
Submitted:
12 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
Cardiotonic steroids (CTS) are a group of steroid compounds derived from certain plants and animals. CTS are selective inhibitors of Na,K-ATPase, so for a long time they were widely used for medical purposes to treat heart failure and some other diseases. However, over time, doctors gradually began to refuse this group of drugs due to their narrow therapeutic range and a number of serious side effects. However, in parallel with the rise and fall of interest in CTS as a drug, interest in them as a single class of high-affinity Na,K-ATPase inhibitors has only increased. Numerous data on the effect of CTS on signaling cascades and cell viability made impelling a search for a rationale for the existence of such a subtle biological regulator. A large number of studies devoted to the potential use of CTS in the treatment of neurodegenerative, oncological, immune and other diseases "revitalized" the already "written off" group of the drugs. If initially the role of exogenous (mainly plant origin) CTS was studied, then by the 90s of the XX century the study of the role of endogenous CTS and their regulatory effects became a popular trend in biology and physiology. The role of endogenous CTS was studied in details in the pathogenesis of cardiovascular diseases [1]. A large number of researchers have unanimously stated that endogenous CTS are very complex regulators of the development of pathological processes; however, the exact cause-and-effect relationship that would unambiguously explain the processes of synthesis, utilization, and the role of endogenous CTS has not yet been established. The effects of CTS in the frame of various regulatory schemes have been described, but the predominant number of studies was devoted to arterial hypertension, heart and renal failure, cancer [2–4], aging [5,6], and neuroinflammation. The effects of both endo- and exogenous CTS on blood cells and have been studied to a much lesser extent.
Keywords:
cardiotonic steroids
; endogenous cardiotonic steroids
; ouabain
; blood
; blood cells
; red blood cells
; Na
; K-ATPase
; Src-kinase
; signalling cascades
; immunogenic effects
1. Introduction
Cardiotonic steroids (CTS) are a group of steroid compounds derived from certain plants and animals. CTS are selective inhibitors of Na,K-ATPase, so for a long time they were widely used for medical purposes to treat heart failure and some other diseases. However, over time, doctors gradually began to refuse this group of drugs due to their narrow therapeutic range and a number of serious side effects. However, in parallel with the rise and fall of interest in CTS as a drug, interest in them as a single class of high-affinity Na,K-ATPase inhibitors has only increased. Numerous data on the effect of CTS on signaling cascades and cell viability made impelling a search for a rationale for the existence of such a subtle biological regulator. A large number of studies devoted to the potential use of CTS in the treatment of neurodegenerative, oncological, immune and other diseases "revitalized" the already "written off" group of the drugs. If initially the role of exogenous (mainly plant origin) CTS was studied, then by the 90s of the XX century the study of the role of endogenous CTS and their regulatory effects became a popular trend in biology and physiology. The role of endogenous CTS was studied in details in the pathogenesis of cardiovascular diseases [1]. A large number of researchers have unanimously stated that endogenous CTS are very complex regulators of the development of pathological processes; however, the exact cause-and-effect relationship that would unambiguously explain the processes of synthesis, utilization, and the role of endogenous CTS has not yet been established. The effects of CTS in the frame of various regulatory schemes have been described, but the predominant number of studies was devoted to arterial hypertension, heart and renal failure, cancer [2,3,4], aging [5,6], and neuroinflammation. The effects of both endo- and exogenous CTS on blood cells have been studied to a much lesser extent.
2. Cardiotonic steroids. Mechanism of action.
2.1. A brief history of CTS physiological effects studies
СTSs were first isolated from the leaves of Digitalis purpurea and Digitalis lanata and were used to treat heart failure [1,7]. The clinical use of them has led to the search for other sources of CTS [1,7]. The first plаnt origin CTSs, digoxin and digitoxin, were classified as cardenolides [7]. Later CTSs were found in amphibians and named after the common toad (lat. Bufa Bufa) – bufadienolides [8]. Currently, endogenous CTCs have also been found in the mammalians [1,7,9,10]. A characteristic feature of CTS is the presence of a steroid core, a lactone ring at position C-17 and a hydroxyl group at C-14. CTSs are divided into two classes: cardenolides and bufadienolides. Cardenolides have a five-membered ring at C17, while bufadienolides have a six-membered lactone ring at C17 with two double bonds [4]. The Figure 1 shows the structures of cardenolides - digoxin and ouabain and bufadienolides - bufalin and marinobufagenin.
2.2. Current view of the mechanisms involved in CTS effects
Currently, the only receptor known to bind CTS is Na,K-ATPase. Na,K- ATPase is an ATP-dependent transmembrane ion transporter of potassium and sodium ions (Na-pump), that was found in all types of animal cells [11]. The main function of Na,K-ATPase is to maintain ionic homeostasis of the cell. Na,K-ATPase is composed of two main subunits: α and β. Complexes of α- and β-subunits form functional dimer (αβ)2. The α-subunit of Na,K-ATPase (~110 kDa) has 10 transmembrane segments (H1-H10). The CTS binding site as well as the binding sites for ATP, sodium and potassium ions are located on the α-subunit. The α-subunit participates in ATP hydrolysis, which leads to a change in its conformation (E1 and E2 that have more high affinity to Na+ and K+, respectively) and transient phosphorylation of aspartate residue in active site by ATP (EP-conformation) that promotes electrogenic transport of sodium and potassium ions (3 intracellular Na+ to 2 extracellular K+ ).
Mammals express four α- (α1, α2, α3, α4), and three β-isoforms (β1, β2, β3) encoding by different genes. The α1 subunit is present in all mammalian cells. Three other α-isoforms are tissue-specific. The α2 was found in astrocytes, cardiomyocytes, skeletal muscle, and glial cells, and α3-isoform in neuronal cells, heart [12]. The α4 subunit has been found in mammalian testes. The β-subunit is a glycoprotein (protein part 35 kDa), represented by 3 isoforms, is required for the transport of newly synthesized Na,K-ATPase into the membrane, as well as for the correct orientation of the α-subunit in the cytoplasmic membrane and for the occlusion of K+ [13]. The γ-subunit of Na,K-ATPase (FXYD) was found at first in kidney cells, but later 6 other FXYD subunit isoforms were identified which contributes to the regulation of enzymatic activity [14].
2.2.1. CTS binding to Na,K-ATPase provides its inhibition and activation of signaling cascades
It was suggested that the therapeutic effect of CTS in the treatment of cardiovascular failure is based on the inhibition of the transport function of Na,K-ATPase. Disruption of Na,K-ATPase functioning leads to impaired ion-transport, in particular, to intracellular Na+ load and K+ depletion, which, in turn, leads to changes in membrane potential, activation of Na+/Ca2+ exchanger and accumulation of intracellular calcium. Thus, inhibition of myocardial Na,K-ATPase results in the augmentation of calcium level in cardiomyocytes and, accordingly, to the increase of cardiac contractions force. In addition, these changes can lead to impaired cell adhesion and intercellular interaction [15,16,17,18]. Apparently, the β-subunit of Na,K-ATPase, which is an adhesion molecule, is involved in this process [19]. Long-term practice of using CTS for the treatment of chronic heart failure has demonstrated that prolonged usage of CTS leads to a number of side effects, such as myocardial hypertrophy, etc., which can be based on the activation of signaling cascades [20].
Activation of signaling cascades occurs because in addition to the function of ion transport,Na,K-ATPase is able to realize a receptor function. The binding of CTS leads to the fixation of Na,K-ATPase in E2P-like conformation, to the binding of protein-partners and to the activation of signaling cascades, most of which include activation of Src kinase [21,22,23]. The interaction of CTS with Na,K-ATPase occurs on extracellular surface of membrane in a narrow “channel” formed by transmembrane segments of α-subunit H1-H2, H5-H6, and H7-H8 [24,25]. The depth of entry into this channel is different for various types of CTS. For example, marinobufagenin binds 5Å closer to the channel exit than ouabain [22,24]. This explains the fact that marinobufagenin is able to bind to any conformation of Na,K-ATPase with equal affinity, while ouabain binds with high affinity only to the E2P conformation [22]. In addition, the binding of these CTSs leads to different conformational changes [22] and, probably, to the activation of different signaling cascades due to binding to various partner proteins. These data allow to explain the reason for the different effects [26] of marinobufagenin and ouabain on cells. It can be assumed that in vivo ouabain and marinobufagenin can modulate each other's actions.
2.2.2. CTS affinity to different isoforms of Na,K-ATPase
The affinity of Na,K-ATPase for CTS depends on its subunit composition. In 1987, it was found that canine cardiac myocytes contain two isoforms of Na,K-ATPase α-subunit which differ in molecular weight and affinity to ouabain: the dissociation constant is 2 nM and about 300 nM for high and low affinity isoform correspondently [27]. At that time, it was also suggested that the toxicity of CTS which were used as drugs was related to their inhibition of the low affinity isoform which is now known as the α1-subunit, and the presumed high-affinity isoform has turned to be, actually, two isoforms, now known as α2 and α3. The α4-subunit which was discovered later also shows low affinity for ouabain [28].
The data on higher affinity of α2, α3 isoforms for ouabain compared to α1 isoform were also found in another study [29]. The α2 isoform is colocalized with Na+/Ca2+ exchanger [30], participating in the regulation of calcium level in muscle cells, and in combination with β2 has a reduced affinity for potassium compared to other isoforms (K0.5 for K+ is 3 mM, other isoforms have K0.5 ~ 1 mM) [31,32] (Table 1). In glia and astrocytes, α2β2 Na,K-ATPase is believed to be well suited for potassium influx after intense neuronal activity. The α3, which is widely represented in neurons, is optimized to pump out high sodium after neuronal excitation [33], and is therefore characterized by a lower Na+ sodium affinity with K0.5 25-50 mM compared to other isoforms (for which K0.5 is about 10 mM) [34,35]. It should also be noted that α2 and α3 isoforms are more sensitive to oxidation and glutathionylation [36,37], than the α1 isoform (Table 1). The number of cysteines does not vary significantly between various α-isoforms (the number of cysteine residues for α1 and α2 Na,K-ATPase subunits of Norway rat is the same, and in α3 there is one more, PubMed NCBI sequence library), however, they are differently arranged in the ternary structure of the α-subunit [38]. Thus, oxidation and glutathionylation sensitivity of α-isoforms is determined by the structural arrangement of cysteine residues and, particularly, by the shift in E2-E1 structural equilibrium of α2-subunit towards E1 conformation, which is more open to the cytosol [38,39,40]. Data on the properties of different isoforms are summarized in Table 1.
The α1-subunit of rodent Na,K-ATPase, also known as CTS-resistant α1R-Na,K-ATPase, has a ~1000-fold lower affinity for ouabain compared to other mammalian species [25]. Its impaired binding to CTS is caused by the replacement of uncharged amino acids Gln111 and Asn122 with charged Arg and Asp [24,47] (Figure 2). This amino acid substitution dramatically reduces the ouabain affinity of the α2- and α3-subunit-Na,K-ATPase and results in a decrease in the depth of CTS entry into the binding channel (Figure 2). Thus, for rat Na,K-ATPase expressed in Sf-9 insect cells, the value of dissociation constant for ouabain are 4.3±1.9×10-5 M for α1β1 ; 1.7±0.1×10-7 M for α2β1; and 3.1±0.3×10-9 M for α3β1 [35]. At the same time, physiological concentrations of ouabain in rodents are approximately the same as in humans (0.34±0.06 nM vs. 0.152-0.53±0.10 nM) [1], that allows to presume other ways of CTS-dependent regulation in rodents.
CTS binds to Na,K-ATPase pseudo-reversibly, leading to its persistent dose-dependent inhibition [24]. In the comparative study, ouabain in the concentration range between 100 nM-10 µM led to the death of the cells that were expressing CTS-sensitive Na,K-ATPase, while, in the case of cells that were expressing CTS-resistant Na,K-ATPase, the ouabain concentrations up to 3 mM did not result in cell death [48].
2.2.3. Endogenous CTS
Both cardenolides and bufadienolides are present in the human body. First, the endogenous ouabain (cardenolide) was detected in blood plasma [49]. Later the endogenous marinobufagenin (bufadienolide) was detected in plasma and urine [49,50,51]. Currently, ouabain [52,53], digoxin [54], bufalin [8], marinobufotoxin [55], marinobufagenin [50,56], telocinobufagin [1,9,57,58]; and several digoxin-immunoreactive compounds [59] have been found in human plasma and urine. The range of CTS concentrations in blood varies from subnanomolar to nanomolar [1]. The most studied are endogenous marinobufagenin and ouabain. Both of them are vasoconstrictors [50]. Their plasma concentrations vary in the subnanomolar range but do not exceed 1 nM [50,60,61,62,63,64,65], yet pathological concentrations can be up to several nM [1]. Marinobufagenin and ouabain effectively inhibit Na,K-ATPase containing ouabain-sensitive α-isoform at concentrations of 2.0 and 0.8 µM, respectively, [24] thus it is assumed that the physiological effects of endogenous CTS, is mediated probably, through Src kinase associated to Na,K-ATPase, or by inhibition of small fraction of Na,K-ATPas [66].
In the mammalian organisms, CTSs acts as hormones. They are produced by the midbrain and adrenal glands in response to various stimuli: angiotensin II, acetylcholine, vasopressin, catecholamines, and even in hypoxia [67]. Pathological CTS concentrations are involved in the development of hypertension [68]. In myocardial [50] and renal ischemia the significant increase in the level of marinobufagenin was observed [69]. It is assumed that CTS mediate between chronic kidney disease and the development of cardiac hypertrophy and fibrosis, and, in particular, increasing levels of marinobufagenia explain the high frequency of cardiovascular disorders and individuals with renal artery stenosis [69].
