
Submitted:
13 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
The asymmetric synthesis of polyunsaturated triene C18:3 n-3 and C18:3 n-6 methoxylated ether lipids (MEL) of the 1-O-alkyl-sn-glycerol type is described as possible structural candidates for a triene C18:3 MEL of an unknown identity found in a mixture of shark and dogfish liver oil. Their C18:3 hydrocarbon chains constitute an all-cis methylene skipped n-3 or n-6 triene framework, along with a methoxyl group at the 2'-position and R-configuration of the resulting stereogenic centre. The methoxylated polyenes are attached by an ether linkage to the pro-S hydroxymethyl group of the glycerol backbone. The syntheses were based on the polyacetylene approach that involves a semi-hydrogenation of the resulting triynes. Both syntheses were started from our previously described enantio- and diastereomerically pure isopropylidene-protected glyceryl glycidyl ether, a double-C3 building block that was designed as a head group synthon for synthesis of various types of MELs.
Keywords:
asymmetric synthesis
; methoxylated ether lipid (MEL)
; semi-hydrogenation
; shark liver oil
1. Introduction
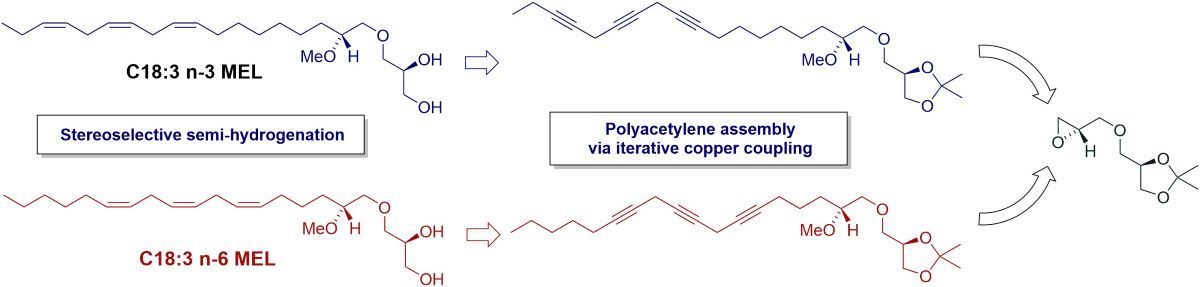
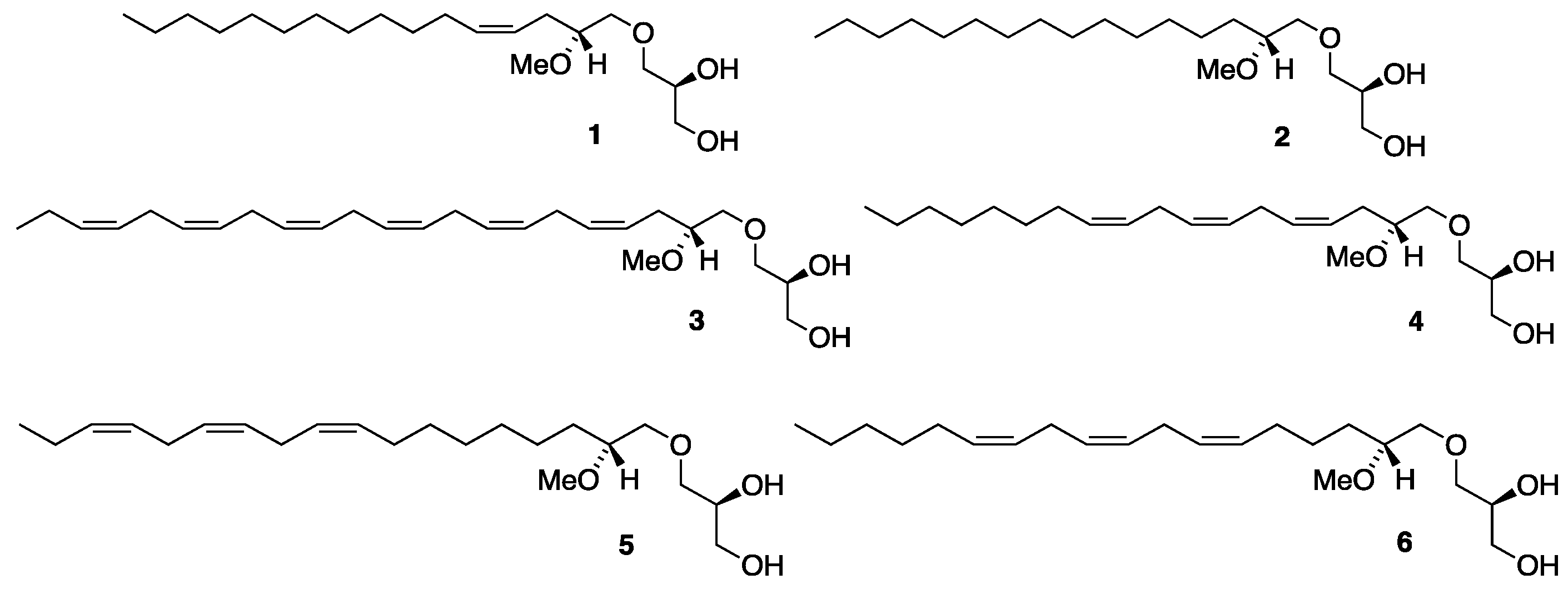
Nonpolar ether lipids (ELs), triacylglycerols and squalene are the three major lipid constituents present in the liver oil of shark and cartilaginous fish [1,2]. The ELs consist of 1-O-alkyl-sn-glycerols to which two fatty acyl groups are linked as carboxylate esters and, accordingly, they are often named as diacylglyceryl ethers (DAGEs). Numerous benefits on human and animal health are attributed to these ELs [3,4,5], but more so when their 1-O-hydrocarbon ether moiety is rendered with monounsaturation [6]. The methoxylated ether lipids (MELs) are quite a noteworthy subclass of the 1-O-alkyl-sn-glycerols. Their 1-O-hydrocarbon ether entity is substituted with a methoxyl group at the 2'-position and the resulting chiral center possesses the R-configuration [2,7,8,9]. The MELs are also present in the liver oil of elasmobranch fish and shark, where they commonly represent 2 – 4% of the 1-O-alkyl-sn-glycerols in the oil. The MELs occur widely in nature in low concentrations including mammals and humans and they have been observed to offer numerous biological activities of importance such as various antimicrobial and anticarcinogenic effects, and to stimulate the immune system [2,10,11,12]. The two most prevalent components of the MEL fraction are the monounsaturated C16:1 MEL 1 with a cis double bond located at the 4'-position (n-12) of the olefin chain, and a saturated C16:0 MEL 2 (see Figure 1). Their C18:1 and C18:0 MEL homologues are also relatively common, and recently we reported the discovery of a novel C18:1 n-9 MEL regioisomer [12]. Polyunsaturated MELs are also known and that includes an amazing docosahexaenoic acid (DHA)-like C22:6 n-3 MEL 3 (Figure 1) that was discovered in shark liver oil [8].
Our investigation of a MEL sample derived from a mixture of shark and dogfish liver oils revealed the presence of all six of the above stated MEL derivatives in the natural oil [12]. They were all synthesized and the identity of the unsaturated MELs from the sample unequivocally confirmed by comparison studies between their MS/MS spectra and those obtained from the synthesized compounds [12]. All syntheses were based on the application of a glyceryl glycidol ether key building block that was recently designed for such MEL asymmetric syntheses and derived from R-solketal as a chiral precursor [13,14]. Besides the six above described MELs there was a triene C18:3 MEL of an unknown identity detected in the sample. We picked out the C18:3 n-8 MEL 4 for synthesis as the most likely structural candidate for the unknown MEL with the aim of having its MS/MS spectrum compared to that of the unknown one. The successful synthesis of the MEL 4 was described in a recent report but since its MS/MS degradation spectrum failed to align with the spectrum obtained from the natural sample other alternative structures had to be considered [15]. Other likely candidates were thought to be the C18:3 n-3 MEL 5, an α-linolenic acid (ALA)-like derivative and the C18:3 n-6 MEL 6, a γ-linolenic acid (GLA)-like derivative. Their asymmetric syntheses are described in the current report as based on the already established polyacetylene approach [14,15]. The structure of the MELs 1 – 6 is shown in Figure 1.
2. Results and Discussion
Hayashi and Takagi in 1982 reported the presence of a C18:3 MEL in cartilaginous fish liver oil without providing further information on its structure [16]. Our initial guess was that the C18:3 MEL most likely had the first double bond of a proposed methylene interrupted triene framework located in the Δ4'-position, thus making it the C18:3 n-8 MEL 4. Its synthesis was reported recently, but when the MS/MS degradation spectrum of the synthesized MEL 4 did not align with the spectrum obtained from the natural sample, alternative structures had to be considered. Other likely candidates were thought to be the C18:3 n-3 MEL 5, possessing the n-3 methylene interrupted triene framework identical to the one present in α-linolenic acid (ALA), or the C18:3 n-6 MEL 6, with the corresponding n-6 triene framework as found in γ-linolenic acid (GLA). The synthesis of MELs 5 and 6 is reported in the current paper. Their convergent syntheses were based on the polyacetylene approach [17,18] already established in the syntheses of the MELs 3 [14] and 4 [15] by iterative copper promoted coupling reactions of propargyl bromides with terminal alkynes [19,20] and stereoselective semi-hydrogenation [21] of the resulting triyne. As in the previous cases the enantio- and diastereopure glyceryl glycidol ether (2R,2'S)-7 [13] was used as a double-chiral precursor to control the stereochemistry of the C6 head group segment of the molecules. It was thought of interest to have the MELs 4 – 6 synthesized with possibilities to have them later screened for some interesting bioactivities, regardless of their structures to fit the undisclosed C18:3 MEL derivative.
