
Submitted:
11 December 2023
Posted:
12 December 2023
You are already at the latest version
Abstract
In the rapidly evolving landscape of genetic engineering, the advent of CRISPR-Cas technologies has catalyzed a paradigm shift, empowering scientists to manipulate the genetic code with unprece-dented accuracy and efficiency. Despite the remarkable capabilities inherent to CRISPR-Cas sys-tems, recent advancements have witnessed the integration of small molecule to augment their functionality, introducing new dimensions to the precision and versatility of gene editing applica-tions. This review delves into the synergy between CRISPR-Cas technologies and small molecule drugs, elucidating the pivotal role of chemicals in optimizing target specificity and editing efficiency. By examining a diverse array of applications, ranging from therapeutic interventions to agricultural advancements, we explore how the judicious use of chemicals enhances the precision of CRISPR-Cas-mediated genetic modifications. In this review we emphasize the significance of small molecule drugs in fine-tuning the CRISPR-Cas machinery, which allows researchers to exert meticulous con-trol over the editing process. We delve into the mechanisms through which these chemicals bolster target specificity, mitigate off-target effects, and contribute to the overall refinement of gene edit-ing outcomes. Additionally, we discuss the potential of chemical integration in expanding the scope of CRISPR-Cas technologies, enabling tailored solutions for diverse genetic manipulation challenges. As CRISPR-Cas technologies continue to evolve, the integration of small molecule drugs emerges as a crucial avenue for advancing the precision and applicability of gene editing techniques. This re-view not only synthesizes current knowledge but also highlights future prospects, paving the way for a deeper understanding of the synergistic interplay between CRISPR-Cas systems and chemical modulators in the pursuit of more controlled and efficient genetic modifications.
Keywords:
Cas9
; Small Molecules
; Genome Editing
Introduction
In bacteria and archaea, an important part of their immune system are the clustered regulatory interspaced short palindromic repeats (CRISPRs) [1]. These are utilized to protect the host organism from invading viruses and plasmids. Within this system, nucleic acids of intruders are silenced by specific small ribonucleic acids (RNAs) originating from the host organism itself [2]. Over recent years, scientific advancements have transformed this system into a practical tool for (epi)genome editing, organismal studies, and the exploration and combat of diseases, particularly hereditary diseases [3]. Playing a pivotal role in the CRISPR system are the CRISPR-associated proteins (Cas) [4]. Among them, Cas9 is the most widely used [5]. It functions as an endonuclease, capable of recognizing specific double-stranded DNA (dsDNA) and, in turn, silencing it through cleavage [2]. Moreover, in scientific applications, after dsDNA cleavage, a new sequence can be inserted to achieve designated genome editing [3]. However, low editing efficiency and unwanted off-target effects largely limit clinical applications of CRISPR/Cas9 system [6]. This overview aims to delineate the potential influence of small molecules on CRISPR/Cas9 system.
Genetic Structure and Function of the CRISPR System
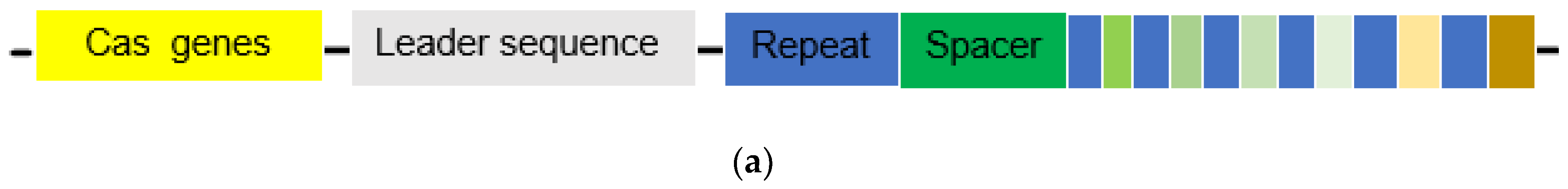
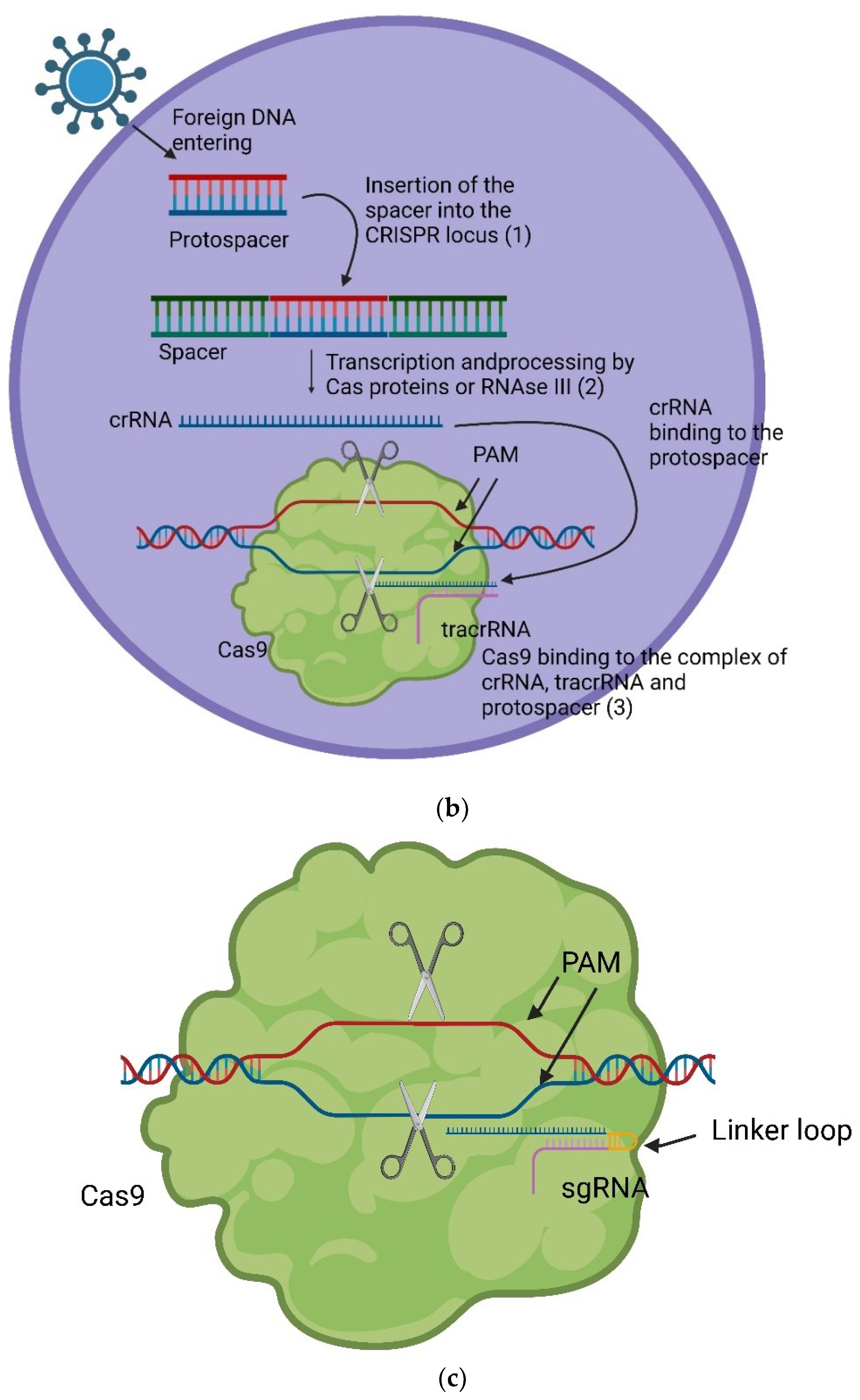
The CRISPR gene locus consists of Cas genes, a leader sequence, short repeats, and similar short spacers positioned between the repeats. The length of the repeats varies from 21 to 47 bp contingent on the species, while remaining constant within a species [7]. Importantly, the spacers do not originate from the host organism, but from intruding viruses or plasmids. These spacers, integral parts of the genetic material of the intruders, are incorporated as proto-spacers between the repeats (Figure 1 a) during the initial adaptive phase (1) of the immune reaction. This integration occurs when a bacterium or archaea encounters an intruder for the first time. The spacer is always introduced at the proximal end of the CRISPR array [2]. Subsequent to this adaptive phase, two additional phases unfold in the immune response. In the expression phase (2), CRISPR RNAs (crRNAs) are expressed, containing both the repeat sequence and the spacer sequence. For instance, a Cpf1 (also known as Cas12a) crRNA contains 42-44 nt, with a 19 nt-repeat and a 23-25 nt-spacer [8]. In the interference phase (3), crRNA pairs with the complementary proto-spacers of an invading virus or plasmid, facilitating the recognition and subsequent silencing of the intruding genetic material by Cas proteins (Figure 1 b) [2]. The position of the double-strand break (DBS) is determined by the complementary pairing and the localisation of the proto-spacer adjacent motive (PAM) [2], with the PAM sequence being 2-5 nt long and specific to certain bacteria [9]. Similar to Cas9, other Cas proteins can act as endonucleases, cutting the DNA strands near the binding position of the proto-spacer [10]. There are three types of systems in which DNA cutting can occur. In systems I and III, pre-crRNA is processed by Cas proteins, and the complementary strand is then detected and cleaved in a multi-enzyme complex. In system II pre-crRNA initially binds to trans-activating crRNA (tracrRNA), which is complementary to the repeats, and processing is conducted by RNase III in presence of Cas9 [2]. By producing a chimera of tracrRNA and crRNA, connected through a linker loop at the 3’ end of the crRNA, a single guiding RNA (sgRNA) can be formed (Figure 1 c). This allows Cas9 to be precisely programmed for inducing a DSB at a specific genomic position using single RNA [2]. The underlying principle closely mimics the naturally occurring process, as illustrated in Figure 1 c.
Figure 1.
(a) The general structure of a CRISPR locus. Consisting of multiple Cas genes organized in operons (shown in yellow), followed by a leader sequence (gray) and afterwards the repeat (blue) array in which the spacers (various colours) are built in. The various colours indicate that the spacers are of different nature while the consistent blue colour indicates the consistent nature of the repeats. Furthermore, the consistency of length is shown [2,7]. (b): The general triad of an immune reaction within the CRISPR system. In the adaptive phase (1) foreign DNA enters the cell and the protospacer is integrated between two repeats. In the expression phase (2) crRNA is transcribed and further processed by Cas proteins (Systems I and III) and then binds onto the protospacer of the foreign DNA. In case of System II this happens with the help of a tracrRNA. The formed complex, in the case of system II, consisting out of Cas9, crRNA bound to the protospacer of the viral DNA and tracrRNA bound to the repeat sequence of the crRNA leads to silencing of the foreign DNA through a DBS at the PAM by the endonuclease activity of Cas9 [1,2]. (c) The sgRNA combines tracrRNA and crRNA into one ribonucleic acid and induce the same effect as the complex shown in Figure 1 b [2].
Figure 1.
(a) The general structure of a CRISPR locus. Consisting of multiple Cas genes organized in operons (shown in yellow), followed by a leader sequence (gray) and afterwards the repeat (blue) array in which the spacers (various colours) are built in. The various colours indicate that the spacers are of different nature while the consistent blue colour indicates the consistent nature of the repeats. Furthermore, the consistency of length is shown [2,7]. (b): The general triad of an immune reaction within the CRISPR system. In the adaptive phase (1) foreign DNA enters the cell and the protospacer is integrated between two repeats. In the expression phase (2) crRNA is transcribed and further processed by Cas proteins (Systems I and III) and then binds onto the protospacer of the foreign DNA. In case of System II this happens with the help of a tracrRNA. The formed complex, in the case of system II, consisting out of Cas9, crRNA bound to the protospacer of the viral DNA and tracrRNA bound to the repeat sequence of the crRNA leads to silencing of the foreign DNA through a DBS at the PAM by the endonuclease activity of Cas9 [1,2]. (c) The sgRNA combines tracrRNA and crRNA into one ribonucleic acid and induce the same effect as the complex shown in Figure 1 b [2].
