
Submitted:
13 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
The T cell composition of the tumor microenvironment (TME) of esophageal adenocarcinoma (EAC) in comparison to the T cell composition of the TME of Barrett esophagus (BE) and in the context of clinicopathologic features and patient survival is unknown. To help fill these knowledge gaps, we used a multiplex immunofluorescence platform to study adaptive T cells and T cell subtypes based on costimulatory/inhibitory markers in EAC (n=54) and matched BE (n=21) samples from 54 patients with EAC and correlated the T cell phenotype distribution with clinicopathologic features and disease-free survival. We observed significant enrichment of cytotoxic T cells (CTCs), memory T cells, effector memory T cells, memory T regulatory cells (MTregs), and T cells expressing costimulatory/inhibitory markers LAG3, TIM3, and ICOS in EAC compared to BE, in the stroma. In EAC, increased TIM3 expression on T cells significantly correlated with increased CTCs but not T regulatory cells (Tregs). In EAC, higher densities of Tregs and MTregs correlated with pT1 disease, low pathologic stage, and absence of lymphovascular invasion. In EAC, patients with higher density of TIM3-expressing T cells had shorter disease-free survival by univariate and multivariate analyses. Our results identify T cell subtypes that are relevant to EAC biology and patient outcome. Furthermore, our results identify TIM3 as a potential target for optimization of immunomodulatory therapy in EAC.
Keywords:
Esophagus
; adenocarcinoma
; Barrett esophagus
; multiplex immunofluorescence
; T regulatory cells
; TIM3
Introduction
Most patients with esophageal adenocarcinoma (EAC) present with advanced disease and have very poor survival outcomes; patients with locoregional disease have a 5-year survival rate of 22%, and those with distant metastasis have a median survival duration of less than 20 months [1,2].
The role of immune checkpoint inhibitors in improving survival in patients with EAC and gastroesophageal junction adenocarcinoma is evolving. A recent randomized phase 3 trial (CheckMate 577) demonstrated significant benefit of adjuvant nivolumab in improving survival of patients with surgically resected stage II-III EAC regardless of tumor PD-L1 expression . In addition, the combination of nivolumab and chemotherapy as first-line therapy resulted in better disease-free survival than chemotherapy alone in patients with unresectable gastric adenocarcinoma, gastroesophageal junction adenocarcinoma, and EAC whose tumors had a PD-L1 combined positive score higher than 1 [3].
Despite progress in understanding of immune checkpoint inhibitor–based therapy and use of PD-L1 expression as a predictive marker in EAC, there is limited information regarding T cell phenotype distribution and its biologic and prognostic relevance in development and progression of EAC. Prior studies have demonstrated differences in T-helper cell subtypes between Barrett esophagus (BE) and EAC and have helped to elucidate the roles of critical immune mediators [e.g., interleukin (IL)-2, IL-4, IL-6, IL-8, tumor necrosis factor-alpha, and interferon-gamma] in the development of BE and progression of BE to EAC [4,5,6,7,8,9]. Higher T cell infiltration, higher CD3:CD163 ratio, and Th2 genotypes were associated with better response of EAC to preoperative chemoradiation [10]. However, achieving success with immune checkpoint inhibitors in the treatment and prevention of EAC will require an improved understanding of the T cell repertoire in the microenvironment of treatment naïve EAC and BE.
In this study, we used multiplex immunophenotyping analysis of EAC and matched BE specimens to compare the T cell composition of the tumor microenvironment in EAC and BE and to study the relationship between the T cell composition of the EAC tumor microenvironment and clinicopathologic features and patient survival. We identified characteristics of T cell distribution unique to EAC. We also found that TIM3 is a prognostic marker in EAC, and that higher density of T regulatory cells (Tregs) signifies favorable tumor biology in EAC.
Material and Methods
The retrospective study was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center with a waiver of informed consent (protocol LAB-04-0979).
Patient Population and Samples
The institutional databases were searched to identify patients with EAC who underwent esophago-gastrectomy without preoperative tumor-targeted therapy from 2000 through 2017 and tumor samples were available for the study. Pretreatment clinical stage was determined by computed tomography and/or positron emission tomography, esophagogastroduodenoscopy, and endoscopic ultrasonography. Endoscopic findings were reviewed to determine the presence of BE; the segment length of BE (long, short, or ultrashort); the epicenter of the tumor; the presence and extent of invasion of the tumor into the stomach beyond the gastroesophageal junction; and the subtype of GE junction adenocarcinoma per the Siewert classification [11]. Patients with emergency surgery, death within 3 months of surgery, limited amount of tumor in the specimen/tissue block, tumor-associated ulceration, extensively (>90%) necrotic tumor or insufficient tumor cellularity for construction of tissue microarrays (TMAs) were excluded.
TMAs of EAC were prepared by obtaining three 1-mm cores composed exclusively of tumor cells and stroma without ulceration or admixed normal tissue. The tissue cores were designed to include both superficial and deep invasive components of the tumor. For each patient with BE, a separate TMA was built by obtaining three 1-mm cores of non-dysplastic BE without ulceration or admixed normal squamous mucosa from surgical resection specimens.
Multiplex Immunofluorescence (mIF) Phenotyping
mIF staining was performed using techniques developed and validated previously [12,13]. Briefly, 4-micrometer-thick formalin-fixed, paraffin-embedded TMA sections were stained using an autostainer (BOND-RX; Leica Biosystems). As previously described [14] all the antibodies were linked with a fluorophore from the Opal 7-color fluorescent immunohistochemistry kit (#NEL797001KT; Akoya Biosciences, Marlborough, MA), including DAPI (4’,6 diamidino-2-phenylindole) and Opal Polaris fluorophores 480, 520, 540, 570, 620, 650, and 690. The slides were incubated with the antibody for 30 to 60 min at room temperature, depending on the antibody, at fixed dilutions (Supplementary Table 1). The slides were washed and then incubated for 10 min at room temperature with polymer horseradish peroxidase conjugated to anti-mouse/rabbit secondary antibody (Akoya Biosciences). After successive washes with BOND wash solution (BOND-RX; Leica Biosystems), the slides were incubated for 10 min with each Opal fluorophore tyramide to detect the different antibodies. After additional washes with BOND wash solution, the slides were counterstained with DAPI. Staining for the different markers of each panel was performed consecutively using the same steps, and detection of each marker was completed before application of the next antibody. Uniplex immunofluorescence staining was performed simultaneously for each fluorophore to make a spectral library in human tonsil formalin-fixed, paraffin-embedded tissues, and the tonsil specimens were used as controls in the multispectral analysis.
The stained slides were scanned using the Vectra Polaris 3.0.3 (Akoya Biosciences), a multispectral imaging system, at 10x magnification (1.0 μm/pixel) through the full emission spectrum, from 440 to 720 nm, to extract fluorescence intensity information, with the tonsil positive controls run in parallel with the samples. The Immunofluorescence markers used were grouped into two antibody panels: panel 1 for conventional adaptive T cells with activation and regulation markers of T cells, including CD3, CD8, CD45RO, Granzyme B, FOXP3, and pan cytokeratin; and panel 2 for T cell costimulatory and inhibitory immune checkpoint markers, including CD3, TIM3, LAG3, VISTA, ICOS, OX40, and pan cytokeratin. Each tissue core with EAC and BE regions was assessed as one region of interest by a pathologist using the phenochart 1.0.9 whole slide viewer (Akoya Biosciences) [13,15].
inForm 2.4.8 image analysis software (Akoya Biosciences) was used to analyze the scanned multispectral component images. Using the tissue segmentation tool, each region of interest was divided into the intraepithelial compartment, defined as glandular structures and nests of tumor cells in EAC and intestinal-type glandular epithelium in BE; and the stromal compartment, defined as the connective tissue between epithelial nests and less than 1 mm from the invasive border of the tumor or deepest focus of BE epithelium. The combined analysis of stroma and epithelium was designated as the total compartment. The inForm algorithm was optimized by a pathologist for tissue segmentation and cell segmentation. For tissue segmentation, areas of intraepithelial and stromal compartment were identified and annotated using pan cytokeratin marker (positive in epithelial and tumor cells). For the cell segmentation, optimization of the algorithm was performed for nuclear and cytoplasmic components. The nuclear segmentation was performed using DAPI counterstain and using minimum nuclear size and nucleus splitting sensitivity as the criteria for optimization. Subsequently, the cytoplasmic segmentation was optimized by adjusting the values for cytoplasmic thickness defined as part of the cell from nuclear membrane to the cell membrane.
Once cell segmentation was optimized, cell phenotyping was performed using inForm’s phenotyping feature, which automatically classifies cell phenotypes to differentiate cell types across a tissue section (Figure 1).