2.1. Synthesis of MEL 5
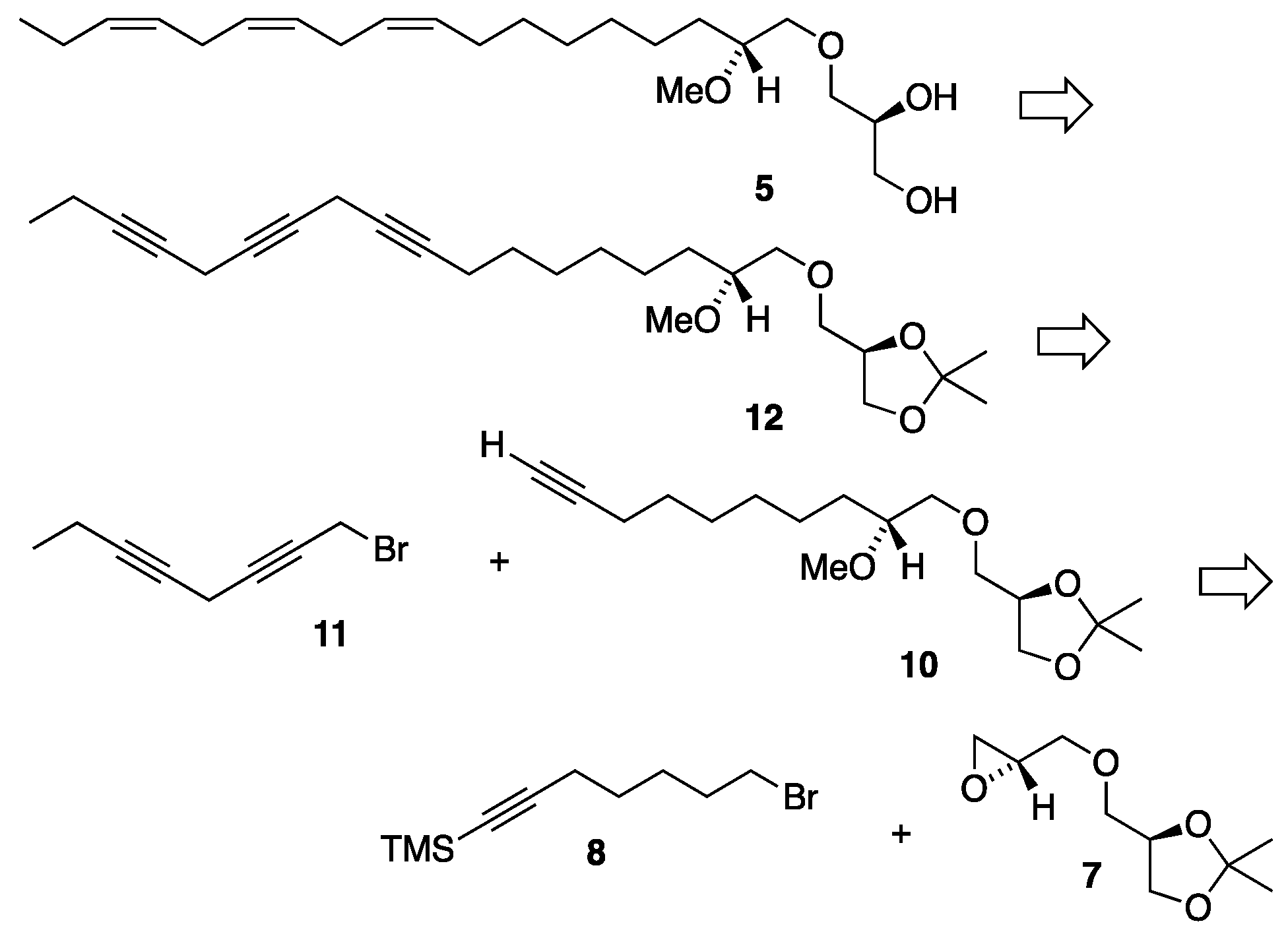
The second proposed structural candidate for the unknown C18:3 MEL was the C18:3 n-3 MEL 5 and its synthetic strategy is disclosed in the retrosynthetic analysis displayed in Scheme 1. This is a convergent synthesis involving a Z-selective semi-hydrogenation of the methylene interrupted omega-3 triyne framework of the isopropylidene protected glyceryl ether 12. Triyne 12 is disconnected into two subunit fragments, the monyne 10 possessing the glyceryl head part and the diyne 11 possessing the hydrocarbon tail segment. Since the first double bond in the 1-O-hydrocarbon chain was located at the Δ9'-position, we made use of the monoyne 10, an already known intermediate from our previous synthesis of the 18:1 n-9 MEL [12]. Furthermore, the diyne 11 was also available from the recently reported synthesis of the n-3 polyunsaturated DHA-like MEL 3 [14]. The monoyne 10 is further disconnected into the key head piece 7 and the TMS protected monoyne bromide linker 8 from the previous synthesis of the 18:1 n-9 MEL [12].
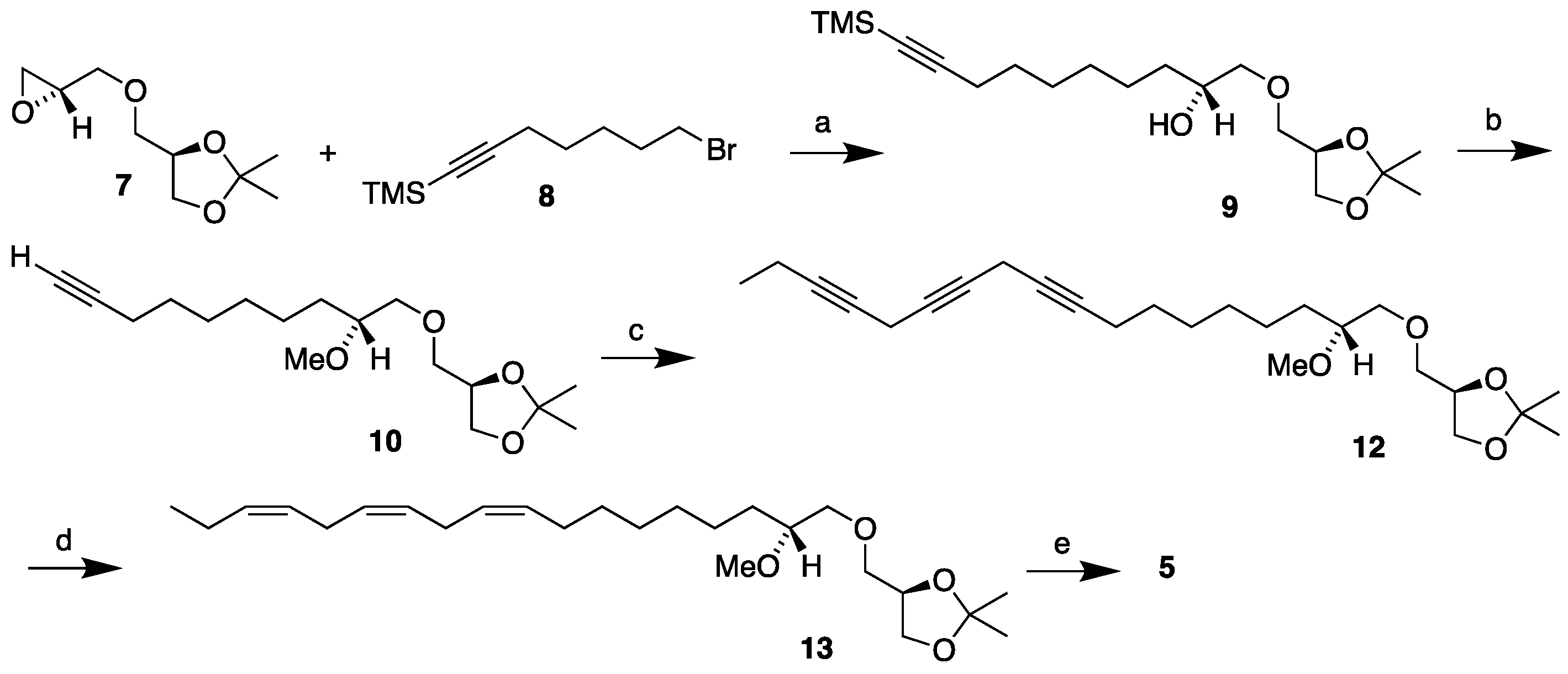
As based on the above retrosynthetic analysis the synthesis of the MEL 5 is depicted in Scheme 2, starting from the head piece 7. The synthesis of monoyne 10 has already been reported as stated above. It was accomplished in 73% overall yield from the reaction of the head piece 7 in its reaction with the monoyne linker 8 and the subsequent one-pot TMS deprotection-methylation of the resulting alcohol [12]. The monoyne 10 was coupled to the diyne 11, accessible from the previously stated synthesis of the polyunsaturated DHA-like MEL 3 [14] that established the desired methylene interrupted triyne structure. This was accomplished by the copper promoted coupling that was described in detail in our previous synthesis of the n-3 polyunsaturated DHA-like MEL with cesium carbonate as a base [14]. This afforded the triyne 12 in 91% yield. The triyne 12 was semi-hydrogenated in the same manner as described for the MEL 4 [15] by use of the Lindlar catalyst with quinoline in toluene at room temperature. The isopropylidene protected triene 13 was afforded in 78% yield after flash chromatography and then in 51% yield after purification by use of argentation chromatography on silver nitrate impregnated silica gel [14,15,22,23]. This means 40% yield in all for the semi-hydrogenation step. Finally, like before, the triene 13 was refluxed in 96% ethanol in the presence of acidic Amberlyst-15® ion exchange resin to remove the isopropylidene group [24] affording the MEL 5 in 89% yield. The MEL 5 was thus obtained in 24% overall yield as based on the head piece 7 over five steps (17% as based on the R-solketal) which is quite comparable to the yield obtained in the MEL 4 synthesis.
The over-hydrogenation of the triyne 12 in the semi-hydrogenation was significantly higher than for the corresponding step in the synthesis of the triene MEL 4 [15]. In that case the isopropylidene protected triene was obtained in 98% yield with 10% contamination of over-hydrogenated byproducts. In the current case the triene 13 was obtained in 78% yield with 14% of over-hydrogenated byproducts present. As described before [14] the level of over- hydrogenation was accurately determined using 1H NMR analysis by comparing the integration of the olefin protons to that of the protons belonging to the glycerol moiety, the olefin protons integrating 5.17 out of 6. After purification by the argentation chromatography the over-hydrogenated byproducts had dropped down to < 0.5% (5.98/6) in the product. This is comparable to what was accomplished in the corresponding step in the synthesis of the MEL 4 where the final product was obtained free of over-hydrogenated byproducts and presumably trans-isomers also [15]. We are unable to accurately measure the trans-isomer content of the triene 12 but from arguments based on comparison studies with n-3 polyunsaturated fatty acids (PUFAs) provided in our previous synthesis of MEL 3 [14] we are confident that it remains very low and in no more than trace amounts in our current synthesis of the purified C18:3 n-8 MEL 5. This will be further clarified below in relation to the synthesis of the MEL 6. Again, the power of the argentation chromatography is nicely demonstrated by these results in terms of removing virtually all the over-hydrogenated and trans-isomer byproducts.
