
Submitted:
12 December 2023
Posted:
13 December 2023
You are already at the latest version
Abstract
Mixtures of ethanol and methanol being synthesized from CO2 and green H2 can serve as sustainable base chemicals for a number of chemical processes. Amongst these processes, the catalytically-supported synthesis of CO2-neutral C4 to C10 alcohols is of increasing importance as, e.g., iso-butanol can be used as a drop-in fuel or after dehydration to produce iso-butene as a feedstock for the synthesis of plastics. 2-ethyl-hexanol can be further refined into solvents, tensides, or monomers. In this respect, NiPt alloys on an activated carbon support were found to be active and stable catalysts for the synthesis of iso-butanol following the Guerbet reaction scheme. In this study, two different routes are applied to the synthesis of these NiPt catalysts: a more conventional one based on the impregnation of Ni and Pt salts and an advanced path with a surface redox reaction between elemental Ni on the support and Pt ions in a polar solution. The experimental evaluation showed that the Pt particles from the surface redox reaction being exposed on the Ni particles are more active than those on the impregnated catalysts due to their high surface energy. Their specific space-time yields were 10–20 times higher.
Keywords:
Mixed alcohols
; catalytic upgrading
; iso-butanol
; NiPt alloy catalysts
; Guerbet reaction
; surface redox reaction
1. Introduction
The human-caused increase in global temperatures had already reached 1.1 K in 2017 compared to the pre-industrial era. If the emissions of the greenhouse gases responsible for global warming – above all CO2 – are not significantly reduced, the temperature increase by 2040 will probably total around 1.5 K [1,2]. Based on previous data and experience, it can be assumed that an increase of this magnitude would lead to major environmental damage worldwide (e.g., floods, forest fires, severe storms, etc.) and to societal tensions in regions of the world that are particularly affected if soils dry out and water resources become scarce or dry up entirely. In response to this problem, many countries, along with the European Union, have established ambitious targets for reducing the emission of greenhouse gases for the coming decades up to around the year 2050. In order to achieve these goals, new concepts, materials and technologies are required in all sectors of the energy economy (heat, transport, and industry). In this context, the currently dominant approach in the transport sector is to completely dispense with carbon-containing substances as energy carriers (gasoline, diesel, natural gas) for vehicle drivetrains and to electrify these using batteries and electric motors. Although this electrification in the mass market of passenger cars will progress in the coming years, there will still be a profound need for green and sustainable fuels wherever large masses must be transported over long distances. This applies, for example, to large cargo ships, long-haul aircraft, long-distance road freight transport, as well as the existing car fleet, as long as the electrification of the transport sector, which will certainly take many years, is not yet complete. In addition, there are a large number of industrial chemical processes that cannot run without platform chemicals containing carbon. These have thus far relied on oil and natural gas. In order to be able to defossilize these processes, CO2-neutral basic chemicals must be made available for further processing on a large scale. In this context, the focus of scientific research in recent past has primarily been on the synthesis of the molecules of methanol, ethanol, and dimethyl ether, which are regarded as suitable basic chemicals for a large number of chemical processes. These can be synthesized from CO2 that has been separated from the air, biogenic sources, or industrial exhaust gases and green hydrogen (from solar or wind-powered electrolysis). Thus, they meet the requirement of CO2-neutrality. In this context, the IEK-14 at the Forschungszentrum Jülich is working on the catalytically-supported further processing (“value upgrading”) of mixtures of green ethanol and green methanol to produce higher-quality products. The main focus is on isobutanol (C4), which can be used as a drop-in fuel for internal combustion engines [3] and alcohols with longer carbon chains (C4-C10). Further prominent representatives of the C4-C10 alcohols are 2-ethyl-1-hexanol, n-octanol, and n-decanol. The principle of the Guerbet reaction is used for this, in which ethanol and methanol are first dehydrogenated and then the resulting aldehydes enter into an aldol condensation with one another. It was found in previous work that PtNi alloy catalysts supported on activated carbon show a promising catalytic behavior for the Guerbet reaction [4]. This is why these catalysts were used for the investigations in this study.
Figure 1.
Reaction pathway for the synthesis of iso-butanol from ethanol and methanol [5].
Figure 1.
Reaction pathway for the synthesis of iso-butanol from ethanol and methanol [5].
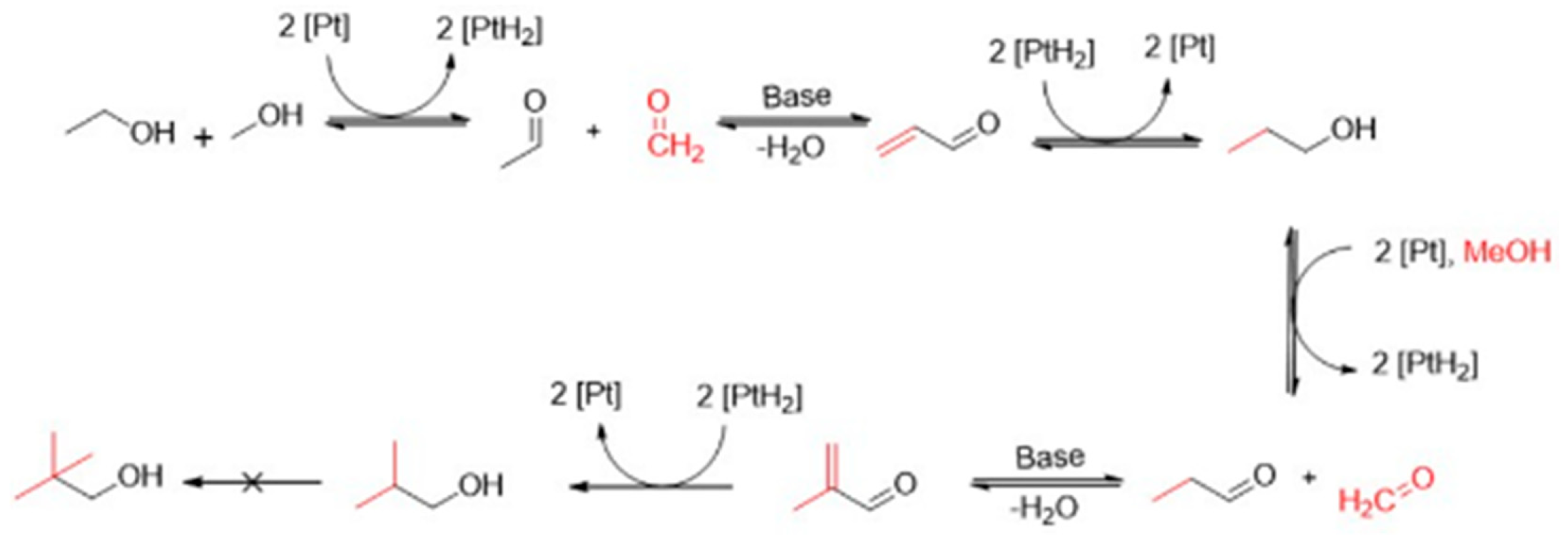
The overall approach of this paper is: (i) to apply two different routes to the synthesis of NiPt alloy catalysts deposited on an activated carbon support; and (ii) to investigate their influences on the catalytic activity for the above-presented Guerbet reaction, yielding iso-butanol. In any case and independent of the synthesis route, a total weight metal loading for Ni plus Pt of 10% was aimed for. The first, more conventional synthesis route provides the impregnation of Ni and Pt salts on the surface of an activated carbon support, followed by subsequent calcination and reduction to form elemental metal particles on the surface of the support. In the case of this route, two different samples with atomic Ni:Pt ratios of 99:1 and 95:5, respectively, were prepared. Their acronyms herein will be IMP Ni99Pt1/C and IMP Ni95Pt5/C, respectively. The second synthesis route, however, starts with a catalyst, for which in the first step only Ni is conventionally deposited on the activated carbon support via impregnation of the Ni salt, followed again by calcination and reduction. Then, this Ni/C catalyst is given to a polar solution, in which Pt is present in ionic form. This experimental set-up now allows for a so-called surface redox reaction (SRR) between elemental Ni and the Pt ions. This synthesis approach – also called galvanic replacement or transmetallation – was fundamentally investigated and introduced by Brankovic et al. [6,7], Sasaki at al. [8], and van Brussel et al. [9,10]. Pt has a standard potential of 1.20 V and Ni of -0.25 V. The difference between the two half-cell potentials is used to calculate the electrochemical potential of the overall redox reaction. In the reaction between Ni and Pt, this is 1.45 V. The difference is large enough for the redox reaction to take place voluntarily. This enables the Pt ions to be reduced to elemental Pt and the Ni to be oxidized to form Ni2+ ions. The electrons migrate from the base Ni to the noble Pt in the polar solution, as is shown in the following reaction equation:
2 Ni + [PtCl6]2- → Pt(Ni) + 2 Ni2+ + 6 Cl-
Hereby, the Ni2+ ions pass into the polar solution. This exchange is assumed to happen at a ratio of 1 [11]. The goal of the SRR procedure aims at generating Pt particles that are exposed on the Ni particles, thus possessing a high surface energy, while, on the contrary, for the conventional Ni-impregnated catalysts, the Pt particles could also be hidden in the Ni bulk phase and so be inaccessible to the reactants. The SRR synthesis exploits the fact that the energy required for the exposed Pt particles to diffuse into the Ni bulk phase cannot be raised under the given reaction conditions. Thereby, it was hypothesized that synthesis via SRR leads to improved activity of the catalysts. In the literature, PtNi catalysts supported on activated carbon supports and synthesized via surface redox reactions were tested for the methanol oxidation reaction against the background of fuel cell applications [12,13,14]. Hu et al. [12] detected a better electrocatalytic performance with improved Pt utilization efficiency for hollow mesoporous PtNi nanospheres, while Tamašauskaitė-Tamašiūnaitė et al. [13] showed that nano-Pt(Ni)/Ti and nano-Pt/Ti catalysts are more active with respect to the oxidation of borohydride, ethanol, and methanol compared with that of pure Pt. Wang et al. [14] found higher activity and stability of their Pt-decorated Ni nanoparticles compared to conventional Pt/C and PtRu/C catalysts. Meanwhile, Burger et al. [15] used the surface redox reaction principle to synthesize Ni–Al catalyst with Fe doping. They observed the formation of Ni–Fe alloys to which they attributed an increase in catalytic activity and better thermal stability.
2. Results and discussion
2.1. NiPt catalysts prepared by conventional wet impregnation
This section presents and discusses the experimental results obtained from the two impregnated catalysts IMP Ni99Pt1/C and IMP Ni95Pt5/C. At first, the findings from the catalyst characterization (X-ray Diffraction (XRD) analysis, Inductively Coupled Plasma combined with Optical Emission Spectroscopy (ICP–OES), Temperature Programmed Desorption of H2 (H2–TPD), Transmission Electron Microscopy (TEM) / Scanning Electron Microscopy (SEM), combined with Energy Dispersive X-ray Spectroscopy (EDX)) are shown, followed by an explanation of the catalytic behaviors of the two samples.
