
Submitted:
12 December 2023
Posted:
13 December 2023
You are already at the latest version
Abstract
Very disparate types of cell populations exhibit similar transcription specificity for certain genes. We identified 20 genes that were expressed preferentially in myoblasts and cerebellum (Myob/Cbl genes) and examined their cell/tissue-specific epigenetics using a combined approach of comparing DNA methylation, chromatin accessibility, and RNA expression in many different cell populations. Some Myob/Cbl genes shared DNA hypo- or hypermethylated regions in myoblasts and cerebellum. Particularly striking was ZNF556, whose promoter is hypomethylated in expressing cells but highly methylated in the many cell populations that do not express the gene. In reporter gene assays, we demonstrated that its promoter’s activity is methylation-sensitive. The atypical epigenetics of ZNF556 may have originated from its promoter’s hypomethylation and selective activation in sperm progenitors and oocytes. Five of the Myob/Cbl genes (KCNJ12, ST8SIA5, ZIC1, VAX2, and EN2) have much higher RNA levels in cerebellum than in myoblasts and displayed myoblast-specific hypermethylation upstream and/or downstream of their promoters that may downmodulate expression. Differential DNA methylation was associated with alternative promoter usage for Myob/Cbl genes MCF2L, DOK7, CNPY1, and ANK1. Myob/Cbl genes PAX3, LBX1, ZNF556, ZIC1, EN2, and VAX2 encode sequence-specific transcription factors, which likely help drive the myoblast and cerebellum specificity of other Myob/Cbl genes. Our study extends our understanding of epigenetic/transcription associations related to differentiation and may help elucidate relationships between epigenetic signatures and muscular dystrophies or cerebellar-linked neuropathologies.
Keywords:
myoblasts
; cerebellum
; epigenetics
; WGBS
; EM-seq
; ZNF556
; CHD15
; TRIM72
; DOK7
; miR-486
1. Introduction
Skeletal muscle (SkM), the largest tissue in human body, has extraordinary regenerative ability and morphological plasticity in response to internal changes and external challenges [1,2]. SkM muscle progenitor cells play a central role in SkM formation and repair. Postnatally, there is a special mechanism of muscle repair involving the activation of muscle satellite cells, SkM stem cells that are usually dormant and lodged under the sarcolemma (the specialized outer membrane of SkM fibers) [3]. In response to SkM damage, satellite cells become activated to form myoblasts (SkM progenitor cells), which then differentiate and fuse with damaged myofibers, whose muscle cells (myocytes) contain hundreds or even thousands of nuclei.
The complexity of SkM formation and repair and the sensitivity of SkM to physiological and clinical changes makes the study of its epigenetics, including differentially methylated DNA regions (DMRs), of special interest. Increases in muscle mass, e.g., due to exercise, rely on hypertrophy (because myocytes cannot divide) to enlarge the fibers radially and longitudinally [4]. Hypertrophy depends on both transcriptional changes in myofiber nuclei and activation of satellite cells [3,5]. Muscle injury can affect DNA methylation profiles of satellite cells [6]. During activation of satellite cells, there are further changes in DNA methylation and in chromatin [6,7]. There is overrepresentation of age-related SkM DMRs in SkM enhancer chromatin and regions around transcription start sites (TSS).
Epigenetics is also implicated in satellite cell, myoblast, and muscle fiber heterogeneity [8,9,10] and in memory effects for strenuous muscle use and possibly for muscle disuse [5,6,11,12]. There are differences in expression profiles and epigenetics for SkM muscle fiber subtypes (e.g., fast or slow), which can interconvert [10]. Muscle memory may be partly due to DNA methylation changes associated with SkM conditioning involving a bout of certain types of exercise followed by an interval of inactivity and a subsequent reinitiation of intense activity [5].
During a recent analysis of genes associated with myoblast DMRs, we found that ZNF556 (Zinc Finger Protein 556) has a strong preference for expression in both myoblasts and cerebellum, as described below. In a previous study of myoblast epigenetics using reduced representation bisulfite sequencing (RRBS) profiles, we identified CDH15 (Cadherin 15), as a gene also displaying myoblast and cerebellum-specific expression [13]. Cerebellum is critical for motor coordination, cognition, and emotional processes [14]. Cerebellum is quite distinct morphologically and functionally from other brain regions and important in neuromuscular disease [15]. It has a transcriptome that is strikingly distinct from those of all the other brain regions [16].
In this study, we systematically explored myoblast/cerebellum transcription associations. We first identified 20 human genes that are preferentially expressed in myoblasts and cerebellum (Myob/Cbl genes). We then investigated transcription and epigenetics relationships for Myob/Cbl genes using our newly available whole-genome bisulfite sequencing (WGBS) or enzymatic methyl-seq (EM-seq) myoblast methylomes [17] and a recent WGBS profile for cerebellum neurons from Loyfer et al. [18] as well as chromatin epigenomics databases [19]. Unlike RRBS, which covers only up to 5% of the CpGs [20], WGBS and EM-seq allow the quantitation of methylation at essentially all the CpGs in the genome. These transcriptomic/epigenomic analyses using data from diverse tissues and cell cultures for comparison to myoblasts and cerebellum elucidate differences and similarities in regulation of genes that are preferentially expressed in myoblasts and cerebellum, two very different kinds of cell populations.
2. Results
2.1. Genome-wide Search for Genes Preferentially Expressed in Both Myoblasts and Cerebellum
Given our preliminary findings of myoblast/cerebellum preferential expression of several genes [13,21], we determined the frequency of such preferences for dual expression in a large set of human protein-coding genes. We examined transcription data for 13847 genes that were in both the GTEx tissue RNA-seq database (52 tissue types [16]) and an ENCODE RNA-seq database for cell cultures (six of the nine cell cultures not derived from cancers) and that had appreciable expression in at least one of the tissues and one of the cell cultures (TPM, transcripts per million, or FPKM, fragments per kilobase million, ≥ 1). This gene set had also been depleted of most noncoding RNA (ncRNA) genes. The six types of cell cultures were myoblasts, lung fibroblasts (NHLF), keratinocytes (NHEK), umbilical vein endothelial cells (HUVEC), a B-cell lymphoblastoid cell line (LCL, GM12878), and embryonic stem cells (ESC, H1). Preferential expression of a gene in myoblasts was defined as an expression ratio of ≥ 5 for myoblast FPKM to the average FPKM of the five heterologous cell cultures and a myoblast TPM ≥1. For cerebellum, preferentially expressed genes were defined as those with the cerebellum TPM vs. the average TPM of 10 other brain regions ≥ 5 and also the cerebellum TPM vs. the average TPM of 41 non-brain tissues ≥ 5. The TPM in cerebellum also had to be ≥ 1.
We found that 422 genes were preferentially expressed in myoblasts (Myob genes; 3.0% of the 13847 genes) and 239 genes were preferentially expressed in cerebellum (Cbl genes; 1.7% of the 13847 genes; Tables S1 and S2). The strongest enrichment in functional or structural terms (DAVID analysis [22]) among the Myob genes was for muscle contraction genes (35 genes; p = 2E-35). Among the Cbl genes, genes involved in neurogenesis were most enriched (16 genes, p = 3E-7). A comparison of preferential expression in each of the brain regions confirmed [16] that cerebellum is clearly the brain region with the largest number of preferentially expressed genes (Table S3). We then determined that ~5% (20) of the Myob genes were also preferentially expressed in cerebellum (Myob/Cbl genes; Table 1 and Table S2). All but two of these genes were assigned Gene Ontology (GO) terms related to the SkM and/or neural lineages (Table S4).
To test whether cell cultures other than myoblasts also share preferential expression with cerebellum, we first identified NHLF-, LCL-, HUVEC-, NHEK-, or ESC-preferentially expressed genes among the 13847-gene dataset. Preferentially expressed genes were again defined as those with expression ratios of ≥ 5 for the FPKM of each of these cell cultures to the average FPKM of five of the heterologous cell cultures (including myoblasts) and an FPKM for the cell culture of interest ≥ 1. The overlap of the resulting five gene sets with the set of Cbl genes gave the following numbers of genes that were preferentially expressed in both cerebellum and ESC, LCL, NHEK, HUVEC, or NHLF: 68, 16, 15, 11, and 4, respectively (Table S5).
Of the genes preferentially expressed in both cerebellum and one of the six cell cultures, only the Myob/Cbl genes were significantly (p < 0.01) enriched in “homeobox” and/or “developmental protein” terms (Table S5; four genes, PAX3, EN2, LBX1 and VAX1, p = 0.001 and six genes, ZIC1, EN2, CHRD, PAX3, LBX1, VAX2, p = 0.004). In contrast, among all 422 genes preferentially expressed in myoblasts, “homeobox” had less enrichment (p = 4E-7) than did muscle contraction genes (p = 2E -35) and were not significantly enriched among all the 239 genes preferentially expressed in cerebellum. Fifteen of 20 Myob/Cbl genes were associated with Myob DMRs (Table 1). We examined the epigenetic/transcription relationships of these 15 genes using RoadMap and ENCODE chromatin state segmentation and DNase-seq profiles, and WGBS methylomes.
CDH15, Which Encodes a Myoblast/Cerebellum Cadherin, Has a Hypomethylated 5’ Region in Myoblasts and Cerebellum
CDH15 (cadherin 15, M-cadherin) displayed the highest specificity for expression in cerebellum vs. other brain regions of all 20 Myob/Cbl genes (Table 1) as well as a high specificity for cerebellum vs. non-brain tissues (Table S2). We confirmed that CDH15 was more highly expressed in myotubes than in myoblasts, as was previously reported [23]. Using RRBS methylomes and reporter gene assays, we previously found that CDH15 has an intragenic cluster of Myob-hypermethylated CpGs that is part of a methylation-sensitive cryptic promoter highly active in myoblast host cells, but not in MCF7 (breast cancer cell line) [13]. With the recent availability of our myoblast WGBS/EM-seq-derived myoblast DMRs [17] and a cerebellum WGBS profile from Loyfer et al. [18], we re-examined the epigenetics of CDH15. It is not only preferentially, but also strongly, expressed in both cerebellum and myoblasts, and to a lesser extent in skeletal muscle (SkM; Figure 1A and Table 1). Its closest gene neighbor, SLC22A31, displays cerebellum- but not myoblast-specific transcription. The chromatin state profiles at CDH15 reflect its expression profile (Figure 1B). Such data is not yet available for cerebellum. Myoblasts exhibited an ~5-kb low methylated region (LMR; a region with significantly lower methylation compared to the rest of the same genome [24]) extending downstream from the promoter region. Cerebellar neurons, myoblasts, and SkM also displayed lower methylation in this region than did most other samples (Figure 1C). This region contained MyoD binding sites in myoblasts, as deduced from two ChIP-seq databases (Figure 1D). It also displayed DNaseI hypersensitivity (DNase-seq) peaks, which denote open/accessible chromatin, specific to either cerebellum or myoblasts (Figure 1E). The intragenic cryptic promoter, which overlaps two CpG islands (CGIs), that is specifically hypermethylated in myoblasts (Figure 1C, purple dotted box) was found to be larger than previously estimated from RRBS. This hypermethylation was absent in cerebellum, perhaps because cerebellum may not need to silence a cryptic promoter most active in myogenic cells. We also found that there was another Myob-hyperm DMR 0.7 kb upstream of the CDH15 TSS, which had very low levels of methylation in cerebellar neurons. Therefore, only some of the presumably cis-acting DNA differentially methylated regions in or adjacent to CDH15 are shared between myoblasts and cerebellum in this gene which is very highly and specifically expressed in both cell populations.
2.2. The Promoter region of Transcription Factor-Encoding ZNF556 Is Hypomethylated Specifically in Tissues and Cells Expressing the Gene
Of the 20 Myob/Cbl genes, ZNF556 (zinc finger protein 556) has the strongest specificity for expression in myoblasts (Table 1). It is a very little studied gene that encodes a protein which contains C2H2 zinc finger domains and a KRAB domain seen in many ZNF transcription factors (TFs). ZNF556 is also specifically expressed in cerebellum, ovary, HepG2 (liver cancer cell line), and testis, with lower expression in SkM (GTEx; Figure 2A and Figure 3A, top). Only expressing cell types and tissues display promoter chromatin and low DNA methylation both upstream and downstream of the TSS (Figure 2B and C, and Figure 3A). In addition, only expressing samples had a prominent DNase-seq peak in this region, which was seen immediately upstream of the TSS (Figure 2D and Figure 3A).
The nearest neighbor to ZNF556 is ZNF555, whose 3’ end is 7 kb upstream of the ZNF556 TSS (Figure 2A). ZNF555 encodes a TF [25] that, like ZNF556, is a member of the KRAB-ZNF family. ZNF555 had a much broader tissue and cell culture expression profile than ZNF556 although its RNA levels in myoblasts were twice that of the average of 5 other cultures. Like ZNF556, ZNF555 was expressed at high levels in testis relative to other tissues (TPM Testis/Average 36 other tissues = 4.3). The three other genes within the 150-kb neighborhood of ZNF556 are also zinc finger protein-encoding genes but none show preferential expression in myoblasts although one (ZNF57) does in testis (Figure S1). Given the lack of myoblast-associated enhancer chromatin overlapping or near ZNF556 and its presence in ZNF555 intragenic regions with overlapping Myob-hypom DMRs (Figure 2B), the intragenic ZNF555 enhancer chromatin may help upregulate ZNF556 expression in myoblasts and myotubes.