2.2.4. Activating effect of CTS on Na,K-ATPase
Importantly, while high doses of CTS inhibit Na,K-ATPase, CTS at concentrations below IC50 are able to increase its activity. Ouabain, digoxin, and marinobufagenin activated Na,K-ATPase transport activity at 120 nM, 10 nM, and 100 pM. The inhibitory effect of these CTSs was exerted at 1200, 100, and 1000 pM, respectively [70]. The activation effect of ouabain has been shown to protect neuronal cell culture from hypoxia-hypoglycemia [70]. The protective effect of activating doses of ouabain was observed in vitro in hippocampal slice cultures when ouabain was administered 30 minutes before or 2 hours after experimental ischemia [70]. Blocking the increase in Na,K-ATPase activity impaired the protective effect of CTS [70]. Under hypoxia conditions, the effect of CTS on cells is altered [71,72]. In particular, ouabain binding to Na,K-ATPase in hypoxia does not induce the activation of Src family kinases, but impairs their activity, that is usually increased under hypoxia [71,72]. CTS increases cell viability in hypoxia, presumably due to the fact that inhibition of some fraction of the enzyme promotes cell survival by saving ATP [71]. At low doses of CTS, the effects of activation on transport function and signaling cascades also cannot be excluded. The role of CTSs as potential antihypoxants remains incompletely understood. However, the increasing CTS levels observed in congestive heart failure and acute myocardial infarction [1,50] may serve as indirect evidence of their antihypoxic effect.
2.2.5. Signaling cascades
Src kinase has been shown to form a complex with α1-containing Na,K-ATPase, and this interaction is highly specific. Accepted model of signal transduction in this complex implies two interaction interfaces: the SH2 domain of Src kinase binds to the actuator domain of the α1-subunit of Na,K-ATPase, while its kinase domain binds to the nucleotide-binding domain, preventing Src kinase from autophosphorylation and activation. When ouabain binds to the extracellular domain of Na,K-ATPase, the interaction between the kinase domain of Src and the nucleotide-binding domain of the pump is disrupted. The kinase domain is released, that leads to its autophosphorylation and activation of the kinase. At the same time, the interaction between the SH2 domain of Src and the actuator domain of Na,K-ATPase is preserved [73,74]. Src can transduct the activating signal to mitogen-activated kinases, Erk1/2-kinase, induce further cascades via phospholipase C or the PI3-kinase- mediated signaling pathway [75].
It was assumed that only the α1 isoform was involved in interaction with Src kinase, since it was the only isoform containing the predicted Src interaction interface [72,74,76]. However, recent evidence suggests that the α2 subunit may also be involved in interaction with Src, and its knockout leads to impaired signaling transmission via CTS [77]. Two "populations" of Na,K-ATPase in cells are distinguished: free, or pumping, Na,K-ATPase, which mainly performs transport activity and accounts for about 30% of the total amount of Na,K-ATPase on the cell surface, and non-pumping Src-associated Na,K-ATPase [78]. Binding CTS to non-pumping Na,K-ATPase leads to Src-dependent transactivation of receptor tyrosine kinases (RTKs) such as the EGF receptor (EGFR), which converts CTS binding to activation of serine/threonine kinases, lipid kinases and lipases [79]. CTS binding has been shown to induce the endocytosis of Na,K-ATPase via the Src-dependent pathway. Src activation is independent of sodium, potassium and calcium ions concentrations and leads to the changes in the expression of more than 100 genes, demonstrating a fourfold increase in c-Fos mRNA within 30 min after ouabain addition.
Direct interaction of Src-kinase and Na,K-ATPase has been shown many times, both in vitro and on cell cultures, and the dissociation constant of this complex was recently measured (Kd=0.21 ± 0.04 µM) [80], All these data makes reliable the mechanism of signal transduction through direct interaction between Src-kinase and Na,K-ATPase.
Src kinase is the most studied Na,K-ATPase protein partner, but not the only one. In cells, about 10 proteins are shown to be associated with Na,K-ATPase. Some of them are able to affect the transport activity of Na,K-ATPase, similarly to CTS: for example, the homolog of the γ-subunit of Na,K-ATPase phospholemman, or agrin, which interacts with α3-subunit of Na,K-ATPase. The caveolin-1 protein together with Src mediates cholesterol-dependent [81] and CTS-dependent [82] regulation of Na,K-ATPase isoformal ratio at the surface of cells, inducing the endocytosis of Na,K-ATPase together with its signaling protein partners.
Curiosly, the receptor function of Na,K-ATPase activated by CTS may also include Src-independent pathways. Namely, ouabain binding to Na,K-ATPase in neurons changes the sensitivity of NMDA receptors, which are colocalized with Na,K-ATPase in cholesterine-rich membrane rafts. In subnanomolar concentrations ouabain mediated the inhibition of NMDA-receptors [83], while in nanomolar concentration it causes their hypersensitization not involving Src kinase activation [84]. Besides, the incubation of cells that contain α3-subunit Na,K-ATPase in low ouabain concentrations leads to the activation of Erk1/2-kinase but not Src-kinase [76,85] implying the existence of other possible proteins that can mediate CTS-dependent signaling pathways. In fibroblasts, PI3K1A and Akt kinases can also be activated in the presence of ouabain via the Src-independent pathway [86]. The molecular mechanisms of these processes remain unclear. For instance, the effect of CTS on NMDA receptor sensitivity may be caused by direct contact between NMDA receptors and Na,K-ATPase in the membrane [83].
The role of CTS in the pathogenesis of various diseases has been reviewed in detail [1]. The highest concerntrations of endogenous ouabain are observed in essential hypertension; the increasing levels of both ouabain and marinobufagenin are observed in primary hyperaldosteronism; marinobufagenin is increased in chronic renal failure. Taking into account the complex development of the pathological patterns in these diseases, the role of CTS in every particular disease requires further study. All diseases mentioned above can be independent clinical entities as well as the links in the pathological chain of metabolic syndrome, thus, at this stage, it is sensible to further study the effect of CTS on different types of cells, organs and tissues. Since blood cells are continuously exposed to circulating endogenous CTS, which change in levels in numerous pathologies, as well as to the therapeutic doses of exogenous CTS, the aim of our work is to systematize and analyze the information on the effect of CTS on various blood cells.
3. Effect of CTS on blood cells
Blood is a complex multicomponent liquid medium that is responsible for various vital needs of the organism. CTS effect on the whole blood is basically determined by their effect on each subsystem of the blood. In a number of pathological conditions (heart failure, chronic renal failure, hypertension, etc.), the concentration of endogenous CTS in the blood is increased [1], exerting various regulatory effects on certain components of the circulatory system. The study of systemic effects in the absence of connection with pathological conditions is a complex and nearly impossible task; therefore, we decided to focus on the individual groups of blood cells and the effects of CTS on these cells.
In general, the effects of CTS on blood cells can be classified as activating, inhibitory and “modulating”, the latter term indicates a combination of activating and inhibitory influences.
3.1. Distribution of Na,K-ATPase isoforms in blood cells
The isoforms of the catalytic α-subunit of Na,K-ATPase differ in their affinity for CTS and sensitivity to oxidative modifications (Table 1), therefore the isoformal composition Na,K-ATPase in the cell largely determines its response to CTS. Table 2 contains the generalized data on α1-α4 isoform protein and their mRNA presence in various blood cells.
Regarding β-isoforms, it should be noted that erythroid cells exhibit a unique expression profile of the β-subunit, having β2 but not β1. At the same time, leukocytes and platelets have β1 but not β2 [32].
3.2. CTS effects on some immune diseases and blood immune cells
Generally CTSs provide mainly anti-inflammatory and immunosuppressive effects, and different CTSs behave similarly in most cases. CTS role in autoimmune diseases (rheumatoid arthritis, Chagrin syndrome, etc.), general inflammatory status and immune status in tumor diseases has been widely described [89,90]. The effects of CTS on different types of immune blood cells are summarized below.
3.2.1. Leukocytes
3.2.1.1. Mononuclear cells/Macrophages
The effect of CTS on macrophages is currently being actively studied, but the mechanism of action of CTS on these cells has not been fully established. It was demonstrated [91] that administration of ouabain in a dose of 0.56 mg/kg to mice infected with Leishmania (L.) amazonensis resulted in a decrease of peritoneal migration of macrophages and in the production of two proinflammatory cytokines interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) and had no cytotoxic effects on macrophages. Intraperitoneal injection of marinobufagenin in a dose of 0.56 mg/kg to mice showed a decrease in zymosan-activated production of proinflammatory cytokines IL-1β and IL-6, without affecting the level of TNF-α [92]. The treatment of peritoneal macrophages with different concentration (10, 100, 1000, and 10000 nM) of marinobufagenin in vitro showed no cytotoxic effect. Usually cytotoxic effect is a result of Na,K-ATPase inhibition. The lack of toxicity is likely due to the presence in mice Na,K-ATPase containing α1-isoform, which is 1000 times less sensitive to CTS than human Na,K-ATPase with α1-isoform. Moreover, marinobufagenin have 70-fold lower potency to inhibit α2/α3 isoforms than ouabain (IC50 = 3.8 ± 0.9 μM vs IC50 = 55 ± 9 nM for ouabain) and failure to inhibit mice α1 (for ouabain IC50 = 25 ± 12 μM) [92]. The reasons for such different actions of these CTS were discussed above in section 2. Notably, the marinobufagenin treatment at non-inhibitory Na,K-ATPase concentrations 10 nM reduced levels of proinflammatory cytokines induced zymozan IL-1β, IL-6, and TNF- α [92]. Investigation of the putative mechanism of of marinobufagenin action demonstrate that the expression of macrophages surface molecules Toll-like receptor 2 (TLR2) and a transmembrane type II C-lectin receptor (CD69) and phosphorylated-p38 mitogen-activated protein kinase (P-p38 MAPK) does not change that means they do not involved in this regulation [92].
Opposed data have been obtained for human monocytes [93]. In vitro study [93] was shown that human monocytes treated with ouabain at a concentration of 10-7 M (100 nM) produced high levels of IL-1β, TNF-α, IL-10, vascular endothelial growth factor (VEGF) and increased expression of surface activation markers such as CD69, a transmembrane type II C-lectin receptor, and HLA-DR (Human Leukocyte Antigen – DR isotype) that are early markers of monocytes activation [94], CD80 and CD86 (B7-1 and B7-2 type I membrane protein in the immunoglobulin superfamily, correspondingly) are considered as a markers of increased cytokine expression (they are costimulating signals for activation and surviving of T-lymphocytes) [95]. Authors concluded that ouabain affects monocytes activity as immunomodulator [93].
An assessment of cytotoxicity against human macrophages [96] showed that ouabain in the concentration range 50 nM < IC50 < 100 nM leads to a dose-dependent toxic effect, while the density of the macrophage mannose receptor CD206 decreases. CD206 is a specific marker for adipose tissue macrophages. Adipose tissue-infiltrating macrophages are responsible for persistent lowgrade inflammationthat leads tosystemic insulin resistance observed in obesity [97]. Application of ouabain (50–100 nM) to the ex vivo cultured white adipose tissue explants results in the increase of sensitivity to insulin [96]. It may be explained by the fact that decrease of the amount of CD206+ macrophages raises insulin sensitivity [98]. Therefore therapy with nanomolar concentrations of CTSs may be considered as perspective approach for treatmet of metabolic syndrome that is characterized by infiltration and activation of macrophages in the white adipose tissue.Treatment human macrophages with ouabain resulted also in the activation of caspases 1, 3, and 7, indicating cell death by apoptosis or pyroptosis. This cytotoxicity has been found to be dependent on the cation homeostasis, becauses ouabain induced intracellular K+ depletion and accumulation of Na+ and Ca2+ [96]. This demonstrates that cytotoxicity reason is Na,K-ATPase inhibition. The cell death caused by this ionic imbalance can be prevented by adding KCl to human monocyte-derived macrophages. At the same time, ouabain had a stronger effect on monocyte-derived macrophages compared to non-adherent peripheral blood mononuclear cell populations. Bufalin and digoxin do not have such cytotoxic effects [96]. The bufadienolides gamabufotalin and arenobufagin (up to 40 ng/ml) demonstrate no cytotoxicity against human peripheral blood mononuclear cell [99].
However, the papers [100] and [93] provide data on an increase in immunoreactivity when human monocytes were exposed to CTSs. Short-term (within 1 hour) exposure to oleandrin (200 ng/ml, 3.5×10-7 M=350 nM) enhances the biological response of human monocytes to IL-8 and increases the number of receptors for IL-8 [100] without cytotoxicity.
The contradictory effects of CTS can be explained by different concentrations used for exposure and by different sensitivity of α1 subunits of human and mice α-isoforms of Na,K-ATPase. In paper [93], where the activating effect of CTS was observed, human monocytes were treated with 100 nM of ouabain in-vitro, while the inhibitory effect was described in-vivo on mice with ouabain concentrations reaching 0.56 mg/kg which can not be bring into correlation with molar concentration CTS used in vitro and in-vitro with 10 nM of marinobufagenin [91].
Telocinobufagin treatment of peritoneal macrophages (10 and 100 nM, 24 h) was shown to induce oxidative burst and enhanced NF-kB activation [101]. These effects were not found after macrophages pretreatment with Src kinase inhibitors PP2 or peptide pNaKtide (both 30 min 1μM), or if macrophages were isolated from Na/K-ATPase α−1 subunit heterozygous null mice (NKA α−1+/−) compared to macrophages isolated from wild type mice [101]. pNaKtide is peptide that specifically inhibits Na/K-ATPase-associated Src signaling [73]. These data demonstrate that telocinobufagin effect is due to activation of Src-kinase associated with α1-subunit of Na,K-АТPase [101].
We can conclude that influence of CTS on monocytes and macrophages is mainly modulating one. Immunomodulation is mainly result in the activation or reduction of proinflamatory cytokines secretion. It may be connected with activaion of signaling cascades (including signaling through Src-kinase) or with activation of Na,K-ATPase, that may induce expression of Na,K-dependent genes. The described results allow us to conclude that the effect of CTS on macrophages essentially depends on the type of CTS, expression of resistant or sensitive α1 isoform and can be caused not by inhibition of Na,K-ATPase, but by activation of signaling cascades.
