Submitted:
06 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
Metagenomic analysis of Aedes aegypti and Ae. albopictus mosquitoes from diverse geographical regions of India revealed presence of several insect viruses of human interest. Most abundant reads found in Ae. aegypti mosquitoes were Phasi-Charoen like virus (PCLV), Choristoneura fumiferana granulovirus (CfGV), Cell fusing agent virus (CFAV) and Wenzhou sobemo-like virus 4 (WSLV4), whereas WSLV4 and CfGV constituted the highest percentage in Ae. albopictus viromes. Other reads which were of low percentage included Hubei mosquito virus 2, Porcine astrovirus 4 and Wild Boar astrovirus. PCLV and CFAV, which were found to be abundant in Ae. aegypti viromes, were absent in Ae. albopictus viromes. Among the viromes analyzed, Ae. aegypti sampled from Pune showed highest percentage (79.82%) of viral reads while Ae. aegypti mosquitoes sampled from Dibrugarh showed lowest percentage (3.47%). Shamonda orthobunyavirus, African swine fever virus, Aroa virus and Ilheus virus, having potential to infect vertebrates including humans, were also detected in both mosquito species, albeit in low read numbers. Reads of gemykibivirus, avian retrovirus, bacteriophages, herpesviruses and viruses infecting protozoans, algae, etc. were also detected in the mosquitoes. A high percentage of reads in the Ae. albopictus mosquito samples remained unclassified and warrant further investigation. Data generated in the present work may not only lead to studies to explain the influence of these viruses on replication and transmission of viruses of clinical importance but also find application as biocontrol agents against pathogenic viruses.
Keywords:
Virome
; Aedes aegypti
; Aedes albopictus
; PCLV
; CFAV
; CfGV
1. Introduction:
Arboviral diseases continue to plague human populations around the world, especially in the tropical and subtropical countries. Mosquito-borne viruses, i.e., dengue virus (DENV), Zika virus (ZIKV), West Nile virus (WNV), Japanese encephalitis virus (JEV), Chikungunya virus (CHIKV) and tick-borne viruses such as Crimean Congo hemorrhagic fever virus (CCHFV), Tick-borne encephalitis virus (TBEV), etc., contribute to the main burden. Aedes aegypti, the day biting mosquito, has emerged as a major concern in the last few decades as it transmits deadly viruses, i.e., DENV, CHIKV, Yellow fever virus (YFV) and ZIKV [1], which have tremendous public health importance. It is estimated that half the global population is under the threat of dengue and ~390 million people contract DENV infection annually, of which 96 million develop symptomatic disease [2].
Mosquitoes also harbour certain viruses that replicate within select arthropod host species, thus earning them the name “insect-specific viruses” (ISVs), which have the potential to modulate replication of arboviruses of human importance. Nhumirim virus, an insect-specific flavivirus (ISF), was not only found to suppress replication of DENV serotype-2 (DENV-2) and ZIKV in Aedes albopictus C6/36 cells but also affect vector competence of Ae. aegypti mosquitoes to DENV-2 and ZIKV [3]. Growing evidence suggests that ISVs may have been the ancestors of currently circulating human-infecting arboviruses, as proven by Alphaviruses belonging to Western equine encephalitis virus complex, viz., Eilat virus (EILV) and Taï Forest alphavirus (TALV) [4]. A genetic surveillance of flavivirus-specific sequences from Culex tritaeniorhynchus mosquitoes in Assam, India, revealed the presence of an ISV with highest sequence identity to Palm Creek virus [5]. Novel control measures to reduce virus transmission are emerging, including usage of Ae. aegypti endosymbiont Wolbachia pipientis. A cluster-randomized trial using Wolbachia-infected Ae. aegypti introgression into natural mosquito populations showed significant decrease in dengue incidence and hospitalizations ([6]. ISVs, similar to Wolbachia, are speculated to have potential to be used as biocontrol agents [7].
Metagenomics is an advancing field that involves genetic sampling of micro-organisms to have insight into their composition and diversity [8]. This reveals a plethora of organisms present in any microbial community in various environmental and clinical samples. Virome is the total of all the virus-specific sequences discovered in an organism, designated to their respective taxonomic virus units. Analysis of viromes as opposed to total metagenomes has revealed a greater richness in viral Operational Taxonomic Units (vOTUs), and this approach can thus reveal vastly diverse viruses that exist in any kind of sample [9]. Virome analysis of haematophagous arthropods (mosquitoes) is a lucrative approach to identify the presence of pathogens of interest, which often consist of human viruses and ISVs. Virome analysis may reveal human pathogens that may have been overlooked by surveillance measures and also discover viruses that might evolve into potential human pathogens [10]. It has been reported that Ae. aegypti and Cx. quinquefasciatus mosquitoes harbour core viromes that are distinct in diversity, with the former having a richer eukaryotic virome while the latter having a more diverse variety of phages [11]. Metaviromic analysis can employ the use of amplification to enhance the detection of different viral species when random anchored primers were used to amplify parts of viral genomes in a sequence-independent manner [12].
In a previous study, we reported abundance of Phasi Charoen-like phasivirus (PCLV) in Ae. aegypti populations throughout India, a country endemic to DENV and CHIKV [13]. Here, we report the metagenomic analysis of virome constituents of Aedes aegypti and Aedes albopictus mosquitoes sampled from geographically diverse regions of India.
2. Materials and Methods
2.1. Collection and storage of wild mosquitoes, larvae and pupae
Ae. aegypti and Ae. albopictus mosquitoes were collected from their natural breeding habitats and resting spots across various locations (Figure 1), using handheld-manual aspirators and brought to the Medical Entomology and Zoology Group, Indian Council of Medical Research – National Institute of Virology (ICMR-NIV), Pune. Ae. albopictus mosquitoes from Alappuzha, Kerala were collected and sent by the ICMR-NIV Kerala unit through a consistent cold chain. Adults were identified using entomological keys, sorted by species and sex, pooled (n=20) and stored at -80°C for further use. Larvae and pupae were reared to adults and processed similarly.
2.2. Viral RNA extraction, double-stranded complementary DNA (cDNA) preparation, and Sequence-Independent Single Primer Amplification (SISPA)
Pools of 20 adult female mosquitoes were processed for sequencing library preparation according to a previously published protocol [13], with a few modifications. In brief, mosquitoes were washed with sterile MilliQ water and homogenized in liquid Nitrogen using sterile mortar and pestle. Homogenized tissue was resuspended in 1X phosphate buffered saline (PBS) containing 5 mM magnesium chloride (MgCl2) and 1.4 mM Dithiothreitol (DTT) (Invitrogen), centrifuged at 17,000 g for 5 min at 4°C. The supernatant was filtered through a 0.22 µm syringe filter (Merck Millipore, USA) and treated with 20 Units (U) each of TURBO DNase and RNase I (Thermo Scientific, Massachusetts, USA) for 1 hour. Viral RNA was extracted using QIAamp Viral RNA Mini kit (Qiagen, Hilden, Germany). The eluted RNA was quantified using Qubit RNA HS assay kit and Qubit Flex Fluorometer (Invitrogen, Massachusetts, USA). A maximum of 100 nanograms (ng) RNA was used for first strand cDNA synthesis followed by RNaseH treatment. Second strand synthesis using Klenow fragment and Sequence-independent single-primer amplification (SISPA) (14) were carried out as described earlier (13). The primers used for SISPA are given in Table 1. Three separate PCR products were generated for each sample using P5, P12 and IDT-K sets of primers. The resulting amplified double-stranded cDNA (ds-cDNA) was purified using AMPure XP Reagent magnetic beads (Beckman Coulter, California, USA).
2.3. Library preparation and sequencing on Oxford Nanopore MinION Mk1C sequencer
Purified amplicons were used to prepare sequencing libraries using the Ligation Sequencing Kit, SQK-LSK109, and the Native Barcoding Kit, EXP-NBD104 (Oxford Nanopore Technologies (ONT), UK), according to manufacturer’s protocol. Briefly, 200 femtomoles (fmol) of the P5, P12 and IDT-K SISPA reaction products were subjected to DNA repair and end preparation using NEBNext Ultra II End repair/dA-tailing Module (New England Biolabs, USA). Equimolar amounts of the end-prepped DNA were subjected to barcoding using Native Barcoding Kit EXP-NBD104 and NEB Blunt/TA Ligase Master Mix (New England Biolabs, USA). The barcoded samples were pooled together and adapter ligation was carried out using adapter mix and the NEBNext Quick Ligation Module (New England Biolabs, USA). At each level, i.e., DNA repair/end preparation, native barcoding, pooling of barcoded samples and adaptor ligation, purification was carried out using AMPure XP Reagent magnetic beads (Beckman Coulter, California, USA) and quantitation using Qubit dsDNA HS assay kit and Qubit Flex fluorometer (Invitrogen, Massachusetts, USA). A final volume of 100-200 fmol of the adaptor ligated library was loaded for sequencing onto SpotON FLO-MIN106D flow cell (version R9.4.1) set on ONT MinION Mk1C device (Oxford, UK), with the MinKNOW interface. For Ae. albopictus samples, a final volume of 20 fmol was loaded onto a SpotON FLO-MIN111 flow cell (version R10.4) set on the Mk1C device. Basecalling was done in real time using Fast Basecalling option. Data acquisition was done keeping a minimum q-score cutoff of 8. Minimum read length was set at 200 bases. Demultiplexed and quality-controlled reads were stored into output files that were acquired in FASTQ format without compression. A maximum of 5000 reads per file was permitted and each run was carried out for at least 14 hours.
2.4. Analysis of raw metagenomics data
For each barcode, the FASTQ files containing raw reads were merged using Commander-NGS software (Genotypic Technology Pvt. Ltd., India) and processed using Porechop v0.2.4 software (https://github.com/rrwick/Porechop) for removal of adapter sequences. Trim Galore! v0.6.4_dev (https://github.com/FelixKrueger/TrimGalore) was used to trim primers from merged FASTQ files and further analyzed using EPI2ME Portal (https://epi2me. nanoporetech.com), a cloud-based algorithm including analytical workflow for taxonomic classification (ONT). All the viral species identified through EPI2ME that were given “no rank” through the classification pipeline and were checked through the “Commander-NGS” software for the authenticity of read classification. The merged FASTQ file for each sample was processed through Long Reads Variant Calling pipeline of the “Commander-NGS” software using RefSeq whole genome sequence for each virus species obtained through EPI2ME. This pipeline follows minimap2, samtools, and bcftools. Viral species that failed to generate any results through this pipeline were excluded from analysis. Percent genome coverage for each virus was calculated using samtools. Information on family and known hosts of the viruses was obtained through National Centre for Biotechnology Information (NCBI) Taxonomy Browser [15] and the Virus-Host DB [16]. To obtain consensus FASTA files of whole genome sequences, the merged FASTQ file representing metagenomics data of each location was uploaded to the Genome Detective online server for analysis by its Virus Tool version 2.49 [17]. Upon report generation, each virus detected by EPI2ME and those with >50% genome coverage were chosen and the corresponding FASTA files were downloaded for further analyses.
2.5. Sanger sequencing to obtain complete Cell fusing agent virus (CFAV) genome sequence
To obtain the complete genome sequence of CFAV, Reverse Transcription-PCR (RT-PCR) followed by Sanger sequencing was carried out for the 5’end of genome. Primers specific for the first ~550 bases of CFAV genome were designed (Table 1) using alignment of CFAV whole genome reference sequences from across the world (Figure 12) (18). Primer specificity was checked using the Basic Local Alignment Search Tool (BLAST, NCBI). Melting temperatures (Tm) and annealing temperature for the primers were checked using Thermo Fisher Scientific Tm Calculator (https://www.thermofisher.com/in/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/tm-calculator.html). Mosquitoes were homogenized in Minimum Essential Medium (HiMedia, India), centrifuged at 17,000g for 5 min at 4°C, and supernatant was used for viral RNA extraction using QIAamp Viral RNA Mini kit (Qiagen, Hilden, Germany). Ten µl of the extracted RNA was used to perform RT-PCR using SuperScript™ III One-Step RT-PCR System with Platinum™ Taq DNA Polymerase kit (Thermo Scientific, USA). Each 50 µl reaction composed of 25.0 µl of 2X Reaction Mix, 50 picomoles each of forward and reverse primers (Table 1), 3.0 µl Nuclease Free Water (NFW), 2.0 µl of the SuperScript III RT/Platinum Taq Mix, and 10 µl of template RNA. The reaction conditions were; 50°C for 45 min, 95°C for 10 min, followed by 35 cycles at 94°C for 1 min, 55°C for 1 min and 68°C for 90 sec, and a final extension at 68°C for 5 min. PCR products were analyzed on 2% agarose gel and purified using MinElute Gel Extraction Kit (Qiagen, Hilden, Germany). Purified PCR product was sequenced from both strands using Applied Biosystems™ BigDye™ Terminator v3.1 Cycle Sequencing Kit and analyzed using Applied Biosystems™ 3130xl Genetic Analyzer (Thermo Scientific, USA).
2.5. Phylogenetic analysis of metagenomics derived whole genome sequences of viruses
Consensus FASTA files were used for phylogenetic analyses using MEGA software, version 5.2. Whole genome reference sequences were downloaded from the NCBI database, and aligned using the ‘Multiple Alignment using Fast Fourier Transform’ (MAFFT) online server [18]. For sequences of yet unclassified viruses, BLAST searches were done to find the most identical nucleotide sequences, which were subsequently used for alignment and analyses. The same was also done for viruses that gave ambiguous results through BLAST, i.e., showing high identities with sequences of multiple taxa. Phylogenetic tree construction was done using MEGA v.5.2 according to the best fit substitution model deduced by the software for each alignment. BLAST searches were done for the generated whole genomes to find respective sequences showing highest percent nucleotide identity (PNI). PNIs for CFAV whole genome sequence were calculated using Clustal2.1 algorithm on MAFFT server.
