
Submitted:
13 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
Background: There is a paucity of evidence on people with thoracic aortic aneurysm and dissection. We aimed to determine the prevalence of genetic variants and associations with phenotypes. Methods: In the current cross-sectional single centre cohort study of consecutive patients who underwent endovascular or open-surgical repair of thoracic aortic aneurysm and dissection, we applied genetic analysis using four-stage Next Generation Sequencing and confirmed findings with Sanger sequencing. We collected personal and family history on comorbidities, clinical examination, anthropometrics, skeletal deformities, joint function ophthalmological measures. Cardiovascular risk- and phenotype scores were calculated. Results: 95 patients were eligible (mean age 54±9 years, 70% males, 56% aortic dissection). One-fifth had a family history of aortic disease, 95% and 54% had a phenotype score of ≤5 and ≤2, respectively. There weren’t significant differences in the distribution of phenotype characteristics according to age, sex, aortic pathology, or performed invasive procedure. Genetic variants were detected in 40% of patients; classic variants comprised 18% of all variants. We observed no significant association with cardiovascular and phenotype score but with higher joint function scores (p=.015). Conclusion: Genetic variants are highly present in clinically-relevant aortic pathologies. Variants appear to play a larger role than previously described. The different variants don’t correlate with specific phenotypes, nor with age, pathology, sex, or family history.
Keywords:
thoracic aortic disease
; phenotype
; genetic mutation
; genetic variant
; aneurysm
; dissection
Introduction
Thoracic aortic aneurysm and dissection (TAAD) is a relatively low frequent disease affecting 5-10 per 100,000 of the global population [1]. Aneurysms stay asymptomatic in up to 95% of cases until a life-threatening dissection or rupture of the aorta occurs [1,2].
Early detection of TAAD and precise classification of underlying aetiology may help to optimise lifelong follow-up and patient tailored therapy plans [3]. Whether it is cardiovascular atherosclerosis, connective tissue disease, trauma, or inflammatory vascular wall changes has an impact on many decision pathways [4,5].
Generally, TAADs can be syndromic (e.g., Marfan syndrome (MFS), Loeys-Dietz syndrome (LDS) or Ehlers-Danlos syndrome (EDS)) or non-syndromic. Despite having different clinical pictures and prognoses, both syndromic and non-syndromic forms are often based on genetic variants [2].
Since the establishment of high-throughput sequencing, also known as next generation sequencing (NGS), knowledge of pathogenic variants has been accumulated [6]. Yet, the prevalence of genetic variants in patients with clinically relevant aortic pathology indicating aortic therapy is not fully studied. Furthermore, in the presence of modern NGS, the current correlation of genetic variants and the presence of phenotypical features typical for syndromic TAAD needs to be reevaluated.
The aim of the present study was to determine the prevalence of genetic variants in patients with TAAD who underwent invasive aortic repair. Furthermore, phenotype-genotype correlation was studied to reveal the prevalence of phenotypical characteristics in patients with confirmed genetic variant. This may help to understand its role in the justification of indication for human genetic testing using NGS.
Material and Methods
Ethical Statement
The study was reviewed and approved by the ethical committee board of the Technische Universität Dresden (decision number EK317082014). The indication for human genetic analysis was made by the Department of Angiology at the University Vascular Centre Dresden and was also confirmed by the cooperating practice for human genetics. The STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) statement for the reporting of observational studies have been complied with [7].
Study Design and Cohort
In this cross-sectional single centre cohort study, patients who were admitted to a tertiary reference centre in a university setting from 1st of January 2008 to 30th of June 2019 were screened for inclusion in the study. The inclusion criteria were age of 18 years or older, undergoing endovascular or open-surgical treatment for an index aortic pathology, and patient informed consent. The exclusion criteria were aortic pathology due to inflammatory or traumatic genesis, and patients above 65 years with no offspring.
Recruitment
A database screening for patients with aortic pathology who underwent an open-surgical or endovascular treatment from January 2008 through June 2019 according to the documented diagnosis code (World Health Organisation International Classification of Diseases, WHO-ICD) was carried out. The list of suitable ICD and procedural codes is provided in the supplement files. Eligible patients were subsequently contacted and asked for inclusion in the study. As part of the comprehensive sensitivity analyses and to determine the risk of selection bias, we used the anonymised data of excluded patients to comparing the demographic data, risk factors, aortic pathology, performed procedures, and medications with the study cohort.
The included patients underwent personal and family history taking, clinical examination including thorough phenotype analysis and subsequent human genetic analysis as follows:
Collection of History Data, Clinical Parameters, and Risk Scores Calculation:
Data Collection for Risk Score Calculation
Demographic data were collected from patients’ files and directly from patients. For age-groups analysis, age was divided into three groups: Patients under 45 years (group 1); between 45 and 60 years (group 2), over 60 years (group 3).
Further history and clinical data with possible associations with connective tissue diseases were collected during a comprehensive clinical examination and anamnesis of medical history:
Family History
Positive history was defined as confirmed connective tissue disease or clinically suspected due to confirmed aortic disease in relatives of the 1st, 2nd or 3rd degree.
Stature and Extremities
Tall stature (defined as >97th percentile of the normal range, where normal range is defined according to age and population group between the 3rd and 97th percentile); arm span/body length ratio >1.05, arachnodactyly, striae, increased skin elasticity.
