
Submitted:
11 December 2023
Posted:
13 December 2023
You are already at the latest version
Abstract
The mitochondrial network (MN) is a dynamic structure undergoing constant remodeling in the cell. It is assumed that the violation of MN may be associated with various pathologies, including Parkinson’s disease (PD). Using automatic image analysis and super-resolution microscopy, we have assessed the MN parameters in fibroblasts from patients with established hereditary PD mutations (associated with PINK1, LLRK2, α-synuclein, PINK1 and Parkin simultaneously proteins) under normal conditions and after hydrogen peroxide-induced stress. Fibroblasts with Pink1/Parkin mutation are the most different in morphology from fibroblasts obtained from conditionally healthy donors: MN is larger, contains longer mitochondria and accumulated individual mitochondria. In addition to MN, we evaluated other cellular parameters, such as: cytosolic and mitochondrial ROS production and mitochondrial membrane potential. It has been shown that mitochondria of fibroblasts with mutations in genes encoding PINK1, α-synuclein and Pink/Parkin tends to hyperpolarization and cytosolic ROS overproduction, while mitochondrial ROS production was higher only in fibroblasts with PINK1 and α-synuclein mutation.
Keywords:
mitochondrial network
; Parkinson's disease
; super-resolution microscopy
; fibroblasts
1. Introduction
Parkinson's disease (PD) is a complex multifactorial neurodegenerative disease, including sporadic and hereditary forms accompanied by the development of mitochondrial dysfunction and oxidative stress [1]. Hereditary forms of PD are only a small proportion of all cases of PD, but studying them allows to understand the fundamental mechanisms of this pathology. Since mitochondria perform a number of important functions in the cell: energy production, intracellular signaling, control of intracellular calcium homeostasis, trigger apoptosis, etc. for the normal functioning of the cell, it is important to maintain functional healthy mitochondria and remove the damaged ones. To eliminate the toxic effects of dysfunctional mitochondria, the organisms have developed a complex system of mitochondrial quality control implemented through the processes of mitochondrial fission/fusion [2] and regulation of mitochondrial biogenesis and mitophagy. In the cell mitochondria exist in the form of a dynamic structure – mitochondrial network (MN), the morphology of which depends on the equilibrium of these opposite processes, as well as mitochondrial dynamics and autophagy (Figure 1).
Changing the MN morphology is also necessary for regulation of metabolic processes, neuronal differentiation, and apoptosis [3]. Mitophagy is the process of selective autophagy of mitochondria by lysosomes. It is known that such proteins as PTEN-induced kinase 1 (PINK1) and Parkin RBR E3 ubiquitin protein ligase (Parkin) participate in mitophagy [4], and disruption of their functions as a result of mutations leads to the development of hereditary PD [5,6].
1.1. Mitochondrial Fusion and Fission
The mitochondrial fusion and fission contribute to the adaptation of cells under various conditions, including oxidative stress [3]. With minor damage, individual mitochondria can merge with the MN to share resources, optimize respiration, and synthesize ATP. For instance, mitochondria with violated mitochondrial DNA (mtDNA) have been shown to fuse with intact mitochondria, allowing them to mix and efficiently redistribute mtDNA, as well as proteins and metabolites [7]. The process of mitochondrial fusion is realized due to the activity of GTPases that bind the outer mitochondrial membranes – Mitofusin-1 (Mfn1) and Mitofusin-2 (Mfn2) and the inner one – Dynamin-like 120 kDa protein (OPA1). Disruption of fusion processes leads to the formation of a highly fragmented MN, which is characteristic of neurodegenerative diseases such as Charcot-Marie-Toute disease and Dominant Optic Atrophy [8]. In the case of severe damage of individual mitochondria, its fusion with MN can be dangerous for the entire MN, therefore such mitochondria must be separated and safely degraded by mitophagy and autophagy. Mitochondrial fission is the opposite of fusion, and is necessary, among other things, for the segregation of dysfunctional mitochondria. Mitochondrial fission is regulated by the GTPase-dynamics-related protein 1 (Drp1), as well as the Fission 1 (Fis1) and Mitochondrial fission factor (Mff) proteins [9]. When the fission processes are disrupted, the MN contains highly elongated mitochondria, which may also be associated with pathological processes. Thus, it was shown that in Purkinje cells, the absence of the main mitochondrial division protein, DRP1, first leads to the elongation of mitochondria, which then swell due to oxidative damage [10].
1.2. Mitochondrial Network Morphology and Diseases
Maintaining a functional MN is especially important for tissues that have a narrow specialization and high metabolic activity, such as neurons, cardiac and skeletal muscles. Since these cells are long-lived and postmitotic, they cannot divide damaged mitochondria between daughter cells and, unlike dividing cells, use more of the fusion/fission mechanism to preserve or restore the functions of damaged mitochondria [3]. At the moment, it is assumed that impairment in the morphology and dynamics of the MN play a significant role not only in Charcot-Marie-Toute disease and Dominant Optic Atrophy, but also in PD. For instance, it has been shown that mtDNA mutations accumulate because of MN disruption in PD dopaminergic neurons [11]. In addition, it has been reported that under the influence of various stress factors on the cell, the length of mitochondria decreases/increases, structures “donuts” (fully looped mitochondria), “drops” (fragmented mitochondria) appear [12]. Assessment MN parameters such as length, connectivity, and the mitochondrial footprint may provide insight into the role of MN in pathology [12,13]. However, there is still no clear idea how various mutations associated with PD can affect the MN morphology in other somatic cells and how this is related, in particular, to the level of oxidative stress. Fibroblasts are a convenient model for studying the MN morphology, since they are large sprawled cells.
In present work, using automatic image analysis and super resolution microscopy, we studied the parameters of the MN in fibroblasts from patients with established hereditary PD, associated with: 1) mutations in PINK1 gene, encoding PTEN-induced kinase 1; 2) simultaneous mutations in genes PINK1 and PARK2, encoding PINK1 and Parkin RBR E3 ubiquitin protein ligase; 3) A53T point mutation in SNCA gene, encoding α-synuclein; 4) G2019S mutation in LRRK2 gene, encoding leucine-rich repeat kinase 2 protein. Moreover, we have analyzed cellular parameters of these fibroblast lines, such as: the rate of mitochondrial and cytosolic ROS production, mitochondrial membrane potential under normal conditions and in response to stress.
2. Materials and Methods
2.1. Materials
H2DCFDA, MitoSOX Red, MitoTracker Red CM-H2XRos were purchased from Invitrogen; DMEM, FBS, Glutamax, Sodium Pyruvate and TrypLE™ Express were purchased from Gibco, USA; HBSS was purchased from PanEco, Russia.
2.2. Cell Lines
All cell cultures were kindly provided by prof. Andrey Y. Abramov (UCL Institute of Neurology, London, Great Britain). Cell cultures of human skin fibroblasts carrying mutations in the genes encoding alpha-synuclein (a-syn, A53T het), PINK1 (homozygous p.Try90Leufsx12), PINK1 and Parkin (PINK1/Parkin, PARK2 R275W/WT+PINK1 p.Try90Leu fs*12/WT), and G2019S mutation in LRRK2 gene, as well as control lines of fibroblasts were used as objects in this study (Table 1). We used the same cell lines as in the previous study [14].
Cells were cultured on 25 cm2 culture flasks in DMEM containing 10% FBS (Invitrogen, USA), 2 mM glutamine, 1 mM pyruvate at 37 °C, 5% CO2, and 100% humidity. Upon reaching 80–85% confluence, the cells were splitted to maintain the culture, or seeded on round 25 mm glass coverslips for the experiment. All cells used for experiments were not older than 18 passage.
2.3. Hydrogen Peroxide-Induced Stress
Hydrogen peroxide was added to the incubation culture medium at 37 degrees for 1 hour, after which it was washed three times and left to rest for another hour, after which the experiment was started. The concentration of hydrogen peroxide was selected from the calculation of the dose leading to the death of 20% of cells for control fibroblast lines.
2.4. ROS Measurements
To assess cytosolic ROS production, the cells were loaded with the H2DCFDA probe (10 μM, 40 min) followed by washing with HBSS. Fluorescence registration was carried out using a Spark10M tablet reader (Tecan Group, Switzerland) or an imaging station based on a Leica DMI6000 B inverted microscope (Leica Microsystems, Germany) using a standard FITC filter set (excitation: 494 ± 10 nm, emission: 535 ± 10 nm) with a 20x objective lens. Recording speed 1 frame/5 sec. To assess ROS production in mitochondria, cells were loaded with a MitoSOX Red probe (5 μM, 15 min) or MitoTracker Red CM-H2XRos (100 nM, 30 min) followed by washing with HBSS. Registration was performed using a standard Texas Red filter set (excitation: 575 ± 10 nm, registration: 624 ± 20 nm) and a 20x objective lens. Recording speed 1 frame/5 sec.