Phenotypes were defined based on the markers in panels 1 and 2, and cells not expressing any of the markers were classified as “others”. Training for phenotype identification involved selecting initial set of approximately five example cells for each marker, followed by iterative addition of additional cells to refine the classifier. When the training was complete, the algorithm was applied to a batch of similarly stained sections. Due to staining variability across tissue samples, the phenotyping results were verified for each case, and manual phenotype edits were made, if necessary, for individual cases [16]. The phenotypes were defined on the basis of co-localization of antibodies (Supplementary Table 2). To determine the co-expression of markers used to define cell phenotypes, the x and y coordinates of the individual markers of interest within each cell were merged using the phenoptr script from R Studio (Akoya Biosciences). The densities of T cell phenotypes were calculated as number of cells per unit area (n/mm2) for all tumor compartments: intraepithelial, stromal, and total.
Statistical Analysis
Statistical analyses were performed using SPSS 23 software (IBM Corp, Armonk, NY). The differences in nonparametric continuous variables were assessed using the Mann-Whitney U test. Overall survival curves were obtained using the Kaplan-Meier method and compared using the log-rank test. Univariable Cox regression analysis for disease-free survival was then performed, and covariates with p less than 0.25 were entered into a multivariable analysis. Stepwise Wald backward elimination was used in multivariable Cox regression analysis to identify variables predictive of the outcome of interest. Statistical significance was defined as p less than 0.05. Spearman’s correlation coefficient was calculated using GraphPad Prism Version 9.0.0 software to calculate monotonic association for quantitative changes between two T cell phenotypes.
Results
Patient and tumor characteristics
A total of fifty-four patients with EAC met the inclusion criteria for the study, and twenty-one of these patients also had BE. Among the twenty-one patients with BE, the area of BE analyzed with mIF was adjacent to EAC in twenty patients and distant from focus of EAC in one patient. Table 1 shows the demographic and clinicopathologic characteristics of the patients in the study.
The characteristics of our study population with respect to male/female ratio, race, and age were similar to characteristics previously reported for EAC [14,17]. The proportions of patients who were smokers and who were obese were higher than previously reported [17]. More patients had pT1 or pN0 disease than had pT2 or higher or node-positive disease. More than half of the patients had poorly differentiated tumors, and more than half had lymphovascular invasion (LVI)
T cell phenotypes in EAC
For all the T cell phenotypes, T cell infiltration in EAC was significantly higher in the stroma than in the intraepithelial compartment (Table 2).
In both the intraepithelial and stromal compartments, memory T cells were the most abundant conventional adaptive T cells, followed by cytotoxic T cells (CTCs) and T regulatory cells (Tregs). The effector memory CTCs and activated CTCs were the least abundant conventional adaptive T cells and constituted a very small proportion of CTCs, and the median number of activated CTCs per square millimeter was 0 or 1, indicating poor activated CTC response. Among the tested costimulatory and inhibitory T cell markers, the most abundantly expressed on T cells was LAG3, a negative regulatory and T cell exhaustion marker, followed by ICOS, a costimulatory marker, and TIM3, a T cell exhaustion marker. There was very low expression of VISTA, a negative regulator of T cell function and absent to minimal expression of OX40, a T cell activation marker on T cells.
Results of analysis of Spearman correlation between conventional adaptive T cell phenotype and costimulatory/inhibitory phenotype are summarized in Figure 2. Moderate correlation was observed between CTCs and TIM3-expressing (TIM3+) T cells in the intraepithelial and stromal compartments and in the total compartment (Figure 2A-C). Moderate correlation was also observed between CTCs and ICOS+ T cells in intraepithelial (r=0.33, p=0.015) and stromal (r=0.43, p=0.0012) compartments and in the total compartment (r=0.50, p=0.0001). Moderate to strong correlation was observed between CTCs and LAG3+ T cells in intraepithelial (r=0.57, p<0.0001) and stromal (r=0.72, p<0.0001) compartments and in the total compartment (r=0.84, p<0.0001). The patterns of correlation between memory T cells and TIM3+, ICOS+, and LAG+ T cells were identical to the patterns of correlation between CTCs and TIM3+, ICOS+, and LAG+ T cells. Moderate correlation was observed between Tregs and ICOS+ T cells in intraepithelial (r=0.36, p=0.07) and stromal (r=0.50, p=0.0001) compartments and the total compartment (r=0.57, p<0.0001). Moderate correlation was observed between Tregs and LAG3+ T cells in the stromal compartment (r=0.49, p=0.0002) and in the total compartment (r=0.50, p=0.0001) but not in the intraepithelial compartment (r=0.24, p=0.08). In contrast, no significant correlation was observed between Tregs and TIM3+ T cells in intraepithelial or stromal compartments or in the total compartment ((Figure 2D–F)
These findings indicated that density of TIM3+ T cells was correlated with density of CTCs but not correlated with density of Tregs.
Comparison of T cell phenotypes between BE and EAC
Table 3 shows the T cell phenotypes in matched BE and EAC samples from twenty-one patients.
For most phenotypes of conventional adaptive T cells, density in the stromal compartment was significantly higher for EAC than for BE (Figure 3). Interestingly, activated CTCs, which are cytotoxic against tumor cells and dysplastic cells did not differ significantly between BE and EAC in stroma or total (Table 3). TIM3+, LAG3+, and ICOS+ T cells were more abundant in EAC, primarily in the stromal compartment, than in BE (Table 3). As in EAC, VISTA+ T cells and OX40+ T cells were absent to minimal in BE.
Correlation of T cell phenotypes with clinicopathologic characteristics of EAC
There was no significant correlation between the densities of the T cell phenotypes with majority of the clinical characteristics of the patients. Density of ICOS+ T cells in the intraepithelial compartment was higher in tumors from obese patients than in tumors from non-obese patients. Density of TIM3+ T cells in the intraepithelial compartment was lower in tumors from smokers than in tumors from non-smokers.
Higher T cell density in the total compartment was significantly associated with lack of LVI (Figure 4A, higher T cell density in the intraepithelial compartment was associated with lack of LVI (Figure 4B) and with pN0 versus pN1-3 disease (Figure 4C); and higher T cell density in the stromal compartment was associated with lack of LVI (Figure 4D) and with pT1 versus pT2-4 disease (Figure 4E).
Higher Treg density in the total compartment, higher Treg density in the intraepithelial compartment, and higher Treg density in the stromal compartment were associated with pT1 versus pT2-4 disease (Figure 5A,C,E, respectively) and with lack of LVI (Figure 5B,D,F, respectively).
Treg density in intraepithelial, stromal, and total compartments was lower in stage II-IV tumors than in stage I tumors [median (range), 14 (0-151) vs. 27 (0-165) n/mm2, p=0.002, 72 (0-237) vs. 214 (2-891) n/mm2, p=0.001, and 47 (0-171) vs. 87 (2-450) n/mm2, p=0.016, respectively]. Memory Treg density in intraepithelial, stromal, and total compartments was also lower in pT2-pT4 tumors than in pT1 tumors [median (range), 5 (0-61) vs. 12 (0-89) n/mm2, p=0.016, 39 (0-231) vs. 124 (0-668) n/mm2, p=0.003, and 22 (0-63) vs. 51 (2-367) n/mm2, p=0.001, respectively]. Memory Treg density in stromal and total compartments was lower in stage II-III tumors than in stage I tumors [median (range), 57 (0-416) vs. 127 (0-668) n/mm2, p=0.005, 27 (0-123) vs. 54 (2-365) n/mm2, p=0.04] and in tumors without LVI than in tumors with LVI [median (range), 63 (0-416) vs. 142 (2-668) n/mm2, p=0.005, 25 (0-86) vs. 66 (2-367) n/mm2, p=0.02]. TIM3+ T cell density in intraepithelial and total compartments was higher in pT2-4 tumors than in pT1 tumors [median (range), 8 (0-42) vs. 4 (0-42), p=0.019, 17 (0-82) vs. 8 (0-49), p=0.056].
Correlation of T cell phenotypes and clinicopathologic features with disease-free survival in EAC.
Figure 6 and Table 4 show the results of univariate and multivariate analyses of the variables that were significant predictors of disease-free survival.
