
Submitted:
11 December 2023
Posted:
12 December 2023
You are already at the latest version
Abstract
Alectinib hydrochloride is an anticancer medication used for the first-line treatment of non-small cell lung cancer. Although it has been approved for medical use ten years ago, and three polymorphs of this substance were proposed based on X-ray diffraction patterns, their crystal structures remained unknown to date. The main problem was preparation of high quality single crystals due to very low solubility of the salt. Herein we report on molecular and crystal structure of form I of alectinib hydrochloride as obtained by powder X-ray diffraction data from laboratory source. The refinement revealed that the nitrogen atom of the morpholinyl ring is protonated. The chloride anion interacts with the H(N) atoms of the morpholine and benzo[b]carbazole rings to form infinite H-bonded chains.
Keywords:
active pharmaceutical ingredient
; alectinib
; half uncertainty window
; polymorphism
; powder X-ray diffraction
1. Introduction
Alectinib (brand name Alecensa, Drug Bank No DB11363) is an inhibitor of activity of anaplastic lymphoma kinase [1]. In 2014, it was approved for treatment of non-small cell lung cancer in Japan; and now it is worldwide used. It has the systematic name 9-ethyl-6,6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile. Its’ solid form used in medications is alectinib hydrochloride for which three solid forms were detected based on powder X-ray diffraction (XRD) data [2], although none of these patterns was indexed and corresponding molecular and crystal structures remained unknown. Unfortunately, this it typical for significant fraction of pharmaceuticals despite the need of pharm industry in high-quality reference powder patterns for phase identification and purity control. However, recent progress in algorithms for structure solution and refinement from powder XRD data significantly increased the number of crystal structures of multi-component API solids obtained from powder XRD (see, for example, besifloxacin hydrochloride [3] and butenafine hydrochloride [4]).
Within our research study of crystal and molecular structures of solid forms of active pharmaceutical ingredients [5,6,7,8,9] powder pattern of alectinib hydrochloride was measured using CuKα radiation. Room temperature was used to overcome effects of thermal contraction and to escape possible phase transitions. It corresponded to type I crystals of alectinib hydrochloride (1) previously patented [2]. The pattern was indexed, and crystal structure of 1 was solved and refined. The two-dimensional molecular diagram of this compound is shown in Scheme 1.
2. Results and Discussion
A sample of alectinib hydrochloride was purchased from Cdymax and used without purification and recrystallization. The sample was characterized using powder X-ray diffraction, FTIR, 1H, 13C and 13C-1H HSQC NMR spectra (see Figures S1 – S5, Supporting Information).
Powder pattern was obtained at r.t., thus, it can be used in industry for phase identification (Figure S5). It was indexed using Topas 5.0 package [10] that indicates sample purity. The systematic absences suggested the space group P21/c, which was confirmed by successful solution and refinement of the structure. A simulated annealing algorithm of the Topas 5.0 was applied to find positions of non-hydrogen atoms of alectinib and an independent chlorine atom in an asymmetric unit. The Rietveld refinement was carried out to refine coordinates of all atoms [11]. The asymmetric unit of 1 is depicted on Figure 1. It contains one alectinib molecule, and an anion. Morpholine and piperidine rings are in the chair conformation. The condensed cycles are nearly coplanar; average deviation of atoms with an exception of (CH3)2C fragment is equal to 0.11(8) Å. The C15 atom of (CH3)2C is situated 0.197(6) Å above the plane of its’ six-membered ring. Although the positions of hydrogen atoms can not be located during refinement of powder XRD data, the most likely system of H-bonds corresponds to location of the additional H(N) proton on the nitrogen atom of the morpholinyl cycle.