CRISPR/Cas9-mediated Genome Editing
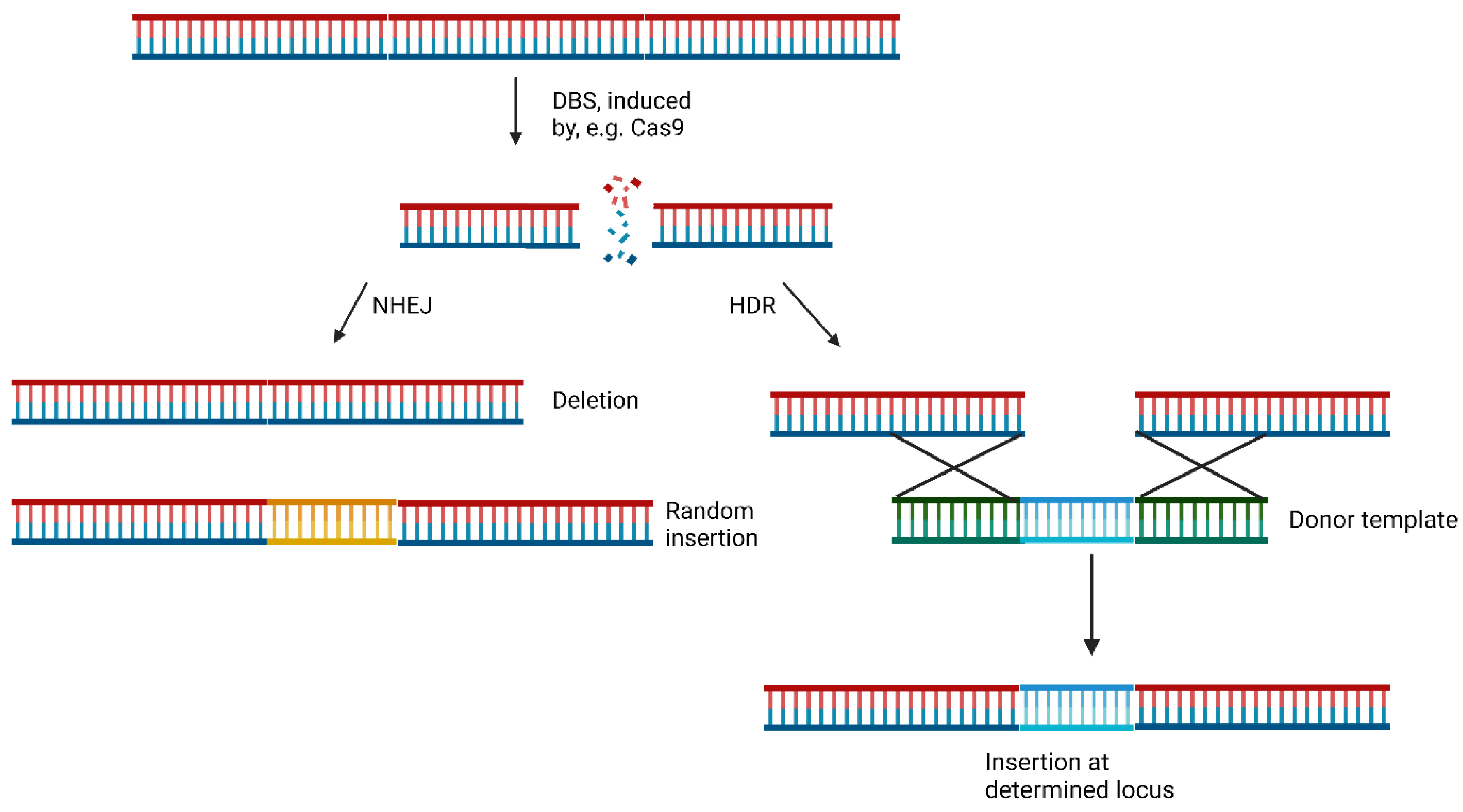
The widely studied CRISPR/Cas system, derived from Streptococcus pyogenes (spCas9), is predominantly employed for genetic editing. This Cas9-based system falls under type II of the CRISPR systems [11]. During the genome editing process, a single-guide RNA (sgRNA) directs Cas9 to a specific location to induce a DSB. Non-homologous end joining (NHEJ), and homology-directed repair (HDR) are two major pathways for DSB repair. The more prevalent and efficient NHEJ pathway doesn't require a template [3]. It leads to either the deletion of genetic information (knockouts) or the introduction of random genetic modifications (collectively termed indels) into the DSB [3,12,13]. In contrast, HDR can facilitate the precise incorporation of genetic information by pairing the broken strands with a new complementary strand at the ends. This allows for the hybridization of strands and the insertion of the strand containing the new genetic information [14]. NHEJ is present in all four cell cycle phases, while HDR is mainly limited to the phases S and G2, when DNA duplication has occurred, and sister chromatids are available for repair [15]. The mechanics of NHEJ and HDR are shown in Figure 2 [3]. Moreover, microhomology-mediated end joining (MMEJ), a subtype of NHEJ, involves annealing of two 3’-ends of a DSB at microhomologies, leading to 3’ flaps that are removed, and the strands are subsequently filled in. Notably, this process leads to significant loss of genetic information due to flap removal, resulting in deletions known as microhomologies containing deletions (MH) [16].
Figure 2.
The pathways of NHEJ (left) and HDR (right). During a NHEJ after a DSB a part of the genetic material is lost, or a random genetic modification (orange) is included. During a HDR a donor template (green and blue) hybridises to the broken DS and new genetic information (blue) can precisely inserted [3,12,13,14].
Figure 2.
The pathways of NHEJ (left) and HDR (right). During a NHEJ after a DSB a part of the genetic material is lost, or a random genetic modification (orange) is included. During a HDR a donor template (green and blue) hybridises to the broken DS and new genetic information (blue) can precisely inserted [3,12,13,14].
To introduce a new gene, a substantial amount of the donor template is required, primarily favoring the homology-directed repair (HDR) pathway when the correct template is available [9]. This process, known as knock-in (event), involves the precise incorporation of a new gene [15]. To deliver the CRISPR/Cas-9 system into cells, various methods have been developed, including physical approaches, such as injection, or viral delivery methods using, for example, adenoviruses or non-viral delivery methods like liposomes [17].
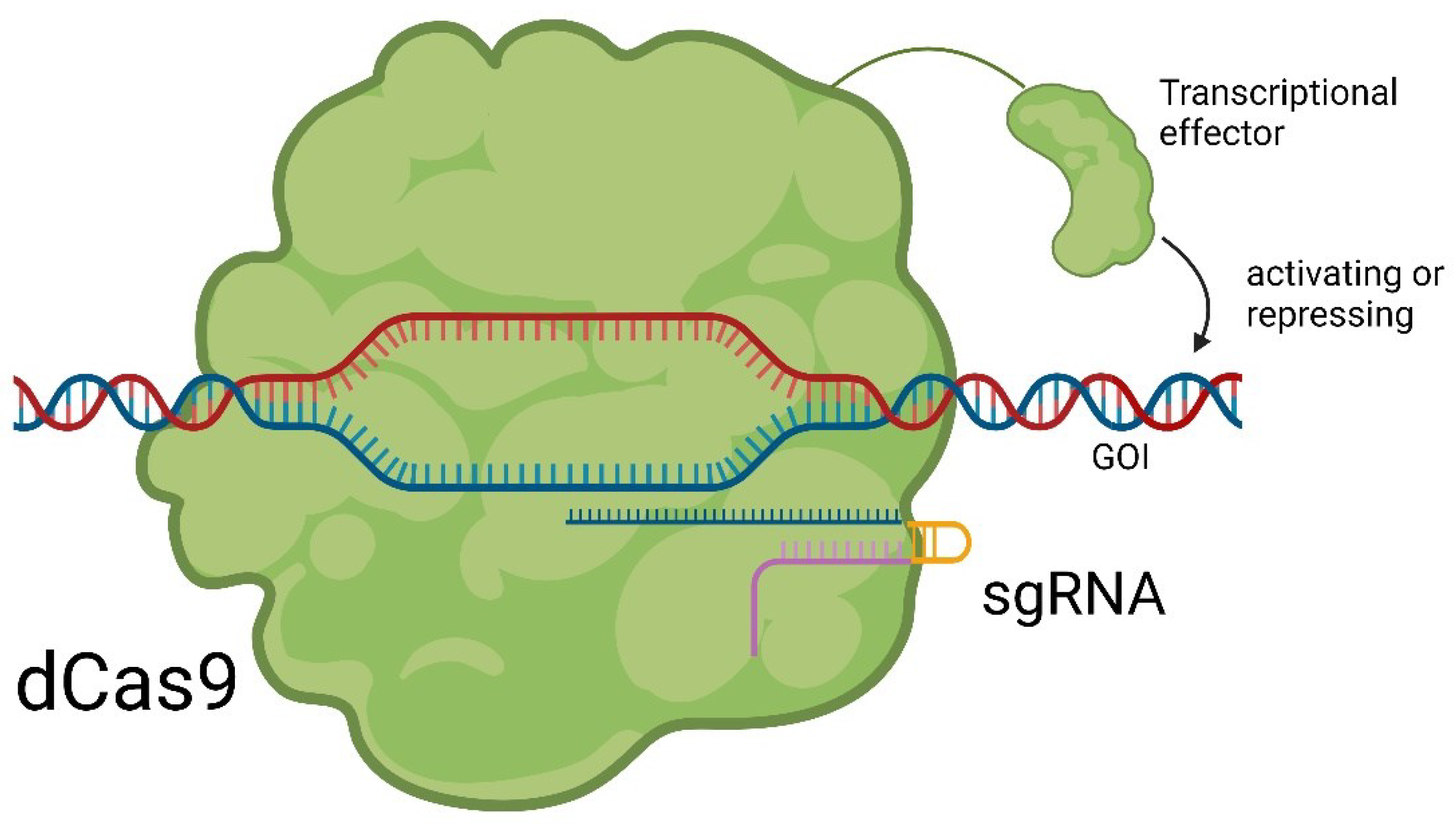
Besides the genomic editing by inserting full donor templates that can be expressed, it is possible to make an endonuclease dead Cas9 (dCas9) by two-point mutations, H840A and D10A, which lead to the loss of the endonuclease activity [18]. In this configuration, dCas9 can still bind to a specific DNA sequence under the guidance of designated sgRNA. However, due to the absence of endonuclease activity, target dsDNA is kept intact. When modified with gene effectors and targeting the promoter/enhancer region of the gene of interest (GOI), dCas9 can either activate or repress the GOI transcription, termed CRISPRi (CRISPR interference) and CRISPRa (CRISPR activation) [19,20]. Examples of gene effectors include Kruppel-associated Box (KRAB) for repression [21] and Herpes simplex viral protein (HSVP) for activation [22]. The principle of epigenome editing with dCas9 is shown in Figure 3 [23].
Figure 3.
Principle of epigenetic editing with dCas9. Catalytic unfunctional dCas9 binds onto the promotor region of a GOI. The effector attached to dCas9 either activates or represses the gene leading to an altered expression [23].
Figure 3.
Principle of epigenetic editing with dCas9. Catalytic unfunctional dCas9 binds onto the promotor region of a GOI. The effector attached to dCas9 either activates or represses the gene leading to an altered expression [23].
Further applications include single nucleotide exchanges or the introduction of new protospacer adjacent motifs (PAMs), enabling broader utilization within the human genome [3].
The applications of genetic editing with CRISPR/Cas9 span from human disease model, diagnosis, to gene therapy [3]. Given the diverse methods in the realm of genes, it is crucial to control Cas9's action in terms of dosage, temporal aspects, and spatial considerations [24,25]. This control is essential for achieving reproducible results and, more importantly, ensuring the safety of Cas9 gene editing in therapeutic applications [6]. The significance lies in the fact that Cas9 has been shown to exhibit several side effects with prolonged activity at high levels, including off-target genome editing, gene toxicity, gene translocation, and more [24]. One approach to inhibit Cas9 involves naturally occurring proteins from bacteriophages, developed as a counter to CRISPR systems [26]. Some of these, such as AcrIE1, have demonstrated the ability to block site-specific DNA cleavage [27]. However, therapeutic proteins come with significant drawbacks, including a short half-life, poor stability, low solubility, and high production costs [28]. This underscores the interest in identifying small molecules that can be employed to control Cas9 activity [6]. Small molecules are often more cost-effective, better soluble, more stable, easier to produce and modify, and cheaper to manufacture [29].