3. Results:
3.1. Length and count statistics of sequencing data generated on ONT platform
Average quality score calculated for all the samples ranged between 10.75 and 12.67 (Tables 2a & 2b) and average read lengths ranged between 280 to 476 base pairs (bp). Viruses with most abundant reads found in Ae. aegypti mosquitoes were PCLV, Choristoneura fumiferana granulovirus (CfGV), CFAV and Wenzhou sobemo-like virus-4 (WSLV4). In Ae. albopictus, WSLV4 and CfGV constituted the highest percentage of virome reads and was conspicuous of the absence of PCLV and CFAV. There appeared to be no correlation between the number of analyzed reads and the final number of classified viral reads, as evidenced from Ae. aegypti samples from Dibrugarh, Assam, having 2,566,367 analyzed reads with 38,089 viral reads, and New Delhi having 1,728,868 analyzed reads with 140,179 viral reads (Table 2a). Among Ae. aegypti samples from different locations, Pune had the highest percentage of viral reads (79.82%) while Dibrugarh had the lowest percentage (3.47%). Ae. aegypti mosquito samples from Jodhpur had the highest percentage of PCLV reads (98.82%), while the mosquito samples from Dibrugarh had the lowest percentage (62.96%). Of the two Ae. albopictus viromes analyzed, the mosquito virome of Alappuzha, Kerala, samples had fewer WSLV4 reads. A high percentage of reads in both Ae. aegypti and Ae. albopictus samples remained unclassified, possibly indicating either an inability of the applied workflow to map the reads or representing potentially uncharacterized organisms.

Table 2.
a. Quality scores and classification of the metagenomics data for Ae. aegypti mosquitoes.
| Sampling location | Average quality score | Average sequence length | Reads analyzed | Reads classified | Reads unclassified | Cumulative reads (Eukaryota, Viruses, Bacteria and Archaea) | Virus reads |
|---|---|---|---|---|---|---|---|
| Dibrugarh | 12.29 | 378 | 2,566,367 | 1,148,796 | 1,409,637 | 1,097,121 | 38,089 |
| Jodhpur | 12.40 | 357 | 1,416,476 | 605,659 | 805,246 | 604,734 | 35,220 |
| Kolkata | 12.29 | 318 | 1,258,839 | 246,022 | 1,008,574 | 214,756 | 52,798 |
| Madurai | 11.91 | 286 | 7,786,762 | 3,749,144 | 3,997,735 | 2,945,046 | 808,370 |
| New Delhi | 12.41 | 280 | 1,728,868 | 761,242 | 960,985 | 595,398 | 140,179 |
| Patiala | 12.48 | 300 | 5,966,644 | 3,494,089 | 2,472,555 | 3,414,206 | 188,311 |
| Pune | 12.67 | 401 | 2,415,355 | 774,139 | 344,458 | 612,756 | 276,367 |
Table 2.
b. Quality scores and classification of the metagenomics data for Ae. albopictus mosquitoes.
Table 2.
b. Quality scores and classification of the metagenomics data for Ae. albopictus mosquitoes.
| Sampling location | Average quality score | Average sequence length | Reads Analysed | Reads classified | Reads unclassified | Cumulative reads (Eukaryota, Viruses, Bacteria and Archaea) | Total virus reads |
|---|---|---|---|---|---|---|---|
| Pune | 11.27 | 476 | 359,883 | 153,303 | 204,490 | 153,303 | 122,369 |
| Alappuzha | 10.75 | 296 | 514,027 | 185,691 | 306,661 | 185,691 | 61,982 |
3.2. Virus taxa identified through metaviromics
Tables 3a & 3b show viral species identified and results for the species-specific reads which were sorted and mapped against reference genomes from the Centrifuge database, through EPI2ME online server. Reads classified as Homo sapiens were the most abundant in viromes of mosquitoes from all locations. There was a wide diversity of viral species in all samples. The mosquito virome belonging to Patiala showed the highest species-wise diversity, while samples collected from Kolkata showed the lowest diversity. There was no correlation between the number of species identified and the region to which each location belonged. Interestingly, Dibrugarh mosquitoes had the maximum number of viral species with high reads of PCLV, CFAV, WSLV4, CfGV, Hubei mosquito virus 2, Porcine astrovirus 4 and Wild Boar astrovirus (Astrovirus wild boar/WBAstV-1/2011/HUN), with the six remaining species having only negligible reads (1-10). The mosquito virome derived from the Kolkata samples, appeared to have a few identified taxa at the species level. Similarly, there appeared to be no correlation of the number of viral taxa identified and the sample location.
Ae. albopictus mosquito viromes were found to lack two most prominent viruses found in viromes of Ae. aegypti mosquitoes, i.e., PCLV and CFAV. Otherwise, multiple viruses were found to be common to both species. Apart from WSLV4 and CfGV, Hubei mosquito virus 2 (HMV2) was the most abundant constituent of the two Ae. albopictus viromes.
Table 3.
a. Virus species and reads in Ae. aegypti viromes from different locations.
| Location | Patiala (Punjab) | Pune (Maharashtra) | Dibrugarh (Assam) | Madurai (Tamil Nadu) | New Delhi | Jodhpur (Rajasthan) | Kolkata (West Bengal) |
|---|---|---|---|---|---|---|---|
|
Species identified 
|
Phasi Charoen-like phasivirus | Phasi Charoen-like phasivirus | Phasi Charoen-like phasivirus | Phasi Charoen-like phasivirus | Phasi Charoen-like phasivirus | Phasi Charoen-like phasivirus | Phasi Charoen-like phasivirus |
| Choristoneura fumiferana granulovirus | Choristoneura fumiferana granulovirus | Cell fusing agent virus | Choristoneura fumiferana granulovirus | Choristoneura fumiferana granulovirus | Wenzhou sobemo-like virus 4 | Cell fusing agent virus | |
| Cell fusing agent virus | Shamonda orthobunyavirus | Wenzhou sobemo-like virus 4 | Shamonda orthobunyavirus | Cell fusing agent virus | Choristoneura fumiferana granulovirus | Choristoneura fumiferana granulovirus | |
| Avianendogenous retrovirus EAV-HP | Lactobacillus virus LP65 | Choristoneura fumiferana granulovirus | Hubei toti-like virus 10 | Shamonda orthobunyavirus | Tobacco mild green mosaic virus | Ilheus virus | |
| Ostreococcus lucimarinus virus 1 | Cyprinid herpesvirus 2 | Hubei mosquito virus 2 | Badu phasivirus | Lactobacillus virus LP65 | Aroa virus | Tobacco mosaic virus | |
| Lactobacillus virus LP65 | Ostreid herpesvirus 1 | Porcine astrovirus 4 | Serratia phage Muldoon | Wenzhou hepe-like virus 1 | Ilheus virus | Aroa virus | |
| Chrysochromulina ericina virus | Bacillus virus G | Astrovirus wild boar/WBAstV-1/2011/HUN | Synechococcus phage S-CAM4 | Drosophila immigrans Nora virus | Culex Flavi-like virus | Shamonda orthobunyavirus | |
| Cotesia congregata bracovirus | Lactobacillus virus LLKu | Ilheus virus | Salinivibrio phage CW02 | Sphingomonas phage PAU | Synechococcus phage S-CAM22 | Shuangao insect virus 7 | |
| Lactobacillus virus LLKu | Chilli ringspot virus | Astrovirus MLB2 | Mycobacterium phage UnionJack | Esparto virus | Chrysochromulina ericina virus | ||
| Badu phasivirus | Aotine betaherpesvirus 1 | Shamonda orthobunyavirus | Jacquemontia yellow vein virus | Trichoplusia ni ascovirus 2c | |||
| Wenzhou sobemo-like virus 4 | Tenacibaculum phage PTm1 | Porcine astrovirus 2 | Esparto virus | Golden Marseillevirus | |||
| Euproctis pseudoconspersa nucleopolyhedrovirus | Lactococcus virus KSY1 | Agrotis ipsilon multiple nucleopolyhedrovirus | Cotesia congregata bracovirus | ||||
| Bacillus virus G | Vibrio phage douglas 12A4 | Cyprinid herpesvirus 2 | |||||
| Rhinolophus associated gemykibivirus 2 | Cotesia congregata bracovirus | ||||||
| Pacmanvirus A23 | Acanthamoeba polyphaga mimivirus | ||||||
| Pandoravirus quercus | Amsacta moorei entomopoxvirus | ||||||
| Shamonda orthobunyavirus | Pandoravirus neocaledonia | ||||||
| Emiliania huxleyi virus 86 |
Table 3.
b. Virus species and reads in Aedes albopictus viromes.
| Location | Alappuzha (Kerala) | Pune (Maharashtra) |
|---|---|---|
| Species identified 
|
Wenzhou sobemo-like virus 4 | Wenzhou sobemo-like virus 4 |
| Choristoneura fumiferana granulovirus | Hubei mosquito virus 2 | |
| Hubei mosquito virus 2 | Choristoneura fumiferana granulovirus | |
| Tobacco mosaic virus | Wenzhou shrimp virus 9 | |
| Shamonda orthobunyavirus | Shamonda orthobunyavirus | |
| Cyprinid herpesvirus 2 | Mythimna unipuncta nucleopolyhedrovirus | |
| Elephant endotheliotropic herpesvirus 4 | ||
| Wilkie partiti-like virus 2 | ||
| Tobacco mild green mosaic virus | ||
| Tomato brown rugose fruit virus | ||
| Tomato mottle mosaic virus | ||
| Hubei picorna-like virus 34 | ||
| Hubei sobemo-like virus 9 | ||
| African swine fever virus | ||
| Pandoravirus quercus |
3.3. Viruses identified have a wide range of hosts
The host type showing the most abundance of viral taxa represented invertebrates, indicating a dominance of ISVs (Figure 2). Among the analysed viromes, mosquitoes collected from Patiala and New Delhi represented most ISVs. There were also viruses having both invertebrates and vertebrates including humans as hosts. Shamonda orthobunyavirus (SHAV) and African swine fever virus (ASFV), known to infect both invertebrate and vertebrate hosts, were detected in Ae. albopictus mosquitoes from Kerala while Ilheus virus (ILHV), capable of infecting both humans and invertebrates, was found in Ae. aegypti mosquitoes obtained from Dibrugarh, Jodhpur and Kolkata, though in low reads. Considering the abundance of RNA viruses identified through our protocol and the low number of reads classified as ILHV, it is unlikely that viable infectious virus was actually present, therefore, warrants further studies for confirmation. Similarly, Aroa virus (AROAV), a virus capable of infecting invertebrates and vertebrate hosts was detected in Ae. aegypti mosquitoes collected from Jodhpur and Kolkata, albeit with very low read numbers.
Several taxa were identified as having host range limited only to vertebrates. Four such viruses were found in mosquito samples obtained from Dibrugarh, i.e., Porcine astrovirus 2 (PoAstV2); Porcine astrovirus 4 (PoAstV4), Wild boar astrovirus (WBAstV), and a fish virus, Cyprinid herpesvirus 2 (CyHV-2). Patiala mosquitoes had two species of vertebrate viruses; the avian endogenous retrovirus EAV-HP and the Rhinolophus associated gemykibivirus 2. Ae. albopictus virome of Kerala had Elephant endotheliotropic herpesvirus-4 and CyHV-2. Interestingly, Astrovirus MLB2, a virus with host range limited only to humans, was found in the samples of Dibrugarh (<10 reads). However, more studies using RT-PCR and sequencing is needed for confirmation.
In terms of viruses having a bacteria-restricted host range, i.e., bacteriophages, all the Ae. aegypti viromes, except of the samples from Dibrugarh and Kolkata harboured different phage populations. These ranged from six phage species detected in Ae. aegypti mosquito samples from Pune to only one from the Jodhpur samples. The phage species in mosquitoes sampled from Pune included Lactobacillus virus LP65, Bacillus virus G, Lactobacillus virus LLKu, Tenacibaculum phage PTm1, Lactococcus virus KSY1, and Vibrio phage douglas 12A4. Lactobacillus virus LP65, Bacillus virus G and Lactobacillus virus LLKu phages were also found in mosquitoes sampled from Patiala. Mosquito samples from New Delhi had Lactobacillus virus LP65 and Sphingomonas phage PAU. The mosquitoes sampled from Madurai harboured a distinct phageome consisting of Serratia phage Muldoon, Synechococcus phage S-CAM4, Salinivibrio phage CW02, and Mycobacterium phage UnionJack. Mosquitoes collected from Jodhpur had only Synechococcus phage S-CAM22. Thus, phages infecting Lactobacillus and Lactococcus were found in the viromes of samples from Pune, Patiala and New Delhi, indicating a possible presence of these bacteria in the mosquitoes. Phages that infect members of the bacterial family Vibrionaceae were found in mosquito viromes of Pune and Madurai. Mosquitoes sampled from Madurai also exhibited phages infecting bacteria belonging to taxa Serratia; Synechococcus and Mycobacterium, indicating a possible circulation of these bacteria in the location.
Viruses infecting protozoa were also detected in all the viromes but in negligible numbers (1-10). Other than the giant viruses Pandoravirus neocaledonia (Pune) and P. quercus (Patiala), Pacmanvirus A23 and Acanthamoeba polyphaga mimivirus (both infecting Acanthamoeba) were found in Patiala and Pune Ae aegypti, respectively. The Pacmanvirus A23 infects Acanthamoeba castellani, which is also a host for viruses of the Pandoravirus genus. P. quercus, was also found in the virome of Ae. albopictus mosquitoes from Kerala, whereas Golden Marseillevirus was found in the mosquito virome of New Delhi. Presence of giant viruses in the mosquitoes could be from aquatic habitats of the mosquito larvae that are often shared by their protozoan hosts.
Viruses having eukaryotic algae as hosts were detected in Jodhpur and Patiala mosquitoes indicating a possible presence of their hosts Ostreococcus lucimarinus, Chrysochromulina ericina and Emiliania huxleyi in the mosquitoes or their larval/pupal stages.
Mosquito viromes also showed presence of plant viruses. Kerala Ae. albopictus mosquitoes had four taxa belonging to genus Tobamovirus, i.e., Tobacco mosaic virus (TMV), Tobacco mild green mosaic virus (TMGMV), Tomato brown rugose fruit virus and Tomato mottle mosaic virus. TMGMV and TMV were also detected in Jodhpur and Kolkata mosquito viromes respectively. Jacquemontia yellow vein virus (JacYVV), a Begomovirus, was found in Madurai mosquitoes, while a Potyvirus, the Chilli ringspot virus (ChiRSV), was found in Pune Ae. aegypti mosquitoes, possibly acquired from nectar of flowers on which male mosquitoes feed and subsequent transmission to their female mating partners
3.4. ISVs dominate the viromes of Ae. aegypti and Ae. albopictus
PCLV belonging to Order Bunyavirales and family Phenuiviridae, represented the highest abundance of reads among all the virus specific reads in viromes of Ae. aegypti across India (Figure 3). PCLV has a three-segmented genome, segment L (NC_038262.1), segment M (NC_038261.1), and segment S (NC_038263.1), corresponding to three Open Reading Frames (ORFs) for RNA-dependent RNA polymerase, glycoprotein, and nucleocapsid respectively. Jodhpur mosquitoes had the highest abundance (98.82%), Dibrugarh had the lowest (62.96%) while Ae. albopictus completely lacked PCLV reads. The Orthobunyavirus SHAV also has a three segmented genome. Similarly, Badu phasivirus (BADUV; Order Bunyavirales) possesses three genomic segments similar to PCLV and SHAV. WSLV4 had the highest abundance of comparative reads in Kerala and Pune Ae. albopictus viromes (Figure 3).
