructose-1-P by ketohexokinase. High fructose phosphorylation rates may cause ATP depletion, leading to purine degradation, uric acid formation and its release to systemic circulation. Cleavage of fructose-1-P forms dihydroxyacetone-P and glyceraldehyde that are then converted to glyceraldehyde-3-P, which participates in glycolytic process to form pyruvate. In mitochondria, pyruvate is converted to acetyl-CoA that enters tricarboxylic acid cycle (TCA cycle), resulting in ATP generation in mitochondrial electron transport chain. However, after intense fructose oxidation, the citrate produced in the mitochondria can be displaced into fatty acid synthesis. Citrate exits mitochondria and is reconverted to acetyl-CoA. In the cytoplasm, acetyl-CoA is converted to malonyl-CoA that ultimately forms triglyceride and VLDL (very low-density lipoprotein) in endoplasmic reticulum. VLDL is exported to systemic circulation. Triglyceride esterification is accentuated by dihydroxyacetone-P conversion to glycerol-3-P.
Figure 1.
Consequences of high fructose intake for hepatic metabolism. After meal, the fructose absorbed and not metabolized in the intestine is directed to liver through portal vein and enter the hepatocyte via GLUT2, to then be phosphorylated to fructose-1-P by ketohexokinase. High fructose phosphorylation rates may cause ATP depletion, leading to purine degradation, uric acid formation and its release to systemic circulation. Cleavage of fructose-1-P forms dihydroxyacetone-P and glyceraldehyde that are then converted to glyceraldehyde-3-P, which