Skeletal Deformities
Thoracic deformities (pectus carinatum / excavatum), scoliosis, foot deformities (pes planus, pes equinovarus), protrusio acetabuli, craniofascial abnormalities (cleft palate, high palate, uvula bifida, narrow lips, dolichocephaly, craniosynostosis, midface hypoplasia, micrognathia, hypertelorism).
Joint function
Positive Murdoch or Steinberg signs, Beighton score (hypermobility), joint dislocations.
Ophthalmologic Pathologies
Lens luxation/ectopia, myopia >3 dioptres, glaucoma, cataract, retinal detachment, enophthalmos, iris hypoplasia, blue sclera.
General Diagnoses
Hernia, spontaneously developed pneumothorax, Dura ectasia.
Cardiovascular Pathology and Cumulative Risk Score
Bicuspid aortic valve, atrial septal defect (ASD).
For each category, a total value from all risk points was created for all patients. As a rule, one point was awarded for each risk characteristic and the total risk score was calculated. In the joint function category, the higher point value from either Murdoch and Steinberg signs with one point each (maximum 2) or the total value of the Beighton score (maximum 9 points) was used (i.e.: If patients had a Beighton score higher than 2, the point value from the Beighton score was automatically considered, even if both finger signs were positive). The higher the cumulative score, the higher the clinical probability of the possible presence of a connective tissue disease.
Further Comorbidities
The following comorbidities were recorded: arterial hypertension, defined as a systolic blood pressure values of 130mmHg or more and/or diastolic blood pressure of 90mmHg or more and/or indicating medical treatment; clinically relevant hyperlipoproteinemia, defined as hyperlipoproteinemia indicating oral medical therapy; Diabetes mellitus indicating medical treatment (either oral antidiabetic or Insulin therapy); renal impairment, defined as a glomerular filtration rate (GFR) <60ml/min; active or past history of smoking; coronary heart disease (CHD); lower extremity peripheral arterial disease (PAD); documented carotid stenosis; history of stroke; chronic obstructive pulmonary disease (COPD) indicating medical treatment.
Molecular Genetic Analysis
The molecular genetic analysis was carried out from obtained ethylenediaminetetraacetic acid (EDTA) blood using the Next Generation Sequencing (NGS) panel analysis method. After capture-based enrichment of the genetic material, the NGS four-stage sequencing method (IMiSeq Desktop Sequencer, llumina Inc, San Diego, CA, USA) was applied. If a gene change was detected, it was confirmed using conventional Sanger sequencing. In addition to Multiplex Ligation-dependent Probe Amplification, the Sanger method was also used if a follow-up analysis became necessary due to ambiguous NGS result.
Due to the genetic heterogenicity of the connective tissue diseases spectrum, nine genes were initially panel examined and evaluated according to the specification of the German health authorities for examining genes associated with connective tissue diseases with possible involvement of the thoracic aorta. The examined gene loci were: ACTA2, COL3A1, FBN1, MYH11, MYLK, SMAD3, TGFB2, TGFBR1 and TGFBR2. In case of presence of phenotypical features or positive family history, and after patient consent, the diagnostics were expanded to include the analysis of following genes: AEBP1, BGN, COL1A1, COL4A5, COL5A1, COL5A2, EF-EM P2, ELN, FBLN5, FBN2, FLNA, FOXE3, GATA5, LOX, MAT2A, M FAP5, NOTCH1, NOTCH3, PLOD1, PRKG1, RPL26, SKI, SLC2A10, SMAD4, SMAD6, TAB2, TGFB3.
Data Acquisition and Cooperation Partners
General patient data as well as diagnoses and findings were recorded using the electronic hospital information system used at the Dresden University Hospital, Dresden, Germany. These included performed interventions, documentation of the clinical and image morphological data, structured follow-up, and the multidisciplinary vascular conferences of the disciplines of angiology, vascular surgery, interventional radiology, and cardiac surgery of the Dresden Heart Centre, in accordance with established standards at the University Vascular Centre of the Medical Clinic and Policlinic III of the Carl Gustav Carus University Hospital.
Cardiac surgical intervention data and the corresponding documentation were acquired from the department of Cardiac Surgery of the Dresden Heart Centre. Human genetic clinical examination as well as routine blood tests and subsequent human genetic analysis were performed by outpatient specialists in human genetics from the group practice for human genetics (Gutenbergstr. 5, 01307 Dresden, Germany; existing cooperation agreement with the University Hospital Carl Gustav Carus).
Statistical Analysis
Descriptive analyses were provided using absolute or relative frequencies. The mean and standard deviation were used to present continuous variables. Frequency differences were analysed using chi-square text for nominal scale variables and Kruskal-Wallis or Mann-Whitney U test for ordinal scale variables. To assess the correlation between age and cardiovascular risk factors, the Kendall's Tau correlation coefficient was calculated. Continuous variables were compared using the t-test. Results were considered statistically significant when p < 0.05. If the probability of error, p, yielded values between 0.05 and 0.1 (0.05 ≤ p < 0.1), the results were considered to be showing a trend. IBM SPSS software (IBM SPSS Statistics Version 28, IBM, Armonk, NY, USA) was used for the statistical analysis.
Results
Patient Cohort, Demographic Data and Representability of the Study Cohort
A total of 1334 patients with aortic disease of any aetiology were initially screened for eligibility. Thereof, 716 patients were eligible according to the inclusion and exclusion criteria. Ultimately, 116 patients gave their full consent while 95 patients provided complete and were included in the current study.