3.2.1.2. Neutrophils
CTS have a general inhibitory effect on neutrophils which was revealed as a decrease of chemotaxis and production of pro-inflammatory cytokines. Previous study [92] has established, that intraperitoneally injection of marinobufagenin (0.56 mg/kg) to mice reduces IL-1β and IL-6 production and neutropiles migration when it was stimulated with zymosan, which induces peritoneal inflammation. Similar results were obtained for ouabain: ouabain treatment (1, 10 and, 100 nM) of mouse neutrophils for 2 hours reduces neutrophil chemotaxis, which was induced by chemotactic peptide fMLP (at concentrations 1, 10 nM the effect was more pronounced), but this substance did not inhibit Akt, ERK, and JNK activation induced by zymosan [102]. However, ouabain at concentrations 1 and 10 nM decreased p38 phosphorylation in zymosan-stimulated neutrophils whereas at a concentration 100 nM, ouabain partially inhibited Na,K-ATPase and did not alter p38-signaling. These results suggest that low doses of ouabain lead to the decrease of neutrophil migration through p38 MAPK inhibition [102]. Similar inflammation-reducing effects have been demonstrated for 21-benzylidene digoxin [103] and bufalin [104]. Notably, the synthetic digoxin derivative compound 21-benzylidene digoxin does not inhibit Na,K-ATPase activity (that eliminates dangerous toxic effects), but in a dose of 0.3 mg/kg effectively suppresses the oedema progression induced by carrageenan [103]. Histologic analysis revealed a decrease in the number of inflammatory cells and inducible nitric oxide synthase (iNOS) expression in the paw pads of mice (6 hours after carrageenan injection). Tumor necrosis factor (TNF) level also was decreased. In the study of [91] it was demonstrated that ouabain (0.56 mg/kg ouabain) is able to reduce the migration peritoneal exudate cells in mice with Leishmania (L.) Amazonensis infection. This infection increased polymorphonuclear cell population. Treatment with ouabain resulted in the percentage of polymorphonuclear cells to return to the baseline. Authors suppose, that decrease of the amount of polymorphonuclear leukocytes observed as result of treatment with ouabain possibly reflects a decrease of neutrophils, because it is typically the predominant cell population at the beginning of the inflammatory process [91].
All this data indicate that the decrease in neutrophil chemotaxis leading to the decrease in their migration under the action of low concentrations of CTS is realized due to changes in signaling pathways.
Thus, CTS can be considered as anti-inflammatory and anti-edematous drugs, as well as under conditions associated with pathological infiltration and activation of macrophages, for example, in metabolic syndrome [92,102,103,104]. It should be noted that the effect of CTS on neutrophil activity is mainly caused by their complex immunosuppressive effect due to inhibition of the cellular component of the immune system.
3.2.1.3. Eosinophils
The effect of CTS on eosinophils has not been studied. However, a study [104] of bufalin effect on asthmatic response in a mouse model, has demonstrated that this CTS (in a dose 5 and 10 mg/kg) significantly attenuated hyperresponsiveness and strongly suppressed the ovalbumin-induced increase of total amount of inflammatory cells including macrophages, eosinophils, lymphocytes, and neutrophils in BALF (bronchoalveolar lavage fluid). Exposure to aeroallergens (1% aerosol of OVA (wt/vol) in 0.9% salin) results in antigen-specific T-helper lymphocytes (Th2 cells) response and the release of OVA-specific immunoglobulin E (IgE) in serum, and accompanied by intrapulmonary production of interleukin (IL)-4, IL-5, and IL-13 by Th2 cells. The levels of IL-4, IL-5, and IL-13 in BALF and ovalbumin-specific (IgE) in serum were significantly reduced by bufalin [104]. NF-κB is a key transcription factor in the modulation of acute inflammatory response and eosinophilic inflammation in this asthmatic model of mice was blocked by a specific NF-κB inhibitor. Since it was found that bufalin inhibit of NF-κB activity in the lung tissues, the authors suggest that this may underlie its anti-inflammatory effects. Notably, under hyperreactive conditions there is a pronounced eosinophilic infiltration of respiratory epithelium, thus the main effects of bufalin are assumably provided by the lowered reactivity of eosinophils.
3.2.1.4. Lymphocytes3.2.1.4.1. NK-cells
The effects of CTS on NK-cells are discussed in detail in [105]. Bufalin directly counterbalances stimulatory and inhibitory receptors on the surface of NK-cells and indirectly activates natural killer (NK)-cells through inhibition of MICA (MHC class I chain-related polypeptide A) shedding, thereby enhancing the cytotoxic activity of the cells against the hepatocellular carcinoma cell line [105].
3.2.1.4.2. T-helpers
Telocinobufagin has also been shown to dramatically enhance the proliferative response of splenocytes to concanavalin A, ovalbumin and substantially increase transcription factors T-bet (Th1) mRNA level and the CD3(+)/CD3(+)CD4(+)/CD3(+)CD8(+) phenotype in splenocytes from ovalbumin-immunized mice [106]. There are contradictory data on the effect of ouabain on the number of T-helpers cells, for example, in a study [107] it was shown that intraperitoneal injection of ouabain in mice significantly decreased the number of CD4+ T-lymphocytes in the spleen, especially regulatory T-cells, while the percentage and total number of lymphocytes in mesenteric lymph nodes remained the same [108]. Reduction of levels of antigen-specific T-helper lymphocytes cytokines (IL-4, IL-5, IL-13) in bronchoalveolar lavage fluid by bufalin was described above [104].
3.2.1.4.3. Regulatory T-cells
Regulatory T-cells (T-regs) are a subpopulation of T-cells that suppresses the immune response and maintaines homeostasis and self-tolerance. T-regs have been shown to be able to suppress T-cell proliferation and cytokine production and play a crucial role in preventing autoimmunity [89]. The effects of various CTS on the functioning of T-reg lymphocytes have been widely studied. Digoxin at a concentration of 5 mg/kg injected in mice with collagen-induced arthritis, bufatolin at a concentration of 100 μg/kg injected in mice with induced Chagrin syndrome and gamabufatolin (8 ng/ml) in vitro show similar inhibitory effects: reducion of T-helper cells expressing IL-17 (Th17) polarization and production of key pro-inflammatory cytokines IL-1β, IL-6, TNF-α and IL-21 by inhibition retinoic acid receptor-related orphan receptor γ thymus (RORγt) translational activity [90,99,109,110]. T-regs themselves can convert into Th17 cells in the presence of IL-6 [111]. One of the main functions of T-regs is regulation of immune system activity, including restriction of hyperergic immune responses. Activation of T-regs results in a general immunosuppressive effect. However, digoxin (1 μM), strophantin and dihydroouabain stimulated IL17A and IL17F expression and enhanced IL17 secretion in human Th17 lymphocytes [112]. Incubation with 8 ng/mL of gamabufothaline, which was a nearly nontoxic concentration for normal human peripheral blood mononuclear cells, effectively reduced the percentage of CD4+CD25+Foxp3+ regulatory T cells in mitogen-activated peripheral blood mononuclear cells [99]
The difference between observed effects can be explained by the choice of model organisms with significantly different α1-subunit Na,K-ATPase affinity to CTS. This difference illustrates that the CTS experiments carried out on rodents cannot be extrapolated to other species of mammalian including humans.
The isoformal composition of Na,K-ATPase in T-regs remains unclear (see Table 2). In addition, digoxin decreased the number of Th17 cells and increased the number of T-Regs, which promoted immunosuppression in mice [109]. Ouabain has been shown to reduce the number of T-regs by decreasing Il-2 production by T lymphocytes [107]. The regulation of lymphocytes by CTS will offer new and modified treatment strategies for rheumatologic and autonomic diseases such as Chagrin syndrome [90], rheumatoid arthritis [109] via promoting T-Regs and suppressing joint inflammation and bone erosion in mice CIA (collagen induced arthritis) arthritis model.
3.2.1.4.4. T- cells CD8+ (cytotoxic T lymphocytes)
It was shown by Li et. al [113] that after administration of oleandrin to BALB/c mice with engrafted murine breast cancer cell line EMT6 and tumor-bearing mice intraperitoneally (0.3 mg/kg and 0.6 mg/kg) every day tumor growth was suppressed and the amount of tumor-infiltrating lymphocytes including dendritic cells and CD8+ T-cells were increased. In another study it was shown that ouabain (0.56 mg/kg with 140 mg/kg sodium succinate of hydrocortisone ) injected intraperitonially induced the death of immature double positive lymphocytes (CD4+CD8+) whereas CD69+ cells survived after both treatments [114]. It was revealed [115] that that combination of anti-vascular agent - (5,6-Dimethylxanthenone-4-acetic Acid; also known as: ASA404, Vadimezan) and HIF-1α inhibitor - digoxin inhibits the growth of melanoma tumors in murine model. It was established that in tumors of mice treated with combination therapy, the number of macrophages M1, CD8+ cytotoxic lymphocytes, NK cells and to a lesser extent CD4+ cells was increased. In the paper [116] digoxin reversed the inability of Cisplatin to trigger calreticulin exposure, and HPMA (N-(2-hydroxypropyl) methacrylamide) copolymer-amplified Cisplatin-induced ATP release in melanoma mice model. These complementary mechanisms induced potent immunogenic cell death that promotes dendritic cell maturation and activates CD8+ T cell responses. The effects of CTS on CD8+ T-cells are mainly described in oncological studies where CTS are presumed as a potential addition to chemotherapy and is its effects are mainly observed in tumor models. Thus it is very difficult to highlight the exact effects of CTS on CD8+ cells.
3.2.1.4.5. B cells
Ouabain was shown to affect the level of B-cells. Intraperitoneal ouabain administration to mice (0.56 mg/kg) for 3 straight days led to the decrease of subpopulation of mature B-cells in bone marrow, spleen and peripheral blood in 24 hours after last injection [117]. Later it was found that in 24 h after ouabain injection for 3 days the amount and percentage content of follicular B-cells in spleen was decreased, in contrast, the amount of B-cells in mesenteric lymph nodes was increased. It is suggested that these events took place as result of the increase of chemokine receptor CXCR5 expression and the decrease of the amount of cell adhesion molecule L-selectin (CD62L) This effect disappeared after 48 hours. Therefore, ouabain regulates the dynamic of В-lymphocyte settling in peripheral organs. It was noted that production of total IgM и IgG in serum of animal injected with ouabain in vivo did not change [118]. Beside this bufalin increased B-cell proliferation from leukemic BALB/c mice compared with the leukemic control group at doses 0.1 and 0.2 mg/kg. It should be noted that a stiff dose of bufalin (0.4 mg/kg) did not lead to this effect [119,120]. In the paper [108] it was shown that the best viability of melanoma-bearing animals, which were treated with ouabain was, particularly, due to the maintenance by ouabain the absolute number of B cells in splenic and mesenteric lymph nodes in melanoma-bearing animals.
3.3. Red blood cells
The effect of CTS on erythrocytes also is poorly understood. It is known that in red blood cells Na,K-ATPase, which generates a sodium-potassium gradient and is assumed to be the only receptor for CTS, is involved in maintaining the charge constancy of the erythrocyte membrane and in regulating the deformability of erythrocytes [121]. Apparently, CTS binding to Na,K-ATPase can influence these parameters. First of all, the pool of Na,K-ATPase on the surface of erythrocytes is scarce: the maximum number of membrane-bound ouabain binding sites, that are located solely on the α-subunit of Na,K-ATPase, is 288 +/- 28 per erythrocyte [122]. This number, as well as the ouabain affinity to the receptor, does not vary by age and sex of the blood donors. The association rate constant and the dissociation rate constant for this complex were measured at 37 °C and are equal to 4-6 x 104 M-1sec-1 1-4 x 10(-4) sec-1, respectively. The dissociation constant of this complex is about 0-28 x 10-8 M [122].
The α1 and α3 isoforms of Na,K-ATPase are expressed in human erythrocytes [123]. In this cells, the digoxin, ouabain and ouabain-like factor (OLF) effect on the Na,K-ATPase transport activity, estimated by 86Rb uptake, was investigated. At ouabain concentrations below 10-9 M, Rb+ uptake was significantly stimulated, reaching its maximum rate of about 18% at ouabain concentration of 10-10 M. The effect of OLF was similar to that of ouabain. At the same time, digoxin did not stimulate the Na,K-ATPase activity. Calcium homeostasis was not altered at ouabain concentrations below 10-9 M. The authors suggest that Na,K-ATPase activation effect may be related to the presence of α3 isoform in erythrocytes [123], consisting with the fact that α3 isoform shows higher affinity for CTS than α1 isoform (Table 1). However as we mentioned above the activation of Na,K-ATPase was observed even on purifed Na,K-ATPase preperation [66] it means that another explanation may be considered.
In 2019, it was shown that the lifespan of red blood cells is reduced in chronic kidney disease (CKD) [125], which is accompanied by the increasing CTS level in the blood [1]. Therefore, Na,K-ATPase and, particularly, CTS-induced signaling were suggested to play an important role in the development of anemia in CKD [126]. The hypothesis is that an increase in CTS in chronic renal failure, as well as oxidative stress, may lead to activation of the Src-dependent signaling cascade through Na,K-ATPase, causing the increase in ROS levels, impaired erythrocyte deformability and lifespan reduction [126].
Enhanced oxidative stress may activate Src kinases in erythrocytes [127]. In addition, activation of erythrocytic p38 MAPK is often observed in hemolysis, especially under oxidative stress. Inhibition of p38 MAPK in erythrocytes treated with diamide, an oxidative stress inducer, may be connected to the decreased phosphorylation of Src tyrosine kinases and Band 3 protein. In addition, inhibition of p38 MAPK prevents sickling and hemolysis in response to diamide treatment and osmotic shock of erythrocytes in experimental hypoxia [128]. Thus, CTS-mediated Src kinase activation may contribute to erythrocyte hemolysis.
Notably, under various stress effects including hypoxia [129], and metabolic stress [130], the redox status of erythrocytes may change significantly, affecting Src kinase/Na,K-ATPase complex. The glutathionylation of Na,K-ATPase caused by hypoxia or other oxidative stress [44] may disrupt its interaction with Src kinase, that has been shown by molecular modeling [72]. This effect is assumed to be connected to the impaired activation of Src kinase in hypoxia [72]. Thus, various types of physiological stress to which erythrocytes are exposed to (metabolic stress, temperature stress, mechanical stress, deoxygenation), may alter their response to circulating CTS in blood by affecting their redox status.