participates in glycolytic process to form pyruvate. In mitochondria, pyruvate is converted to acetyl-CoA that enters tricarboxylic acid cycle (TCA cycle), resulting in ATP generation in mitochondrial electron transport chain. However, after intense fructose oxidation, the citrate produced in the mitochondria can be displaced into fatty acid synthesis. Citrate exits mitochondria and is reconverted to acetyl-CoA. In the cytoplasm, acetyl-CoA is converted to malonyl-CoA that ultimately forms triglyceride and VLDL (very low-density lipoprotein) in endoplasmic reticulum. VLDL is exported to systemic circulation. Triglyceride esterification is accentuated by dihydroxyacetone-P conversion to glycerol-3-P.
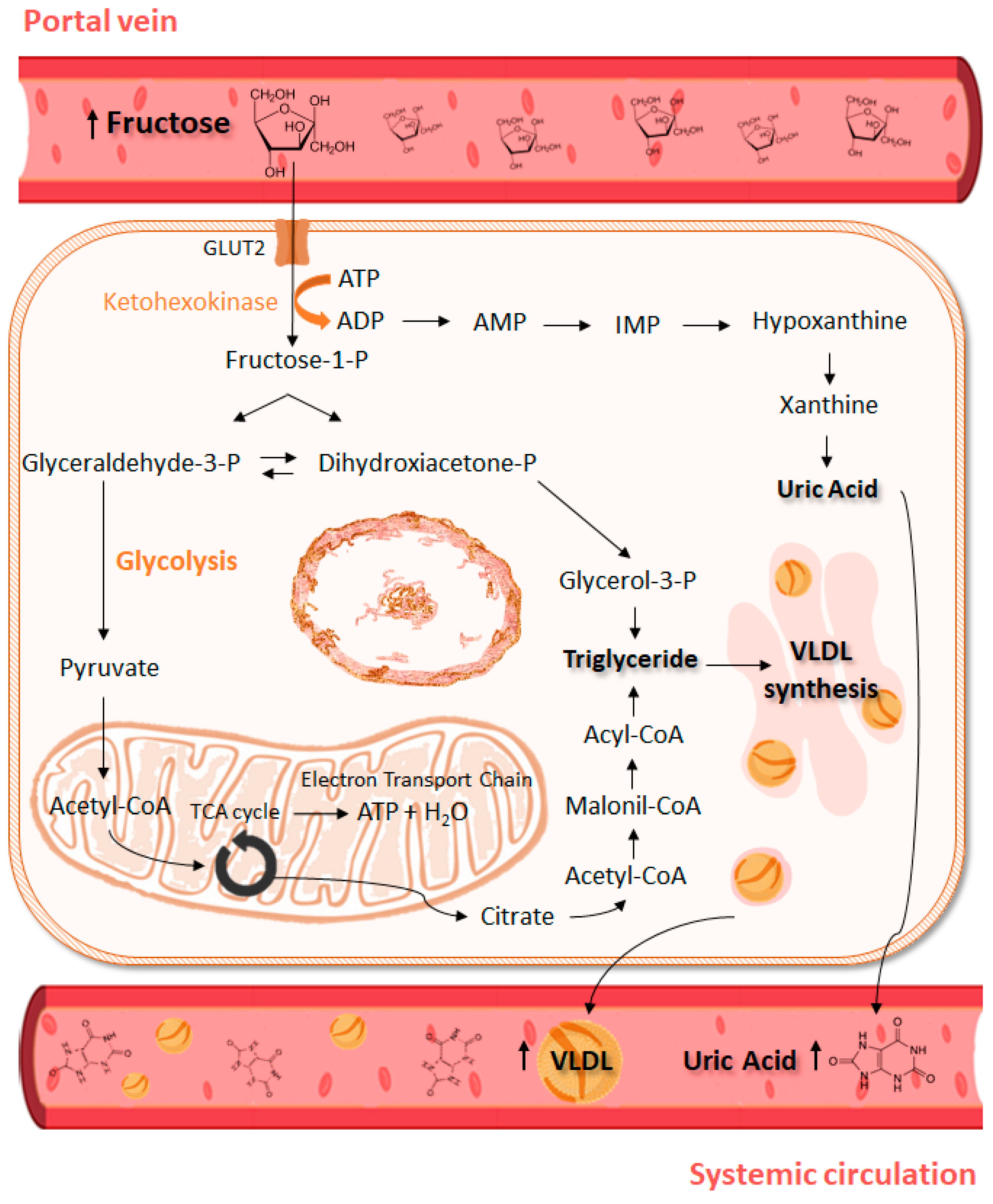
Figure 2.
Effects of excessive fructose intake on different organs and tissues. High fructose intake affects metabolic process through several pathways in organs such as hypothalamus, skeletal muscle, liver, BAT and WAT, contributing to an obesity phenotype and metabolic syndrome development. BAT, brown adipose tissue; WAT, white adipose tissue; UCP1, uncoupling protein 1; VLDL, very low-density lipoprotein.
Figure 2.
Effects of excessive fructose intake on different organs and tissues. High fructose intake affects metabolic process through several pathways in organs such as hypothalamus, skeletal muscle, liver, BAT and WAT, contributing to an obesity phenotype and metabolic syndrome development. BAT, brown adipose tissue; WAT, white adipose tissue; UCP1, uncoupling protein 1; VLDL, very low-density lipoprotein.