The comparative analysis between the study cohort and the patients who met the inclusion criteria and were excluded due to failure of patient consent or incomplete data set showed no significant differences between both cohorts regarding age, sex, performed procedures, arterial hypertension, history of smoking, hyperlipoproteinemia, diabetes mellitus, coronary heart disease (CHD), carotid stenosis, peripheral artery disease (PAD; supplement Table S1). The analysis showed that the study cohort had more aortic dissections than the excluded cohort (56% vs 45%, p=.012). Both cohorts showed no significant differences in medical therapy with Angiotensin Converting Enzyme inhibitors, Calcium antagonists, Beta blockers, antiplatelet drugs, and other antihypertensives. More study cohort patients were on oral anticoagulants than the excluded cohort (37% vs 30%, p=.021)
Data analysis of the study cohort showed a mean age of 54 ±9 years with 70.5% male quota. Arterial hypertension was documented in 83.2% of patients, history of smoking in 33.7%, and hypercholesterolemia in 32.6%. Fifty-six percent of patients suffered aortic dissections and 41.1% had aortic aneurysms. Further distribution analysis of pathology according to age groups are depicted in Supplement Figure S1. Sixty-eight percent of patients underwent open aortic surgery and 31.6% were treated endovascularly.
Family History Analysis and Distribution of Phenotypical Characteristics
A positive family history was documented in 20% of patients. A positive history in first-degree relatives was confirmed in 4.2% of aneurysm patients and 2.1% of dissection patients.
Fifty-four percent of patients showed a total phenotypic score of 2 or less and 92.6% showed a score of 5 or less (the detailed score distribution across the study population is depicted in Supplement Figure S2). The phenotypical category analysis (Figure 1, Supplement Table S2) showed that tall stature was present in 6.3% of patients.
The arm span to body length ratio was above normal in 14.7% of patients. In skeletal deformities, pes planus was the most present feature with a prevalence of 20.0%; followed by scoliosis in 13.7% of patients. Thin lips were the most prevalent craniofacial anomaly and was present in 25.3% of patients; followed by high palate with 14.7% prevalence. All joint function signs were present in less than 10%, with the highest prevalence of 9.5% documented for Murdoch sign. Myopia was present in 14.7% of patients and enophthalmos in 11.6%. Twenty-three percent of patients showed a positive history of hernia and 17.9% had bicuspid aortic valve. Further subgroup analysis showed no significant difference in the distribution of phenotypical characteristics according to age group, sex, aortic pathology, or performed procedure (Table 1).
Analysis of Genetic Variants and Correlation with Phenotype
The genetic analysis confirmed gene variants in 40% of all patients. Classic variants comprised 18.4% of all variants, while other variants constituted 81.6% of detected variants. The distribution of genetic variants did not differ significantly across age or sex. The analysis showed a trend toward a higher prevalence of genetic variants in aortic dissections and in patients undergoing open surgery (p=.054 and p=.072 respectively, Figure 2 and Table 2).
Genetic variants were found in thirteen genes. FBN1 gene variants comprised 32.5% of total variants; 7.5% of which were classic variants and 25% were other variants. MYH11 and TGFB2 followed and comprised 20% and 10% of total variants, respectively. Other gene variants percentages were under 10% each (Table 2). Simultaneous variants in two genes were detected in three patients (FGFB2 and MYH11 genes, in MYH11 and NOTCH1 gene, and in FBN1 and SMAD3 gene).
The distribution of different risk scores by genetic variant is depicted in Figure 3.
For each risk score, a total value from all risk points was created for each patient. One point was awarded for each risk characteristic and the total risk score was calculated. The percentage of each patient group with the same score is depicted with a different color in each column.
Cardiovascular scores and phenotype scores did not show a significant difference in distribution between patients with or without genetic variants (p=.140 and p=.110 respectively). A significant difference was found in joint function score analysis, with higher joint function score in patients with genetic variants (p=.015). The detailed results of the analysis are listed in Table 3.
Genetic Variants and Association with Genetic Disease
Two patients with FBN1 gene variants showed the characteristics of Marfan syndrome. Both patients carried heterozygous variants. Two patients showed a high probability of Loeys-Dietz syndrome class 4; one of which carried a heterozygous TGFBR1 gene variant and the other carried a heterozygous deletion of TGFB2 gene. An Ehlers-Danlos syndrome of the vascular type was documented in one patient with missense variant in COL2A1 gene.
Furthermore, in one patient with heterozygous duplication of MYH11 gene and heterozygous variant in NOTCH1 gene showed a micro-duplication syndrome 16p13.11., a patient with heterozygous variant of NOTCH3 gene showed CADASIL syndrome, a non-syndromal craniosynostosis was documented in a patient with heterozygous variant of NOTCH1 gene, and a patient with karyotype 45 X0 suffered Turner syndrome.
All other detected gene variants were unclassified variants, variants with low clinical relevance or variants without known clinical relevance (class 1 to 3 according to Plon et al. [8]).
Discussion
In the current study, human genetic analysis was abnormal in 40% of all cases, with 87.5% of all identified variants assigned to category A genes, which represent a relevant risk for thoracic aortic aneurysm and dissection (TAAD) according to the classification published by Renard et al [9]. Multiple syndromes were detected, including Marfan syndrome, Loeys-Dietz syndrome, and Ehlers-Danlos syndrome of the vascular type. To be noted that most of the variants were variants of unclear significance. This is, however, comparable to similar studies, whose design were similar to the presented study with their inclusion of non-selected TAAD patients [10,11]. Studies which showed higher prevalence of pathologic variants were those, whose design only considered TAAD patients under certain inclusion criteria [12,13,14,15,16,17,18,19]. The high prevalence of variants of unclear significance in non-selected TAAD patients denotes that variants of unclear significance might play a larger role than is currently known and this should be thoroughly examined in future studies.