Both acidic H(N) atoms of the five-membered cycle and morpholinyl cycle take part in N–H⋯Cl interactions. Parameters of H-bonds are listed in Table 1. As both cation and anion take part in two hydrogen bonds, infinite H-bonded chains are observed in solid 1. The chains are depicted on Figure 2. Patent [2] contains information about different polymorphs of alectinib hydrochloride, thus, it is of interest to reveal if type I crystals correspond to a stable or metastable polymorph. Crystal structures of other polymorphs remain unknown yet; however, the H-bond propensity tool can be used to estimate if the most likely hydrogen bonds are present in a solid [12]. Within this method it is assumed that the most stable polymorph contains also the most likely hydrogen bonds. Stable polymorphs are characterized by points in the right bottom corner of the H-bond Coordination / H-bond Propensity Plot. In accord with such calculations, the experimentally observed N–H⋯Cl interactions are more likely than any of theoretically possible N–H⋯O or N–H⋯N bonds. All donors of H-bonds take part in H-bonding; and the solid is characterized by the point in the right bottom corner of the Plot (Figure 3). Thus, based on this approach, the system of H-bonds in 1 is the most likely, and the structure is expected to be the most stable polymorph.
Rietveld plot of 1 depicted on Figure S5 and convergence factors demonstrate that some experimental data are insufficiently described by the model obtained. This is probably effect of preferred orientation. Thus, we used HUW (half uncertainty window [13]) parameter in order to estimate quality of our model. HUW parameter identifies the range where the restraints in the model can be varied without the refined bond lengths deviating from the target values in a statistically significant way. For highly crystalline powders and synchrotron data HUW can be lower than 0.05 Å, while unacceptable models are characterized by HUW > 0.3 Å. For our model and data, HUW = 0.082 Å. This is indicative of good refinement.
To sum up, the first crystal and molecular structure of a solid containing alectinib was obtained. It corresponds to type I crystals patented in 2017. The model was obtained by the Rietveld refinement of powder XRD data taken from laboratory diffractometer at room temperature. Nevertheless, HUW parameter indicates good quality of refinement.
3. Materials and Methods
Fine powder of 1 was obtained from Cdymax Pharmaceuticals Co Ltd (Jiangsu, China) and used without further purification. NMR spectra in dimethyl sulfoxide solution were obtained for 1H at 400 MHz, for 13C at 100 MHz using Bruker AVANCE III WB 400 spectrometer (Bruker, Billerica, MA, USA). FTIR spectrum was recorded on an IR spectrometer with a Fourier transformer Shimadzu IRTracer100 (Kyoto, Japan) in the range of 4000–600 cm−1 at a resolution of 1 cm−1 (Nujol mull, KBr pellets). The powder XRD data were recorded at Bruker D8 Advance diffractometer (Bruker, Billerica, MA, USA) equipped a LynxEye detector and Ge(111) monochromator in a transmission mode. CuKα radiation with a wavelength of 1.544493 Å was used. The 2θ range was 3.50–90.0° with a step size of 0.1431°.
3.1. X-Ray diffraction
The indexing of the powder pattern and the subsequent structure solution were performed with the Topas 5.0 software [10]. The pattern was indexed in the P-centred monoclinic unit cell. The systematic absences suggested the space group P21/c, which was confirmed by successful solution and refinement of the structure. A model of alectinib was taken from PubChem [14]. A simulated annealing algorithm of the Topas 5.0 was applied to find positions of non-hydrogen atoms of alectinib and an independent chlorine atom in an asymmetric unit. The solution result was used as a starting geometry for the periodic DFT calculations at PBE exchange–correlation functional level with fixed unit cell using VASP 5.4.1 [15,16,17]. Atomic cores were described using PAW potentials [18,19]. Valence electrons were described in terms of a plane-wave basis set.
Optimization result with the fixed unit cell was used as the starting geometry and the sources of bond and angle restraints in the Rietveld refinement. Anisotropic displacement parameters were refined equal for all of carbon atoms, all nitrogen, al oxygen, all hydrogen atoms and a chlorine atom. The positions of the hydrogen atoms were calculated geometrically and refined in the riding model. The Rietveld plot recorded is given on Figure S5 as Supplementary Material.