Small Molecules Modulate Wild-type Cas9 Protein
Through high-throughput screening, Maji et. al., identified several small molecules that could disrupt Cas9 binding to DNA, preventing DNA double strand breaks (DSBs). Testing eGFP further confirmed that some of these Cas9 inhibitors, such as BRD0539 1, worked reversibly [30,31]. Recently, using a high-content fluorescence-based approach, we identified valproic acid 2 (VPA) as a Cas9 degrader from a chemical library consisting of nearly 300 drug-like compounds and natural products [32]. VPA, a well-known histone deacetylase inhibitor (HDACi), demonstrated significant Cas9WT degradation under hyperthermia conditions generated either in the presence of a photothermal agent, indocyanine green, upon irradiation by a near-infrared laser, or heating with an external heat bag. This degradation effect was independent of its HDAC inhibitory effect. However, off-target effects of BRD0539 1 or VPA 2 remain unknown.
For a clearer understanding of off-target effects, Yang et. al. searched for small molecules that inhibit Cas9WT. In their studies, the most effective Cas9 inhibitor were shown to be SP24 3 with an IC50 to Cas9WT of approximately 14 µM and to the Cas9-sgRNA complex of about 7 µM. This was shown during an FP assay, where SP24 3 significantly decreased fluorescence polarization. Furthermore, SP24 3 and SP2 4 were proven to enhance precision of Cas9-mediated genome editing (Figure 4) [30,31].
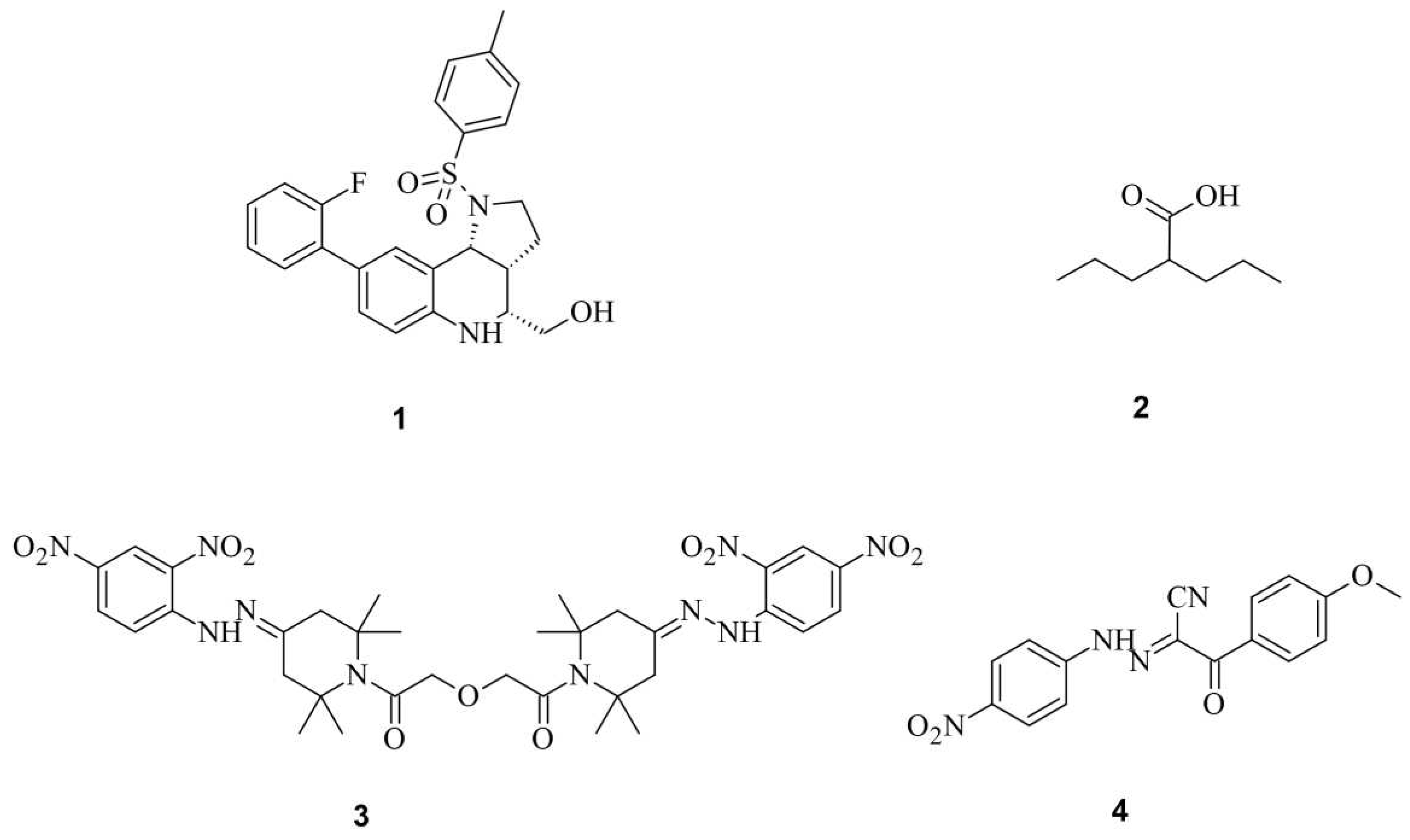
Small Molecules Modulate Engineered Cas9 Protein
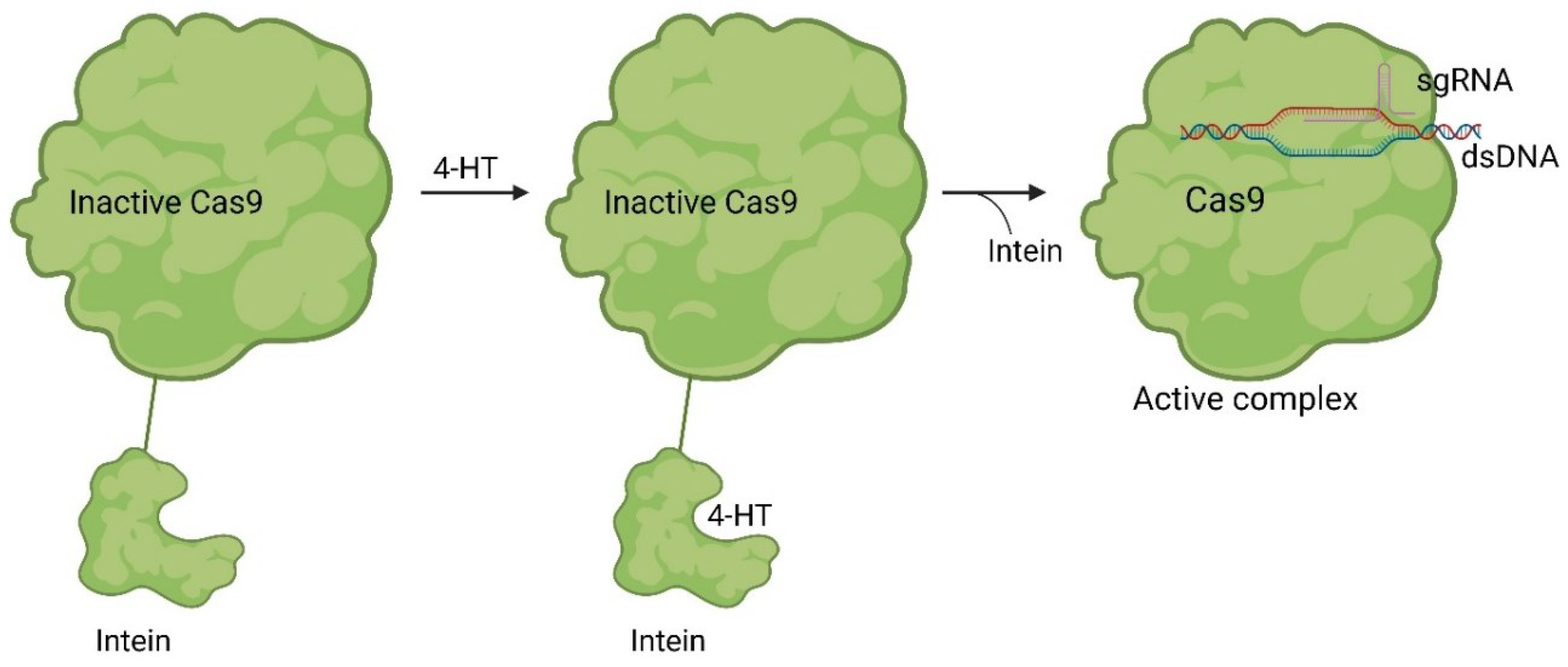
While only a few small molecules have been identified to function with Cas9WT, the primary Cas9 chemical modulators interact with engineered Cas9. A way to activate Cas9 with a small molecule was demonstrated by Davis et. al. [33]. As shown in Figure 5, Buskirk et al. successfully evolved inteins that could only self-remove in the presence of 4-hydroxytamoxifen (4-HT) 5 [34].
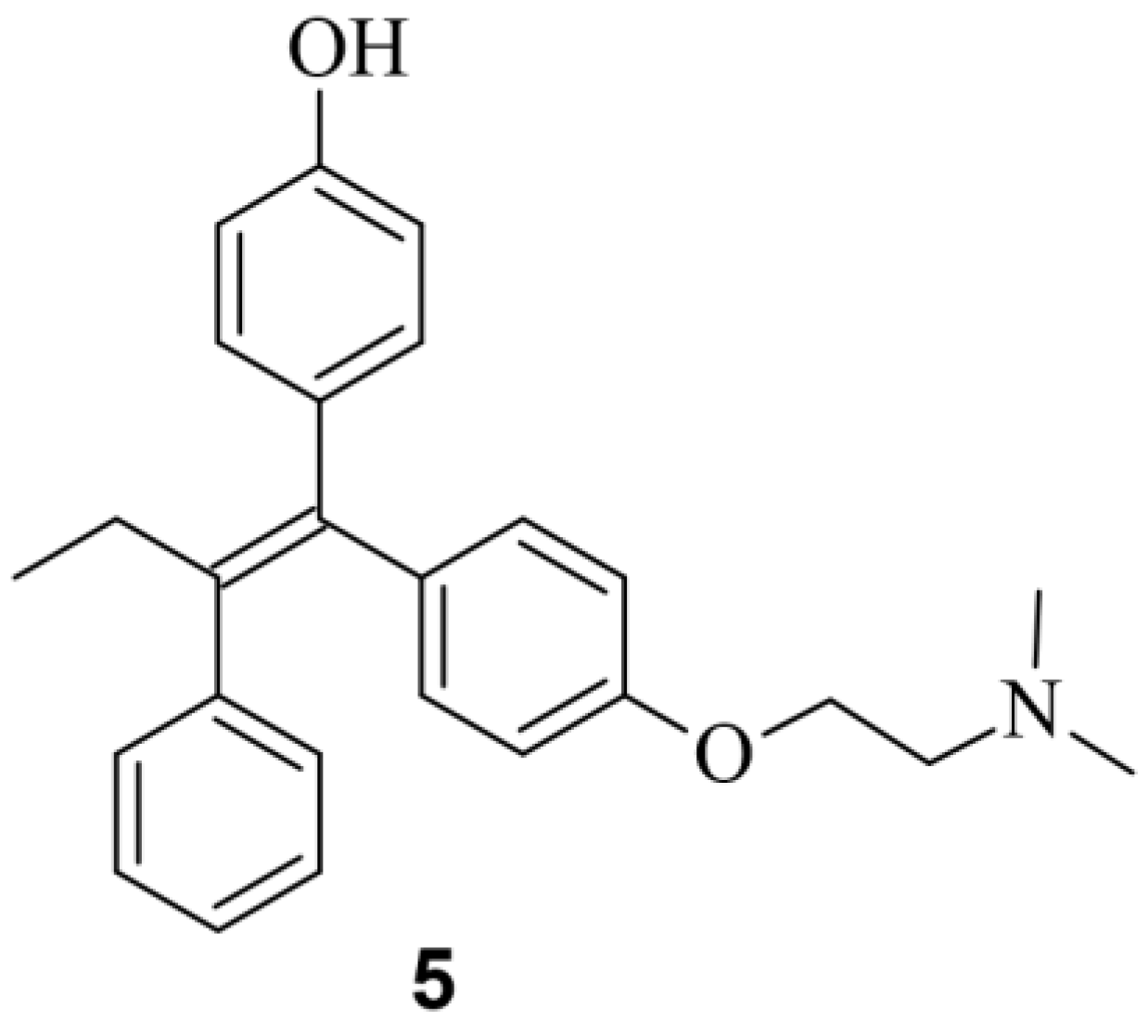
Figure 5.