2.5. Mitochondrial Membrane Potential Measurements
The mitochondrial membrane potential (Δψm) was measured by incubating cells with 25 nM tetramethylrhodamine methyl ester (TMRM) fluorescent dye in a buffered saline solution (HBSS) for 40 minutes at room temperature. Fluorescent images were obtained on Zeiss 900 CLSM (Carl Zeiss Microscopy GmbH, Jena, Germany) confocal microscope equipped with a × 63 oil immersion objective and during the measurements, the TMRM remained in the HBSS solution. Illumination intensity was kept to a minimum (0.1–0.2 % of laser output) to avoid phototoxicity. The cells were excited with a laser at 561 nm, and the fluorescence was detected above 580 nm. FCCP, which is uncouplers of oxidative phosphorylation, were used to assess the mitochondrial function. When FCCP is added, TMRM fluorescence is quenched. The mitochondrial membrane potential was estimated as the difference in TMRM fluorescence (maximum signal minus the signal with the addition of 2 μM FCCP) and was taken as 100% in control fibroblasts without treatment.
2.6. Mitochondrial Network Morphology Analysis
To analyze the MN morphology, images of cells loaded with TMRM (25 nM), which constantly remained in the working solution, were taken on Zeiss 900 CLSM (Carl Zeiss Microscopy GmbH, Jena, Germany) confocal microscope equipped with a × 63 oil immersion objective. Resolution is 23.4 pixels per micron. The mitochondrial area may depend on the cell volume. In order to take into account this effect on the MN area, we have previously estimated the volume of cells using Calcein AM fluorescence. Calcein at a concentration of 5 μM was loaded into cells for 30 minutes at room temperature, after which Z-stacks images were obtained on a confocal microscope and the fluorescence area was calculated. It was found that there is no difference in this parameter in the studied cell lines. Next, we used a self-written Fiji plugin for automatic batch processing. The protocol for assessing the MN morphology was based on the approaches described earlier [12,15,16]. Detailed analysis steps are shown in Figure 2.
1. At the first stage, the image is preprocessed, which included background subtraction (rolling ball radius set on 50 pixels), median filter and local contrast enhance (CLACHE) functions. 3. Next, the image was binarized using threshold by “Otsu” method. 3.1 Next, the image was copied and the mitochondrial footprint was calculated by the sum of the mitochondrial areas in the “Particle analyzer” Fiji plugin. 4. Further, the image was skeletonized using the “Skeletonize” function and processed using the built-in “Skeleton Analysis” function. 5. Next, the table of the received data was transmitted to the R-programming environment. The data table was filtered using the “filter” function from “dplyr” package into two data subsets: “mitochondrial network” and “individual mitochondria” according to the described scheme (mitochondrial network were considered as the object what consist of more one branch and non-zero junction, whereas individual mitochondria consist of only one branch and zero junction).
2.7. Statistical Analysis
Computer processing and data images analysis of cell cultures were carried out using Fiji and R-Studio. OriginPro2018 was used for plotting. For experiments with the assessment of mitochondrial membrane potential and the rate of cytosolic and mitochondrial ROS production, two control lines of fibroblasts were combined and taken as 100%. When processing data on the measurement of ROS production, curves were obtained, then were linearly approximated, and the rate of the fluorescent signal increase was calculated. Statistical analysis was performed using “dplyr” package and OriginPro2018 programs using one-way ANOVA (post Tukey test with Bonferroni correction) parametric analysis. The results were presented as mean ± standard deviation of the mean (SD). Differences were considered statistically significant at: * p < 0.05, ** p < 0.01.
3. Results
3.1. Mitochondrial Network Morphology of Fibroblasts
We investigated the MN morphology using an automatic plugin self-written in Fiji and R programing language. We evaluated 5 different parameters of MN: the total mitochondrial area (mitochondrial footprint) in µm2, the size of mitochondria in the MN, the ratio of MN and individual mitochondria in cell, size of individual mitochondria and the connectivity of MN in fibroblasts with mutations from PD patients and conditionally healthy individuals under normal conditions and after H2O2-induced stress. As is known, hydrogen peroxide can increase the production of superoxide by mitochondria in a positive feedback loop according to the RIRR (ROS-Induced ROS Release) effect [17,18].
We estimated the mitochondrial footprint by the total fluorescence area of TMRM (Figure 3a). Staining mitochondria with mitochondrial potential-depended TMRM giving confidence that it only healthy mitochondria. A preliminary assessment of the cell volume was carried out using Calcein AM fluorescence and no difference was found in the cell volume of the studied cell lines. The mitochondrial footprint (Figure 3b) in all fibroblasts, except Pink/Parkin, does not significantly differ from both controls (for control1 is 635±196 µm2 for control2 – 709±190 µm2; for LLRK2 – 873±399 µm2; PINK1 – 639±279 µm2; А53Т 765±468 µm2). Pink/Parkin mitochondrial footprint significantly higher by 2.2 times than in the two controls and is 1474±793 µm2. When exposed to hydrogen peroxide, the mitochondrial footprint for all fibroblasts tends to decrease by an average of 1.7 times (the reduction coefficient for control1 is 1.7; control2 – 2.8; LLRK2 – 1.5; PINK1 – 1.7; Pink/Parkin – 1.36) however, a significant decrease occurred only for control2 line. After hydrogen peroxide stress, the mitochondrial footprint was still the largest in Pink/Parkin fibroblasts.
Next, we evaluated the length of mitochondria (Figure 3c) in the MN (branch length). The mitochondria branch length can be an important parameter showing the adaptation of the cell to substrate deprivation, stress, and may change in pathology. As mentioned above, a MN is considered to be a structure containing strictly more than one branch and a non-zero connections. Since the mitochondrial branch lengths have an abnormal distribution, we use the median, not the mean. The median mitochondrial branch lengths of fibroblasts with Pink/Parkin (2.28±0.19 µm) and A53T (1.66±0.6 µm) mutation were characterized by higher values than control lines (for control1 – 1.46±0.55 and for control2 – 1.2±0.18) and all other fibroblasts with mutations (LLRK2 – 1.37±0.63 µm; PINK1 – 1.19±0.36 µm;). As expected, hydrogen peroxide addition activated mitochondrial fission, which led to a significant decrease in the mitochondrial branch length almost in all fibroblasts except A53T (the coefficient of length reduction for control1 is 1.58; for control2 – 1.5; for LLRK2 – 1.65; PINK1 – 1.7; Pink/Parkin – 1.79).
The accumulation of individual mitochondria may indicate an imbalance of the fission/fusion, as well as mitophagy. Interestingly, early reports suggest that mitophagy level in these mutated fibroblasts is comparable with conditionally healthy controls [14]. In addition, since TMRM dye stains mitochondria with mitochondrial membrane potential, we believe that in our case it is possible to analyze fission/fusion balance. Figure 4a shows the ratio of individual mitochondria to MN, since the absolute values of individual mitochondria vary in a wide range even in the same culture analyzed. Thus, the greater this value, the more individual mitochondria in the cell. Under normal conditions, the ratio of individual mitochondria for fibroblasts with PINK1 (6.1±1.5) and Pink/Parkin (7.6±3.3) mutations are significantly higher by 1.9 and 2.4 times, respectively, than in control cells (for control1 it is 3.4±1 and for control2 it is 2.9±1.2). For fibroblast with mutations LLRK2 it is 4.1±1.6, for A53T it is 4.3±2.9 and after hydrogen peroxide stress it is 2.7±1.1 и 4.5±2.8 respectively. Interesting, that after hydrogen peroxide stress the ratio of individual mitochondria of PINK1 cells significantly decreases by 2.1 times, whereas for the Pink/Parkin mutation, on the contrary, it does not change and remains significantly higher than in controls.
Next, we estimated the median length of individual mitochondria (Figure 4b), since we were interested in which mitochondria make up the population of non-MN mitochondria in cells with various mutations. We assumed that the length of individual mitochondria should be less than mitochondrial branch in the MN. Indeed, the length of individual mitochondria for all the studied lines was lower than the network ones (control1 – 0.73±0.41; control2 – 0.77±0.31; LLRK2 – 0.64±0.44; PINK1 – 0.39±0.21; Pink/Parkin – 0.82±0.2; A53T – 0.99±0.47). At the same time, in cells with the PINK1 mutation, it is significantly lower by 1.9 times than in control cells. After the peroxide addition, there is a tendency to decrease the individual mitochondria length for all studied fibroblasts, however, this parameter significantly decreases only in control cells (2.2 times for the first control) and Pink/Parkin cells (2 times), possibly due to higher initial values.
Further, we evaluated the MN connectivity (Figure 5) by calculating the average number of junctions in the MN per cell. A change in mitochondrial connectivity may indicate the adaptation of cells to a substrate decrease, a violation of bioenergetics and mitochondrial fission/fusion. Under normal conditions, the average number of junctions in the MN for all cell lines does not differ significantly (for control1 – 17.3±7.5; control2 – 17.1±7.9; LLRK2 – 15.6±7.1; PINK1 – 17.9±6.8; A53T – 19.3±13.9), except Pink/Parkin, which were characterized by large values of this parameter (25.4±13.8). When peroxide is added, the average number of junctions in the MN decreases for control cells and fibroblasts with LLRK2 mutation significantly, (the reduction coefficient for control 1 is 2.9; control2 – 3; LLRK2 – 2.1), whereas in cells with PINK1, Pink/Parkin and A53T mutation, connectivity does not change and is significantly higher than in control cells (for PINK1 by 1.2 times and Pink/Parkin by 1.5 times).