2.2. Synthesis of MEL 6
As neither of the n-8 and n-3 18:3 MELs 4 and 5 did align with the MS/MS spectra of the natural compound (see below) a third and final 18:3 MEL synthesis of an unusual n-6 variant was performed. The strategy for the C18:3 n-6 MEL 6 synthesis is disclosed in the retrosynthetic analysis shown in Scheme 3. This is also a convergent synthesis involving a Z-selective semi-hydrogenation of the methylene interrupted omega-6 triyne framework of the isopropylidene protected glyceryl ether 21. Triyne 21 is disconnected into two subunit fragments, the monyne 17 possessing the glyceryl head part and the diyne bromide 20 possessing the hydrocarbon tail segment. The monoyne 17 is further disconnected into the key head piece 7 and the TMS protected monoyne bromide linker 15.
To accomplish the intended n-6 methylene skipped triene structure a shorter linker was required as the first double bond in the 1-O-hydrocarbon chain was located at the Δ6'-position. Our synthesis of the monoyne linker 15, which is a commercially available compound, is depicted in Scheme 4.
But-3-yn-1-ol was allowed to react with two equivalents of TMS-chloride and n-butyllithium in dry THF to afford the monoyne homopropargyl alcohol 14 in 74% yield after addition of 2.0 M hydrochloric acid and vigorous stirring. The alcohol was then brominated in an Appel type reaction [25] affording the monoyne linker 15 in 96% yield.
The synthesis of the n-6 diyne tail piece 20 is depicted in Scheme 5. Its synthesis was accomplished by the Appel type bromination of oct-2-yn-1-ol, which afforded the monoyne 18 in quantitative yield after flash chromatography. Copper coupling of the resulting monoyne 18 and propargyl alcohol gave the diyne alcohol 19 in 74% yield. The diyne 20 was then afforded in a second Appel bromination reaction in 89% yield after flash chromatography. This compound was prepared earlier in relation to the synthesis of arachidonic acid by a different approach [26].
The overall synthetic route to the n-6 MEL 6 is demonstrated in Scheme 6. Like the synthesis of the MEL 5, the linker 15 was reacted with magnesium metal to obtain the corresponding Grignard reagent which in turn was reacted with the head piece 7 in the presence of cuprous bromide to obtain the monoyne 16. Initially, this reaction gave poor yields. This was because some amount of bromoform eluted with the linker 15 off the silica column. To resolve that, some further flash chromatography was applied until no bromoform residue was left and the reaction then proceeded smoothly, affording the monoyne 16 in 76% yield.
The monoyne 16 was then, like in the cases of the MEL 3 synthesis [14] and monoyne 10 above, deprotected and methylated in the same operation using methyl iodide and potassium hydroxide. And, as before, the substrate would only methylate in part but with the addition of molecular sieves the monoyne 17 was afforded in 83% yield. The monoyne 17 and diyne 20 were then allowed to undergo copper coupling with cesium carbonate as a base, to furnish the triyne 21 in 94% yield.
The high yield in the coupling reaction involving a monoyne head part and a diene tail part is noteworthy and warrants a comment. In previous cases involving a monoyne head part with the triple bond located at the Δ4'-position in a close vicinity to the isopropylidene protected glyceryl head part, far lower yields were obtained as was evident in the syntheses of both the hexaene MEL 3 [14] and triene MEL 4 [15] where a diyne head part was required for high yields. In the reactions of monoyne head groups 17 and 10 with the corresponding diyne tail parts this was not a problem, and the reactions took place in excellent yields.
The key semi-hydrogenation step was conducted in the same manner as for the previous 18:3 MELs 4 and 5 and the triene 22 was accomplished in 78% yield after flash chromatography. Then the argentation chromatography gave 43% yield, in all 33% yield from the semi-hydrogenation. The semi-hydrogenation overall yields were lower for this product than the corresponding trienes of the other 18:3 MEL syntheses. This may be explained by poorer condition of the labile triyne 21 when it was submitted to the semi-hydrogenation, but the reaction was not optimized further. The final isopropylidene deprotection by aid of the acidic Amberlyst-15® ion exchange resin in refluxing ethanol afforded the MEL 6 in quantitative yield. Thus, the MEL 6 was obtained in 20% yield from the key head piece 7 over five steps (14% total yield as based on the R-solketal).
The over-hydrogenation content of the crude product from the semi-hydrogenation obtained from the flash chromatography was 10%, significantly lower than obtained for MEL 5 above, but comparable to that obtained in the MEL 4 synthesis. After the argentation chromatography purification the over-hydrogenation contamination had dropped to 2% (5.86/6) which is significantly higher than obtained for MELs 4 and 5. The yield of the purified triene 22 was significantly lower than obtained for the corresponding 13, despite the lower purity.
This is clearly less favorable than was accomplished in the corresponding step in the synthesis of the MEL 4, where the final product was obtained free of over-hydrogenated and trans-isomer byproducts [15], and MEL 5 also. The over-hydrogenation level of 2% indicates that there may still be some trans-isomers present (0.5 - 1%) in the purified triene 22 and therefore in the MEL 6 final product. This is estimated as based on our previous results obtained from comparison of various long-chain n-3 PUFAs (C18:4, C20:5 and C22:6) prepared under identical semi-hydrogenation conditions as in the MEL syntheses and accurately determined by GLC analysis. This was described and used to estimate the trans-isomer byproducts in the synthesis of the hexaene MEL 3 [14] as well as evaluating the purity of the triene MEL 4 [15].
2.3. HPLC-MS/MS comparison of C18:3 MELs
Surprisingly, none of the above C18:3 MEL structures seemed to correspond to the one found in the natural shark and dogfish liver oil sample, since none of their MS/MS spectra matched. Figure S1 of the Supplementary Material shows the [M+Li]+ MS/MS fragmentation of the synthesized MELs 4, 5 and 6 as compared to the corresponding unknown C18:3 MEL obtained from the shark and dogfish liver oil mixture. Therefore, based on this comparison it is evident that the true structure of the polyunsaturated C18:3 MEL remains unknown.
2.4. Trends in specific optical rotation values among unsaturated MELs
The specific optical rotation obtained for MELs 5 and 6 warrants a special comment. It is noteworthy that all the unsaturated MEL derivatives obtained from our syntheses so far, except one, displayed negative specific rotation values in ethanol. This includes C16:1 n-12 MEL 1, C18:1 n-14 MEL, C22:6 n-3 MEL 3 and C18:3 n-8 MEL 4 where there commonly is a carbon-carbon double bond located in the D4' position of the 1-O-hydrocarbon ether entity. The C18:1 n-9 MEL on the other hand displayed specific optical rotation opposite in direction in ethanol. In that case the double bond is in the D9' position of the hydrocarbon chain. Similarly, the C18:3 n-3 MEL 5 and the C18:3 n-6 MEL 6 displayed positive specific optical rotation values. They possess the double bond closest to the chiral centre located in the D9' and D6' positions of their hydrocarbon moiety, respectively. Table 1 reveals the specific optical rotation values for all our unsaturated MEL derivatives that have been synthesized.
3. Materials and Methods
3.1. General Information
1H and 13C nuclear magnetic resonance spectra were recorded on a Bruker Avance 400 spectrometer in deuterated chloroform as a solvent at 400.12 and 100.61 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) and the coupling constants (J) in Hertz (Hz). The following abbreviations are used to describe the multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; dd, doublet of doublets; m, multiplet. Structural assignments were made with additional information from COSY and HSQC experiments. Infrared spectra were conducted on a Thermo Nicolet FT-IR iS10 Spectrophotometer on a KBr pellet (crystalline material) or as a neat liquid (oils). Melting points were determined on a Büchi 520 melting point apparatus and are uncorrected. The high-resolution mass spectra (HRMS) were acquired on a Bruker micrOTOF-Q mass spectrometer. All data analysis was done on Bruker software. Optical activity measurements were performed on an AutopolR V Automatic Polarimeter from Rudolph Research Analytical using a 40T-2.5-100-0.7 Temp Trol™ polarimetric cell with 2.5 mm inside diameter, 100 mm optical path length and 0.7 mL volume with c referring to g sample/100 mL. The reactions that required heating were conducted in an aluminum block for round-bottom flask installed on a magnetic stirrer hotplate equipped with a contact thermometer.
All chemicals and solvents were used without further purification unless otherwise stated. Quinoline (98%), propargyl alcohol (99%), but-3-yn-1-ol (97%), oct-2-yn-1-ol (97%), cuprous iodide (>99.5%), cuprous bromide (98%), sodium iodide (>99%), cesium carbonate (99.9%), tetrabromomethane (99%), triphenylphosphine (99%), trimethylsilyl chloride (≥99%), n-butyl lithium in cyclohexane (2.0 M), palladium on calcium carbonate, 5% wt, poisoned with lead (Lindlar catalyst) were from Sigma-Aldrich (Steinheim). Molecular sieves 4A were obtained from Acros Organics (New Jersey, USA). Amberlyst-15® ion exchange resin was obtained from Fluka (Buchs, Switzerland). Methyl iodide and potassium hydroxide were purchased from Merck (Darmstad, Germany). Dichloromethane and ethyl acetate were obtained HPLC grade, toluene, acetone (≥99.8%), and N,N-dimethylformamide (anhydrous, 99,8%) from Sigma-Aldrich (Steinheim, Germany), petroleum ether, boiling range 40-60 °C, tetrahydrofuran, diethyl ether, methanol, and ethanol (99.8%) from Honeywell (Seelze, Germany). Dichloromethane was dried over CaH2 under dry nitrogen atmosphere. Tetrahydrofuran HPLC grade was obtained from Sigma-Aldrich (Steinheim) and dried over Na wire in presence of benzophenone under dry nitrogen atmosphere. Column chromatography was performed on Silica gel 60 (Silicycle, Ontario). Reactions were monitored by TLC on Silica gel 60 F254 (Silicycle, Ontario), with detection by quenching of fluorescence and/or with phosphomolybdic acid in methanol. Argentation column chromatography was performed with 10% Silver nitrate impregnated silica gel (R23530B) (Silicycle, Ontario).