2.1.1. XRD analysis
Figure 2 presents the XRD patterns of the catalysts IMP Ni99Pt1/C and IMP Ni95Pt5/C, respectively, that were prepared via the above-described impregnation method. All XRD recordings from this figure show reflections between 2Θ = 20° and 30°, and at 80°. These reflections have already been observed in previous measurements of various carbon-supported catalysts [4]. They represent the peaks of the amorphous activated carbon support itself.
For the catalyst IMP Ni99Pt1/C, no Pt phase can be seen in the XRD pattern. At a Ni:Pt ratio of 95:5, however, the Pt can be recognized by weak reflections at 2Θ = 40° and 46°, which are superimposed by Ni reflections at 2Θ = 45° and 52°. For both catalysts, there is no NiO-phase detectable, which would have had signals at 2Θ = 37° and 43°. These results suggest that the reduction of the catalyst in the tube furnace following impregnation was successful and that the metals were subsequently available in reduced form. The absence of the Pt reflections of the catalyst with a 99:1 Ni:Pt ratio does not rule out the presence of Pt as the closely spaced, significantly broader and more intense Ni reflections might have overlaid the Pt signals.
2.1.2. ICP–OES
Table 1 depicts a comparison of values for the weight and atomic percentages of the two impregnated NiPt catalysts, when the values were determined, on the one hand, by ICP–OES measurements and, on the other, when they were calculated on the basis of the numbers from the experimental syntheses.
It becomes obvious that in the case of the IMP Ni99Pt1/C catalyst, there is a significant difference between the two values for wt% Ni and wt% Pt, respectively. However, the intended atomic ratio of 99:1 was achieved. In contrast, for the catalyst IMP Ni95Pt5/C, the intended atomic ratio of 95:5 could not be set and amounted to only 98:2. The observed differences in the values for the weight percentages result from the fact that some portions of the impregnation solution were retained on the beaker glass walls and in the pipette instead of being deposited on the activated carbon support.
2.1.3. H2–TPD
Figure 3 presents the results of an H2–TPD experiment with an IMP Ni95Pt5/C catalyst. The sample was heated in a stream of Ar from 35 °C to 800 °C, where it was held for one hour. The corresponding thermal conductivity detector (TCD) signal is plotted on the left Y-axis, whereas the mass spectrometer signals for H2 and CO2 can be seen on the right Y-axis. Figure 3 shows a significant increase in the TCD signal normalized to the sample weight between 400 °C and 500 °C. When looking at the signals measured using a mass spectrometer, this increase was due to the desorption of substances with the m/z values of 2 for H2, and 44 for CO2. CO2 achieved the highest signal intensity. The H2 peak extends over the entire temperature range between 450 °C and 800°C and reaches its maximum signal intensity at around 470 °C. The occurrence of the signal between 400 °C and 500 °C is explained by H2 spillover from Pt to the activated carbon support at around 350 °C. In this spillover process, H2 on the Pt surface is dissociated by carboxyl and lactate groups and migrates to the surface of the activated carbon support. There, it is more strongly bound, which is why the peak only occurs at higher temperatures [16,17]. A very similar behavior during H2–TPD was observed with the IMP Ni99Pt1 catalyst.
2.1.4. TEM / SEM combined with EDX
Figure 4 shows the TEM images of the impregnated catalysts IMP Ni99Pt1/C (above) and IMP Ni95Pt5/C (below) at different magnifications. In both cases, the carbon particles shown are evenly coated with metal nanoparticles. There are no local clusters. The particles are separate from each other and primarily round. The largest particles have a diameter of approximately 10 nm, whereas the value for the smallest particles is a few nanometers.
Figure 5 depicts the SEM images of the impregnated catalysts IMP Ni99Pt1/C (above) and IMP Ni95Pt5/C (below) at different magnifications. It can be seen that the largest activated carbon particles have dimensions of 80 x 60 µm. They are elongated and T-shaped with partition walls 10 µm apart. The partition walls are two µm-wide. No pore structures are visible. The particles have a smooth surface and an irregular edge structure. The EDX analysis demonstrated an even signal intensity for both the Ni and Pt metals over the entire activated carbon particle.
2.1.5. Catalytic activity
Table 2 presents the yields (Y) of, and selectivities (S) towards, the products of the reaction from Figure 1 on the two impregnated catalysts. A final concentration of iso-butanol of 14.72 mmoles l-1 was measured after four hours in the case of IMP Ni99Pt1/C, which corresponds to a yield of 2.45% based on the inlet concentration of 600 mmoles l-1 ethanol with a selectivity of 61.03% towards iso-butanol. In addition, the specific space-time yield (STY) was calculated from the final iso-butanol concentration. For this purpose, the number of moles of generated iso-butanol molecules were related to the sum of the masses of Pt and Ni over the entire reaction period according to equation (2). The masses of Pt and Ni were in turn calculated using equation (3). Here, the weighed catalyst mass was divided by weight percentages for Pt and Ni being experimentally determined via ICP–OES (cf. Table 1). For the impregnated catalyst with a Ni:Pt atomic ratio of 99:1, the specific STY is 1.02 mole g-1 h-1. A final concentration of 18.04 mmoles l-1 of iso-butanol was obtained with the impregnated Ni95Pt5/C catalyst. This corresponds to a yield of 3.01%, with a selectivity towards iso-butanol of 69.49%. The STY was 1.35 mole g-1 h-1. As can be seen from Table 2, the primary by-product of the Guerbet condensation is 1-hexanol with both catalysts, with a selectivity towards 1-hexanol of 15.32% and 15.47%, respectively. In addition, 2-methylpropanal is synthesized with both catalysts, with a selectivity of almost 9%. 2-ethylhexane-1-ol was only synthesized on the IMP Ni99Pt1/C catalyst.
2.2. NiPt catalysts prepared by surface redox reactions
To investigate the possible benefit of the surface redox reaction for the Pt doping onto the Ni-impregnated activated carbon support, a series of catalysts based on the conventionally-synthesized Ni/C sample was prepared. An important criterion in optimizing the SRR catalysts is the quantity of Pt on the surface of the Ni particles supported on the activated carbon support and the amount of Ni dissolved in the polar solution. Ni is then no longer available for catalyzing the Guerbet reaction. For this reason, Mintsouli et al. [11] chose a 0.1 M HCl polar solution for their investigations. As they observed, however, a large quantity of 46 wt% Ni dissolved in the 0.1 M HCl and then formed NiCl2; for this work, an HCL/KCl buffer was also applied, which reduces the number of Cl ions in the solution and thus aims to minimize the Ni quantities that get lost. A third route for this study was applying a further diluted 0.01 M HCl solution with plainly fewer Cl ions and a potentially reduced formation of NiCl2 in the solution.
The different catalysts are abbreviated as follows in the next sections:
NiPt/C synthesized in an HCl/KCl buffer
Stirring for 15 min → SRR 15 buffer
Stirring for 30 min → SRR 30 buffer
Stirring for 60 min → SRR 60 buffer
NiPt/C synthesized in a 0.1 M HCl solution
Stirring for 15 min → SRR 15 0.1 HCl
Stirring for 30 min → SRR 30 0.1 HCl
NiPt/C synthesized in a 0.01 M HCl solution
Stirring for 5 min → SRR 5 0.01 HCl
For these catalysts, the initial findings from the catalyst characterization (VIS spectroscopy/ICP–OES, XRD, Thermal Gravimetric Analysis (TGA), and TEM / SEM combined with EDX, and H2–TPD) are shown, followed by an explanation of their catalytic behaviors.
2.2.1. VIS spectroscopy/ICP–OES
The quantities of Ni in the three different buffer solutions were determined by means of VIS spectroscopy and ICP–OES. The photometric determination was possible due to the green color of NiCl2 in the buffer solution. Table 2 summarizes the obtained values for the Ni quantities in the buffer solutions after 15, 30, and 60 min of stirring. It makes clear that VIS spectroscopy is a precise, quick, and reliable method for determining the Ni loss, as there is only a very small difference in the range of 1% to the values obtained via ICP–OES. It also becomes evident that the Ni values in the buffer solutions are significant and independent of the reaction time.

Table 2.
Comparison of the quantities of Ni being dissolved in the polar buffer solutions measured by means of (i) Vis spectroscopy and (ii) ICP–OES.
Table 2.
Comparison of the quantities of Ni being dissolved in the polar buffer solutions measured by means of (i) Vis spectroscopy and (ii) ICP–OES.
| Catalyst | βVIS [mg l-1] | βICP-OES [mg l-1] | |
|---|---|---|---|
| SRR 15 buffer | 942 | 930 | |
| SRR 30 buffer | 931 | 930 | |
| SRR 60 buffer | 919 | 910 |
As a complement to table 2, Table 3 shows the values for Ni and Pt weight percentages on the three buffer-based NiPt SRR catalysts, when the values were determined, on the one hand, via ICP–OES measurements and, on the other, when they were calculated based on the numbers derived from the experimental syntheses. It is obvious from the table that the reaction time did not have a significant influence on the weight values for the Ni and Pt metals. In the case of Pt, the difference between the values determined via ICP–OES and the calculated values based on weighed portions of the educt chemicals was in the range of approximately 25%. In any case, the calculated values were higher than the experimentally-determined ones. These results mean that the deposition of Pt on the activated carbon support proceeded to significant extents. However, when analyzing the corresponding Ni values, it becomes obvious that there is a huge difference between the calculated numbers and data from ICP–OES, which is due to the fact that the Ni particles dissolved into the buffer solution in large amounts. This is in good agreement with the results from Table 2. It can be concluded that the SRR reaction using the KCl/HCl buffer is not yet the optimal solution.
For the measurements depicted in Table 4, the buffer solution was no longer used but rather different HCl solutions, as proposed by Mintsouli et al. [11]. It can be seen that the differences in the Ni values were comparably high when a 0.1 M HCL solution was used and the reaction time amounted to 15 min. However, when further reducing the reaction time to 5 min and diluting the HCl solution to a value of 0.01 M, 7.33 wt% for Ni were measured via ICP–OES. This is fairly close to the calculated value of 8.59 wt% and much higher than the data previously presented in tables 3 and 4, which were in the range of 0.10 wt% to 0.40 wt%. These reaction conditions are much more promising, as the value for Pt was also reasonable compared to the calculated one (0.24 wt% vs. 0.38).