Because of the strong association of ZNF556 expression with its promoter hypomethylation, we cloned several fragments from its 5’ end (Figure 3A) to test the effect of in vitro CpG methylation at the cloned sequences on promoter activity. Surprisingly, we found that DNA sequences from 0.6 kb upstream of the TSS to 0.1 kb downstream (Figure 3A, -0.6 to +0.1) had negligible promoter activity in C2C12 myoblasts despite their overlap with the myoblast-associated DNase-seq peak, the Myob-hypom DMR, and an apparent nucleosome-free region as seen in the myoblast histone H3 lysine-27 acetylation profile (H3K27ac; Figure 3A and B). However, moderately strong activity was seen in C2C12 myoblasts when the cloned DNA sequences were extended upstream to -1.3 to +0.1 kb relative to the TSS or when a shorter region from -0.2 to +0.3 kb (instead of -0.6 to +0.1 kb) were tested for promoter activity. No activity or lower activity was observed in MCF7 cells with these constructs (Figure 3B). Therefore, the +0.1 to +0.3 and the -0.6 to -1.3 kb sequences may contribute to promoter function in vivo even though the latter are normally methylated in myoblasts at their low CpG-density DNA sequence (Figure 3A). When the two reporter constructs that contained promoter activity were CpG-methylated in vitro only at the cloned sequences by M.SssI, about half to 60% of the promoter activity was lost (Figure 3C). Our findings also indicate cooperative interactions to establish promoter activity involving sequences outside of DNase-seq (open chromatin) peaks interacting with DNA sequences in the DNase-seq peak region.
Although the role of ZNF556 in development is unclear, there are indications of its importance from additional aspects of its transcriptional and epigenetic specificity. First, its expression in human fetal cells is highest in SkM myocytes and satellite cells, as determined from single-cell RNA-seq (scRNA-seq) profiles (UCSC Genome Browser, hg38, data not shown [26]). Among various non-brain mid-gestation tissues, the expression of ZNF556 (but not ZNF555) is much higher in muscle than in other examined tissues (Figure 4A). From our previous bulk RNA-seq data for myoblasts and myotubes [27], ZNF556 RNA levels in myotubes were twice as high as in myoblasts. Importantly, in an extensive scRNA-seq analysis of many post-natal human tissues (not including cerebellum) and cell types [28], ZNF556 RNA levels are highest in ovarian stromal cells, oocytes, and spermatogonia, as well as in skeletal myocyte, esophageal squamous epithelial cells, and plasmacytoid dendritic cells. Transcriptome analysis of early embryos as part of the EmAtlas collection [29] indicates the presence of ZNF556 and ZNF555 in zygotes, 2-cell, and 4-cell human embryos (Figure 4A and B). In addition, WGBS profiles of sperm, oocytes, zygotes, 2-cell, 4-cell, 8-cell and morula embryos [29,30] show a loss of promoter region hypomethylation at the 8-cell stage, when ZNF556 RNA is no longer detectable (Figure 4C). However, much RNA detected at the earliest stages in the pre-implantation embryo is carried over from the oocyte to the zygote.
2.3. Differential Methylation of the Extended Promoter Regions of TRIM72 and Its Intronic Gene, PYDC, Correlates with Differential Expression in Myoblasts and Cerebellum
TRIM72 (tripartite motif containing 72) encodes a ubiquitin ligase that is involved in muscle regeneration, calcium homeostasis, excitation-contraction coupling, and mitochondrial autophagy [31]. It has a very high specificity for SkM and myoblasts as seen in RNA-seq profiles and chromatin epigenetic profiles (Figure 5A and B). TRIM72 is a myokine, a SkM secreted protein, with systemic effects on membrane repair and skin repair [32,33]. It is one of the unusual genes that harbors an antisense coding gene, PYDC1 (which encodes an inhibitor of NFκB [34]), within one of its introns. Both PYDC1 and TRIM72 are specifically expressed in cerebellum but only TRIM72 is also expressed in myoblasts and SkM. PYDC1 is also strongly expressed in skin and suprabasal keratinocytes in skin [28] and in a primary keratinocyte cell culture (NHEK, Figure 5A, purple signal). Correlated with the transcription profiles, myoblasts and SkM displayed hypermethylation at the PYDC1 promoter and throughout this 1-kb gene (Figure 5C). Although the lack of methylation in this region in PYDC1-expressing cerebellum and skin was consistent with expression in these tissues, there also was little or no methylation in tissues not expressing this gene. Therefore, the lack of methylation was necessary but not sufficient for expression. In contrast, only TRIM72-expressing samples (myoblasts, SkM, and to a lesser extent, cerebellum) displayed hypomethylation in the promoter region of the TRIM72 as an extension of a constitutively unmethylated CGI originating at its exon 2.
2.4. Alternate ANK1 Promoter Usage in Myoblasts vs. Cerebellum Is Associated with Differential Promoter Methylation
ANK1 (ankyrin 1) codes for a protein that links integral membrane proteins to the cytoskeleton and is involved in cell motility, proliferation, and the maintenance of specialized membrane domains [34]. It is very highly expressed in erythroblasts and also preferentially transcribed in cerebellum, SkM, and, to a lesser extent heart (Figure 6A) [26,34]. It has many tissue-specific RNA isoforms (see Figure 6A for several of them) involving alternative promoter usage as well as alternative splicing. SkM [35], myoblasts, and heart transcribe predominantly one of the shortest isoforms (ENST00000314214) while cerebellum transcribes predominantly one of the longest isoforms (ENST00000289734, Figure 6A and GTEx isoform expression profiles [16]). Myoblasts express this gene at the highest levels compared to 17 other non-transformed cell cultures in the ENCODE database (Figure 6A and data not shown). Some cell cultures unrelated to the muscle lineage express ANK1 at moderate levels, e.g., embryonic stem cells (ESC), bone marrow stem/stromal cells, and osteoblasts, but they do not use myoblasts’ proximal promoter, as indicated by 5’ cap analysis gene expression (CAGE) [36] of cell cultures (data not shown). The 5’ ends of the large ANK1 isoforms can be difficult to ascertain. For example, CAGE profiles show that ESC primarily use the promoter of the ANK1 isoform ENST00000705522, and GTEx indicates that cerebellum predominantly initiates transcription at a more distal promoter although this is not clear from RNA-seq profiles (Figure 6A). In the last intron of all the ANK1 isoforms, MIR486 is found. It encodes miR-486, a miRNA that is essential for myoblast proliferation and differentiation, normal myogenesis, and normal SkM and heart formation [37].
Tissue-specific differential ANK1 promoter usage was reflected in DNA methylation profiles and chromatin state profiles (Figure 6B and C). Only SkM had a long LMR that overlapped the gene body of the main striated muscle lineage-associated short isoform, its promoter, and a super-enhancer [38] (>5 kb cluster of enhancer and promoter chromatin with strong H3K27ac; Figure 6B, dotted box). Heart also displays a super-enhancer in this region but, consistent with its much lower ANK1 expression than in SkM, it has less extensive DNA hypomethylation (Figure 6A and B). At the muscle/myoblast promoter, myoblasts exhibited a smaller LMR than seen in SkM (Figure 6C)) and, under less stringent conditions of hypom DMR assignment (methylation difference of ≤-0.20 instead of our standard ≤-0.35), a Myob-hypom DMR that coincided with the myoblast LMR (data not shown). In contrast, ANK1-expressing cerebellum as well as tissues/cells that do not express ANK1 were highly methylated in this region (Figure 6C). The cerebellum promoter for ANK1 overlaps enhancer chromatin in SkM and heart and a CGI (Figure 6A and C). Importantly, most of this CGI overlapped a Myob/SkM-hyperm DMR, which has a Myob-hypom DMR upstream (Figure 6C). These results suggest that the Myob-hyperm DMR represses formation of the cerebellum promoter but also permits this region to be harnessed as an enhancer. Consistent with this interpretation, a strong DNase-seq peak and a MyoD site in myoblasts overlaps the far-upstream Myob-hypom DMR (Figures 6D and E).
2.4. Myoblast DNA Hypermethylation of PAX3 May Be Downmodulating Gene Expression by Suppressing Super-Enhancer Formation
PAX3, which encodes a developmental TF that plays critical roles in embryogenesis, including in neural development and myogenesis [34,39,40], is preferentially, but lowly, expressed in skin fibroblasts, myoblasts, and cerebellum (Figure 7A). Among postnatal somatic cells, it is much more highly expressed in melanocytes than in other cell types (Figure 7B and [28]), which is consistent with its role in melanocyte development and homeostasis [41]. Myoblasts exhibit hypermethylation at the 5’ end of the gene and upstream through the adjacent CCDC140 ncRNA gene, as was seen previously in RRBS profiles [42]. A cluster of Myob-hypermeth DMRs, which were deduced from WGBS and EM-seq, extended from +0.4 to +9.7 kb and -0.5 to -14.6 kb relative to the PAX3 TSS. RRBS data indicated that this region was mostly unmethylated in melanocytes (with the exception of a far-upstream subregion (Figure 7D, dotted box) as well as in 15 primary cell cultures and 13 tissues that show little or no expression of this gene (Figure 7C and D and data not shown [43]). This long unmethylated region in PAX3-nonexpressing cells/tissues overlaid repressed chromatin. In cerebellum, foreskin fibroblasts, and SkM, as well as in myoblasts, all of which express this gene (Figure 7A; [28]), there was a cluster of highly methylated subregions in this region (Figure 7C). The Myob-hyperm DMRs and high-methylation subregions in cerebellum were interspersed with methylation valleys that correspond to tissue/cell-specific DNase-seq peaks in myoblasts and cerebellum and, sometimes also, MyoD binding sites in myoblasts (Figure 7C and E). These findings suggest that a major function of the hypermethylation in myoblasts and cerebellum is to downmodulate expression of this gene in myoblasts and cerebellum by helping to prevent the formation of the 5’ super-enhancer seen in highly expressing melanocytes. In addition, DNA hypermethylation may suppress expression of the very weakly expressed CCDC140 ncRNA gene which shares a bidirectional promoter with PAX3. Because of the two genes’ strikingly similar transcription profiles and shared preference for expression in melanocytes, skin fibroblasts, and myoblasts, this very little studied ncRNA gene, which overlaps Myob-hyperm DMRs, may help regulate PAX3 expression.
2.5. Other Genes Preferentially Expressed in Myoblasts and Cerebellum Profiles Display Myoblast- or Cerebellum-Associated Hypomethylation and Overlapping Transcription Factor Binding Sites
Ten additional Myob/Cbl genes, which are associated with Myob DMRs were examined for epigenetic/transcriptional associations (Figures S2 – S10). Seven of these (ZIC1, CHRD, KCNJ12, VAX2, and ST8SIA5, and the gene neighbors EN2 and CNPY1) are expressed much more highly in cerebellum than in myoblasts (Table 1). All but one of the seven genes, CHRD (chordin), had Myob-hyperm DMRs, which were missing in cerebellum, near promoter regions and/or DNA hypomethylation at or near the promoter specifically in cerebellum. This myoblast- or cerebellum-specific differential methylation can help explain the differences in the extent of expression of these genes. CHRD had its only Myob-DMR as a hypomethylated DMR overlapping Myob-associated enhancer chromatin but had multiple intragenic regions of hypomethylation in cerebellum (Figure S3). The methylomes for LBX1 (ladybird homeobox 1) indicate that the multiple Myob-hyperm DMRs are likely downregulating its expression in myoblasts (Figure S8). However, the lack of chromatin state profiles for cerebellum at these regions that are largely unmethylated regions in cerebellum (LBX1 expressing) and most tissues (LBX1 silent) makes interpreting their DNA methylation/transcription relationships difficult (Figure S8).
MCF2L displayed associations between differential DNA methylation at alternative promoters and alternate promoter usage in myoblasts vs. cerebellum (Figure S9). DOK7 (docking protein 7) displays a Myob-hyperm DMR as well as DNA hypermethylation in cerebellum immediately downstream of an alternative intragenic promoter (Figure S10). The canonical upstream promoter rather than the downstream one is used preferentially in myoblasts and cerebellum and some other expressing cell cultures and tissues (Figure S10 and GTEx isoform profiles [16]). In contrast, mammary epithelial cells (HMEC, Figure S10) and B cells (data not shown) use the downstream promoter and are hypomethylated in this TSS-downstream region. DOK7 is, therefore, an example of Myob/Cbl gene with regions displaying similar cell/tissue-specific DNA methylation and alternative promoter usage in myoblasts and cerebellum.
We examined the seven Myob/Cbl genes (KCNH1, ZNF556, TRIM72, ANK1, NRXN2, LBX1, and MCF2L) that had Myob-hypom DMRs to ascertain whether cerebellum also had an overlapping region of unusually low DNA methylation. This was the case for ZNF556, ANK1, TRIM72, and MCF2L. Within three of these Myob-hypom DMRs, we found predicted TF binding sites (TFBS) in the JASPAR database for TFs that are encoded by Myob/Cbl genes (LBX1, EN2, and ZIC1) (Figure S11) but none of the six TFs encoded by Myob/Cbl genes was in the UniBind ChIP-seq database. We also found predicted or ChIP-seq-detected TFBS for TFs that are highly specific for either myoblasts (MYOD, MYF5, MYF6, and MYOG) or cerebellum (ETV1, NEUROD1, NEUROG1, OTX2, or PAX6; Figure S11).
Lastly, for the Myob/Cbl genes that encode TFs, we also examined relative transcription profiles in human fetuses and embryos using EmAtlas [29] (which does not have brain data other than spinal cord). Four of these genes are expressed preferentially in fetal SkM and spinal cord (ZIC1, PAX3, VAX2, and LBX1). EN2, like ZNF556 (Figure 4), is preferentially expressed in fetal SkM, oocytes, and pre-implantation embryos (Figure S12).