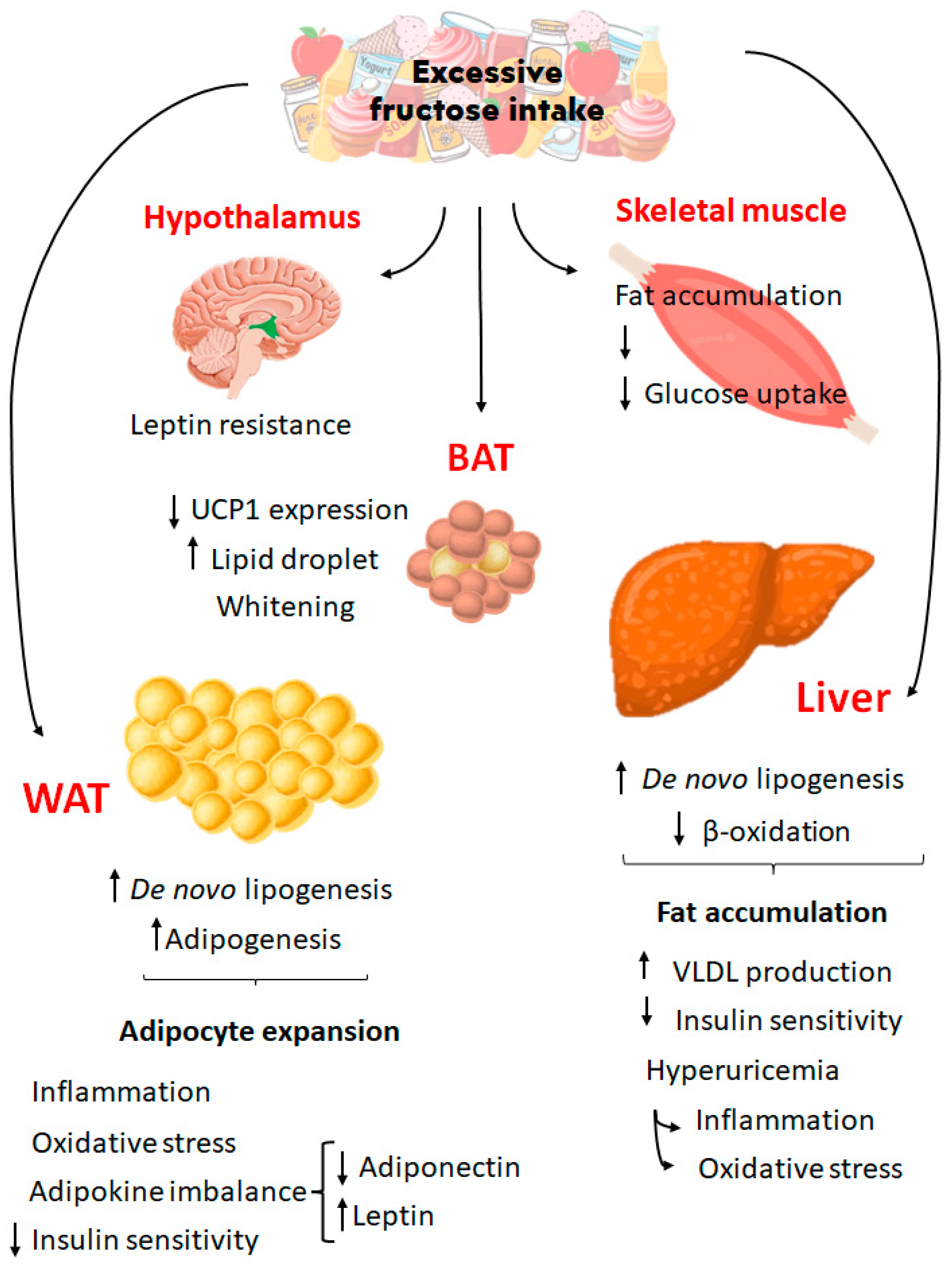
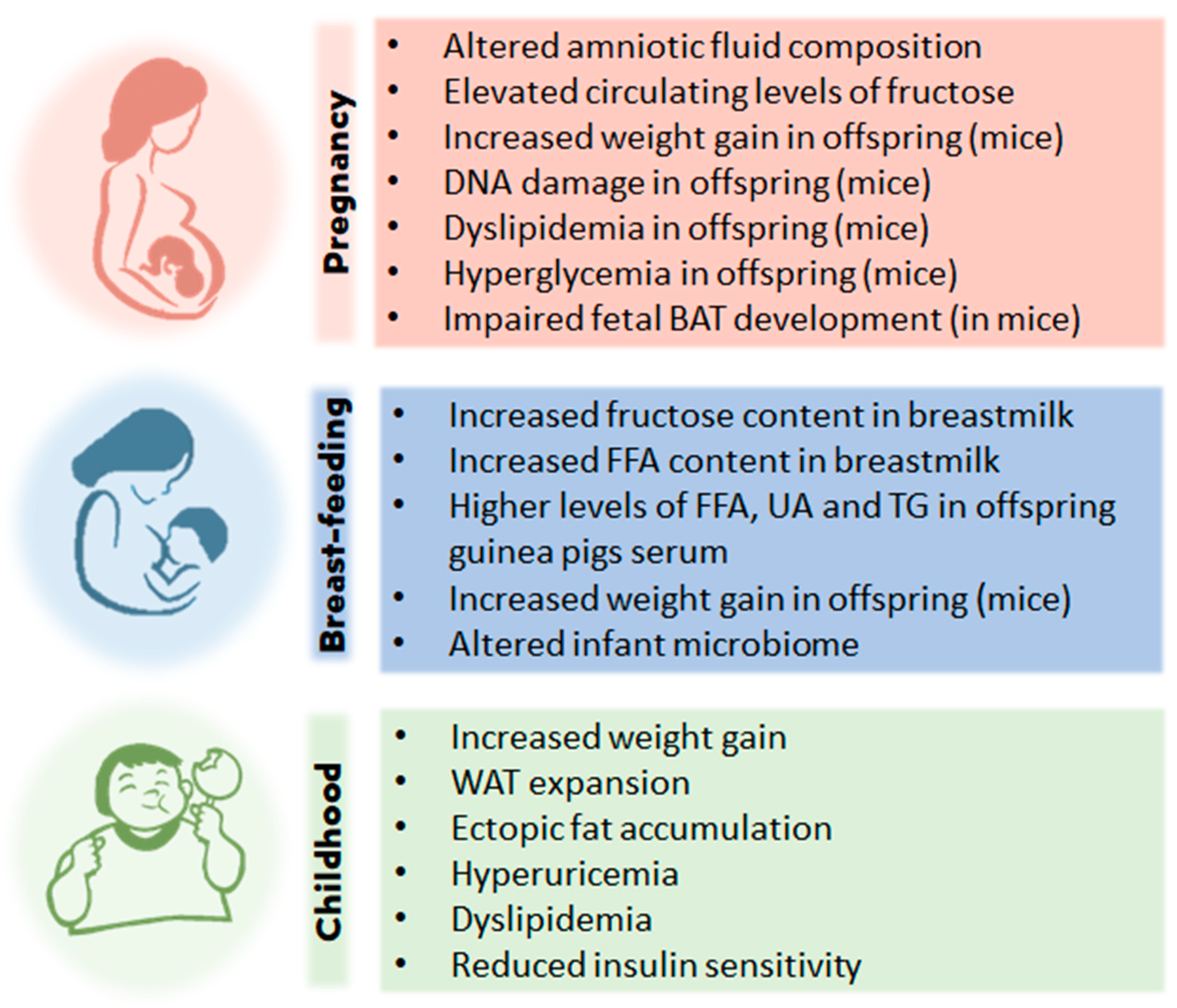
Figure 3.
Effects of high fructose intake and childhood obesity. Studies with humans and other animal models support the hypothesis that at different stages of life, from the fetal stage to childhood, excessive consumption of fructose can cause effects that will lead to overweight and/or childhood obesity. BAT, brown adipose tissue; FFA, free fatty acids; UA, uric acid; TG, triglyceride; WAT, white adipose tissue.
Figure 3.
Effects of high fructose intake and childhood obesity. Studies with humans and other animal models support the hypothesis that at different stages of life, from the fetal stage to childhood, excessive consumption of fructose can cause effects that will lead to overweight and/or childhood obesity. BAT, brown adipose tissue; FFA, free fatty acids; UA, uric acid; TG, triglyceride; WAT, white adipose tissue.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