2.2.2. XRD analysis
Figure 6 depicts the XRD patterns of the three samples SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl, each of them representing the synthesis via the three different polar solutions. In the case of the SRR 5 0.01 HCl catalyst, two sharp reflections at 2Θ = 45° and 52° can be observed, indicating crystalline Ni phases, while in parallel no Pt signal was found for this sample. As Table 4 shows high values for the Ni weight loading on the surface of 7.33% in comparison to 0.24% for Pt, it is assumed that the supposable Pt reflections at 2Θ = 40° and 46° are superimposed by the described signals for Ni. For catalysts SRR 15 buffer and SRR 15 0.1 HCl, which both have very small quantities of Ni weight loading on their surfaces (0.32% and 0.10%, respectively), broad reflections again at 2Θ = 45° and 52° were found together with very small signals for Pt at 2Θ = 40° and 46°. The lower intensities and broader appearances of the Ni reflections are due to the small quantities of Ni on the carbon support.
2.2.3. TGA
The TGA measurements revealed for all catalysts a major mass loss up to a temperature of 150 °C, which can be explained by the desorption of physisorped H2O. These measurements allowed the calculation of the mass proportions of H2O of each SRR catalyst, which amounted to 5–10% related to the total mass of the catalyst.
2.2.4. TEM / SEM combined with EDX
Figure 7 shows TEM images of the Ni/C catalyst and of the three samples, namely the SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl, each of them again representing the synthesis via the three different polar solutions. In the two upper images of the Ni/C catalyst, many small, darker points can be seen, which are stochastically distributed over the entire support. A total of 150 nanoparticles with an average diameter of 8.7 nm were identified. However, nanoparticles were not found on each of the activated carbon particles. The second line shows TEM images of the SRR 15 buffer catalyst. In this case, in comparison to the Ni/C catalyst, significantly fewer but also stochastically distributed nanoparticles can be seen. The average diameter of these is 7.7 nm and the total number of nanoparticles measured in the section was 15. Several stochastically-distributed nanoparticles can also be seen in the TEM images of the SRR catalyst with a reaction time of 15 minutes in 0.1 M HCl. The total number of nanoparticles in the examined section was determined to be 25. These have an average diameter of 7.9 nm. Again, nanoparticles were not found on all of the examined sections.
Both TEM images of the SRR 5 0.01 HCl catalyst display many stochastically-distributed nanoparticles. The number of nanoparticles of 150 corresponds to that of the Ni/C catalyst, whereas the other two SRR variations have significantly fewer nanoparticles. The nanoparticles have an average particle diameter of 9.5 nm. The exact size distributions are shown in the boxplot diagram in Figure 8, which shows the maximum, minimum, and mean values determined by the TEM measurements for the four different catalysts. Overall, the nanoparticle sizes of the SRR 5 0.01 HCl catalyst are the largest, with an average diameter of 9.5 nm, whereas those of the catalyst with a stirring time of 15 minutes in the KCl/HCl buffer solution are the smallest at 7.7 nm.
In the SEM images in the left column of Figure 9 for the Ni/C catalyst, many elongated layer structures can be seen at lower magnifications. At the highest magnification, in the third image of this column, however, nanoparticles can be seen as small, distributed dots. As can be determined from the lowest image of this column, the Ni distribution analyzed using EDX is predominantly stochastic. A strong green coloration can be seen in the middle-left-hand part of the EDX image. In the central segment of the EDX image, Ni is shown to be slightly less green. It must be pointed out here that in addition to the element concentrations, the color intensities in the EDX measurements also depend on the particles’ depth within the support structure. In the second column from the left, which deals with the sample SRR 15 buffer, a few clearly lighter particles that have higher atomic masses stand out. Most particles have an elongated, partially stacked shape. In addition to a few particles that are brighter overall, small bright dots can also be seen on and between the stacked structures, as is apparent in the second image from the top of this column. In this section, most of the layer structures have a length of about 100 μm, with a distance between the layers of 10–20 μm. At the highest magnification in the third image from the top, a relatively uniform surface is visible. The element mapping exhibits a non-homogeneous distribution of Ni, which accumulates on individual particles. Pt, in turn, appears to be homogeneously distributed over the entire activated carbon surface. The SEM images of the SRR catalyst, with a reaction time of 15 minutes in 0.1 M HCl in the third column from the left, again show tubular structures. Small, bright particles are spread across their surfaces. The second image from the top in this column also exhibits a very bright particle with a length of 14 μm. In the third image, which is stochastically-distributed, bright particles can be seen. These particles have different sizes of up to one micrometer. The smallest have a size of less than 50 nm. A locally-centered distribution of Ni cannot be seen from the EDX measurements. Both Ni and Pt appear to be stochastically-distributed on the catalyst surface. In the SEM image of the SRR 5 0.01 HCl catalyst (fourth column), many layered structures can be seen, as has already been established for the other catalysts. It can be seen in the middle of the upper part of the image that these have a size of up to 200 μm in both length and width. In addition, a relatively large number of bright spots can be seen, which are mostly stochastically-distributed, but sometimes accumulate. These seem to lie more on the surface of the layer structures. The second image from the top shows many similar-looking, very small, bright particles and a few much larger ones, about one micron in width. These particles are less stochastically-distributed and tend to accumulate in individual places. Four of the small particles can be seen in the third image. These look very similar, are round or spherical, and have a diameter of 75–100 nm. As can be seen in the EDX measurement in the lowest image of the fourth column, Ni can be found on almost the entire catalyst surface. In some places, such as in the middle of the right-hand section of the image, the image exhibits a much stronger green coloration, which can be attributed to the concentration of Ni or its particle depth in the structure of the carbon support. Pt is primarily distributed stochastically on the surface. However, in some places, such as the lower left quarter of the figure, there is hardly any Ni and virtually only Pt. Overall, the SEM–EDX measurements of the various SRR catalysts show all tube/layer structures of the activated carbon support. The distribution of Pt by the SRR is stochastic for the catalysts studied. Meanwhile, the Ni loading is homogeneous for the Ni/C catalyst and the SRR 15 0.1 HCl sample. The other two catalysts do not have a homogeneous Ni distribution.
2.2.5. H2–TPD
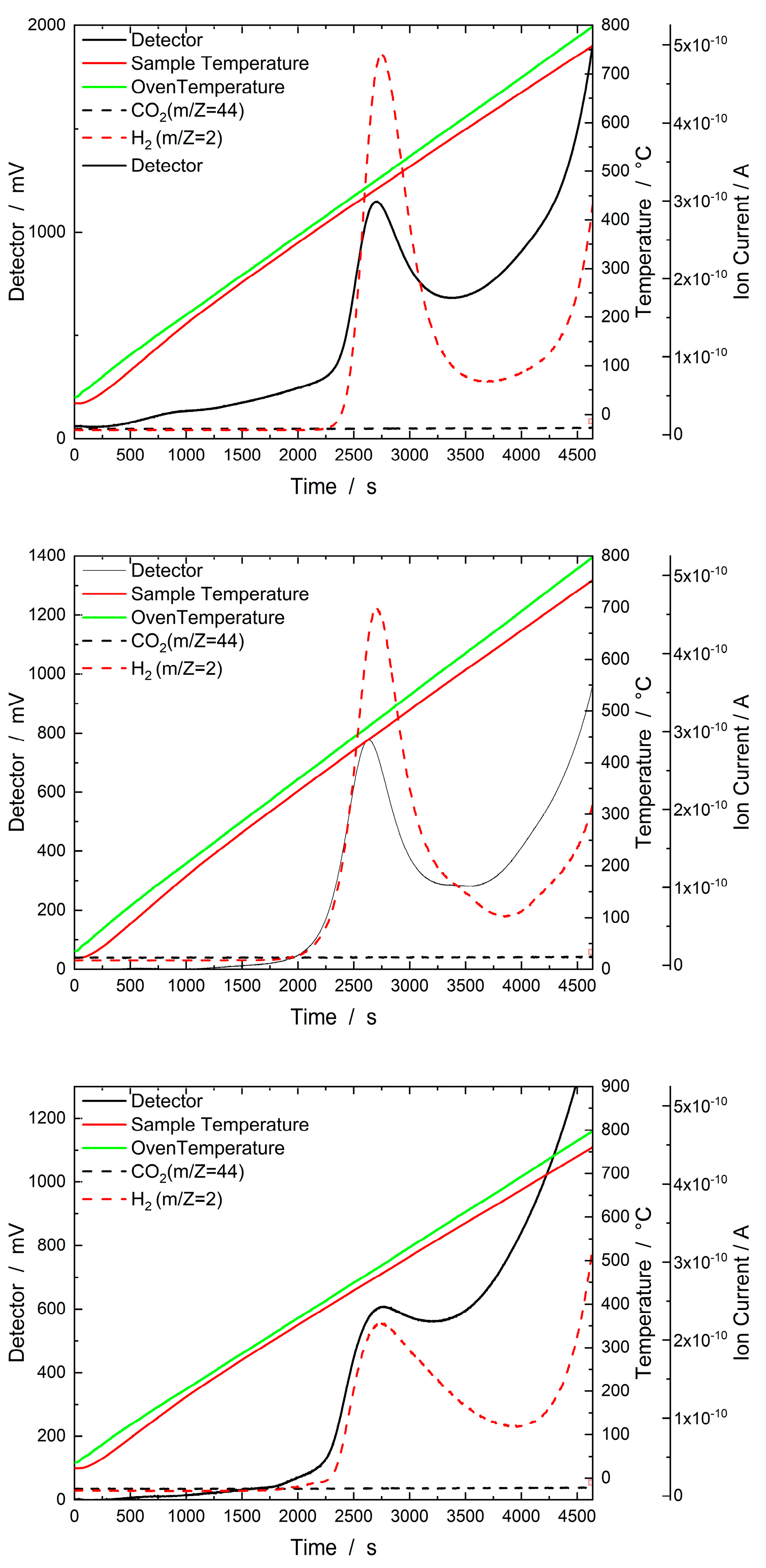
All H2–TPD measurements of the SRR catalysts show TCD signals at temperatures between 400 °C and 550 °C. In this range, a strong MS signal at an m/z value of 2 is detected for all catalysts. The signal at an m/z value of 2 is due to the desorption of H2. A CO2 peak with an m/z value of 44 was not detected for any of the SRR catalysts.
Figure 10.
Results of the H2–TPD experiment with the three samples, SRR 15 buffer (top), SRR 15 0.1 HCl (middle), and SRR 5 0.01 HCl (bottom). The samples were heated in a stream of Ar (30 ml min-1) from 35 °C to 800 °C, which was then held for one hour. The corresponding thermal conductivity detector signal is plotted on the left Y-axis, the sample and oven temperatures on the first right Y-axis, and the mass spectrometer signals (ion currents) on the second right Y-axis.
Figure 10.
Results of the H2–TPD experiment with the three samples, SRR 15 buffer (top), SRR 15 0.1 HCl (middle), and SRR 5 0.01 HCl (bottom). The samples were heated in a stream of Ar (30 ml min-1) from 35 °C to 800 °C, which was then held for one hour. The corresponding thermal conductivity detector signal is plotted on the left Y-axis, the sample and oven temperatures on the first right Y-axis, and the mass spectrometer signals (ion currents) on the second right Y-axis.