3. Discussion
From a genome-wide examination of transcriptomics and epigenomics, we identified 20 genes (Myob/Cbl genes) that have a strong preference for transcription in myoblasts (mesodermally-derived SkM progenitor cells) and cerebellum, a highly dissimilar cell population (ectodermally derived). The co-expression of genes in myoblasts and cerebellum, rather than in other brain regions (Table S3), reflects the much higher number of genes preferentially expressed in cerebellum than elsewhere in the brain ([16,44] and Table S2). In contrast, myoblasts were not unusual compared to several other progenitor cell strains in having a small subset of genes preferentially expressed in both that cell culture (HUVEC, NHEK, or NHLF) and cerebellum (Table S5). However, only Myob/Cbl genes had a significant ontological association with TFs (Table S5, ZIC1, EN2, PAX3, VAX2, LBX1, and ZNF556), which suggest a special transcriptional relationship for myoblasts and cerebellum, although for only a very small percentage of the genome. Among the 20 Myob/Cbl genes, the strongest association of myoblast DNA hypomethylation with gene expression was seen for ZNF556. Very little is known about its function, but the encoded protein has the structural hallmarks of KRAB zinc finger TFs, which often act as repressor proteins [34,45]. The ZNF556 promoter overlaid a Myob-hypom DMR, which was highly methylated in all 17 examined non-expressing ENCODE cell populations and hypomethylated in promoter chromatin specifically in all ZNF556-expressing cell populations (Figure 2 and Figure 3). Its hypomethylation and expression in HepG2, a hepatocarcinoma cell line, provides an example of cancer DNA hypomethylation coupled with expression of a gene normally active in a small number of very different cell types.
In predicting the relationship of promoter DNA methylation to gene expression, the CpG content of the promoter region is critical [46]. The ZNF556 Myob-hypom DMR (TSS -0.7 to +1.3 kb) almost fits the UCSC Genome Browser’s definition of a CGI except that its observed CpG density/expected CpG density was 0.58 instead of >0.6. This promoter DMR would be classified as an intermediate CpG promoter by the definition of Weber et al. [46]. CGI promoters are predominantly constitutively unmethylated or very lowly methylated. Normal or disease-acquired methylation of CGI promoters and intermediate-to-high CpG promoters is strongly associated with transcription repression in human cells [46,47,48]. Therefore, it is likely that methylation of the ZNF556 DMR in vivo helps silence it. To test this, we determined the effect of DNA methylation on ZNF556 promoter activity in reporter gene assays. In vitro methylation of cloned ZNF556 DMR sequences reduced promoter activity by about half (Figure 3). Downregulation in vivo by DNA methylation might be much stronger because we tested the effect of DNA methylation on constructs that did not include the further downstream, CpG-enriched parts of the DMR (Figure 3). We conclude that the ZNF556 DNA hypomethylation in vivo coupled with promoter chromatin at the promoter region enables tissue/cell-specific transcription of this gene.
Unexpectedly, the region immediately upstream of the ZNF556 TSS (TSS -0.6 to +0.1 kb) did not suffice for appreciable promoter activity in the reporter gene assays even though it overlapped the only prominent DNase-seq peak at the promoter region in ZNF556-expressing samples. This expression-linked open-chromatin region likely cooperates with adjacent upstream or downstream sequences, which are hypomethylated in myoblasts and cerebellum (Figure 3). The unusually high concentration of DNA repeats in the TSS-upstream region, especially the LTRs (Long Terminal Repeats, Figure 3A, bottom), might influence the activity and/or the methylation status of the promoter [49].
The hypomethylation of the ZNF556 promoter region in germline cells can help explain the finding that high methylation of its promoter is the default state in non-expressing cells. The ZNF556 promoter region was hypomethylated in ovarian stromal cells, oocytes, spermatogonia, early spermatids as well as zygotes, 2-cell embryos, and 4-cell embryos (but not morula), and all of these cell populations contain ZNF556 RNA (Figure 4; [28,29]). The high level of methylation of the ZNF556 CGI promoter in most somatic cell types is an exception to the strong correlation of CpG-rich promoters having little or no DNA methylation in normal somatic cell types [50]. The best characterized exception to this general finding is for the set of genes specifically expressed in the germline [46]. Some CGI promoters are highly methylated in somatic tissues and hypomethylated in sperm [51] as a result of large losses in DNA methylation from many parts of the genome in mammalian primordial germ cells, which ultimately give rise to both sperm and oocytes [52]. For some of the above-mentioned cell types, e.g., zygotes, ZNF556 promoter hypomethylation might be a memory of former transcriptional activity and possibly a poised state for future activation. This relationship of ZNF556 expression to gametogenesis and pre-implantation embryos suggests a role for ZNF556 in pre-implantation embryos that might be shared by its neighbor ZNF555, as evidenced by the even higher enrichment of ZNF555 RNA in zygote, 2-cell, and 4-cell embryos than was seen for ZNF556 (Figure 4). Therefore, we propose that during evolution, first, ZNF556 was expressed just in the germline and possibly the preimplantation embryo and only later was there repurposing of the associated epigenetics in myoblasts and cerebellum to extend the tissue/cell specificity of this TF and its functionality.
Two other Myob/Cbl genes, CDH15 and TRIM72, had DNA hypomethylation in the promoter region that is likely to help contribute to the high transcription of these genes in myoblasts and cerebellum (Table 1). Previously, we demonstrated that a region of RRBS-detected myoblast hypermethylation at an intragenic CGI in CDH15 is a methylation-inhibited cryptic promoter [13]. However, because of the limited coverage by RRBS, that study did not find the long region of low DNA methylation that stretches from upstream of the CDH15 TSS to far downstream into intron 1 in myoblasts and cerebellum, as seen in WGBS profiles (Figure 1). The myoblast- and cerebellum-hypomethylated DNA regions overlapping the CDH15 and TRIM72 promoters are transcription-associated extensions of adjacent, constitutively unmethylated regions that overlap CGIs (Figure 1 and Figure 5).
TRIM72 presents the unusual example of a gene with a Myob-hypom DMR at its promoter and a Myob-hyperm DMR overlapping the promoter of its intronic, protein-coding gene, PYDC1 (Figure 5). PYDC1 resides in intron 1 of TRIM72 and is positioned antisense to it. Unlike its TRIM72 host gene, PYDC1 was silenced in myoblasts and SkM but selectively expressed in cerebellum (TPM ratio 4.7 for cerebellum vs. the average of 10 other brain regions). RNA levels for TRIM72 are almost 4-fold lower in cerebellum than in SkM, which might be due, in part, to dampening of TRIM72 transcription in cerebellum due to the clashing movement of RNA polymerase complexes from the PYDC1 TSS. This dichotomous expression of two overlapping genes in cerebellum and myoblasts is probably achieved, in part, by the Myob/SkM-hyperm DMR that extended from the PYDC1 promoter region. In contrast, cerebellum lacked DNA methylation at and around PYDC1 as did most tissues with a silent PYDC1 gene. This indicates that the PYDC1 CGI promoter is constitutively unmethylated regardless of expression status with the exceptions of myoblasts and SkM (Figure 5). This SkM-lineage hypermethylation overlaps enhancer chromatin or mixed enhancer/promoter chromatin. The hypermethylation probably helps establish or maintain an intragenic TRIM72 enhancer in the SkM lineage at a region that is an active, unmethylated PYDC1 promoter in cerebellum and repressed chromatin in most other cell populations.
Among the Myob/Cbl genes, Myob-hyperm DMRs were more frequent than Myob-hypom DMRs (Table 1). Some of this differential methylation may help direct alternative promoter usage for ANK1, CNPY1, DOK7, and MCF2L (Figure 6, Figure S7, 8, and Figure S10). The alternate promoter usage for these genes not only changes the polypeptide that the gene encodes, but also, in the case of ANK1, might affect the efficiency of production of a co-transcribed intronic miRNA miR-486 (Figure 6). This miRNA plays pivotal roles in myogenesis and is important for normal heart formation in mice [37]. Moreover, it is being considered for therapeutic disease modulation because its upregulation in dystrophic mouse models can reduce symptoms of muscular dystrophy [37]. In addition, in some cancers, miR-486 is an oncogenic marker and may play a role in oncogenesis [53,54]. DNA hypermethylation at multiple ANK1 CGI promoter regions is associated with cancer-linked changes in miRNA levels [53]. However, the epigenetics of the myoblast/SkM/heart promoter in cancers has received little attention probably because it does not overlap a CGI. There might be cancer-associated hypomethylation of this ANK1 promoter, which could influence miR-486 levels as well as ANK1 isoform abundance ratios.
Some Myob/Cbl genes exhibit only moderately low steady-state levels of RNA in myoblasts but much higher levels in cerebellum (Table 1). Five of these genes (KCNJ12, ST8SIA5, ZIC1, EN2, and VAX2) displayed Myob-hyperm DMRs upstream and/or downstream of their core promoter region that was missing in cerebellar neurons (Table 1, Figures S2, S4-S6) and was probably downmodulating expression of these gene in myoblasts vs. cerebellum. In a previous study of TBX15, a TF-encoding gene preferentially expressed in myoblasts and SkM but not in any brain region, we used reporter gene assays and in vitro methylation to demonstrate that enhancer and promoter activity of Myob-hyperm DMR sequences upstream or downstream of its TSS was strongly suppressed by DNA methylation [17]. Myoblast and cerebellum hypermethylation upstream and downstream of the PAX3 promoter in both myoblasts and cerebellum is probably helping to keep transcription low in both cell populations (Table 1, Figure 7).
In addition to regulating usage of alternative promoters, directing usage of intronic promoters, and silencing cryptic promoters, intragenic hypermethylation in transcribed genes can facilitate movement of the RNA polymerase complex across the gene body of actively transcribed genes to regulate alternate splicing [55,56]. It has also been proposed that such intragenic methylation associated with transcription may be simply a consequence of the recruitment of DNMT3 enzymes’ PWWP domain by H3K36me3. Twelve of the Myob/Cbl genes had intragenic Myob-hyperm DMRs but only three of them (CDH15, ANK1, and MCF2L) had DMRs that overlapped H3K36me3-enriched chromatin in myoblasts (Txn-chrom, Figure 1 and Figure 6, and Figure S9). Another mechanism for intragenic or intergenic DNA hypermethylation being positively associated with gene expression is that DNA methylation may decrease the spread of repressive H3K27me3-enriched chromatin in many chromatin/DNA contexts [57,58]. Although epigenetic profiles suggested that this is not the function of most of the examined DNA hypermethylation at Myob/Cbl genes, it might be the case for the VAX2 TSS-downstream DNA hypermethylation in myoblasts and cerebellum (Figure S5).
A caveat in our study is that DNA methylation levels at cis-regulatory elements can vary with the exact nature of the cells or tissues studied (physiology, cell composition, age, and health status of the donor, and for SkM, the muscle location, and fiber type [8,59,60,61]. However, these changes are usually less than the strong differences in DNA methylation that are tissue-specific. Another caveat is that WGBS does not distinguish between genomic 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), which often have different biological effects [62,63]. This is not a complication for Myob-hyperm DMRs because we found that myoblast cell strains have little or no 5hmC at tested CpG sites in 13 examined myoblast RRBS-delineated DMRs, including one in the CDH15 intragenic Myob-hyperm DMR at its cryptic intragenic promoter [13]. This is consistent with the loss of genomic 5hmC reported upon passage of mouse embryonic fibroblast cell strains [64]. In the case of tissues, especially cerebellum, how much of the WGBS signal for DNA methylation is actually 5hmC is an important consideration [13,63,65]. For example, we previously found that SkM and cerebellum at a tested CpG in the CDH15 Myob/SkM-hyperm DMR has about twice as much 5hmC as 5mC [13].
An illustration of the need to consider the 5hmC content of hypermethylated regions in cerebellum comes from the important findings of James et al. [59,66]. Their studies suggest a role for the homeobox TF EN2 (encoded by a Myob/Cbl gene) in autism spectrum disorder in addition to its pivotal contributions to cerebellum development [67]. They found that a 0.15-kb region ~ 3 kb upstream of the EN2 TSS had more 5hmC as well as 5mC in patient samples than in matched controls. These epigenetic changes were positively associated with more EN2 RNA and protein. We observed that this region is partially methylated in control cerebellum in contrast to little or no methylation in other studied tissues and cell types (Figure S7E and G). Moreover, Szulwach et al. [68] found that the ~ 4 – 8 kb region upstream of the mouse En2 gene in cerebellum contains peaks of 5hmC overlapping a previously identified embryonic enhancer for the En2 gene. Several of the human cerebellum-hypermethylated regions that we observed upstream of the EN2 promoter are adjacent to DNase-seq peaks. They might help demarcate enhancers or, if they contain sufficient 5hmC, counteract binding of the repressive MECP2 protein [59,67,69].
For some of the Myob/Cbl genes there are apparent functional relationships between myoblasts and cerebellum. Nine of these genes (ANK1, CDH15, DOK7, FNDC5, MCF2L, TRIM72, CHRD, KCNJ12, and PTPRR) encode proteins localized mostly or in part to the plasma membrane, and the first six of these were expressed at moderate to high levels in both myoblasts and cerebellum (Table 1 and Table S5). Regulation of cell-cell interactions is critical for controlling neuronal function [70] as well as for regulating myoblast fusion [71,72]. An example of a Myob/Cbl gene with known myoblast and brain functions for one of these plasma membrane-associated Myob/Cbl is the above-mentioned CDH15. Its encoded protein is a cadherin implicated in fusion of myoblasts to form multinucleated myotubes via its role in cell-cell interactions [23,71] and in intellectual function from studies of mutationally linked intellectual disability syndromes for which the mutations alter cell-cell contacts [73,74].
Another functional relationship shared by multiple Myob/Cbl genes is that six of them (ZNF556, EN2, ZIC1, PAX3, LBX1, and VAX2) encode TFs, four of which are homeobox-containing TFs. Both “transcription factor activity” and “homeobox” categories were significantly overrepresented among the 20 Myob/Cbl genes (Table S5). Precise modulation of levels of expression at different times in development or in response to physiological changes is especially important for such proteins [67,75] and often requires changes in epigenetics. Some of the regulation of promoters and enhancers precisely modulating expression of pivotal Myob/Cbl genes is likely to involve binding of TFs that are specific for either the SkM or the neural lineages (Figure S11). In contrast, TFs encoded by Myob/Cbl genes have dual myoblast and cerebellum specificities. Two Myob/Cbl genes, PAX3 and LBX1, code for TFs that are involved in both skeletal muscle development and neuronal differentiation (Table S5; [72,76,77,78]). PAX3 is implicated in the regulation of transcription of three other Myob/Cbl genes, EN2, ANK1, and LBX1 [79,80]. LBX1, EN2, and VAX2 TFs were predicted to bind to Myob-hypom DMRs at ZNF556 and ANK1 promoters. We propose that epigenetic regulation of expression of TF-encoding Myob/Cbl genes in these two dissimilar cell populations not only helps regulate their expression, but also indirectly regulates the tissue/cell-specificity of other Myob/Cbl genes.