Thus, we investigated 5 different parameters describing MN morphology in fibroblasts with PD-associated mutations. The greatest difference in the MN parameters from the control lines was found for cells with a double mutation associated with mitophagy – Pink/Parkin.
3.2. Mitochondrial Membrane Potential
The maintenance of the mitochondrial potential is necessary for efficient ATP synthesis, while changes in the mitochondrial membrane potential may indicate possible impairment of mitochondrial function. We evaluated the values of the mitochondrial potential using a potential-sensitive probe TMRM (Figure 6). It was found that the mitochondrial membrane is significantly hyperpolarized in fibroblasts with Pink/Parkin (137.6±21.2%) and A53T (154.5±10.4%) mutations relative to the control (100%). After hydrogen peroxide-induction stress (150 μM 30 min), there is significant decrease of mitochondrial membrane potential in fibroblasts with PINK1 (by 34.8±8.3) and Pink/Parkin (by 90.7±7.5) mutations, while in cells with the A53T mutation, the mitochondrial membrane potential remains hyperpolarized.
3.3. Cytosolic and Mitochondrial ROS Production Rate
We measured the rate of cytosolic ROS-production under normal conditions using the time kinetics of DCF fluorescence (chemically reduced and acetylated forms of 2′,7′-dichlorofluorescein H2DCFDA probe) and calculated the rate as a differential (Figure 7a). A significant increase in the rate of ROS production was found almost for all fibroblasts with mutations: PINK1 by 1.9 times (191.5±36.6%), Pink/Parkin by 1.8 times (180.3±31.1%); and 1.9 times (199.4±21.4%) in A53T relative to the control taken as 100% (with the exception of LLRK2 mutation fibroblast, where only a trend was observed).
Besides to mitochondrial membrane potential, an important indicator of mitochondrial function is the rate of mitochondrial ROS production. Since we did not find any difference under normal conditions, we assumed that exogenous stress could reveal a change in the rate of mitochondrial ROS production in studied fibroblasts. The rate of mitochondrial ROS production (Figure 7b) was measured after the hydrogen peroxide addition (50 µM, for 1 hour) by the time kinetics of MitoSOX Red and MitoTracker Red CM-H2XRos probes (their fluorescent derivatives after oxidation). After the addition of hydrogen peroxide, a significant increase in the rate of mitochondrial production was found for PINK1 fibroblasts (by 1.6 times – 163.5±32.1%) and A53T in (by 1.4 times – 147.6±21.3%) relative to the control taken as 100%.
4. Discussion
This work is a quantitative analysis of the mitochondrial network (MN) morphology (parameters such as: mitochondrial footprint, length of mitochondria in NW, the ratio of individual mitochondria and their length, as well as mitochondrial connectivity) in fibroblasts obtained from patients with hereditary forms of PD and established mutations, and also their response to hydrogen peroxide-induced stress. Two fibroblast lines studied in this work are associated with a mitophagy-related genes PINK1 and PARK2 malfunction and one line contains a mutation in a Leucine-rich repeat kinase 2 (LRRK2), a protein involved in autophagy. Another fibroblast line contains a point mutation in the gene encoding α-synuclein (A53T, SNCA), the aggregates of which are found in the brains of PD patients. As a control, we used fibroblasts from conditionally healthy individuals comparable in age to patients with PD, between which in most cases there were no significant differences. This fact is important for such an analysis, since cells from donors are highly heterogeneous. In addition to the MN morphology, we evaluated the mitochondrial membrane potential and the rate of cytosolic and mitochondrial ROS production in these fibroblasts. A change in the stress response may indicate pathology. In our study, hydrogen peroxide was used as a stress model, since it is known that it causes ROS-dependent ROS production and in some cases [17], it has revealed differences in the cells parameters that were not present under normal conditions. It is believed that MN morphology may depend on the external stress on the cell, substrate deprivation, etc., and the rearrangement of MN morphology is the most important for cell adaptation under various conditions [3,10,18].
The MN may be a potential target for the therapy of neurodegenerative diseases, including PD [19]. It is shown that with aging, mitochondria undergo fragmentation and lose area [20], which is also may be associated with the development of neurodegeneration [21]. However, in this work we showed a significant change in the mitochondrial footprint only for the Pink/Parkin mutation fibroblasts, which, on the contrary, led to an increase in this area of MN (Figure 3b), probably, due to a violation of mitophagy. Interestingly, when exposed to stress, there was indeed a decrease in the mitochondrial footprint of fibroblasts, but for the most part it was not significant and mitochondrial footprint in Pink/Parkin mutation fibroblasts remained the largest. The change in the mitochondria length may occur due to increased fusion or disruption of fission. An increase in the mitochondria length may indicate, for instance, an intensification of the ATP production processes [22]. In our work, we found an increase in the median mitochondria length of MN (mitochondrial branch) in cells with Pink/Parkin and A53T mutations (Figure 3c). Interestingly, although it was previously shown that the expression of α-synuclein with A53T mutation in mammalian cells leads to fragmentation of mitochondria, we found the opposite effect [23]. Since Mfn1 and Mfn2 involved in mitochondrial fusion are known to be substrates for Parkin ubiquitination [24], it is not surprising that impaired Parkin function leads to an increase in the length of mitochondria due intense fusion. Moreover, fusion of damaged mitochondria with a network has been shown to separate intact mDNA between these mitochondria [7] and possibly this process took place in the fibroblast in cells with Pink/Parkin and A53T mutations. At the same time, it is worth noting that the peroxide-induced stress led to a significant decrease in the mitochondria length in all the studied cells, except for fibroblasts with the A53T mutation. It is possible that α-synuclein somehow involved in the mitochondrial fission in fibroblasts, which remains to be elucidated in future studies.
The accumulation of individual mitochondria may also indicate a violation of mitochondrial dynamics and mitophagy [16,15]. We found that for fibroblasts with Pink/Parkin and PINK1 mutations, the ratio of individual mitochondria/to network ones is higher compared to the control (Figure 4a), which may indicate increased division or/and inefficiency of mitophagy. Interestingly, despite mutations in Parkin and PINK1 involved in the recruitment of mitochondria into mitophagy 15 [15,25,26], early reports of these cells with mutations showed the levels of mitophagy in fibroblast similar to control cells, studied by double staining of lysosomes and mitochondria [14]. Perhaps from our results it can be concluded that when analyzing mitophagy, it is necessary to look not only at the colocalized lysosomes and mitochondria, but also the ratio of individual mitochondria in cells. It is noteworthy that under the action of hydrogen peroxide, the ratio of individual mitochondria to mitochondrial networks in fibroblasts with PINK1 cell mutation decreased, whereas for the double Pink/Parkin mutation it did not change. A decrease in the ratio of individual mitochondria may be associated with the activation of transcription factor, which is known to be involved in the launch of mitophagy and is dependent on stress [27,28,29]. Perhaps such stress can cause mitophagy activation, but only in the case of a single PINK1 mutation. This is consistent with data where it was previously shown mild and sustained hydrogen peroxide (H2O2) stimulation induces Parkin-dependent mitophagy accompanied by downregulation of the mitophagy-associated proteins OPTN, NDP52, and MFN2 [31]. This protective effect of moderate stress during exercise or starvation may also play a role in increasing mitochondrial biogenesis via PGC-1α and DRP1 [22,31,32].
In addition to ratio of individual mitochondria we made an attempt to study which size of individual mitochondria mainly represents in fibroblasts. To do this, we estimated the median length of individual mitochondria (Figure 4b). It was found that in control fibroblasts and fibroblasts with mutations, individual mitochondria are predominantly up to 1 µm in length. Surprisingly, the PINK1 mutation is characterized by shorter individual mitochondria, while the A53T mutation, on the contrary, is longer compared to the control. The impact of stress had an ambiguous effect on the size of individual mitochondria: in control1 and in cells with the Pink/Parkin mutation, they became shorter, although there were no significant changes in other fibroblasts.
The MN connectivity can also change in pathology and as a response to physiological conditions, for example, starvation [16]. We have shown, that the MN connectivity does not differ in fibroblasts with mutations and control cells under normal conditions (Figure 5). Interestingly, after hydrogen peroxide addition, the number of junctions in the MN of controls fibroblasts and fibroblasts with the LLRK2 mutation significantly decreases, while for PINK1, A53T and Pink/Parkin fibroblasts it does not significantly change in response to stress. Possibly this may be due to a violation in these cells of the mitochondrial fission machinery, because fission requires the coordinated work of the GTPase separating the inner and outer mitochondrial membranes [33]. When only the inner membrane separates, the mitochondria can still be connected to the rest of the MN [13].