As mentioned above, the levels of endogenous ouabain and marinobufagenin may increase in various pathologies, and the increase in marinobufagenin level is often more pronounced [1]. The intravenous administration of 0.9% NaCl saline solution (1000 ml within an hour) to patients stimulates systolic and diastolic blood pressure, increases the marinobufagenin concentration in blood plasma by more than 1.6 times and inhibits Na,K-ATPase transport activity by 1.67 times [131]. This inhibition is prevented by the antibodies against marinobufagenin. The data demonstrate that marinobufagenin may by released in response of hypervolemia, and the increase of cicrculation blood volume led to secretion of suffecient amount of marinobufagenin to inhibit Na,K-ATPase Similar decrease in erythrocyte Na,K-ATPase activity under rising marinobufagenin concentration has been observed in rats [132]. A 2-fold increase in endogenous marinobufagenin in rats with inducted 2 diabetes mellitus leads to a 30% decrease in erythrocyte Na,K-ATPase activity. Antibodies against marinobufagenin abolish this effect [132].
The binding constant of ouabain to Na,K-ATPase of human erythrocytes is (1.30±0.17)×10-8 M for healthy Europeans [133]. In diabetic patients, the development of neuropathy is strongly related to decreased Na,K-ATPase activity. In erythrocytes of European patients with insulin-dependent diabetes and North African subjects predisposed to diabetic neuropathy Na,K-ATPase activity is reduced by 30%. Additionally, its affinity to ouabain is nearly unchanged, and the number of ouabain binding sites on erythrocytes reflecting the amount of Na,K-ATPase on the red blood cell surface, was reduced in patients with insulin-dependent diabetes [133]. For North African subjects predisposed to the development of diabetic neuropathy, when the diabetes develops, the decrease in Na,K-ATPase activity correlates with a decrease in the maximum number of ouabain binding sites on erythrocytes, whereas no such correlation has been found for patients with insulin-dependent diabetes. The absence of the correlation between the decrease in Na,K-ATPase activity and the number of ouabain binding sites in diabetic patients allows to assume the impaired availability of ouabain binding sites on erythrocytes, presumably caused by the altered structure of the lipid membrane [133].
All in all, despite the scarcity of Na,K-ATPase, this enzyme is crucial for erythrocytes, and its role as a receptor for CTS is also essential for normal erythrocyte functionality and viability. This must be taken into account both when prescribing CTS drugs and when infering the effects of altered endogenous CTS levels on the body.
3.4. Clotting and platelet dependent effects
The hemostatic system is a complex multicomponent set of interacting components with electrostatic, biochemical and cellular interactions. Numerous studies have shown that some CTSs have a procoagulant effect. For example, in patients taking digoxin, the expression level of P-selectin (CD62P, a marker of platelet activation) in platelets and platelet-leukocyte conjugates increased significantly, as were indicated by the markers of endothelial activation, EMP62E (endothelial microparticles (EMP) identified by anti-E-selectin antibodies) and EMP31 (endothelial microparticles identified by anti-CD31). After adjustment for confounders (including age, congestive heart failure, coronary heart disease, ejection fraction, use of antiaggregants, beta-blockers, and calcium channel blockers), the differences persisted [134]. Digoxin (2.4 ng/ml) induced calcium mobilization, PAC-1 (procaspase-activating compound 1) and platelet aggregation in patients with atrial fibrillation but not in healthy subjects.
After pretreatment of thrombocytes with collagen in vitro, digoxin induced calcium mobilization, arachidonic acid release, TxB2 biosynthesis, platelet PAC-1 and P-selectin expression, and promoted platelet aggregation in a dose-dependent manner. Antibodies to digoxin abolished these effects. Serum digoxin concentrations above therapeutic levels increased platelet aggregation in vitro via phosphorylation of calcium-bound phospholipase A2 [135]. However, procoagulant activity was not observed in healthy volunteers taking digoxin (at a dose of 0.6 mg on day 1, 0.4 mg on day 2, then 0.1 mg daily for 10 days), and no difference between digoxin and placebo was observed [136]. Notably, ouabain also has procoagulant activity. It has been shown that treatment of human platelets in vitro with ouabain (20-200 μM for 20-60 min) is associated with intracellular accumulation of sodium, generation of a weak calcium signal, and induced procoagulation. According to thromboelastography data, increasing ouabain concentration to nanomolar range (50-1000 nM, 15 min) in human whole blood results in an increasing rate of clot formation initiated by contact and high extracellular calcium concentration. Thus, the inhibition of platelet Na,K-ATPase by ouabain leads to an increase in intracellular Na+ The increase in Na+ and associated cell swelling may in turn lead to phosphatidylserine exposure and increased membrane curvature, resulting in procoagulant activity [137]. The procoagulant effect induced by ouabain is dose- and time-dependent, less pronounced than the collagen-induced response, and significantly reduced in the absence of extracellular Na+ or in hyperosmolality [137].
All the effects of different CTS on blood cells are summarized in Table 3.
The pathways of influence of different concentrations of CTS on the state of cells upon binding to Na,K-ATPase are presented in Figure 3.
The effect of CTS depends on their concentration (Figure 3). At high concentrations comparable to the enzyme inhibition constant or higher (High Doses), interaction of CTS with Na,K-ATPase inhibits the transport activity of the enzyme, adversely affecting cell viability, and leads to the development of toxic effects at prolonged exposure. At lower concentrations (5×10-7-5×10-10 M) [1], activation of signaling cascades is observed [124,138,139]. Activation of signaling cascades is mostly caused by the Src kinase interaction with Na,K-ATPase [73,74]. Src kinase has been shown to interact directly to α1 subunit, and this interaction is disrupted by CTS binding to Na,K-ATPase, that leads to autophosphorylation of Src and its activation [72,74,76,80]. In turn, Src kinase induces EGFR activation [79] , resulting in Ras/Raf/MEK/ERK signaling cascade and affecting the regulation of cell cycle and proliferation [140]. CTS binding to α2 subunit also leads to Src kinase activation, while α3 induces Erk1/2 activation in a Src-independent way mediated by PI3K and PKC [76,85]. α4 subunit may also activate Erk1/2 signaling cascades and stimulate integrins expression through CREB and ATF-1 [141]. Ras and JAK-Stat molecules [142], as well as PLC-IP3-IP3R [75,143] pathways, can also be activated via Na,K-ATPase. Lower concentrations of CTS about 3×10-10-10-11 М [1] stimulate Na,K-ATPase transport activity [66,70,124].
The analyzed data allow us to conclude that cytotoxic effects of CTS on blood cells are observed at high CTS concentrations and are caused by partial inhibition of Na,K-ATPase of cells. Low (below Kd) concentrations of CTS often have the opposite effect on blood cells, which may be associated with activation of signaling cascades and/or activating effect of low concentrations of CTS on Na,K-ATPase activity.
4. Discussion
Cardiotonic steroids are important regulators of cellular processes with unclear up to now mechanism of action. In recent years, their role in various pathologies has been actively studied on using different cell lines. It is known that the human body maintains a rather low but constant rate of CTS synthesis in concentrations insufficient for inhibition of Na,K-ATPase. However, their concentration increases in a number of diaseses and pathologies. This may indicate that CTS play the role of an adaptogen and a regulator, allowing the organism to survive under acute pathological conditions. This concept is supported by the fact that CTS level increase is observed in diseases affecting a large number of organs and systems, that are connected with circulation system and water-salt metabolism, such as arterial hypertension, myocardial infarction, heart failure, pre-eclampsia, chronic renal failure and aldosteronism [1]. All these diseases have in common a violation of tissue perfusion and an increased load on the cardiovascular system. As it was shown in a number of studies, the inhibition of Na,K-ATPase in itself contributes to the conservation of ATP and the protection of cells under adverse conditions; the limitation of potassium supply and the accumulation of sodium in the cells of excitable tissues leads to an increase in intracellular calcium and a change in its transients, which, in turn, realizes a positive inotropic effect, contributing to the preservation of systolic myocardial function [144]. This observation suggests that this mechanism is realized in arterial hypertension, heart failure and myocardial infarction and allows the body to survive under adverse conditions. At the same time, ouabain has mainly inhibitory effect on Na,K-ATPase, while digoxin distinctly induces its receptor effects. Not less important is the study of the level of endogenous CTS in other systemic pathologies, including autoimmune diseases. For example, CTS have a regulatory effect on the cells of the immune system, preferentially preventing the development of hyperergic (superphysiological) responses and contributing to the limitation of inflammatory damage to organs and tissues. The effect of CTS on blood vessels and their ability to cause fibrosis [131,145,146] is probably an uncontrolled side effect associated with the growth of endogenous CTS; this hypothesis is supported by the fact that high CTS concentrations are toxic and, when their concentration increases in supraphysiological doses, they adversely affect cells and tissues.
It is also known that the Na,K-ATPase-mediated signaling transduction changes the expression of a large number of genes and triggers Src and PI3K-Art dependent signaling cascades, which, in turn, are regulators of vital cellular processes, including the organization of intercellular interactions, protein transport, cell cycle regulation and regulation of the immune response [147]. It is noteworthy that sometimes the CTS effect is not associated with Na,K-ATPase inhibition. On the contrary, at low concentrations (nanomolar and subnanomolar) CTS are able to activate Na,K-ATPase. In oxygen deficiency, the effect of CTS on cells is drastically altered, which should be taken into account when considering the mechanism of their action under hypoxic and ischemic conditions [71,72]. Thus, it can be assumed that CTS are complex adaptive regulators that allow the organism to survive short-term adverse conditions but they exert numerous toxic effects at a prolonged high-dose exposure. In summary, it can be concluded that, with the universality of the action mechanism (interaction with Na,K-ATPase present in all cells), CTS have various tissue-specific effects that contribute to the maintenance of homeostasis and the formation of appropriate responses to external and internal stimuli. It is interesting to note that a deoxygenation leads to an increase CTS in the blood [1,67], and at the same time, at the molecular level, hypoxia changes the cell response to the action of CTS [71,72]. Possible reasons for the different effects of CTS on different cell types are the expression of tissue-specific isoforms of Na,K-ATPase and the peculiarities of regulation of signaling cascades in different cell types. Our analysis of the effect of CTS on blood cells demonstrated that the change in the level of endogenous CTS and the effect of exogenous CTS in nanomolar concentrations affect almost all blood cells, being a modulator of the immune response, affecting blood coagulation, the functionality of erythrocytes, as well as vascular endothelial cells (Table 3). Thus, the alterations of CTS levels in blood, induced by various external factors or pathologies, essentially affect the functioning of blood cells and vessels, contributing to the conditional adaptation of the organism.
5. Conclusion
CTS are complex physiological regulators that provide the maintenance of homeostasis of the organism under various pathological conditions. CTS have specific effects on different blood cells. The expression levels of different Na,K-ATPase isoforms with different properties and different sensitivity to CTS, which vary in different cell types, determine the general CTS effect on each cell type of blood. At high concentrations, comparable or higher than the dissociation constant of the ubiquitous α1-isoform of Na,K-ATPase, CTS exert an inhibitory effect on ion transporters, resulting in a cytotoxic effect at long-term exposure. At low concentrations (nanomolar and subnanomolar), insufficient to inhibit the pump function, the signaling function of CTS and/or Na,K-ATPase overactivation are observed, stimulating cell proliferation and altering cell functionality. Consideration of endogenous CTS as a marker of compensation in various pathological conditions will allow to create approaches for promising diagnostic technique.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, Y.P. and I.P.; methodology, Y.P.; writing—original draft preparation, Y.P., I.P., O.D., M.S., I.K.; writing—review and editing, I.P., Y.P., O.D., A.M.; visualization, I.P., Y.P.; supervision, A.M.; project administration, I.P..; funding acquisition, I.P. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by the Russian Science Foundation, grant #19-14-00374.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
CTS — Cardiotonic steroids, DGX — digoxin, OBN — ouabain, MBG — marinobufagenin, BUF— bufalin, RTKs — receptor tyrosine kinases, EGFR — epidermal growth factor receptor, NMDA — N-methyl-D-aspartic acid, VEGF — vascular endothelial growth factor, Akt —RAC-alpha serine/threonine-protein kinase, PI3K1 - Phosphatidylinositol 3-kinase, ERK —Extracellular signal-regulated kinases, JNK — c-Jun N-Terminal Kinase, MAPK— mitogen-activated protein kinase, TNF— Tumor necrosis factor, BALF — bronchoalveolar lavage fluid, NK — natural killer, MICA — MHC class I chain-related polypeptide A, IFN — interferon, IL — interleukin, T-bet transcription factor, T-regs — regulatory T-cells, RORγt — RAR-related orphan receptor gamma, CKD — chronic kidney disease, CD62P— P-selectin, TxB2 — Thromboxane B2, PAC-1 — first procaspase activating compound, TLR2- Toll-like receptor 2, CD69 -transmembrane type II C-lectin receptor, P-p38 MAPK- phosphorylated-p38 mitogen-activated protein kinase, HLA-DR - Human Leukocyte Antigen – DR isotype, CD206 - mannose receptor, CD80 - (B7-1) is a B7 type I membrane protein in the immunoglobulin superfamily, CD86 – (B7-2) is a B7 type I membrane protein in the immunoglobulin superfamily, PP2- inhibitor for Src-family kinases (1-tert-Butyl-3-(4-chlorophenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine), pNaKtide - peptide (part of Na,K-ATPase, interacting with Src-kinase), Th cells – T helper lymphocytes, Ig= immunoglobulin, NF-κB - Nuclear Factor Kappa B, OVA – ovalbumin, EMP62E -endothelial microparticles (EMP) identified by anti-E-selectin antibodies, EMP31 -endothelial microparticles identified by anti-CD31.