In univariate analysis, higher than median TIM3+ T cell density in the total compartment was associated with shorter disease-free survival than lower TIM3+ T cell density (mean, 93 months vs. 154 months, p=0.063). Higher than median TIM3+ T cell density in the intraepithelial compartment was associated with significantly shorter disease-free survival than lower TIM3+ T cell density (mean, 92 months vs. 156 months, p=0.049) (Figure 6B). Higher than median TIM3+ T cell density in the stromal compartment was associated with significantly shorter disease-free survival than lower TIM3+ T cell density (mean, 94 months vs. 163 months, p=0.032) (Figure 6A–E). In the multivariate analysis higher than median TIM3+T cell densities as compared to lower than median TIM3+T cell densities in total (p=0.01), intraepithelial (p=0.013) and stromal (p=0.042) were associated with shorter disease-free survival. 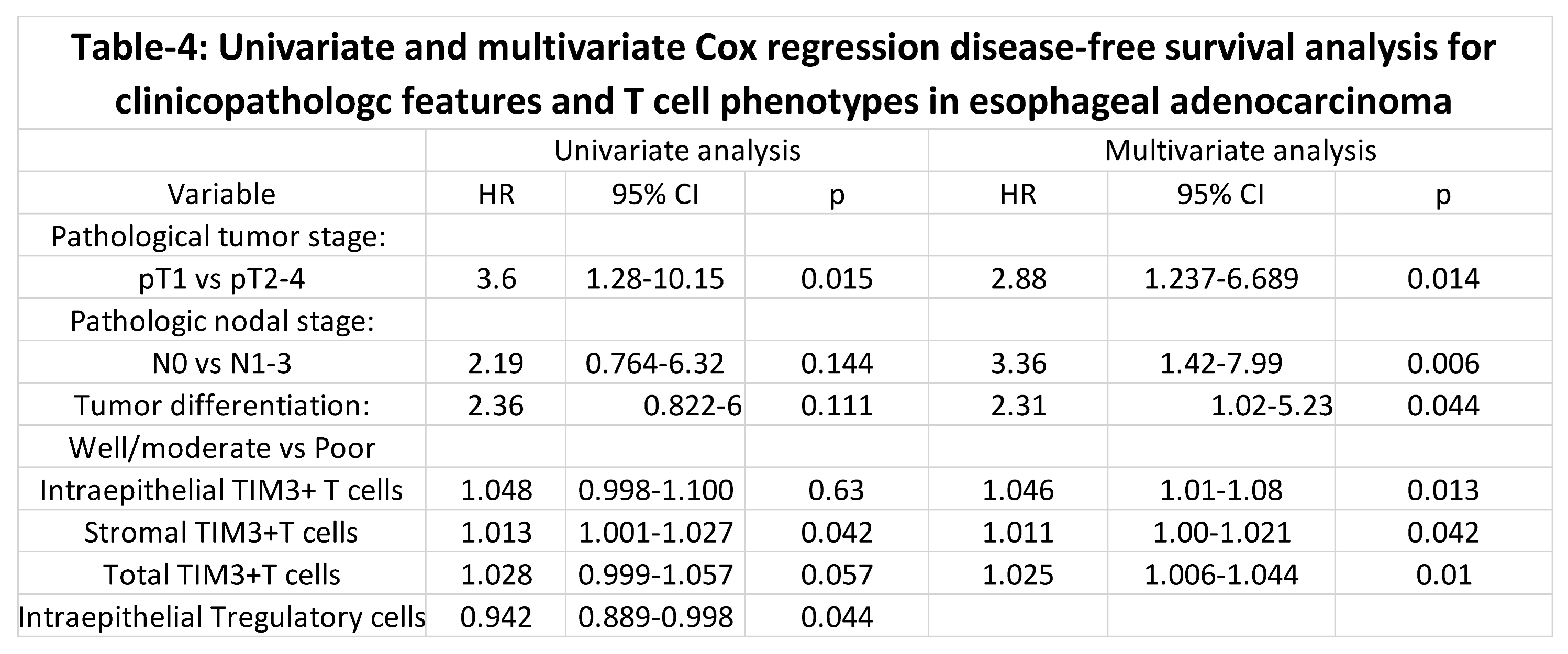
Other significant variables of shorter disease-free survival in the multivariate analysis included regional lymph node metastasis, higher pathologic tumor stage and poorly differentiated histology. Patients with regional lymph node metastasis had shorter disease-free survival than patients without regional lymph node metastasis (mean, 64 months vs. 89 months, p=0.006). Patients with tumor invading into or beyond the muscularis propria (pT2-pT4) had shorter disease-free survival than patients with tumor invasion limited to the mucosa or submucosa (mean, 40 months vs. 104 months, p=0.014). Patients with poorly differentiated tumors had shorter disease-free survival than patients with well or moderately differentiated tumors (mean, 64 months vs. 94 months, p=0.044). Higher Treg density in the intraepithelial compartment was associated with longer disease-free survival than lower Treg density (mean, 135 months vs. 99 months, p=0.044) in the univariate analysis but not in the multivariate analysis.
Discussion
In this quantitative analysis based on multiplex immunophenotyping of several T cell phenotypes in EAC and matched BE, we observed higher T cell response in EAC than in BE. The more pronounced increase in T cells in stroma compared to epithelium in EAC than in BE raises the question of how much of the T cell response is directly targeting tumor cells. Higher density of stromal and/or intraepithelial CD3+ T cells correlated with lower T category, lower N category, and absence of LVI, suggesting higher T cell response is associated with better tumor biology. Among the tested adaptive T cell subtypes, higher FOXP3+ Tregs and memory regulatory T cell responses were associated with pathologic features of better tumor biology and with a trend toward improved survival. Other than correlation of higher intraepithelial CTC density with absence of LVI, neither CTC density nor activated CTC density correlated with pathologic parameters or patient outcome. These findings, along with the very low to absent activated CTC response, suggest that functionally effective T cell response in EAC is biased towards FOXP3+ Tregs and not towards CTCs.
Using immunohistochemistry analysis, Svensson et al demonstrated that higher T cell and FOXP3+ T cell responses were associated with better survival outcomes in patients with esophageal cancer treated with neoadjuvant chemoradiation followed by surgery [18]. The findings from our current study, taken together with the findings of Svensson et al indicate the need to further explore the role of Tregs in the tumor microenvironment of EAC. Different FoxP3+ Treg subpopulations are associated with different outcomes in patients with colon cancer: high-FOXP3-expressing CD4+CD45RA- Tregs are associated with poor outcome, while low-FOXP3-expressing CD4+CD45RA- Tregs are associated with better outcome [19]. A proposed subtyping of Foxp3-positive T cells [19,20] into Foxp3lowCD45RA+CD25low resting Tregs (fraction 1), Foxp3highCD45RA-CD25high effective Tregs (fraction 2), and Foxp3lowCD45RA-CD25low non-Tregs (fraction 3) in EAC may provide further insight into the functional role of Tregs in EAC.
Association of higher intraepithelial TIM3+ T cell density with higher T category and significant correlation of higher TIM3+ T cell density with shorter disease-free survival in univariate and multivariate analyses indicate a role of T cell exhaustion marker TIM3 in tumor behavior and patient outcome. Our findings of stronger correlations between CTCs and memory T cells and TIM3 expression than between Tregs and TIM3 expression suggest higher likelihood of T cell exhaustion through TIM3+CTCs than through FOXP3+ Tregs in EAC. TIM3+ T cell subsets have been shown to be of prognostic significance in other epithelial tumors, including esophageal squamous carcinoma and gastric and ovarian serous carcinomas (22-25). Humphries et al (23), using single-plex immunohistochemistry, demonstrated an association of high density of TIM3+ T cells with poor overall survival and poor response to 5-fluorouracil-based adjuvant chemotherapy in gastric cancer. The range and median values of TIM3+ T cell density were not significantly different between our study and the study of Humphries et al Given the similarity in mucin profile and other intracellular proteins between esophageal and gastric adenocarcinoma, findings in these studies strongly support further exploration of TIM3 as a target of immune modulation in preclinical and clinical settings.
In our current study, LAG3 expression was identified in more than two-thirds of T cells in EAC and was the dominant marker among all tested costimulatory and inhibitory markers. Prior gene expression–based analysis identified higher T cell TIM3 and LAG3 expression than PD-1 or PD-L1 expression in EAC [21]. De Klerk et al [22], in a phase 2 trial of pembrolizumab in esophageal carcinoma, observed lower frequency of expression of LAG3 in EAC and squamous carcinoma than we observed in our present study. A likely explanation for this difference is that tumor samples in the de Klerk et al study were EAC samples obtained after treatment whereas samples in our present study were from patients who did not receive any preoperative tumor-targeted therapy. In a study of a large cohort of patients with EAC by Gebauer et al [23], LAG3 expression by single-plex and limited multiplex immunophenotyping correlated with overall survival. High frequency of LAG3 expression in our study and prognostic relevance of T cell LAG3 expression shown by Gebauer et al support exploration of LAG3 as a potential target of immune modulation in EAC.
Humphries et al found significant presence of CD45RO-positive T cells in EAC and found that higher ICOS+CD45RO+ T cells were associated with better patient survival [24]. Conroy et al studied T cell subtypes by single-plex immunohistochemistry and found that higher level of CD8+CD45RO+ T cells (memory T cells) was not significantly correlated with patient outcome [14]. The difference in findings between our study and other studies can be partly explained by differences in the methodology used to analyze T cell subtypes (multiplex immunofluorescence vs. single-plex immunohistochemistry).