According to modern concepts, mitochondrial dysfunction is a key factor in the pathogenesis of hereditary and sporadic PD. The mitochondrial function can be indicated by the potential of the mitochondrial membrane. We evaluated this parameter by TMRM fluorescence (Figure 6). It was found that fibroblasts with Pink/Parkin and A53T mutations are characterized by a significant increase – hyperpolarization of the mitochondrial membrane, and such a trend is observed for PINK1 mutation. Hyperpolarization of the mitochondrial membrane has been shown for aging cells and may be associated with oxidative stress and also as consequence of increased energy requirements, substrate deprivation or disruption of the ETC [34,35]. Interestingly, the cells with hyperpolarized membranes turned out to be sensitive to stress – in the control lines, the addition of hydrogen peroxide did not lead to any significant differences, whereas for A53T and Pink/Parkin and PINK1 cells, a significant decrease in mitochondrial potential occurred.
Oxidative stress is a hallmark of neurodegenerative diseases, in addition, mitochondrial dysfunction also causes oxidative stress [1]. Though it is believed that oxidative stress in PD is characteristic of brain neurons, we found an increase in the rate of cytosolic ROS production in almost all fibroblasts with mutations (Figure 7a). Despite we cannot estimate the contribution of any particular form of ROS, using mitochondrial-oriented probes, it is possible to measure whether mitochondria are sources of ROS overproduction in fibroblasts with mutations. Since under normal conditions it was not possible to detect the difference in mitochondrial ROS production between control cells and fibroblasts with mutations, then we applied a model with peroxide-induced stress (Figure 7b). After the hydrogen peroxide addition, a significant increase in mitochondrial ROS production was detected in fibroblasts with PINK1 and A53T mutations, that may also indicate ETC disruption and mitochondrial dysfunction [1]. It should also be noted that we did not find an increase in mitochondrial ROS production in fibroblasts with Pink/Parkin mutation, possibly due to a decrease in mitochondrial potential after the stress, which can disrupt the accumulation of fluorescent probe near the mitochondrial membrane.
To summarize, in fibroblasts from patients with PD and established mutations, we found various changes of MN morphology compared to control cells from conditionally healthy donors. Fibroblasts with a double Pink/Parkin mutation have the most different morphology, whose MN is larger and contains longer mitochondria. Despite the fact that the A53T mutation in SNCA gene is not directly related to mitochondrial dynamics, the fibroblasts that contain it also showed significant differences in the MN morphology, similar to Pink/Parkin cells. It is worth noting that a new parameter was introduced in this work – the ratio of individual mitochondria and MN, using which the accumulation of individual mitochondria in cells with the PINK1 and Pink/Parkin mutation was revealed. Remodeling of the MN in response to stress also differed in conditionally healthy control cells and fibroblasts with mutations and, to the greatest extent, in cells with a double Pink/Parkin mutation.
It is important to note that the morphology of the mitochondrial network does not always correlate with function, but may be a consequence of cell adaptation to conditions. In addition, it is necessary to investigate not only the MN morphology, but also its relationship with metabolic signatures and the state of the cell [36]. Impairment in the mitochondrial membrane potential and the rate of mitochondrial and cytosolic ROS production were also estimated in this work, but we have not established a connection whether this is the cause or consequence of changes in the MN morphology in these fibroblasts.
Surprisingly, cells with the c mutation in the gene encoding LLRK2 kinase showed no significant differences in the studied parameters, although it is believed that this protein is also associated with autophagy and mitochondrial function [37,38].
We believe that such violation in fibroblasts with mutations do not have critical consequences as in neurons, since fibroblasts are relatively rapidly dividing cells, whereas neurons live throughout the life of an individual. However, as we have shown, these cells can be a promising and convenient model for studying the relationship between MN morphology and pathology associated with neurodegeneration [39]. Furthermore, a recent article has shown that the MN can be a target for regulation of stem cell differentiation [40]. This is of a great interest in light of the fact that neurons can be obtained from the fibroblasts of patients with PD to replace dead cells in the brain.
5. Conclusions
Thus, the present work can be useful for understanding the role of the MN morphology of the in the development of the hereditary PD form by the example of fibroblasts from patients with an established disease.
Author Contributions
Conceptualization, A.V.B., and K.A.K.; methodology, A.V.B., E.I.F. and K.A.K.; Formal analysis, K.A.K. and E.I.F.; Investigation K.A.K. and E.I.F; Resources, A.V.B.; Writing – original draft preparation, K.A.K.; Writing – review and editing, A.V.B.; Visualization, K.A.K. and E.I.F; Funding Acquisition, A.V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (grant no. 22-24-01043).
Institutional Review Board Statement
All experimental protocols in this study were approved by the Commission on Biosafety and Bioethics of ICB RAS (Permission No. 2, 12 June 2020). Experimental protocols were carried out according to Act708n (23 August 2010) of the Russian Federation National Ministry of Public Health, which states the rules of laboratory practice for the care and use of laboratory animals, and the Council Directive 2010/63 EU of the European Parliament (22 September 2010) on the protection of animals used for scientific purposes.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu Rev Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnauné-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; Rojo, M.; Belenguer, P. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Goh, J.-Y.; Xiao, L.; Xian, H.; Lim, K.-L.; Liou, Y.-C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Spinazzi, M.; Cazzola, S.; Bortolozzi, M.; Baracca, A.; Loro, E.; Casarin, A.; Solaini, G.; Sgarbi, G.; Casalena, G.; Cenacchi, G.; Malena, A.; Frezza, C.; Carrara, F.; Angelini, C.; Scorrano, L.; Salviati, L.; Vergani, L. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum Mol Genet. 2008, 17, 3291–3302. [Google Scholar] [CrossRef]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature. 2021, 590, 57–66. [Google Scholar] [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; Bindoff, L.A.; Tzoulis, C. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Aggarwal, K.; Pattnaik, B.; Mukherjee, S.; Sethi, T.; Tiwari, B.K.; Kumar, M.; Micheal, A.; Mabalirajan, U.; Ghosh, B.; Roy, S.S.; Agrawal, A. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013, 4, e461. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hajnóczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Komilova, N.R.; Angelova, P.R.; Berezhnov, A.V.; Stelmashchuk, O.A.; Mirkhodjaev, U.Z.; Houlden, H.; Gourine, A.V.; Esteras, N.; Abramov, A.Y. Metabolically induced intracellular pH changes activate mitophagy, autophagy, and cell protection in familial forms of Parkinson’s disease. FEBS J. 2022, 289, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, M.; Guillebaud, G.; Gervais, V.; St-Pierre, D.L.; Germain, M. A novel algorithm identifies stress-induced alterations in mitochondrial connectivity and inner membrane structure from confocal images. PLoS Comput Biol. 2017, 13, e1005612. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 2010, 6, e1000657. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. , Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Nadais, A.; Carvalho, A.; Morgado, B.; Santos, F.; Nóbrega-Pereira, S.; da Cruz e Silva, O.A.B. , Mitochondria dysfunction and impaired response to oxidative stress promotes proteostasis disruption in aged human cells. Mitochondrion 2023, 69, 1–9. [Google Scholar] [CrossRef]
- Hung, C.H.-L.; Cheng, S.S.-Y.; Cheung, Y.-T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.-S.; Lee, S.M.-Y.; Chang, R.C.-C. , A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, M.; Gunnarsson, T.P.; Hostrup, M.; Iaia, F.M.; Schena, F.; Pilegaard, H.; Bangsbo, J. , Metabolic stress-dependent regulation of the mitochondrial biogenic molecular response to high-intensity exercise in human skeletal muscle. J Physiol. 2018, 596, 2823–2840. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; Sesaki, H.; Cheng, Y.; Finkbeiner, S.; Nussbaum, R.L.; Masliah, E.; Edwards, R.H. , Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-associated Protein α-Synuclein *♦ Journal of Biological Chemistry 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. , Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Gegg, M.; Grunewald, A.; Wood, N.W.; Klein, C.; Schapira, A.H.V. , Bioenergetic consequences of PINK1 mutations in parkinson disease. PLoS One. 2011, 6. [Google Scholar] [CrossRef]
- Piccoli, C.; Sardanelli, A.; Scrima, R.; Ripoli, M.; Quarato, G.; D’Aprile, A.; Bellomo, F.; Scacco, S.; De Michele, G.; Filla, A.; Iuso, A.; Boffoli, D.; Capitanio, N.; Papa, S. , Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem Res. 2008, 33, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. , Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. , The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. , Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Zhang, C.; Nie, P.; Zhou, C.; Hu, Y.; Duan, S.; Gu, M.; Jiang, D.; Wang, Y.; Deng, Z.; Chen, J.; Chen, S.; Wang, L. , Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 2021, 12, 209. [Google Scholar] [CrossRef]
- Safdar, A.; Little, J.P.; Stokl, A.J.; Hettinga, B.P.; Akhtar, M.; Tarnopolsky, M.A. , Exercise Increases Mitochondrial PGC-1α Content and Promotes Nuclear-Mitochondrial Cross-talk to Coordinate Mitochondrial Biogenesis *. Journal of Biological Chemistry 2011, 286, 10605–10617. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. , Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2012, 1823, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. , Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Bustamante-Barrientos, F.A.; Luque-Campos, N.; Araya, M.J.; Lara-Barba, E.; de Solminihac, J.; Pradenas, C.; Molina, L.; Herrera-Luna, Y.; Utreras-Mendoza, Y.; Elizondo-Vega, R.; Vega-Letter, A.M.; Luz-Crawford, P. , Mitochondrial dysfunction in neurodegenerative disorders: Potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J Transl Med. 2023, 21, 613. [Google Scholar] [CrossRef]
- Rottenberg, H. , The Reduction in the Mitochondrial Membrane Potential in Aging: The Role of the Mitochondrial Permeability Transition Pore. Int J Mol Sci. 2023, 24, 12295. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. , Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Flinkman, D.; Hong, Y.; Gnjatovic, J.; Deshpande, P.; Ortutay, Z.; Peltonen, S.; Kaasinen, V.; James, P.; Coffey, E. , Regulators of proteostasis are translationally repressed in fibroblasts from patients with sporadic and LRRK2-G2019S Parkinson’s disease. NPJ Parkinsons Dis. 2023, 9, 20. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.S.; Pedro, J.M.B.-S.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Rodríguez-Arribas, M.; Climent, V.; Aiastui, A.; de Munain, A.L.; Fuentes, J.M.; González-Polo, R.A. , G2019S LRRK2 mutant fibroblasts from Parkinson’s disease patients show increased sensitivity to neurotoxin 1-methyl-4-phenylpyridinium dependent of autophagy. Toxicology 2014, 324, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhou, M.; Li, H.; Liu, D.; Liao, P.; Zong, Y.; Zhang, C.; Zou, W.; Gao, J. , Mitochondrial heterogeneity in diseases. Signal Transduct Target Ther. 2023, 8, 311. [Google Scholar] [CrossRef]
- Iwata, R.; Casimir, P.; Vanderhaeghen, P. , Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science (1979) 2020, 369, 858–862. [Google Scholar] [CrossRef]
Figure 1.