3.2. Synthesis of MEL 5
3.2.1. Synthesis of (2'R)-1-O-(2'-methoxyocta-9',12',15'-triyn-1-yl)-2,3-O-isopropylidene-sn-glycerol, 12
To a stirred suspension of cuprous iodide (0.360g, 1.89 mmol), sodium iodide (0.703g, 4.73 mmol) and cesium carbonate (1.230g, 3.78 mmol) in dry DMF (5 mL) under nitrogen atmosphere was added 1-bromoocta-2,5-diyne 11 [14] (0.350g, 1.89 mmol) in DMF (2 mL), followed by the methoxylated monoyne 10 [12] (0.282g, 0.945 mmol) in DMF (2 mL). The resulting mixture was stirred vigorously for 41 hours at room temperature. Then, the reaction mixture was quenched with a saturated ammonium chloride solution and stirred for 10 min. The mixture was then extracted three times with diethyl ether and the combined ether extracts washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude concentrate was purified by flash column chromatography using ethyl acetate/petroleum ether (1:3) as eluent affording the triyne product 12 as a yellow oil (0.344g, 91% yield). [α]D20 = -3.80 (c 2.0, ethanol). IR (NaCl, νmax / cm-1): 2981, 2935, 2860, 2213. 1H NMR (400 MHz, CDCl3) δH: 4.29 – 4.23 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.75 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.52 (dd, J = 10.3, 5.8 Hz, 1H, CH2-1'), 3.49 (dd, J = 10.0, 5.7 Hz, 1H, CH2 sn-1), 3.47 (dd, J = 10.3, 4.2 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.35 – 3.28 (m, 1H, CH-2'), 3.15 – 3.12 (m, 4H, ≡CCH2C≡), 2.20 – 2.12 (m, 4H, ≡CCH2CH2/CH3), 1.53 – 1.25 (m, 10H, CH2), 1.42 (s, 3H, C(CH3)2), 1.35 (s, 3H, C(CH3)2), 1.12 (t, J = 7.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 109.5, 82.3, 80.9, 80.3, 75.01, 74.96, 74.8, 73.94, 73.91, 73.3, 67.0, 57.8, 31.5, 29.4, 29.0, 28.8, 26.9, 25.6, 25.4, 18.8, 14.0, 12.5, 10.0, 9.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H38O4Na 425.2668; found, 425.2674.
3.2.2. Synthesis of (2'R)-1-O-[(9'Z,12'Z,15'Z)-2'-methoxyocta-9',12',15'-trien-1-yl]-2,3-O-isopropylidene-sn-glycerol, 13
Lindlar catalyst (0.500g) was placed into a dry two-necked round-bottom flask equipped with a magnetic stirrer under nitrogen at room temperature and the flask sealed with a septum. Toluene (20 mL) was added to the flask with a syringe. A balloon filled with hydrogen gas was then mounted on a syringe and stuck through the septum. The mixture was then stirred while the hydrogen gas was blown through the flask to replace the nitrogen atmosphere with hydrogen. Then quinoline (0.055g, 0.438 mmol) and triyne 12 (0.353g, 0.877 mmol) dissolved in toluene (2 mL) were added with a syringe and the reaction stirred vigorously while being monitored with TLC. When the reaction came to completion according to the TLC (75 min), the flask was promptly opened, and the reaction mixture filtered through a celite layer on a fritted disk using dichloromethane as eluent. After removing the solvent in vacuo, the crude concentrate was purified by flash column chromatography using ethyl acetate/petroleum ether (1:4) as eluent affording the crude product 13 as a light-yellow liquid (0.280g, 78% yield). Part of the product (0.043g) was then applied to a 10% silver nitrate impregnated silica gel column using acetone/petroleum ether (1:4) as eluent resulting in the purified product 12 as a faintly yellow liquid (0.022g, 51% yield; overall in 40% yield). [α]D20 = -4.25 (c 1.6, ethanol). IR (NaCl, νmax / cm-1): 3011, 2932, 2857, 846. 1H NMR (400 MHz, CDCl3) δH: 5.43 – 5.28 (m, 6H, =CH), 4.29 – 4.23 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.76 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.52 (dd, J = 10.3, 5.8 Hz, 1H, CH2-1'), 3.49 (dd, J = 10.0, 5.8 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.3, 4.4 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.35 – 3.28 (m, 1H, CH-2'), 2.81 (t, J = 6.0 Hz, 4H, =CHCH2CH=), 2.11-2.03 (m, 4H, =CHCH2CH2/=CHCH2CH3), 1.52 – 1.45 (m, 2H, CH2-3'), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2), 1.40 – 1.25 (m, 8H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 132.1, 130.4, 128.43, 128.41, 127.9, 127.3, 109.5, 80.3, 74.8, 74.0, 72.5, 67.0, 57.7, 31.6, 29.9, 29.8, 29.4, 27.4, 26.9, 25.8, 25.7, 25.6, 25.5, 20.7, 14.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H44O4Na 431.3137; found, 431.3132.
3.2.3. Synthesis of (2'R)-1-O-[(9'Z,12'Z,15'Z)-2'-methoxyocta-9',12',15'-trien-1-yl]-sn-glycerol, 5
Triene 13 (0.020g, 0.049 mmol) in ethanol (2.7 mL) was loaded into a two-necked round-bottom flask equipped with a magnetic stirrer and a reflux condenser under nitrogen. To that solution wet Amberlyst-15® (0.007g) was added and the reaction heated to reflux. After refluxing for 3 hours the mixture was allowed to cool, the Amberlyst-15® filtered off and the solvent removed in vacuo. The crude concentrate was then purified by flash column chromatography, eluting first with ethyl acetate/petroleum ether (1:2) and then with pure ethyl acetate to afford the final product MEL 5 as a yellow oil (0.016g, 89%). [α]D20 = +4.27 (c 1.1, ethanol). IR (NaCl, νmax / cm-1): 3403, 3010, 2926, 2855, 1093, 928. 1H NMR (400 MHz, CDCl3) δH: 5.43 – 5.28 (m, 6H, =CH), 3.89-3.84 (m, 1H, CH sn-2), 3.71 (dd, J = 11.4, 3.9 Hz, 1H, CH2 sn-3), 3.64 (dd, J = 11.4, 5.2 Hz, 1H, CH2 sn-3), 3.61 (dd, J = 10.0, 4.0 Hz, 1H, CH2 sn-1), 3.56 (dd, J = 10.5, 3.5 Hz, 1H, CH2-1'), 3.54 (dd, J = 10.0, 6.4 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.5, 6.0 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.35 – 3.29 (m, 1H, CH-2'), 2.98 (br s, 1H, OH), 2.81 (t, J = 6.1 Hz, 4H, =CHCH2CH=), 2.32 (br s, 1H, OH), 2.11 – 2.03 (m, 4H, =CHCH2CH2/=CHCH2CH3), 1.54 – 1.42 (m, 2H, CH2-3'), 1.41 – 1.22 (m, 8H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 132.1, 130.4, 128.44, 128.39, 127.9, 127.3, 80.5, 73.8, 73.5, 70.7, 64.2, 57.5, 31.1, 29.8, 29.7, 29.4, 27.4, 25.8, 25.7, 25.5, 20.7, 14.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H40O4Na 391.2824; found, 391.2819.
3.3. Synthesis of MEL 6
3.3.1. Synthesis of 4-(trimethylsilyl)but-3-yn-1-ol, 14
Into a dried round-bottom flask under nitrogen atmosphere containing but-3-yn-1-ol (0.385g, 5.50 mmol) in tetrahydrofuran (18 mL) at -78°C n-butyllithium in cyclohexane (6.05mL, 2M, 12.10 mmol) was added with a syringe. After 60 minutes at -78°C trimethylsilyl chloride (1.315g, 12.10 mmol) was added dropwise from a syringe. 5 minutes after that the reaction was allowed to reach room temperature. After 6 hours 2M HCl (12mL) was added and the reaction stirred vigorously for 10 minutes, then a saturated solution of sodium bicarbonate was added and the solution extracted three times with diethyl ether, dried over MgSO4, filtered and the solvent evaporated under reduced pressure. The extract was then applied to a silica gel chromatography using ethyl acetate/petroleum ether (1:3) as eluent affording the product 14 as a colourless liquid (0.579g, 74% yield). IR (NaCl, νmax / cm-1): 3347, 2960, 2900, 2177. 1H NMR (400 MHz, CDCl3) δH: 3.71 (q, J = 6.3 Hz, 2H, HOCH2), 2.51 (t, J = 6.2 Hz, 2H, ≡CCH2), 1.75 (t, J = 6.4 Hz, 1H, OH), 0.16 (s, 9H, TMS) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 103.5, 87.2, 61.0, 24.4, 0.20 (3) ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C7H14OSiNa 165.0712; found, 165.0706.
3.3.2. Synthesis of 4-bromo-1-(trimethylsilyl)but-1-yne, 15
Into a dry round-bottom flask equipped with a magnetic stirrer TMS-protected but-3-yn-1-ol 14 (0.202g, 1.60 mmol) dissolved in dichloromethane (20mL) was placed. The flask was then cooled to 0 – 4°C (ice/water), carbon tetrabromide (1.427g, 4.30 mmol) added and the resulting mixture stirred for 10 minutes. Then triphenylphosphine (0.939g, 3.58 mmol) was added, the reaction stirred for further 10 minutes and then more triphenylphosphine (0.376g, 1.42 mmol) added in small doses every 30 minutes until the reaction turned yellow. The reaction mixture was then diluted with diethyl ether (50 mL) and the white precipitate filtered through a short silica layer (2 cm) using diethyl ether as eluent. The solvent was removed in vacuo resulting in a colourless oil which was then applied to a silica gel flash chromatography using ethyl acetate/petroleum ether (1:6) as eluent affording the product 15 as a colourless oil (0.710g, 96% yield). IR (NaCl, νmax / cm-1): 3019, 2960, 2924, 2899, 2854, 2178. 1H NMR (400 MHz, CDCl3) δH: 3.43 (t, J = 7.5 Hz, 2H, BrCH2), 2.77 (t, J = 7.5 Hz, 2H, ≡CCH2), 0.16 (s, 9H, TMS) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 103.3, 87.2, 29.3, 24.5, 0.1 (3) ppm. Too volatile for HRMS measurement.