CFAV reads were found to be the most abundant in Dibrugarh (16.17%) and Kolkata (20.25%) while the lowest in Delhi (0.01%) Ae. aegypti mosquitoes. However, its abundance remained less as compared to PCLV. CfGV (Betabaculovirus) showed an almost comparable percent abundance in viromes of both Ae. aegypti and Ae. albopictus, with an average value of 4.97%. Mosquito samples of New Delhi showed the highest average value (11.35%), whereas Jodhpur had the lowest (0.12%) abundance of CfGV reads. Despite having highest abundance, we could not generate a consensus genomic sequence of >50% coverage, probably due to its relatively large virus genome (approximately 104,000 bp). BLAST analysis of generated CfGV sequences from this study showed highest nucleotide identity (99.97%) with CfGV reference sequence (NC_008168.1) from USA. Overall, the viruses with most abundant reads among viromes had invertebrates as their specific hosts, including PCLV, CfGV, CFAV, WSLV4 and HMV2, suggestive of a dominance of ISVs in the mosquito populations of India. Three (CfGV, WSLV4 & HMV2) out of the five viral species were found commonly in both the Aedes species. Reads corresponding to viruses of vertebrate host, i.e., SHAV, ILHV & CyHV-2 were very low and hence the genomes of these viruses could not be assembled (Figure 3).
3.5. Read depths and genome coverage
Figure 4 depicts the percent genomic coverage and depth, i.e., the number of times the entire genome or sequenced part of the genome was covered in the analysed viromes. Nearly full genome coverage was achieved for PCLV from the sequences generated in this study, with an average of 96.7%, 97.4% and 99.8% for L, M and S segments, respectively. While the coverage values were relatively uniform throughout the analysed viromes, there were intra-location and intra-segment variation in the depths of coverage, i.e., L segment of PCLV had a read depth varying from 982 in the Jodhpur mosquito virome to 21,494.6 in the Madurai virome. Similarly, Pune mosquito virome yielded a depth of 5149.21, 6159.9 and 19840.7 for segments L, S and M, respectively. The varying trend of read depth may be due to varying efficiency of the SISPA protocol. For CfGV, despite being present in all analysed Aedes viromes, genomic coverage was almost negligible (0.2% on an average), albeit with an average depth of sequence coverage of 51.7 (with Dibrugarh and Pune Ae. albopictus mosquitoes showing the lowest depth values). WSLV4 interestingly had an almost complete genome coverage in Ae. albopictus virome data with Pune and Kerala having depths of 10,732 and 20,405, respectively. Jodhpur and Dibrugarh Ae. aegypti viromes showing hits of WSLV4 had depths of coverage of 11.08 and 801.22, respectively. For CFAV, mosquitoes from Kolkata and Dibrugarh showed genome coverage depth of 443.69 and 277.78, respectively, with nearly 100% coverage for genomes. For New Delhi and Patiala mosquitoes, it was not possible to derive a complete consensus genomic sequence.
3.6. Aedes albopictus viromes were dominated by unclassified viruses
WSLV4 and HMV2 are both unclassified members of the realm Riboviria, which includes viruses possessing an RNA genome. Due to WSLV4 and HMV2 constituting a large proportion of the reads in both the Ae. albopictus viromes, we observed a dominance of unclassified virus families in terms of viral reads. The next most dominant virus family is Baculoviridae, owing to the dominance of CfGV reads in these viromes. Virgaviridae, constituted by viruses of genus Tobamovirus in the Kerala Ae. albopictus virome, has a notable percentage due to the total number of reads from all its members.
Members of family Phenuiviridae dominated the Ae. aegypti viromes from all the locations (Figure 5) and its abundance varied from 62.96% (Dibrugarh) to 98.83% (Jodhpur). The major contributors to this family included PCLV, BADUV among others. SHAV belonging to family Peribunyaviridae contributed to reads in the viromes of Ae. aegypti from Pune, Madurai and Delhi. A large number of reads matching to CfGV was present in all the analysed viromes. Reads representing Avian endogenous retrovirus EAV-HP (Family: Retroviridae), were found in Patiala mosquitoes indicating feeding of the mosquitoes on avian species. Members of family Astroviridae, PoAstV4 and the WBAstV, were detected in Dibrugarh Ae. aegypti mosquitoes. High numbers of reads of both the viruses enabled generation of consensus genomes with considerable coverage. These are unclassified mamastroviruses of genus Mamastrovirus. Family Flaviviridae had viral read contributions from several species. CFAV and ILHV was detected in Kolkata and Dibrugarh mosquitoes, while AROAV was found in Kolkata mosquitoes. Overall, Dibrugarh Ae. aegypti mosquitoes were found to possess the highest family-level diversity of viruses, however, comparatively low percentage of Phenuiviridae reads in these mosquitoes indicated competition among the viruses.
It appeared that Ae. albopictus mosquitoes had a far higher percentage of unclassified viruses than Ae. aegypti mosquitoes. Pune Ae. albopictus mosquitoes had a marginally higher percentage of unclassified viruses (96.57%) than Alappuzha mosquitoes (93.09%). Unclassified viruses contribute to the sequences of viruses that have not yet been assigned to any taxon. One can observe a uniform presence of such viruses throughout all the viromes described here, with read numbers greater than 10.
Details of unclassified viruses discovered in this study are given in Figure 6. Kerala Ae. albopictus mosquito virome displayed a wide variety of unclassified viruses, with hosts including mosquitoes (WSLV4, HMV2 and Wilkie partiti-like virus-2), beetles (Hubei picorna-like virus-34) and spiders (Hubei sobemo-like virus-9) while no unclassified viruses were detected in the virome of Ae. aegypti from Pune, despite having considerable viral reads (276,367). WSLV4 was the most frequently occurring unclassified virus found among all the viromes, followed by HMV2. The remaining viruses were specific to either Ae. aegypti (Hubei toti-like virus-10, Pacmanvirus A23, Shuangao insect virus-7 (SAIV7), Wenzhou hepe-like virus-1 and D. immigrans Nora virus) or Ae. albopictus (Wenzhou shrimp virus-9, Wilkie partiti-like virus-2, Hubei picorna-like virus-34 and Hubei sobemo-like virus-9). This shows distinct viromes of unclassified viruses were found this study and could possibly be due to host ranges limited to the respective mosquito species. HMV2 had more reads in both the Ae. albopictus viromes than Dibrugarh Ae. aegypti virome. However, BLAST results for HMV2 sequences generated from Ae. albopictus metagenomics data were confounding. Reads matching to WSLV4 were numerous in all the viromes except for Patiala mosquitoes, but with a large variation for each virome (in the order of 102, 103, 104 and 105 for Jodhpur & Dibrugarh Ae. aegypti and Kerala & Pune Ae. albopictus respectively). It is possible that the number of total viral reads or cumulative classified reads may be an influencing factor for the numbers of virus species or family-specific reads.
3.7. Phylogenetic analysis of virus sequences derived from Aedes viromes
Raw metagenomics data in the form of a merged FASTQ file was uploaded onto the Genome Detective online server for generating consensus sequences. Viral genomic sequences or sequences of genomic segments that gave a coverage of >50% were used for phylogenetic analysis. These sequences were processed for BLAST search to find highest PNI hits. Some FASTA sequences showed ambiguous results through BLAST searches, e.g., showed high PNI values with viruses of distantly related taxa, but with low query coverage. Some sequences also showed comparably high PNIs with more than one taxon at the species level, which further added to the ambiguity towards species specificity. These sequences were subsequently excluded and the remaining ones were used for phylogenetic analyses (Figure 7). For that, sequences were aligned using MAFFT and alignment were used to infer the best DNA nucleotide substitution model that could be used for constructing a phylogenetic tree. Tamura 3-Parameter was found to be the best fit model and hence used to construct Maximum Likelihood (ML) phylogenetic trees with 1000 bootstrap replicates. The alignment was also used to construct a Neighbour-Joining (NJ) phylogenetic tree (data not shown), resulting in the same clade arrangement as the respective ML trees.
The L, M and S segments formed three distinct clades (Figure 7). There appeared to be no subclades, except for the repeated clustering together of sequences from New Delhi and Kolkata. The M and L segment sequences yielded query coverage of 98-100%, whereas the S segment had a slightly lower coverage, ranging from 90% to 100%. PCLV sequences from Kolkata and New Delhi appeared to have some distinct evolutionary characteristics. The host range of PCLV limited only to Ae. aegypti is also supported by data presented in our study (Figure 3). PCLV formed a distinct clade from all the other viral consensus FASTA sequences analyzed in this study (Figure 7), with the sole exception of Wilkie partiti-like virus-2. Notably, Wilkie partiti-like virus-2 sequence from Kerala Ae. albopictus mosquitoes clustered along with the PCLV L and M sequences. However, a paired BLAST of both sequences gave no significant similarity, even with the least stringency parameters. Global Alignment using Needleman-Wunsch algorithm also yielded only 50% identity [19]. It showed a high query coverage of 91%, but a relatively low PNI (73.97%) with GenBank reference sequence, MF176346.1 (Wilkie partiti-like virus-2 strain mosWSCP53020, complete genome).
Distinct clades were also formed by WSLV4 and CFAV, as well as TMV and the astroviruses PoAstV4 and WBAstV. Notably, despite constituting only 0.056% of the total viral reads in the virome of Ae. albopictus from Kerala, nearly complete genome of TMV could be generated, indicating possibility of presence of infectious virions in mosquitoes.
3.8. PCLV sequences form two distinct clades separated by location and time
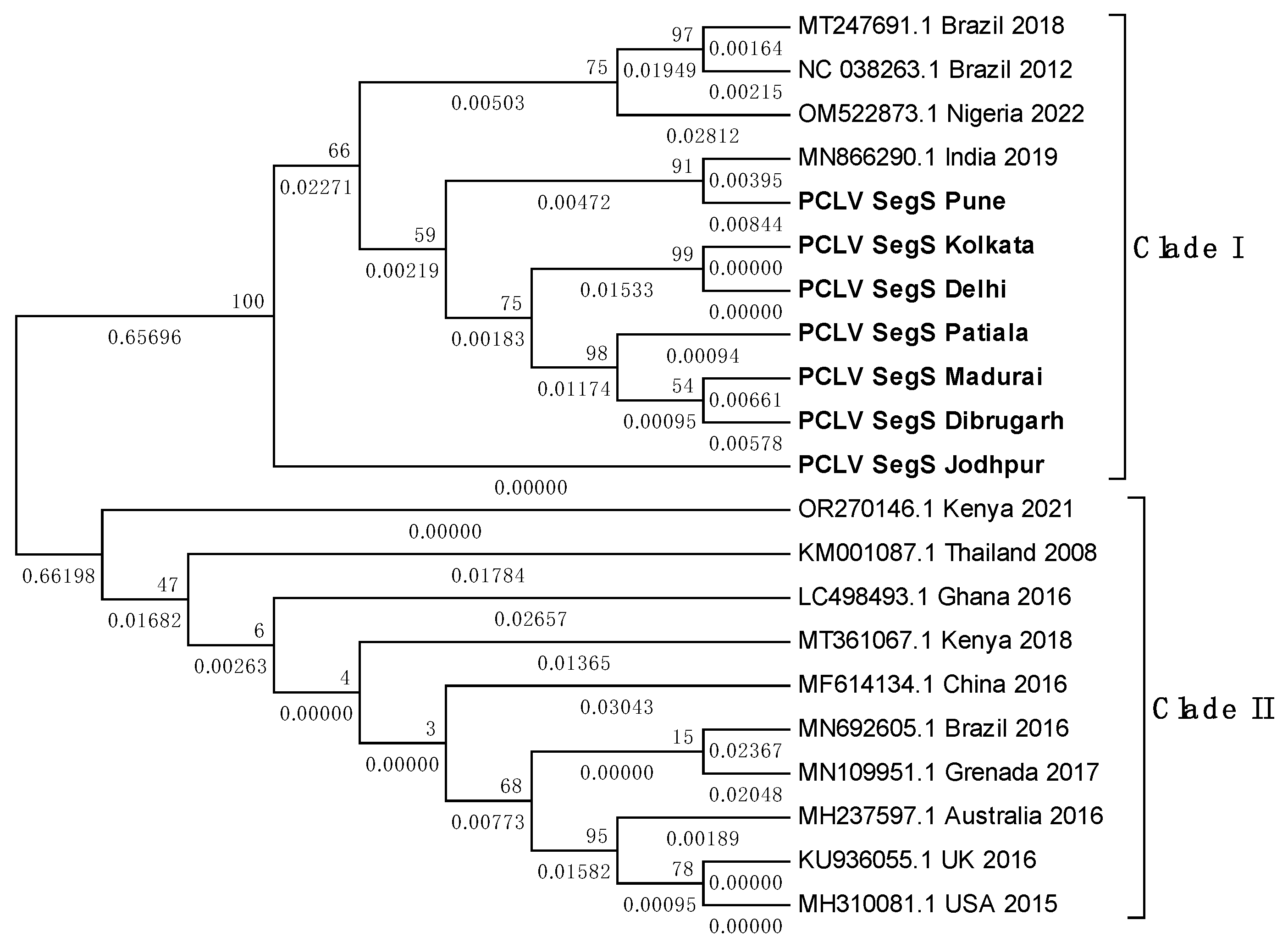
Overall, PCLV L, M and S segment sequences formed two distinct clades. Indian PCLV sequences clustered together in a single clade which also included sequences from Nigeria (for L segment), Nigeria, Brazil, and Kenya (for M segment) and Nigeria & Brazil (for S segment). Clade II of L segment sequences included sequences from Thailand, Kenya, China, Ghana, Trinidad & Tobago, Guadeloupe, Australia, USA and Brazil (2016) (Figure 8a); of M segment sequences (Figure 8b) included Brazil, China, Thailand, Kenya, Australia, USA, Trinidad & Tobago, Guadeloupe; and of S segment sequences (Figure 8c) included Kenya, Thailand, Ghana, China, Brazil, Grenada, Australia, United Kingdom and USA.