Crystal Data for C30H35ClN4O2 (M = 519.06 g/mol): monoclinic, space group P21/n (no. 14), a = 20.3941(9), b = 10.4451(4), c = 12.6842(5) Å, α = 90, β = 93.113(1), γ = 90°, V = 2698.0(2) Å3, Z = 4, µ = 1.278 mm–1, Dcalc = 1.278 g cm-3, F(000) = 276, 3.5° ≤ 2Θ ≤ 90.0°range was used in all calculations. The final Rp/RWP/Rwp’/RBragg/GOF were 7.25/9.95/1.37/6.38/7.24.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, NMR and FTIR spectra, Rietveld plot, crystallographic data in Crystallographic Information File (CIF) format.
Author Contributions
Conceptualization, A.A.K.; methodology, P.A.B.; investigation, R.A.N. and P.A.B.; writing—A.A.K. and A.V.V.; funding acquisition, A.V.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 23-73-00027.
Data Availability Statement
The X-ray data are available at CCDC under ref. code CCDC 2310527.
Acknowledgments
Ministry of Science and Higher Education of the Russian Federation is acknowledged for providing access to scientific literature. NMR experiments were performed using NMR facility at the Department of Structural Studies of N. D. Zelinsky Institute of Organic Chemistry of the RAS, Moscow.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McKeage, K. Alectinib: A Review of Its Use in Advanced ALK-Rearranged Non-Small Cell Lung Cancer. Drugs 2015, 75, 75–82. [CrossRef]
- Tanaka, K.; Ueto, T. Crystal of Tetracyclic Compound 2017. Patent US9714229B2. https://patents.google.com/patent/US9714229B2/en?oq=alectinib++WO2015163447 (accessed on 29 November, 2023).
- Kaduk, J.A.; Gates-Rector, S.; Blanton, T.N. Crystal Structure of Besifloxacin Hydrochloride, C19H22ClFN3O3Cl. Powder Diffr. 2023, 38, 43–52. [CrossRef]
- Kaduk, J.A.; Gates-Rector, S.; Blanton, T.N. Crystal Structure of Butenafine Hydrochloride, C23H28NCl. Powder Diffr. 2023, 38, 30–36. [CrossRef]
- Goloveshkin, A.S.; Korlyukov, A.A.; Vologzhanina, A.V. Novel Polymorph of Favipiravir—An Antiviral Medication. Pharmaceutics 2021, 13, 139. [CrossRef]
- Korlyukov, A.A.; Buikin, P.A.; Dorovatovskii, P.V.; Vologzhanina, A.V. Synthesis, NoSpherA2 Refinement, and Noncovalent Bonding of Abiraterone Bromide Monohydrate. Struct. Chem. 2023, 34, 1927–1934. [CrossRef]
- Buikin, P.; Vologzhanina, A.; Novikov, R.; Dorovatovskii, P.; Korlyukov, A. Abiraterone Acetate Complexes with Biometals: Synthesis, Characterization in Solid and Solution, and the Nature of Chemical Bonding. Pharmaceutics 2023, 15, 2180. [CrossRef]
- Korlyukov, A.A.; Dorovatovskii, P.V.; Vologzhanina, A.V. N-(4-Methyl-3-((4-(Pyridin-3-Yl)Pyrimidin-2-Yl)Amino)Phenyl)-4-((4-Methylpiperazin-1-Yl)Methyl)Benzamide. Molbank 2022, 2022, M1461. [CrossRef]
- Volodin, A.D.; Vologzhanina, A.V.; Peresypkina, E.V.; Korlyukov, A.A. Conformational Polymorphism of Solidum Salt of Elsulfavirin. J. Struct. Chem. 2024, 65, 123238. [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Cryst. 2018, 51, 210–218. [CrossRef]
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Cryst. 1969, 2, 65–71. [CrossRef]
- Galek, P.T.A.; Allen, F.H.; Fábián, L.; Feeder, N. Knowledge-Based H-Bond Prediction to Aid Experimental Polymorph Screening. CrystEngComm 2009, 11, 2634–2639. [CrossRef]
- Dmitrienko, A.O.; Bushmarinov, I.S. Reliable Structural Data from Rietveld Refinements via Restraint Consistency. J. Appl. Cryst. 2015, 48, 1777–1784. [CrossRef]
- PubChem Alectinib Available online: https://pubchem.ncbi.nlm.nih.gov/compound/49806720 (accessed on 29 November 2023).