An important finding of the study was that there was no significant correlation between phenotyping and genetic variants. Only the joint-movement score was significantly correlated with genetic variants. Further analysis confirmed that the genetic variant distribution was independent of age, sex, pathology, or cardiovascular risk. This confirms the importance of genetic testing irrespective of phenotype, demographic data, or cardiovascular risk. To the best of our knowledge, this is the first report systemically investigating the phenotypical features of all-comer TAAD patients and its correlation to the genotype. In the study by Pope et al., only people with suspected hereditary TAAD, not those with sporadic TAAD, were included in the study. Clinical data were collected but not systematically. A clinical study of their study participants was carried out by Duan et al. To rule out hereditary connective tissue disease, this study specifically included people with marfanoid characteristics or lens ectopy, not just those with TAAD, of which only 68.2% were affected. This could explain higher proportions of striking clinical features [19]. A correlation to the genotype was not recorded. However, the authors found a significant association of striae distensae with TAADs. The presence of striae distensae could therefore be a clinical clue to look for TAAD in individuals at risk [19]. In the study cohort of Wooderchak-Donahue et al., the characteristics of lens ectopia and some musculoskeletal findings (duraectasia, reduced elbow extension, marfanoid habitus) were more conspicuous in persons with the genetic variant. In contrast, skin changes and musculoskeletal features such as hypermobile joints, enlarged limbs, pes planus and hindfoot deformities were more common in the negative cohort [14]. In the study by Campens et al., the presence of syndromic features significantly increased (up to three-fold) the likelihood of genetic variant detection [15]. As a conclusion, we believe that although the presence of typical phenotypical feature increases the probability of genetic variants, its absence should not be an exclusion criterion in the decision-making algorithm for indicating genetic testing.
Although familial studies have shown that there is a ten-fold increased incidence rate in first-degree relatives with a family history of thoracic aortic aneurysm [20], other studies report that even sporadic TAADs without evidence of a hereditary association could be based on genetic mechanisms and state that a gene analysis could be indicated with these patients [2]. In a study by Guo et al. of subjects with sporadic thoracic aortic dissection and aged ≤ 56 years, 28% presented at least one variants of unclear significance, and 9.3% carried a genetic variant in any of eleven syndromic or familial TAAD genes, significantly more than controls [21]. In the study published by Renner et al., the diagnostic yield was not significantly higher in people with a positive family history than in those without [18]. This concurs with the results of our study that also showed a comparable genetic variant prevalence with no correlation with family history. An explanation might be the fact that most of the detected genetic variants were of unclear significance. Here it is important to denote that although to date, over thirty-seven genes are known to be associated with hereditary TAADs [22], only around 30% of familial non-syndromic TAAD cases have a genetic variant in these genes; which in turn suggests that most of the genetic basis of these thoracic aortic aneurysms and dissections remains undiscovered [22,23]. Further genetic testing of family members to examine the prevalence of detected genetic variants and its clinical relevance is crucially needed.
Limitations
Besides many strengths, there were also limitations to the study. First, the study was limited to a single centre and was cross-sectional by design. The completeness of data included in such epidemiological studies on genetic variants depends on voluntary participation. Due to the high value of biogenetic data and informational self-determination in the global discussion about individual privacy, it cannot be taken-for-granted that people always consent. This challenge applied to numerous cohort studies in the past. Although we applied comprehensive sensitivity analyses and are confident that excluded patients were not systematically different from the study cohort, a selection bias and residual confounding cannot be totally excluded; however. The solicitation of family history came only from the participants themselves and did not include additional information from relatives. Self-reported medical information remains another challenge of epidemiological studies while we designed the variables in a robust way to avoid another bias. Finally, the gene palate that was examined was limited. Future considerations for research include expanding the participant base to include more than one centre, expanding the gene palate, and involving participant relatives in the family history solicitation process.
Conclusion
Genetic variants are highly present in clinically relevant aortic pathology. Variants of unknown relevance seem to play a larger role than was previously known. The genetic variants do not correlate with a specific phenotype, age group, pathology, sex, or family history. Therefore, extension of genetic testing to all patients with clinically relevant aortic pathology should be considered irrespective of these factors.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.M. and T.G.; Methodology, Formal analysis, Investigation, Data curation, Writing & editing, all authors; Project administration, A.M and T.G. All authors have read and agreed to the published version of the manuscript.
Funding
Open Access funding provided by Open Access Publishing Fund of Philipps-University Marburg.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the local ethics committee of the Technical Universität Dresden, (File number: EK317082014).
Informed Consent Statement
The study does not include identifying information of individual participants. All data was achieved by daily ENT routine.