2.2.6. Catalytic activity
This subsection describes the catalytic behaviors of the different SRR catalysts and compares them with those of the conventionally impregnated samples IMP Ni99Pt1/C and IMP Ni95Pt5/C. The points of comparison are the specific space-time yields, the iso-butanol yields, and the selectivities towards iso-butanol. It becomes obvious that the specific STYs for the samples, IMP Ni99Pt1/C and IMP Ni95Pt5/C, amount to 1.02 and 1.35 mole g-1 h-1, respectively, and increase by factors of between 10 and 20 to 15.16 mole g-1 h-1 for the SRR 30 buffer and even 21.26 mole g-1 h-1 for the SRR 15 one. Although it can be clearly seen in the TEM images in figures 4 and 7, respectively, that the SRR samples have much fewer Ni and Pt nanoparticles on their activated carbon supports than the two impregnated catalysts, all four of these catalysts provide comparable iso-butanol yields of between 2% and 3%, as well as selectivities towards iso-butanol of between 60 and 70%. From these findings, the authors conclude for the SRR buffer catalysts that the Pt particles being exposed on the Ni particles are more active than those on the impregnated catalysts due to their high surface energy. When analyzing the STYs of the catalysts, which were synthesized using the HCl solution, the value of 79.87 mole g-1 h-1 for SRR 15 0.1 HCl is outstanding and partially due to the very low quantities of Ni and Pt being deposited on the activated carbon surface (cf. Table 4). The sample SRR 5 0.01 HCl, however, represents the exact opposite in terms of the quantities of Ni, with 7.33 wt% (cf. Table 4) on its activated carbon surface. According to equations (2) and (3), this then led to a comparably low value for the specific STY of only 3.30 mole g-1 h-1. However, it can be pointed out that the number for the iso-butanol yield is highest with this sample and amounts to approximately 7% at a selectivity towards iso-butanol of 76%. This is further evidence for the higher activity of the metal particles of the SRR catalysts and demonstrates the potential of this synthesis route over the conventional one. Table 5 also shows the specific surface areas of the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl, as well as those of IMP Ni99Pt1/C and IMP Ni95Pt5/C. They are comparable and amount to between 671 m2 g-1 and 780 m2 g-1. Thus, the specific surface areas are not decisive for the observed differences in the catalytic activities of the different samples.
As a complement to the values in Table 5, Table 6 summarizes the yields and selectivities with respect to the by-products of the Guerbet reaction for the samples, SRR 15 buffer, SRR 15 0.1 HCl, and SRR 5 0.01 HCl. The table shows that the main by-products of all catalysts were 2-methylpropanal and 1-hexanol. These results are comparable to those obtained with the impregnated catalysts, which are displayed in Table 2.
3. Materials and methods
3.1. Synthesis of the impregnated cataylsts
In order to produce the impregnated catalysts, the required amounts of Ni and Pt salts were first calculated for 10 wt% loading with atomic ratios of Ni:Pt of 99:1 and 95:5, respectively. To impregnate the activated carbon, the calculated amounts of Ni(NO₃)₂ · 6 H₂O and [Pt(NH3)4](NO3)2 were first weighed in a test tube. The salts were then dissolved in a predefined amount of demineralized water. The solution was prepared by stirring and, if necessary, slight heating in an oil bath. After the salts were dissolved, the green solution was added dropwise to a 100 ml Schott bottle containing the activated carbon to be impregnated. After about three drops were added, the Schott bottle was closed and the solution homogenized by tapping on a rubber ring. This was repeated until the complete solution was added to the activated carbon. The impregnated activated carbon was first dried in a rotary evaporator at 40 °C and a pressure of 40 mbar. The catalysts were then calcined at 500 °C in N2 and reduced at 250 °C in 2 vol% H2 in N2. After the reduction phase, the material was cooled to room temperature overnight in the tube furnace.
3.2. Synthesis of the catalysts via the surface redox reaction
Three different synthesis routes were applied that differ from each other with respect to the polar solution in which the surface redox reaction took place.
3.2.1. KCl/HCl buffer as a polar solution
The synthesis of the impregnated Ni/C base catalyst was performed analogously to the experimental procedure described in subsection 3.1. The difference is that only Ni (10 wt% loading) was impregnated onto the activated carbon. Calcination and reduction were performed in the same way as described in subsection 3.1. For the subsequent surface redox reaction, K2PtCl6 was dissolved in a KCl/HCl buffer solution. For this buffer, 100 ml of 0.2 M HCL was added to 400 ml of 0.2 M KCl. In the next step, the Ni/C catalyst, with a particle size smaller than 75 μm, was added to the buffer solution that also contained K2PtCl6. The suspension was first stirred at room temperature for 15, 30, and 60 minutes and afterwards centrifuged for 5 min at 0 °C and then decanted. For means of washing, the residues were slurried three times with demineralized water, centrifuged, and then decanted. The washing solutions were stored and their absorbances determined with a Vis spectrometer at 394 nm (see subsection 3.4). The washed catalyst was then transferred to a round-bottom flask, which was placed in a water bath overnight at 65 °C. The catalyst was dried in an Ar flow within a Schlenk line, which was connected to the round-bottom flask. A small flow of Ar was passed via a cannula through an attached septum into the round-bottom flask. A second cannula was then inserted into the septum to equalize the pressure. The finished catalyst was stored under an Ar atmosphere.
3.2.2. 0.1 M HCl as a polar solution
In this variation, 0.1 M HCl is used as the reaction solution instead of the KCl/HCl buffer. The impregnated Ni/C catalyst and K2PtCl6 were added to the HCl solution. Two different batches were stirred at room temperature for 15 and 30 min, respectively. The suspensions were then centrifuged, decanted and washed three times with demineralized water. The catalyst was then dried in a rotary evaporator at 40 °C and 30 mbar until no more water could be rotated off.
3.2.3. 0.01 M HCl as a polar solution
The only difference from the experimental procedure outlined in subsection 3.2.2 was that in this case, 0.01 M HCl was applied instead of 0.1 M HCl. 5 ml of 0.2 M HCL was diluted with 95 ml of demineralized water.
3.3. Catalytic experiments
To examine the catalytic activity, 250 mg of the catalysts with a grain size ≤ 75 μm were weighed in a 100 ml autoclave. The autoclave used was a batch reactor with a reactor pot made of an Inconell 600 alloy manufactured by the Parr company. The reaction temperature, stirring speed, and switch-off pressure can be controlled by software. The actual temperature, pressure, and stirring speed were recorded by the software throughout the entire reaction. In addition to the catalyst, 70 ml of a methanolic reaction solution with 0.45 mole l-1 NaOH, 0.6 mole l-1 ethanol, and 0.015 mole l-1 n-decane was added to the reactor pot. The autoclave was then closed and exposed to an N2 atmosphere. The reaction was carried out at 165 °C. The reaction time was four hours and started when the target temperature of 165 °C was reached. A sample with a volume of 1 ml was taken every 30 min for the gas–chromatographic analysis. To take a sample, the shut-off valve of the capillary tube was opened. Then, three μl of the samples taken during the reaction were transferred into headspace vials (Thermo Scientific) using a 10 μl Eppendorf pipette. The vials were closed immediately afterwards. The components within the vials were separated using a gas chromatograph (Agilent 8890 GC System) and analyzed with a mass spectrometer (GC–MS, Agilent 5977B GC7MSD). The analysis was carried out using the full evaporation method to avoid NaOH and catalyst particles getting onto the chromatography column. The chromatographic measurement took place on a 30 m long DB–Wax column with an inner diameter of 0.25 mm and a film thickness of 0.25 μm. This served as the carrier gas with a flow rate of 1.2 ml min-1. The GC program started at a temperature of 35 °C, which held for 4 min. The mixture was then heated to 200 °C at a heating rate of 20 °C min-1. This temperature was held again for 4 min. Calibration solutions made from the pure substances were used to quantify the reaction products. N-decane was used as an internal standard.
3.4. Vis spectroscopy
To determine the NiCl2 content in the reaction solutions, the respective extinctions were measured with a VWR Visible Spectrophotometer PV4 at a wavelength of 394 nm. For quantification, six different calibration solutions with mass concentrations of NiCl2 from 1400 mg l-1 to 100 mg l-1 were prepared and measured.
3.5. TGA
A PerkinElmer STA 6000 was used to determine the water content of the catalysts. Approximately 10 mg of the sample was weighed into an Al2O3 crucible and heated in two stages. First, the sample was heated from 30 °C to 150 °C at a heating rate of 15 K min-1. This temperature was held for 15 min. The sample was then heated from 150 °C to 500 °C again with a heating rate of 15 K min-1 and held at 500 °C for 15 min. It was then cooled to 100 °C at a rate of 40 K min-1. During the entire measurement, the N2 flow was 20 ml min-1. To determine the background, an empty Al2O3 crucible was measured using the same temperature program.
3.6. H2–TPD
H2–TPD was performed in three steps. First, during the rinsing phase, Ar flowed over the catalyst surface at a temperature of 35 °C with a rate of 30 ml min-1. H2 was then adsorbed on the catalyst surface in single pulses. 20 pulses of H2 with a fixed volume were pulsed at a constant temperature of 35 °C. There was a time span of 2 min between the pulses. In the third step, H2–TPD takes place. The sample was heated from 35 °C to 800 °C under an Ar flow of 30 ml min-1 at a heating rate of 100 K min-1. The temperature was maintained at 800 °C for one hour. A TPDRO 1100 Series apparatus from Thermo Fischer Scientific was used.
3.7. ICP–OES
For the ICP–OES measurements, 4–30 mg of the catalyst samples were leached with 4 ml of aqua regia at 80 °C for two hours and then diluted to a total volume of 50 ml. Each of the solutions was diluted between 10- and 100-fold. In addition, the reaction solutions from the surface redox reactions were analyzed by diluting each sample 1000-fold, 100-fold, and 10-fold. The samples were measured using a Thermo Scientific iCAP 7600.
3.8. XRD
A Bruker D8 DISCOVER with a LYNXEYE XE–T detector was used for the XRD measurements. The measurements were carried out with Cu–Kα radiation (λ = 0.15419 nm). The distance between the sample and radiation source was 430 mm. The measurements were carried out with a step size of 0.08° at 0.5 steps per second in the range of 10° ≤ 90°. The voltage was 40 kV and the current 40 A. A 2.5° Soller slit was used in the primary beam path.
3.9. N2 sorption
3.10. TEM
The TEM measurements for Figure 7 were carried out using a Philips CM 200 field emission gun transmission electron microscope. Thereby, the catalysts were suspended in iso-propanol, a drop of which was placed on a 3 mm Cu grid. The grid was coated with an ultra-thin carbon film and air dried overnight at room temperature before the analysis was undertaken. The acceleration voltage was 200 kV. The TEM images from Figure 4 were recorded using a FEI Technai G2 F20 transmission electron microscope with an acceleration voltage of 200 kV. The samples were dissolved in an iso-propanol/water-mixture and then homogenized in an ultra-sonic bath for 15 min. Two droplets of the solution were then deposited on a Cu grid. The grid was dried overnight in an oven at 60 °C.