3.3.3. (2'R)-1-O-[7'-(trimethylsilyl)-2'-hydroxyhept-6'-yn-1-yl]-2,3-O-isopropylidene-sn-glycerol, 16
To a dried two-necked round-bottom flask equipped with a reflux condenser, magnesium (0.078g, 3.20 mmol) was added under nitrogen atmosphere. To the flask, dry THF (8 mL) and TMS-protected monoyne bromide 15 (0.586g, 2.86 mmol) in THF (2 mL) was then added via syringe. Heat was applied and the reaction allowed to reflux until most of the magnesium metal had disappeared. The reaction was then cooled to about -10°C (ice/acetone) and cuprous bromide (0.046g, 0.32 mmol) and the head piece 7 [13] (0.301g, 1.60 mmol) in THF (1 mL) were added. The stirred reaction was allowed to warm up overnight and finally quenched with a saturated aqueous ammonium chloride solution, extracted three times with diethyl ether and the combined ether extracts washed once with a saturated sodium chloride solution and dried over MgSO4. After filtering and evaporation of the solvent the resulting concentrate was applied to a silica gel column chromatography using ethyl acetate/petroleum ether (1:2) as eluent affording the TMS-protected monoyne 16 as a colourless liquid (0.385g, 76% yield). [α]D20 = +1.7 (c 0.1, ethanol). IR (NaCl, νmax / cm-1): 3474, 2986, 2956, 2936, 2900, 2872, 2174. 1H NMR (400 MHz, CDCl3) δH: 4.33 – 4.22 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.5 Hz, 1H, CH2 sn-3), 3.86 – 3.77 (m, 1H, CH-2'), 3.74 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.56 (d, J = 5.3 Hz, 2H, CH2 sn-1), 3.54 (dd, J = 9.7, 3.0 Hz, 1H, CH2-1'), 3.35 (dd, J = 9.7, 7.9 Hz, 1H, CH2-1'), 2.42 (d, J = 3.4 Hz, 1H, OH), 2.26 (t, J = 6.8 Hz, 2H, ≡CCH2), 1.77 – 1.48 (m, 4H, CH2-3' and ≡CCH2CH2), 1.43 (s, 3H, C(CH3)2), 1.37 (s, 3H, C(CH3)2), 0.14 (s, 9H, TMS) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 109.7, 107.2, 100.1, 85.0, 76.2, 74.9, 72.5, 70.0, 69.9, 66.7, 32.2, 26.9, 25.5, 24.8, 20.0, 0.3 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H30O4SiNa 337.1811; found, 337.1806.
3.3.4. (2'R)-1-O-(2'-methoxydec-6'-yn-1-yl)-2,3-O-isopropylidene-sn-glycerol, 17
Into a flame-dried round-bottom 25 mL flask equipped with a magnetic stirrer, the TMS protected alkyne alcohol 16 (0.093g, 0.296 mmol) dissolved in dry DMF (6 mL) was placed and molecular sieves added (0.2 g). Into the stirred mixture potassium hydroxide (0.066g, 1.18 mmol) was added and stirred for 10 min. Subsequently methyl iodide (0.420g, 2.96 mmol) was added. The reaction was monitored with TLC and after two hours more potassium hydroxide (0.015g, 0.27 mmol) was added and then every 40 min until the reaction was nearly completed according to TLC. When the reaction came to a stop according to TLC the reaction mixture was quenched with a saturated ammonium chloride solution and extracted three times with diethyl ether. The combined extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The resulting concentrate was purified by flash silica gel column chromatography using ethyl acetate/petroleum ether (1:1) as eluent affording the methoxylated monoyne product 16 as a colorless liquid (0.063g, 83% yield). [α]D20 = -3.29 (c 0.7, ethanol). IR (NaCl, νmax / cm-1): 3290, 2986, 2934, 2872, 2828, 2117. 1H NMR (400 MHz, CDCl3) δH: 4.30 – 4.22 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.75 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.54 (dd, J = 10.3, 5.6 Hz, 1H, CH2-1'), 3.49 (dd, J = 10.0, 5.7 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.3, 5.7 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.37 – 3.31 (m, 1H, CH-2'), 2.25 – 2.14 (m, 2H, ≡CCH2), 1.94 (t, J = 2.6 Hz, 1H, ≡CH), 1.71 – 1.51 (m, 4H, CH2-3' and ≡CCH2CH2), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 109.5, 84.4, 79.7, 74.8, 73.8, 72.6, 68.6, 67.0, 57.8, 30.7, 26.9, 25.6, 24.5, 18.7 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C14H24O4Na 279.1572; found, 279.1567.
3.3.5. Synthesis of 1-bromooct-2-yne, 18
Into a dry two-necked round-bottom flask equipped with a magnetic stirrer oct-2-yn-1-ol (0.500g, 3.96 mmol) dissolved in dichloromethane (22mL) was placed. The flask was then cooled to 0 – 4°C (ice/water), carbon tetrabromide (1.58g, 4.75 mmol) added and the resulting mixture stirred for 10 minutes. Then triphenylphosphine (1.040g, 3.97 mmol) was added, the reaction stirred for further 10 minutes. After 15 minutes and 30 minutes more triphenylphosphine (0.208g, 0.79 mmol) was added at which point the reaction mixture turned yellow (in all 1.456g, 5.55 mmol). The reaction mixture was then diluted with diethyl ether (50 mL) and the white precipitate filtered through a short silica layer (2 cm) using diethyl ether as eluent. The solvent was removed in vacuo resulting in a colourless oil which was then applied to a silica gel flash chromatography using ethyl acetate/petroleum ether (1:6) as eluent affording the product 18 as a colourless oil (0.745g, 100% yield). IR (NaCl, νmax / cm-1): 2957, 2932, 2871, 2860, 2311, 2234. 1H NMR (400 MHz, CDCl3) δH: 3.93 (t, J = 2.4 Hz, 2H, BrCH2), 2.23 (tt, J = 7.2, 2.4 Hz, 2H, ≡CCH2CH2), 1.58 – 1.46 (m, 2H, ≡CCH2CH2), 1.41 – 1.23 (m, 4H, CH2CH2CH3), 0.90 (t, J = 7.0 Hz, 3H, CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 88.5, 75.4, 31.2, 28.2, 22.3, 19.1, 16.0, 14.1 ppm. Too volatile for HRMS measurement.
3.3.6. Synthesis of undeca-2,5-diyn-1-ol, 19
To a stirred suspension of cuprous iodide (0.236 g, 1.24 mmol), sodium iodide (0.184 g, 1.24 mmol) and cesium carbonate (0.403 g, 1.24 mmol) in dry DMF (5 mL) under nitrogen atmosphere was added 1-bromooct-2-yne 18 (0.234 g, 1.24 mmol) in DMF (2 mL), followed by propargyl alcohol (0.069 g, 1.24 mmol). The resulting mixture was stirred vigorously for 24 hours at room temperature. Then, the reaction mixture was quenched with a saturated ammonium chloride solution and stirred for 10 min. The mixture was then extracted three times with diethyl ether and the combined ether extracts washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude concentrate was purified by flash column chromatography using ethyl acetate/petroleum ether (1:3) as eluent affording the diyne alcohol product 19 as a yellow oil (0.150 g, 74% yield). IR (NaCl, νmax / cm-1): 3358, 2957, 2932, 2871, 2860, 2259. 1H NMR (400 MHz, CDCl3) δH: 4.26 (t, J = 2.2 Hz, 2H, HOCH2), 3.19 (quin, J = 2.3 Hz, 2H, ≡CCH2C≡), 2.15 (tt, J = 7.2, 2.4 Hz, 2H, ≡CCH2CH2), 1.55 (br s, 1H, OH) 1.49 (quin, J = 7.3 Hz, 2H, ≡CCH2CH2), 1.40 – 1.25 (m, 4H, CH2CH2CH3), 0.89 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 81.4, 81.1, 78.5, 73.4, 51.5, 31.2, 28.6, 22.4, 18.8, 14.1, 10.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C11H16ONa 187.1099; found, 187.1093.
3.3.7. Synthesis of 1-bromoundeca-2,5-diyne, 20
Into a dry two-necked round-bottom flask equipped with a magnetic stirrer undeca-2,5-diyn-1-ol 19 (0.168g, 1.02 mmol) dissolved in dichloromethane (20 mL) was placed. The flask was then cooled to 0 – 4°C (ice/water), carbon tetrabromide (0.407g, 1.23 mmol) added and the resulting mixture stirred for 10 minutes. Triphenylphosphine (0.268g, 1.02 mmol) was added and the reaction stirred for further 40 minutes. Then more triphenylphosphine (0.080g, 0.30 mmol) was added after which the reaction turned yellow within 20 minutes and showed completion on TLC. The reaction mixture was then diluted with diethyl ether (50 mL) and the precipitate filtered through a short silica layer (2 cm) using diethyl ether as eluent. The solvent was removed in vacuo resulting in a yellow oil which was applied to a silica gel flash chromatography using ethyl acetate/petroleum ether (1:6) as eluent affording the diyne product 20 as a yellow oil (0.207g, 89% yield). IR (NaCl, νmax / cm-1): 3003, 2957, 2931, 2870, 2859, 2268, 2234. 1H NMR (400 MHz, CDCl3) δH: 3.91 (t, J = 2.3 Hz, 2H, BrCH2), 3.21 (quin, J = 2.4 Hz, 2H, ≡CCH2C≡), 2.15 (tt, J = 7.2, 2.4 Hz, 2H, ≡CCH2CH2), 1.49 (quin, J = 7.2 Hz, 2H, ≡CCH2CH2), 1.40 – 1.27 (m, 4H, CH2CH2CH3), 0.90 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 82.2, 81.5, 75.2, 72.8, 31.1, 28.4, 22.2, 18.7, 14.9, 14.0, 10.1 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C11H15BrNa 249.0255; found, 249.0249.