Data Availability Statement
Study data is unavailable due to privacy or ethical restrictions.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Bossone E, Eagle KA. Epidemiology and management of aortic diseases: Aortic aneurysms and acute aortic syndromes. Nature Rev Card 2020;18:331-348. [CrossRef]
- Ostberg N, Zafar M, Ziganshin B, Elefteriades J. The genetics of thoracic aortic aneurysms and dissection: A clinical perspective. Biomolecules 2020;10:182. [CrossRef]
- Brownstein A, Kostiuk V, Ziganshin B, Zafar M, Kuivaniemi H, Body S, et al. Genes associated with thoracic aortic aneurysm and dissection: 2018 update and clinical implications. AORTA 2018;06:013-020. [CrossRef]
- Yuan X, Mitsis A, Nienaber CA. Current understanding of aortic dissection. Life 2020;12:1606. [CrossRef]
- Zhou Z, Cecchi AC, Prakash SK, Milewicz DM. Risk factors for thoracic aortic dissection. Genes 2022;13:1814. [CrossRef]
- Czerny M, Schmidli J, Adler S, van den Berg JC, Bertoglio L, Carrel T, et al. Current options and recommendations for the treatment of thoracic aortic pathologies involving the aortic arch: An expert consensus document of the European Association for Cardio-Thoracic Surgery (EACTS) and the European Society for Vascular Surgery (ESVS). Eur J Cardio-Thorac Surg 2019;55:133-162. [CrossRef]
- Von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: Guidelines for reporting observational studies. J Clin Epidemiol. 2008;61:344-349. [CrossRef]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Human Mutation 2008;29:1282-1291. [CrossRef]
- Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adés LC, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol 2018;72:605-615. [CrossRef]
- Proost D, Vandeweyer G, Meester JAN, Salemink S, Kempers M, Ingram C, et al. (2015). Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Human Mutation 2015;36:808-814. [CrossRef]
- Fang M, Yu C, Chen S, Xiong W, Li X, Zeng R, et al. (2017). Identification of novel clinically relevant variants in 70 Southern Chinese patients with thoracic aortic aneurysm and dissection by Next-Generation Sequencing. Sci Rep 2017;7:10035. [CrossRef]
- Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH, et al. (2015). Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg 2015;100:1604-1611. [CrossRef]
- Poninska JK, Bilinska ZT, Franaszczyk M, Michalak E, Rydzanicz M, Szpakowski E, et al. (2016). Next-generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: Diagnostic yield, novel mutations, and genotype-phenotype corrections. J Transl Med 2016;14:115. [CrossRef]
- Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T, Plant P, Lewis T, Stocks J, et al. (2015). Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet 2015;167:1747-1757. [CrossRef]
- Campens L, Callewaert B, Muiño Mosquera L, Renard M, Symoens S, De Paepe AS, et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities – results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis 2015;10:9. [CrossRef]
- Yang H, Luo M, Fu Y, Cao Y, Yin K, Li W, et al. (2016). Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci Rep 2016;6:33002. [CrossRef]
- Overwater E, Marsili L, Baars MJH, Baas AF, van de Beek I, Dulfer E, et al. (2018). Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Human Mutation 2018;39:1173-1192. [CrossRef]
- Renner S, Schüler H, Alawi M, Kolbe V, Rybczynski M, Woitschach R, et al. Next-generation sequencing of 32 genes associated with hereditary aortopathies and related disorders of connective tissue in a cohort of 199 patients. Gen Med 2019;21:1832-1841. [CrossRef]
- Duan D-M, Chiu H-H, Chen P-L, Yeh P-T, Yu C-W, Yang K-C, et al. Clinical manifestations and genetic characteristics in the Taiwan thoracic aortic aneurysm and dissection cohort – A prospective cohort study. J Form Med Assoc 2022;121:1093-1101. [CrossRef]
- Keravnou A, Bashiardes E, Michailidou K, Soteriou M, Moushi A, & Cariolou M. (2018). Novel variants in the ACTA2 and MYH11 genes in a Cypriot family with thoracic aortic aneurysms: A case report. BMC Med Gen 2018;19:208. [CrossRef]
- Guo D, Hostetler EM, Fan Y, Kulmacz RJ, Zhang D, Nickerson DA, et al. Heritable thoracic aortic disease genes in sporadic aortic dissection. J Am Coll Cardiol 2017;70:2728-2730. [CrossRef]
- Faggion Vinholo T, Brownstein AJ, Ziganshin BA, Zafar MA, Kuivaniemi H, Body SC, et al. Genes associated with thoracic aortic aneurysm and dissection: 2019 update and clinical implications. AORTA 2019;07:099-107. [CrossRef]
- Acharya M, Masselli D, Mariscalco G. Genetic screening in heritable thoracic aortic disease – Rationale, potentials, and pitfalls. Ind J Thorac Cardiovasc Surg 2022;38:24-35. [CrossRef]
Figure 1.
Distibution of patients‘ phenotype scores: For each category, a total value from all risk points was created for every patient. One point was awarded for each risk characteristic and the total risk score was calculated. In the joint function category, the higher point value from either Murdoch and Steinberg signs with one point each (maximum 2) or the total value of the Beighton score (maximum 9 points) was used.
Figure 1.
Distibution of patients‘ phenotype scores: For each category, a total value from all risk points was created for every patient. One point was awarded for each risk characteristic and the total risk score was calculated. In the joint function category, the higher point value from either Murdoch and Steinberg signs with one point each (maximum 2) or the total value of the Beighton score (maximum 9 points) was used.