Chemical structure of the small molecule 4-hydroxytamofen 5.
Upon binding to the intein, 4-HT 5 induces the self-splicing of the intein [34]. In the case of Cas9, it is inactivated when the intein is attached to it. However, treatment with 4-HT 5 led to the self-splicing of the intein from Cas9, reactivating its functionality (Figure 6) [33].
Figure 6.
Working mode of intein inhibited and activated Cas9. Through a modification with an intein Cas9 is inactivated (left). Through treatment with the small molecule 4-HT 5 self-splicing of the intein is induced (middle). After splicing Cas9 can form an active sgRNA complex and the desired gene can be modified [33].
Figure 6.
Working mode of intein inhibited and activated Cas9. Through a modification with an intein Cas9 is inactivated (left). Through treatment with the small molecule 4-HT 5 self-splicing of the intein is induced (middle). After splicing Cas9 can form an active sgRNA complex and the desired gene can be modified [33].
Davis et. al. modified spCas9 at 15 different positions and expressed the modified Cas9 variants in HEK293-GFP cells with sgRNA targeting the EGFP locus. They determined the loss of function in expressing GFP after treatment with 1 µM 4-hydroxytamoxifen (4-HT) 5 in 8 cases. In a more in-depth analysis with Cas9 variants modified at S219 and C574, respectively, in comparison to Cas9wt, it was demonstrated that the modified variants exhibited higher specificity at similar on-target cleavage rates. For instance, the C574-modified Cas9 and Cas9wt had similar on-target DNA cleavage rates of 6.4%. However, the modified variant resulted in a fourfold lower frequency at the four critical off-target sites [33]. By precisely activating Cas9, the precision of Cas9-mediated genome editing could be significantly enhanced.
A similar approach was undertaken by Wu et al., where Cas9 was modified with a small molecule-assisted shut-off (SMASh) tag. Cas9 was fused with a degron from the Hepatitis C virus (NS4A) and a protease domain. Under non-treatment conditions, the protease self-cleaves the SMASh tag, converting Cas9 into an active species. However, upon adding the protease inhibitor Asunaprevir (ASV) 6, the protease activity is inhibited, preventing the cleavage of the SMASh tag. This results in the recognition of the degron by the proteasome or lysosome, leading to the degradation of Cas9 and rendering it inactive. The mode of action of this system, as well as ASV 6, are shown in Figure 7 [35].
Figure 7.
Working principle of SMASh Tag controlled Cas9. Under non-treatment conditions modified Cas9 will be processed into active Cas9 by self-cleavage of the SMASh tag. The tag will be degraded by the proteasome or lysosome. If, however treatment with ASV 6 (shown on the left) is applied, the protease activity is inhibited and Cas9 marked with a SMASh tag is degraded as whole unit [35].
Figure 7.
Working principle of SMASh Tag controlled Cas9. Under non-treatment conditions modified Cas9 will be processed into active Cas9 by self-cleavage of the SMASh tag. The tag will be degraded by the proteasome or lysosome. If, however treatment with ASV 6 (shown on the left) is applied, the protease activity is inhibited and Cas9 marked with a SMASh tag is degraded as whole unit [35].
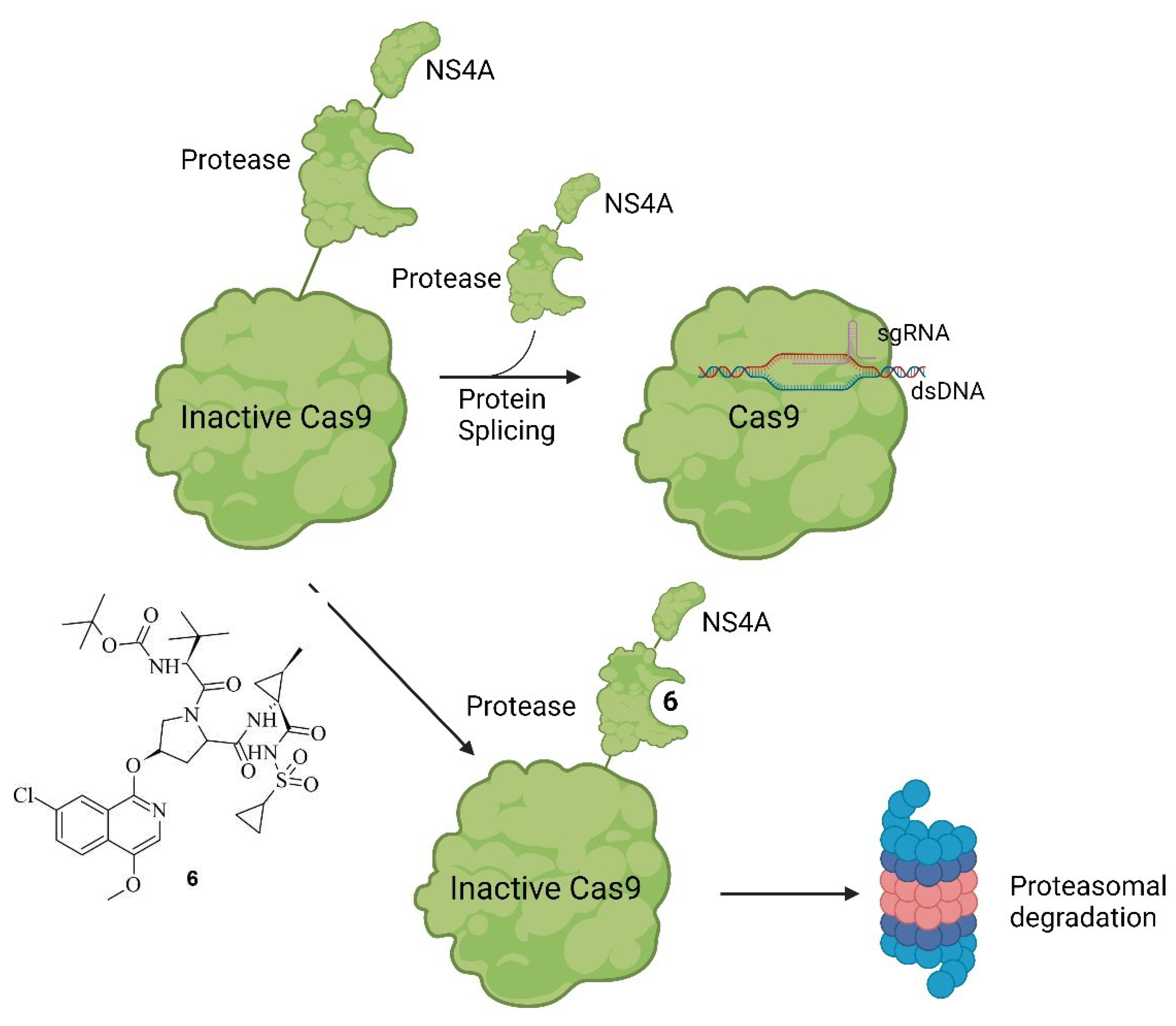
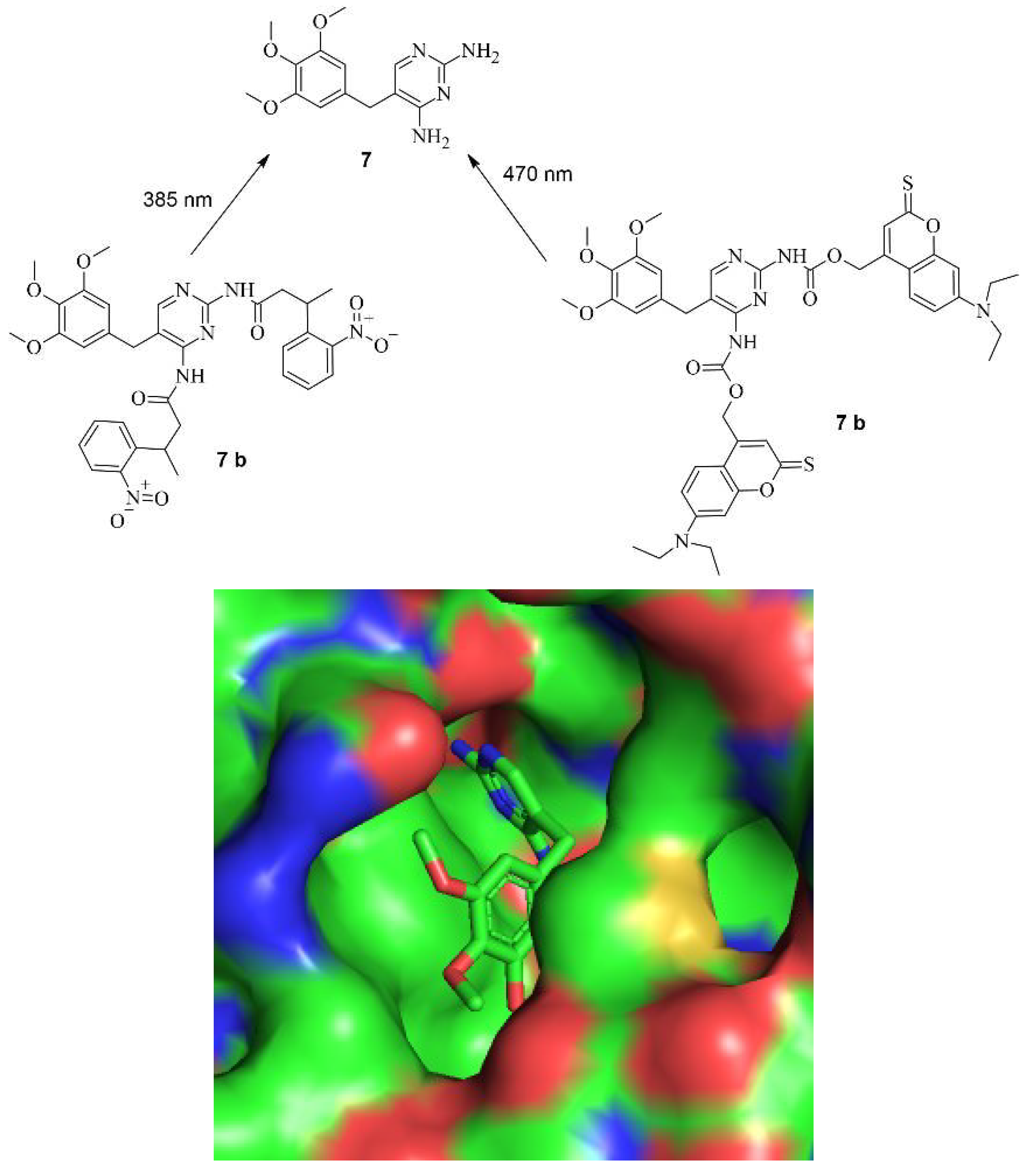
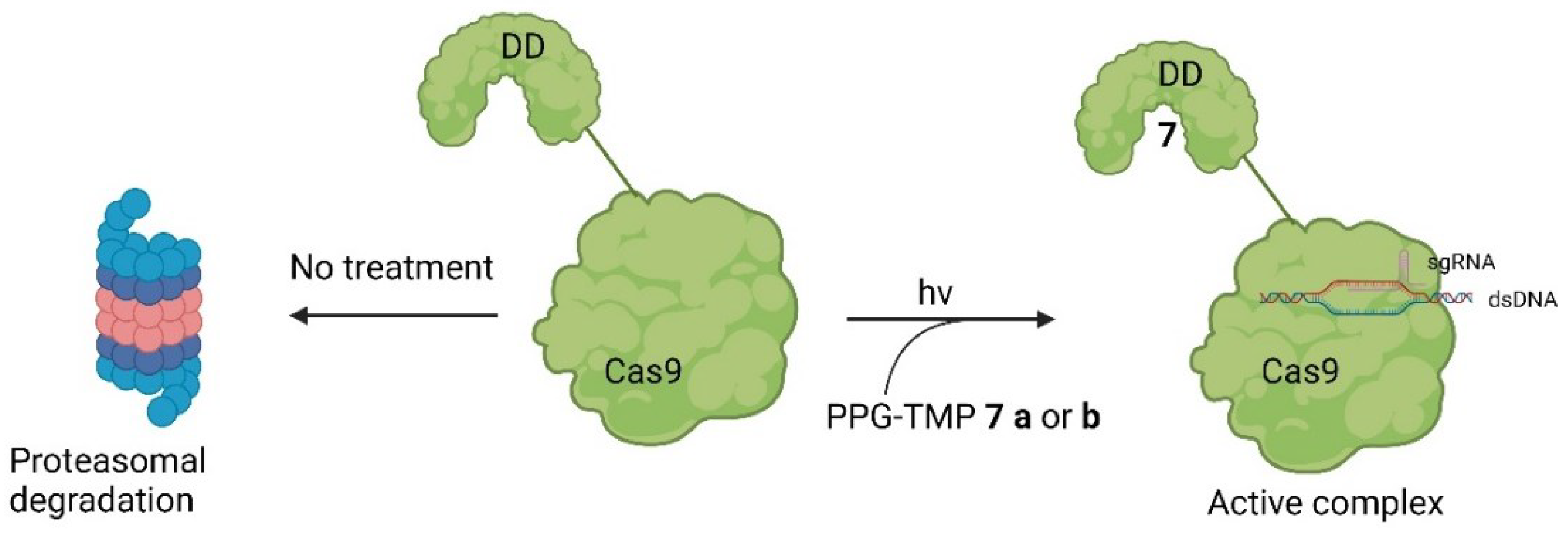
The working principle is exactly inverted in comparison to the system described earlier. In the first case, the addition of a small molecule activates Cas9 through cleavage, as demonstrated by Davis et al. [33]. In the second case, the addition of the small molecule inhibitor prevents self-cleavage and keeps Cas9 inevitably inactive because it is degraded by the proteasome or lysosome [35]. Notably, the removal of ASV 6 by washing with uncontaminated media allowed the newly expressed Cas9 to become active again since self-cleavage wasn't hindered anymore. This reversibility is valuable to prevent Cas9 from re-editing previously edited loci [36]. Often, the editing of multiple genes is necessary [37], and in such cases, Cas9 can be inactivated after editing a particular gene and re-activated when editing the next gene. Such a light-switch-like system is extremely useful for controlling the effects of Cas9, making it safer and more efficient [3,35]. Speaking of light, it represents a very useful tool for spatial control of the activity of small molecules [38]. Based on photoactivable protecting groups (PPGs) Manna et. al. designed a “fused” system of the ones described by Davis et. al. and Wu et. Al. [33,35]. Cas9 was modified with destabilized domains (DDs) of dihydrofolate reductase (DHFR). Through these unstable domains, the fused Cas9 is recognized and degraded rapidly by the proteasome, making it inactive, similar to the activated SMASh tag [35]. However, if treatment with trimethoprim (TMP) 7 is applied, the DDs get stabilized, averting the degradation of the fused Cas9, thus keeping it active [39]. This is like the system of Davis et al. in the way that by binding a small molecule, Cas9 is activated [33]; however, in this case, it occurs through the inhibition of degradation of Cas9 so that it can provide its nuclease activity [35]. By such systems, only the dosage and timing of Cas9 can be controlled using the concentration and temporal exposure of the small molecules, e.g., ASV 6 or TMP 7 [39]. To add spatial controllability to the Cas9 activity, two PPGs were added to TMP 7 [25]. The