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal--Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Compt. Mat. Sci. 1996, 6, 15–50. [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [CrossRef]
Scheme 1.
Schematic representation of alectinib hydrochloride.
Figure 1.
Asymmetric unit of 1.
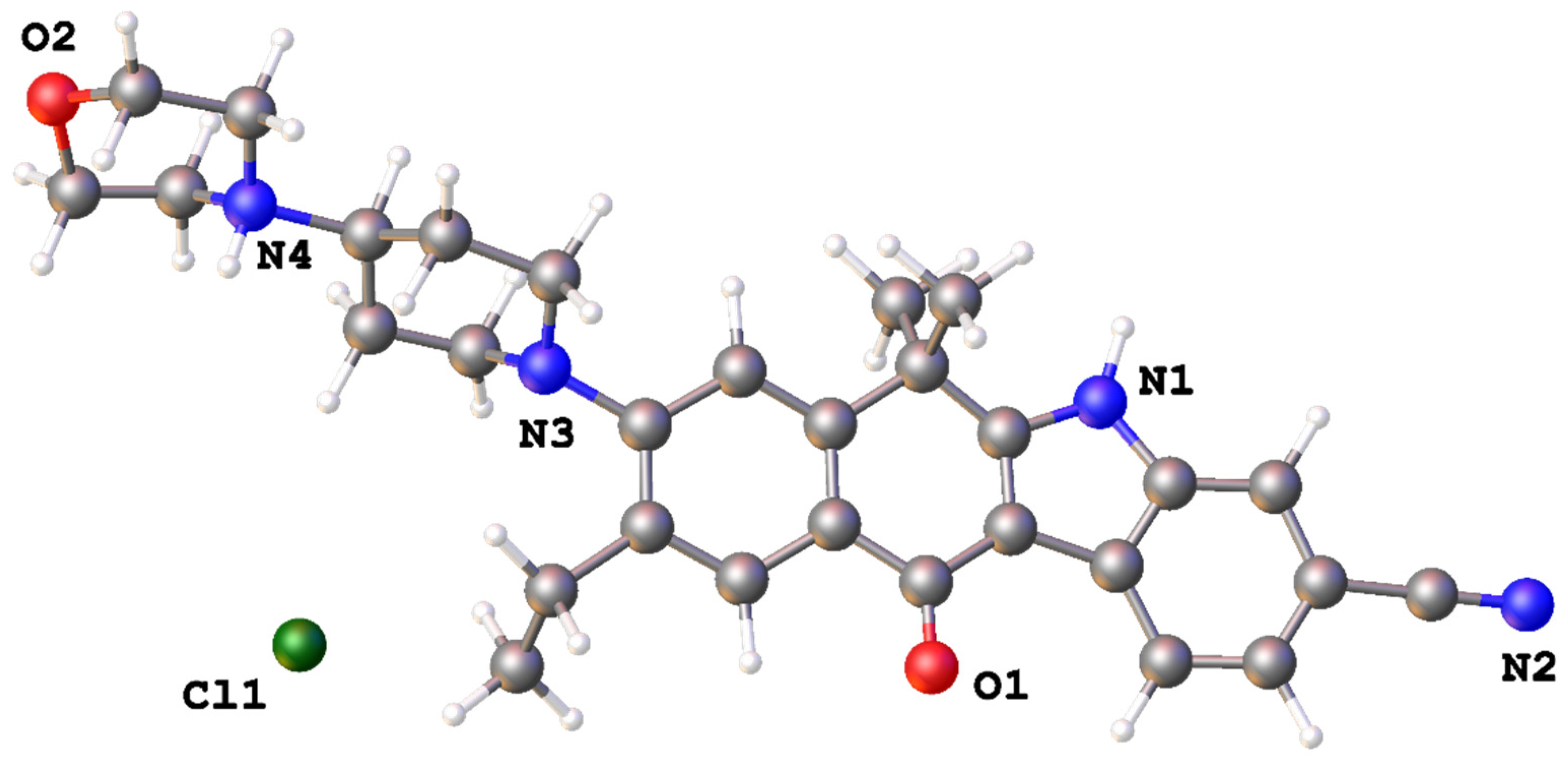
Figure 2.