Despite the MEGA software deducing three different models for construction of the three different PCLV segments’ phylogenetic trees, the clade formation remained constant throughout. The Jodhpur sequence clustered separately from other Indian sequences and formed its own distinct subclade within clade I. The 2021 sequence from Kenya showed the least evolutionary distance (ED) from the common root of all the S segment sequences, and this could have implied that there is no temporal distribution of PCLV S segment mutation and evolution throughout the world. Instead, we see the three segments with independent evolutionary characteristics, with the L segment seemingly following a temporal increase of evolutionary divergence, while M and S segments apparently not following such a trend. The sequences from Brazil 2018 (MT247691.1) and Brazil 2012 clustered together in clade I, whereas the Brazil 2016 sequence (MN692605.1) clustered in clade II.
Figure 8.
a. Maximum Likelihood phylogenetic tree of Phasi Charoen-like phasivirus segment L sequences: The tree is displayed in the topology only format, and was constructed using the Tamura Nei model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/sequence submission are mentioned along with GenBank accession numbers for reference sequences.
Figure 8.
a. Maximum Likelihood phylogenetic tree of Phasi Charoen-like phasivirus segment L sequences: The tree is displayed in the topology only format, and was constructed using the Tamura Nei model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/sequence submission are mentioned along with GenBank accession numbers for reference sequences.
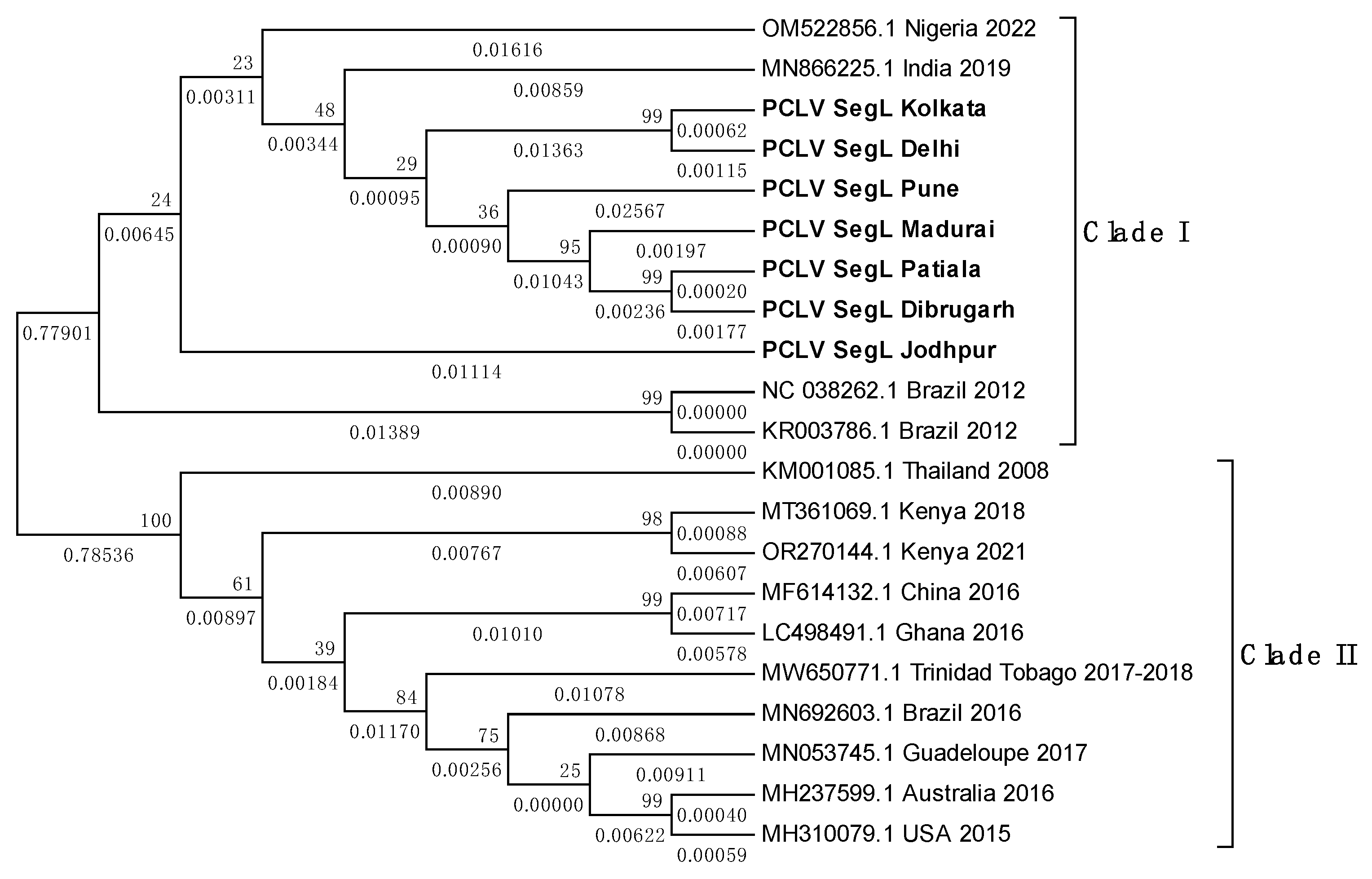
Figure 8.
b. Maximum Likelihood phylogenetic tree of PCLV segment M sequences: The tree is displayed in the topology only format, and was constructed using the General Time Reversible model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/sequence submission are mentioned along with GenBank accession numbers for reference sequences.
Figure 8.
b. Maximum Likelihood phylogenetic tree of PCLV segment M sequences: The tree is displayed in the topology only format, and was constructed using the General Time Reversible model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/sequence submission are mentioned along with GenBank accession numbers for reference sequences.
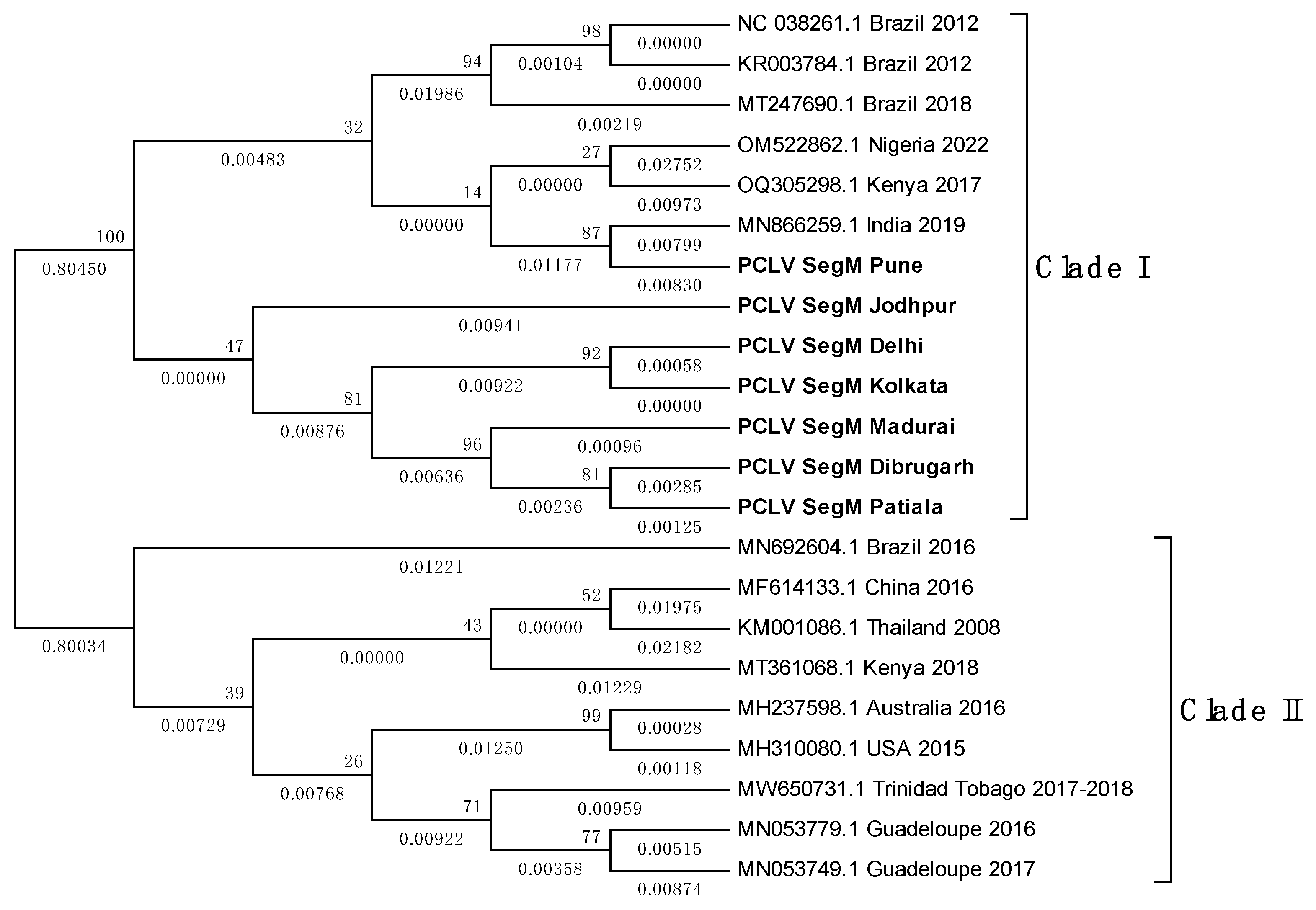
Figure 8.
c. Maximum Likelihood phylogenetic tree of PCLV segment S sequences: The tree is displayed in the topology only format, and was constructed using the Tamura 3-parameter, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/sequence submission are mentioned along with GenBank accession numbers for reference sequences.
Figure 8.
c. Maximum Likelihood phylogenetic tree of PCLV segment S sequences: The tree is displayed in the topology only format, and was constructed using the Tamura 3-parameter, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/sequence submission are mentioned along with GenBank accession numbers for reference sequences.
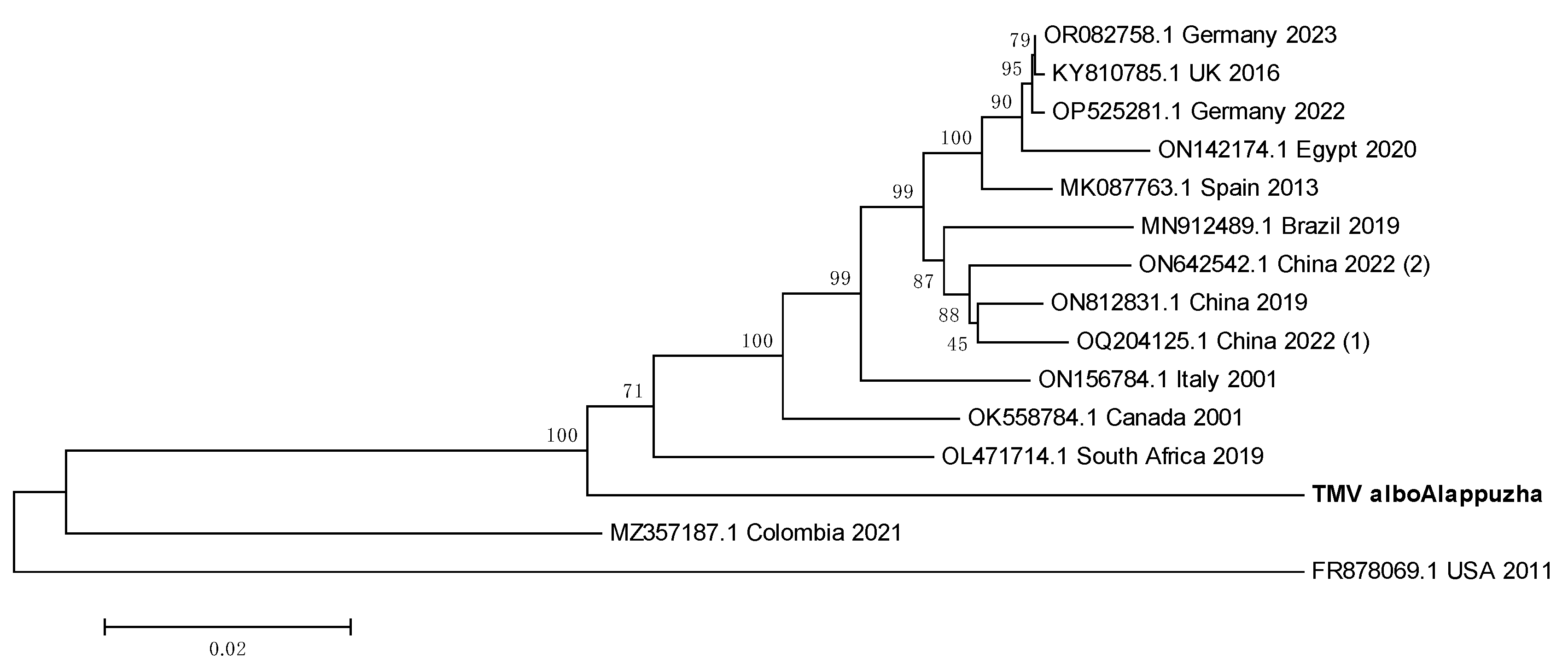
3.9. TMV sequence from Ae. albopictus is closely related to South African TMV sequence
The whole genome sequence of TMV from Kerala Ae. albopictus mosquitoes was used to construct a phylogenetic tree (Figure 9). The Indian sequence seemed to have the least ED with the South African sequence from 2019 (OL471714.1) and may thus be the most evolutionarily related to it.
Figure 9.
Maximum Likelihood phylogenetic tree of the Tobacco mosaic virus genomic sequence: The tree was constructed using the General Time Reversible model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/ sequence submission are mentioned along with GenBank accession numbers for reference sequences.
Figure 9.
Maximum Likelihood phylogenetic tree of the Tobacco mosaic virus genomic sequence: The tree was constructed using the General Time Reversible model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate the number of substitutions per site, and the values at nodes represent the bootstrap support. Names of countries and year of sample collection/ sequence submission are mentioned along with GenBank accession numbers for reference sequences.
3.10. WBAstV from Belgium is the likely introduction source of the virus into Assam
It was possible to generate consensus FASTA sequences of the two astroviruses; PoAstV4 and WBAstV with genome coverages of 72% and 58% respectively (Figure 7). These showed varying PNIs with multiple sequences of various lengths from multiple astroviruses. Therefore, all the sequences with highest PNIs found through BLAST were downloaded and a phylogenetic tree was constructed (Figure 10). The WBAstV sequence showed highest PNI with a PoAstV4 sequence from Belgium 2015 (KY214437.1) (Figure 7). PoAstV4 and WBAstV sequences from Dibrugarh clustered together with a 99.58% PNI and a query cover of 28% when run on a paired BLAST. This indicate that the WBAstV, as classified by EPI2ME and Genome Detective, belongs to the same taxon as the Porcine Astrovirus and was closest to Belgium 2015 sequence of PAstV4.