4. Conclusions
We identified 20 human genes (Myob/Cbl genes) preferentially expressed in myoblasts and cerebellum, two highly divergent cell populations. Similarities in cell/tissue-specific promoter hypomethylation between myoblasts and cerebellum vs. other cell cultures or tissue types correlate with the similar cell/tissue specificity for several of the genes. In addition, differences in DNA methylation between myoblasts and cerebellum may contribute to modulating relative expression levels or directing alternative promoter usage for some Myob/Cbl genes. The six Myob/Cbl genes that encode transcription factors may help drive the specific transcription profiles of the other genes preferentially expressed in myoblasts and cerebellum. Our study shows how epigenetic analyses in many different cell populations for genes that share highly specific and unexpected cell/tissue specificity can help in understanding normal differentiation and disease-linked changes in gene expression.
5. Materials and Methods
5.1. Transcriptomics
The set of human genes considered for identifying Myob/Cbl genes came from the intersection of the GTEx RNA-seq database set for tissues and an ENCODE dataset for cell cultures [16,42,43]. Genes without at least one tissue and at least one cell culture having a TPM or FPKM of ≥ 1 as well as mitochondrially located genes and most non-coding genes were removed to give 13847 genes. These were first sorted for myoblast specificity using non-strand specific cell culture RNA-seq data from ENCODE (ENCODE Regulation Transcription Track Settings (ucsc.edu)) for the six available cell cultures that were not derived from cancers. These are myoblasts, NHLF, LCL (GM12878), HUVEC, NHEK, and ESC (H1). Of these cultures, only the LCL was a transformed cell line (EBV-transformed B cells). Myoblast-preferential expression was defined as genes with a myoblast FPKM divided by the average of the FPKM for the other cell-types of ≥ 5 and an FPKM for myoblast ≥ 1. Cerebellum-preferential expression was assessed with RNA-seq data for human tissues in the GTEx database (GTEx Portal). The criteria were an expression ratio of ≥ 5 for cerebellum TPM (a median value from 241 biological replicates) vs. the median TPM of 10 tissues from other brain regions, a ratio of ≥ 5 for cerebellum vs. the average of 41 non brain tissues, and a TPM of ≥ 1 for cerebellum (Table S2). For the 12 genes where the average TPM of non-brain tissues was 0, two of those genes had their maximum TPM in cerebellum and were added to the set of cerebellum-preferentially transcribed genes. Another bulk RNA-seq database used to characterize selected genes was the strand-specific ENCODE RNA-seq for cell cultures ((ENC RNA-seq CSHL Long RNA-seq Track Settings (ucsc.edu)). CAGE profiles of cell cultures available at the UCSC Genome Browser RIKEN CAGE Loc Track Settings (ucsc.edu) were examined where indicated. For comparison of myoblast and myotube expression our RNA-seq data were used [27]. In addition, three single-cell RNA-seq (scRNA-seq) databases used for analysis of Myob/Cbl genes were HPA (The Human Protein Atlas, [28]) for postnatal cells, Fetal Gene Atlas Tracks (ucsc.edu) for fetal cells [26], and EmAtlas, a compilation of scRNA-seq data from human early embryonic cells, oocytes, and mid-gestation fetal tissues (EmAtlas (imu.edu.cn)) [29].
5.2. Epigenomics
The chromatin epigenomics data (chromatin state segmentation and H3K27ac) were from the Roadmap Epigenomics Project [19], as previously described [17], and visualized in the UCSC Genome Browser (https://www.genome.ucsc.edu/ ). Coordinates are in hg19, unless otherwise stated; tracks only in hg38 coordinates were lifted over to hg19. DNase-seq data was from ENCODE ((ENC DNase/FAIRE Duke DNaseI HS Track Settings (ucsc.edu)). TF binding analyses used UCSC Genome Browser track hubs for JASPAR TFBS prediction (JASPAR CORE 2022, minimum score, 400) and the UniBind database (for TFBS of interest, including CTCF) based upon ChIP-seq results and TFBS site location within the predicted region (UniBind 2021 Permissive). C2C12 MyoD sites are sequences homologous C2C12 mouse myoblast sequences shown to bind MyoD upon ChIP-seq [81]. WGBS data were obtained from different sources available on the UCSC Genome Browser, with low methylated regions displayed, where available (http://smithlabresearch.org/software/methbase/ ). We generated myoblast WGBS and EM-seq profiles from well-characterized primary myoblast cultures derived from gastrocnemius muscle as previously described [17,42] and included them as well as a publicly available cerebellum WGBS profile [18] (GSM5652231_Cerebellum-Neuron-Z000000TB.hg38.bigwig) as custom tracks at the UCSC Genome Browser. SkM and heart refer to psoas muscle and left ventricle, respectively, unless otherwise specified. Myoblast DMRs were determined by comparing the EM-seq profiles from three biological replicates to WGBS profiles of foreskin fibroblasts, HMEC, IMR90 (fetal lung fibroblast cell line), ESC, and adipose-derived mesenchymal stem cells induced to differentiate to adipocytes, prostate epithelial cells as previously described [17]. SkM DMRs were determined from psoas SkM WGBS vs. WGBS from heart (left ventricle), aorta, monocytes, lung, and subcutaneous adipose tissue as previously described [17,82]. The threshold for DMR attribution was absolute methylation differences of ≥0.35. Myoblast methylome profiles from WGBS and EM-seq from myoblast samples were very similar, as illustrated in Figure S1C.
5.3. Reporter gene assays
Reporter gene constructs were prepared by overlap extension PCR (Table S6) or by using the Gibson assembly kit (NEBuilder HiFi Assembly, New England Biolabs) and a CpG-free plasmid vector (pCpGfree-Lucia, InvivoGen) as previously described [17]. Recombinant plasmid structure was checked by partial DNA sequencing and restriction site analysis. Transfection into C2C12 or MCF-7 cells utilized a lipid-based reagent (Fast-forward protocol, Effectene reagent, Qiagen). As a reference for transfection efficiency, pCMV-CLuc 2 (New England Biolabs) encoding the Cypridina luciferase was co-transfected with the test construct. About 48 h after the transfection, Lucia and Cypridina luciferase activity was quantified by bioluminescence from aliquots of the cell supernatant (BioLux Cypridina Luciferase assay kit, New England Biolabs; Quanti-Luc, InvivoGen). Reference plasmid-normalized luciferase activity was from the average of three independent transfections. Methylation of the plasmids was targeted just to the ZNF556 inserts, which were the only CpG-containing sequences, by incubating the DNA construct (1 μg) with 4 units of SssI methylase and 160 μM S-adenosylmethionine (New England Biolabs) for 4 h at 37°C or mock-methylating by similar incubation but in the absence of S-adenosylmethionine. A plasmid construct that contained three BstUI CGCG sites was similarly methylated and shown thereafter to be fully resistant to BstUI cleavage.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. The ZNF556 neighborhood contains other KRAB-ZNF genes that, unlike ZNF556, are not expressed preferentially in myoblasts or cerebellum. Figure S2. Promoter hypomethylation in cerebellum is associated with upregulated expression of ZIC1 while, in myoblasts, hypermethylation of the neighboring gene, ZIC4, is associated with repression of its expression. Figure S3. Intragenic hypomethylation in CHRD is associated with its upregulation in cerebellum. Figure S4. Cerebellar hypomethylation and myoblast hypermethylation in KCNJ12 are associated with up- or down regulation of this gene. Figure S5. Hypermethylation upstream of the VAX2 promoter in myoblasts and in intron 1 in myoblasts and cerebellum is associated with VAX2 preferential expression both cell populations. Figure S6. The promoter region of ST8SIA5 in cerebellum is more extensively hypomethylated than that of brain prefrontal cortex. Figure S7. EN2 and CNPY1 have differential DNA methylation associated with tissue-specific expression levels and display cerebellar DNA hypermethylation in a region previously associated with autism-linked hypermethylation. Figure S8. Extensive upstream hypermethylation in myoblasts and SkM and hypomethylation in cerebellum is associated with enhanced LBX1 expression in muscle. Figure S9. Myoblast DMRs in MCF2L likely affect promoter usage in cerebellum and myoblasts. Figure S10. An intragenic region of myoblast and cerebellum hypermethylation is associated with repression of alternative promoter usage for DOK7 in these cell populations. Figure S11. Transcription factor (TF) binding at four Myob/Cbl gene regions showing hypomethylation in both myoblasts and cerebellum. Figure S12. Relative mRNA levels for five of the Myob/Cbl genes in embryonic tissues or pre-implantation embryos. Table S1. Genes preferentially expressed in myoblasts. Table S2. Genes with preferential expression in cerebellum. Table S3. Genes preferentially expressed in various brain regions. Table S4. Gene Ontology (GO) associations for individual genes preferentially expressed in both myoblasts and cerebellum. Table S5. Gene ontology analyses on the sets of genes with cerebellum preferential expression as well as preferential expression in one of each of six cell cultures. Table S6. Oligonucleotides used for fusion cloning of 5’ ZNF556 sequences into CpG-free Luciferase reporter vectors.
Author Contributions
KCE and ME conceived the study, made the reporter constructs, did the transfection experiments, and wrote the manuscript. ML determined the DMRs for myoblasts and CB determined the LMRs from myoblast methylomes generated by SS and POE, who were under the direction of SP. The myoblast DNA was previously isolated from primary cells grown and characterized by immunocytochemistry under the direction of ME.
Funding
This research was funded in part by grants from the National Institutes of Health (NS04885) and the Louisiana Cancer Center. This research was also supported in part using high performance computing (HPC) resources and services provided by Information Technology at Tulane University, New Orleans, LA.