In summary, ours is the first study to comprehensively analyze adaptive T cells and candidate costimulatory and inhibitory T cell markers in EAC and BE. Our results contribute to identifying candidate T cell subtypes with relevance to tumor biology and patient outcome in EAC and support further studies to better understand roles of Tregs and TIM3+ T cells in EAC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Isha Khanduri and Dipen Maru; Data curation, Isha Khanduri, Renganayaki Pandurengan, Arlene M. Correa, Riham Katkhuda, Anuj Verma, Alicia Mejia, Zhimin Tong, Akash Mitra, Luisa Solis Soto, Wayne Hofstetter and Edwin Parra; Formal analysis, Isha Khanduri, Renganayaki Pandurengan and Arlene M. Correa; Funding acquisition, Dipen Maru; Investigation, Isha Khanduri; Methodology, Luisa Solis Soto, Edwin Parra and Dipen Maru; Project administration, Isha Khanduri and Dipen Maru; Resources, Riham Katkhuda, Anuj Verma, Alicia Mejia, Zhimin Tong, Luisa Solis Soto, Ignacio Wistuba, Wayne Hofstetter and Edwin Parra; Supervision, Dipen Maru; Writing – original draft, Isha Khanduri and Dipen Maru; Writing – review & editing, Isha Khanduri, Akshaya Jadhav, Edwin Parra and Dipen Maru.
Funding
This work was funded by the Charles B. Swank Endowed Chair funds given to Dr. Dipen Maru. Part of this research was performed at MD Anderson’s core facilities supported in part by the NIH through Cancer Center Support Grant CA016672.
Informed Consent Statement
The study was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center with a waiver of informed consent (protocol no: LAB-04-0979).
Data Availability Statement
Not applicable
Acknowledgements
We thank Kim-Anh-Vu in MD Anderson’s Department of Anatomic Pathology for helping with the figures. We also thank Ms. Stephanie Deming, Research Medical Library, MD Anderson Cancer Center, for copyediting the manuscript.
Conflicts of Interest
The authors report no conflict of interest.
References
- Rosemurgy, A.; Wilfong, C.; Craigg, D.; Co, F.; Sucandy, I.; Ross, S., The Evolving Landscape of Esophageal Cancer: A Four-Decade Analysis. Am Surg 2019, 85 (9), 944-948. [CrossRef]
- Kelly, R. J.; Ajani, J. A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lievre, A.; Uronis, H.; Elimova, E.; Grootscholten, C.; Geboes, K.; Zafar, S.; Snow, S.; Ko, A. H.; Feeney, K.; Schenker, M.; Kocon, P.; Zhang, J.; Zhu, L.; Lei, M.; Singh, P.; Kondo, K.; Cleary, J. M.; Moehler, M.; CheckMate, I., Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N Engl J Med 2021, 384 (13), 1191-1203. [CrossRef]
- Janjigian, Y. Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; Liu, T.; Schenker, M.; Yanez, P.; Tehfe, M.; Kowalyszyn, R.; Karamouzis, M. V.; Bruges, R.; Zander, T.; Pazo-Cid, R.; Hitre, E.; Feeney, K.; Cleary, J. M.; Poulart, V.; Cullen, D.; Lei, M.; Xiao, H.; Kondo, K.; Li, M.; Ajani, J. A., First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 2021, 398 (10294), 27-40. [CrossRef]
- Smith, K. J.; O'Brien, S. M.; Green, A. C.; Webb, P. M.; Whiteman, D. C.; Study of Digestive, H., Current and past smoking significantly increase risk for Barrett's esophagus. Clin Gastroenterol Hepatol 2009, 7 (8), 840-8. [CrossRef]
- Kavanagh, M. E.; O'Sullivan, K. E.; O'Hanlon, C.; O'Sullivan, J. N.; Lysaght, J.; Reynolds, J. V., The esophagitis to adenocarcinoma sequence; the role of inflammation. Cancer Lett 2014, 345 (2), 182-9. [CrossRef]
- Kavanagh, M. E.; Conroy, M. J.; Clarke, N. E.; Gilmartin, N. T.; O'Sullivan, K. E.; Feighery, R.; MacCarthy, F.; O'Toole, D.; Ravi, N.; Reynolds, J. V.; O'Sullivan, J.; Lysaght, J., Impact of the inflammatory microenvironment on T-cell phenotype in the progression from reflux oesophagitis to Barrett oesophagus and oesophageal adenocarcinoma. Cancer Lett 2016, 370 (1), 117-24. [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J. A.; Marache, F.; Good, P.; Lee, M. D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; Mahmood, U.; Figueiredo, J. L.; Kitajewski, J.; Shawber, C.; Lightdale, C. J.; Rustgi, A. K.; Wang, T. C., Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21 (1), 36-51. [CrossRef]
- Buas, M. F.; He, Q.; Johnson, L. G.; Onstad, L.; Levine, D. M.; Thrift, A. P.; Gharahkhani, P.; Palles, C.; Lagergren, J.; Fitzgerald, R. C.; Ye, W.; Caldas, C.; Bird, N. C.; Shaheen, N. J.; Bernstein, L.; Gammon, M. D.; Wu, A. H.; Hardie, L. J.; Pharoah, P. D.; Liu, G.; Iyer, P.; Corley, D. A.; Risch, H. A.; Chow, W. H.; Prenen, H.; Chegwidden, L.; Love, S.; Attwood, S.; Moayyedi, P.; MacDonald, D.; Harrison, R.; Watson, P.; Barr, H.; deCaestecker, J.; Tomlinson, I.; Jankowski, J.; Whiteman, D. C.; MacGregor, S.; Vaughan, T. L.; Madeleine, M. M., Germline variation in inflammation-related pathways and risk of Barrett's oesophagus and oesophageal adenocarcinoma. Gut 2017, 66 (10), 1739-1747. [CrossRef]
- Souza, R. F.; Huo, X.; Mittal, V.; Schuler, C. M.; Carmack, S. W.; Zhang, H. Y.; Zhang, X.; Yu, C.; Hormi-Carver, K.; Genta, R. M.; Spechler, S. J., Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 2009, 137 (5), 1776-84. [CrossRef]
- Goedegebuure, R. S. A.; Harrasser, M.; de Klerk, L. K.; van Schooten, T. S.; van Grieken, N. C. T.; Eken, M.; Grifhorst, M. S.; Pocorni, N.; Jordanova, E. S.; van Berge Henegouwen, M. I.; Pouw, R. E.; Verheul, H. M. W.; van der Vliet, J. J.; van Laarhoven, H. W. M.; Thijssen, V.; Bass, A. J.; De Gruijl, T. D.; Derks, S., Pre-treatment tumor-infiltrating T cells influence response to neoadjuvant chemoradiotherapy in esophageal adenocarcinoma. Oncoimmunology 2021, 10 (1), 1954807. [CrossRef]
- Siewert, J. R.; Stein, H. J., Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998, 85 (11), 1457-9. [CrossRef]
- Parra, E. R.; Uraoka, N.; Jiang, M.; Cook, P.; Gibbons, D.; Forget, M. A.; Bernatchez, C.; Haymaker, C.; Wistuba, II; Rodriguez-Canales, J., Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci Rep 2017, 7 (1), 13380. [CrossRef]