Figure 10.
Maximum Likelihood phylogenetic tree of the Porcine astrovirus-4 and Wild Boar astrovirus genomic sequences: The tree is displayed in the topology only format, and was constructed using Tamura 3-parameter, with 1000 bootstrap replicates, on MEGA v.5.2 software. Values at nodes represent the bootstrap support. The scale represents evolutionary distance in terms of base substitutions per site. Names of countries from which the samples were collected and year of sample collection (or sequence submission) are mentioned along with GenBank accession number for each reference. ORF: open reading frame, cds: coding DNA sequence, PoAstV/PAstV: porcine astrovirus, WBAstV: wild boar astrovirus, sp.: species.
Figure 10.
Maximum Likelihood phylogenetic tree of the Porcine astrovirus-4 and Wild Boar astrovirus genomic sequences: The tree is displayed in the topology only format, and was constructed using Tamura 3-parameter, with 1000 bootstrap replicates, on MEGA v.5.2 software. Values at nodes represent the bootstrap support. The scale represents evolutionary distance in terms of base substitutions per site. Names of countries from which the samples were collected and year of sample collection (or sequence submission) are mentioned along with GenBank accession number for each reference. ORF: open reading frame, cds: coding DNA sequence, PoAstV/PAstV: porcine astrovirus, WBAstV: wild boar astrovirus, sp.: species.

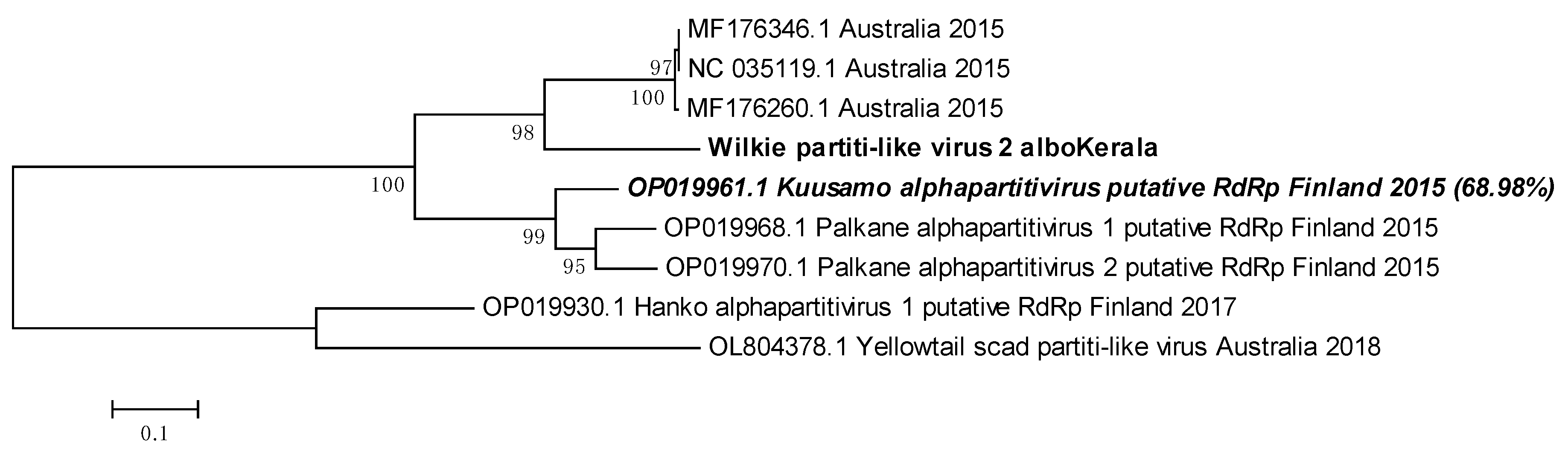
3.11. Wilkie partiti-like virus- 2 is related to the Kuusamo alphapartitivirus
Wilkie partiti-like virus-2 genomic sequence with 91% coverage was obtained from Kerala Ae. albopictus mosquitoes. Since the virus is unclassified, we used the next most identical nucleotide sequences from viruses as found through BLAST for phylogenetic tree building (Figure 11). PNIs of our sequence with already available Wilkie partiti-like virus-2 sequences were relatively low, with none above 73.97% in the phylogenetic tree analysis. Wilkie partiti-like virus-2 sequence from Kerala Ae. albopictus clustered together with three other sequences in the same clade. The other viruses used in the analysis were found using the Discontiguous Megablast option in the BLAST suite. Palkane alphapartitivirus-2 (OP019970.1) and Kuusamo alphapartitivirus (OP019961.1) had similar PNIs as the Kerala virus sequence (68.99% and 68.98% respectively). Nevertheless, it was difficult to draw a meaningful conclusion regarding the taxonomic placement of this virus.
Figure 11.
Maximum Likelihood phylogenetic tree of the Wilkie partiti-like virus-2 genomic sequence: The tree was constructed using Tamura 3-parameter, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values at nodes represent the bootstrap support. The scale represents evolutionary distance in terms of base substitutions per site. Names of countries from which the samples were collected and year of sample collection/sequence submission are mentioned along with GenBank accession number for each reference.
Figure 11.
Maximum Likelihood phylogenetic tree of the Wilkie partiti-like virus-2 genomic sequence: The tree was constructed using Tamura 3-parameter, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values at nodes represent the bootstrap support. The scale represents evolutionary distance in terms of base substitutions per site. Names of countries from which the samples were collected and year of sample collection/sequence submission are mentioned along with GenBank accession number for each reference.
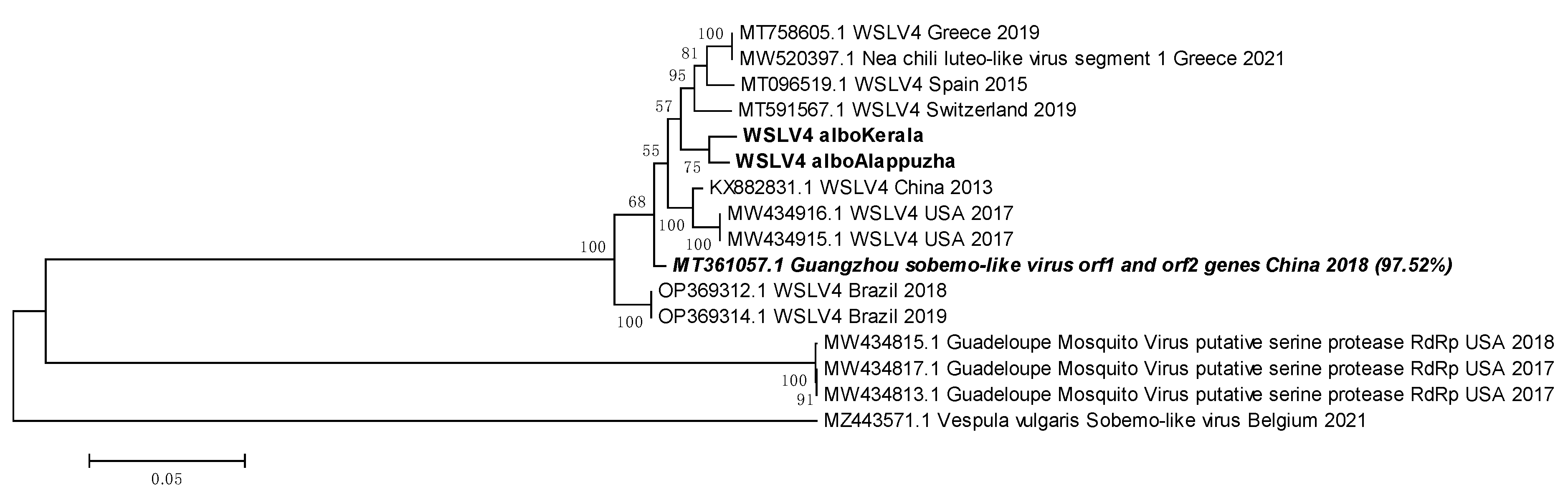
3.12. WSLV4 belongs to either the Solemoviridae family or the Tolivirales order
WSLV4 sequences from Ae. albopictus mosquitoes of Kerala and Pune were used to construct a phylogenetic tree (Figure 12). The Indian sequences clustered tightly. The least ED was shown with a sequence from China, 2013 (KX882831.1). China has the closest geographic proximity to India out of all the source locations of the WSLV4 reference sequences in the tree. While Indian WSLV4 sequences showed close evolutionary relationships and low EDs with existing WSLV4 sequences, sequences from two other viruses, Nea chili luteo-like virus and Guangzhou sobemo-like virus also clustered with the WSLV4 sequences. This made deduction of possible taxonomic placement of WSLV4 ambiguous. BLAST analysis of the sequence derived from Pune Ae. albopictus mosquitoes showed PNIs of 97.52% with Guangzhou sobemo-like virus and 97.24% with Nea chili luteo-like virus. Despite the PNI difference being marginal, we may infer that the WSLV4 is more closely related to the Guangzhou sobemo-like virus, which belongs to family Solemoviridae and Nea chili luteo-like virus, belonging to Order Tolivirales. While this is an inference drawn from nucleotide sequences, more investigation based on morphology, ultrastructure and antigenicity is needed for confirmation.
Figure 12.
Maximum Likelihood phylogenetic tree of Wenzhou Sobemo-like virus-4 genomic sequences: The tree was constructed using Tamura Nei model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values at nodes represent the bootstrap support. The scale represents evolutionary distance in terms of base substitutions per site. Names of countries from which the samples were collected and year of sample collection (or sequence submission) are mentioned along with GenBank accession number for each reference. orf: open reading frame, albo: albopictus, RdRp: RNA dependent RNA polymerase.
Figure 12.
Maximum Likelihood phylogenetic tree of Wenzhou Sobemo-like virus-4 genomic sequences: The tree was constructed using Tamura Nei model, with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values at nodes represent the bootstrap support. The scale represents evolutionary distance in terms of base substitutions per site. Names of countries from which the samples were collected and year of sample collection (or sequence submission) are mentioned along with GenBank accession number for each reference. orf: open reading frame, albo: albopictus, RdRp: RNA dependent RNA polymerase.
3.13. Indian CFAV sequences clustered together to form a single clade
CFAV sequences from Ae. aegypti mosquitoes of Dibrugarh and Kolkata were used to construct a phylogenetic tree with CFAV whole genome reference sequences from across the world (Figure 13). A list of PNIs of Indian CFAV sequences with reference sequences from across the world is given in Table 4. Sequences from Dibrugarh and Kolkata had a PNI of 99.88%, indicating their close relationship. Australia 2014 reference sequence (LR694075.1) showed highest PNIs with the Indian sequences, suggestive of an introduction from Ae. aegypti mosquitoes of Australia or vice versa. The least PNI was noted with a 2015 sequence from Cambodia (LR694078.1). Despite these two reference sequences were collected only one year apart, they have low evolutionary relatedness, indicating that location of a mosquito population may decide the characteristics of a CFAV population. Sequences from Cambodia (2013 and 2015) and Thailand clustered together, implying that the evolution of the virus is slow (Figure 13). Sequences from Brazil (2017) and USA (2016) grouped in the same clade, indicating a minimal ED between the two, possibly due to circulation of the same virus. It may be interesting to analyse CFAV sequences available from Mexico, to check for any spatial relationship. Interestingly, sequences from Ghana and Uganda clustered together to form an independent clade despite being on the western and eastern sides of Africa.
Figure 13.
Maximum Likelihood phylogenetic tree of Cell fusing agent virus: genomic sequences: The tree was constructed using the General Time Reversible model, with 1000 bootstrap replicates, on the MEGA v.5.2 software.
Figure 13.
Maximum Likelihood phylogenetic tree of Cell fusing agent virus: genomic sequences: The tree was constructed using the General Time Reversible model, with 1000 bootstrap replicates, on the MEGA v.5.2 software.
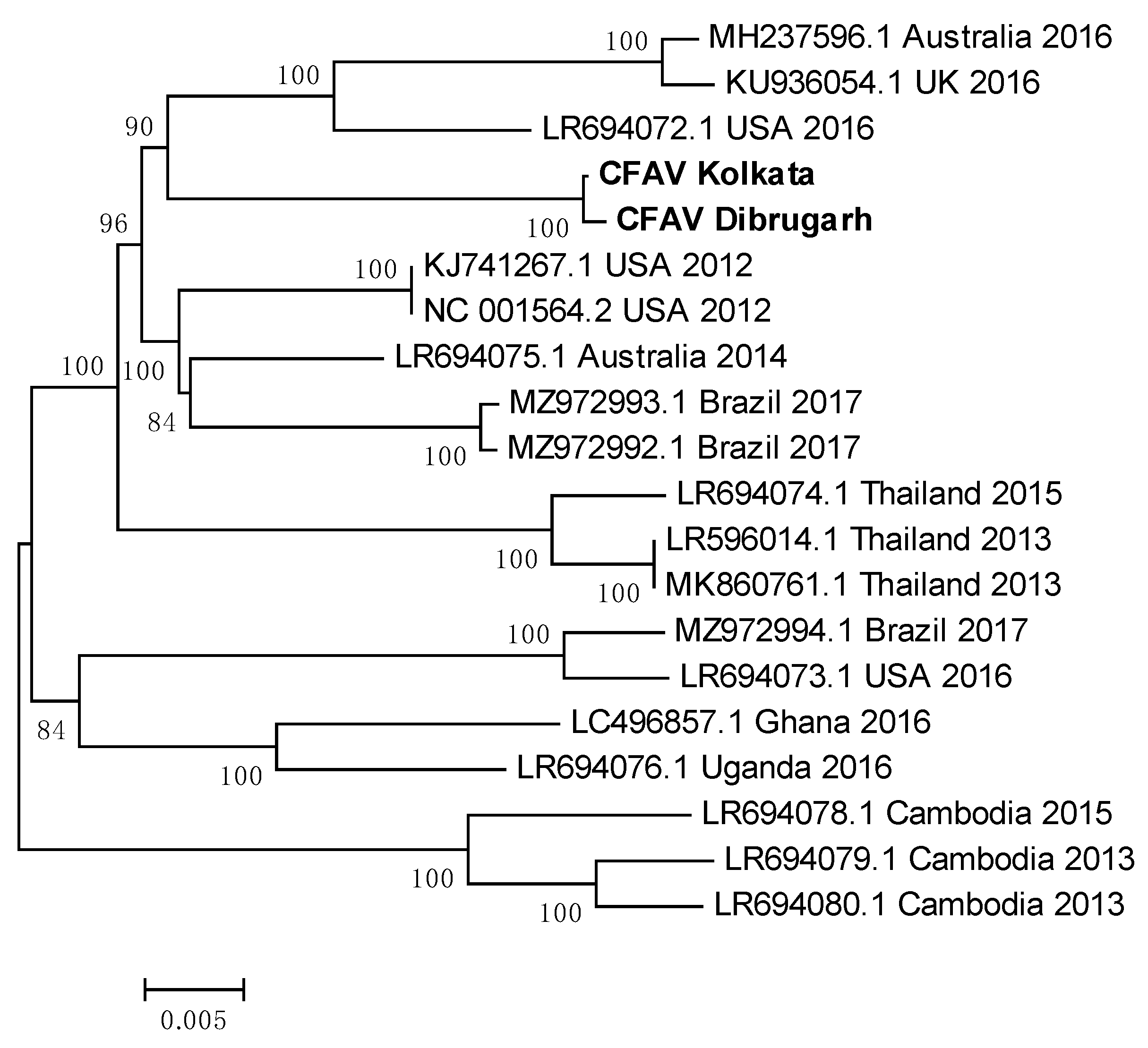
Table 4.