3.11. SEM/EDX
An in-lens detector was used to measure the signal from the scanning electron microscope. An acceleration voltage of 20 kV and a working distance of 8.5 mm were utilized in all measurements.
4. Conclusions
The technique of surface redox reactions in a polar solution is described in the literature as a promising route for preparing catalysts with exposed metal particles possessing high surface energies. Thereby, the electrochemical potential of the overall redox reaction must be large enough for the noble metal to be reduced and the less noble one to be oxidized. In this context, this work concentrated on the concerted deposition of Pt particles on the surface of Ni particles supported on activated carbon via the surface redox reaction. The experimentally-determined specific space-time yield is defined as the measure of these Pt particles’ catalytic activity. It was observed in this work that the specific space-time yield of the NiPt catalysts for the iso-butanol synthesis from methanol and ethanol mixtures increased by a factor of between 10 and 20 compared to a conventional impregnation synthesis for NiPt/C. The experiments also make it clear that undesired side reactions like the formation of NiCl2 from Ni and Cl ions must be suppressed by choosing appropriate synthesis conditions. Otherwise, active catalyst species will finally be lost in the polar solution.
Author Contributions
Conceptualization, Johannes Häusler; Funding acquisition, Ralf Peters; Investigation, Friederike Woltmann; Methodology, Johannes Häusler; Supervision, Joachim Pasel and Ralf Peters; Writing – original draft, Joachim Pasel; Writing – review & editing, Joachim Pasel, Johannes Häusler and Ralf Peters. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 491111487.
Acknowledgments
Special thanks are due to the fuel synthesis team at Jülich and all project and cooperation partners. The authors would also like to thank Birgit Schumacher and Denise Günther for recording the SEM and TEM images.
Conflicts of Interest
The authors have no conflicts of interest to disclose. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- M.R. Allen, O.P. Dube, W. Solecki, IPCC Special Report Global Warming of 1.5°C, Chapter 1 - Framing and Context, FAQ 1.2, Figure 1.
- R. Rohde, Global temperature report for 2020, http://berkeleyearth.org/global-temperature-report-for-2020/.
- Y.-l. Li, Y.-c. Yu, Z.-w. Wang, J.-f. Wang, Physical and chemical properties of isobutanol-gasoline blends, 34 (2015) 908-914. [CrossRef]
- J. Häusler, Synthese von höheren Alkoholen als zukünftige Kraftstoffe für den Verkehrssektor, PhD thesis, Fakultät für Maschinenwesen, RWTH Aachen, in preparation.
- J. Pasel, J. Häusler, D. Schmitt, H. Valencia, M. Meledina, J. Mayer, R. Peters, Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products, 10 (2020) 1151. [CrossRef]
- S.R. Brankovic, J.X. Wang, R.R. Adžić, Metal monolayer deposition by replacement of metal adlayers on electrode surfaces, Surface Science 474 (2001) L173-L179. [CrossRef]
- S.R. Brankovic, J.X. Wang, Y. Zhu, R. Sabatini, J. McBreen, R.R. Adžić, Electrosorption and catalytic properties of bare and Pt modified single crystal and nanostructured Ru surfaces, Journal of Electroanalytical Chemistry 524-525 (2002) 231-241. [CrossRef]
- K. Sasaki, Y. Mo, J.X. Wang, M. Balasubramanian, F. Uribe, J. McBreen, R.R. Adzic, Pt submonolayers on metal nanoparticles—novel electrocatalysts for H2 oxidation and O2 reduction, Electrochimica Acta 48 (2003) 3841-3849. [CrossRef]
- M. Van Brussel, G. Kokkinidis, A. Hubin, C. Buess-Herman, Oxygen reduction at platinum modified gold electrodes, Electrochimica Acta 48 (2003) 3909-3919. [CrossRef]
- M. Van Brussel, G. Kokkinidis, I. Vandendael, C. Buess-Herman, High performance gold-supported platinum electrocatalyst for oxygen reduction, Electrochemistry Communications 4 (2002) 808-813. [CrossRef]
- Mintsouli, J. Georgieva, E. Valova, S. Armyanov, A. Kakaroglou, A. Hubin, O. Steenhaut, J. Dille, A. Papaderakis, G. Kokkinidis, S. Sotiropoulos, Pt–Ni carbon-supported catalysts for methanol oxidation prepared by Ni electroless deposition and its galvanic replacement by Pt, Journal of Solid State Electrochemistry 17 (2013) 435-443. [CrossRef]
- Y. Hu, Q. Shao, P. Wu, H. Zhang, C. Cai, Synthesis of hollow mesoporous Pt–Ni nanosphere for highly active electrocatalysis toward the methanol oxidation reaction, Electrochemistry Communications 18 (2012) 96-99. [CrossRef]
- L. Tamašauskaitė-Tamašiūnaitė, A. Balčiūnaitė, A. Vaiciukevičienė, A. Selskis, V. Pakštas, Investigation of nanostructured platinum–nickel supported on the titanium surface as electrocatalysts for alkaline fuel cells, Journal of Power Sources 208 (2012) 242-247. [CrossRef]
- X. Wang, H. Wang, R. Wang, Q. Wang, Z. Lei, Carbon-supported platinum-decorated nickel nanoparticles for enhanced methanol oxidation in acid media, Journal of Solid State Electrochemistry 16 (2012) 1049-1054. [CrossRef]
- T. Burger, H.M.S. Augenstein, F. Hnyk, M. Döblinger, K. Köhler, O. Hinrichsen, Targeted Fe-Doping of Ni−Al Catalysts via the Surface Redox Reaction Technique for Unravelling its Promoter Effect in the CO2 Methanation Reaction, 12 (2020) 649-662. [CrossRef]
- P. Badenes, L. Daza, I. Rodriguez-Ramos, A. Guerrero-Ruiz, Mechanism of hydrogen spillover over carbon supported metal catalysts, in: C. Li, Q. Xin, (Eds.), Studies in Surface Science and Catalysis, Elsevier, 1997, pp. 241-250.
- J.T. Miller, B.L. Meyers, M.K. Barr, F.S. Modica, D.C. Koningsberger, Hydrogen Temperature-Programmed Desorptions in PlatinumCatalysts: Decomposition and Isotopic Exchange by SpilloverHydrogen of Chemisorbed Ammonia, Journal of Catalysis 159 (1996) 41-49. [CrossRef]
- S. Brunauer, P.H. Emmett, E. Teller, Adsorption of Gases in Multimolecular Layers, Journal of the American Chemical Society 60 (1938) 309-319.
Figure 2.
XRD patterns of the catalysts prepared via the impregnation method. Catalysts were calcined at 500 °C in N2 and reduced at 250 °C in 2 vol% H2 in N2, with the following measuring conditions: 2Ө = 10° to 90°, and Cu Kα-radiation (λ = 1.5419 Å).
Figure 2.
XRD patterns of the catalysts prepared via the impregnation method. Catalysts were calcined at 500 °C in N2 and reduced at 250 °C in 2 vol% H2 in N2, with the following measuring conditions: 2Ө = 10° to 90°, and Cu Kα-radiation (λ = 1.5419 Å).
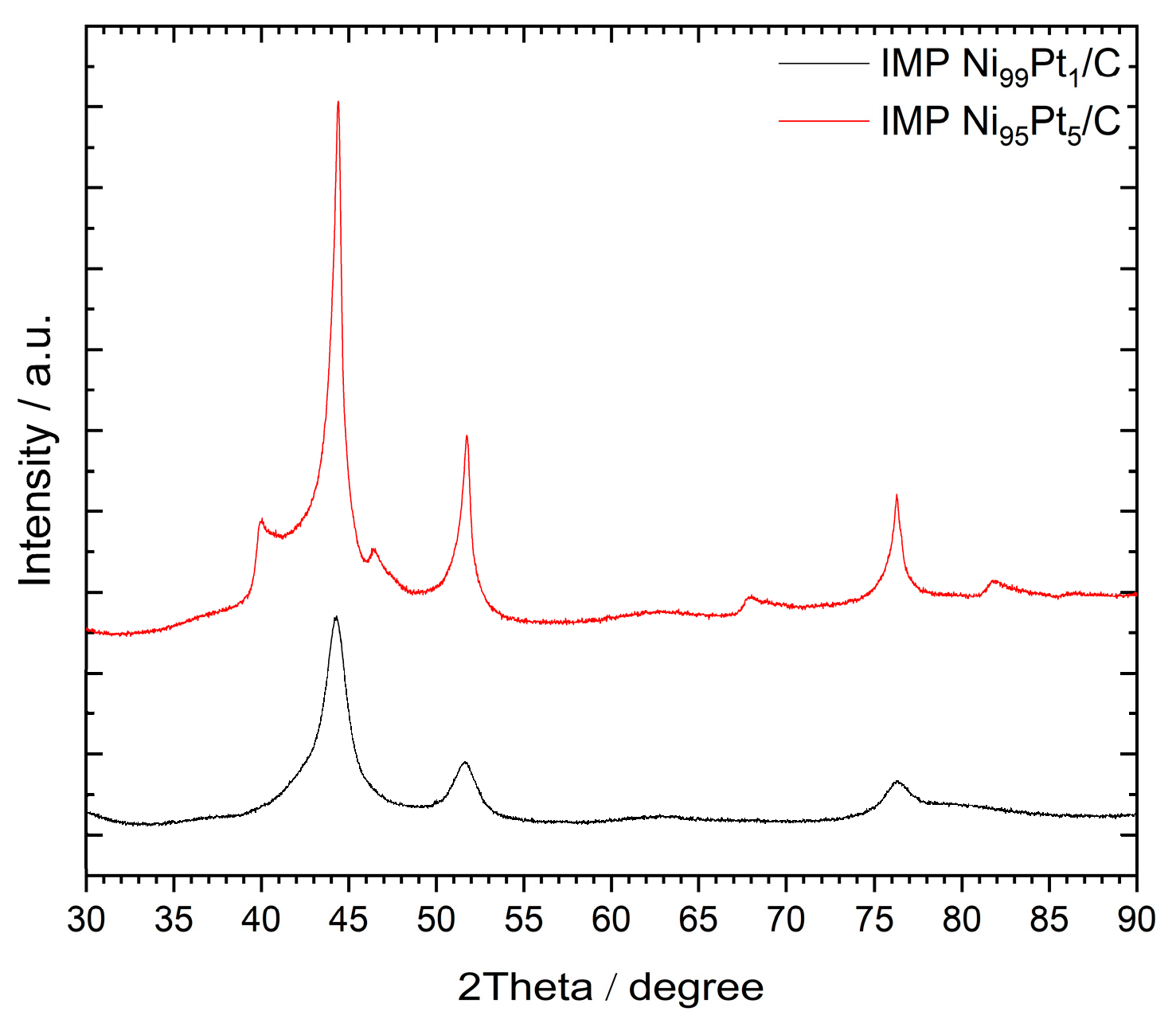
Figure 3.