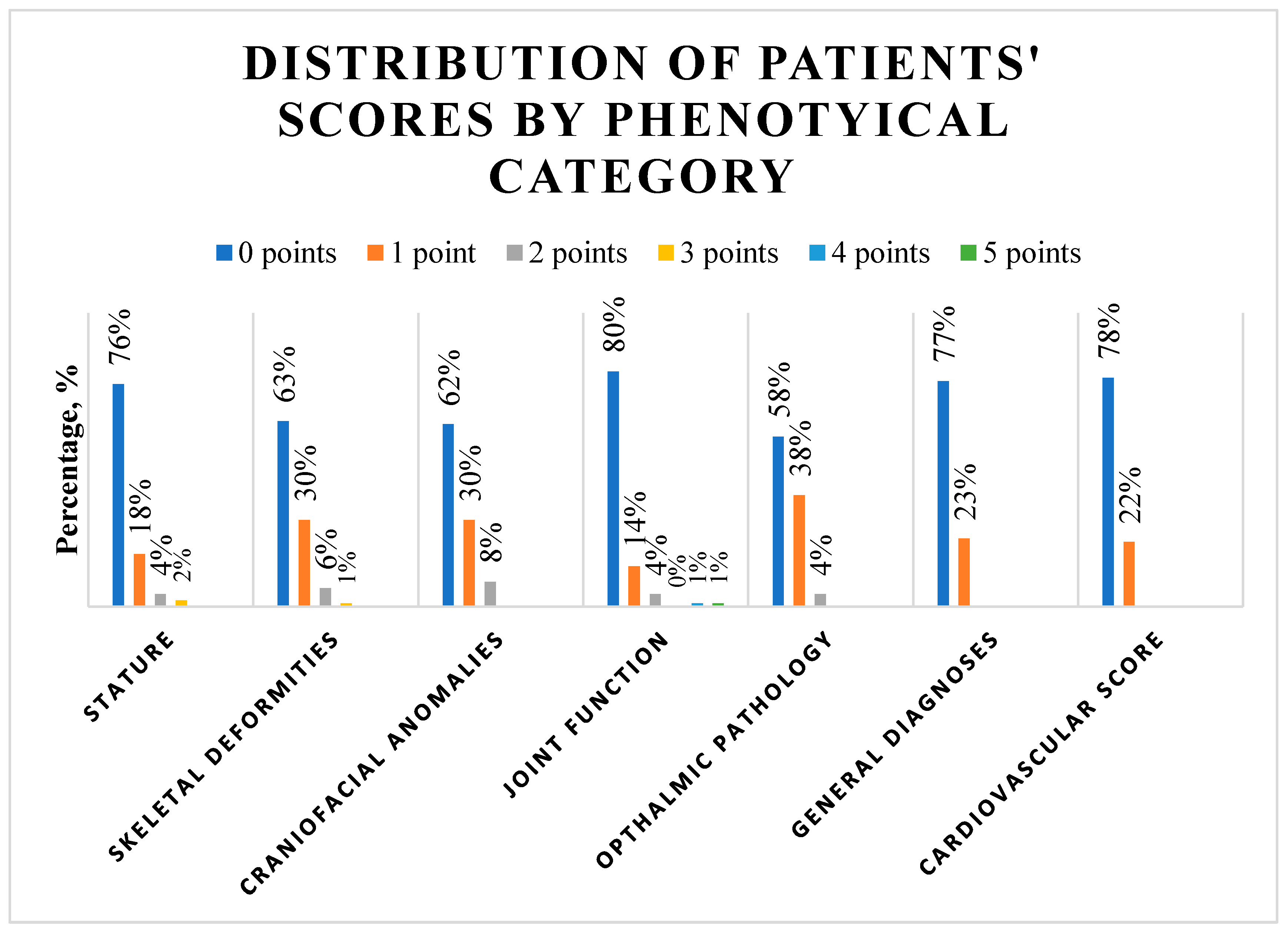
Figure 2.
Distribution of Gene Mutation across demographic and clinical patients` subgroups.