Percent nucleotide identities of CFAV whole genome sequences obtained in this study with reference genomes from across the world.
Table 4.
Percent nucleotide identities of CFAV whole genome sequences obtained in this study with reference genomes from across the world.
| Sequence/Reference | Country | Year of sample collection/sequence submission | CFAV Kolkata | CFAV Dibrugarh |
|---|---|---|---|---|
| CFAV Kolkata | India | 2022 | 100 | 99.88 |
| CFAV Dibrugarh | 99.88 | 100 | ||
| LR694075.1 | Australia | 2014 | 96.75 | 96.54 |
| KJ741267.1 | USA | 2012 | 96.7 | 96.51 |
| NC 001564.2 | 96.7 | 96.51 | ||
| MZ972993.1 | Brazil | 2017 | 96.22 | 95.98 |
| MZ972992.1 | 96.22 | 95.98 | ||
| MH237596.1 | Australia | 2016 | 95.56 | 95.34 |
| KU936054.1 | UK | 95.51 | 95.29 | |
| LR694072.1 | USA | 96.31 | 96.11 | |
| LR596014.1 | Thailand | 2013 | 95.41 | 95.22 |
| MK860761.1 | 95.41 | 95.22 | ||
| LR694074.1 | 2015 | 95.35 | 95.16 | |
| LC496857.1 | Ghana | 2016 | 95.28 | 95.04 |
| LR694076.1 | Uganda | 95.48 | 95.25 | |
| MZ972994.1 | Brazil | 2017 | 94.81 | 94.56 |
| LR694073.1 | USA | 2016 | 94.86 | 94.61 |
| LR694079.1 | Cambodia | 2013 | 94.5 | 94.26 |
| LR694080.1 | 94.52 | 94.28 | ||
| LR694078.1 | 2015 | 94.38 | 94.15 |
4. Discussion
We report metaviromic characterization of Ae. aegypti mosquitoes from seven geographically diverse locations of India, and Ae. albopictus mosquitoes from two geographically distinct regions. Of these, one location, Pune, Maharashtra, had mosquitoes of both the species analysed, thus allowing for comparison of viromes between the two prominent anthropophilic Aedes species. We employed NGS using the ONT platform, taking advantage of the long reads generated through Nanopore sequencing. The aim was to identify viruses inhabiting mosquito populations and try finding the known human pathogenic viruses, which makes this, to our knowledge, the first report of its kind from India.
Akin to our previous report [13], a large proportion of the reads was assigned to Homo sapiens, ranging from 4,566 reads to 167,340 reads across all the viromes. In the previous study (15) we used the Centrifuge v1.0.4 database for metagenomic taxa assignment, whereas in this one the EPI2ME Portal’s classification system (Kraken2) was used. A report from 2018 elucidates occurrence of high numbers of H. sapiens reads obtained from RNA sequencing (RNA-seq) data of mosquitoes fed on blood meals [20]. Some reads also matched to DNA from mice/rats, cows and dogs, implying that blood meals can introduce RNA and DNA from hosts to mosquitoes sampled for metagenomics. In fact, Avian endogenous retrovirus EAV-HP was found to be integrated in the genomes of Gallus spp. (chicken) in a non-random manner, as evidenced from Sequence Read Archive (SRA) dataset analyses [21], and full genomes sequenced from whole blood of village chickens of Ethiopia [22]. We detected EAV-HP in the virome of Patiala mosquitoes. While DNase and RNaseI treatment was done for all homogenized mosquito samples before viral RNA extraction, these findings indicated that the treatment might have been insufficient. Furthermore, low genomic coverage of CfGV, i.e., DNA viruses with large genomes, implies that they may not be well-represented in the metaviromics data generated by us, and our protocol has worked better on RNA viruses.
Detection of Serratia phage Muldoon, which infects the bacterium Serratia marcescens in Madurai Ae. aegypti led us to look for S. marcescens reads. A total of 7,097 reads assigned to S. marcescens was found in Madurai data, in addition to 7,572 reads from Patiala, 1,124 reads from Dibrugarh and 877 reads from Jodhpur (data not shown). This was coupled with the presence of Serratia phage Muldoon in the Madurai virome, which strongly indicated presence of S. marcescens in the mosquitoes sampled. This bacterium is known to influence modulation of midgut genes in mosquitoes and inhibit development of the parasite Plasmodium berghei [23]. Importantly, S. marcescens has been reported to act as a commensal in Ae. aegypti and enhance vector competence for DENV [24]. We did not detect any medically important arboviruses in any of our viromes. It would be worthwhile to study the presence of S. marcescens in mosquitoes and vector competence of Indian mosquitoes as S. marcescens not only causes opportunistic nosocomial infections in humans [25], but also possesses resistance mechanisms to antibiotics. Reports of S. marcescens infection was reported from Punjab, and its source was found to have been a soap dispenser in the Intensive Care Unit (ICU) [26]. Similarly, an outbreak of S. marcescens was reported in a tertiary care hospital in Chennai, Tamil Nadu, near to Madurai in 2019 [27]. Anopheles spp. mosquitoes can also transmit S. marcescens and P. berghei through its saliva [28]. This raises the question of whether the Ae. aegypti of Madurai, which seemingly harboured S. marcescens, can transmit it to humans.
The Dibrugarh mosquito virome had the highest percentage (~12%) of unclassified virus reads, amongst a total of 38,089 viral reads, while the virome of Pune Ae. aegypti mosquitoes lacked any reads matching to unclassified viruses despite having relatively high viral reads (276,367) (Table 2a). In contrast, Pune Ae. albopictus virome harboured the highest percentage of unclassified virus reads (~97%) with total viral reads of 122,369. This makes the absence of unclassified virus reads in Pune Ae. aegypti virome an exceptional occurrence and needs further investigation.
Our observations include a trend of increasing viral read counts with increasing cumulative (Eukaryota, Viruses, Bacteria and Archaea) read counts. When ONT sequencing and the EPI2ME portal, coupled with usage of a Taq-based polymerase, was used for testing the read assignment efficiency of samples spiked with Bovine herpesvirus 1 (BoHV-1), there appeared to be a positive correlation between the number of amplification cycles, cumulative read counts and BoHV-1 specific reads [29]. We used SISPA mediated by Taq polymerase, and thus a similar occurrence may have happened here, as observed by the vastly differing WSLV4 read numbers for Jodhpur and Dibrugarh Ae. aegypti viromes, and Pune and Kerala Ae. albopictus viromes. A report from 2015 describes how varying viral titres resulted in varying read numbers for different viruses such as Ebola virus, Hepatitis C virus and CHIKV [30]. Another report mentions the correlation between initial viral load and the number of reads mapped to Influenza A H1N1 virus [31]. Viral load dependence of read numbers for virus spiking experiments has also been reported in yet another study [32]. These studies point towards the correlation of viral load with read numbers, and can thus be used to explain multiple observations in our study that hints at this relationship. We have, however, noted a variation in the number of PCLV- L, M and S segment-specific reads from the same samples. This could be probably due to either replication intermediates contributing as templates, or virions may not have necessarily encapsulated single molecules of each segment [33].
There arises the question of whether long-read sequencing has an advantage over short-read sequencing. Indeed, a comparative analysis of both kinds of sequencing has shown that homologs that are more distant may not be detected by short-read sequencing, i.e., when read lengths are between 100-200 bp [34]. Sequencing of relatively small reads may lead to incorrect taxon assignments. Whether such a phenomenon has occurred in our experiments and analyses is unclear, but the ambiguous results of sequences run on BLAST become more explicable if viewed through this lens. We found multiple consensus FASTA sequences generated through Genome Detective and assigned independently by the EPI2ME portal giving comparably high nucleotide identities to multiple taxa. Such a confounding occurrence may be explained by the still relatively short read length that resulted from the SISPA generated amplicons in our experiments.
Another observation was the occurrence of porcine astroviruses PoAstV4 and WBAstV in the virome of Dibrugarh mosquitoes. Detection of a porcine virus in mosquitoes will not be the first, as there is experimental evidence of mechanical transmission of porcine reproductive and respiratory syndrome virus from infected pigs to naïve ones through Aedes vexans mosquitoes [35]. Similarly, Porcine circovirus-2 has been detected from mosquitoes from pig farms in Hubei and Anhui provinces of China [36]. Thus, it is likely for PoAstV4 and WBAstV to occur in mosquitoes collected from Dibrugarh, though there are no reports of porcine astrovirus infections in Assam. Another possibility could be association of the virus with mosquito larval or pupal stages, which thrive in aquatic environments infested with enteric viruses such as adenovirus and circovirus [37]. Importantly, these findings indicate the possibility of porcine astrovirus circulation in Assam.
Sequences of all the three segments of PCLV were generated from Ae. aegypti samples of all locations. Phylogenetic trees displayed a divergence of sequences into two distinct clades; clade-I and clade-II. PCLV appears to have undergone different evolutionary processes for its three segments as evidenced by varying placements of the sequences from the same location but from different time periods in different clades. This imply that PCLV may experience events similar to Influenza virus or Rotavirus, wherein the multi-segmented genomes are mixed and assembled when two strains infect the same host. PCLV is an ISV that solely infects Ae. aegypti, with no occurrence in Ae. albopictus reported so far. Attempts to isolate the virus in our laboratory from PCR-positive mosquito samples using Ae. albopictus C6/36 cell line, were unsuccessful (unpublished data). Thus, seemingly inexplicable phylogenetic characteristics may be explained by considering the possibility of exchanges of PCLV genomic segments when Ae. aegypti from different locations mate. Whether this really happens in nature or not need confirmation using in vivo experiments. The evolutionary trend of PCLV may also be correlated with the spread of Ae. aegypti throughout the world. Publications have taken into account both mosquito-transmitted virus outbreak history and population genomics to guess at the probable historic spread of Ae. aegypti [38]. To correlate both the phenomena, i.e., spread of Ae. aegypti and that of PCLV, knowledge of origin of PCLV will also be required.
Aedes aegypti is a highly anthropophilic mosquito, and Ae. albopictus is only second to it as a major vector for arboviruses. This human-centred behaviour of Ae. aegypti arose initially due to climate change, and as of the past 20-40 years, due to rapid urbanization [39]. As the mosquitoes started becoming more anthropophilic, higher emergence of arboviral disease spread was observed. A role of this, if played, in the global spread of CFAV needs to be investigated. In this study, we found that Indian CFAV sequences are most identical to a 2015 Australian CFAV sequence. Interestingly, there is evidence of CFAV integration into Ae. albopictus genome. The integrated segments exist as nonretroviral endogenous viral elements (nrEVEs) and contain intact viral protein coding regions [40]. These are speculated to be remnants of the virus that had infected and settled into Ae. albopictus population a long time ago, and has since integrated itself into its genome. However, CFAV was not found in both the Ae. albopictus viromes in the present study. Genomic characteristics of CFAV may thus be interesting to study, and could shed light on its possible influence on arboviral pathogens.
Our study also had a few drawbacks. Despite numerous reads having been mapped to certain viruses, for example, the CfGV, there was a poor genomic coverage, and thus whether the viable, infectious virions of the virus are present in the mosquitoes remains unclear. The reads generated by our protocol were on an average of ~476 bases, and is longer than that generated through short-read technologies, this may not be informative or specific enough to correctly assign taxa. Due to long length of eukaryotic or prokaryotic genes, and the presence of introns in the former, there arises the chance of erroneous assignments occurring due to relatively short reads. One report mentions how the EPI2ME pipeline made use of poor-quality bacterial and viral genome assemblies available on RefSeq, which led to incorrect assignment of BoHV-1 sequences to other viral and bacterial taxa [29].
In conclusion, we were able to discover viruses circulating in mosquito populations from geographically diverse sampling locations across India, and through this we were able to detect certain viruses that have implications on major pathogens. Circumstantial evidence generated by other studies on the probability of S. marcescens circulation in mosquitoes of Tamil Nadu (and the possibility of human infections resulting from it), the experimental transmission of porcine viruses through mosquitoes, and the valid presence of the CFAV in mosquitoes of Assam and West Bengal, has relevance in terms of animal and human diseases. This is the first report of a countrywide viral surveillance through the use of untargeted metagenomics from India, and these results may lead to further studies to explain the influence of these viruses on replication and transmission of viruses of clinical importance.
Funding
Source of support: This work was supported by the Indian Council of Medical Research, New Delhi, India [Grant number: 6/9-7(209)/2019-ECD-II].
Declarations
Competing Interests. The authors have no relevant financial or non-financial interests to disclose.