3.3.8. Synthesis of (2'R)-1-O-(2'-methoxyocta-6',9',12'-triyn-1-yl)-2,3-O-isopropylidene-sn-glycerol, 21
To a stirred suspension of cuprous iodide (0.119g, 0.62 mmol), sodium iodide (0.232g, 1.56 mmol) and cesium carbonate (0.407g, 1.25 mmol) in dry DMF (3 mL) under nitrogen atmosphere was added 1-bromoundeca-2,5-diyne 20 (0.207g, 0.91 mmol) in DMF (2 mL), followed by the methoxylated monoyne 17 (0.080g, 0.312 mmol) in DMF (2 mL). The resulting mixture was stirred vigorously for 43 hours at room temperature. Then, the reaction mixture was quenched with a saturated ammonium chloride solution and stirred for 10 min. The mixture was then extracted three times with diethyl ether and the combined ether extracts washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude concentrate was purified by flash column chromatography using ethyl acetate/petroleum ether (4:11) as eluent affording the triyne product 21 as a yellow oil (0.119g, 94% yield). [α]D20 = -3.14 (c 0.7, ethanol). IR (NaCl, νmax / cm-1): 2984, 2933, 2872, 2211, 1088. 1H NMR (400 MHz, CDCl3) δH: 4.30 – 4.22 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.76 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.53 (dd, J = 10.3, 5.8 Hz, 1H, CH2-1'), 3.49 (dd, J = 10.0, 5.7 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.3, 4.3 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.37 – 3.30 (m, 1H, CH-2'), 3.15 – 3.09 (m, 4H, ≡CCH2C≡), 2.22 – 2.10 (m, 4H, ≡CCH2CH2), 1.68 – 1.44 (m, 6H, CH2), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2), 1.38 – 1.23 (m, 4H, CH2CH2CH3), 0.89 (t, J = 7.1 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 109.5, 81.1, 80.5, 79.8, 75.1, 74.9, 74.8, 74.3, 73.84, 73.81, 72.6, 67.0, 57.8, 31.2, 30.8, 28.6, 26.9, 25.6, 24.7, 22.4, 19.0, 18.8, 14.1, 9.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H38O4Na 425.2668; found, 425.2662.
3.3.9. Synthesis of (2'R)-1-O-[(6'Z,9'Z,12'Z)-2'-methoxyocta-6',9',12'-trien-1-yl]-2,3-O-isopropylidene-sn-glycerol, 22
Lindlar catalyst (0.117g) was placed into a dry two-necked round-bottom flask equipped with a magnetic stirrer under nitrogen at room temperature and the flask sealed with a septum. Toluene (5 mL) was added to the flask with a syringe. A balloon filled with hydrogen gas was then mounted on a syringe and stuck through the septum. The mixture was then stirred while the hydrogen gas was blown through the flask to replace the nitrogen atmosphere with hydrogen. Then quinoline (0.018g, 0.145 mmol) and triyne 21 (0.109g, 0.290 mmol) dissolved in toluene (2 mL) were added with a syringe and the reaction stirred vigorously while being monitored with TLC. When the reaction came to completion according to the TLC (40 min), the flask was promptly opened, and the reaction mixture filtered through a celite layer on a fritted disk using dichloromethane as eluent. After removing the solvent in vacuo, the crude concentrate was purified by flash column chromatography using ethyl acetate/petroleum ether (1:4) as eluent affording the crude product 22 as a light-yellow liquid (0.087g, 78% yield). The crude product was then applied to a 10% silver nitrate impregnated silica gel column using an ethyl acetate/petroleum ether gradient from 1:7 to 7:9 as eluent resulting in the purified triene product 22 as a faintly yellow liquid (0.037g, 43% yield; overall in 33% yield). [α]D20 = -6.88 (c 0.9, ethanol). IR (NaCl, νmax / cm-1): 3010, 2985, 2930, 2860, 1652, 975. 1H NMR (400 MHz, CDCl3) δH: 5.45 – 5.28 (m, 6H, =CH), 4.32 – 4.21 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.76 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.57 (dd, J = 10.0, 5.3 Hz, 1H, CH2 sn-1), 3.55 – 3.48 (m, 2H, CH2-1' and CH2 sn-1), 3.48 (dd, J = 10.3, 4.4 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.36 – 3.29 (m, 1H, CH-2'), 2.80 (t, J = 5.8 Hz, 4H, =CHCH2CH=), 2.13-2.01 (m, 4H, =CHCH2), 1.55 – 1.43 (m, 4H, CH2-3' and CH2-4'), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2), 1.40 – 1.24 (m, 6H, CH2), 0.89 (t, J = 6.8 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 130.5, 129.9, 128.4, 128.3, 128.1, 127.6, 109.5, 80.1, 74.7, 73.8, 72.4, 66.9, 57.7, 31.5, 31.1, 29.4, 27.3, 27.2, 26.8, 25.7 (2), 25.5, 25.4, 22.6, 14.1 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H44O4Na 431.3137; found, 431.3132.
3.3.10. Synthesis of (2'R)-1-O-[(6'Z,9'Z,12'Z)-2'-methoxyocta-6',9',12'-trien-1-yl]-sn-glycerol, 6
Triene 22 (0.017g, 0.042mmol) in ethanol (3 mL) was loaded into a two-necked round-bottom flask equipped with a magnetic stirrer and a reflux condenser under nitrogen. To that solution wet Amberlyst-15® (0.007g) was added and the reaction heated to reflux. After refluxing for 3 hours the mixture was allowed to cool, the Amberlyst-15® filtered off and the solvent removed in vacuo. The crude concentrate was then purified by flash column chromatography, eluting first with ethyl acetate/petroleum ether (1:2) and then with pure ethyl acetate to afford the final product MEL 6 as a yellow oil (0.015g, 100%). [α]D20 = +3.26 (c 1.5, ethanol). IR (NaCl, νmax / cm-1): 3404, 3011, 2927, 2858, 1651. 1H NMR (400 MHz, CDCl3) δH: 5.44 – 5.28 (m, 6H, =CH), 3.89 – 3.84 (br m, 1H, CH sn-2), 3.73 – 3.69 (br dd, 1H, CH2 sn-3), 3.66 – 3.63 (br dd, 1H, CH2 sn-3), 3.61 (dd, J = 10.0, 3.9 Hz, 1H, CH2 sn-1), 3.56 (dd, J = 10.5, 3.6 Hz, 1H, CH2-1'), 3.54 (dd, J = 10.0, 6.3 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.5, 6.0 Hz, 1H, CH2-1'), 3.40 (s, 3H, OCH3), 3.36 – 3.30 (m, 1H, CH-2'), 2.95 (br s, 1H, OH), 2.80 (t, J = 5.7 Hz, 4H, =CHCH2CH=), 2.28 (br s, 1H, OH), 2.14 – 2.00 (m, 4H, =CHCH2CH2), 1.58 – 1.22 (m, 10H, CH2), 0.89 (t, J = 6.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 130.6, 129.8, 128.6, 128.5, 128.2, 127.7, 80.4, 73.8, 73.5, 70.7, 64.2, 57.6, 31.7 30.7, 29.5 27.4, 27.4, 25.8 (2), 25.5, 22.7, 14.2 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H40O4Na 391.2824; found, 391.2819.
4. Conclusions
The successful asymmetric synthesis of the trienes C18:3 n-3 MEL 5 and C18:3 n-6 MEL 6 is described. Both syntheses were based on the use of the head group synthon (2R,2'S)-7 which is a double-C3 building block. The syntheses were brought about by the polyacetylene approach involving a crucial stereoselective semi-hydrogenation of the triynes 12 and 21. The n-3 C18:3 MEL 5 was obtained virtually free of over-hydrogenation and trans-isomer byproducts after flash chromatography and a subsequent argentation chromatography treatment of the triene 13 intermediate obtained from the semi-hydrogenation key step of the synthesis. The n-6 C18:3 MEL 6 was obtained in acceptable but somewhat lower purity after similar purification treatment of the triene 22. Neither of the products were observed to match with the unknown C18:3 MEL that was found in a mixture of shark and dogfish liver oil in terms of their MS/MS spectra, so its identity remains undisclosed.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Comparison of the MS/MS fragmentation spectra of the unknown C18:3 MEL from the natural dogfish and shark liver oil mixture with MEL 4 from a previous synthesis and MELs 5 and 6 obtained from the current synthesis; Spectral data of all compounds synthesized 5, 6, 12 – 14 and 16 – 22 (1H, 13C, COSY and HSQC NMR); for 15 (1H and 13C NMR).
Author Contributions
Conceptualization, GGH and SS; methodology, SS and GGH; validation, SS and GGH; formal analysis, SS and GGH; investigation, SS and GGH; writing - original draft preparation, SS and GGH; writing – review and editing, GGH and SS; funding acquisition GGH and SS; supervision, GGH. The authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Icelandic Research Fund (120023 – 021-023 and 141595 – 051-053) that is also acknowledged for a doctorate student grant (SS) (174396 – 051-053).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.