H-bonded dimers in 1.
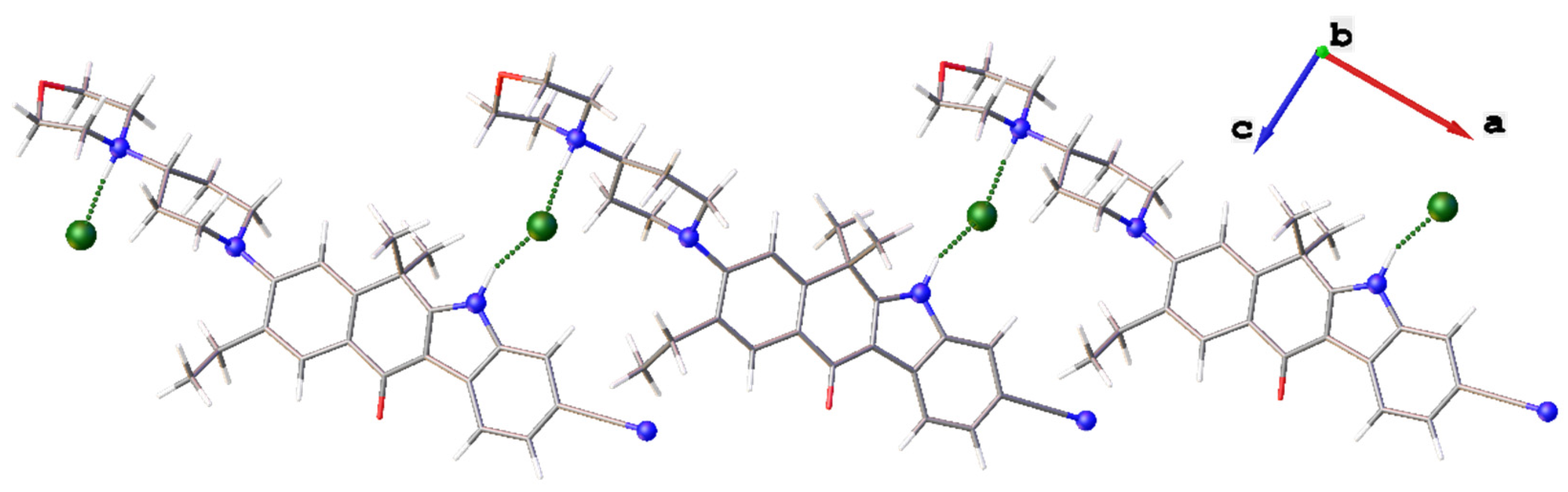
Figure 3.
H-bond Propensity/Coordination plot for theoretically possible systems of H-bonds in 1. Fuchsia circle denotes experimentally observed data.
Figure 3.
H-bond Propensity/Coordination plot for theoretically possible systems of H-bonds in 1. Fuchsia circle denotes experimentally observed data.
