
Submitted:
14 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
Colony-stimulating factor 1 receptor (CFS-1R) is a myeloid receptor with a crucial role in monocyte survival and differentiation. Its overexpression is associated with aggressive tumor characterized by an immunosuppressive microenvironment and poor prognosis. CSF-1R ligands, IL-34 and M-CSF, are produced by many cells in the tumor microenvironment (TME), suggesting a key role for the receptor in the crosstalk between tumor, immune and stromal cells in the TME. Recently, CSF-1R expression was reported at the cell membrane of cancer cell from different solid tumors, capturing the interest of various research group interested in investigating the role of this receptor in non-myeloid cells. This review summarizes current data available on the expression and activity of CSF-1R in different tumor types. Notably, CSF-1R+ cancer cells have been shown to produce CSF-1R ligands, indicating that CSF-1R signaling is positively regulated in autocrine manner in cancer cells. Recent research demonstrated that CSF-1R signaling enhances cell transformation by supporting tumor cell proliferation, invasion, stemness and drug resistance. In addition, this review covers recent therapeutic strategies, including monoclonal antibodies and small molecule inhibitors, targeting the CSF-1R and designed to block the pro-oncogenic role of CSF-1R in cancer cell.
Keywords:
CSF-1R
; cancer cell signaling
; cell proliferation
; stemness
; cell migration
; chemoresistance
1. Introduction
Colony-Stimulating Factor 1 Receptor (CSF-1R) is a tyrosine kinase receptor expressed primarily on the surface of monocytes and tissue macrophage, where it plays a central role in the regulation of immune responses and cell differentiation [1]. The receptor is activated by the binding of its cognate ligands, which induce receptor dimerization, autophosphorylation and activation of downstream signaling pathways. CSF-1/M-CSF and IL-34 are the known ligands of CSF-1R and bind different regions in the Ig-like domain of the extracellular region of CSF-1R [2,3]. This receptor-ligand interaction is critical for immune development and function, as well as tissue homeostasis [4]. Dysregulation of CSF-1R expression was associated with the development of an immunosuppressive TME in different cancer types [5], and CSF-1R favored TME interactions and the recruitment of TAMs at cancer sites [6,7,8]. In human immune cells, genetic mutations in the extracellular region of CSF-1R induce tumorigenesis and constitutive receptor activation [9]. Additionally, mutations in the c-fms codon 301, leading to constitutive tyrosine kinase activity, associated with neoplastic transformation [10]. Additionally, CSF-1R expression was found in the cancer cell membrane on different tumor types, where it is crucial for the development of the disease [11,12,13,14]. In this review we summarize the evidence of CSF-1R expression in cancer tissue and cancer cell lines. Moreover, we deeply described the pro-tumoral activity of CSF-1R in tumor cells and the mechanisms leading to its activation. The therapeutic strategies targeting CSF-1R were also discussed; however, it should be noted that the primary focus of these studies was on CSF-1R-expressing TAMs. The expression of CSF-1R on cancer cells has only recently been studied, and there is still much to learn regarding the area. However, as will be discussed later, evidence indicated that CSF-1R is an important factor in the development of tumor cell aggressiveness. Therefore, additional studies on different tumor types are required to determine whether CSF-1R is similarly involved in all malignancies and to emphasize the significance of this receptor in TME crosstalk as well as cancer cell signaling.
2. CSF-1R in Cancer
In cancer, prolonged exposure to CSF-1R ligands leads to recruitment of macrophages at tumor sites and their differentiation into pro-tumorigenic macrophages, indicated as M2-like tumor-associated macrophages (M2-TAMs) [15]. Indeed, a correlation between increased circulating CSF-1 and enhanced CSF-1R+ TAMs was observed in breast cancer [16]. The development of invasive phenotype in breast, ovarian and endometrial tumors was also associated with elevated levels of CSF-1R and circulating CSF-1 [17]. Similarly, gene expression profiling revealed increased Csf-1 and Csf-1r mRNA expression levels in leiomyosarcoma (LMS) patient-derived samples. According to this study, LMS tumor cells secreted CSF-1 that, in turn, induced macrophages recruitment at tumor sites. Additionally, in situ hybridization analysis in a set of LMS cases demonstrated that LMS cells produced both CSF-1 and CSF-1R, while macrophages did not secrete CSF-1, indicating an autocrine receptor activation at the tumor cell membrane [18]. The same mechanism was observed in ovarian cancer, where blockade of this autocrine loop reversed the malignant phenotype [19]. Ide et al. reported the involvement of CSF-1R in prostate cancer carcinogenesis [20]. IHC analysis of prostatic adenocarcinoma indicated that CSF-1R expression was higher in metastatic tissue compared to non-metastatic controls and the receptor was expressed by both cancer and stromal cells [21]. High levels of serum CSF-1 were considered cancer biomarkers in certain tumors, including squamous cell carcinoma of the head and neck and colorectal cancer [22,23]. Li et al. described new roles for CSF-1R in hTERT immortal human epithelial ovarian cell lines. According to this study, CSF-1R was up-regulated and involved in survival and growth of these cell lines. Additionally, CSF-1R represented an important component for hTERT cellular localization, as in fact CSF-1 stimulation induced phosphorylation of NFkBp65 and the formation of a NFkBp65-hTERT complex. On the contrary, genetic silencing of CSF-1R reduced nuclear levels of hTERT and complex formation. Lastly, they showed that CSF-1R activation regulated the transcription of hTERT via c-Myc activity [24].