Results of the H2–TPD experiment with the IMP Ni95Pt5/C catalyst. The sample was heated in a stream of Ar (30 ml min-1) from 35 °C to 800 °C, where it was held for one hour. The corresponding thermal conductivity detector signal is plotted on the left Y-axis, with the sample and oven temperatures on the first right Y-axis and mass spectrometer signals (ion currents) on the second right Y-axis.
Figure 3.
Results of the H2–TPD experiment with the IMP Ni95Pt5/C catalyst. The sample was heated in a stream of Ar (30 ml min-1) from 35 °C to 800 °C, where it was held for one hour. The corresponding thermal conductivity detector signal is plotted on the left Y-axis, with the sample and oven temperatures on the first right Y-axis and mass spectrometer signals (ion currents) on the second right Y-axis.
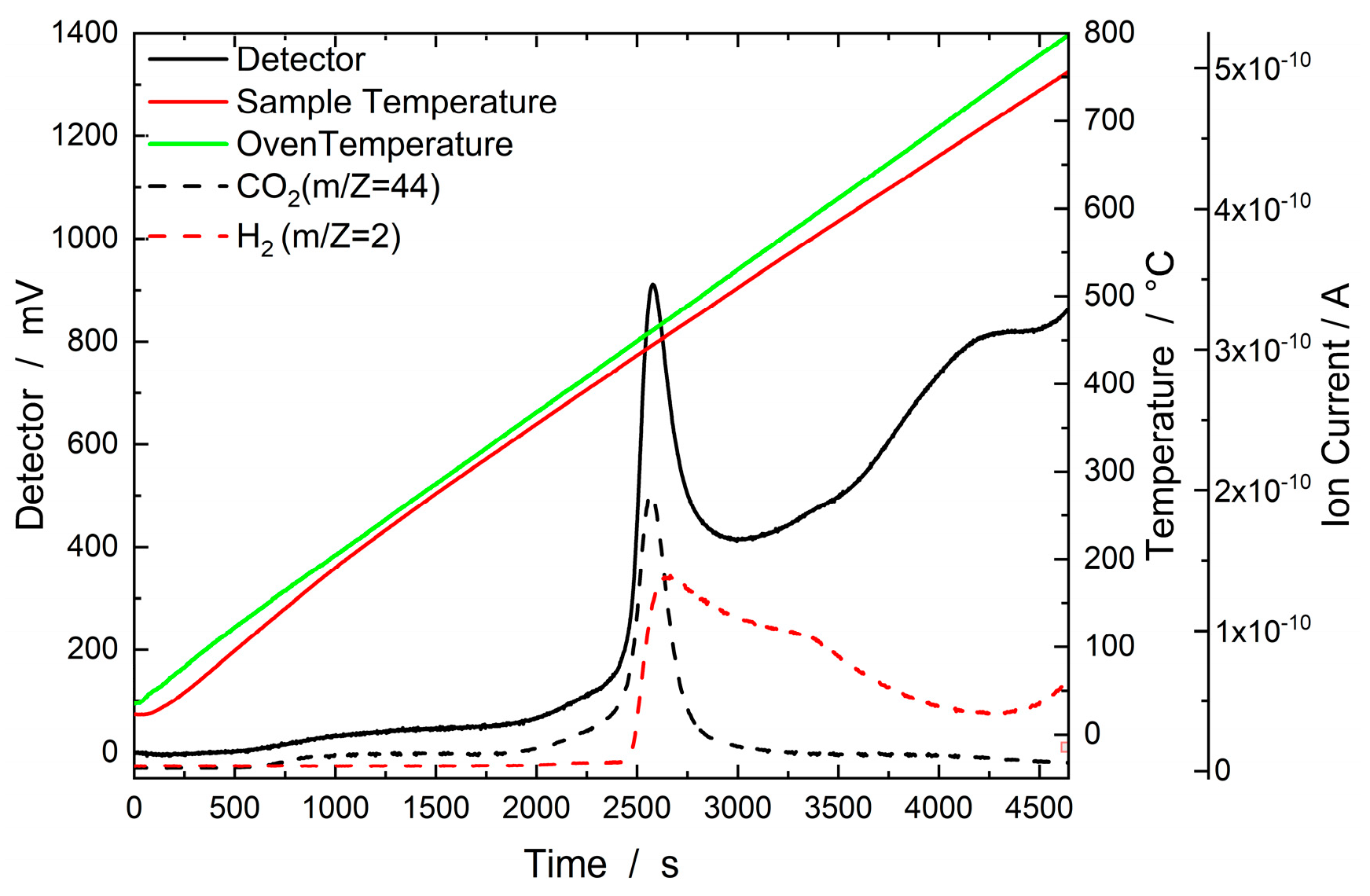
Figure 4.
TEM images of the IMP Ni99Pt1/C (above) and IMP Ni95Pt5/C (below) catalysts at different magnifications.
Figure 4.
TEM images of the IMP Ni99Pt1/C (above) and IMP Ni95Pt5/C (below) catalysts at different magnifications.
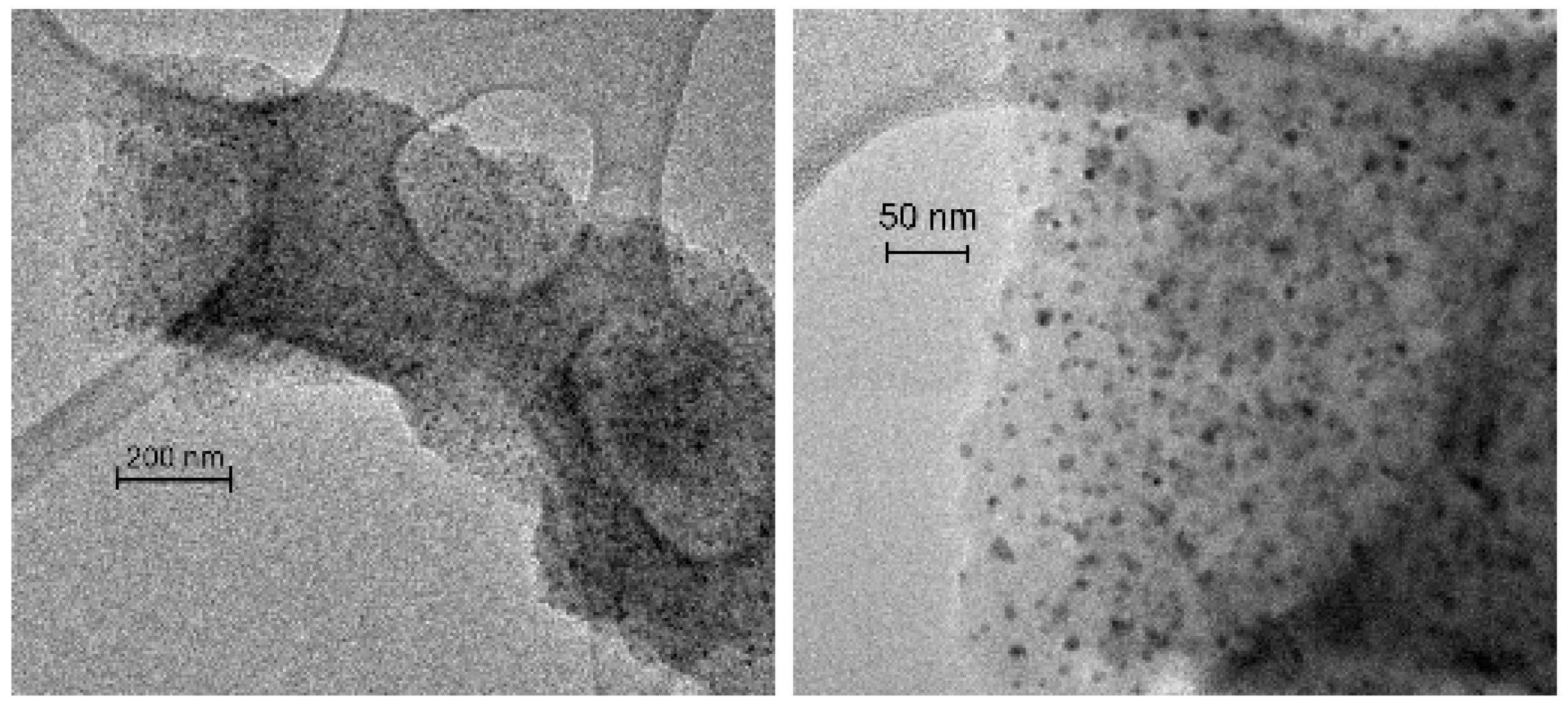
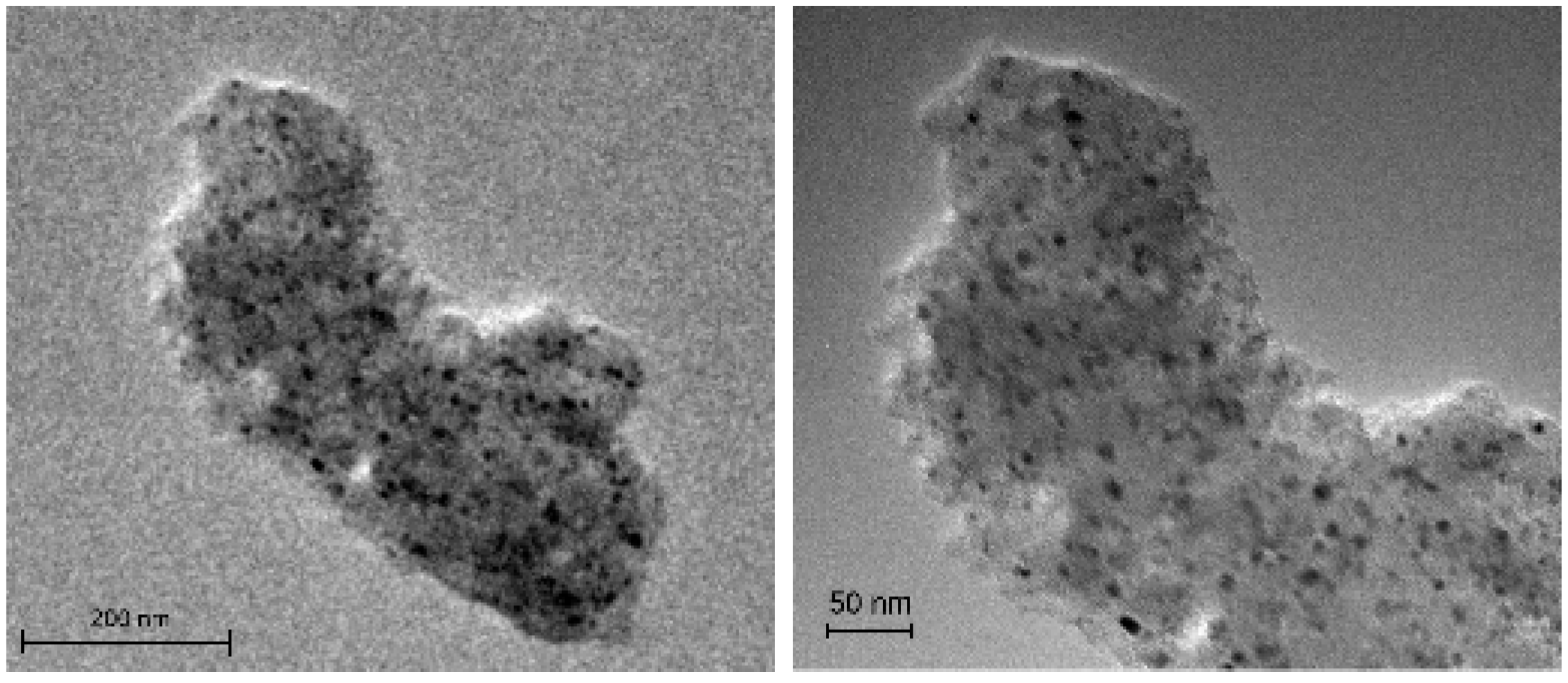
Figure 5.