PPGs were introduced at the amine groups because in the co-crystal structure of TMP 7 and DHFR (PDB: 7R6G), it is visible that the amine groups of TMP 7 are buried in the binding pocket of DHFR [40]. In an eGFP disruption assay with a sgRNA Plasmid targeting the eGFP gene and DHFR fused Cas9, treatment with PPG-modified TMP 7 b or c did not induce an observable loss of fluorescence. This leads to the conclusion that protected TMP 7 b or c indeed can't bind to the DDs. Modified Cas9 is therefore left unstable and is quickly degraded by the proteasome. However, after an irradiation treatment of just a couple of minutes, loss of fluorescence was observable, meaning that the deprotection of the protected TMP 7 b or c into TMP 7 was possible, and the binding ability to the DDs was restored. Following irradiation, the inhibition of proteasomal degradation was gained, and Cas9 was kept active. Apart from controlling double-strand breaks and silencing genes with DHFR-modified Cas9, the expression of IL1RN could be influenced. For that, a dCas9 was modified with DHFR, and the transcriptional activator domains (VP64 and PP7) were attached. When treatment with protected TMP 7 b or c was applied, no leverage of expression was observable, meaning that protected TMP 7 b or c could not bind to the modified dCas9, leading to dCas9 being degraded. After treatment with light for 12 minutes, however, unprotected TMP 7 was formed, which led to a binding of it to the modified dCas9. By the binding, the degradation of dCas9 was prevented, resulting in a noticeable increase of IL1RN expression [25]. In total, the findings of Manna et al. provide a similar control of Cas9 activity like the system of Wu et al. by turning Cas9 active through treatment with a small molecule [35]. However, the system of Manna et al. not only provides temporal and dosage control but also spatial control through the inclusion of a light-activable pathway. Furthermore, they did not only gain control of double-strand breaks but also of dCas9-mediated gene activation [25]. The structure of TMP 7 and its PPG-modified version 7 b and c are shown in Figure 8 a, the co-crystal structure of TMP 7 and DHFR in Figure 8 b, and the working principle in Figure 8 c [25,40].
Figure 8.
a (above, left): The structure of TMP 7 and the protected derivates 7 b and c including the used wave lengths for deprotection [25]. b (above, right): The structure of TMP 7 in the binding pocket of DHFR. The amines are deep in the binding pocket, leaving them as a good target for protection (PDB: 7R6G) [25,40]. c (below): The principle of the system. Through introduction of the DDs Cas9 is quickly degraded by the proteosome. By treatment with PPG-TMP 7 b or c and irradiation TMP 7 can be formed, bind to the DDs, stabilizing it and thus turning Cas9 active [25].
Figure 8.
a (above, left): The structure of TMP 7 and the protected derivates 7 b and c including the used wave lengths for deprotection [25]. b (above, right): The structure of TMP 7 in the binding pocket of DHFR. The amines are deep in the binding pocket, leaving them as a good target for protection (PDB: 7R6G) [25,40]. c (below): The principle of the system. Through introduction of the DDs Cas9 is quickly degraded by the proteosome. By treatment with PPG-TMP 7 b or c and irradiation TMP 7 can be formed, bind to the DDs, stabilizing it and thus turning Cas9 active [25].
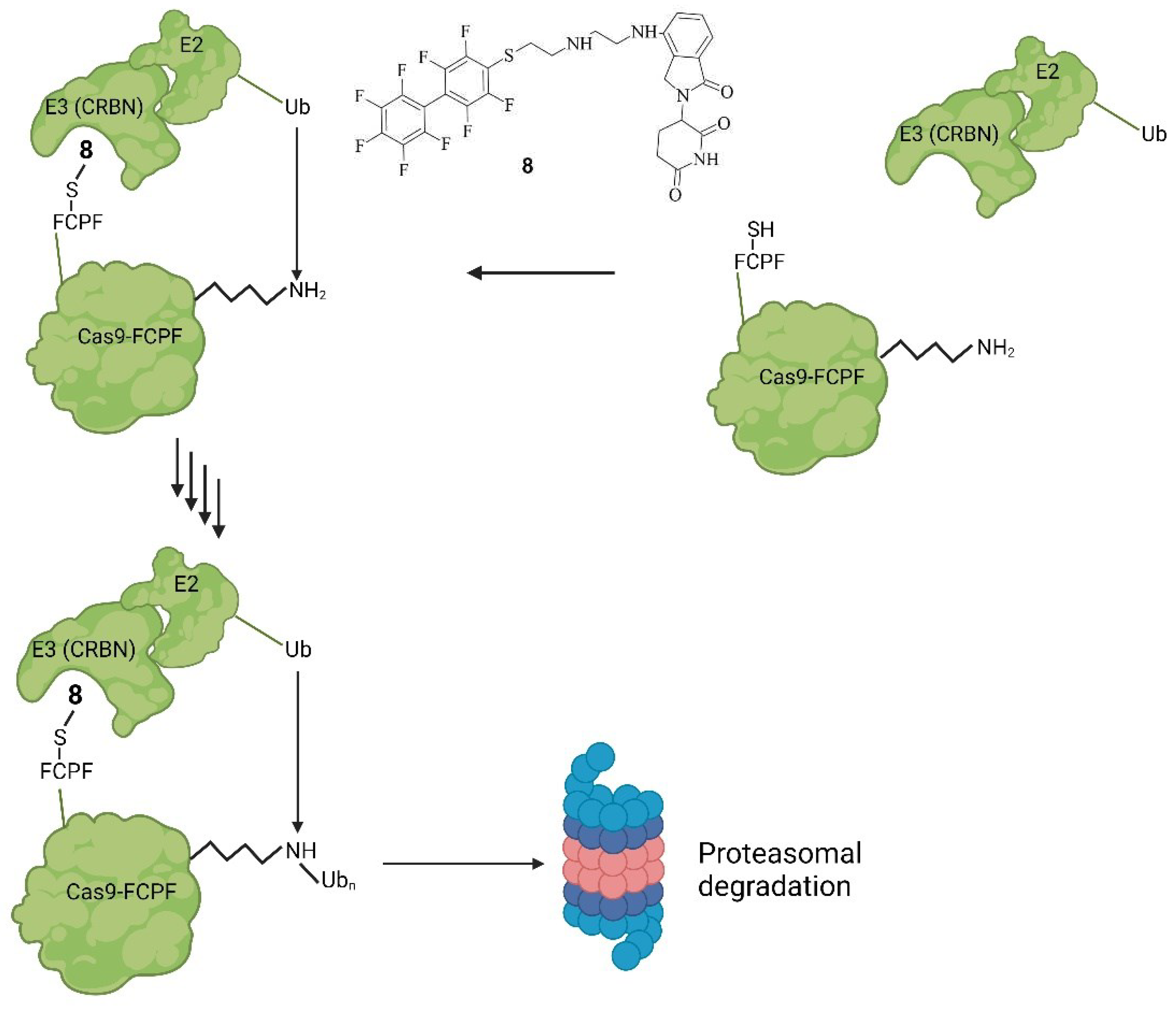
In the systems described so far, rather substantial sequence modifications were needed to make Cas9 targetable by a small molecule. Recently, we modified Cas9 with an amino acid sequence consisting of phenylalanine, cysteine, proline, and phenylalanine (FCPF) [41]. This so called π-clamp leads to a specific reactivity of the cysteine, which then reacts with perfluoro aromatic moieties [42]. With the FCPF modification of Cas9 (Cas9FCPF), small molecules could precisely recognize Cas9FCPF [42]. This was used for labeling strategies but most importantly for a proteolysis targeting chimera (PROTAC) [41]. PROTACs are hetero-bifunctional molecules with a ligand that binds an E3-ligase. The E3 ligand is connected via a linker to another ligand on the other side that can bind to the protein of interest (POI). By binding on both sides simultaneously, they can catalyze the transfer of ubiquitin (Ub) onto the POI. Ub is transferred from an E2 ligase, bound to the E3 ligase, which itself is bound to the POI. The PROTAC serves as an enhancer of the binding between the E3 ligase and POI. Through the ubiquitination of the POI at a lysine residue or the N-terminal, it is marked for the 26S proteasome and is then degraded by the ubiquitin-proteasome system (UPS) [43]. We generated perfluoro derivative conjugated with lenalidomide, a ligand of the E3-ligase Cereblon (CRBN), called PROTAC-FCPF 8. 8-FCPF-Cas9 could be degraded in HeLa cells at a concentration of 10 µM after 6 h [41]. Via T7E1 [44,45] assay it was further proven, that the biologic activity of Cas9FCPF was comparable to unmodified Cas9. Degradation was further proven for dCas9FCPF, Cas12FCPF, and Cas13FCPF [41]. In short, a similar system to that of Wu et. al. was established. In both systems, Cas9’s stability is control by small molecule-induced proteasomal degradation [35,41]. However instead of introducing two domains as a SMASh-tag [35], only a peptide consisting of four amino acids was needed for Cas9FCPF [41]. The mode of action of PROTAC-FCPF 8 and its structure are shown in Figure 9 [41,43].