Acknowledgments
Dr. Sigrídur Jónsdóttir University of Iceland is acknowledged for NMR and accurate MS measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ether Lipids. Biochemical and Biomedical Aspects, Mangold, H.K., Palthauf, F., Eds.; Academic: New York, 1983; p. 439. [Google Scholar]
- Magnusson, C.D.; Haraldsson, G.G. Ether lipids. Chem. Phys. Lipids 2011, 164, 315–340. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Palmieri, B. An update on the therapeutic role of alkylglycerols. Mar. Drugs 2010, 8, 2267–2300. [Google Scholar] [CrossRef] [PubMed]
- Deniau, A.L.; Mosset, P.; Pedrono, F.; Mitre, R.; Le Bot, D.; Legrand, A.B. Multiple beneficial effects of natural alkylglycerols from shark liver oil. Mar. Drugs 2010, 8, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Huynh, K.; Giles, C.; Mellett, N.A.; Duong, T.; Nguyen, A.; Lim, W.L.F.; Smith, A.A.T.; Olshansky, G.; Cadby, G.; Hung, J.; Hui, J.; Beilby, J.; Watts, G.F.; Chatteriee, P.; Martins, I.; Laws, S.M.; Bush, A.; Rowe, C.C.; Villemagne, V.L.; Ames, D.; Masters, C.L.; Taddei, K.; Doré, V.; Fripp, J.; Arnold, M.; Kastenüller, G.; Nho, K.; Saykin, A.J.; Baillie, R.; Han, X.; Martins, R.N.; Moses, E.K.; Kaddurah-Daouk, R.; Meikle, P.J. APOE e2 resilience for Alzheimer’s disease is mediated by plasma lipid species: Analysis of three independent cohort studies. Alzheimer’s Dement. 2022; 1-16. [Google Scholar]
- Pemha, R.; Le Bot, D.; Mosset, P.; Legrand, A.B. Anti-angiogenic and cytotoxicity effects of selachyl alcohol analogues. Anti-Cancer Agents Med. Chem. 2022, 22, 1913–1920. [Google Scholar]
- Hallgren, B.; Stallberg, G. Methoxy-substituted glycerol ethers isolated from Greenland shark liver oil. Acta Chem. Scand. 1967, 21, 1519–1529. [Google Scholar] [CrossRef]
- Hallgren, B.; Stallberg, G.; Thorin, H. The separation and identification of a methoxy-substituted polyunsaturated glycerol ether from Greenland shark liver oil. Acta Chem. Scand. 1971, 25, 3781–3784. [Google Scholar] [CrossRef]
- Stallberg, G. Synthesis of the stereoisomers of 1-O-(2’-Methoxyhexadecyl)-glycerol and some phosphocholine derivatives. Acta Chem. Scand. 1990, 44, 368–376. [Google Scholar] [CrossRef]
- Pemha, R.; Pegnyemb, D.E.; Mosset, P. Synthesis of (Z)-(2'R)-1-O-(2'-methoxynonadec-10'-enyl)-sn-glycerol, a new analog of bioactive ether lipids. Tetrahedron 2012, 68, 2973–2983. [Google Scholar]
- Pemha, R.; Kuete, V.; Pages, J.-M.; Pegnyemb, D.E.; Mosset, P. Synthesis and biological evaluation of four new ricinoleic acid-derived 1-O-alkylglycerols. Mar. Drugs 2020, 18, 113. [Google Scholar] [CrossRef]
- Sigurjonsson, S.; Luthersson, E.; Albertsdottir, A.D.; Rögnvaldsdottir, E.K.; Haraldsson, G.G. Asymmetric synthesis of methoxylated ether lipids: Total synthesis of two monounsaturated C18:1 and a saturated C18:0 methoxylated ether lipid derivatives. Tetrahedron 2023, 134, 133304. [Google Scholar] [CrossRef]
- Sigurjonsson, S.; Luthersson, E.; Gudmundsson, H.G.; Haraldsdottir, H.; Kristinsdottir, L.; Haraldsson, G.G. Asymmetric synthesis of methoxylated ether lipids: A glyceryl glycidyl ether key building block design, preparation and synthetic application. J. Org. Chem. 2022, 87, 12306–12314. [Google Scholar] [CrossRef] [PubMed]
- Sigurjonsson, S.; Luthersson, E.; Magnusson, C.D.; Gudmundsson, H.G.; Das, E.; Haraldsson, G.G. Asymmetric synthesis of methoxylated ether lipids: Total synthesis of n-3 polyunsaturated docosahexaenoic acid-like methoxylated ether lipid. J. Org. Chem. 2022, 87, 14623–14635. [Google Scholar] [CrossRef] [PubMed]
- Sigurjonsson, S.; Luthersson, E.; Haraldsson, G.G. Asymmetric synthesis of methoxylated ether lipids: Total synthesis of a triene C18:3 omega-8 MEL derivative. ChemistrySelect 2023, 134, e202301228. [Google Scholar] [CrossRef]
- Hayashi, K.; Takagi, T. Characteristics of methoxyglyceryl ethers from some cartilaginous fish liver lipids. Nippon Suisan Gakkaishi 1982, 48, 1345–1351. [Google Scholar] [CrossRef]
- Durand, S.; Parrain, J.-L.; Santelli, M. Construction of (Z,Z) skipped 1,4-dienes. Application to the synthesis of polyunsaturated fatty acids and derivatives. J. Chem. Soc., Perkin Trans. I, 2000; 253–273. [Google Scholar]
- Tedeschi, C.; Saccavini, C.; Maurette, L.; Soleilhavoup, M.; Chauvin, R. 1,4-Diynes from alkynyl-propargyl coupling reactions. J. Organomet. Chem. 2003, 670, 151–169. [Google Scholar] [CrossRef]
- Lapitskaya, M.A.; Vasiljeva, L.L.; Pivnitsky, K.K. A chemoselective synthesis of functionalized 1,4-alkadiynes (skipped diacetylenes). Synthesis 1993, 65–66. [Google Scholar] [CrossRef]
- Caruso, T.; Spinella, A. CsCO3 Promoted coupling reactions for the preparation of skipped diynes. Tetrahedron 2003, 59, 7787–7790. [Google Scholar] [CrossRef]
- Oger, C.; Balas, L.; Durand, T.; Galano, J.M. Are alkyne reductions chemo-, regio-, and stereoselective enough to provide pure (Z)-olefins in polyfunctionalized bioactive molecules? Chem. Rev. 2013, 113, 1313–1350. [Google Scholar] [CrossRef]
- Morris, L.J. Separation of Lipids by silver ion chromatography. J. Lipid Res. 1966, 7, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Damyanova, B. Retention of lipids in silver ion high-performance liquid chromatography: Facts and assumptions. J. Chromatography A 2009, 1216, 1815–1824. [Google Scholar] [CrossRef]
- Yu, C.C.; Lee, Y.S.; Cheon, B.S.; Lee, S.H. Synthesis of glycerol monostearate with high purity. Bull. Korean Chem. Soc. 2003, 24, 1229–1231. [Google Scholar]
- Appel, R. Tertiary phosphane/tetrachloromethane, a versatile reagent for chlorination, dehydration, and P-N linkage. Angewandte Chemie, Int. Ed. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Ege, S.N.; Wolovsky, R.; Gensler, W.J. Synthesis of methyl 5,8,11,14-eicosatetraenoate (methyl arachidonate). J. Am. Chem. Soc. 1961, 83, 3080–3085. [Google Scholar] [CrossRef]
Figure 1.
The structure of MELs 1 - 6.
Scheme 1.
Retrosynthetic analysis for the synthesis of MEL 5.
Scheme 2.
Synthesis of the MEL 5 starting from the head piece 7. Reagents and conditions: Reagents and conditions: (a) Mg, CuBr, THF, -10 °C (92%); (b) KOH, MeI, DMF, r.t. (79%); (c) 1-bromo-2,5-octadiyne 11, CuI, NaI, Cs2CO3, DMF, r.t. (91%); (d) H2, Lindlar cat., quinoline, toluene, r.t. (40%); (d) Amberlyst-15®, 96% EtOH, reflux (89%).
Scheme 2.
Synthesis of the MEL 5 starting from the head piece 7. Reagents and conditions: Reagents and conditions: (a) Mg, CuBr, THF, -10 °C (92%); (b) KOH, MeI, DMF, r.t. (79%); (c) 1-bromo-2,5-octadiyne 11, CuI, NaI, Cs2CO3, DMF, r.t. (91%); (d) H2, Lindlar cat., quinoline, toluene, r.t. (40%); (d) Amberlyst-15®, 96% EtOH, reflux (89%).
Scheme 3.
Retrosynthetic analysis for the synthesis of MEL 6.
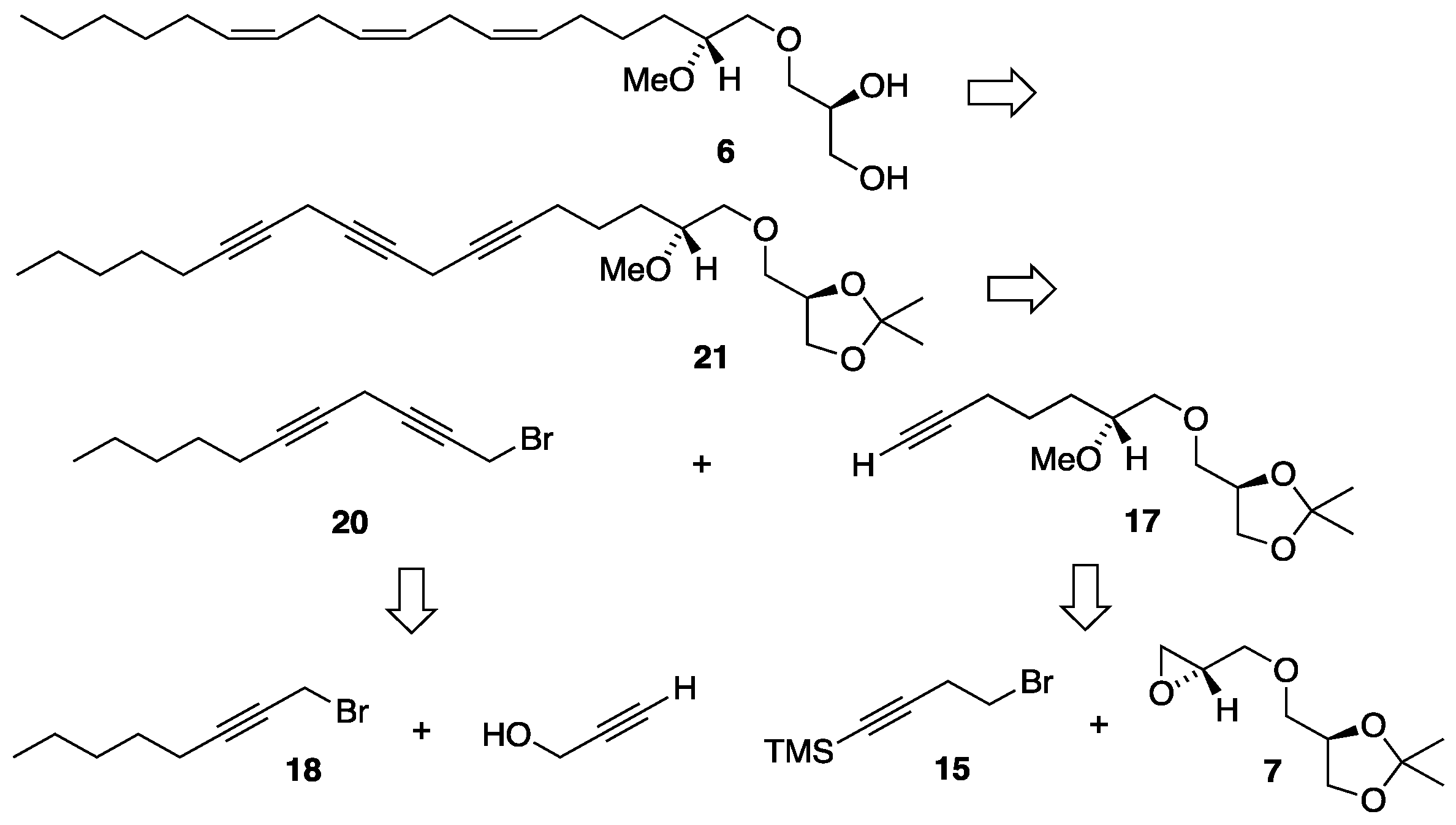
Scheme 4.