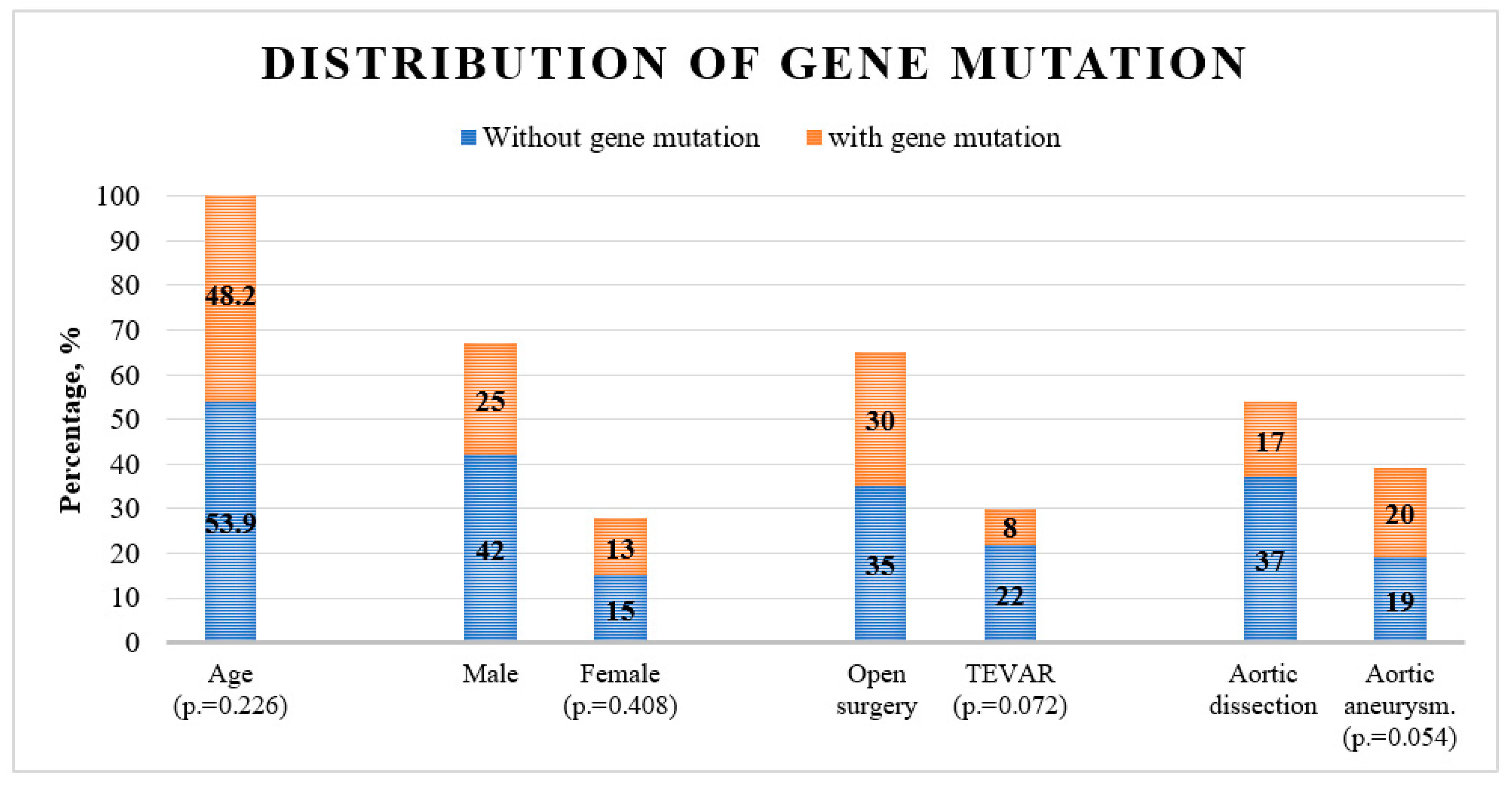
Figure 3.
Distribution of risk scores by genetic mutation:.
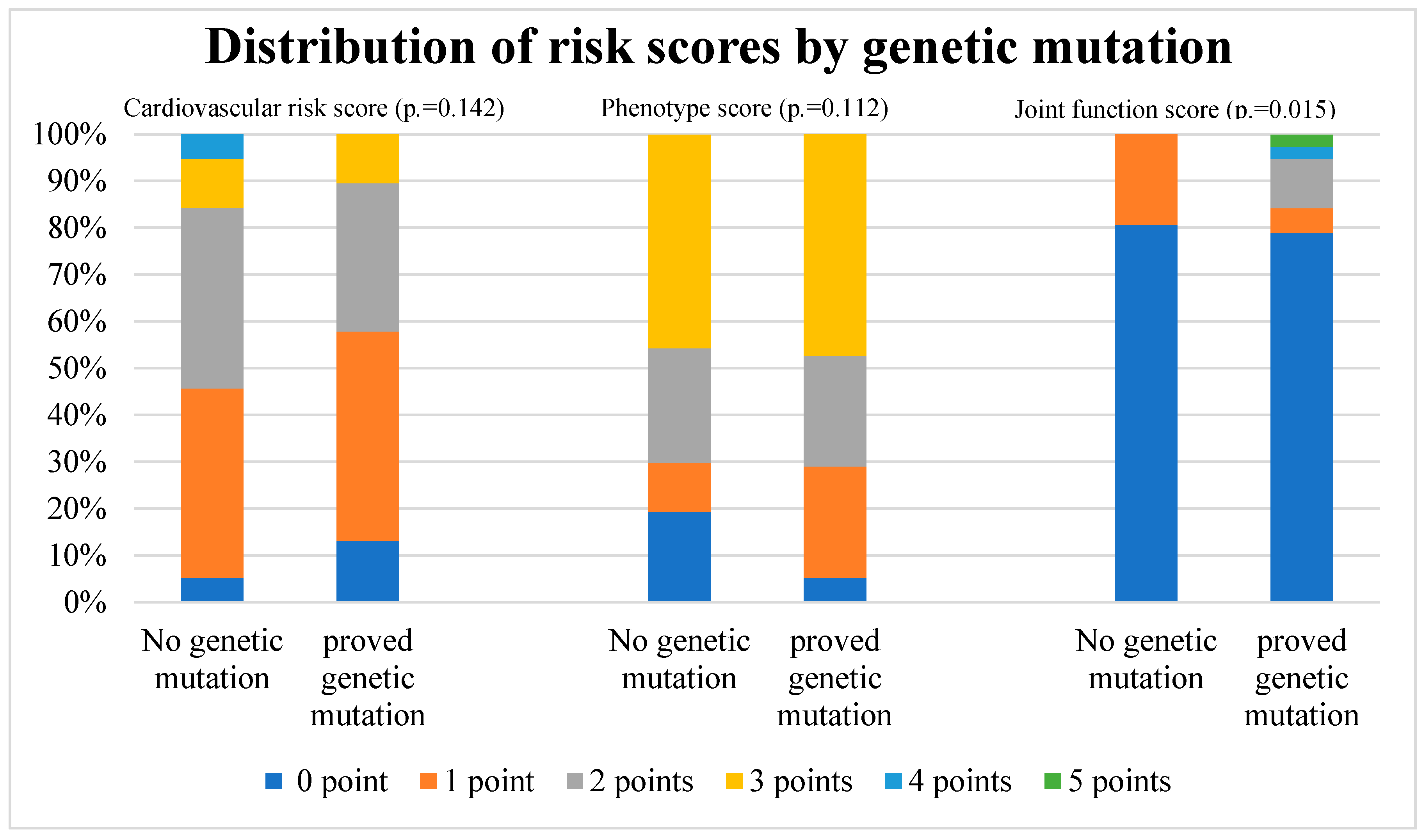

Table 1.