Processes involved in remodeling of the mitochondrial network morphology. The junction formation and mitochondria elongation occur by mitochondrial fusion, while separation of mitochondria from the network occurs by mitochondrial fission. Elimination of damaged mitochondria occurs through mitophagy and autophagy. In addition to these processes, mitochondrial morphology is also affected by oxidative stress, in which abnormal forms of mitochondria appear, such as “donuts” and “drops”.
Figure 1.
Processes involved in remodeling of the mitochondrial network morphology. The junction formation and mitochondria elongation occur by mitochondrial fusion, while separation of mitochondria from the network occurs by mitochondrial fission. Elimination of damaged mitochondria occurs through mitophagy and autophagy. In addition to these processes, mitochondrial morphology is also affected by oxidative stress, in which abnormal forms of mitochondria appear, such as “donuts” and “drops”.
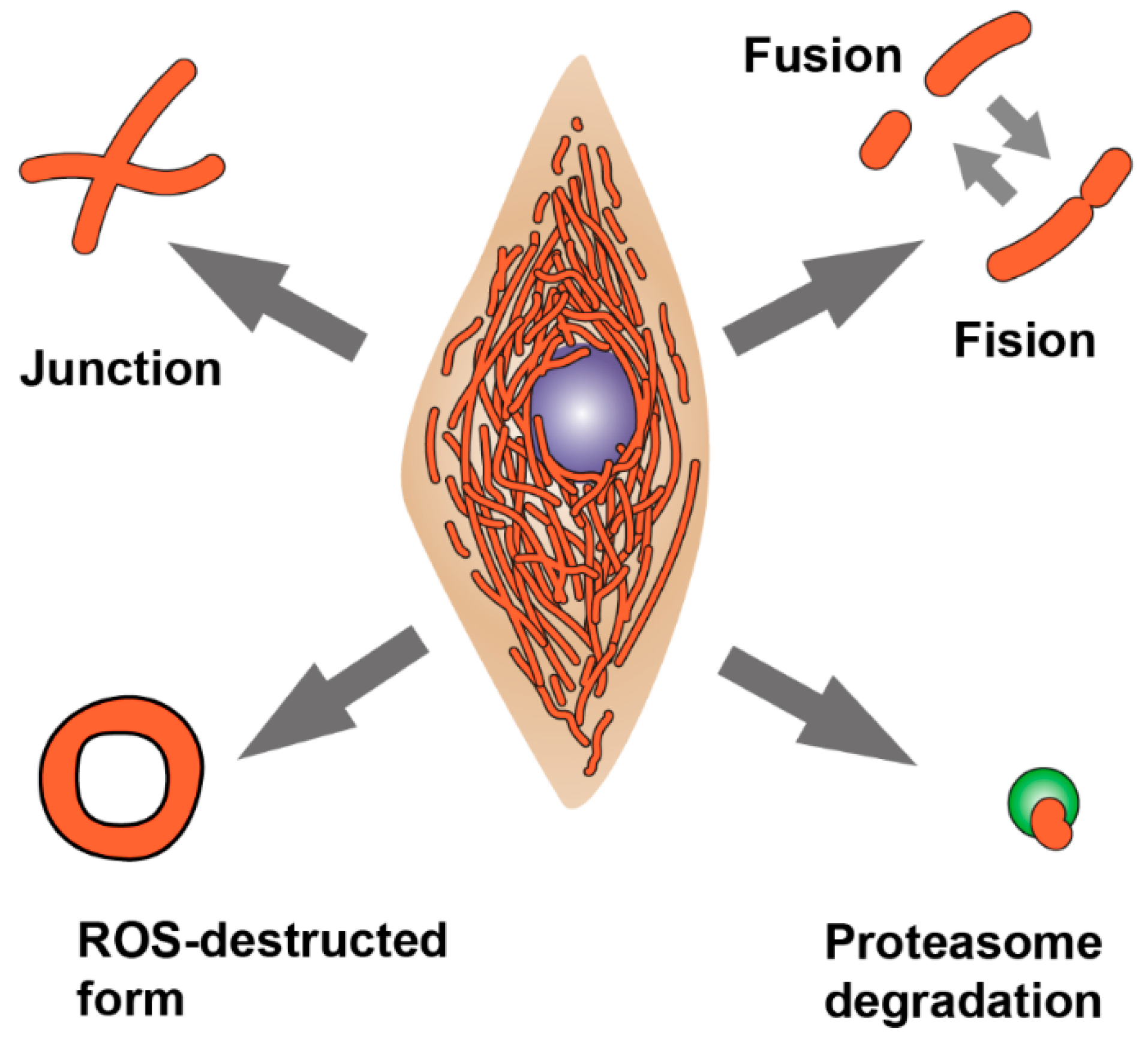
Figure 2.
Mitochondrial network (MN) morphology analysis used in this work.