References
- Fukada, S.I.; Higashimoto, T.; Kaneshige, A. Differences in muscle satellite cell dynamics during muscle hypertrophy and regeneration. Skelet Muscle 2022, 12, 17. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making muscle: skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; García-Prat, L.; Muñoz-Cánoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat Rev Mol Cell Biol 2022, 23, 204–226. [Google Scholar] [CrossRef]
- Murach, K.A.; Fry, C.S.; Dupont-Versteegden, E.E.; McCarthy, J.J.; Peterson, C.A. Fusion and beyond: Satellite cell contributions to loading-induced skeletal muscle adaptation. Faseb j 2021, 35, e21893. [Google Scholar] [CrossRef]
- Sharples, A.P.; Turner, D.C. Skeletal muscle memory. Am J Physiol Cell Physiol 2023, 324, C1274–c1294. [Google Scholar] [CrossRef]
- Falick Michaeli, T.; Sabag, O.; Fok, R.; Azria, B.; Monin, J.; Nevo, Y.; Gielchinsky, Y.; Berman, B.P.; Cedar, H.; Bergman, Y. Muscle injury causes long-term changes in stem-cell DNA methylation. Proc Natl Acad Sci U S A 2022, 119, e2212306119. [Google Scholar] [CrossRef]
- Robinson, D.C.L.; Dilworth, F.J. Epigenetic Regulation of Adult Myogenesis. Curr Top Dev Biol 2018, 126, 235–284. [Google Scholar] [CrossRef]
- Biressi, S.; Molinaro, M.; Cossu, G. Cellular heterogeneity during vertebrate skeletal muscle development. Dev Biol 2007, 308, 281–293. [Google Scholar] [CrossRef]
- Relaix, F.; Marcelle, C. Muscle stem cells. Curr Opin Cell Biol 2009, 21, 748–753. [Google Scholar] [CrossRef]
- Bengtsen, M.; Winje, I.M.; Eftestøl, E.; Landskron, J.; Sun, C.; Nygård, K.; Domanska, D.; Millay, D.P.; Meza-Zepeda, L.A.; Gundersen, K. Comparing the epigenetic landscape in myonuclei purified with a PCM1 antibody from a fast/glycolytic and a slow/oxidative muscle. PLoS Genet 2021, 17, e1009907. [Google Scholar] [CrossRef]
- Murach, K.A.; Dungan, C.M.; von Walden, F.; Wen, Y. Epigenetic evidence for distinct contributions of resident and acquired myonuclei during long-term exercise adaptation using timed in vivo myonuclear labeling. Am J Physiol Cell Physiol 2022, 322, C86–c93. [Google Scholar] [CrossRef]
- Wen, Y.; Dungan, C.M.; Mobley, C.B.; Valentino, T.; von Walden, F.; Murach, K.A. Nucleus type-specific DNA methylomics reveals epigenetic "memory" of prior sdaptation in skeletal muscle. Function (Oxf) 2021, 2, zqab038. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.; Ehrlich, K.C.; Zhang, G.; Lacey, M.; Johnston, D.; Pradhan, S.; Ehrlich, M. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics 2016, 1–16. [Google Scholar] [CrossRef]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front Cell Neurosci 2014, 8, 450. [Google Scholar] [CrossRef]
- Rudolph, S.; Badura, A.; Lutzu, S.; Pathak, S.S.; Thieme, A.; Verpeut, J.L.; Wagner, M.J.; Yang, Y.M.; Fioravante, D. Cognitive-Affective Functions of the Cerebellum. J Neurosci 2023, 43, 7554–7564. [Google Scholar] [CrossRef]
- The_GTEx_Consortium Human genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Ehrlich, K.C.; Lacey, M.; Baribault, C.; Sen, S.; Esteve, P.O.; Pradhan, S.; Ehrlich, M. Promoter-Adjacent DNA Hypermethylation Can Downmodulate Gene Expression: TBX15 in the Muscle Lineage. Epigenomes 2022, 6. [Google Scholar] [CrossRef]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A DNA methylation atlas of normal human cell types. Nature 2023, 613, 355–364. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef]
- Baribault, C.; Ehrlich, K.C.; Ponnaluri, V.K.C.; Pradhan, S.; Lacey, M.; Ehrlich, M. Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription. Epigenetics 2018, 13, 275–289. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Donalies, M.; Cramer, M.; Ringwald, M.; Starzinski-Powitz, A. Expression of M-cadherin, a member of the cadherin multigene family, correlates with differentiation of skeletal muscle cells. Proc Natl Acad Sci U S A 1991, 88, 8024–8028. [Google Scholar] [CrossRef]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS One 2013, 8, e81148. [Google Scholar] [CrossRef]
- Kim, E.; Rich, J.; Karoutas, A.; Tarlykov, P.; Cochet, E.; Malysheva, D.; Mamchaoui, K.; Ogryzko, V.; Pirozhkova, I. ZNF555 protein binds to transcriptional activator site of 4qA allele and ANT1: potential implication in Facioscapulohumeral dystrophy. Nucleic Acids Res 2015, 43, 8227–8242. [Google Scholar] [CrossRef]
- Cao, J.; O’Day, D.R.; Pliner, H.A.; Kingsley, P.D.; Deng, M.; Daza, R.M.; Zager, M.A.; Aldinger, K.A.; Blecher-Gonen, R.; Zhang, F.; et al. A human cell atlas of fetal gene expression. Science 2020, 370. [Google Scholar] [CrossRef]
- Terragni, J.; Zhang, G.; Sun, Z.; Pradhan, S.; Song, L.; Crawford, G.E.; Lacey, M.; Ehrlich, M. Notch signaling genes: Myogenic DNA hypomethylation and 5-hydroxymethylcytosine. Epigenetics 2014, 9, 842–850. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci Adv 2021, 7. [Google Scholar] [CrossRef]
- Zheng, L.; Liang, P.; Long, C.; Li, H.; Li, H.; Liang, Y.; He, X.; Xi, Q.; Xing, Y.; Zuo, Y. EmAtlas: a comprehensive atlas for exploring spatiotemporal activation in mammalian embryogenesis. Nucleic Acids Res 2023, 51, D924–d932. [Google Scholar] [CrossRef]
- Hammoud, S.S.; Low, D.H.; Yi, C.; Carrell, D.T.; Guccione, E.; Cairns, B.R. Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell 2014, 15, 239–253. [Google Scholar] [CrossRef]
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum Mol Genet 2015, 24, 2873–2883. [Google Scholar] [CrossRef]
- Park, S.H.; Han, J.; Jeong, B.C.; Song, J.H.; Jang, S.H.; Jeong, H.; Kim, B.H.; Ko, Y.G.; Park, Z.Y.; Lee, K.E.; et al. Structure and activation of the RING E3 ubiquitin ligase TRIM72 on the membrane. Nat Struct Mol Biol 2023, 30, 1695–1706. [Google Scholar] [CrossRef]
- Benissan-Messan, D.Z.; Zhu, H.; Zhong, W.; Tan, T.; Ma, J.; Lee, P.H.U. Multi-Cellular Functions of MG53 in Muscle Calcium Signaling and Regeneration. Front Physiol 2020, 11, 583393. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinformatics 2016, 54, 1.30.31–31.30.33. [Google Scholar] [CrossRef]
- Gallagher, P.G.; Forget, B.G. An alternate promoter directs expression of a truncated, muscle-specific isoform of the human ankyrin 1 gene. J Biol Chem 1998, 273, 1339–1348. [Google Scholar] [CrossRef]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M.; et al. CAGE: cap analysis of gene expression. Nat Methods 2006, 3, 211–222. [Google Scholar] [CrossRef]
- Samani, A.; Hightower, R.M.; Reid, A.L.; English, K.G.; Lopez, M.A.; Doyle, J.S.; Conklin, M.J.; Schneider, D.A.; Bamman, M.M.; Widrick, J.J.; et al. miR-486 is essential for muscle function and suppresses a dystrophic transcriptome. Life Sci Alliance 2022, 5. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Wallén, A.; Perlmann, T. Transcriptional control of dopamine neuron development. Ann N Y Acad Sci 2003, 991, 48–60. [Google Scholar] [CrossRef]
- Buckingham, M. Gene regulatory networks and cell lineages that underlie the formation of skeletal muscle. Proc. Natl. Acad. Sci. U S A 2017, 114, 5830–5837. [Google Scholar] [CrossRef]
- Medic, S.; Rizos, H.; Ziman, M. Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochemical and biophysical research communications 2011, 411, 832–837. [Google Scholar] [CrossRef]
- Tsumagari, K.; Baribault, C.; Terragni, J.; Varley, K.E.; Gertz, J.; Pradhan, S.; Badoo, M.; Crain, C.M.; Song, L.; Crawford, G.E.; et al. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics 2013, 8, 317–332. [Google Scholar] [CrossRef]
- Myers, R.M.; Stamatoyannopoulos, J.; Snyder, M.; Dunham, I.; Hardison, R.C.; Bernstein, B.E.; Gingeras, T.R.; Kent, W.J.; Birney, E.; Wold, B.; et al. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS biology 2011, 9, e1001046. [Google Scholar]
- Roth, R.B.; Hevezi, P.; Lee, J.; Willhite, D.; Lechner, S.M.; Foster, A.C.; Zlotnik, A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 2006, 7, 67–80. [Google Scholar] [CrossRef]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Suzuki, M.; Sato, S.; Arai, Y.; Shinohara, T.; Tanaka, S.; Greally, J.M.; Hattori, N.; Shiota, K. A new class of tissue-specifically methylated regions involving entire CpG islands in the mouse. Genes Cells 2007, 12, 1305–1314. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef]
- Rebollo, R.; Miceli-Royer, K.; Zhang, Y.; Farivar, S.; Gagnier, L.; Mager, D.L. Epigenetic interplay between mouse endogenous retroviruses and host genes. Genome Biol 2012, 13, R89. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Shen, L.; Kondo, Y.; Guo, Y.; Zhang, J.; Zhang, L.; Ahmed, S.; Shu, J.; Chen, X.; Waterland, R.A.; Issa, J.P. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 2007, 3, 2023–2036. [Google Scholar] [CrossRef]
- Gruhn, W.H.; Tang, W.W.C.; Dietmann, S.; Alves-Lopes, J.P.; Penfold, C.A.; Wong, F.C.K.; Ramakrishna, N.B.; Surani, M.A. Epigenetic resetting in the human germ line entails histone modification remodeling. Sci Adv 2023, 9, eade1257. [Google Scholar] [CrossRef]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Ryba, T.; Lin, Y.; Bungum, A.O.; Edell, E.S.; Spira, A.; Belinsky, S.A. ANK1 Methylation regulates expression of MicroRNA-486-5p and discriminates lung tumors by histology and smoking status. Cancer Lett 2017, 410, 191–200. [Google Scholar] [CrossRef]
- Chou, S.T.; Peng, H.Y.; Mo, K.C.; Hsu, Y.M.; Wu, G.H.; Hsiao, J.R.; Lin, S.F.; Wang, H.D.; Shiah, S.G. MicroRNA-486-3p functions as a tumor suppressor in oral cancer by targeting DDR1. J Exp Clin Cancer Res 2019, 38, 281. [Google Scholar] [CrossRef]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene body methylation in cancer: molecular mechanisms and clinical applications. Clin Epigenetics 2022, 14, 154. [Google Scholar] [CrossRef]
- Janssen, S.M.; Lorincz, M.C. Interplay between chromatin marks in development and disease. Nat Rev Genet 2022, 23, 137–153. [Google Scholar] [CrossRef]
- Meehan, R.R.; Pennings, S. Shoring up DNA methylation and H3K27me3 domain demarcation at developmental genes. Embo j 2017, 36, 3407–3408. [Google Scholar] [CrossRef]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl Psychiatry 2014, 4, e460. [Google Scholar] [CrossRef]
- Wang, M.; Xie, H.; Shrestha, S.; Sredni, S.; Morgan, G.A.; Pachman, L.M. Methylation alterations of WT1 and homeobox genes in inflamed muscle biopsies from untreated juvenile dermatomyositis suggests self-renewal capacity. Arthritis and rheumatism 2012. [Google Scholar] [CrossRef]
- Van Dyck, L.; Güiza, F.; Derese, I.; Pauwels, L.; Casaer, M.P.; Hermans, G.; Wouters, P.J.; Van den Berghe, G.; Vanhorebeek, I. DNA methylation alterations in muscle of critically ill patients. J Cachexia Sarcopenia Muscle 2022, 13, 1731–1740. [Google Scholar] [CrossRef]
- Wu, F.; Li, X.; Looso, M.; Liu, H.; Ding, D.; Günther, S.; Kuenne, C.; Liu, S.; Weissmann, N.; Boettger, T.; et al. Spurious transcription causing innate immune responses is prevented by 5-hydroxymethylcytosine. Nat Genet 2023, 55, 100–111. [Google Scholar] [CrossRef]
- Xie, J.; Xie, L.; Wei, H.; Li, X.J.; Lin, L. Dynamic Regulation of DNA Methylation and Brain Functions. Biology 2023, 12. [Google Scholar] [CrossRef]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S.; et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol 2015, 16, 11. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry 2013, 3, e232. [Google Scholar] [CrossRef]
- Soltani, A.; Lebrun, S.; Carpentier, G.; Zunino, G.; Chantepie, S.; Maïza, A.; Bozzi, Y.; Desnos, C.; Darchen, F.; Stettler, O. Increased signaling by the autism-related Engrailed-2 protein enhances dendritic branching and spine density, alters synaptic structural matching, and exaggerates protein synthesis. PLoS One 2017, 12, e0181350. [Google Scholar] [CrossRef]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 2011, 14, 1607–1616. [Google Scholar] [CrossRef]
- Ehrlich, M.; Ehrlich, K.C. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics 2014, 6, 563–566. [Google Scholar] [CrossRef]
- Angiari, S.; D’Alessandro, G.; Paolicelli, R.C.; Prada, I.; Vannini, E. Editorial: Cell-Cell Interactions Controlling Neuronal Functionality in Health and Disease. Front Integr Neurosci 2022, 16, 968029. [Google Scholar] [CrossRef]
- Kang, J.S.; Feinleib, J.L.; Knox, S.; Ketteringham, M.A.; Krauss, R.S. Promyogenic members of the Ig and cadherin families associate to positively regulate differentiation. Proc Natl Acad Sci U S A 2003, 100, 3989–3994. [Google Scholar] [CrossRef]
- Krauss, R.S.; Joseph, G.A.; Goel, A.J. Keep Your Friends Close: Cell-Cell Contact and Skeletal Myogenesis. Cold Spring Harb Perspect Biol 2017, 9. [Google Scholar] [CrossRef]
- Bhalla, K.; Luo, Y.; Buchan, T.; Beachem, M.A.; Guzauskas, G.F.; Ladd, S.; Bratcher, S.J.; Schroer, R.J.; Balsamo, J.; DuPont, B.R.; et al. Alterations in CDH15 and KIRREL3 in patients with mild to severe intellectual disability. Am J Hum Genet 2008, 83, 703–713. [Google Scholar] [CrossRef]
- Redies, C.; Hertel, N.; Hübner, C.A. Cadherins and neuropsychiatric disorders. Brain Res 2012, 1470, 130–144. [Google Scholar] [CrossRef]
- Jankowski, J.; Holst, M.I.; Liebig, C.; Oberdick, J.; Baader, S.L. Engrailed-2 negatively regulates the onset of perinatal Purkinje cell differentiation. J Comp Neurol 2004, 472, 87–99. [Google Scholar] [CrossRef]
- Collins, C.A.; Gnocchi, V.F.; White, R.B.; Boldrin, L.; Perez-Ruiz, A.; Relaix, F.; Morgan, J.E.; Zammit, P.S. Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PLoS One 2009, 4, e4475. [Google Scholar] [CrossRef]
- Nakamura, H.; Katahira, T.; Matsunaga, E.; Sato, T. Isthmus organizer for midbrain and hindbrain development. Brain Res Brain Res Rev 2005, 49, 120–126. [Google Scholar] [CrossRef]
- Schinzel, F.; Seyfer, H.; Ebbers, L.; Nothwang, H.G. The Lbx1 lineage differentially contributes to inhibitory cell types of the dorsal cochlear nucleus, a cerebellum-like structure, and the cerebellum. J Comp Neurol 2021, 529, 3032–3045. [Google Scholar] [CrossRef]
- Barber, T.D.; Barber, M.C.; Tomescu, O.; Barr, F.G.; Ruben, S.; Friedman, T.B. Identification of target genes regulated by PAX3 and PAX3-FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics 2002, 79, 278–284. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: from somite to limb. Journal of anatomy 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Cao, Y.; Yao, Z.; Sarkar, D.; Lawrence, M.; Sanchez, G.J.; Parker, M.H.; MacQuarrie, K.L.; Davison, J.; Morgan, M.T.; Ruzzo, W.L.; et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Developmental cell 2010, 18, 662–674. [Google Scholar] [CrossRef]
- Ehrlich, K.C.; Lacey, M.; Ehrlich, M. Epigenetics of Skeletal Muscle-Associated Genes in the ASB, LRRC, TMEM, and OSBPL Gene Families. Epigenomes 2020, 4. [Google Scholar] [CrossRef]
Figure 1.