- Parra, E. R.; Ferrufino-Schmidt, M. C.; Tamegnon, A.; Zhang, J.; Solis, L.; Jiang, M.; Ibarguen, H.; Haymaker, C.; Lee, J. J.; Bernatchez, C.; Wistuba, II, Immuno-profiling and cellular spatial analysis using five immune oncology multiplex immunofluorescence panels for paraffin tumor tissue. Sci Rep 2021, 11 (1), 8511. [CrossRef]
- Conroy, M. J.; Kennedy, S. A.; Doyle, S. L.; Hayes, B.; Kavanagh, M.; van der Stok, E. P.; O'Sullivan, K.; Cathcart, M. C.; Reynolds, J. V.; Lysaght, J., A study of the immune infiltrate and patient outcomes in esophageal cancer. Carcinogenesis 2021, 42 (3), 395-404. [CrossRef]
- Parra, E. R.; Francisco-Cruz, A.; Wistuba, II, State-of-the-Art of Profiling Immune Contexture in the Era of Multiplexed Staining and Digital Analysis to Study Paraffin Tumor Tissues. Cancers (Basel) 2019, 11 (2). [CrossRef]
- Mori, H.; Bolen, J.; Schuetter, L.; Massion, P.; Hoyt, C. C.; VandenBerg, S.; Esserman, L.; Borowsky, A. D.; Campbell, M. J. , Characterizing the Tumor Immune Microenvironment with Tyramide-Based Multiplex Immunofluorescence. J Mammary Gland Biol Neoplasia 2020, 25(4), 417–432. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A. P.; Shaheen, N. J.; Gammon, M. D.; Bernstein, L.; Reid, B. J.; Onstad, L.; Risch, H. A.; Liu, G.; Bird, N. C.; Wu, A. H.; Corley, D. A.; Romero, Y.; Chanock, S. J.; Chow, W. H.; Casson, A. G.; Levine, D. M.; Zhang, R.; Ek, W. E.; MacGregor, S.; Ye, W.; Hardie, L. J.; Vaughan, T. L.; Whiteman, D. C., Obesity and risk of esophageal adenocarcinoma and Barrett's esophagus: a Mendelian randomization study. J Natl Cancer Inst 2014, 106 (11). [CrossRef]
- Christina Svensson, M.; Linden, A.; Nygaard, J.; Borg, D.; Hedner, C.; Nodin, B.; Leandersson, K.; Jirstrom, K., T cells, B cells, and PD-L1 expression in esophageal and gastric adenocarcinoma before and after neoadjuvant chemotherapy: relationship with histopathological response and survival. Oncoimmunology 2021, 10 (1), 1921443. [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; Nagase, H.; Nishimura, J.; Yamamoto, H.; Takiguchi, S.; Tanoue, T.; Suda, W.; Morita, H.; Hattori, M.; Honda, K.; Mori, M.; Doki, Y.; Sakaguchi, S., Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016, 22 (6), 679-84. [CrossRef]
- Ohue, Y.; Nishikawa, H., Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci 2019, 110 (7), 2080-2089. [CrossRef]
- Wagener-Ryczek, S.; Schoemmel, M.; Kraemer, M.; Bruns, C.; Schroeder, W.; Zander, T.; Gebauer, F.; Alakus, H.; Merkelbach-Bruse, S.; Buettner, R.; Loeser, H.; Thelen, M.; Schlosser, H. A.; Quaas, A., Immune profile and immunosurveillance in treatment-naive and neoadjuvantly treated esophageal adenocarcinoma. Cancer Immunol Immunother 2020, 69 (4), 523-533. [CrossRef]
- de Klerk, L. K.; Patel, A. K.; Derks, S.; Pectasides, E.; Augustin, J.; Uduman, M.; Raman, N.; Akarca, F. G.; McCleary, N. J.; Cleary, J. M.; Rubinson, D. A.; Clark, J. W.; Fitzpatrick, B.; Brais, L. K.; Cavanaugh, M. E.; Rode, A. J.; Jean, M. G.; Lizotte, P. H.; Nazzaro, M. J.; Severgnini, M.; Zheng, H.; Fuchs, C. S.; Enzinger, P. C.; Bass, A. J., Phase II study of pembrolizumab in refractory esophageal cancer with correlates of response and survival. J Immunother Cancer 2021, 9 (9). [CrossRef]
- Gebauer, F.; Kramer, M.; Bruns, C.; Schlosser, H. A.; Thelen, M.; Lohneis, P.; Schroder, W.; Zander, T.; Alakus, H.; Buettner, R.; Loeser, H.; Quaas, A., Lymphocyte activation gene-3 (LAG3) mRNA and protein expression on tumour infiltrating lymphocytes (TILs) in oesophageal adenocarcinoma. J Cancer Res Clin Oncol 2020, 146 (9), 2319-2327. [CrossRef]
- Humphries, M. P.; Craig, S. G.; Kacprzyk, R.; Fisher, N. C.; Bingham, V.; McQuaid, S.; Murray, G. I.; McManus, D.; Turkington, R. C.; James, J.; Salto-Tellez, M., The adaptive immune and immune checkpoint landscape of neoadjuvant treated esophageal adenocarcinoma using digital pathology quantitation. BMC Cancer 2020, 20 (1), 500. [CrossRef]
Figure 1.
Workflow for multiplex immunofluorescence image analysis. A) Spectrally unmixed composite image of a single esophageal adenocarcinoma TMA core imported into InForm software. B) Tumor tissue segmentation into tumor (red) and stroma (green) using InForm’s trainable tissue segmentation algorithms. C) Individual cell segmentation performed using InForm. D) Cell phenotyping performed based on individual cell markers using InForm’s trainable cell phenotyping algorithm (cytokeratin- cyan, CD3- red, others- blue). E to O) Microphotographs of representative examples of multiplex IF markers in Panel 1 and 2. E and F - Cytokeratin (CK) and CD3 which are common makers for both the panels. G to J – Panel 1 specific markers (CD8, CD45RO, FOXP3 and Granzyme B). K to O - Panel 2 specific markers (TIM3, LAG3, ICOS, VISTA and OX40).
Figure 1.
Workflow for multiplex immunofluorescence image analysis. A) Spectrally unmixed composite image of a single esophageal adenocarcinoma TMA core imported into InForm software. B) Tumor tissue segmentation into tumor (red) and stroma (green) using InForm’s trainable tissue segmentation algorithms. C) Individual cell segmentation performed using InForm. D) Cell phenotyping performed based on individual cell markers using InForm’s trainable cell phenotyping algorithm (cytokeratin- cyan, CD3- red, others- blue). E to O) Microphotographs of representative examples of multiplex IF markers in Panel 1 and 2. E and F - Cytokeratin (CK) and CD3 which are common makers for both the panels. G to J – Panel 1 specific markers (CD8, CD45RO, FOXP3 and Granzyme B). K to O - Panel 2 specific markers (TIM3, LAG3, ICOS, VISTA and OX40).

Figure 2.
Spearman correlation curves of TIM3 expressing T cells (TIM3+TC) with T regulatory cells (Tregs) and cytotoxic T cells (CTC) in the intraepithelial compartment (A and D), stromal compartment (B and E) and the entire EAC (C and F). EAC: Esophageal adenocarcinoma.
Figure 2.
Spearman correlation curves of TIM3 expressing T cells (TIM3+TC) with T regulatory cells (Tregs) and cytotoxic T cells (CTC) in the intraepithelial compartment (A and D), stromal compartment (B and E) and the entire EAC (C and F). EAC: Esophageal adenocarcinoma.
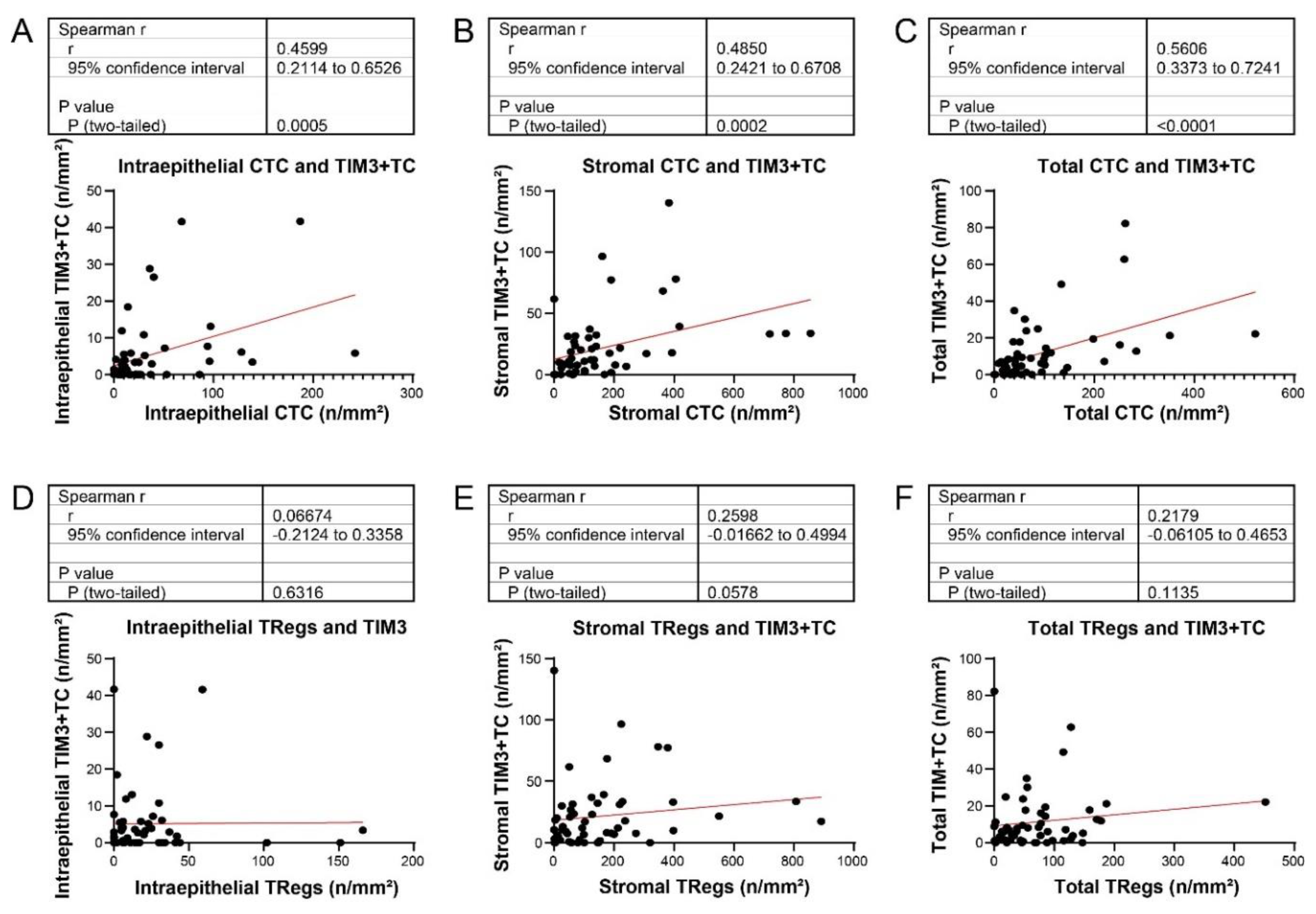
Figure 3.