References
- Orlov, S.N.; Tverskoi, A.M.; Sidorenko, S. V; Smolyaninova, L. V; Lopina, O.D.; Dulin, N.O.; Klimanova, E.A. Na, K-ATPase as a target for endogenous cardiotonic steroids: What’s the evidence? Genes Dis. 2021, 8, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Dufrasne, F.; Kiss, R. Cardiotonic steroids-mediated targeting of the Na(+)/K(+)-ATPase to combat chemoresistant cancers. Curr. Med. Chem. 2012, 19, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Kiss, R. Cardiotonic steroids-mediated Na+/K+-ATPase targeting could circumvent various chemoresistance pathways. Planta Med. 2013, 79, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Van Quaquebeke, E.; Delest, B.; Debeir, O.; Darro, F.; Kiss, R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Biophys. Acta 2007, 1776, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Hollman, A. Plants and cardiac glycosides. Br. Heart J. 1985, 54, 258. [Google Scholar] [CrossRef] [PubMed]
- Lichtstein, D.; Gati, I.; Ovadia, H. Digitalis-like compounds in the toad Bufo viridis: interactions with plasma proteins. J. Cardiovasc. Pharmacol. 1993, 22, S102–S105. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Blaustein, M.P.; Bova, S.; DuCharme, D.W.; Harris, D.W.; Mandel, F.; Mathews, W.R.; Ludens, J.H. Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. 1991, 88, 6259–6263. [Google Scholar] [CrossRef] [PubMed]
- Kieval, R.S.; Butler Jr, V.P.; Derguini, F.; Bruening, R.C.; Rosen, M.R. Cellular electrophysiologic effects of vertebrate digitalis-like substances. J. Am. Coll. Cardiol. 1988, 11, 637–643. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of na, K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M. V; Hilbers, F.; Poulsen, H. The structure and function of the Na, K-ATPase isoforms in health and disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Forte, J.G. Functional significance of the beta-subunit for heterodimeric P-type ATPases. J. Exp. Biol. 1995, 198, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.J.; Bijlani, S.; de Sautu, M.; Spontarelli, K.; Young, V.C.; Gatto, C.; Artigas, P. FXYD protein isoforms differentially modulate human Na/K pump function. J. Gen. Physiol. 2020, 152, e202012660. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, T.; Beitel, G.J. Unexpected roles of the Na-K-ATPase and other ion transporters in cell junctions and tubulogenesis. Physiology 2009, 24, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Larre, I. The Na+-K+-ATPase as self-adhesion molecule and hormone receptor. Am. J. Physiol. Physiol. 2012, 302, C473–C481. [Google Scholar] [CrossRef] [PubMed]
- Vagin, O.; Dada, L.A.; Tokhtaeva, E.; Sachs, G. The Na-K-ATPase α1β1 heterodimer as a cell adhesion molecule in epithelia. Am. J. Physiol. Physiol. 2012, 302, C1271–C1281. [Google Scholar] [CrossRef] [PubMed]
- Larre, I.; Ponce, A.; Franco, M.; Cereijido, M. The emergence of the concept of tight junctions and physiological regulation by ouabain. In Proceedings of the Seminars in cell & developmental biology; Elsevier, 2014; Vol. 36; pp. 149–156. [Google Scholar]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The role of the β1 subunit of the Na, K-ATPase and its glycosylation in cell-cell adhesion. J. Biol. Chem. 2006, 281, 39573–39587. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, J.; Kennedy, D.J. Regulation of cardiac remodeling by cardiac Na+/K+-ATPase isoforms. Front. Physiol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Li, X.; Xie, Z. Regulation of renal function and structure by the signaling Na/K-ATPase. IUBMB Life 2013, 65, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Klimanova, E.A.; Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Orlov, S.N.; Makarov, A.A.; Lopina, O.D. Binding of ouabain and marinobufagenin leads to different structural changes in Na, K-ATPase and depends on the enzyme conformation. FEBS Lett. 2015, 589, 2668–2674. [Google Scholar] [CrossRef]
- El-Mallakh, R.S.; Brar, K.S.; Yeruva, R.R. Cardiac glycosides in human physiology and disease: update for entomologists. Insects 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Tverskoi, A.M.; Poluektov, Y.M.; Klimanova, E.A.; Mitkevich, V.A.; Makarov, A.A.; Orlov, S.N.; Petrushanko, I.Y.; Lopina, O.D. Depth of the steroid core location determines the mode of Na, K-ATPase inhibition by cardiotonic steroids. Int. J. Mol. Sci. 2021, 22, 13268. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B.; Croyle, M.L.; Woo, A.L.; Argüello, J.M. Ligand binding sites of Na, K-ATPase. Acta Physiol. Scand. Suppl. 1998, 643, 69–77. [Google Scholar] [PubMed]
- Akimova, O.A.; Bagrov, A.Y.; Lopina, O.D.; Kamernitsky, A. V; Tremblay, J.; Hamet, P.; Orlov, S.N. Cardiotonic steroids differentially affect intracellular Na+ and [Na+] i/[K+] i-independent signaling in C7-MDCK cells. J. Biol. Chem. 2005, 280, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Maixent, J.M.; Charlemagne, D.; De La Chapelle, B.; Lelievre, L.G. Two Na, K-ATPase isoenzymes in canine cardiac myocytes. Molecular basis of inotropic and toxic effects of digitalis. J. Biol. Chem. 1987, 262, 6842–6848. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Melton, R.J.; Sánchez, G.; Mercer, R.W. Functional Characterization of a Testes-Specific α-Subunit Isoform of the Sodium/Potassium Adenosinetriphosphatase†. Biochemistry 1999, 38, 13661–13669. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Avila, J.; Cózar-Castellano, I.; Brownleader, M.D.; Trevan, M.; Francis, M.J.O.; Lamb, J.F.; Martín-Vasallo, P. Na+, K+-ATPase isozyme diversity; comparative biochemistry and physiological implications of novel functional interactions. Biosci. Rep. 2000, 20, 51–91. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Blaustein, M.P. Distinct Distribution of Different Na+ Pump α Subunit Isoforms in Plasmalemma: Physiological Implications a. Ann. N. Y. Acad. Sci. 1997, 834, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Crambert, G.; Hasler, U.; Beggah, A.T.; Yu, C.; Modyanov, N.N.; Horisberger, J.-D.; Lelievre, L.; Geering, K. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J. Biol. Chem. 2000, 275, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.R.; Assentoft, M.; Cotrina, M.L.; Hua, S.Z.; Nedergaard, M.; Kaila, K.; Voipio, J.; MacAulay, N. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4. 1 to hippocampal K+ clearance and volume responses. Glia 2014, 62, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Azarias, G.; Kruusmägi, M.; Connor, S.; Akkuratov, E.E.; Liu, X.-L.; Lyons, D.; Brismar, H.; Broberger, C.; Aperia, A. A specific and essential role for Na, K-ATPase α3 in neurons co-expressing α1 and α3. J. Biol. Chem. 2013, 288, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Zahler, R.; Zhang, Z.-T.; Manor, M.; Boron, W.F. Sodium kinetics of Na, K-ATPase α isoforms in intact transfected HeLa cells. J. Gen. Physiol. 1997, 110, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. - Ren. Physiol. 1998, 275, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V. a.; Kamanina, Y. V.; Ziganshin, R.H.; Meng, X.; Anashkina, A. a.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic α subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- Xianyu, M.; Petrushanko, I.Y.; Klimanova, E.A.; Dergousova, E.A.; Lopina, O.D. Glutathionylation of the alpha-subunit of Na, K-ATPase from rat heart by oxidized glutathione inhibits the enzyme. Biochem. 2014, 79, 158–164. [Google Scholar] [CrossRef]
- Bogdanova, A.; Petrushanko, I.; Boldyrev, A.; Gassmann, M. Oxygen- and redox-induced regulaton of the Na/K ATPase. Curr. Enzym. Inhib. 2006, 2, 37–59. [Google Scholar] [CrossRef]
- Segall, L.; Javaid, Z.Z.; Carl, S.L.; Lane, L.K.; Blostein, R. Structural basis for α1 versus α2 isoform-distinct behavior of the Na, K-ATPase. J. Biol. Chem. 2003, 278, 9027–9034. [Google Scholar] [CrossRef]
- Poluektov, Y.M.; Dergousova, E.A.; Lopina, O.D.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. Na,K-ATPase α-subunit conformation determines glutathionylation efficiency. Biochem. Biophys. Res. Commun. 2019, 510. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Mascarenhas, S.; De Oliveira, A.D.S.; Amoedo, N.D.; Affonso-Mitidieri, O.R.; Rumjanek, F.D.; Rumjanek, V.M. Modulation of the immune system by ouabain. Ann. N. Y. Acad. Sci. 2009, 1153, 153–163. [Google Scholar] [CrossRef]
- Segall, L.; Lane, L.K.; Blostein, R. Insights into the structural basis for modulation of E1↔E2transitions by cytoplasmic domains of the Na,K-ATPase α subunit. In Proceedings of the Annals of the New York Academy of Sciences; 2003; Vol. 986; pp. 58–62. [Google Scholar]
- Xie, Z.; JackHays, M.; Wang, Y.; Periyasamy, S.M.; Blanco, G.; Huang, W.H.; Askari, A. Different oxidant sensitivities of the α1 and α2 isoforms of Na+/K+-ATPase expressed in baculovirus-infected insect cells. Biochem. Biophys. Res. Commun. 1995, 207, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y. V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic α subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.L.; James, P.F.; Lingrel, J.B. Characterization of the fourth α isoform of the Na, K-ATPase. J. Membr. Biol. 1999, 169, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Wagoner, K.; Sanchez, G.; Nguyen, A.N.; Enders, G.C.; Blanco, G. Different expression and activity of the α1 and α4 isoforms of the Na, K-ATPase during rat male germ cell ontogeny. Reproduction 2005, 130, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na, K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Akimova, O.A.; Tverskoi, A.M.; Smolyaninova, L. V; Mongin, A.A.; Lopina, O.D.; La, J.; Dulin, N.O.; Orlov, S.N. Critical role of the α1-Na+, K+-ATPase subunit in insensitivity of rodent cells to cytotoxic action of ouabain. Apoptosis 2015, 20, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Ludens, J.H.; Clark, M.A.; DuCharme, D.W.; Harris, D.W.; Lutzke, B.S.; Mandel, F.; Mathews, W.R.; Sutter, D.M.; Hamlyn, J.M. Purification of an endogenous digitalislike factor from human plasma for structural analysis. Hypertension 1991, 17, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Fedorova, O. V; Dmitrieva, R.I.; Howald, W.N.; Hunter, A.P.; Kuznetsova, E.A.; Shpen, V.M. Characterization of a urinary bufodienolide Na+, K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension 1998, 31, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O. V; Doris, P.A.; Bagrov, A.Y. Endogenous marinobufagenin-like factor in acute plasma volume expansion. Clin. Exp. Hypertens. 1998, 20, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, A.; Guo, J.; Itagaki, Y.; Bell, C.; Wang, Y.; Haupert Jr, G.T.; Magil, S.; Gallagher, R.T.; Berova, N.; Nakanishi, K. On the structure of endogenous ouabain. Proc. Natl. Acad. Sci. 1999, 96, 6654–6659. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Wray, V.; Nimtz, M.; Lehmann, W.D.; Kirch, U.; Antolovic, R.; Schoner, W. Bovine adrenals contain, in addition to ouabain, a second inhibitor of the sodium pump. J. Biol. Chem. 1998, 273, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Ishiguro, T.; Yamada, K.; Ishii, M.; Yoshioka, M.; Eguchi, C.; Shimora, M.; Sugimoto, T. Isolation of a urinary digitalis-like factor indistinguishable from digoxin. Biochem. Biophys. Res. Commun. 1990, 173, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Yoshika, M.; Komiyama, Y.; Konishi, M.; Akizawa, T.; Kobayashi, T.; Date, M.; Kobatake, S.; Masuda, M.; Masaki, H.; Takahashi, H. Novel digitalis-like factor, marinobufotoxin, isolated from cultured Y-1 cells, and its hypertensive effect in rats. Hypertension 2007, 49, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Fedorova, O. V Effects of two putative endogenous digitalis-like factors, marinobufagenin and ouabain, on the Na+, K+-pump in human mesenteric arteries. J. Hypertens. 1998, 16, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Dong, X.H.; Nishimura, N.; Masaki, H.; Yoshika, M.; Masuda, M.; Takahashi, H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin. Biochem. 2005, 38, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Haddy, F.J.; Overbeck, H.W. The role of humoral agents in volume expanded hypertension. Life Sci. 1976, 19, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, U.; Dolev, S.; Werber, M.M.; Shapiro, M.S.; Shilo, L.; Shenkman, L. Identification and preliminary characterization of two human digitalis-like substances that are structurally related to digoxin and ouabain. Biochem. Biophys. Res. Commun. 1992, 188, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.K.; Yandle, T.G.; Lewis, J.G.; Richards, A.M.; Pidgeon, G.B.; Kaaja, R.J.; Nicholls, M.G. Ouabain is not detectable in human plasma. Hypertension 1994, 24, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manunta, P.; Hamlyn, J.M.; Pavan, E.; De Toni, R.; Semplicini, A.; Pessina, A.C. Immunoreactive endogenous ouabain primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J. Hypertens. 