Figure 9.
Mode of action of PROTAC-FCPF 8. Binding of 8 onto Cas9-FCPF leads to a complex with the E3-ligase CRBN. Attached to that is an E2-ligase. The ubiquitination of Cas9-FCPF is then catalysed which ultimately leads to a degradation of Cas9-FCPF by the UPS rendering Cas9-FCPF inactive [41,43].
Figure 9.
Mode of action of PROTAC-FCPF 8. Binding of 8 onto Cas9-FCPF leads to a complex with the E3-ligase CRBN. Attached to that is an E2-ligase. The ubiquitination of Cas9-FCPF is then catalysed which ultimately leads to a degradation of Cas9-FCPF by the UPS rendering Cas9-FCPF inactive [41,43].
Small molecules regulate DSB Repair Mechanisms
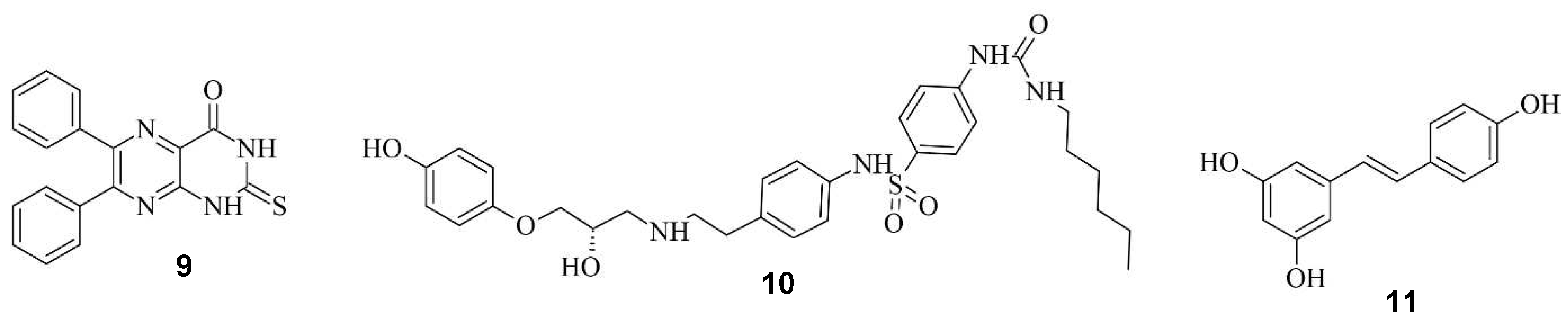
Aside from regulating the activity of engineered Cas9, there is the possibility to modulate CRISPR/Cas9 system by regulating the DNA repair system. Li et. al. investigated three compounds regarding their ability to enhance HDR or down-regulating NHEJ towards a more precise editing [15]. In their study, Scr7 9, L755507 10, and Resveratrol 11 were found to inhibit DNA ligase IV and thereby reduce NHEJ-mediated repair [15], showing that the efficiency of knock-ins can be enhanced in the presence of chemical inhibitors targeting NHEJ-influencing factors [46]. The compounds are illustrated in Figure 10.
Figure 10.
Scr7 9, L755507 10 and Resveratrol 11 from left to right.
A similar approach was undertaken by Bermudez Cabrera et al. Multiple small molecules were investigated, with a focus on the Ataxia-telangiectasia mutated (ATM) inhibitor KU60019 12, as illustrated in Figure 11 [47].
Figure 11.
The ATM inhibitor KU60019 12.
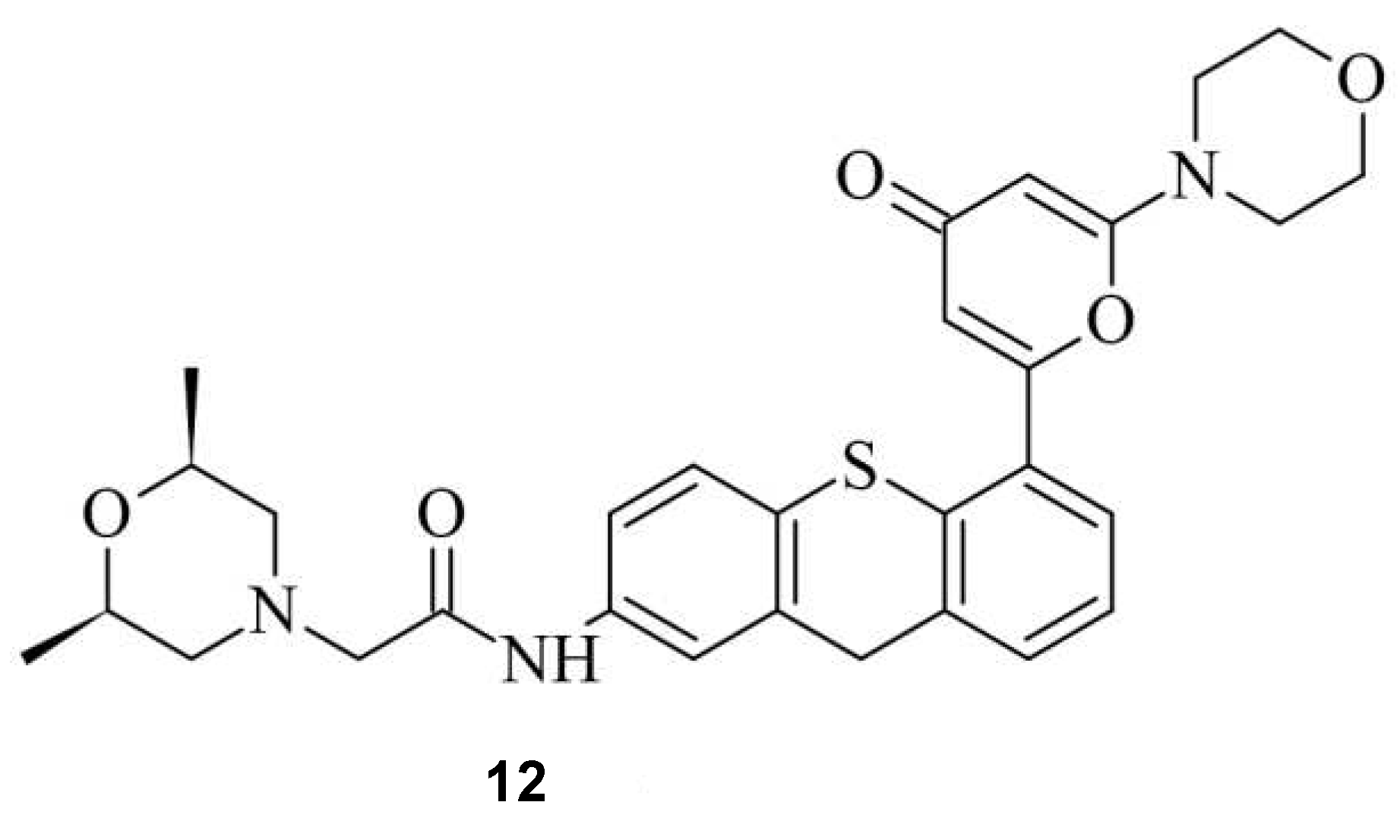
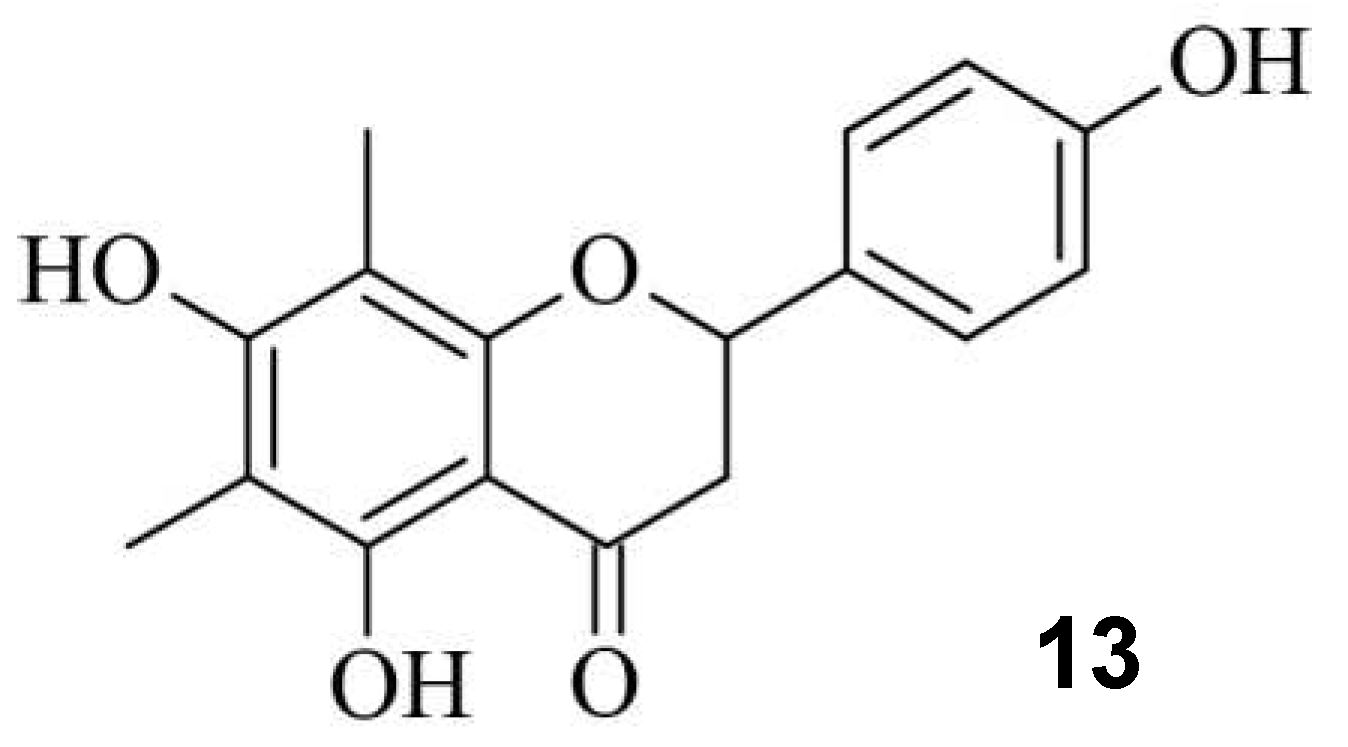
ATM, a serine/threonine kinase, plays a determining role in the initiation of DSB repair mechanism by recruiting necessary repairing factors and determining whether HDR or NHEJ occurs [48]. During a study a dose-dependent downshift from MH deletions (down to 0.74-fold) in the presence of various chemical compounds, including 11 was observed. Also, an increase editing efficiency was noticeable [47]. However, as the main repair mechanism of DSB, downregulating NHEJ is associated with increased potential in tumorigenesis [49,50,51]. To avoid side effects, Zhang et. Al. screened 722 small molecules and identified farrerol 13, which increased knock-in efficiency, while NHEJ was clearly not affected (Figure 12) [52,53].
Figure 12.
The small molecule farrerol 13.