Comparison of T cell phenotypes in BE and EAC. (A, B) Composite mIF low-power (x200) images showing higher density of immune cells in EAC stroma (A) than in BE stroma (B). (C-N) Cell density was higher in EAC than in BE for CD3+ T cells (C and I), CD3+CD8+ CTCs (D and J), CD3+CD45RO+ memory T cells (E and K), ICOS+ T cells (F and L), LAG3+ T cells (G and M), and TIM3+ T cells (H and N).
Figure 3.
Comparison of T cell phenotypes in BE and EAC. (A, B) Composite mIF low-power (x200) images showing higher density of immune cells in EAC stroma (A) than in BE stroma (B). (C-N) Cell density was higher in EAC than in BE for CD3+ T cells (C and I), CD3+CD8+ CTCs (D and J), CD3+CD45RO+ memory T cells (E and K), ICOS+ T cells (F and L), LAG3+ T cells (G and M), and TIM3+ T cells (H and N).
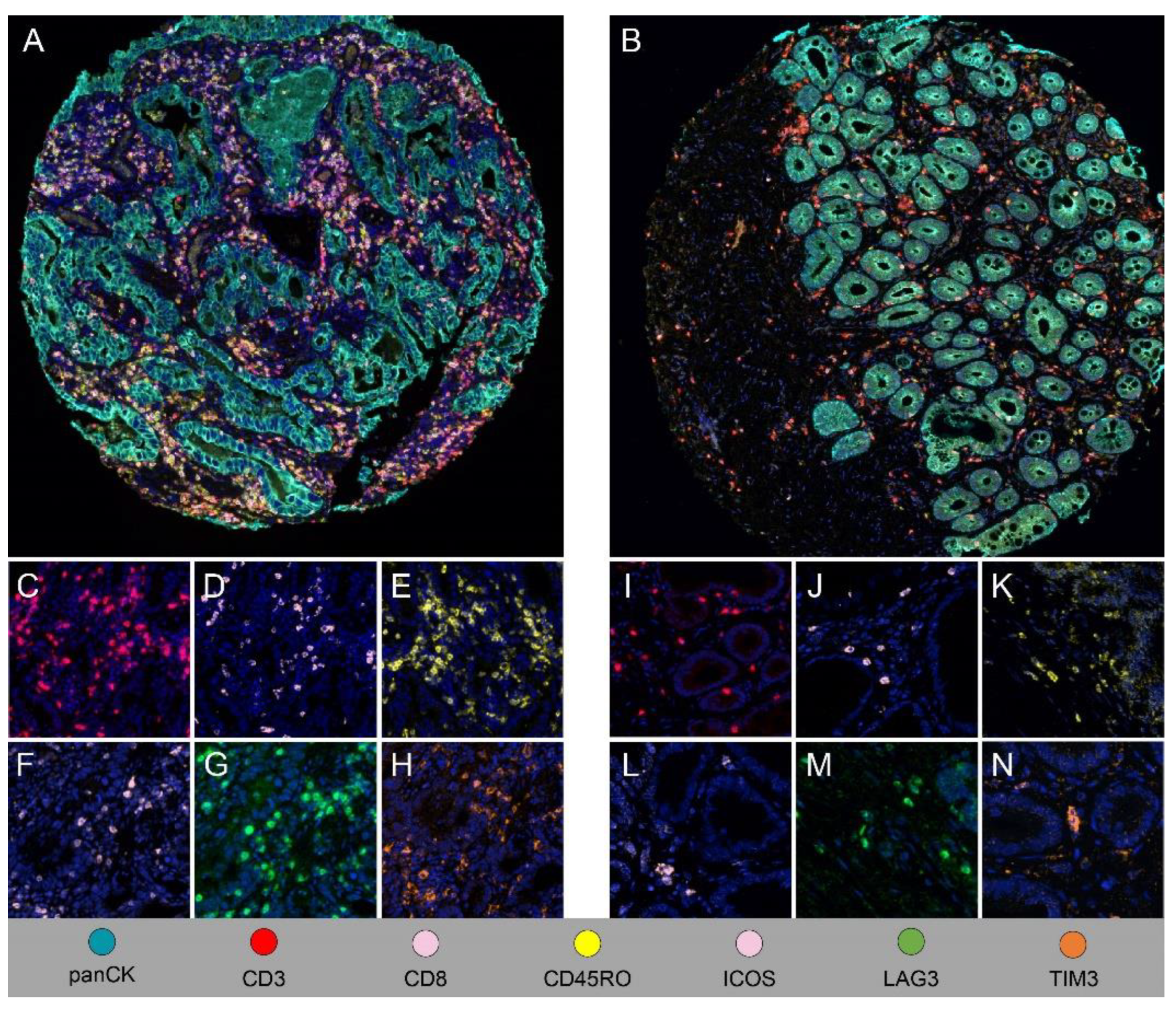
Figure 4.
Correlation of T cell density with pathologic characteristics of EAC. (A-E) Box plots show relationships between densities of total T cells and lymphovascular invasion (LVI) (A), intraepithelial T cells and LVI (B) and pN category (C), and stromal T cells and LVI (D) and pT category (E). (F, G) mIF images (x200) show lower density of T cells (seen as red membranous/cytoplasmic staining) in LVI-positive EAC (F) than in LVI-negative EAC (G).
Figure 4.
Correlation of T cell density with pathologic characteristics of EAC. (A-E) Box plots show relationships between densities of total T cells and lymphovascular invasion (LVI) (A), intraepithelial T cells and LVI (B) and pN category (C), and stromal T cells and LVI (D) and pT category (E). (F, G) mIF images (x200) show lower density of T cells (seen as red membranous/cytoplasmic staining) in LVI-positive EAC (F) than in LVI-negative EAC (G).