References
- Fredericks AC, Russell TA, Wallace LE, Davidson AD, Fernandez-Sesma A, Maringer K. Aedes aegypti (Aag2)-derived clonal mosquito cell lines reveal the effects of pre-existing persistent infection with the insect-specific bunyavirus Phasi Charoen-like virus on arbovirus replication. PLoS Negl Trop Dis. 2019;13: e0007346. [CrossRef]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. 2013;496: 504–507. [CrossRef]
- Romo H, Kenney JL, Blitvich BJ, Brault AC. Restriction of Zika virus infection and transmission in Aedes aegypti mediated by an insect-specific flavivirus. Emerg Microbes Infect. 2018;7: 1–13. [CrossRef]
- Hermanns K, Zirkel F, Kopp A, Marklewitz M, Rwego IB, Estrada A, et al. Discovery of a novel alphavirus related to Eilat virus. J Gen Virol. 2017;98: 43–49. [CrossRef]
- Datta S, Gopalakrishnan R, Chatterjee S, Veer V. Phylogenetic Characterization of a Novel Insect-Specific Flavivirus Detected in a Culex Pool, Collected from Assam, India. Intervirology. 2015;58: 149–154. [CrossRef]
- Utarini A, Indriani C, Ahmad RA, Tantowijoyo W, Arguni E, Ansari MR, et al. Efficacy of Wolbachia-Infected Mosquito Deployments for the Control of Dengue. N Engl J Med. 2021;384: 2177–2186. [CrossRef]
- Öhlund P, Lundén H, Blomström A-L. Insect-specific virus evolution and potential effects on vector competence. Virus Genes. 2019;55: 127–137. [CrossRef]
- Thomas T, Gilbert J, Meyer F. Metagenomics: a guide from sampling to data analysis. Microb Inform Exp. 2012;2: 3. [CrossRef]
- Santos-Medellin C, Zinke LA, ter Horst AM, Gelardi DL, Parikh SJ, Emerson JB. Viromes outperform total metagenomes in revealing the spatiotemporal patterns of agricultural soil viral communities. ISME J. 2021;15: 1956–1970. [CrossRef]
- Hameed M, Wahaab A, Shan T, Wang X, Khan S, Di D, et al. A Metagenomic Analysis of Mosquito Virome Collected From Different Animal Farms at Yunnan–Myanmar Border of China. Front Microbiol. 2021;11. Available: https://www.frontiersin.org/articles/10.3389/fmicb.2020.591478.
- Shi C, Beller L, Deboutte W, Yinda KC, Delang L, Vega-Rúa A, et al. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome. 2019;7: 121. [CrossRef]
- Xiao P, Han J, Zhang Y, Li C, Guo X, Wen S, et al. Metagenomic Analysis of Flaviviridae in Mosquito Viromes Isolated From Yunnan Province in China Reveals Genes From Dengue and Zika Viruses. Front Cell Infect Microbiol. 2018;8. Available: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00359.
- Lole K, Ramdasi A, Patil S, Thakar S, Nath A, Ghuge O, et al. Abundance of Phasi-Charoen-like virus in Aedes aegypti mosquito populations in different states of India. PLOS ONE. 2022;17: e0277276. [CrossRef]
- Chrzastek K, Lee D, Smith D, Sharma P, Suarez DL, Pantin-Jackwood M, et al. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology. 2017;509: 159–166. [CrossRef]
- Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Khovanskaya R, et al. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database J Biol Databases Curation. 2020;2020: baaa062. [CrossRef]
- Mihara T, Nishimura Y, Shimizu Y, Nishiyama H, Yoshikawa G, Uehara H, et al. Linking Virus Genomes with Host Taxonomy. Viruses. 2016;8: 66. [CrossRef]
- Vilsker M, Moosa Y, Nooij S, Fonseca V, Ghysens Y, Dumon K, et al. Genome Detective: an automated system for virus identification from high-throughput sequencing data. Bioinformatics. 2019;35: 871–873. [CrossRef]
- Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022;50: W276–W279. [CrossRef]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25: 3389–3402. [CrossRef]
- Zakrzewski M, Rašić G, Darbro J, Krause L, Poo YS, Filipović I, et al. Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci Rep. 2018;8: 4690. [CrossRef]
- Leinonen R, Sugawara H, Shumway M, on behalf of the International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011;39: D19–D21. [CrossRef]
- Wragg D, Mason AS, Yu L, Kuo R, Lawal RA, Desta TT, et al. Genome-wide analysis reveals the extent of EAV-HP integration in domestic chicken. BMC Genomics. 2015;16: 784. [CrossRef]
- Bai L, Wang L, Vega-Rodríguez J, Wang G, Wang S. A Gut Symbiotic Bacterium Serratia marcescens Renders Mosquito Resistance to Plasmodium Infection Through Activation of Mosquito Immune Responses. Front Microbiol. 2019;10: 1580. [CrossRef]
- Wu P, Sun P, Nie K, Zhu Y, Shi M, Xiao C, et al. A Gut Commensal Bacterium Promotes Mosquito Permissiveness to Arboviruses. Cell Host Microbe. 2019;25: 101-112.e5. [CrossRef]
- Hejazi A, Falkiner FR. Serratia marcescens. J Med Microbiol. 1997;46: 903–912. [CrossRef]
- Khanna A, Khanna M, Aggarwal A. Serratia marcescens- A Rare Opportunistic Nosocomial Pathogen and Measures to Limit its Spread in Hospitalized Patients. J Clin Diagn Res JCDR. 2013;7: 243–246. [CrossRef]
- Rohit A, Suresh Kumar D, Dhinakaran I, Joy J, Vijay Kumar D, Kumar Ballamoole K, et al. Whole-genome-based analysis reveals multiclone Serratia marcescens outbreaks in a non-Neonatal Intensive Care Unit setting in a tertiary care hospital in India. J Med Microbiol. 2019;68: 616–621. [CrossRef]
- Accoti A, Damiani C, Nunzi E, Cappelli A, Iacomelli G, Monacchia G, et al. Anopheline mosquito saliva contains bacteria that are transferred to a mammalian host through blood feeding. Front Microbiol. 2023;14: 1157613. [CrossRef]
- Esnault G, Earley B, Cormican P, Waters SM, Lemon K, Cosby SL, et al. Assessment of Rapid MinION Nanopore DNA Virus Meta-Genomics Using Calves Experimentally Infected with Bovine Herpes Virus-1. Viruses. 2022;14: 1859. [CrossRef]
- Greninger AL, Naccache SN, Federman S, Yu G, Mbala P, Bres V, et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015;7: 99. [CrossRef]
- Jia X, Hu L, Wu M, Ling Y, Wang W, Lu H, et al. A streamlined clinical metagenomic sequencing protocol for rapid pathogen identification. Sci Rep. 2021;11: 4405. [CrossRef]
- Regnault B, Bigot T, Ma L, Pérot P, Temmam S, Eloit M. Deep Impact of Random Amplification and Library Construction Methods on Viral Metagenomics Results. Viruses. 2021;13: 253. [CrossRef]
- Phenuiviridae ~ ViralZone. [cited 17 Sep 2023]. Available: https://viralzone.expasy.org/7101.
- Wommack KE, Bhavsar J, Ravel J. Metagenomics: Read Length Matters. Appl Environ Microbiol. 2008;74: 1453–1463. [CrossRef]
- Otake S, Dee SA, Rossow KD, Moon RD, Pijoan C. Mechanical transmission of porcine reproductive and respiratory syndrome virus by mosquitoes, Aedes vexans (Meigen). Can J Vet Res. 2002;66: 191–195.
- Yang X, Hou L, Ye J, He Q, Cao S. Detection of Porcine Circovirus Type 2 (PCV2) in Mosquitoes from Pig Farms by PCR.
- Garcia LAT, Viancelli A, Rigotto C, Pilotto MR, Esteves PA, Kunz A, et al. Surveillance of human and swine adenovirus, human norovirus and swine circovirus in water samples in Santa Catarina, Brazil. J Water Health. 2012;10: 445–452. [CrossRef]
- Powell JR, Gloria-Soria A, Kotsakiozi P. Recent History of Aedes aegypti: Vector Genomics and Epidemiology Records. Bioscience. 2018;68: 854–860. [CrossRef]
- Rose NH, Badolo A, Sylla M, Akorli J, Otoo S, Gloria-Soria A, et al. Dating the origin and spread of specialization on human hosts in Aedes aegypti mosquitoes. Perry GH, Lou N, editors. eLife. 2023;12: e83524. [CrossRef]
- Palatini U, Alfano N, Carballar RL, Chen X-G, Delatte H, Bonizzoni M. Virome and nrEVEome diversity of Aedes albopictus mosquitoes from La Reunion Island and China. Virol J. 2022;19: 190. [CrossRef]
Figure 1.
Map of India showing collection sites. Red indicates site for only Ae. albopictus collection. 1: Patiala, Punjab; 2: New Delhi; 3: Dibrugarh, Assam; 4: Jodhpur, Rajasthan; 5: Kolkata, West Bengal; 6: Pune, Maharashtra, 7: Madurai, Tamil Nadu, 8: Alappuzha, Kerala. Map source: vecteezy.com.
Figure 1.
Map of India showing collection sites. Red indicates site for only Ae. albopictus collection. 1: Patiala, Punjab; 2: New Delhi; 3: Dibrugarh, Assam; 4: Jodhpur, Rajasthan; 5: Kolkata, West Bengal; 6: Pune, Maharashtra, 7: Madurai, Tamil Nadu, 8: Alappuzha, Kerala. Map source: vecteezy.com.
Figure 2.
Host distribution for the virus taxa identified for the viromes: The host type information was obtained from the NCBI Taxonomy database and Virus-Host DB, and the number of taxa identified in each virome for each host type was plotted. The graph was plotted using GraphPad Prism v.8.4.2. Wherever not mentioned, viromes are from Ae. aegypti mosquitoes, While Ae. albopictus are mentioned as albo.
Figure 2.
Host distribution for the virus taxa identified for the viromes: The host type information was obtained from the NCBI Taxonomy database and Virus-Host DB, and the number of taxa identified in each virome for each host type was plotted. The graph was plotted using GraphPad Prism v.8.4.2. Wherever not mentioned, viromes are from Ae. aegypti mosquitoes, While Ae. albopictus are mentioned as albo.
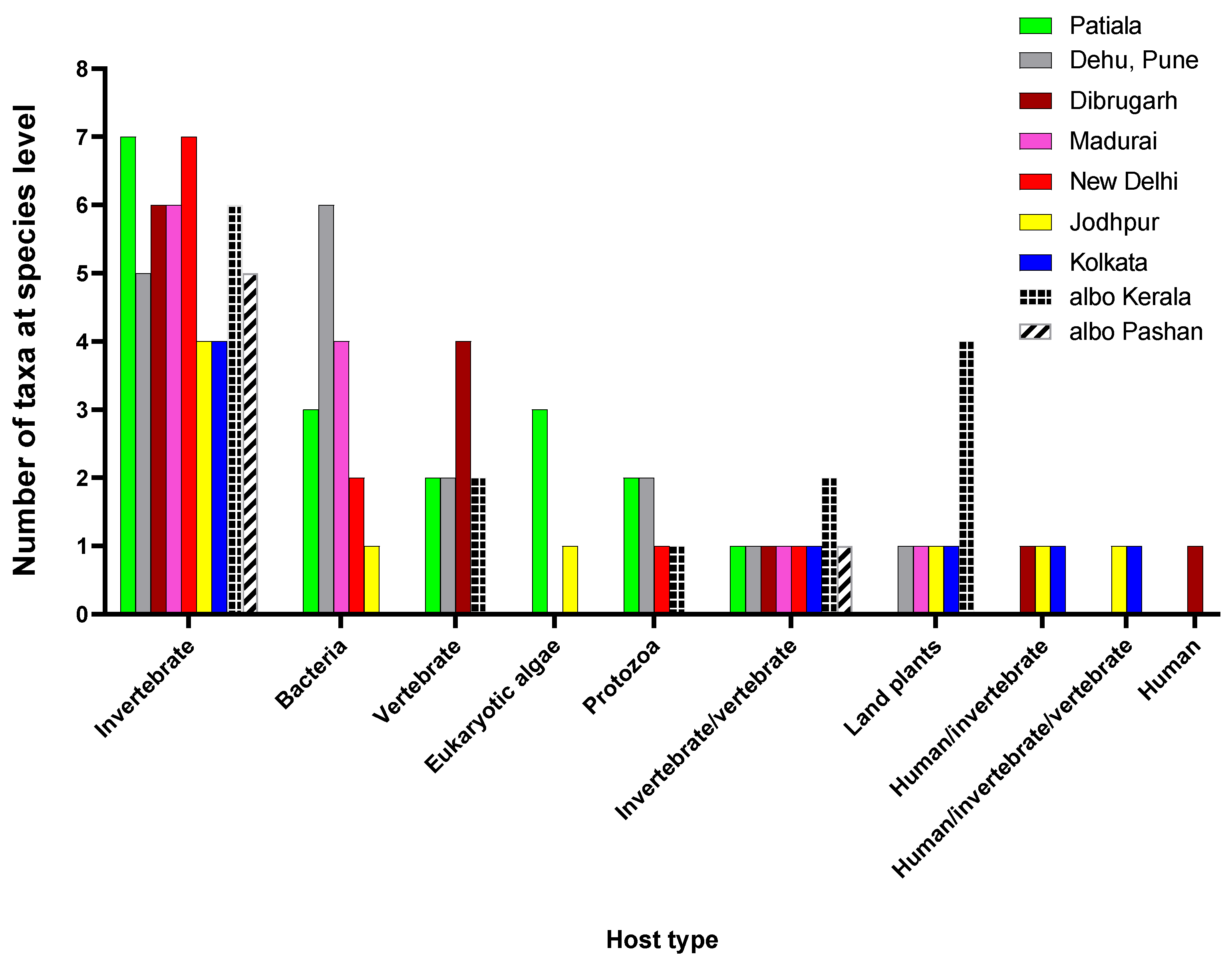
Figure 3.
Abundance of reads corresponding to viruses found in Aedes viromes: The percentage of reads was calculated with respect to the total viral reads for each virome. Only viruses having more than 1 read were considered. The graph was plotted using GraphPad Prism v.8.4.2.
Figure 3.
Abundance of reads corresponding to viruses found in Aedes viromes: The percentage of reads was calculated with respect to the total viral reads for each virome. Only viruses having more than 1 read were considered. The graph was plotted using GraphPad Prism v.8.4.2.