Synthesis of the TMS protected monoyne bromide linker 15. Reagents and conditions: (a) n-BuLi (2 eq), TMSCl (2 eq), THF, -78 °C, then 2M HCl (74%); (b) PPh3, CBr4, CH2Cl2, 0 °C (96%).
Scheme 4.
Synthesis of the TMS protected monoyne bromide linker 15. Reagents and conditions: (a) n-BuLi (2 eq), TMSCl (2 eq), THF, -78 °C, then 2M HCl (74%); (b) PPh3, CBr4, CH2Cl2, 0 °C (96%).
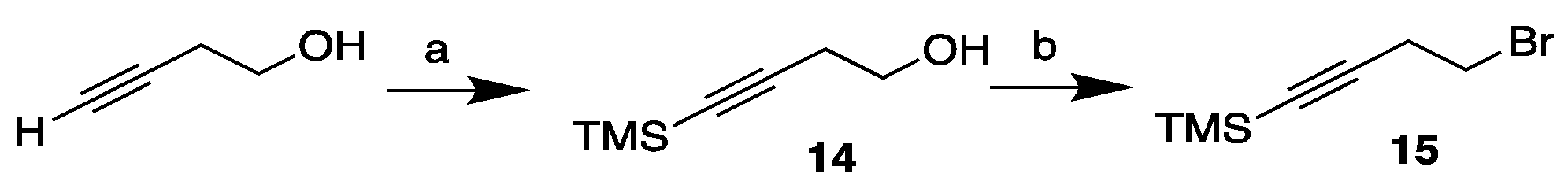
Scheme 5.
Synthesis of the diyne bromide tail fragment 20. Reagents and conditions: (a) PPh3, CBr4, CH2Cl2, 0 °C (100%); (b) Propargyl alcohol, CuI, NaI, Cs2CO3, DMF, r.t. (74%); (c) PPh3, CBr4, CH2Cl2, 0 °C (89%).
Scheme 5.
Synthesis of the diyne bromide tail fragment 20. Reagents and conditions: (a) PPh3, CBr4, CH2Cl2, 0 °C (100%); (b) Propargyl alcohol, CuI, NaI, Cs2CO3, DMF, r.t. (74%); (c) PPh3, CBr4, CH2Cl2, 0 °C (89%).
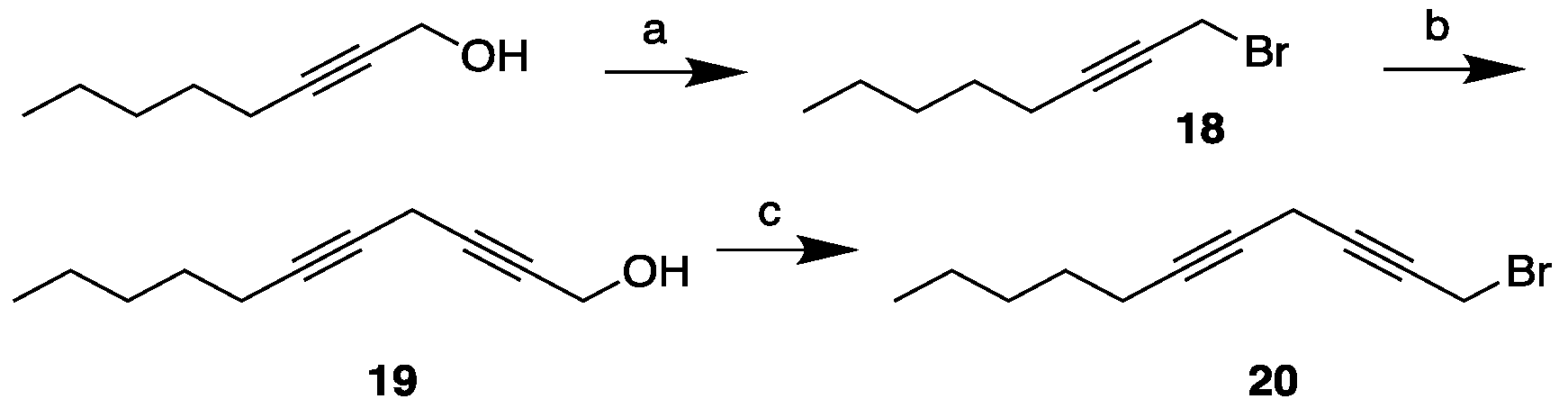
Scheme 6.
Synthesis of the MEL 6 starting from the head piece 7. Reagents and conditions: Reagents and conditions: (a) Mg, CuBr, THF, -10 °C (76%); (b) KOH, MeI, DMF, r.t. (83%); (c) CuI, NaI, Cs2CO3, DMF, r.t. (94%); (d) H2, Lindlar cat., quinoline, toluene, r.t. (33%); (d) Amberlyst-15®, 96% EtOH, reflux (100%).
Scheme 6.
Synthesis of the MEL 6 starting from the head piece 7. Reagents and conditions: Reagents and conditions: (a) Mg, CuBr, THF, -10 °C (76%); (b) KOH, MeI, DMF, r.t. (83%); (c) CuI, NaI, Cs2CO3, DMF, r.t. (94%); (d) H2, Lindlar cat., quinoline, toluene, r.t. (33%); (d) Amberlyst-15®, 96% EtOH, reflux (100%).
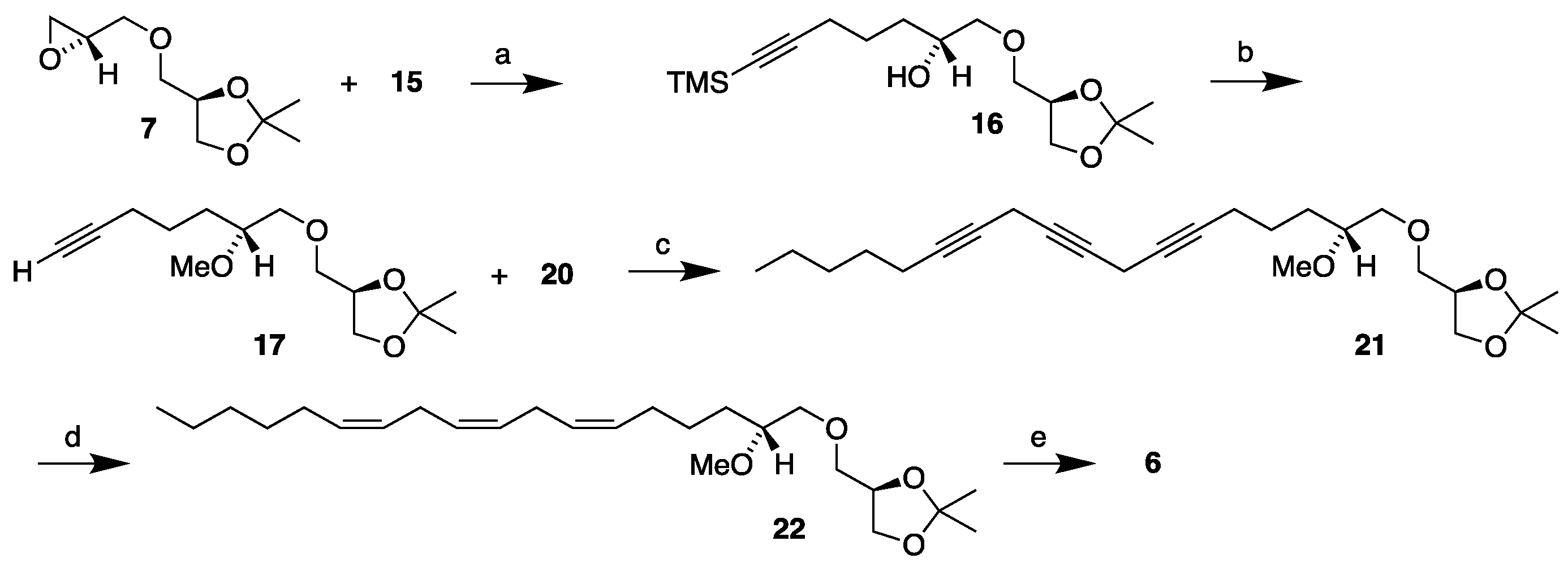

Table 1.
Comparison of optical activity data for unsaturated MEL derivatives.
| MEL derivative | Location of first double bond | [a]20D | Conditions* |
|---|---|---|---|
| C16:1 n-12 (1) | D4' | -4.9 | c, 1.42, ethanol [13] |
| C18:1 n-14 | D4' | -4.2 | c, 2.94, ethanol [12] |
| C18:3 n-8 (4) | D4' | -4.7 | c, 1.7, ethanol [15] |
| C22:6 n-3 (3) | D4' | -3.7 | c, 0.13, ethanol [14] |
| C18:1 n-9 | D9' | +3.7 | c, 2.36, ethanol [12] |
| C18:3 n-3 (5) | D9' | +4.3 | c, 1.1, ethanol* |
| C18:3 n-6 (6) | D6' | +3.3 | c, 1.5, ethanol* |
* Current work.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