Subgroup analysis and its correlation with phenotype.
| Variable | No phenotypical signs | Present phenotypical signs | p-value |
|---|---|---|---|
| Number (percentage) | Number (percentage) | ||
| Age | 0.783 | ||
| under 45 years | 3 (11.5 %) | 23 (88.5 %) | |
| 45-65 years | 8 (16.0 %) | 42 (84.0 %) | |
| over 65 years | 2 (10.5 %) | 17 (89.5 %) | |
| Sex | 0.712 | ||
| males | 9 (13.4 %) | 58 (86.6 %) | |
| females | 4 (14.3 %) | 24 (85.7 %) | |
| Aortic pathology | 0.74 | ||
| Aortic aneurysm | 6 (15.4 %) | 33 (84.6 %) | |
| Aortic dissection | 7 (12.9 %) | 47 (87.1 %) | |
| Treatment | 0.478 | ||
| surgical | 10 (15.4 %) | 55 (84.6 %) | |
| endovascular | 3 (10.0 %) | 27 (90.0 %) | |
- Table 2. lists further the detailed results of gene variant distribution across the cohort.
- Table 2. Distribution of genetic mutations across the study cohort.
- Table 2. lists further the detailed results of gene variant distribution across the cohort.
- Table 2. Distribution of genetic mutations across the study cohort.
|
Name of the gene |
Number of mutation detections (n =) | Proportion of the total number of mutation detections (in %) | Classic mutation (n =) |
Percentage of classic mutations to total number of Evidence of mutation (in %) |
Mutation variant (n =) |
Percentage of mutation variants in total number of mutation detections (in %) |
|---|---|---|---|---|---|---|
| FBN 1 | 13 | 32.5 | 3 | 7.5 | 10 | 25 |
| COL3A1 | 1 | 2.5 | 1 | 2.5 | 0 | 0 |
| SMAD3 | 1 | 2.5 | 0 | 0 | 1 | 2.5 |
| TGFB2 | 4 | 10.0 | 1 | 2.5 | 3 | 7.5 |
| TGFBR1 | 2 | 5.0 | 0 | 0 | 2 | 5.0 |
| MYLK | 2 | 5.0 | 0 | 0 | 2 | 5.0 |
| MYH11 | 8 | 20.0 | 0 | 0 | 8 | 20.0 |
| PRKG1 | 1 | 2.5 | 0 | 0 | 1 | 2.5 |
|
NOTCH3 |
1 | 2.5 | 1 | 2.5 | 0 | 0 |
|
NOTCH1 |
3 | 7.5 | 0 | 0 | 3 | 7.5 |
| TGFBR2 | 1 | 2.5 | 0 | 0 | 1 | 2.5 |
| ACTA2 | 2 | 5.0 | 0 | 0 | 2 | 5.0 |
| SMAD6 | 1 | 2.5 | 0 | 0 | 1 | 2.5 |
Table 3.
Correlation of demographic data and risk factors with gene mutation.
| Variable | Without gene mutation | With gene mutation | p-value |
|---|---|---|---|
| Mean (± standard deviation) | Mean (± standard deviation) | ||
| Age (in years) | 53.9 ± 10.5 | 48.2 ± 13.7 | 0.226 |
| Number (percentage) | Number (percentage) | ||
| Males | 42 (62.7 %) | 25 (37.3 %) | 0.408 |
| Females | 15 (53.6 %) | 13 (46.4 %) | |
| Open surgery | 35 (53.8 %) | 30 (46.2 %) | 0.072 |
| TEVAR | 22 (73.3 %) | 8 (26.7 %) | |
| Aortic aneurysm | 19 (48.7 %) | 20 (51.3 %) | 0.054 |
| Aortic dissection | 37 (68.5 %) | 17 (31.5 %) | |
| Cardiovascular risk score | 0.142 | ||
| 0 point | 3 (5.3%) | 5 (13.2%) | |
| 1 point | 23 (40.4%) | 17 (44.7%) | |
| 2 points | 22 (38.6%) | 12 (31.6%) | |
| 3 points | 6 (10.5%) | 4 (10.5%) | |
| 4 points | 3 (5.3%) | 0 (0%) | |
| Phenotype score category | 0.122 | ||
| Score =0 | 11 (19.3 %) | 2 (5.3 %) | |
| Score =1 | 6 (10.5 %) | 9 (23.7 %) | |
| Score =2 | 14 (24.5 %) | 9 (23.7 %) | |
| Score >= 3 | 26 (45.6 %) | 18 (47.4 %) | |
| Joint function score | 0.015 | ||
| Score =0 | 46 (80.7%) | 30 (78.9%) | |
| Score =1 | 11 (19.3%) | 2 (5.3%) | |
| Score =2 | 0 (0%) | 4 (10.5%) | |
| Score =3 | 0 (0%) | 0 (0%) | |
| Score =4 | 0 (0%) | 1 (2.6%) | |
| Score =5 | 0 (0%) | 1 (2.6 %) | |
| Abbreviations: TEVAR = Thoracic Endovascular Aortic Repair. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