Small Molecules regulate sgRNAs
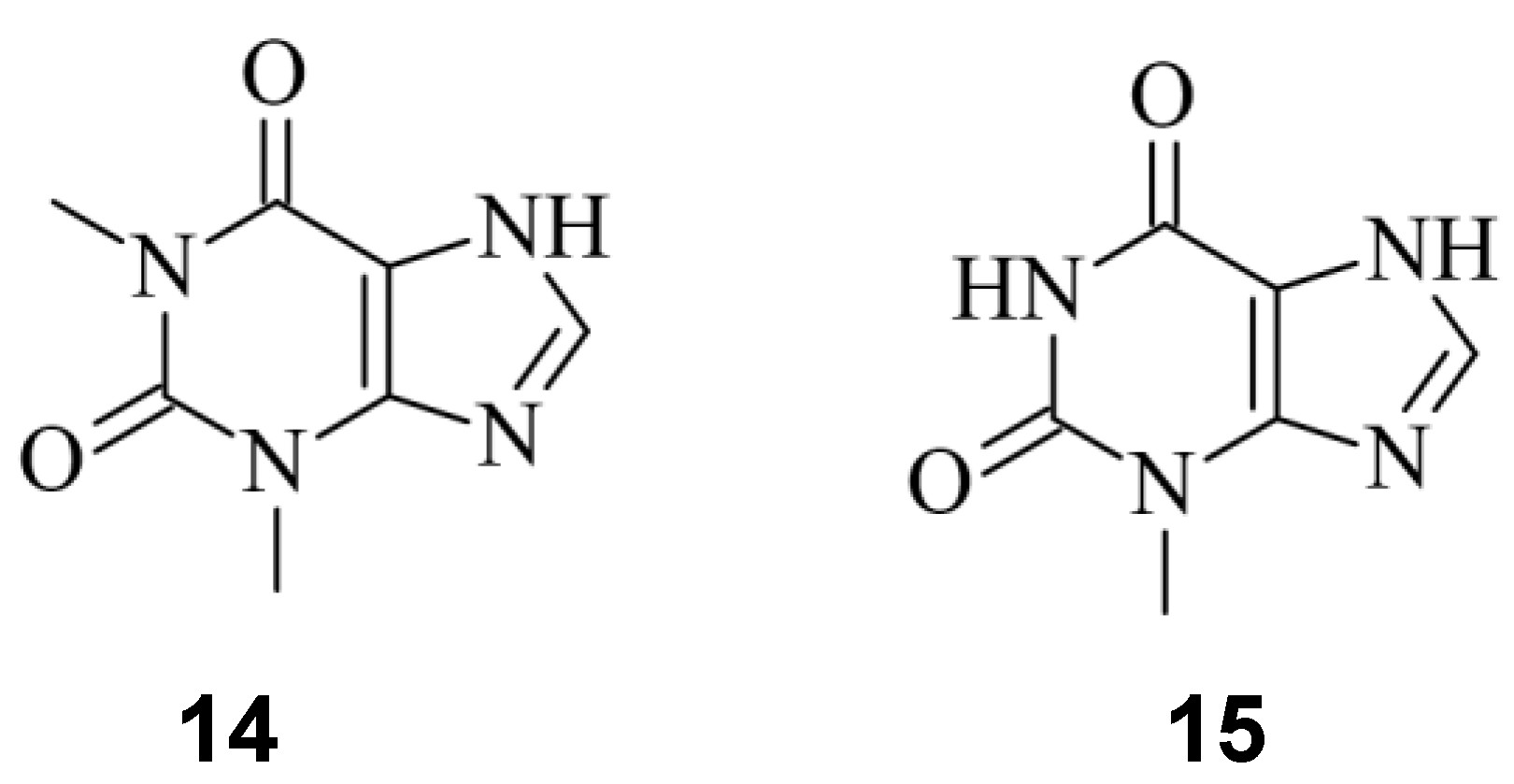
Given the important role of sgRNAs in CRISPR/Cas9 [2], chemicals capable to control the activity of the sgRNA offers another possibility to regulate the process of genome editing [33,35]. Aptamers are short nucleic acids, which can specifically bind ligands, and are commonly employed to regulate nucleic acids, such as sgRNA [54]. Iwasaki et. al. applied this concept to develop so-called aptamer-sgRNA (agRNA), in which the theophylline 14 or 3MX 15 aptamer was used and introduced into sgRNA at different positions, leading to an endonuclease active Cas9 only upon treatment with 14 or 15 respectively. An up to 104-fold increase of transformants in comparison to wild type sgRNA was observed (Figure 13) [55].
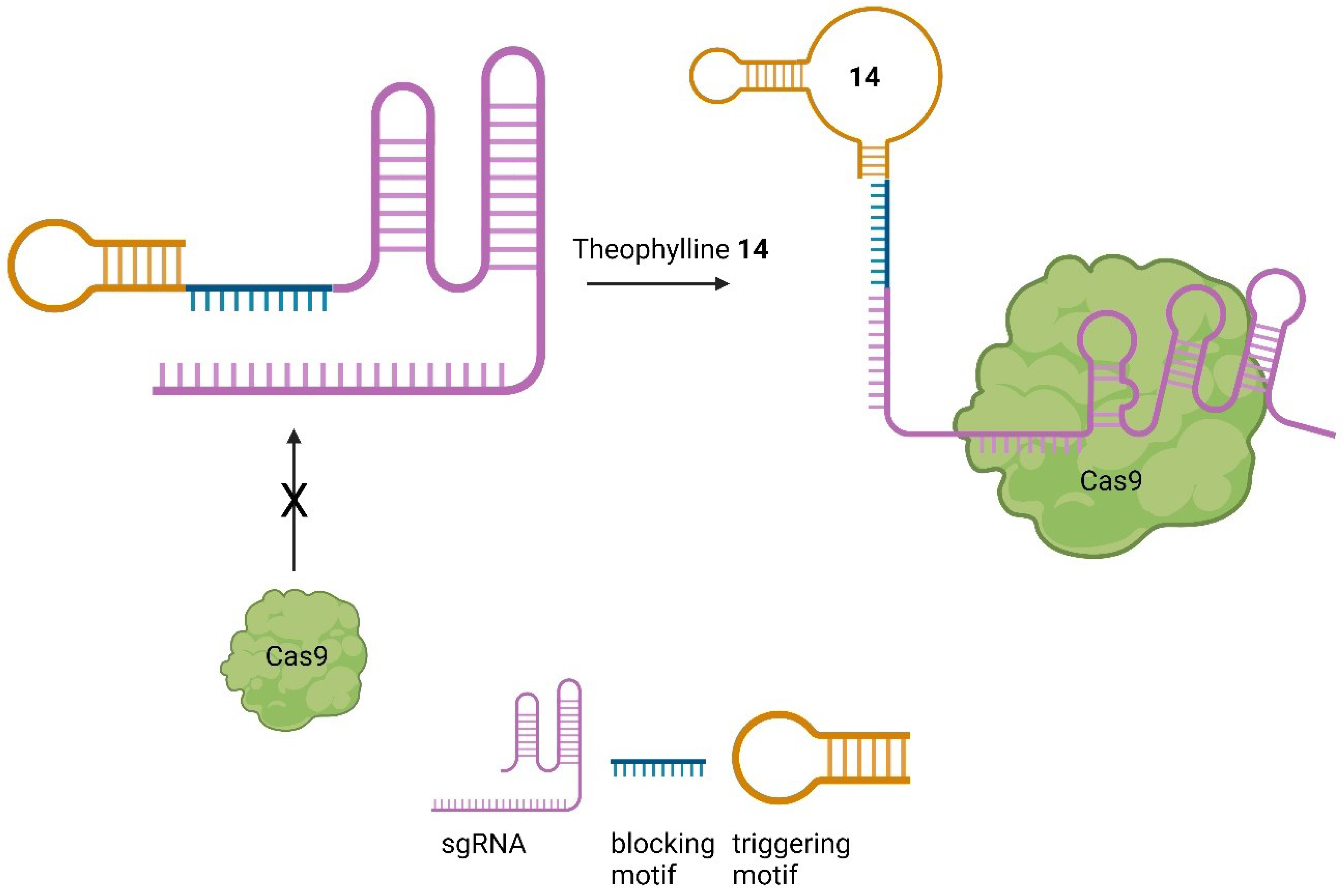
Based on the theophylline 14 aptamer Bingqian et. al. designed small molecule-activated allosteric aptamer regulating (SMART)-sgRNAs [58]. Instead of inhibiting the nuclease activity of Cas9 as demonstrated by Iwasaki et. al. [55]. they used a blocking motif to inhibit binding of Cas9 in the first place. Further, a triggering motif at the sgRNA 3’ end was attached, containing the theophylline 14 aptamer. In the absence of theophylline 14 the blocking and triggering motifs form a loop to block Cas9 from binding. After treatment with theophylline 14, the structure is changed, which allows Cas9 to bind to the sgRNA, leading to the DSB (Figure 14) [58].
By employing this system, the cleavage of EGFP could be reduced in the absence of theophylline 14. Moreover, the system was successfully applied to the firefly luciferase and TurboRFP genes by modifying the sgRNA while retaining the blocking and triggering motif. After treatment with theophylline 14, a SMART-sgRNA activation of up to 61% was observed. This system has been demonstrated to be effective both in vitro and in vivo, providing a versatile tool for activating sgRNA and thereby controlling Cas9 [58].
Figure 14.
Mode of action of SMART-sgRNAs [58].
Figure 14.
Mode of action of SMART-sgRNAs [58].
Conclusion
The CRISPR/Cas9 system is a promising tool in gene editing with multiple thinkable applications. The therapeutics applications may be the most interesting [3]. To use the system in the best way possible in terms of e.g., efficiency and non-toxicity a control over dosage, timing and spatial action is unavoidable [6,25]. Small molecules offer a great possibility to regulate Cas9s activity because of their advantages of relatively low price, often oral bioavailability, simple chemical synthesis, broad modification possibilities and so on [29]. Especially in comparison to the relatively expensive and otherwise complicated to handle Cas9-controlling therapeutic proteins [28]. In this review a broad spectra of small molecules that could influence Cas9 mediated gene editing were presented. A relatively big proportion of them interacts with modified Cas9 variants and can either activate or deactivate Cas9 [25,33,35,41]. Further, control over the activity and interaction with modified sgRNA [55,58] was achieved. Also, regulation of the repair mechanisms following a DSB was shown to be possible [15,47,53]. But not only the activity of modified Cas9 could be altered by small molecules. Some molecules were proven to even downregulate Cas9wt [31,39]. However, not only indels with Cas9 are controllable [12,13]. Gene activation using endonuclease lacking dCas9 with the use of small molecules was further achievable [25]. All in all, multiple way of controlling Cas9s activity effects were found. All these methods provide promising tools in making Cas9 based technologies safer and more efficient. Either by decreasing unwanted effects e.g., off-target editing or enhancing on-target efficiency through e.g., more precise activation [6,24,39]. In the future such systems can have big impacts in medicine and chemistry. Perhaps by even combining the ideas and principles of the individual techniques.
Figures: Figures were drawn with Microsoft Word, ACD/ChemSketch and Biorender.