1995, 13, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, S.S.; Rogowski, A.C.; Weinberg, M.; Krichten, C.M.; Hamilton, B.P.; Hamlyn, J.M. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation 1992, 86, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Gonick, H.C.; Ding, Y.; Vaziri, N.D.; Bagrov, A.Y.; Fedorova, O. V Simultaneous measurement of marinobufagenin, ouabain, and hypertension-associated protein in various disease states. Clin. Exp. Hypertens. 1998, 20, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Lopatin, D.A.; Ailamazian, E.K.; Dmitrieva, R.I.; Shpen, V.M.; Fedorova, O. V; Doris, P.A.; Bagrov, A.Y. Circulating bufodienolide and cardenolide sodium pump inhibitors in preeclampsia. J. Hypertens. 1999, 17, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-G.; Staessen, J.A.; Messaggio, E.; Nawrot, T.; Fagard, R.; Hamlyn, J.M.; Bianchi, G.; Manunta, P. Salt, endogenous ouabain and blood pressure interactions in the general population. J. Hypertens. 2003, 21, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Tverskoi, A.M.; Sidorenko, S. V; Klimanova, E.A.; Akimova, O.A.; Smolyaninova, L. V; Lopina, O.D.; Orlov, S.N. Effects of ouabain on proliferation of human endothelial cells correlate with Na+, K+-ATPase activity and intracellular ratio of Na+ and K+. Biochem. 2016, 81, 876–883. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, C.; Haupert Jr, G.T. Hypoxia triggers release of an endogenous inhibitor of Na+-K+-ATPase from midbrain and adrenal. Am. J. Physiol. Physiol. 1998, 274, F182–F188. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. drugs 2007, 7, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Haller, S.; Periyasamy, S.; Brewster, P.; Zhang, H.; Adlakha, S.; Fedorova, O. V; Xie, Z.; Bagrov, A.Y.; Shapiro, J.I. Renal ischemia regulates marinobufagenin release in humans. Hypertension 2010, 56, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Oselkin, M.; Tian, D.; Bergold, P.J. Low-dose cardiotonic steroids increase sodium–potassium ATPase activity that protects hippocampal slice cultures from experimental ischemia. Neurosci. Lett. 2010, 473, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Lakunina, V.A.; Burnysheva, K.M.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. Changes in the receptor function of Na, K-ATPase during hypoxia and ischemia. Mol. Biol. 2017, 51, 148–154. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Mitkevich, V.A.; Lakunina, V.A.; Anashkina, A.A.; Spirin, P. V; Rubtsov, P.M.; Prassolov, V.S.; Bogdanov, N.B.; Hänggi, P.; Fuller, W. Cysteine residues 244 and 458–459 within the catalytic subunit of Na, K-ATPase control the enzyme’s hydrolytic and signaling function under hypoxic conditions. Redox Biol. 2017, 13, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Duan, Q.; Xie, Z. SH2 ligand-like effects of second cytosolic domain of Na/K-ATPase α1 subunit on Src kinase. PLoS One 2015, 10, e0142119. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflügers Arch. J. Physiol. 2009, 457, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, L.; Tidow, H.; Clausen, M.J.; Nissen, P. Na+, K+-ATPase as a docking station: protein–protein complexes of the Na+, K+-ATPase. Cell. Mol. Life Sci. 2013, 70, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Karpova, L. V; Bulygina, E.R.; Boldyrev, A.A. Different neuronal Na+/K+-ATPase isoforms are involved in diverse signaling pathways. Cell Biochem. Funct. Cell. Biochem. its Modul. by Act. agents or Dis. 2010, 28, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Rognant, S.; Kravtsova, V. V.; Bouzinova, E. V.; Melnikova, E. V.; Krivoi, I.I.; Pierre, S. V.; Aalkjaer, C.; Jepps, T.A.; Matchkov, V. V. The microtubule network enables Src kinase interaction with the Na,K-ATPase to generate Ca2+ flashes in smooth muscle cells. Front. Physiol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Oh, D.; Dubey, A.K.; Yao, M.; Yang, B.; Groves, J.T.; Sheetz, M. EGFR family and Src family kinase interactions: mechanics matters? Curr. Opin. Cell Biol. 2018, 51, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Tverskoi, A.M.; Barykin, E.P.; Petrovskaya, A. V; Strelkova, M.A.; Leonova, O.G.; Anashkina, A.A.; Tolstova, A.P.; Adzhubei, A.A.; Bogdanova, A.Y. Na, K-ATPase acts as a beta-amyloid receptor triggering src kinase activation. Cells 2022, 11, 2753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Yu, H.; Larre, I.; Dube, P.R.; Kennedy, D.J.; Wilson Tang, W.H.; Westfall, K.; Pierre, S. V.; Xie, Z.; et al. Regulation of Na/K-ATPase expression by cholesterol: isoform specificity and the molecular mechanism. Am. J. Physiol. Cell Physiol. 2020, 319, C1107–C1119. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Ouabain-induced endocytosis and signal transduction of the Na/K-ATPase. Front. Biosci. 2005, 10, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Zhuravleva, Z.D.; Ilina, M.A.; Boikov, S.I.; Stepanenko, Y.D.; Karelina, T. V.; Antonov, S.M. Unveiling the Role of Cholesterol in Subnanomolar Ouabain Rescue of Cortical Neurons from Calcium Overload Caused by Excitotoxic Insults. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Akkuratov, E.E.; Westin, L.; Vazquez-Juarez, E.; de Marothy, M.; Melnikova, A.K.; Blom, H.; Lindskog, M.; Brismar, H.; Aperia, A. Ouabain Modulates the Functional Interaction Between Na,K-ATPase and NMDA Receptor. Mol. Neurobiol. 2020, 57, 4018–4030. [Google Scholar] [CrossRef] [PubMed]
- Madan, N.; Xu, Y.; Duan, Q.; Banerjee, M.; Larre, I.; Pierre, S. V; Xie, Z. Src-independent ERK signaling through the rat α3 isoform of Na/K-ATPase. Am. J. Physiol. Physiol. 2017, 312, C222–C232. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Akkuratov, E.E.; Bai, Y.; Gaskill, C.M.; Askari, A.; Liu, L. Cell signaling associated with Na(+)/K(+)-ATPase: activation of phosphatidylinositide 3-kinase IA/Akt by ouabain is independent of Src. Biochemistry 2013, 52, 9059–9067. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015. [Google Scholar] [CrossRef] [PubMed]
- Kondělková, K.; Vokurková, D.; Krejsek, J.; Borská, L.; Fiala, Z.; Ctirad, A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta medica (Hradec Kral. 2010, 53, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, G.; Fei, J.; Wu, Y.; Yan, J. Bufotalin ameliorates experimental Sjögren’s syndrome development by inhibiting Th17 generation. Naunyn. Schmiedebergs. Arch. Pharmacol. 2020, 393, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Jacob, P.L.; Leite, J.A.; Alves, A.K.A.; Rodrigues, Y.K.S.; Amorim, F.M.; Néris, P.L.N.; Oliveira, M.R.; Rodrigues-Mascarenhas, S. Immunomodulatory activity of ouabain in Leishmania leishmania amazonensis-infected Swiss mice. Parasitol. Res. 2013, 112, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.C.M.; Cavalcante-Silva, L.H.A.; De A Lima, É.; Galvão, J.G.F.M.; De A Alves, A.K.; Feijó, P.R.O.; Quintas, L.E.M.; Rodrigues-Mascarenhas, S. Marinobufagenin Inhibits Neutrophil Migration and Proinflammatory Cytokines. J. Immunol. Res. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.P.; Rumjanek, V.M. Ouabain affects the expression of activation markers, cytokine production, and endocytosis of human monocytes. Mediators Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Ramsdell, F.; Alderson, M.R. The activation antigen CD69. Stem Cells 1994, 12, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, I.M.; Tiniakos, D.; Kouloulias, V.; Zygogianni, A. The molecular basis of immuno-radiotherapy. Int. J. Radiat. Biol. 2023, 99, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Olona, A.; Hateley, C.; Guerrero, A.; Ko, J.H.; Johnson, M.R.; Anand, P.K.; Thomas, D.; Gil, J.; Behmoaras, J. Cardiac glycosides cause cytotoxicity in human macrophages and ameliorate white adipose tissue homeostasis. Br. J. Pharmacol. 2022, 179, 1874–1886. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, A.; Aminuddin, A.; Kado, T.; Takikawa, A.; Yamamoto, S.; Tsuneyama, K.; Igarashi, Y.; Ikutani, M.; Nishida, Y.; Nagai, Y. CD206+ M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat. Commun. 2017, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Nawaz, A.; Kado, T.; Bilal, M.; Kuwano, T.; Yamamoto, S.; Sasahara, M.; Jiuxiang, X.; Inujima, A.; Koizumi, K. Partial depletion of CD206-positive M2-like macrophages induces proliferation of beige progenitors and enhances browning after cold stimulation. Sci. Rep. 2018, 8, 14567. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; He, J.; Kisoh, K.; Hayashi, H.; Tanaka, S.; Si, N.; Zhao, H.Y.; Hirano, T.; Bian, B.; Takagi, N. Effects of active bufadienolide compounds on human cancer cells and CD4+CD25+Foxp3+ regulatory T cells in mitogen-activated human peripheral blood mononuclear cells. Oncol. Rep. 2016, 36, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Raviprakash, N.; Manna, S.K. Short-term exposure to oleandrin enhances responses to IL-8 by increasing cell surface IL-8 receptors. Br. J. Pharmacol. 2014, 171, 3339–3351. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, F.K.; Dube, P.; Kleinhenz, A.L.; Malhotra, D.; Gohara, A.; Drummond, C.A.; Tian, J.; Haller, S.T.; Xie, Z.; Kennedy, D.J. Proinflammatory effects of cardiotonic steroids mediated by NKA α-1 (Na+/K+-ATPase α-1)/Src complex in renal epithelial cells and immune cells. Hypertension 2019, 74, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante-Silva, L.H.A.; Carvalho, D.C.M.; de Almeida Lima, É.; Rodrigues-Mascarenhas, S. Ouabain inhibits p38 activation in mice neutrophils. Inflammopharmacology 2021, 29, 1829–1833. [Google Scholar] [CrossRef]
- Vieira, L.; Saldanha, A.A.; Moraes, A.M.; Oliveira, F.M. de; Lopes, D.O.; Barbosa, L.A. de O.; Ribeiro, R.I.M. de A.; Thomé, R.G.; Santos, H.B. dos; Villar, J.A.F.P.; et al. 21-Benzylidene digoxin, a novel digoxin hemi-synthetic derivative, presents an anti-inflammatory activity through inhibition of edema, tumour necrosis factor alpha production, inducible nitric oxide synthase expression and leucocyte migration. Int. Immunopharmacol. 2018, 65, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Zhakeer, Z.; Hadeer, M.; Tuerxun, Z.; Tuerxun, K. Bufalin Inhibits the Inflammatory Effects in Asthmatic Mice through the Suppression of Nuclear Factor-Kappa B Activity. Pharmacology 2017, 99, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yu, F.; Wu, W.; Liu, J.; Li, J.; Guo, F.; Xu, L.; Wang, F.; Cui, X. Bufalin enhances the killing efficacy of NK cells against hepatocellular carcinoma by inhibiting MICA shedding. Int. Immunopharmacol. 2021, 101. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Fu, B.D.; Shen, H.Q.; Yi, P.F.; Zhang, L.Y.; Lv, S.; Guo, X.; Xia, F.; Wu, Y.L.; Wei, X. Bin Telocinobufagin enhances the Th1 immune response and protects against Salmonella typhimurium infection. Int. Immunopharmacol. 2015, 25, 353–362. [Google Scholar] [CrossRef]
- Da Silva, J.M.C.; Azevedo, A.D.N.; Barbosa, R.P.D.S.; Teixeira, M.P.; Vianna, T.A.G.; Fittipaldi, J.; Cabral, V.R.; De Paiva, L.S. Ouabain Decreases Regulatory T Cell Number in Mice by Reducing IL-2 Secretion. Neuroimmunomodulation 2019, 26, 188–197. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.M.C.; Campos, M.L.A.; Teixeira, M.P.; da Silva Faustino, R.; Aleixo, R.C.; Cavalcante, F.J.P.; Gomes, L.R.O.; de Albuquerque, L.Z.; das Neves Azevedo, A.; Cabral, V.R.; et al. Ouabain pre-treatment modulates B and T lymphocytes and improves survival of melanoma-bearing animals. Int. Immunopharmacol. 2020, 86. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Baek, S.; Lee, J.; Lee, J.; Lee, D.G.; Park, M.K.; Cho, M. La; Park, S.H.; Kwok, S.K. Digoxin ameliorates autoimmune arthritis via suppression of Th17 differentiation. Int. Immunopharmacol. 2015, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.R.; Leung, M.W.L.; Huang, P.; Ryan, D.A.; Krout, M.R.; Malapaka, R.R.V.; Chow, J.; Manel, N.; Ciofani, M.; Kim, S. V.; et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 2011, 472, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kitani, A.; Fuss, I.; Strober, W. Cutting edge: regulatory T cells induce CD4+ CD25− Foxp3− T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-β. J. Immunol. 2007, 178, 6725–6729. [Google Scholar] [CrossRef]
- Karaś, K.; Sałkowska, A.; Walczak-Drzewiecka, A.; Ryba, K.; Dastych, J.; Bachorz, R.A.; Ratajewski, M. The cardenolides strophanthidin, digoxigenin and dihydroouabain act as activators of the human RORγ/RORγT receptors. Toxicol. Lett. 2018, 295, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, J.; Chen, S.; Meng, F.; Ning, J.; Sun, S. Oleandrin Induces Immunogenic Cell Death Via the PERK/elF2α/ATF4/CHOP Pathway in Breast Cancer. 2020.