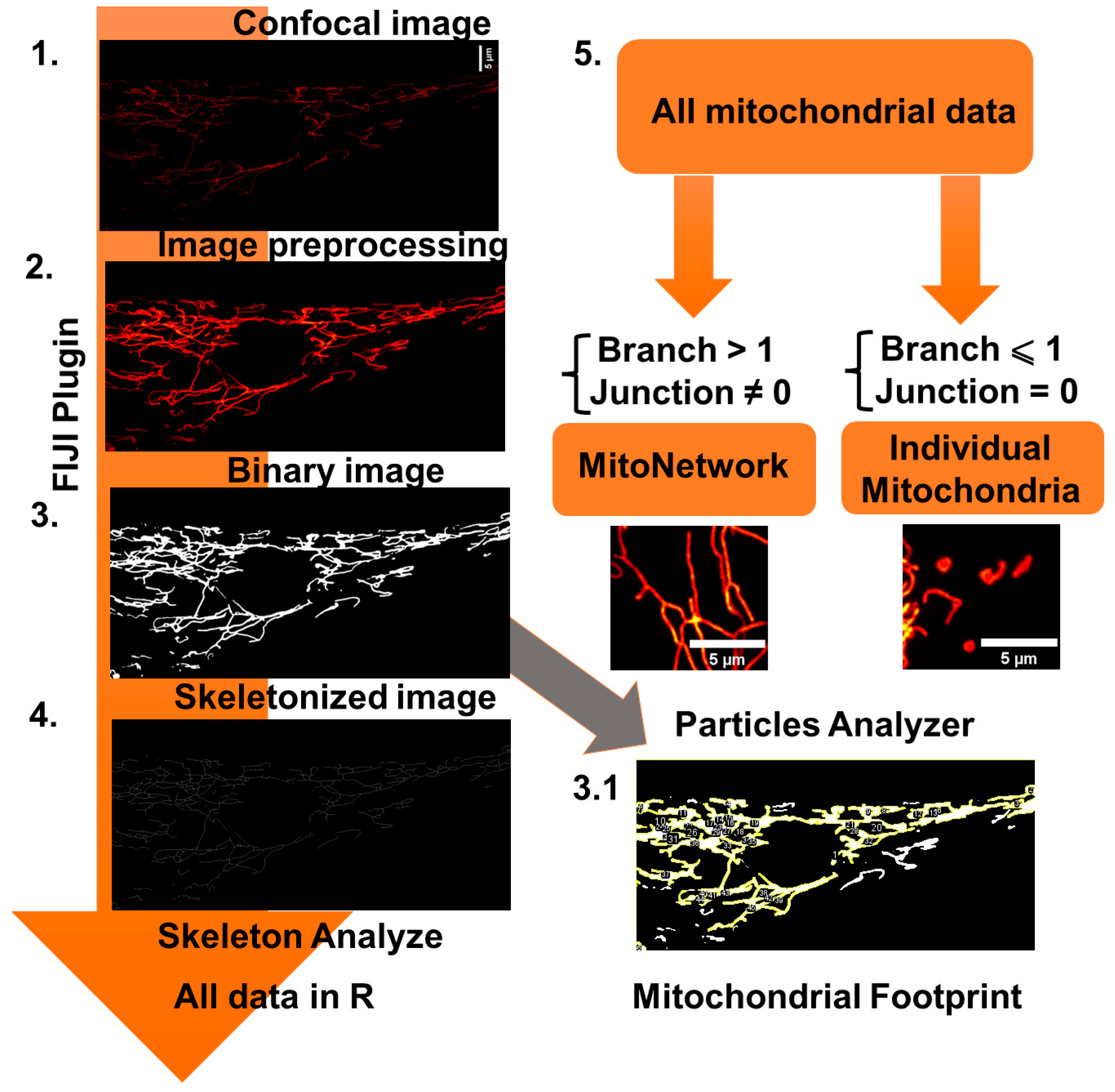
Figure 3.
Mitochondrial network morphology in fibroblasts carrying PD-related mutations. (a) Representative microphotographs of fibroblasts stained with TMRM and obtained by a super-resolution confocal microscopy (applied Fiji “Red hot” LUT). (b) Mitochondrial footprint measured by TMRM fluorescence using the plugin in Fiji “particle analyzer” under normal conditions and after hydrogen peroxide (150 µM for 1 hour) stress. (c) The median branch length in the mitochondrial network (MN), measured using plugin in Fiji “skeleton analyzer” under normal conditions and after H2O2-induced stress (150 µM for 1 hour). n = 12 cells in 3 independent experiments. Scalebar 10 μm.
Figure 3.
Mitochondrial network morphology in fibroblasts carrying PD-related mutations. (a) Representative microphotographs of fibroblasts stained with TMRM and obtained by a super-resolution confocal microscopy (applied Fiji “Red hot” LUT). (b) Mitochondrial footprint measured by TMRM fluorescence using the plugin in Fiji “particle analyzer” under normal conditions and after hydrogen peroxide (150 µM for 1 hour) stress. (c) The median branch length in the mitochondrial network (MN), measured using plugin in Fiji “skeleton analyzer” under normal conditions and after H2O2-induced stress (150 µM for 1 hour). n = 12 cells in 3 independent experiments. Scalebar 10 μm.
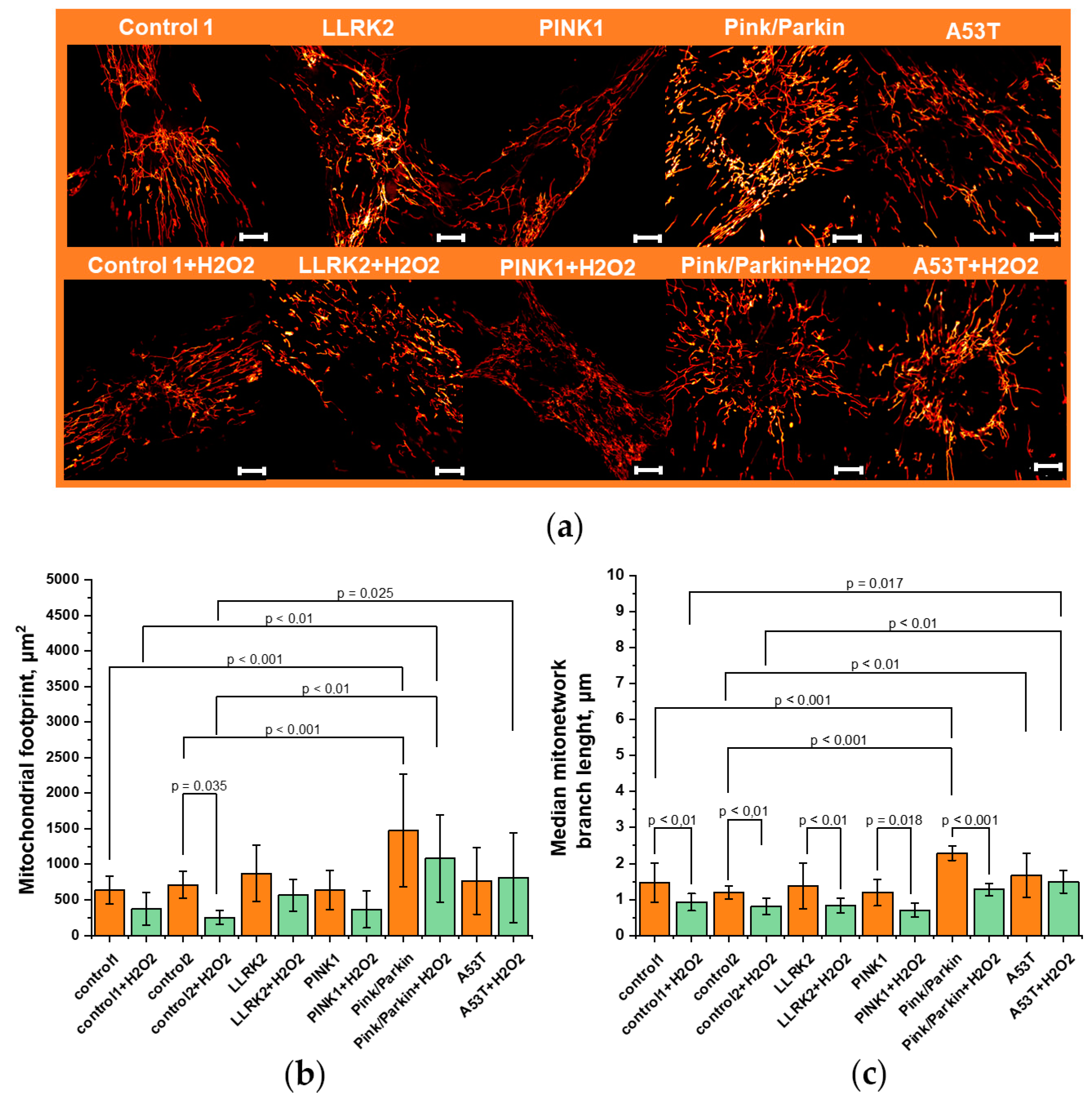
Figure 4.
The ratio and length of individual mitochondria in fibroblasts of patients with Parkinson's disease and established mutations and healthy individuals. (a) The ratio of individual mitochondria to mitochondrial networks (MN) under normal conditions and after hydrogen peroxide (150 µM for 1 hour) stress. MN is considered to be a structure containing strictly more than one branch and a non-zero connections, whereas individual mitochondria contain only one branch and zero junction. (b) The median length of individual mitochondria before and after treatment with hydrogen peroxide (150 µM for 1 hour) stress. n = 12 cells in 3 independent experiments.
Figure 4.
The ratio and length of individual mitochondria in fibroblasts of patients with Parkinson's disease and established mutations and healthy individuals. (a) The ratio of individual mitochondria to mitochondrial networks (MN) under normal conditions and after hydrogen peroxide (150 µM for 1 hour) stress. MN is considered to be a structure containing strictly more than one branch and a non-zero connections, whereas individual mitochondria contain only one branch and zero junction. (b) The median length of individual mitochondria before and after treatment with hydrogen peroxide (150 µM for 1 hour) stress. n = 12 cells in 3 independent experiments.