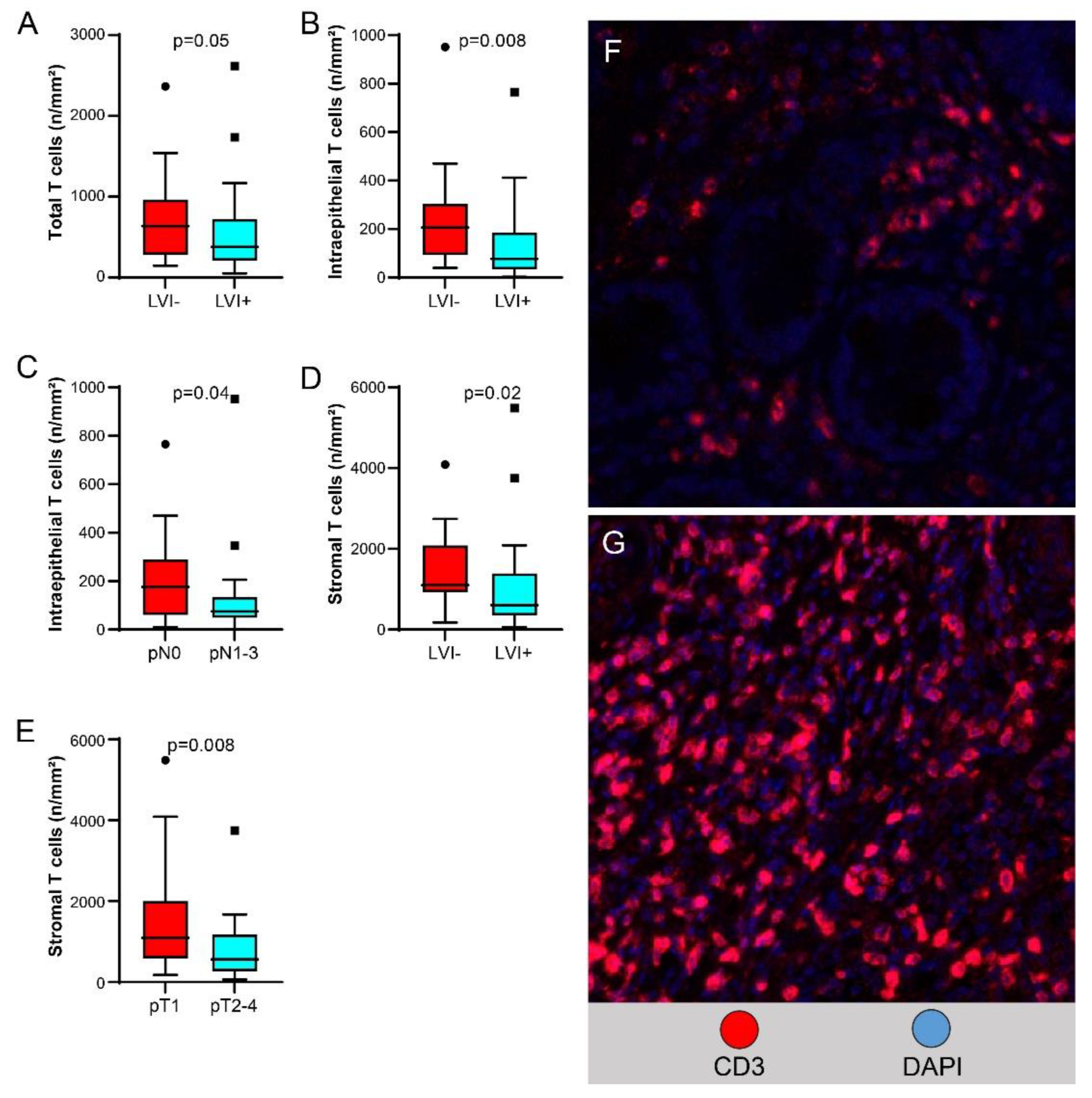
Figure 5.
Correlation of Treg density with pathologic characteristics of EAC. (A-F) Box plots show relationships between densities of total Tregs and pT category (A) and LVI (B), intraepithelial Tregs and pT category (C) and LVI (D), and stromal Tregs and pT category (E) and LVI (F). mIF images (x200) show lower density of Tregs (seen as green nuclear staining) in LVI-positive EAC (G) than in LVI-negative EAC (H).
Figure 5.
Correlation of Treg density with pathologic characteristics of EAC. (A-F) Box plots show relationships between densities of total Tregs and pT category (A) and LVI (B), intraepithelial Tregs and pT category (C) and LVI (D), and stromal Tregs and pT category (E) and LVI (F). mIF images (x200) show lower density of Tregs (seen as green nuclear staining) in LVI-positive EAC (G) than in LVI-negative EAC (H).
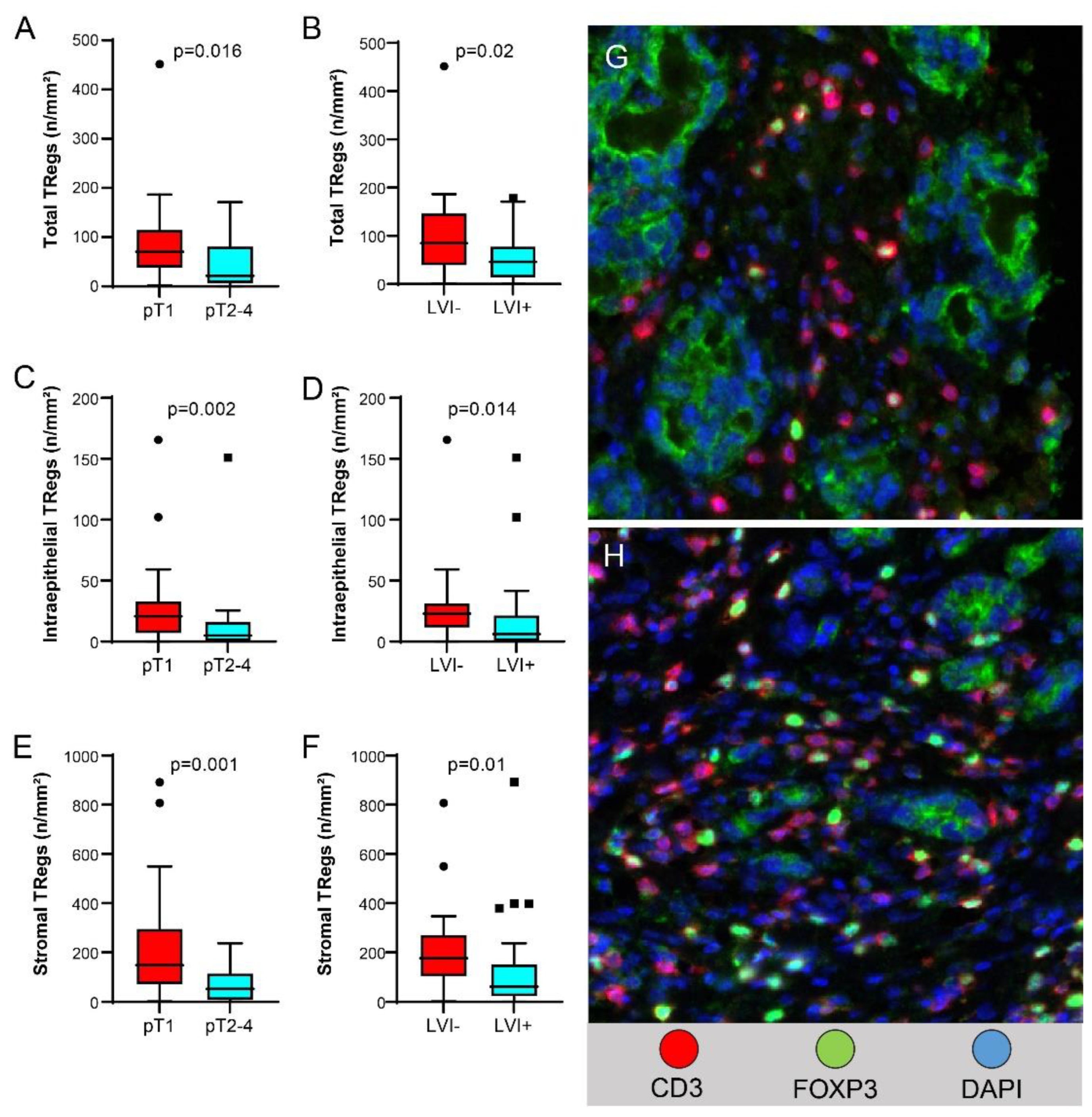
Figure 6.
Relationship between TIM3+ T cell density in EAC and disease-free survival. (A-C) Kaplan-Meier disease-free survival curves of patients with lower or equal to the median or higher than the median density of TIM3+ T cells in the total compartment (A), the intraepithelial compartment (B), and the stromal compartment (C) of EAC. The cutoff values shown in the labels are median values. (D-E) Representative mIF images (x200) showing lower than or equal to median (D) and higher than median (E) density of TIM3+ T cells in EAC. Inset in E shows a higher magnification of TIM3+ T cells.
Figure 6.
Relationship between TIM3+ T cell density in EAC and disease-free survival. (A-C) Kaplan-Meier disease-free survival curves of patients with lower or equal to the median or higher than the median density of TIM3+ T cells in the total compartment (A), the intraepithelial compartment (B), and the stromal compartment (C) of EAC. The cutoff values shown in the labels are median values. (D-E) Representative mIF images (x200) showing lower than or equal to median (D) and higher than median (E) density of TIM3+ T cells in EAC. Inset in E shows a higher magnification of TIM3+ T cells.