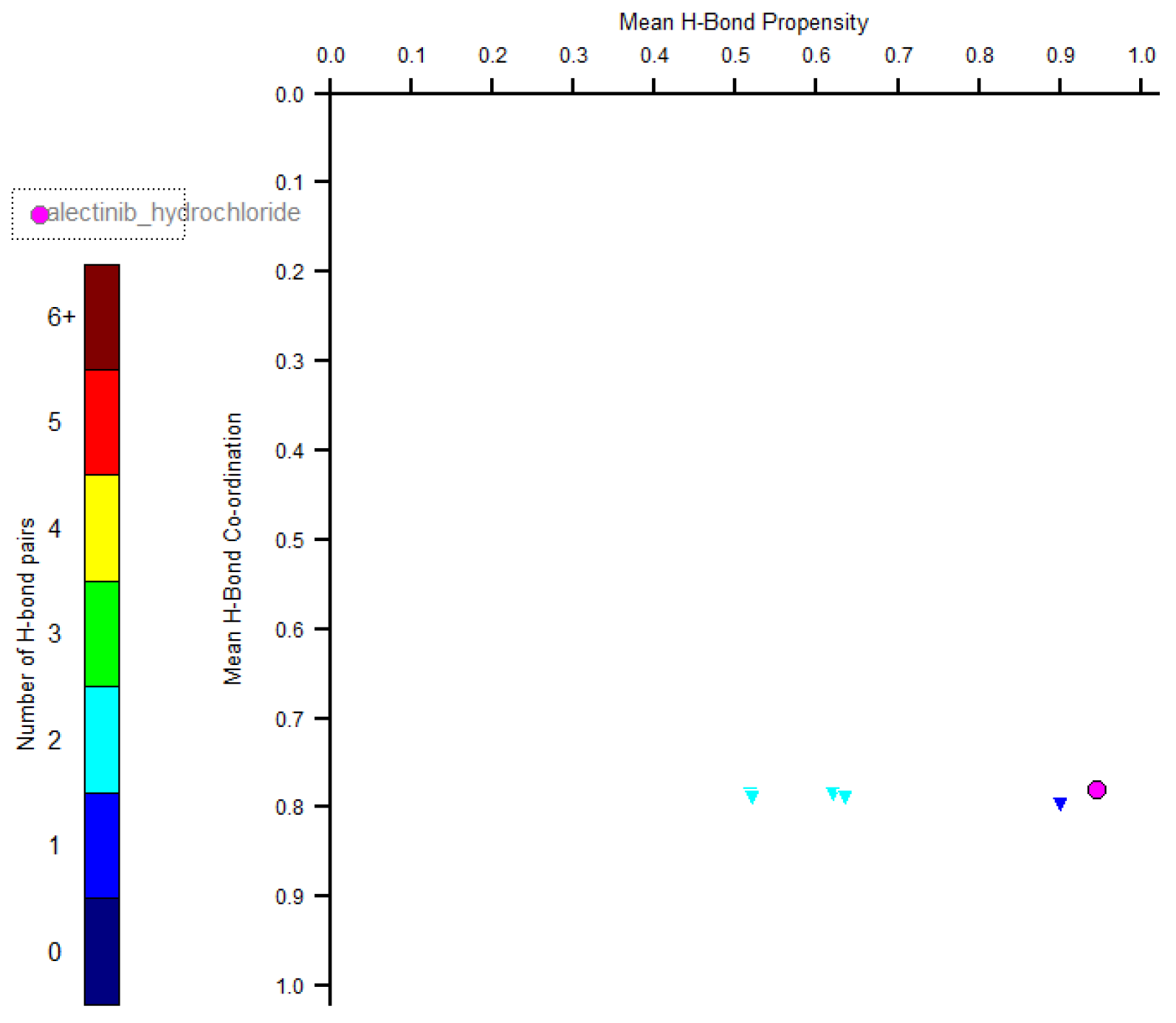

Table 1.
Parameters of H-bonds in 1 (Å, °).
| D–H…A | D–H, Å | H…A, Å | D…A, Å | ∠ (DHA), ° |
| N1–H12⋯Cl1i | 1.04 | 2.28 | 3.224(12) | 149 |
| N4–H8⋯Cl1ii | 0.91 | 2.03 | 2.929(9) | 167 |
Symmetry codes: (i) 1 – x, 1 – y, 1 – z; (ii) 1/2 – x, 1/2 + y, 3/2 – z.
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