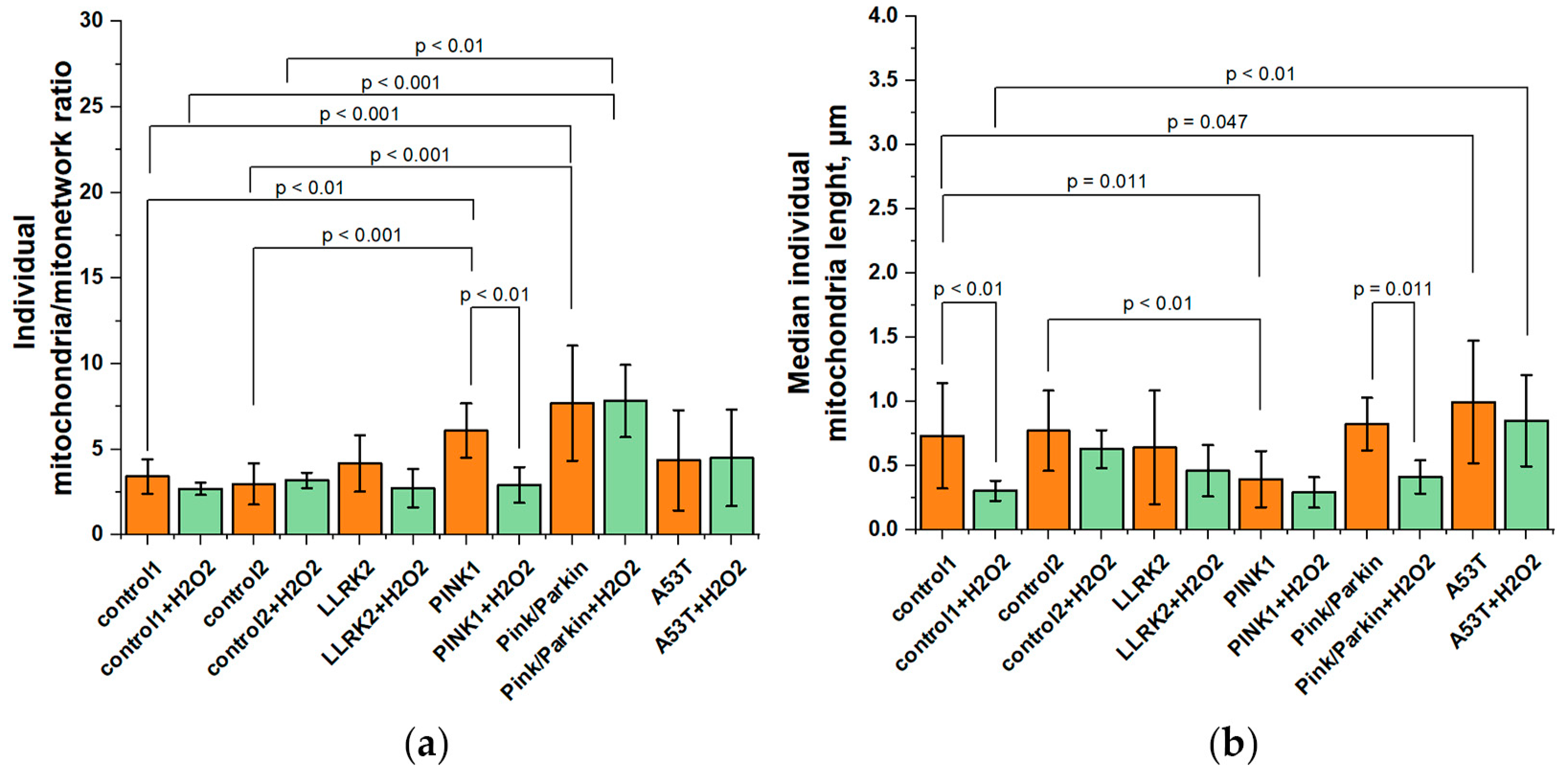
Figure 5.
The average number of junctions in mitochondrial networks (MN connectivity) in fibroblasts of patients with Parkinson's disease and established mutations and healthy individuals. The MN connectivity was evaluated under normal conditions and after hydrogen peroxide-induced stress (150 µM, 1 hour). The comparison was made relatively to two control lines and each cell line before and after hydrogen peroxide treatment. n = 12 cells in 3 independent experiments.
Figure 5.
The average number of junctions in mitochondrial networks (MN connectivity) in fibroblasts of patients with Parkinson's disease and established mutations and healthy individuals. The MN connectivity was evaluated under normal conditions and after hydrogen peroxide-induced stress (150 µM, 1 hour). The comparison was made relatively to two control lines and each cell line before and after hydrogen peroxide treatment. n = 12 cells in 3 independent experiments.
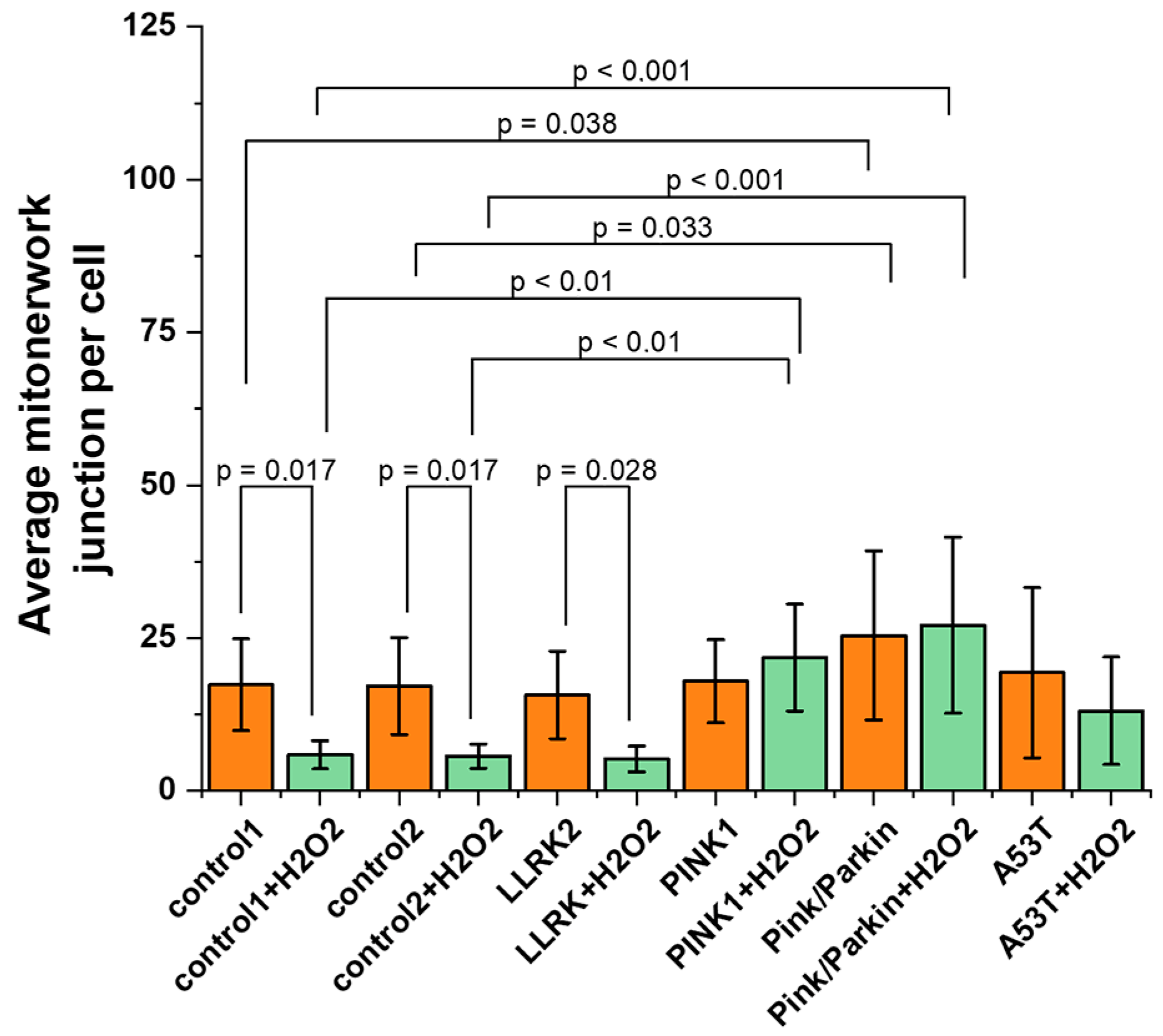
Figure 6.
Mitochondrial membrane potential in control fibroblasts and in cells with PD-related mutations. The mitochondrial membrane potential was evaluated under normal conditions and after hydrogen peroxide-induced stress (50 µM, 1 hour) as a percentage of the control TMRM fluorescence. Two controls are combined, percentages are calculated from the mean in two control cell lines. n = 60 cells in 3 independent experiments.
Figure 6.
Mitochondrial membrane potential in control fibroblasts and in cells with PD-related mutations. The mitochondrial membrane potential was evaluated under normal conditions and after hydrogen peroxide-induced stress (50 µM, 1 hour) as a percentage of the control TMRM fluorescence. Two controls are combined, percentages are calculated from the mean in two control cell lines. n = 60 cells in 3 independent experiments.