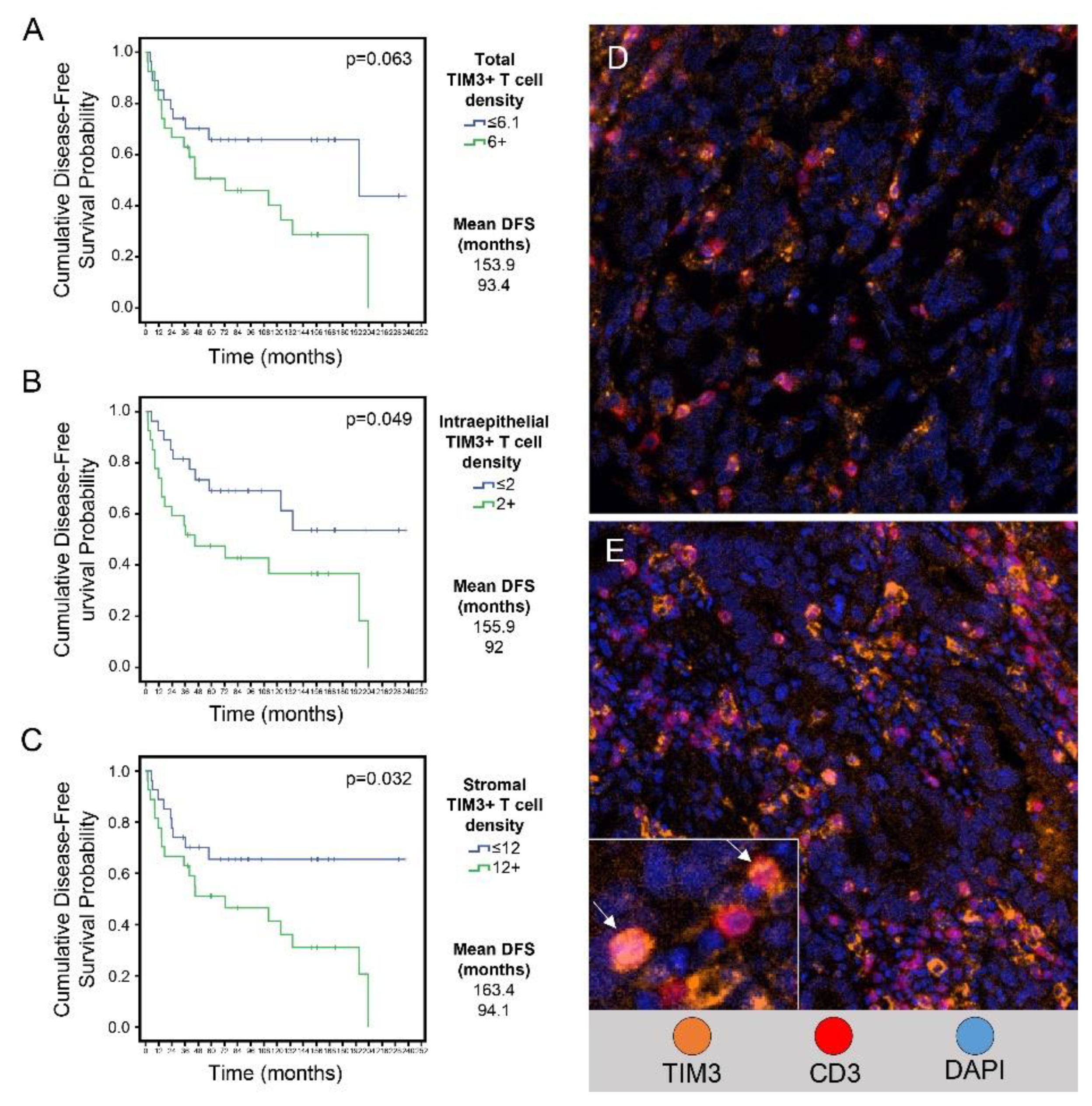

Table 1.
Demographic and clinicopathologic characteristics of the patients (n=54).
| Characteristic | Valuea |
|---|---|
| Age y, median (range), y | 65 (42-80) |
| Sex | |
| Male | 47 (87) |
| Female | 7 (13) |
| Race and ethnicity | |
| White | 50 (93) |
| Hispanic | 4 (7) |
| Smoking status | |
| Smoker | 37 (68) |
| Nonsmoker | 17 (32) |
| Body mass index, kg/m2 | |
| <30 | 26 (48) |
| ≥30 | 28 (52) |
| Surgery type | |
| Ivor-Lewis esophagogastrectomy | 32 (59) |
| Transhiatal esophagectomy | 13 (24) |
| Minimally invasive esophagectomy | 8 (15) |
| Tumor epicenter | |
| Mid-esophagus | 2 (4) |
| Distal esophagus | 18 (33) |
| Gastroesophageal junction type I | 24 (44) |
| Gastroesophageal junction type II | 9 (17) |
| Gastroesophageal junction type III | 1 (2) |
| Pathological tumor category | |
| pT1 | 33 (61) |
| pT2-4 | 21 (39) |
| Pathological nodal category | |
| pN0 | 33 (61) |
| pN1-3 | 21 (39) |
| AJCC stage | |
| I | 27 (50) |
| II | 14 (26) |
| III | 12 (22) |
| IV | 1 (2) |
| Tumor differentiation | |
| Well or moderately differentiated | 25 (46) |
| Poorly or undifferentiated | 29 (54) |
| Lymphovascular invasion present | |
| Yes | 35 (65) |
| No | 19 (35) |
Abbreviation: AJCC, American Joint Committee on Cancer. aValues in table are number of patients (percentage) unless otherwise indicated.
Table 2.
T cell phenotypes in EAC (n=54).
| Cell density, median (range), n/mm2 | |||
|---|---|---|---|
| Cell subtype | Epithelium | Stroma | Total |
| T cells | 111 (3-951) | 967 (64-5488) | 411 (75-2620) |
| Cytotoxic T cells (CTCs) | 17 (0-242) | 102 (0-855) | 53 (0-522) |
| Activated CTCs | 0 (0-12) | 1 (0-36) | 1 (0-14) |
| Memory T cells | 22 (0-177) | 241 (20-4220) | 129 (17-1441) |
| Effector memory CTCs | 3 (0-103) | 31 (0-662) | 17 (0-336) |
| T regulatory cells (Treg) | 13 (0-166) | 102 (0-891) | 53 (0-452) |
| Memory Tregs | 4 (0-89) | 50 (0-668) | 21 (0-367) |
| TIM3+ T cells | 2 (0-42) | 12 (0-140) | 6 (0-82) |
| LAG3+ T cells | 77 (0-407) | 810 (7-2747) | 345 (10-1132) |
| VISTA+ T cells | 0 (0-14) | 4 (0-101) | 2 (0-21) |
| ICOS+ T cells | 11 (0-139) | 84 (0-732) | 46 (0-307) |
| OX40+ T cells | 0 (0-3) | 0 (0-35) | 0 (0-18) |
Table 3.
Comparison of T cell phenotypes in matched BE and EAC samples (n=21).
| Epithelium | Stroma | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell density, median (range), n/mm2 | Cell density, median (range), n/mm2 | Cell density, median (range), n/mm2 | |||||||
| Cell subtype | BE | EAC | p | BE | EAC | p | BE | EAC | p |
| T cell | 186 (30-358) | 114 (19-951) | 0.47 | 419 (89-1159) | 998 (111-5488) | 0.009 | 318 (89-715) | 398 (96-1540) | 0.082 |
| CTC | 19 (0-181) | 25 (0-242) | 0.51 | 33 (0-113) | 119 (0-773) | 0.005 | 29 (0-110) | 61 (9-351) | 0.018 |
| Activated CTC | 0 (0-19) | 0 (0-12) | 0.047 | 1 (0-20) | 0 (0-36) | 0.256 | 2 (0-16) | 0 (0-12) | 0.54 |
| Memory T cell | 16 (0-95) | 37 (0-177) | 0.084 | 41 (0-291) | 233 (20-4200) | 0.041 | 40 (0-180) | 126 (17-585) | 0.012 |
| Effector memory CTC | 3 (0-43) | 6 (0-39) | 0.36 | 2 (0-73) | 40 (0-662) | 0.031 | 3 (0-60) | 20 (0-90) | 0.014 |
| Treg | 18 (0-63) | 21 (0-166) | 0.3 | 60 (7-247) | 125 (0-550) | 0.065 | 41 (8-163) | 48 (0-187) | 0.84 |
| Memory Treg | 3 (0-21) | 8 (0-89) | 0.021 | 9 (0-61) | 51 (0-416) | 0.006 | 6 (0-43) | 20 (0-123) | 0.005 |
| TIM3+ T cell | 0 (0-19) | 1 (0-42) | 0.8 | 5 (0-41) | 22 (0-78) | 0.002 | 5 (0-27) | 8 (0-49) | 0.123 |
| LAG3+ T cell | 81 (13-273) | 136 (19-837) | 0.3 | 197 (52-490) | 893 (7-2455) | <0.001 | 142 (43-327) | 353 (10-917) | 0.001 |
| VISTA+ T cell | 0 (0-4) | 0 (0-4) | 0.66 | 1 (0-16) | 5 (0-78) | 0.11 | 1 (0-10) | 2 (0-16) | 0.28 |
| ICOS+ T cell | 11 (0-103) | 9 (0-139) | 0.28 | 53 (0-148) | 92 (0-589) | 0.009 | 43 (6-87) | 4 (2-307) | 0.125 |
| OX40+ T cell | 0 (0-3) | 0 (0-3) | 0.081 | 1 (0-11) | 1 (0-29) | 0.06 | 1 (0-7) | 1 (0-18) | 0.049 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