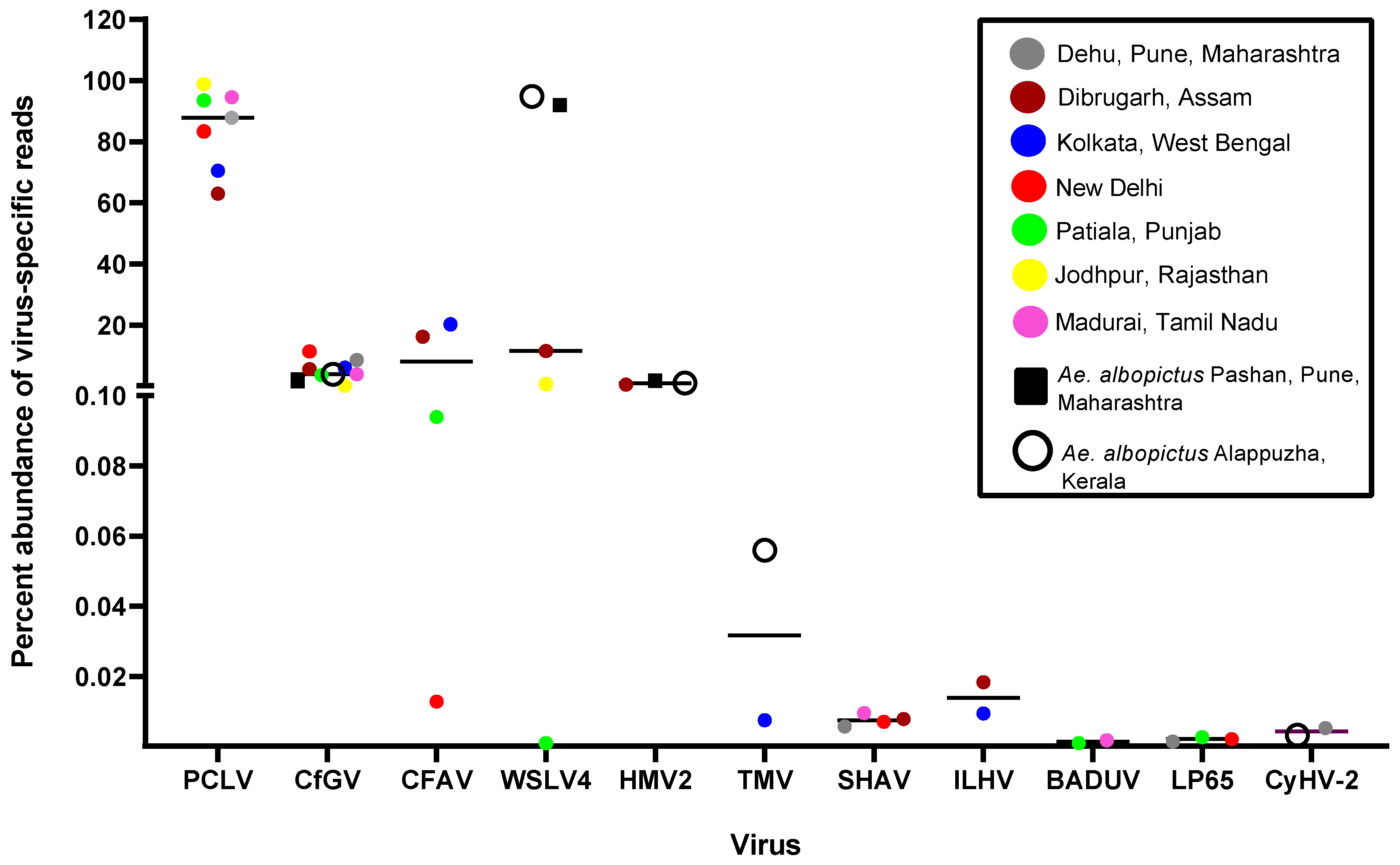
Figure 4.
Percent sequence coverage and mean depth for viruses with most abundant reads: Coverage percentage and mean depth were calculated using Samtools. The graphs were plotted using GraphPad Prism v.8.4.2.
Figure 4.
Percent sequence coverage and mean depth for viruses with most abundant reads: Coverage percentage and mean depth were calculated using Samtools. The graphs were plotted using GraphPad Prism v.8.4.2.
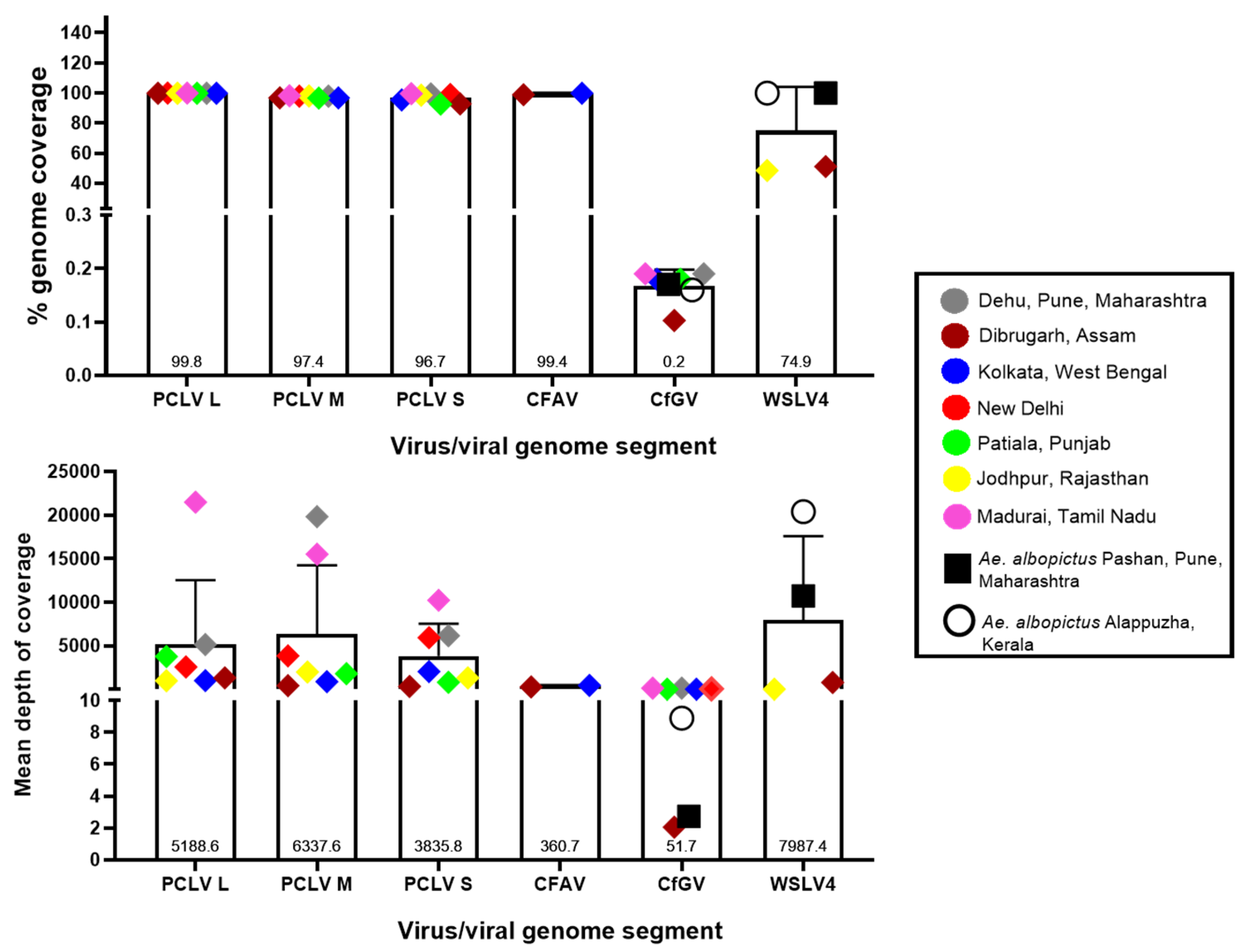
Figure 5.
Distribution for the taxa identified in all viromes: Only viruses that had more than 10 reads were considered for assigning taxa. Remaining are referred as ‘Others’. Information on virus families was obtained from NCBI Taxonomy database. The graphs were plotted using GraphPad Prism v.8.4.2.
Figure 5.
Distribution for the taxa identified in all viromes: Only viruses that had more than 10 reads were considered for assigning taxa. Remaining are referred as ‘Others’. Information on virus families was obtained from NCBI Taxonomy database. The graphs were plotted using GraphPad Prism v.8.4.2.

Figure 6.
Unclassified viruses: a) Heatmap showing distribution with respect to location of mosquitoes, b) Number of reads corresponding to each identified unclassified virus that had >10 reads in any virome. The graphs were plotted using GraphPad Prism v.8.4.2.
Figure 6.
Unclassified viruses: a) Heatmap showing distribution with respect to location of mosquitoes, b) Number of reads corresponding to each identified unclassified virus that had >10 reads in any virome. The graphs were plotted using GraphPad Prism v.8.4.2.

Figure 7.
Maximum Likelihood phylogenetic trees: The tree is displayed in the topology only mode, and construction was done using the Tamura 3-Parameter model with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate number of substitutions per site, and values at nodes represent the bootstrap support. Each sequence was obtained from corresponding raw data using the Genome Detective online platform and BLAST searches were performed to find the GenBank nucleotide sequences having the highest percent nucleotide identity, with the highest possible query cover (albo: albopictus, PCLV: Phasi Charoen-like phasivirus, Seg: segment, L: large, M: medium, S: small, PoAstV: Porcine astrovirus, WBAstV: Wild boar astrovirus, TMV: Tobacco mosaic virus, CFAV: Cell fusing agent virus, WSLV: Wenzhou sobemo-like virus).
Figure 7.
Maximum Likelihood phylogenetic trees: The tree is displayed in the topology only mode, and construction was done using the Tamura 3-Parameter model with 1000 bootstrap replicates, on the MEGA v.5.2 software. Values below branches indicate number of substitutions per site, and values at nodes represent the bootstrap support. Each sequence was obtained from corresponding raw data using the Genome Detective online platform and BLAST searches were performed to find the GenBank nucleotide sequences having the highest percent nucleotide identity, with the highest possible query cover (albo: albopictus, PCLV: Phasi Charoen-like phasivirus, Seg: segment, L: large, M: medium, S: small, PoAstV: Porcine astrovirus, WBAstV: Wild boar astrovirus, TMV: Tobacco mosaic virus, CFAV: Cell fusing agent virus, WSLV: Wenzhou sobemo-like virus).
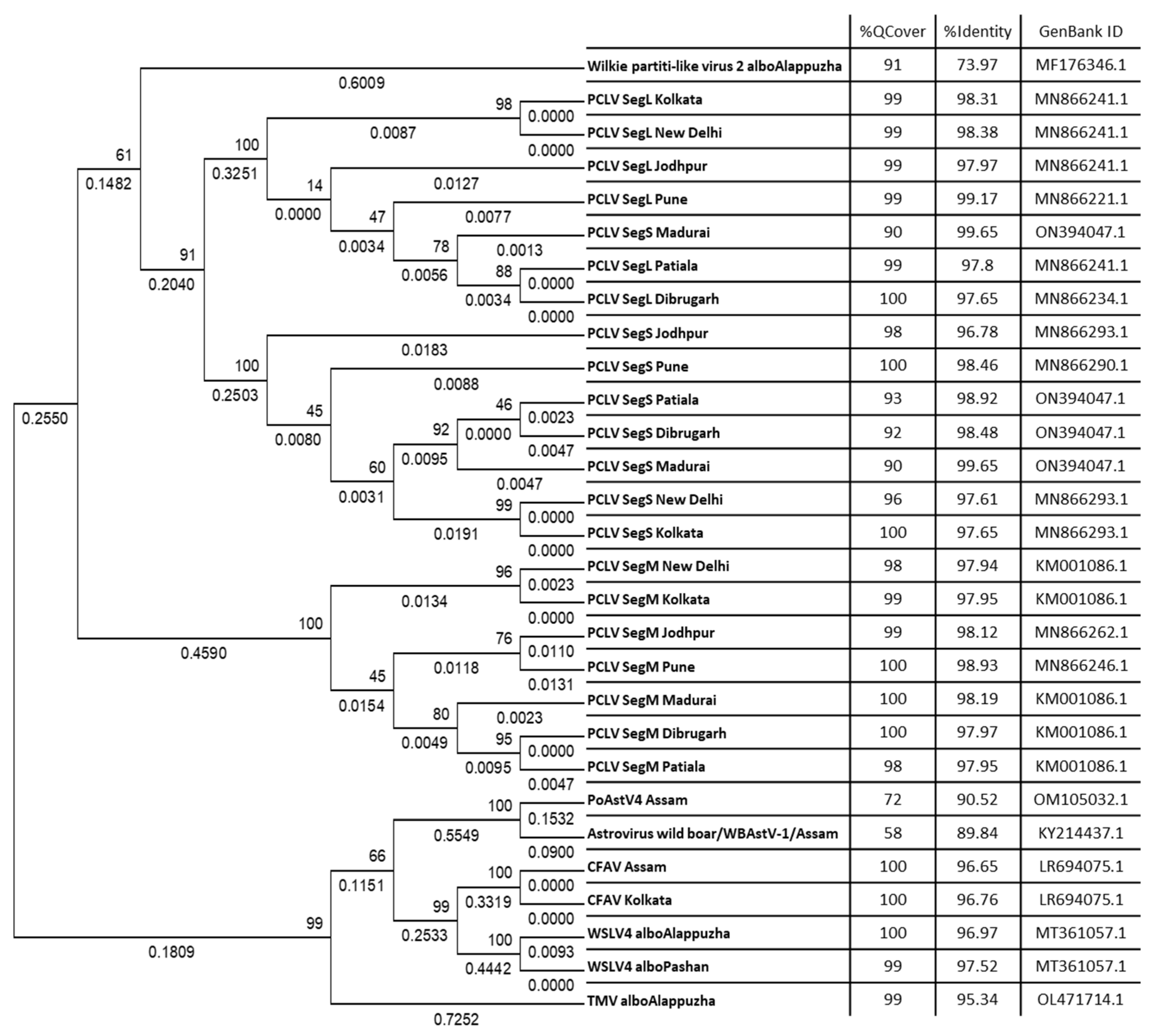
Table 1.
Details of primers used for SISPA and CFAV capsid region amplification.
| Primer name | Sequence | Reference |
|---|---|---|
| Random anchored reverse primer RT5 | CATCACATAGGCGTCCGCTGNNNNNN | [12] |
| Anchored forward primer P5 | CATCACATAGGCGTCCGCTG | |
| Random anchored reverse primer RT12 | GGTGGGCGTGTGAAATCGACNNNNNN | |
| Anchored forward primer P12 | GGTGGGCGTGTGAAATCGAC | |
| Random anchored reverse primer IDT-K-8N | GACCATCTAGCGACCTCCACNNNNNNNN | [14] |
| Anchored forward primer IDT-K | GACCATCTAGCGACCTCCAC | |
| CFAV-Capsid-32F | CAGTTTGGGTCACGCTTA | This study |
| CFAV-Capsid-536R | ATGTCAATCACCACGCAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