The 5’ end of CDH15, a cadherin gene expressed highly in both myoblasts and cerebellum, is hypomethylated in both cell populations. (A) The RefSeq isoform at chr16:89,230,125-89,270,723 (hg19). RNA-seq for cell cultures is depicted as the log transformed overlay signal and for 37 tissues as bar graphs with relative heights (linear scale) for median TPMs (GTEx; see Tables S1 and S2, for detailed data). (B) Chromatin state segmentation (18-state; Roadmap Epigenomics Project) is based on key histone methylation and acetylation profiles. (C) DMRs and WGBS. For WGBS tracks, the blue bars are significantly low methylated regions (LMRs) relative to the whole genome for that sample. DMRs for SkM (psoas) vs. five other tissues and myoblasts vs. six other types of cell cultures are shown. (D) CTCF and MyoD binding sites are from the UniBind ChIP-seq database or from sequences homologous C2C12 mouse myoblast sequences shown to bind MyoD upon ChIP-seq. (E) DNaseI hypersensitivity from ENCODE. Unless otherwise stated, all tracks are from the UCSC Genome Browser. Tracks are aligned except for the GTEx bar graphs. The dotted boxes in Panel C indicate the Myob-hyperm DMR described in the text. Abbreviations: Str, strong; Wk, weak; Prom, promoter; Enh, enhancer; Txn-chrom, chromatin with the H3K36me3 indicative of active transcription; Low signal, negligible signal for H3K27ac or me3, H3K4me1 or 3, H3K9me3, or H3K36me3; Repr, repressed; Myob, myoblasts; LCL, lymphoblastoid cell line (GM12848); ESC, human embryonic stem cells (H1); NHLF, lung fibroblasts; NHEK, normal human erythroid keratinocytes; HUVEC, human umbilical cord epithelial cells; SkM, skeletal muscle (psoas) except for GTEx data, where it indicates gastrocnemius SkM, PFC, pre-frontal cortex; fib, fibroblast; HMEC, human mammary epithelial cells.
Figure 1.
The 5’ end of CDH15, a cadherin gene expressed highly in both myoblasts and cerebellum, is hypomethylated in both cell populations. (A) The RefSeq isoform at chr16:89,230,125-89,270,723 (hg19). RNA-seq for cell cultures is depicted as the log transformed overlay signal and for 37 tissues as bar graphs with relative heights (linear scale) for median TPMs (GTEx; see Tables S1 and S2, for detailed data). (B) Chromatin state segmentation (18-state; Roadmap Epigenomics Project) is based on key histone methylation and acetylation profiles. (C) DMRs and WGBS. For WGBS tracks, the blue bars are significantly low methylated regions (LMRs) relative to the whole genome for that sample. DMRs for SkM (psoas) vs. five other tissues and myoblasts vs. six other types of cell cultures are shown. (D) CTCF and MyoD binding sites are from the UniBind ChIP-seq database or from sequences homologous C2C12 mouse myoblast sequences shown to bind MyoD upon ChIP-seq. (E) DNaseI hypersensitivity from ENCODE. Unless otherwise stated, all tracks are from the UCSC Genome Browser. Tracks are aligned except for the GTEx bar graphs. The dotted boxes in Panel C indicate the Myob-hyperm DMR described in the text. Abbreviations: Str, strong; Wk, weak; Prom, promoter; Enh, enhancer; Txn-chrom, chromatin with the H3K36me3 indicative of active transcription; Low signal, negligible signal for H3K27ac or me3, H3K4me1 or 3, H3K9me3, or H3K36me3; Repr, repressed; Myob, myoblasts; LCL, lymphoblastoid cell line (GM12848); ESC, human embryonic stem cells (H1); NHLF, lung fibroblasts; NHEK, normal human erythroid keratinocytes; HUVEC, human umbilical cord epithelial cells; SkM, skeletal muscle (psoas) except for GTEx data, where it indicates gastrocnemius SkM, PFC, pre-frontal cortex; fib, fibroblast; HMEC, human mammary epithelial cells.
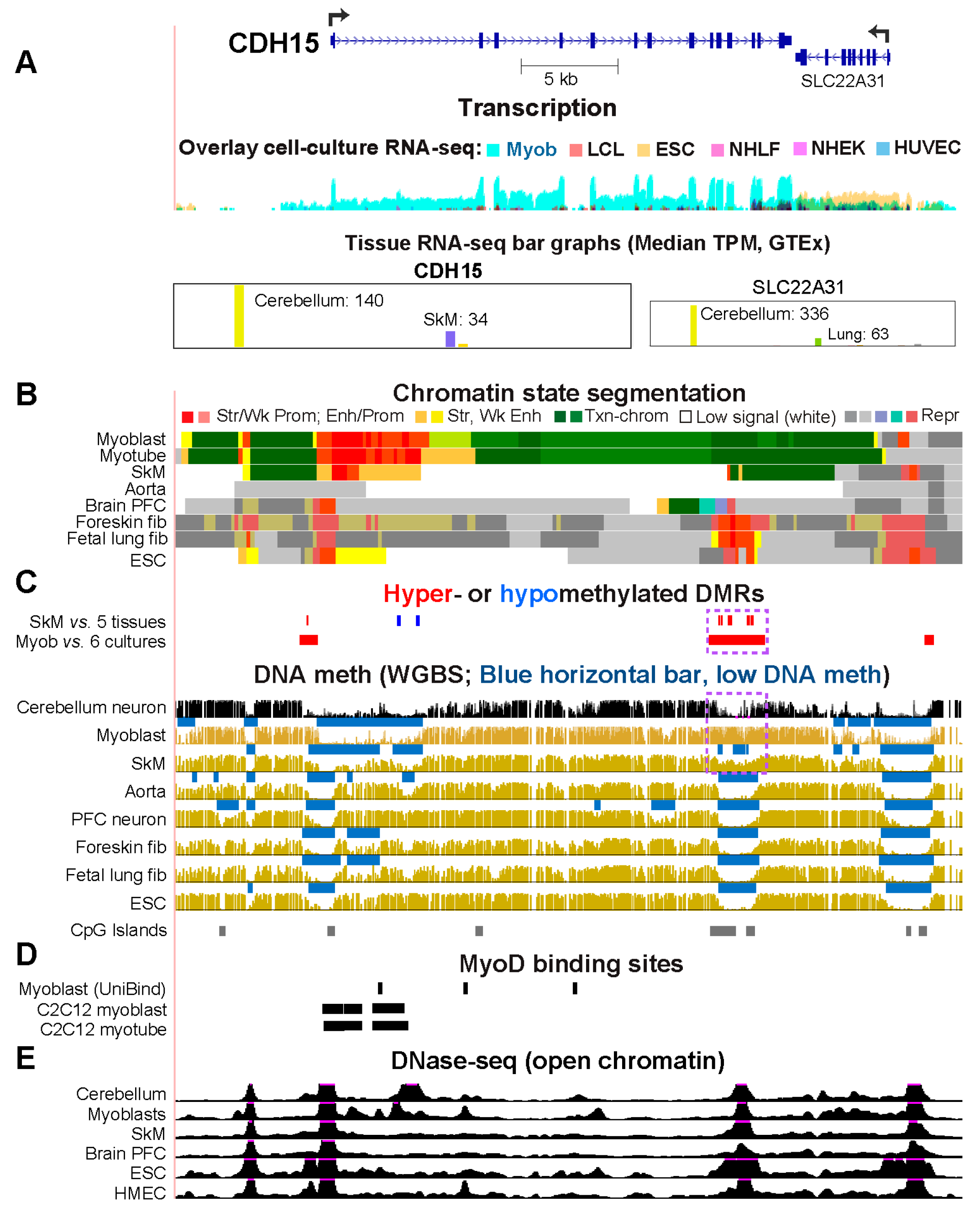
Figure 2.
ZNF556, a KRAB-C2H2 zinc finger protein gene, exhibited a particularly strong association of myoblast DNA hypomethylation with gene expression. The region shown (chr19:2,838,723-2,891,821) illustrates the RefSeq Select isoforms for ZNF556 and ZNF555. Panels are similar to those in Figure 1. The dotted boxes in Panels B, C, and D indicate a hypomethylated region that overlaps the ZNF556 promoter and a prominent DNase-seq peak seen in cells or tissues most strongly expressing ZNF556. HepG2, hepatocellular carcinoma cell line. Blue-green color for repressed chromatin in Panel B, a chromatin segment enriched in both H3K9me3 and H3K36me3, that is often seen in gene bodies of active or inactive ZNF family genes.
Figure 2.
ZNF556, a KRAB-C2H2 zinc finger protein gene, exhibited a particularly strong association of myoblast DNA hypomethylation with gene expression. The region shown (chr19:2,838,723-2,891,821) illustrates the RefSeq Select isoforms for ZNF556 and ZNF555. Panels are similar to those in Figure 1. The dotted boxes in Panels B, C, and D indicate a hypomethylated region that overlaps the ZNF556 promoter and a prominent DNase-seq peak seen in cells or tissues most strongly expressing ZNF556. HepG2, hepatocellular carcinoma cell line. Blue-green color for repressed chromatin in Panel B, a chromatin segment enriched in both H3K9me3 and H3K36me3, that is often seen in gene bodies of active or inactive ZNF family genes.
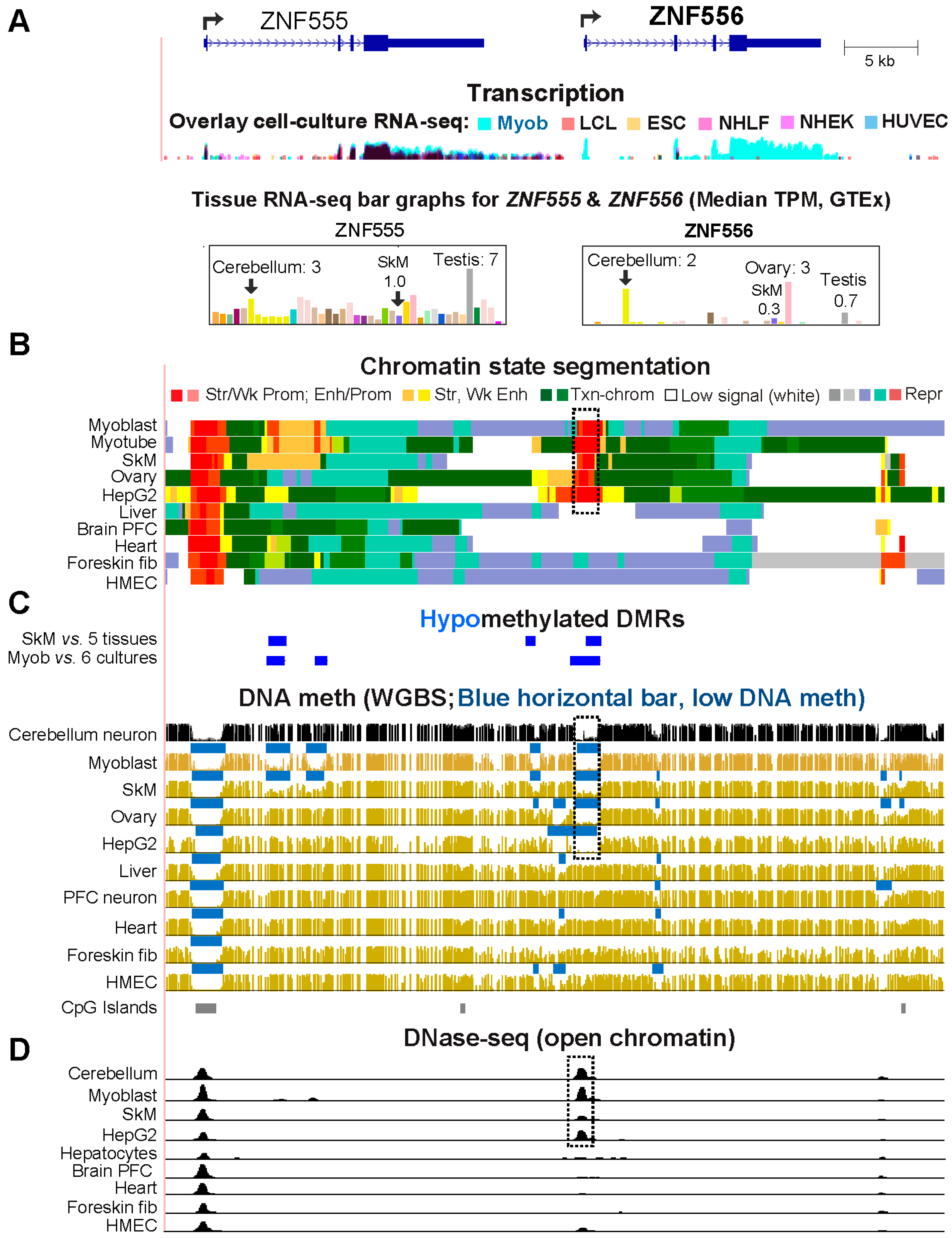
Figure 3.
Hypermethylated sequences in the ZNF556 promoter region have methylation-sensitive promoter activity in transfection assays. (A) The 5’ region of ZNF556 (chr19:2,865,524-2,868,704) depicting the cloned sequences used in reporter gene assays and some of their epigenetic features. H3K27ac, H3K27 acetylation. DNA repeats are from the UCSC Genome Browser with the intensity of gray color indicating the extent of homology to consensus sequences; SINE (short interspersed repeats; Alu repeats), and LTR (long terminal repeat elements; ERV1 or ERVL families). (B) Normalized reporter gene activity for the indicated cloned DNA sequences transiently transfected as part of reporter constructs into C2C12 myoblasts or MCF7 cells. (C) Loss of promoter activity upon in vitro CpG methylation targeted to the cloned sequence.
Figure 3.
Hypermethylated sequences in the ZNF556 promoter region have methylation-sensitive promoter activity in transfection assays. (A) The 5’ region of ZNF556 (chr19:2,865,524-2,868,704) depicting the cloned sequences used in reporter gene assays and some of their epigenetic features. H3K27ac, H3K27 acetylation. DNA repeats are from the UCSC Genome Browser with the intensity of gray color indicating the extent of homology to consensus sequences; SINE (short interspersed repeats; Alu repeats), and LTR (long terminal repeat elements; ERV1 or ERVL families). (B) Normalized reporter gene activity for the indicated cloned DNA sequences transiently transfected as part of reporter constructs into C2C12 myoblasts or MCF7 cells. (C) Loss of promoter activity upon in vitro CpG methylation targeted to the cloned sequence.