2.1. CSF-1R in Cancer Cells
CSF-1R mRNA is expressed in different cancer cell lines (Figure 1) and various studies reported that CSF-1R mRNA upregulation correlated with poor prognosis in tumors [25,26]. Furthermore, oncogenic mutations in c-fms correlated with worse prognosis in myelodysplastic syndrome [27]. Up-regulation of c-fms in cervical cancer cells was associated with the activation of TGF-β receptor signaling, and, in turn, TGF-1 receptor activation enhanced c-fms mRNA levels [28]. Barbetti et al. reported for the first time CSF-1-dependent nuclear localization of CSF-1R in breast cancer cell lines, promoting the transcription of different proliferative genes such as CCND1, c-JUN, and c-MYC [29]. Soares et al. demonstrated high CSF-1R expression in renal carcinoma compared to normal tissues. FISH analysis indicated copy number gain of CSF1-R in 59% of clear cell renal cell carcinoma samples. Moreover, three genetic changes in CSF-1R sequence, including mutations in exon 7 and exon 22, were identified in clear cell renal cell cancer [30]. Previous work demonstrated CSF-1R expression at the membrane and in the nucleus of CSF-1-stimulated cervical, ovarian and breast cancer cell lines [31]. Epigenetic alterations in the CSF-1R promoter region induced CSF-1R upregulation in melanoma cells with the BRAF mutation. Interestingly, these cancer cells did not express the transcriptional factor PU.1, a key regulator of CSF-1R expression in myeloid cells, thereby suggesting that CSF-1R expression is regulated by different transcriptional mechanisms in melanoma cells [13]. An earlier study demonstrated that CSF-1R+ breast cancer cells were associated with worse prognosis and tumor spread, mostly in ER+ breast cancer [26]. In pancreatic cancer, tumor cells expressed various CSF-1R levels, which were high in nerve-invasive PCCs, indicating that CSF-1R expression correlated with perineural migration and worse prognosis [32].
2.2. Role of CSF-1R Ligands in TME Crosstalk
The communication between tumor, stromal and immune cells in the tumor microenvironment (TME) is necessary for the establishment of an environment that is suitable for tumor cells growth [33], proliferation and invasion to secondary tissues [34]. In this scenario, CSF-1R ligands, IL-34 and M-CSF, play an important role in the crosstalk between immune and tumor cells. As in fact immune and cancer cells secreted cytokines and interleukins, which supported tumor development and progression [33,35]. In colorectal cancer (CRC), cancer cells to secrete IL-34 and express CSF-1R, suggesting the presence of an autocrine positive feedback loop on the receptor. Moreover, IL-34-dependent CSF-1R activation increased proliferative index of tumor cells [36]. In ovarian cancer, the production of IL-34 by tumor cells was associated with the development of a malignant microenvironment and tumor progression [37]. According to Shao-Lai Z. [38], IL-34, produced by hepatocellular carcinoma (HCC) cells, induced the recruitment of TAMs at tumor sites. In vivo assays showed that increased IL-34-related TAMs infiltration was correlated with metastasis and poor prognosis in HCC [38]. In addition, in malignant pleural mesothelioma (MPM) high levels of IL-34 were detectable in MPM patients. The same research group demonstrated that tumoral IL-34 strongly supported the development of immunosuppressive phenotypes [39]. Recently, it was discovered that IL-34 stimulated CRC cells proliferation and migration through the activation of ERK1/2 signaling pathway [36,40]. Additional studies demonstrated the involvement of MEK/ERK pathway and PI-3K signaling in CSF-1-mediated proliferation, invasion, and survival of lung cancer cell lines [41]. IL-34 and M-CSF-enriched TME was associated with aggressive tumors and low overall survival in a group of lung cancer patients [42]. In endometrial cancer, tumor cells drove macrophages infiltration via CSF-1 secretion and CSF-1 silencing reduced the migratory ability of macrophages [43]. Kirma et al. demonstrated that cervical cancer cells produced high levels of CSF-1 and CSF-1R blockade reduced the migratory capacity of cancer cells, suggesting the involvement of CSF-1 in tumor malignancy via the autocrine receptor activation [28].