- Rodrigues-Mascarenhas, S.; Dos Santos, N.F.; Rumjanek, V.M. Synergistic effect between ouabain and glucocorticoids for the induction of thymic atrophy. Biosci. Rep. 2006, 26, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Smolarczyk, R.; Cichoń, T.; Pilny, E.; Jarosz-Biej, M.; Poczkaj, A.; Kułach, N.; Szala, S. Combination of anti-vascular agent-DMXAA and HIF-1α inhibitor-digoxin inhibits the growth of melanoma tumors. Sci. Rep. 2018, 8, 7355. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Chen, L.; Li, L.; Huang, Y. Restoration and enhancement of immunogenic cell death of cisplatin by coadministration with digoxin and conjugation to HPMA copolymer. ACS Appl. Mater. Interfaces 2019, 12, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- de Paiva, L.S.; da Costa, K.M.; do Canto, F.B.; Cabral, V.R.; Fucs, R.; Nobrega, A.; Rumjanek, V.M. Modulation of mature B cells in mice following treatment with ouabain. Immunobiology 2011, 216, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.M.C.; das Neves Azevedo, A.; dos Santos Barbosa, R.P.; Vianna, T.A.G.; Fittipaldi, J.; Teixeira, M.P.; do Canto, F.B.; da Costa, K.M.; Pozzatti, R.R.; Cabral, V.R. Dynamics of murine B lymphocytes is modulated by in vivo treatment with steroid ouabain. Immunobiology 2016, 221, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-L.; Chou, J.-S.; Chen, Y.-L.; Hsueh, S.-C.; Chung, H.-Y.; Lee, M.-H.; Chen, C.-P.; Lee, M.-Z.; Hou, H.-T.; Lu, H.-F. Bufalin enhances immune responses in leukemic mice through enhancing phagocytosis of macrophage in vivo. In Vivo (Brooklyn). 2018, 32, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L.; Smith, C.I.E.; Persson, U. Functional Characterization of Lanatoside-C-Responsive Cells. Scand. J. Immunol. 1978, 8, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Radosinska, J.; Vrbjar, N. Erythrocyte deformability and Na, K-ATPase activity in various pathophysiological situations and their protection by selected nutritional antioxidants in humans. Int. J. Mol. Sci. 2021, 22, 11924. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.; Hasse, W. Quantitative aspects of ouabain binding to human erythrocyte and cardiac membranes. J. Physiol. 1975, 251, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; D’Urso, G.; Nicolini, G.; Forini, F.; Pellegrino, M.; Montali, U. Erythrocyte sodium pump stimulation by ouabain and an endogenous ouabain-like factor. Cell Biochem. Funct. 2007, 25, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Lopina, O.D.; Tverskoi, A.M.; Klimanova, E.A.; Sidorenko, S.V.; Orlov, S.N. Ouabain-induced cell death and survival. Role of α1-Na, K-ATPase-mediated signaling and [Na+] i/[K+] i-dependent gene expression. Front. Physiol. 2020, 11, 1060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-D.; Ma, Y.-J.; Liu, Q.-F.; Ye, T.-Z.; Meng, F.-Y.; Zhou, Y.-W.; Yu, G.-P.; Yang, J.-P.; Jiang, H.; Wang, Q.-S. Human erythrocyte lifespan measured by Levitt’s CO breath test with newly developed automatic instrument. J. Breath Res. 2018, 12, 36003. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.D.; Chuang, J.; Chaudhry, M.; Nie, Y.; Bai, F.; Sodhi, K.; Liu, J.; Shapiro, J.I. The potential role of Na-K-ATPase and its signaling in the development of anemia in chronic kidney disease. Am. J. Physiol. Physiol. 2021, 320, F234–F242. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Mallozzi, C.; Di Stasi, A.M.M.; Minetti, M. Peroxynitrite-dependent upregulation of SRC kinases in red blood cells: strategies to study the activation mechanisms. Methods Enzymol. 2005, 396, 215–229. [Google Scholar] [PubMed]
- Hazegh, K.; Fang, F.; Kelly, K.; Sinchar, D.; Wang, L.; Zuchelkowski, B.E.; Ufelle, A.C.; Esparza, O.; Davizon-Castillo, P.; Page, G.P. Erythrocyte mitogen-activated protein kinases mediate hemolytic events under osmotic and oxidative stress and in hemolytic diseases. Cell. Signal. 2022, 99, 110450. [Google Scholar] [CrossRef] [PubMed]
- Fenk, S.; Melnikova, E. V; Anashkina, A.A.; Poluektov, Y.M.; Zaripov, P.I.; Mitkevich, V.A.; Tkachev, Y. V; Kaestner, L.; Minetti, G.; Mairbäurl, H. Hemoglobin is an oxygen-dependent glutathione buffer adapting the intracellular reduced glutathione levels to oxygen availability. Redox Biol. 2022, 58, 102535. [Google Scholar] [CrossRef] [PubMed]
- Zaripov, P.I.; Kuleshova, I.D.; Poluektov, Y.M.; Sidorenko, S. V; Kvan, O.K.; Maksimov, G. V; Mitckevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. Metabolic Stress of Red Blood Cells Induces Hemoglobin Glutathionylation. Mol. Biol. 2023, 6, 1188–1198. [Google Scholar] [CrossRef]
- Emelyanov, I. V.; Konradi, A.O.; Lakatta, E.G.; Fedorova, O. V.; Bagrov, A.Y. Acute salt loading and cardiotonic steroids in resistant hypertension. Curr. Top. Membr. 2019, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O. V; Fadeev, A. V; Grigorova, Y.N.; Marshall, C.A.; Zernetkina, V.; Kolodkin, N.I.; Agalakova, N.I.; Konradi, A.O.; Lakatta, E.G.; Bagrov, A.Y. Cardiotonic Steroids Induce Vascular Fibrosis Via Pressure-Independent Mechanism in NaCl-Loaded Diabetic Rats. J. Cardiovasc. Pharmacol. 2019, 74, 436. [Google Scholar] [CrossRef]
- Raccah, D.; Dadoun, F.; Coste, T.; Vague, P. Decreased Na/K ATPase ouabain binding sites in red blood cells of patients with insulin-dependent diabetes and healthy north African control subjects: relationship with diabetic neuropathy. Horm. Metab. Res. 1996, 28, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Chirinos, J.A.; Castrellon, A.; Zambrano, J.P.; Jimenez, J.J.; Jy, W.; Horstman, L.L.; Willens, H.J.; Castellanos, A.; Myerburg, R.J.; Ahn, Y.S. Digoxin use is associated with increased platelet and endothelial cell activation in patients with nonvalvular atrial fibrillation. Hear. Rhythm 2005, 2, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Carnevale, R.; Nocella, C.; Bartimoccia, S.; Novo, M.; Cammisotto, V.; Piconese, S.; Santulli, M.; Vasaturo, F.; Violi, F.; et al. Digoxin and Platelet Activation in Patients With Atrial Fibrillation: In Vivo and In Vitro Study. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.; Hagberg, I.; Lyberg, T.; Gjesdal, K. Do cardiac glycosides affect platelet function? A flow cytometric study in healthy volunteers. Eur. J. Clin. Pharmacol. 2002, 58, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Tomasiak, M.; Stelmach, H.; Rusak, T.; Ciborowski, M.; Radziwon, P. The involvement of Na+/K(+)-ATPase in the development of platelet procoagulant response. Acta Biochim. Pol. 2007, 54, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase signaling: from specific ligands to general reactive oxygen species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef] [PubMed]
- Paula, S.; Tabet, M.R.; Ball, W.J. Interactions between cardiac glycosides and sodium/potassium-ATPase: three-dimensional structure− activity relationship models for ligand binding to the E2-pi form of the enzyme versus activity inhibition. Biochemistry 2005, 44, 498–510. [Google Scholar] [CrossRef]
- Škubník, J.; Pavlíčková, V.; Rimpelová, S. Cardiac glycosides as immune system modulators. Biomolecules 2021, 11, 659. [Google Scholar] [CrossRef] [PubMed]
- Upmanyu, N.; Dietze, R.; Kirch, U.; Scheiner-Bobis, G. Ouabain interactions with the α4 isoform of the sodium pump trigger non-classical steroid hormone signaling and integrin expression in spermatogenic cells. Biochim. Biophys. Acta (BBA)-Molecular Cell Res. 2016, 1863, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Rakhmetova, V.S.; Kapanova, G.; Tashenova, G.; Tulebayeva, A.; Akhenbekova, A.; Ibekenov, O.; Turgambayeva, A.; Xu, B. Bufalin-Mediated Regulation of Cell Signaling Pathways in Different Cancers: Spotlight on JAK/STAT, Wnt/β-Catenin, mTOR, TRAIL/TRAIL-R, and Non-Coding RNAs. Molecules 2023, 28, 2231. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Cai, T. Na+-K+–ATPase-mediated signal transduction: from protein interaction to cellular function. Mol. Interv. 2003, 3, 157. [Google Scholar] [CrossRef] [PubMed]
- Poluektov, Y.M.; Petrushanko, I.Y.; Undrovinas, N.A.; Lakunina, V.A.; Khapchaev, A.Y.; Kapelko, V.I.; Abramov, A.A.; Lakomkin, V.L.; Novikov, M.S.; Shirinsky, V.P.; et al. Glutathione-related substances maintain cardiomyocyte contractile function in hypoxic conditions. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O. V.; Ishkaraeva, V. V.; Grigorova, Y.N.; Reznik, V.A.; Kolodkin, N.I.; Zazerskaya, I.E.; Zernetkina, V.; Agalakova, N.I.; Tapilskaya, N.I.; Adair, C.D.; et al. Antibody to Marinobufagenin Reverses Placenta-Induced Fibrosis of Umbilical Arteries in Preeclampsia. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O. V.; Emelianov, I. V.; Bagrov, K.A.; Grigorova, Y.N.; Wei, W.; Juhasz, O.; Frolova, E. V.; Marshall, C.A.; Lakatta, E.G.; Konradi, A.O.; et al. Marinobufagenin-induced vascular fibrosis is a likely target for mineralocorticoid antagonists. J. Hypertens. 2015, 33, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Espada, J.; Martín-Pérez, J. An update on Src family of nonreceptor tyrosine kinases biology. Int. Rev. Cell Mol. Biol. 2017, 331, 83–122. [Google Scholar] [PubMed]
- De Paiva, L.S.; Costa, K.M. da; Canto, F.B. do; Cabral, V.R.; Fucs, R.; Nobrega, A.; Rumjanek, V.M. Modulation of mature B cells in mice following treatment with ouabain. Immunobiology 2011, 216, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.M.C.; Campos, M.L.A.; Teixeira, M.P.; da Silva Faustino, R.; Aleixo, R.C.; Cavalcante, F.J.P.; Gomes, L.R.O.; de Albuquerque, L.Z.; das Neves Azevedo, A.; Cabral, V.R. Ouabain pre-treatment modulates B and T lymphocytes and improves survival of melanoma-bearing animals. Int. Immunopharmacol. 2020, 86, 106772. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Formulas of the main cardenolides (digoxin (DGX) and ouabain (OBN)) and bufadienolides (bufalin (BUF) and marinobufagenin (MBG)).
Figure 1.
Formulas of the main cardenolides (digoxin (DGX) and ouabain (OBN)) and bufadienolides (bufalin (BUF) and marinobufagenin (MBG)).
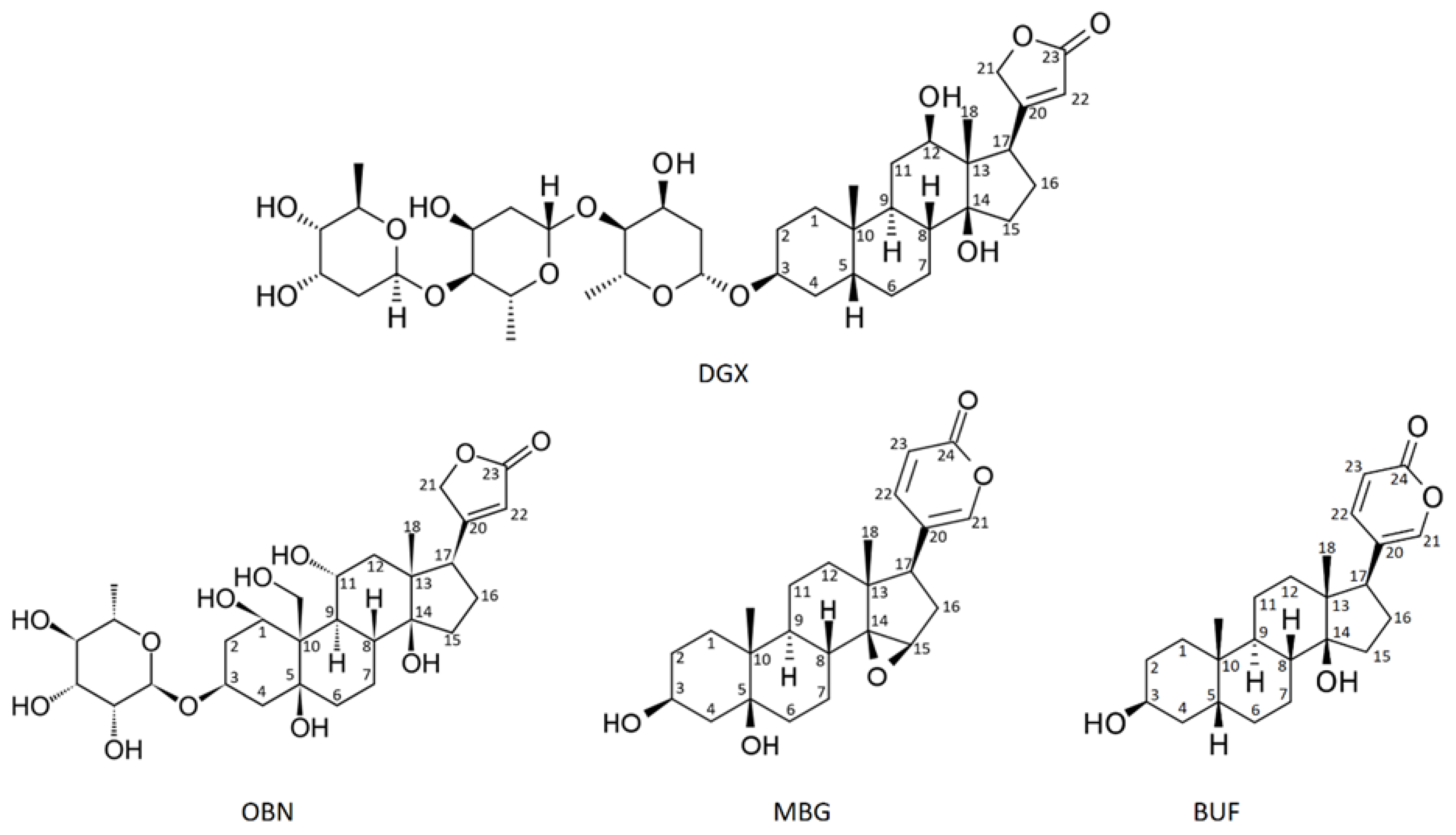
Figure 2.
CTS binding to ouabain-sensitive and ouabain-resistant Na,K-ATPase, according to [24]. The ouabain (OBN) binding localization in sensitive Na,K-ATPase is colored in blue, ouabain binding localization in resistant (rat) Na,K-ATPase is colored in red.
Figure 2.
CTS binding to ouabain-sensitive and ouabain-resistant Na,K-ATPase, according to [24]. The ouabain (OBN) binding localization in sensitive Na,K-ATPase is colored in blue, ouabain binding localization in resistant (rat) Na,K-ATPase is colored in red.