SEM images of the IMP Ni99Pt1/C (above) and IMP Ni95Pt5/C (below) catalysts at different magnifications.
Figure 5.
SEM images of the IMP Ni99Pt1/C (above) and IMP Ni95Pt5/C (below) catalysts at different magnifications.

Figure 6.
XRD patterns of the catalysts prepared via the SRR method. The measuring range was from 20° to 95° 2Ө with Cu Kα-radiation (λ = 1.5419 Å).
Figure 6.
XRD patterns of the catalysts prepared via the SRR method. The measuring range was from 20° to 95° 2Ө with Cu Kα-radiation (λ = 1.5419 Å).
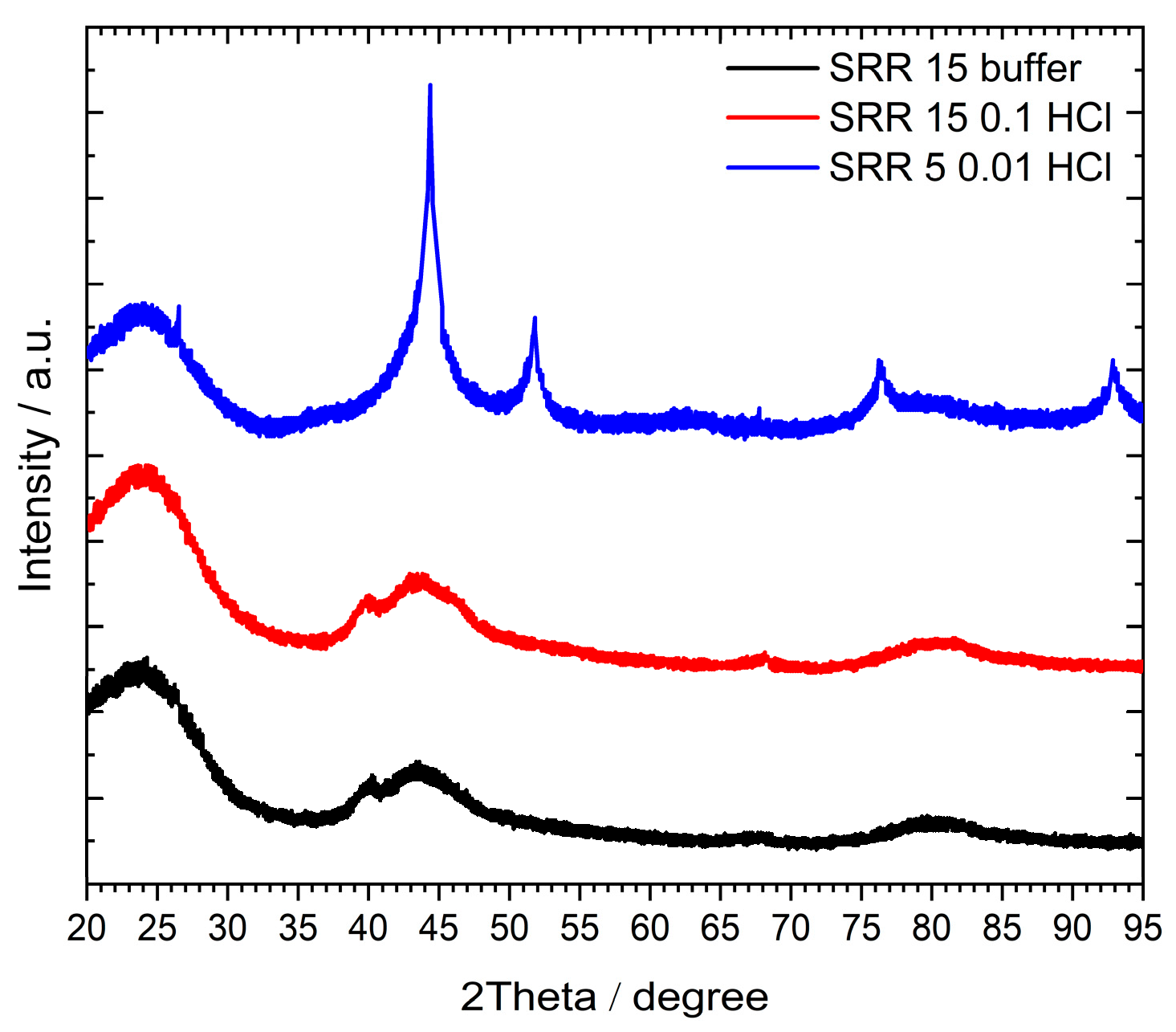
Figure 7.
TEM images of the Ni/C catalyst and of the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl, with the scales being in the range of 20–200 nm.
Figure 7.
TEM images of the Ni/C catalyst and of the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl, with the scales being in the range of 20–200 nm.

Figure 8.
Boxplot diagram for the sizes of the nanoparticles on the Ni/C catalyst and on the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl.
Figure 8.
Boxplot diagram for the sizes of the nanoparticles on the Ni/C catalyst and on the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl.
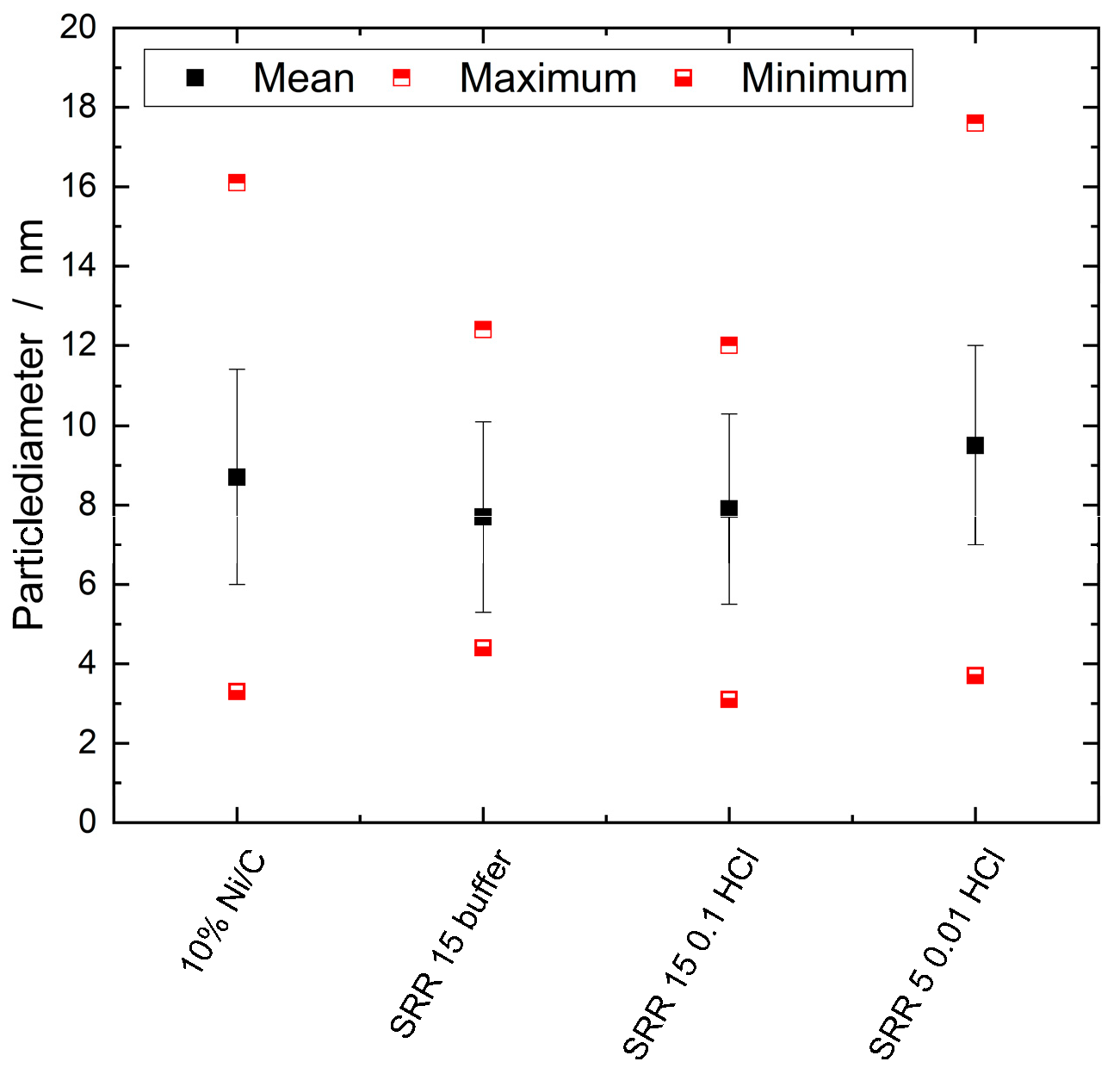
Figure 9.
SEM and EDX images of the Ni/C catalyst and of the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl; the magnifications are between 200 and almost 250,000. In the EDX measurements, Ni is shown in green and Pt in blue.
Figure 9.
SEM and EDX images of the Ni/C catalyst and of the three samples, SRR 15 buffer, SRR 15 0.1 HCL, and SRR 5 0.01 HCl; the magnifications are between 200 and almost 250,000. In the EDX measurements, Ni is shown in green and Pt in blue.