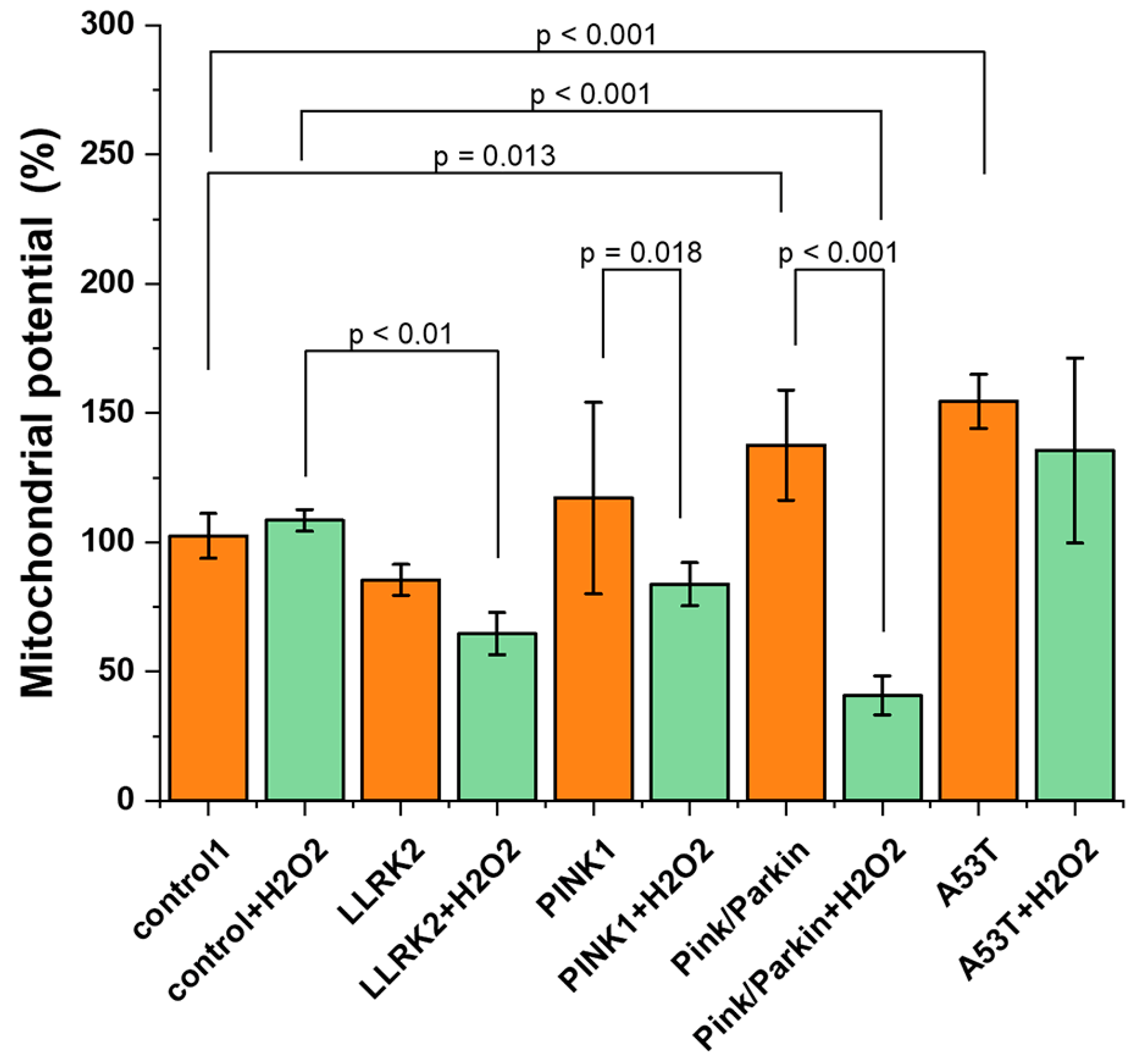
Figure 7.
ROS production in controls fibroblasts and in fibroblasts with Parkinson's disease-related mutations. (a) Cytosolic ROS production rate estimated by the time kinetics of DCF fluorescence (chemically reduced and acetylated forms of 2′,7′-dichlorofluorescein H2DCFDA probe) as a percentage of the control. (b) Mitochondrial ROS production rate estimated by the time kinetics of MitoSOX Red and MitoTracker Red CM-H2XRos probes (their fluorescent derivatives after oxidation) after hydrogen peroxide-induced stress (50 µM, 1 hour) as a percentage of the control. Two controls are combined, percentages are calculated from the mean in two control cell lines. n = 60 cells in 3 independent experiments.
Figure 7.
ROS production in controls fibroblasts and in fibroblasts with Parkinson's disease-related mutations. (a) Cytosolic ROS production rate estimated by the time kinetics of DCF fluorescence (chemically reduced and acetylated forms of 2′,7′-dichlorofluorescein H2DCFDA probe) as a percentage of the control. (b) Mitochondrial ROS production rate estimated by the time kinetics of MitoSOX Red and MitoTracker Red CM-H2XRos probes (their fluorescent derivatives after oxidation) after hydrogen peroxide-induced stress (50 µM, 1 hour) as a percentage of the control. Two controls are combined, percentages are calculated from the mean in two control cell lines. n = 60 cells in 3 independent experiments.
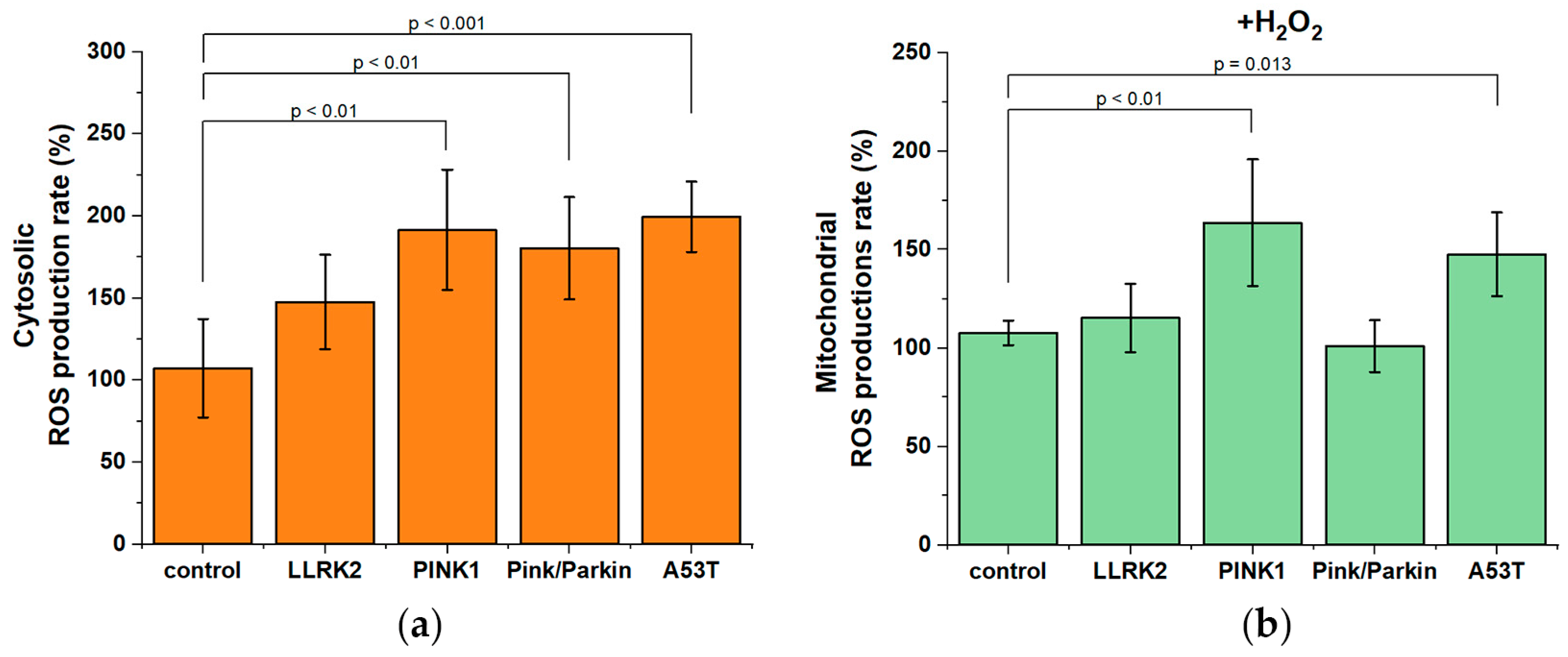

Table 1.
Cell lines used in the study.
| Fibroblasts cell line | Mutation | Diagnosis | Age | Sex |
|---|---|---|---|---|
| Control 1 | – | Healthy donor | 56 | Male |
| Control 2 | – | Healthy donor | 49 | Female |
| LRRK2 | G2019S | PD | 55 | Male |
| PINK1 | Homozygous p. Try90Leufsx12 in PINK1 | PD | 52 | Female |
| PINK1/Parkin | Park2/Pink1 double heterozygous PARK2 R275W/WT + PINK1 p. Try90Leu fs*12/WT | PD | 72 | Male |
| A53T | A53T het in SNCA | Severe PD | 52 | Female |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