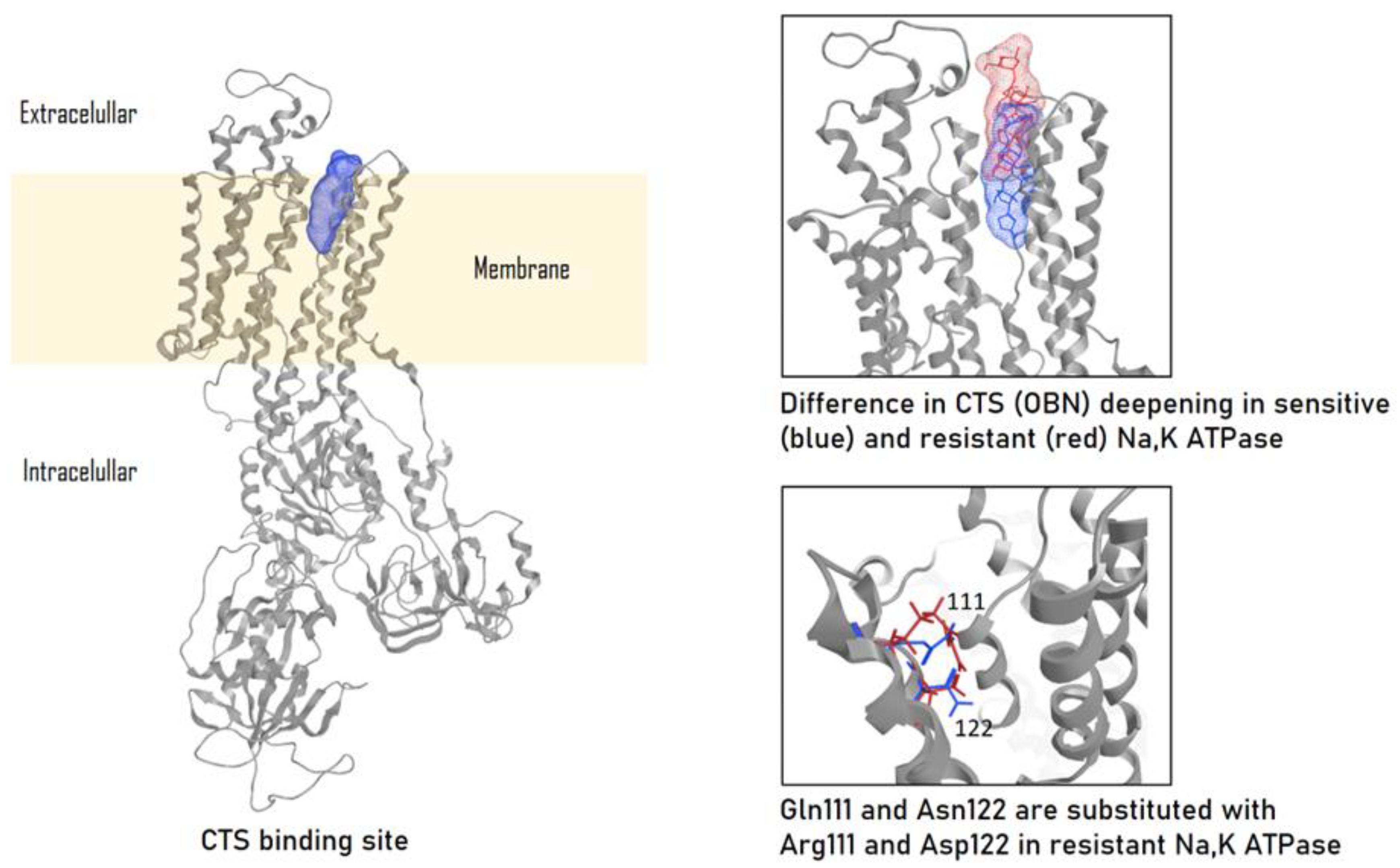
Figure 3.
Scheme of the CTS effects on the Na,K-ATPase functioning.
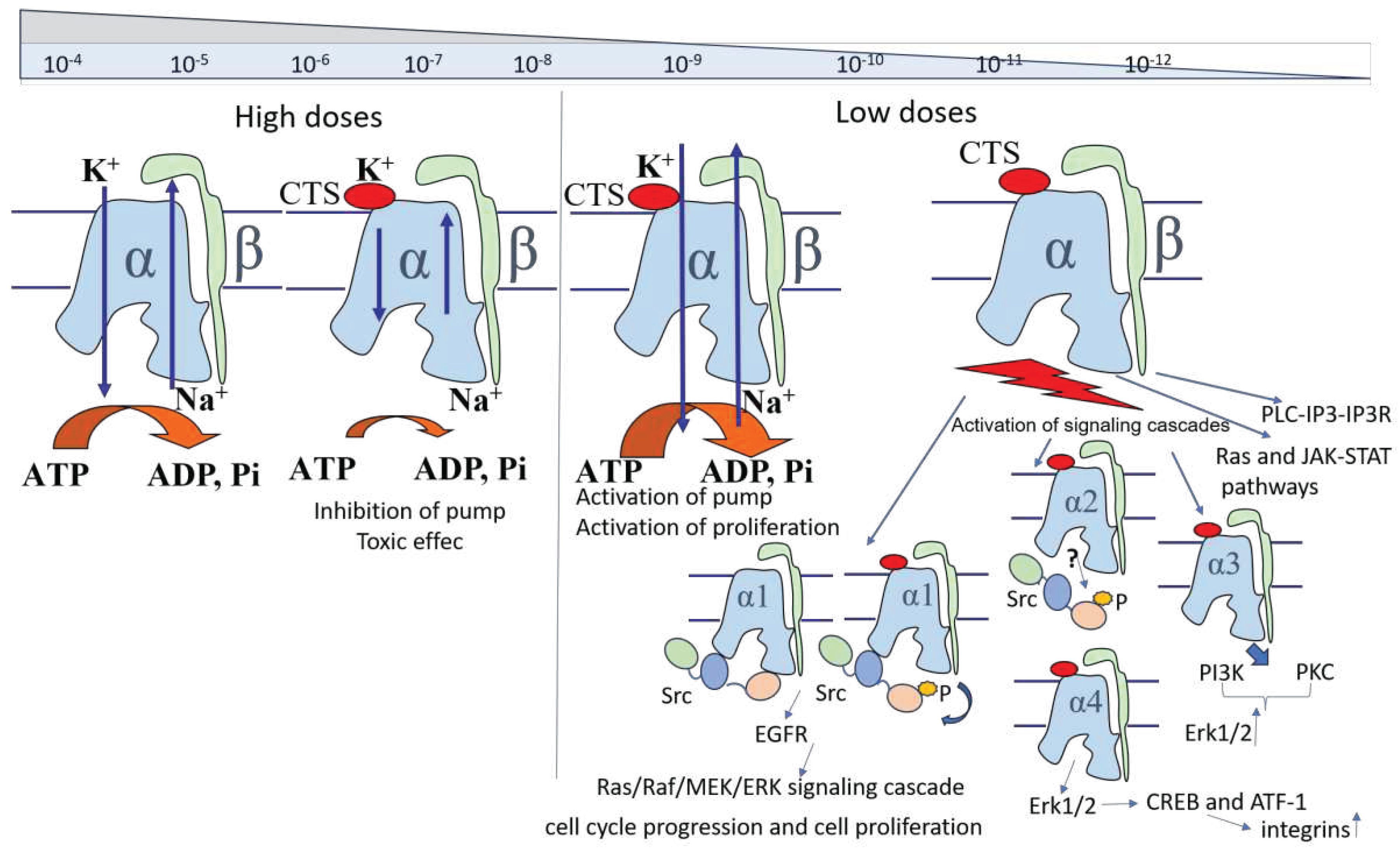

Table 1.
Properties of different isoforms of the catalytic alpha subunit of Na,K-ATPase.
| Isoforms | Ouabain Sensitivity [29,41] |
K+ K 0.5 mM [31,32] |
Na+ K 0.5 mM [31,34,35] | Oxidation Sensitivity [37,38,42,43,44] |
|---|---|---|---|---|
| α 1 | low | 1 | 10 | |
| low | ||||
| α 2 | 3 | 10 | high | |
| intermediate[29,41]/high [29,41] | ||||
| α 3 | 1 | 25-50 | high | |
| high | ||||
| α 4 | 2.14 ±1.14*[45] | 9.13 ± 1.81*[45] | ||
| Low (300 нМ Кd) [45] Ki 1.6 ± 1 × 10− 9 M [46] |
5.9 +/- 1.1 [28] |
13.5 +/- 1.3 [28] |
* Na+ inhibition of [3 H+] ouabain binding and K+ inhibition of [3 H+] ouabain binding.
Table 2.
Distribution of Na,K-ATPase isoforms in the blood cells.
| Cells/Alpha subunit | α1 | α2 | α3 | α4 |
|---|---|---|---|---|
| Red blood cells | +++ [87] | - [87,88] | ++ [87] | - [88] |
| Leucocytes | ||||
| Mononucleacytes/macrophages | +++ [87,88] | ++ [87] | ++ [87] | + [87] |
| Granular leucocytes | +? [88] | +- [88] | +- [88] | - [88] |
| Lymphocytes | ||||
| NK-cells | ++ [87] | + [87] | + [87] | + [87] |
| CD4 Cells | ++ [87] | ++ [87] | ++ [87] | + [87] |
| CD8 Cells | ++ [87] | ++ [87] | ++ [87] | + [87] |
| T-reg | +? [88] | +- [88] | +? [88] | +- [88] |
| Platelets | ++ [87] | + [87] | + [87] | + [87] |
Designations: - - no mRNA or protein was detected; +- - negligible isosyme mRNA content can be detected, isozyme not detected; +? - mRNA detected, no detected isozyme reported; + - negligible isosyme content can be detected; ++ - both mRNA and protein detected; +++ - predominant isoform.
Table 3.
Cellular and systemic effects of cardiotonic steroids on different blood cells.
| Cell type | CTS | Cellular and systematic effects |
|---|---|---|
| Erythrocytes | OBN | (1) Play an important role in the development of anemia in chronic kidney disease [126] through the increase in ROS levels, impaired erythrocyte deformability and lifespan reduction |
| MBG | (1) Released in response of hypervolemia [132] (2) An increase in endogenous marinobufagenin in rats with inducted 2 diabetes mellitus leads to a 30% decrease in erythrocyte Na,K-ATPase activity [132] |
|
| DGX | (1) Did not stimulate the Na,K-ATPase activity [123] | |
| Neutrophils |
OBN | (1) Inhibiting migration [97] (2) Reducing chemotaxis induced by chemotactic peptide fMLP [102] |
| MBG | (1) Inhibiting migration [92] | |
| 21-Benzylidene digoxin | (1) Inhibiting of TNF–α production release (anti-inflammatory and edema inhibiting effects) [103]. Decrease in the inducible nitric oxide synthase (iNOS) expression in the paw pads of mice | |
| BUF | (1) Attenuation of hyperresponsiveness. Decreased total number of inflammatory cells [104] | |
| Eosinophils | BUF | (1) Attenuation of hyperresponsiveness. Decreased total number of inflammatory cells [104] |
| Macrophages |
OBN | (1) Reduced TNF-α and IFN-γ levels [91] (2) Cell death of monocyte-derived macrophages [100] (dose-dependent toxic effect on human macrophages) (3) Produced higher levels of IL-1 β and TNF- α, IL-10 and VEGF. Increased expression of surface activation markers [99] (4) Decrease of macrophage mannose receptor CD206 (marker for adipose tissue macrophages) [97] |
| MBG | (1) Attenuation of proinflammatory cytokines [92] | |
| Oleandrin | (1) Enhanced biological responses to IL-8, without cytoxicity [102] | |
| Telocinobufagin | (1) Induce oxidative burst and enhanced NF-KB activation. [101] | |
| Monocytes | MBG | (1) Reduced levels of proinflammatory cytokines IL-1β, IL-6, and TNF- α [92]. |
| NK-cells | Oleandrin | (1) Balancing stimulating and inhibitory receptors on the surface of NK cells and indirectly activates NK cells by inhibiting MICA shedding [105] |
| B-lymphocytes | OBN | (1) Decrease in the bone marrow cellularity with diminution of the mature B cell subpopulation while the other B cell subpopulations were preserved [148] (2) Maintenance of the number and percentage of B lymphocytes in peripheral organs of melanoma-injected mice [108] |
| T-killers | OBN | (1) Did not alter the percentage and absolute numbers of CD8+T lymphocytes [108] |
| T-helpers | OBN | (1) Reduced number of CD4+ T-lymphocytes in the spleen [107] (2) Did not alter the percentage and absolute numbers of CD4+T lymphocytes [108] |
| Telocinobufagin | (1) Enhancing a Th1 immune response to control intracellular infections [106] | |
| T-regs | OBN | (1) Reduced number by decreased Il-2 production by T-lymphocytes [107] (2) Did not alter the percentage and absolute numbers of CD4+T lymphocytes [108] (3) |
| DihydroOBN | (1) Upregulation of IL17A and IL17F expression and enhanced IL17 secretion [112] | |
| DGX | (1) Reduced expression of proinflammatory cytokines. Can regulate Th17 and reciprocally promote Treg cells and suppress joint inflammation and bone erosion in CIA. [109] (2) Reduced in vitro differentiation and LPS-stimulated IgG production. Suppression of joint inflammation and bone erosion in CIA [109] (3) Upregulation of IL17A and IL17F expression and enhanced IL17 secretion [112] (4) Inhibiting RORγt translational activity. [110] |
|
| BUF | (1) Inhibiting polarization [90] (2) Inhibiting secretion of cytokines IL-17 and IFN-γ [90] |
|
| Strophantin | (1) Upregulation of IL17A and IL17F expression and enhanced IL17 secretion [112] | |
| Gamabufotalin | (1) Downregulation of the percentages of CD4+CD25+Foxp3+ Treg cells in mitogen-activated PBMCs [99] | |
| CD8+ cells | OBN | (1) Induce the death of immature double positive lymphocytes (CD4+CD8+) [114]. |
| DGX | (1) Inhibition of the growth of melanoma tumors in murine model [115] (2) Reversed the inability of Cisplatin to trigger calreticulin exposure, and HPMA copolymer-amplified Cisplatin-induced ATP release in melanoma mice model [116] |
|
| Oleandrin | (1) Inhibite tumor growth and increase tumor-infiltrating lymphocytes including dendritic cells and T cells [113] | |
| B cells | OBN | (1) Decrease the level of B-cells in bone marrow, spleen and peripheral blood in 24 hours Immunobiology. [117] (2) Regulate the dynamic of В-lymphocyte settling in peripheral organs. [118]. (3) Pre-treatment modulates B lymphocytes and improves survival of melanoma-bearing animals.[149] |
| BUF | (1) Increased B-cell proliferation from leukemic BALB/c mice [119,120] | |
| Platelets | OBN | (1) Rise in membrane curvature leading to the generation of a procoagulant activity [137] (due to inefficiently operating Na+/K(+)-ATPase and increased expression of phosphatidylserine) |
| DGX | (1) Activation of platelets in thrombosis-prone patients with heart failure and/or atrial fibrillation [136] (2) Induced calcium mobilization [134] (3) Increased levels of endothelial and platelet activation [134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