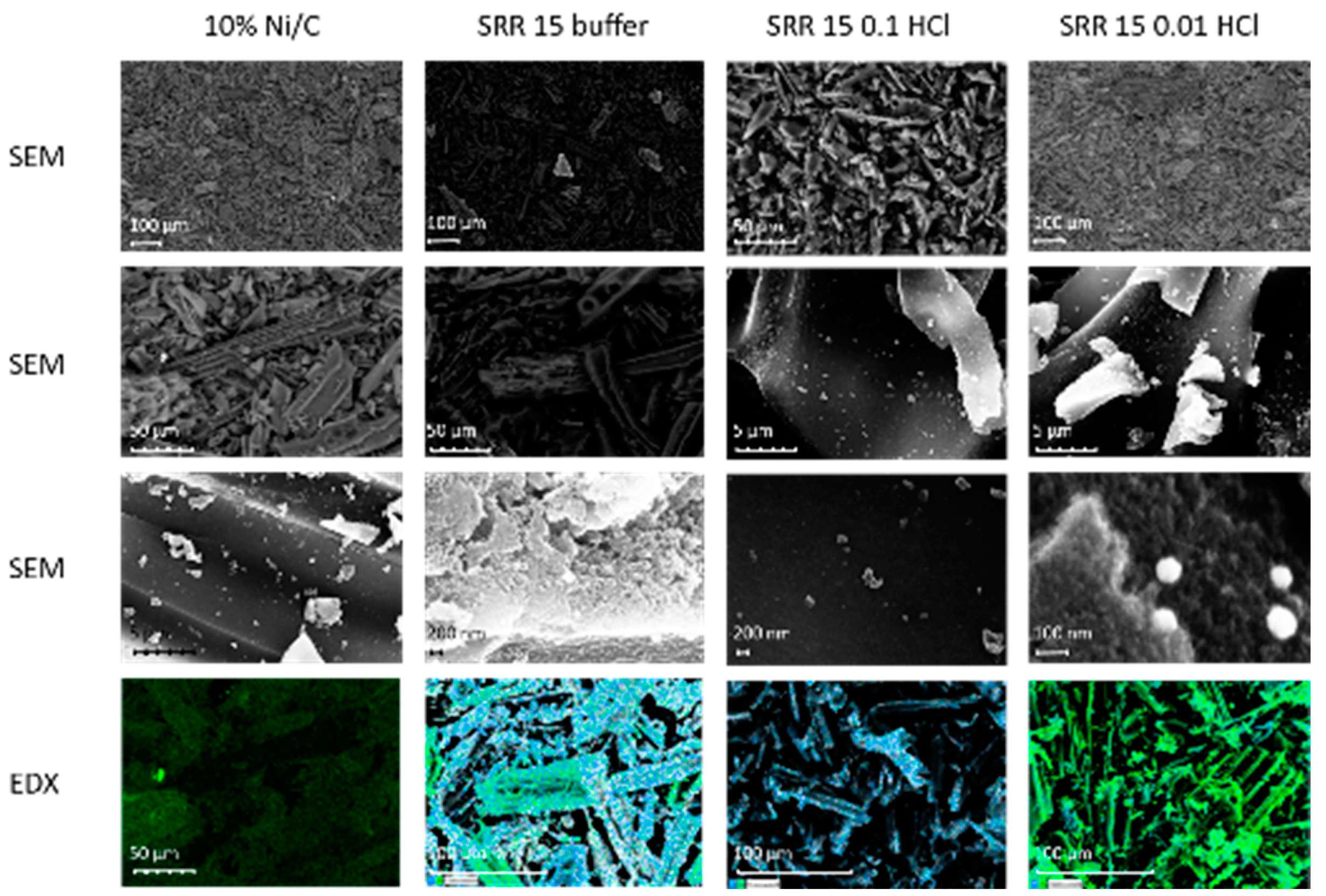
Table 1.
Comparison of weight and atomic percentage values determined via: (i) ICP–OES measurements; and (ii) when they were calculated based on the numbers from the experimental syntheses.
Table 1.
Comparison of weight and atomic percentage values determined via: (i) ICP–OES measurements; and (ii) when they were calculated based on the numbers from the experimental syntheses.
| Percentage values | IMP Ni99Pt1/C | IMP Ni95Pt5/C | |||
|---|---|---|---|---|---|
| ICP-OES | calculated | ICP-OES | calculated | ||
| wt% Ni | 8.4 | 11.2 | 7.7 | 10.0 | |
| wt% Pt | 0.1 | 0.3 | 0.4 | 1.5 | |
| at% Ni | 1 | 1 | 2 | 5 | |
| at% Pt | 99 | 99 | 98 | 95 | |
Table 2.
Yields of, and selectivities towards, the products of the reaction from Figure 1 with the two impregnated catalysts; mCat = 250 mg; reaction temperature = 165 °C; reaction time = 4 h; inlet conc. ethanol = 0.6 mole l-1; inlet conc. NaOH = 0.45 mole l-1.
Table 2.
Yields of, and selectivities towards, the products of the reaction from Figure 1 with the two impregnated catalysts; mCat = 250 mg; reaction temperature = 165 °C; reaction time = 4 h; inlet conc. ethanol = 0.6 mole l-1; inlet conc. NaOH = 0.45 mole l-1.
| Reaction products | IMP Ni99Pt1/C Y [%] | IMP Ni99Pt1/C S [%] | IMP Ni95Pt5/C Y [%] | IMP Ni95Pt5/C S [%] | |
|---|---|---|---|---|---|
| Iso-butanol | 2.45 | 61.03 | 3.01 | 69.49 | |
| 2-methylpropanal | 0.34 | 8.55 | 0.39 | 8.94 | |
| 1-propanol | 0.01 | 0.20 | 0.02 | 0.36 | |
| 2-methylbutane-1-ol | 0.03 | 1.28 | 0.05 | 2.19 | |
| 2-ethylbutane-1-ol | 0.02 | 1.20 | 0.03 | 2.20 | |
| 1-hexanol | 0.21 | 15.32 | 0.22 | 15.47 | |
| 2-ethylhexane-1-ol | 0.12 | 15.32 | 0 | 0 |
Table 3.
Comparison of the weight percentage values for Ni and Pt determined via: (i) the ICP–OES measurements; and (ii) when they were calculated based on the weighed portions of the educt chemicals.
Table 3.
Comparison of the weight percentage values for Ni and Pt determined via: (i) the ICP–OES measurements; and (ii) when they were calculated based on the weighed portions of the educt chemicals.
| Percentage Values | SRR 15 buffer ICP-OES |
SRR 15 buffer calculated |
SRR 30 buffer ICP-OES |
SRR 30 buffer calculated |
SRR 60 buffer ICP-OES |
SRR 60 buffer calculated |
|---|---|---|---|---|---|---|
| wt% Ni | 0.32 | 8.59 | 0.40 | 8.59 | 0.40 | 8.59 |
| wt% Pt | 0.25 | 0.34 | 0.24 | 0.33 | 0.24 | 0.33 |
Table 4.
Comparison of weight percentage values for Ni and Pt on the different catalysts determined via (i) ICP–OES measurements; and (ii) when they were calculated based on the weighted portions of the educt chemicals.
Table 4.
Comparison of weight percentage values for Ni and Pt on the different catalysts determined via (i) ICP–OES measurements; and (ii) when they were calculated based on the weighted portions of the educt chemicals.
| Percentage Values | SRR 15 buffer ICP–OES |
SRR 15 buffer calculated |
SRR 15 0.1 HCl ICP–OES |
SRR 15 0.1 HCl calculated |
SRR 5 0.01 HCl ICP–OES |
SRR 5 0.01 HCl calculated |
|---|---|---|---|---|---|---|
| wt% Ni | 0.32 | 8.59 | 0.10 | 8.59 | 7.33 | 8.56 |
| wt% Pt | 0.25 | 0.34 | 0.19 | 0.33 | 0.24 | 0.38 |
Table 5.
Specific space-time yields, iso-butanol yields, selectivities towards iso-butanol, and specific surface areas of the different catalysts; mCat = 250 mg; reaction temperature = 165 °C; reaction time = 4 h; inlet conc. ethanol = 0.6 mole l-1; and inlet conc. NaOH = 0.45 mole l-1.
Table 5.
Specific space-time yields, iso-butanol yields, selectivities towards iso-butanol, and specific surface areas of the different catalysts; mCat = 250 mg; reaction temperature = 165 °C; reaction time = 4 h; inlet conc. ethanol = 0.6 mole l-1; and inlet conc. NaOH = 0.45 mole l-1.
| Catalyst | Specific STY [mole g-1 h-1] | Y(iso-butanol) [%] | S to iso-butanol [%] | Specific surface areas [m2 g-1] | |
|---|---|---|---|---|---|
| IMP Ni99Pt1/C | 1.02 | 2.45 | 61.03 | 706 | |
| IMP Ni95Pt5/C | 1.35 | 3.01 | 69.49 | 703 | |
| SRR 15 buffer | 21.26 | 2.40 | 68.68 | 780 | |
| SRR 30 buffer | 15.16 | 3.01 | 67.11 | - | |
| SRR 60 buffer | 17.52 | 2.04 | 70.48 | - | |
| SRR 15 0.1 HCl | 79.87 | 3.49 | 74.49 | 758 | |
| SRR 30 0.1 HCl | 42.07 | 6.02 | 80.09 | - | |
| SRR 5 0.01 HCl | 3.30 | 6.92 | 76.19 | 671 |
Table 6.
Yields of and selectivities towards the products of the reaction from Figure 1 on the catalysts, SRR 15 buffer, SRR 15 0.1 HCl, and SRR 5 0.01 HCl; mCat = 250 mg; reaction temperature = 165 °C; reaction time = 4 h; inlet conc. ethanol = 0.6 mole l-1; inlet conc. NaOH = 0.45 mole l-1.
Table 6.
Yields of and selectivities towards the products of the reaction from Figure 1 on the catalysts, SRR 15 buffer, SRR 15 0.1 HCl, and SRR 5 0.01 HCl; mCat = 250 mg; reaction temperature = 165 °C; reaction time = 4 h; inlet conc. ethanol = 0.6 mole l-1; inlet conc. NaOH = 0.45 mole l-1.
| Reaction products | SRR 15 buffer Y [%] | SRR 15 buffer S [%] | SRR 15 0.1 HCl Y [%] | SRR 15 0.1 HClS [%] | SRR 5 0.01 HCl Y [%] | SRR 5 0.01 HCl S [%] |
|---|---|---|---|---|---|---|
| Iso-butanol | 2.40 | 68.67 | 3.49 | 74.49 | 6.92 | 76.19 |
| 2-methylpropanal | 0.49 | 14.01 | 0.60 | 12.75 | 1.23 | 13.56 |
| 1-propanol | 0.00 | 0.04 | 0.00 | 0.05 | 0.02 | 0.23 |
| 2-methylbutane-1-ol | 0.01 | 0.83 | 0.01 | 0.39 | 0.06 | 1.35 |
| 2-ethylbutane-1-ol | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 | 1.01 |
| 1-hexanol | 0.19 | 16.36 | 0.19 | 8.68 | 0.23 | 7.65 |
| 2-ethylhexane-1-ol | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