References
- B. Wiedenheft, S. H. Sternberg, and J. A. Doudna, ‘RNA-guided genetic silencing systems in bacteria and archaea’, Nature 2012 482:7385, vol. 482, no. 7385, pp. 331–338, Feb. 2012. [CrossRef]
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna, and E. Charpentier, ‘A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity’, Science (1979), vol. 337, no. 6096, 2012. [CrossRef]
- Y. Xu and Z. Li, ‘CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy’, Comput Struct Biotechnol J, vol. 18, pp. 2401–2415, Jan. 2020. [CrossRef]
- E. Shrock and M. Güell, ‘CRISPR in Animals and Animal Models’, Prog Mol Biol Transl Sci, vol. 152, pp. 95–114, 2017. [CrossRef]
- M. Adli, ‘The CRISPR tool kit for genome editing and beyond’, Nat Commun, vol. 9, pp. 1–13, 2018. [CrossRef]
- S. W. Lee et al., ‘Identification and Optimization of Novel Small-Molecule Cas9 Inhibitors by Cell-Based High-Throughput Screening’, J Med Chem, vol. 65, no. 4, pp. 3266–3305, Feb. 2022. [CrossRef]
- F. V. Karginov and G. J. Hannon, ‘The CRISPR System: Small RNA-Guided Defense in Bacteria and Archaea’, Mol Cell, vol. 37, no. 1, pp. 7–19, Jan. 2010. [CrossRef]
- F. Safari, K. Zare, M. Negahdaripour, M. Barekati-Mowahed, and Y. Ghasemi, ‘CRISPR Cpf1 proteins: structure, function and implications for genome editing’, Cell & Bioscience 2019 9:1, vol. 9, no. 1, pp. 1–21, May 2019. [CrossRef]
- M. A. Mengstie and B. Z. Wondimu, ‘Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing’, Biologics, vol. 15, p. 353, 2021. [CrossRef]
- K. S. Makarova and E. V. Koonin, ‘Annotation and classification of CRISPR-Cas systems’, Methods in Molecular Biology, vol. 1311, pp. 47–75, 2015. [CrossRef]
- F. Jiang and J. A. Doudna, ‘CRISPR–Cas9 Structures and Mechanisms’, https://doi.org/10.1146/annurevbiophys-062215-010822, vol. 46, pp. 505–529, May 2017. [CrossRef]
- E. Weterings and D. J. Chen, ‘The endless tale of non-homologous end-joining’, Cell Research 2008 18:1, vol. 18, no. 1, pp. 114–124, Jan. 2008. [CrossRef]
- M. R. Lieber, ‘The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway’, Annu Rev Biochem, vol. 79, pp. 181–211, Jul. 2010. [CrossRef]
- J. San Filippo, P. Sung, and H. Klein, ‘Mechanism of Eukaryotic Homologous Recombination’. https://doi.org/10.1146/annurev.biochem.77.061306.125255, vol. 77, pp. 229–257, Jun. 2008. [CrossRef]
- G. Li et al., ‘Small molecules enhance CRISPR/ Cas9-mediated homology-directed genome editing in primary cells OPEN’, Scientific Reports , vol. 7, pp. 1–11, 2017. [CrossRef]
- H. Wang and X. Xu, ‘Microhomology-mediated end joining: New players join the team’, Cell Biosci, vol. 7, no. 1, pp. 1–6, Jan. 2017. [CrossRef]
- C. A. Lino, J. C. Harper, J. P. Carney, and J. A. Timlin, ‘Delivering CRISPR: a review of the challenges and approaches’. https://doi.org/10.1080/10717544.2018.1474964, vol. 25, no. 1, pp. 1234–1257, 2018. [CrossRef]
- M. Lee and H. Kim, ‘Therapeutic application of the CRISPR system: current issues and new prospects’, Hum Genet, vol. 138, no. 6, pp. 563–590, Jun. 2019. [CrossRef]
- A. Mahas, C. Neal Stewart, and M. M. Mahfouz, ‘Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation’, Biotechnol Adv, vol. 36, no. 1, pp. 295–310, Jan. 2018. [CrossRef]
- A. A. Dominguez, W. A. Lim, and L. S. Qi, ‘Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation’, Nat Rev Mol Cell Biol, vol. 17, 2015. [CrossRef]
- R. Urrutia, ‘KRAB-containing zinc-finger repressor proteins’, Genome Biol, vol. 4, no. 10, pp. 1–8, Sep. 2003. [CrossRef]
- J. Armando Casas-Mollano, M. H. Zinselmeier, S. E. Erickson, and M. J. Smanski, ‘CRISPR-Cas Activators for Engineering Gene Expression in Higher Eukaryotes’, CRISPR J, vol. 3, no. 5, pp. 350–364, Oct. 2020. [CrossRef]
- C. K. S. Karlson, S. N. Mohd-noor, N. Nolte, and B. C. Tan, ‘CRISPR/dCas9-Based Systems: Mechanisms and Applications in Plant Sciences’, Plants, vol. 10, no. 10, Oct. 2021. [CrossRef]
- S. A. Gangopadhyay et al., ‘Precision Control of CRISPR-Cas9 Using Small Molecules and Light’, Biochemistry, vol. 58, no. 4, p. 234, Jan. 2019. [CrossRef]
- D. Manna et al., ‘A Singular System with Precise Dosing and Spatiotemporal Control of CRISPR-Cas9’, Angewandte Chemie International Edition, vol. 58, no. 19, pp. 6285–6289, May 2019. [CrossRef]
- J. Bondy-Denomy, ‘Protein inhibitors of CRISPR-Cas9’, ACS Chem Biol, vol. 13, no. 2, pp. 417–423, 2018. [CrossRef]
- P. Vyas and Harish, ‘Anti-CRISPR proteins as a therapeutic agent against drug-resistant bacteria’, Microbiol Res, vol. 257, p. 126963, Apr. 2022. [CrossRef]
- S. E. Franklin and S. P. Mayfield, ‘Expert Opinion on Biological Therapy Recent developments in the production of human therapeutic proteins in eukaryotic algae Recent developments in the production of human therapeutic proteins in eukaryotic algae’, Expert Opin. Biol. Ther, vol. 5, no. 2, pp. 225–235, 2005. [CrossRef]
- H. Beck, M. Härter, B. Haß, C. Schmeck, and L. Baerfacker, ‘Small molecules and their impact in drug discovery: A perspective on the occasion of the 125th anniversary of the Bayer Chemical Research Laboratory’, Drug Discov Today, vol. 27, no. 6, pp. 1560–1574, Jun. 2022. [CrossRef]
- B. Maji et al., ‘A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9’, Cell, vol. 177, no. 4, pp. 1067-1079.e19, May 2019. [CrossRef]
- Y. Yang et al., ‘Identification and Analysis of Small Molecule Inhibitors of CRISPR-Cas9 in Human Cells’, Cells, vol. 11, no. 22, p. 3574, Nov. 2022. [CrossRef]
- X. Cheng, ‘Valproic Acid Thermally Destabilizes and Inhibits SpyCas9 Activity’, Molecular Therapy, vol. 28, no. 12, pp. 2635–2641, Dec. 2020. [CrossRef]
- K. M. Davis, V. Pattanayak, D. B. Thompson, J. A. Zuris, and D. R. Liu, ‘Small molecule–triggered Cas9 protein with improved genome-editing specificity’, 2015. [CrossRef]
- R. Buskirk, Y. C. Ong, Z. J. Gartner, and D. R. Liu, ‘Directed evolution of ligand dependence: Small-molecule-activated protein splicing’, Proc Natl Acad Sci U S A, vol. 101, no. 29, pp. 10505–10510, Jul. 2004. [CrossRef]
- Y. Wu, L. Yang, T. Chang, F. Kandeel, and J. K. Yee, ‘A Small Molecule-Controlled Cas9 Repressible System’, Mol Ther Nucleic Acids, vol. 19, pp. 922–932, Mar. 2020. [CrossRef]
- J. C. P. N. A. , R. C. D. Rose, ‘Suppression of unwanted CRISPR-Cas9 editing by co-administration of catalytically inactivating truncated guide RNAs.’, Nat Commun, vol. 11, no. 2697, pp. 1–11, 2020.
- R. Sekine, T. Kawata, and T. Muramoto, ‘CRISPR/Cas9 mediated targeting of multiple genes in Dictyostelium’, Sci Rep, vol. 8, no. 1, 2018. [CrossRef]
- R. Weinstain, T. T. Slanina, D. Kand, and P. K. Klán, ‘Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials’, Chemicals Reviews, vol. 120, no. 24, pp. 13135–13272, 2020. [CrossRef]
- B. Maji et al., ‘Multidimensional chemical control of CRISPR–Cas9’, Nature Chemical Biology 2016 13:1, vol. 13, no. 1, pp. 9–11, Oct. 2016. [CrossRef]
- J. Krucinska et al., ‘Structure-guided functional studies of plasmid-encoded dihydrofolate reductases reveal a common mechanism of trimethoprim resistance in Gram-negative pathogens’, Communications Biology 2022 5:1, vol. 5, no. 1, pp. 1–14, May 2022. [CrossRef]
- R. A. Gama-Brambila et al., ‘A Chemical Toolbox for Labeling and Degrading Engineered Cas Proteins’, vol. 1, pp. 777–785, 2021. [CrossRef]
- C. Zhang et al., ‘π-Clamp-mediated cysteine conjugation’, Nat Chem, vol. 8, pp. 120–128, 2016. [CrossRef]
- M. Békés, D. R. Langley, and C. M. Crews, ‘PROTAC targeted protein degraders: the past is prologue’, Nat Rev Drug Discov, vol. 21, pp. 181–200, 2022. [CrossRef]
- S. Kim, D. Kim, S. W. Cho, J. Kim, and J. S. Kim, ‘Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins’, Genome Res, vol. 24, no. 6, pp. 1012–1019, Jun. 2014. [CrossRef]
- Bubeck et al., ‘Engineered anti-CRISPR proteins for optogenetic control of CRISPR–Cas9’, Nature Methods 2018 15:11, vol. 15, no. 11, pp. 924–927, Oct. 2018. [CrossRef]
- M. Srivastava et al., ‘An Inhibitor of Nonhomologous End-Joining Abrogates Double-Strand Break Repair and Impedes Cancer Progression’, Cell, vol. 151, no. 7, pp. 1474–1487, Dec. 2012. [CrossRef]
- H. C. Bermudez-Cabrera et al., ‘Small molecule inhibition of ATM kinase increases CRISPR-Cas9 1-bp insertion frequency’, Nat Commun, vol. 12, pp. 1–10, 2021. [CrossRef]
- S. Ueno, T. Sudo, and A. Hirasawa, ‘ATM: Functions of ATM Kinase and Its Relevance to Hereditary Tumors’, International Journal of Molecular Sciences 2022, Vol. 23, Page 523, vol. 23, no. 1, p. 523, Jan. 2022. [CrossRef]
- D. B. Lombard, K. F. Chua, R. Mostoslavsky, S. Franco, M. Gostissa, and F. W. Alt, ‘DNA Repair, Genome Stability, and Aging’, Cell, vol. 120, no. 4, pp. 497–512, Feb. 2005. [CrossRef]
- Z. Mao, M. Bozzella, A. Seluanov, and V. Gorbunova, ‘Comparison of nonhomologous end joining and homologous recombination in human cells’, DNA Repair (Amst), vol. 7, no. 10, pp. 1765–1771, Oct. 2008. [CrossRef]
- Y. Chen et al., ‘A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites’, Nucleic Acids Res, vol. 47, no. 16, pp. 8563–8580, Sep. 2019. [CrossRef]
- I. E. Wassing et al., ‘The RAD51 recombinase protects mitotic chromatin in human cells’, Nature Communications 2021 12:1, vol. 12, no. 1, pp. 1–17, Sep. 2021. [CrossRef]
- W. Zhang et al., ‘A high-throughput small molecule screen identifies farrerol as a potentiator of CRISPR/Cas9-mediated genome editing’, Elife, vol. 9, pp. 1–25, Jul. 2020. [CrossRef]
- K. N. Meek, A. E. Rangel, and J. M. Heemstra, ‘Enhancing aptamer function and stability via in vitro selection using modified nucleic acids’, Methods, vol. 106, pp. 29–36, Aug. 2016. [CrossRef]
- R. S. Iwasaki, B. A. Ozdilek, A. D. Garst, A. Choudhury, and R. T. Batey, ‘Small molecule regulated sgRNAs enable control of genome editing in E. coli by Cas9’, Nature Communications 2020 11:1, vol. 11, no. 1, pp. 1–9, Mar. 2020. [CrossRef]
- A. Wrist, W. Sun, and R. M. Summers, ‘The Theophylline Aptamer: 25 Years as an Important Tool in Cellular Engineering Research’, ACS Synth Biol, vol. 9, no. 4, pp. 682–697, Apr. 2020. [CrossRef]
- S. W. Lee, L. Zhao, A. Pardi, and T. Xia, ‘Ultrafast dynamics show that the theophylline and 3-methylxanthine aptamers employ a conformational capture mechanism for binding their ligands’, Biochemistry, vol. 49, no. 13, pp. 2943–2951, Apr. 2010. [CrossRef]
- B. Lin et al., ‘Control of CRISPR-Cas9 with small molecule-activated allosteric aptamer regulating sgRNAs †’, Chem. Commun, vol. 55, p. 12223, 2019. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