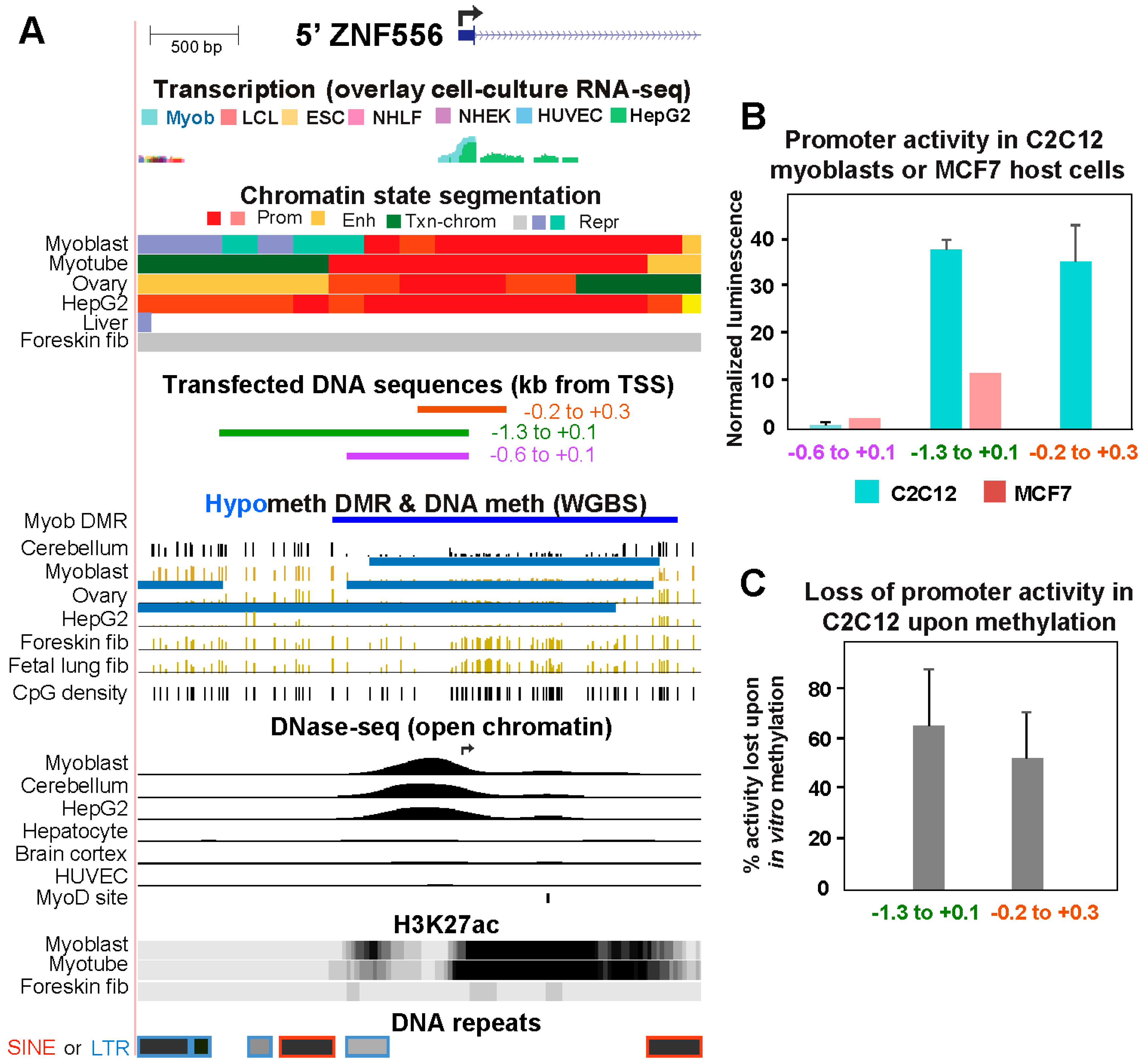
Figure 4.
ZNF555 and ZNF556 RNAs are present in cells from early pre-implantation embryos, in which their genes display ZNF556 promoter hypomethylation. (A) Relative levels of ZNF556 and (B) ZNF555 RNAs in early embryo and fetal tissues, adapted from the EmAtlas’ compiled data [29]. For clarity, the inner sectors for fetal skeletal muscle and early embryo are colored blue and purple, respectively. In the outer ring, yellow indicates no detectable expression, and the intensity of the other colors is a reflection of the relative scRNA-seq signal. The maximum nTPM for ZNF556 was 32 in fetal arm muscle and for ZNF555 was 38 in the zygote. (C) WGBS profiles from the EmAtlas and, for sperm, from the UCSC Genome Browser for the hg38 coordinates chr19:2838727-2891825.
Figure 4.
ZNF555 and ZNF556 RNAs are present in cells from early pre-implantation embryos, in which their genes display ZNF556 promoter hypomethylation. (A) Relative levels of ZNF556 and (B) ZNF555 RNAs in early embryo and fetal tissues, adapted from the EmAtlas’ compiled data [29]. For clarity, the inner sectors for fetal skeletal muscle and early embryo are colored blue and purple, respectively. In the outer ring, yellow indicates no detectable expression, and the intensity of the other colors is a reflection of the relative scRNA-seq signal. The maximum nTPM for ZNF556 was 32 in fetal arm muscle and for ZNF555 was 38 in the zygote. (C) WGBS profiles from the EmAtlas and, for sperm, from the UCSC Genome Browser for the hg38 coordinates chr19:2838727-2891825.
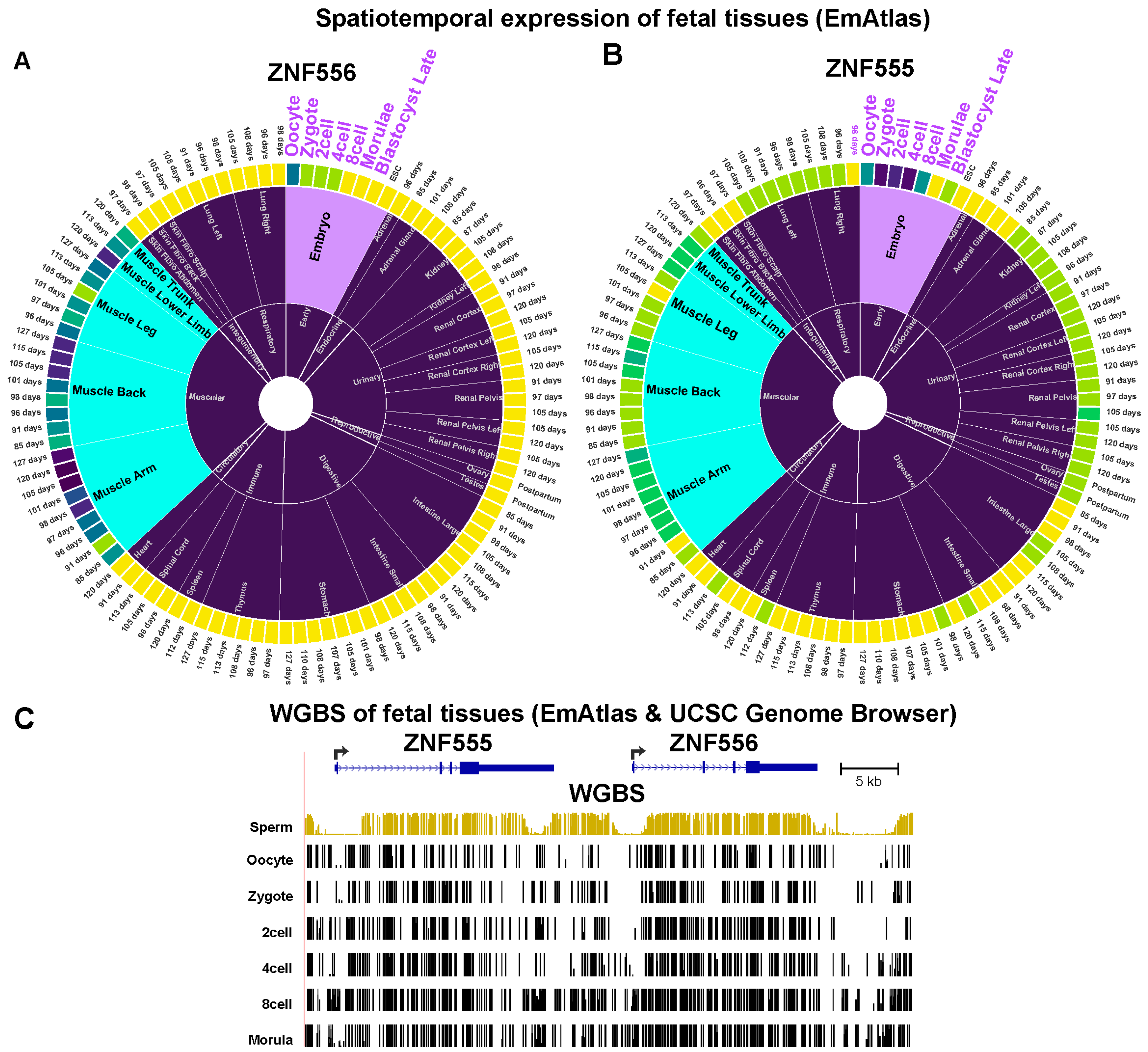
Figure 5.
TRIM72, a myokine-encoding gene, harbors an antisense coding gene, PYDC1, that is hypermethylated, and not expressed, in myoblasts but hypomethylated in cerebellum. The only TRIM72 and PYDC1 RefSeq Curated isoforms are shown at chr16:31,223,125-31,244,961. Panels (A) – (C) are similar to panels in Figure 1.
Figure 5.
TRIM72, a myokine-encoding gene, harbors an antisense coding gene, PYDC1, that is hypermethylated, and not expressed, in myoblasts but hypomethylated in cerebellum. The only TRIM72 and PYDC1 RefSeq Curated isoforms are shown at chr16:31,223,125-31,244,961. Panels (A) – (C) are similar to panels in Figure 1.
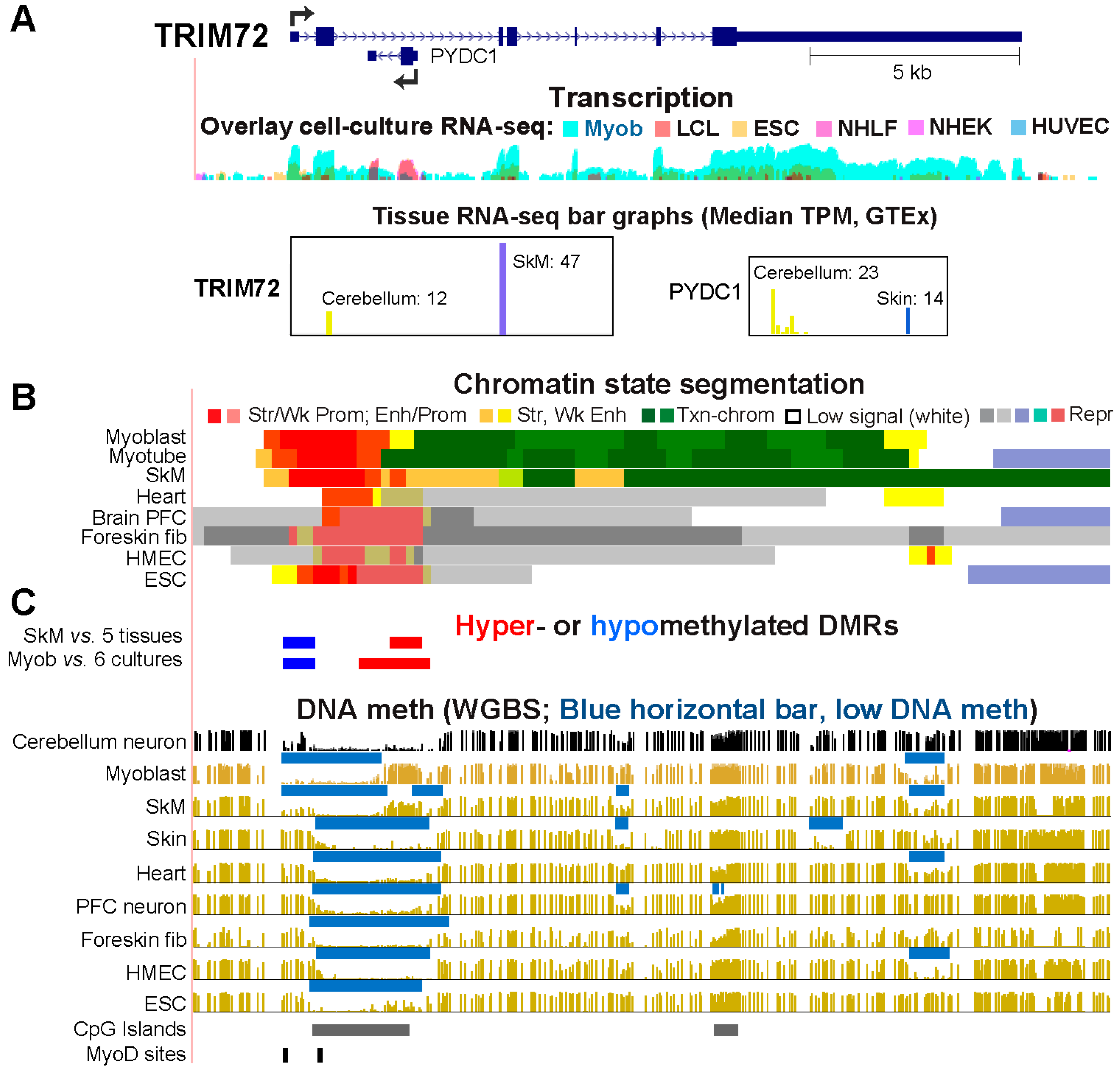
Figure 6.
Hypomethylation at the myoblast/skeletal muscle/heart-associated promoter for ankyrin-encoding ANK1. Most of the many RefSeq Curated or Ensembl isoforms, including ENST00000265709 which has the most distal promoter (GTEx database), are not in the depicted coordinates, chr8: 41,508,221-41,660,626. (A) – (E) are similar to panels depicted in Figure 1 with the addition of CTCF binding sites from Unibind; blue, the CTCF site was seen in myoblasts but not in HMEC or ESC; black, the CTCF binding sites that were seen in myoblasts and ESC or HMEC. In addition to the ENCODE RNA-seq overlay profiles, Panel A shows ENCODE or Roadmap strand-specific RNA-seq signal intensities. Dotted box in Panel B, super-enhancer seen in SkM and heart.
Figure 6.
Hypomethylation at the myoblast/skeletal muscle/heart-associated promoter for ankyrin-encoding ANK1. Most of the many RefSeq Curated or Ensembl isoforms, including ENST00000265709 which has the most distal promoter (GTEx database), are not in the depicted coordinates, chr8: 41,508,221-41,660,626. (A) – (E) are similar to panels depicted in Figure 1 with the addition of CTCF binding sites from Unibind; blue, the CTCF site was seen in myoblasts but not in HMEC or ESC; black, the CTCF binding sites that were seen in myoblasts and ESC or HMEC. In addition to the ENCODE RNA-seq overlay profiles, Panel A shows ENCODE or Roadmap strand-specific RNA-seq signal intensities. Dotted box in Panel B, super-enhancer seen in SkM and heart.
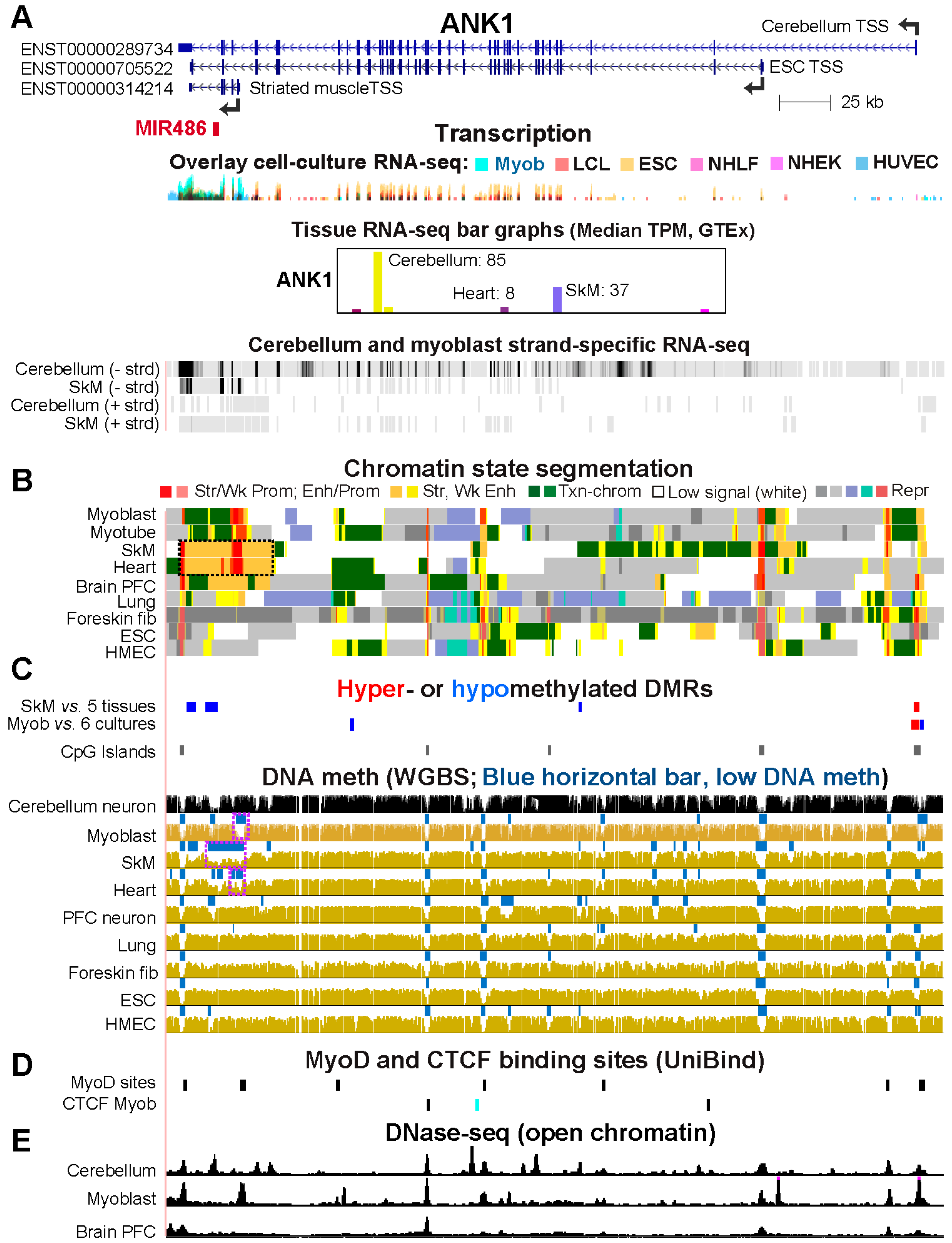
Figure 7.
Hypermethylated DMRs upstream and downstream of the promoter region of the TF-encoding PAX3 may downmodulate expression of this gene in both myoblasts and cerebellum. The RefSeq Select isoform is shown at chr2:223,058,677-223,192,231. (A) – (C) and (E) are similar to panels in Figure 6. (D) RRBS DNA methylation data from ENCODE with the indicated color coding; dotted box, the melanocyte-specific hypermethylated region.
Figure 7.
Hypermethylated DMRs upstream and downstream of the promoter region of the TF-encoding PAX3 may downmodulate expression of this gene in both myoblasts and cerebellum. The RefSeq Select isoform is shown at chr2:223,058,677-223,192,231. (A) – (C) and (E) are similar to panels in Figure 6. (D) RRBS DNA methylation data from ENCODE with the indicated color coding; dotted box, the melanocyte-specific hypermethylated region.
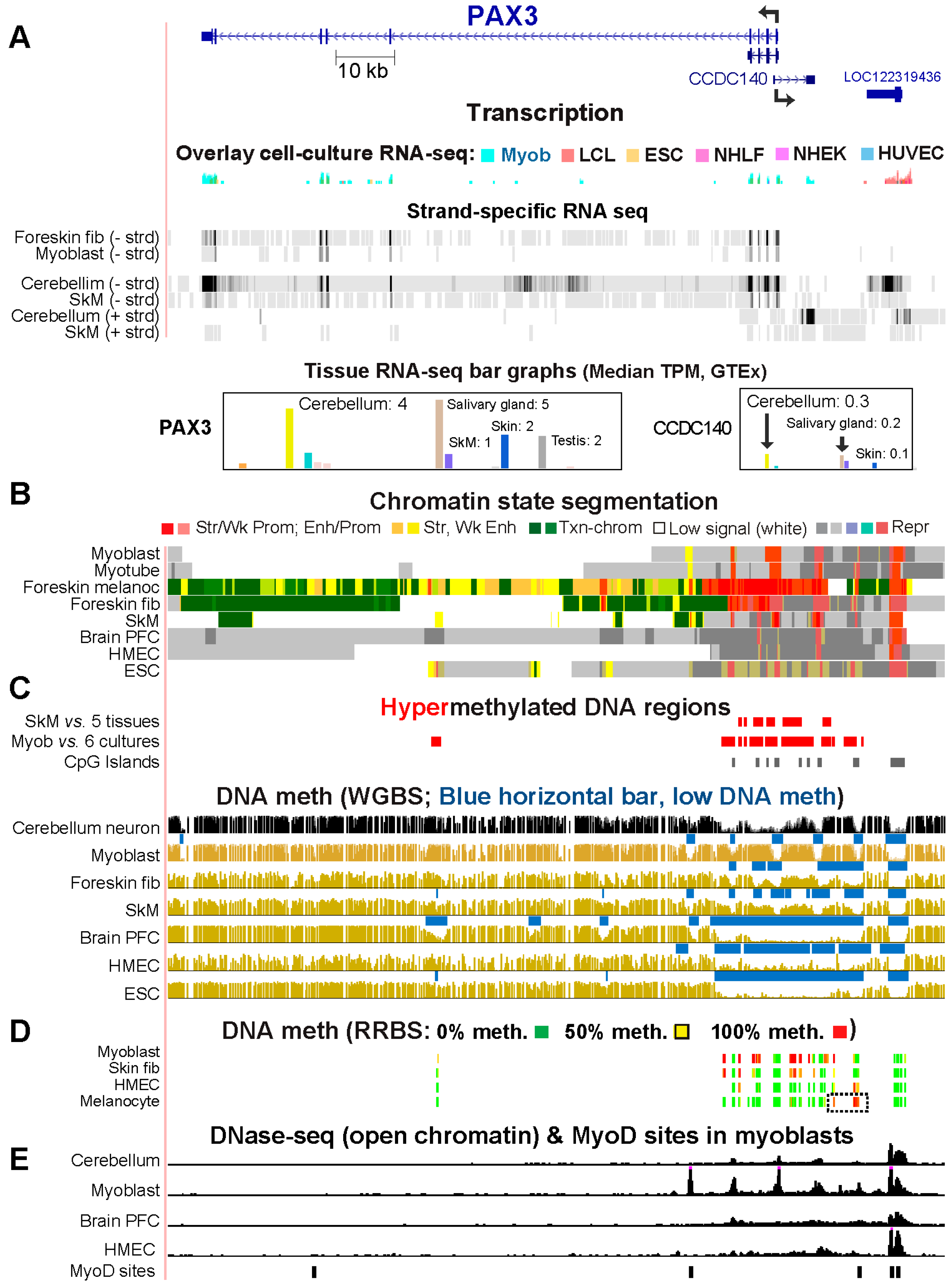

Table 1.
Genes preferentially expressed in myoblasts and cerebellum.
| FPKM or TPM (Expression ratio ≥5)a | No. of Myob DMRsb | Probable function of differential methylationc | ||||
| Gene | Myob | Cbl | Hypom | Hyperm | Myob DMRs | Cbl hypo- or hyperm |
| ZNF556 | 12 (1076) | 2 (27) | 1 | 0 | Prom hypom allowing txnd | Prom hypom allowing txnd |
| CDH15 | 164 (560) | 141 (1441) | 1 | 2 | Prom dnstm hypom ↑ txnd | Prom dnstm hypom ↑ txnd |
| TRIM72 | 43 (127) | 12 (53) | 1 | 1 | Prom hypom ↑ txnd; hyperm repr intronic PYDC1 |
Prom hypom ↑ TRIM72 txnd & intronic PYDC1 |
| ANK1 | 29 (15) | 85 (25) | 2 | 1 | Alt prom usage | Different prom use f‘rom Myob |
| MCF2L | 21 (5) | 110 (9) | 2 | 3 | Alt prom usaged; many RNA splicing isoforms |
Alt prom & splicingd; hypom ↑ txn from enhs |
| DOK7 | 45 (220) | 21 (27) | 0 | 1 | Alt prom usaged | Alt prom usaged |
| CNPY1 | 2 (39) | 26 (158) | 0 | 2 | Alt prom usage | Alt prom usage |
| KCNJ12 | 5 (6) | 81 (40) | 0 | 3 | Prom-dnstm hyperm ↓ txn | Prom-adjacent hypom ↑ txn |
| ST8SIA5 | 4 (6) | 51 (14) | 0 | 1 | Prom-upstm hyperm ↓ txn | Prom-dnstm hypom ↑ txn |
| ZIC1 | 5 (39) | 311 (57) | 0 | 14 | Prom-upstm/dnstm hyperm ↓ txn & repressing adj ZIC4 |
Whole-gene hypom ↑ txn of both ZIC1 & ZIC4 |
| VAX2 | 4 (6) | 19 (15) | 0 | 7 | Prom-upstm hyperm ↓ txn; Intron-1 hyperm may block formation of repr chromd | Intron-1 hyperm may block formation of repr chromd |
| EN2 | 3 (56) | 68 (201) | 0 | 4 | Prom-upstm/dnstm hyperm may ↓ txn |
Hyperm far upstm & dnstm of 7-kb EN2 may ↑ txn |
| LBX1 | 1 (230) | 3 (78) | 0 | 4 | Hyperm upstm/dnstm of 2-kb gene may ↑ txn |
Methylation profile similar to those of most tissues |
| PAX3 | 1 (23) | 4 (28) | 0 | 11 | Prom-upstm/dnstm hyperm ↓ txnd |
Prom-upstm/dnstm hyperm ↓ txnd |
| CHRD | 4 (7) | 243 (22) | 1 | 0 | Intergenic hypom may precede Enh formation |
Hypom at different intergenic regions ↑ txn |
| FNDC5 | 64 (88) | 109 (8) | 0 | 0 | NA | 3’ gene hypom ↑ txn |
| PLCB4 | 54 (22) | 57 (18) | 0 | 0 | NA | Prom-dnstm hypom ↑ txn |
| MPP4 | 9 (9) | 3 (16) | 0 | 0 | NA | Uncertain |
| PTPRR | 2 (6) | 22 (9) | 0 | 0 | NA | Alt prom usage |
| IL11 | 6 (6) | 6 (14) | 0 | 0 | NA | Uncertain |
aExpression ratio, FPKM for myoblasts vs. average FPKM for other cell cultures or TPM for cerebellum vs. average for 10 other non-cerebellum brain regions (Tables S1 and S2). Myob, myoblasts; Cbl, cerebellum; hypom, hypomethylation; hyperm, hypermethylation; Prom, promoter; txn, transcription; ↑, upregulates; ↓, downmodulates; alt, alternative; Enh, enhancer chromatin; dnstm, downstream; upstm, upstream; adj, adjacent; repr chrom, repressive chromatin. bNumber of DMRs; the promoter hypomethylation in myoblasts for CDH15 was seen as a long specific LMR (Figure 1C); the cryptic promoter activity of the intragenic CDH15 hyperm DMR was previously reported [17]. cUp arrow, upregulation; down arrow, downregulation dBoth myoblast and cerebellum share hypomethylation or hypermethylation & similar associations with gene expression.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