2.3. CSF-1R in Cancer Cell Proliferation
The first evidence of CSF-1R expression in tumor cell line was from Kaciniski group [17]. This work demonstrated the expression of CSF-1R transcripts in endometrial, ovarian and breast cancer cells. In addition, they discovered that glucocorticoids and lactogenic steroid enhanced CSF-1R expression at messenger RNA and protein levels in mammary epithelial cells [17]. In CSF-1R-overexpressing MCF-7 cancer cell line, CSF-1 induced cell cycle arrest associated with increased p21 levels and the formation of p21/CDK complexes preventing cell cycle progression. In contrast, in T47-D cells CSF-1 had pro-mitogenic functions and induced low levels of p21, suggesting a cell background-dependent role of CSF-1R [44]. Notably, another study conducted in metastatic breast cancer cells showed that in vivo CSF-1R upregulation was mediated by TGF-β levels in the microenvironment, as in fact, the CSF-1R levels decreased upon TGF-β inhibition [45]. The authors also demonstrated that claudin-low cells expressed higher CSF-1R expression than luminal ones. In contrast to reports showing that CSF-1R suppression increased the proliferation rate of claudin-low cancer cells in vivo [46], Rovida et al. demonstrated that the inhibition of the receptor and autocrine M-CSF reduced cell growth of claudin-low breast cancer cells [11]. Additionally, the proliferative action of CSF-1R correlated with the activation of ERK1/2, c-Jun, Cyclin D and c-Myc downstream signaling pathways [11]. In vitro and in vivo studies indicated that IL-34/CSF-1R activation increased BrdU incorporation, colony forming abilities of breast cancer cells and tumor growth in mouse models. Moreover, the tumorigenic role of IL-34 is mediated by CSF-1R-dependent activation of MEK/ERK and JNK/c-Jun pathways [47]. This study also demonstrated the involvement of PIN1 in IL-34-mediated phosphorylation of MEK, ERK, JNK and c-JUN [47], while the pro-tumorigenic role of CSF-1R was mediated by its interaction with the transforming growth factor-β-stimulated clone-22 (TSC-22) protein in cervical cancer cells. TSC-22 acts as onco-suppressor, and it is expressed at low levels in tumors including cervical cancer. CSF-1R was found as a target of TSC-22, that interacted with tyrosine domain of its intracellular region by blocking its activity [14]. In melanoma cell lines, pharmacological and genetic targeting of the receptor inhibited proliferation of 3D cultures and increased apoptotic rate, suggesting a pro-survival role for CSF-1R [13]. CSF-1R overexpressing glioma cells showed increased cell viability, ki-67-positivity and enhanced colony forming ability [46]. Interestingly, CSF-1R suppression affected cell cycle progression by enhancing p27 expression, thereby preventing cells entering the S phase. Moreover, CSF-1R overexpression did not induce AKT activation, but only ERK1/2 activity in glioma cells [48]. On the contrary, in T-cell lymphoma cells, CSF-1R activation led to AKT phosphorylation in a PI3K-dependent manner [49]. Rattanaburee et al. identified CSF-1R as a possible target of kusunokinin, a lignan-derived molecule with demonstrated anti-cancer efficacy. They demonstrated that kusunokinin down-regulated CSF-1R expression levels by binding to the juxta-membrane domain (JMD) of the receptor. The anti-proliferative activity of kusunokinin observed in breast cancer cells was associated with the suppression of the CSF-1R and AKT pathways. Kusunokinin-mediated CSF-1R suppression also decreased the expression levels of G2-M markers including Cyclin B, CDK1, and c-Myc [50]. In canine mammary cancer cells, CSF-1R inhibition increased apoptosis and decreased Ki-67 positivity, suggesting that CSF-1R supported survival and growth of cancer cells [51]. (Table 1).
2.4. CSF-1R in Cancer Cell Migration
Cancer cell migration is a complex process involving different molecular components and several evidence support the role of CSF-1R in this process [52,53]. Sapi et al. showed that mammary epithelial cell invasiveness was associated with CSF-1R expression, and the invasive capacity of these cells was inhibited by a dominant negative mutant of the transcriptional factor Ets2, suggesting the involvement of Ets2 in modulating CSF-1R signaling [54]. In mammary epithelial MCF-10A cells, constitutive activation of CSF-1R led to hyperproliferation and destruction of the acinar structures. In addition, expression of Y561F CSF-1R mutant in normal human breast cells failed to activate Src family members, indicating that Src signaling is required for CSF-1R action in modulating acinar structure integrity [52]. Intriguingly, autocrine activation of CSF-1R enhanced intracellular localization of E-cadherin and early wound closure [52]. In vivo study demonstrated that CSF-1R inhibition negatively affected the invasiveness potential of claudin-low breast cancer cells: tumor derived from mice orthotopically injected with CSF-1R-depleted breast cancer cells showed decreased invasive ability and reduced lung metastases as compared to controls, thereby suggesting a crucial role of CSF-1R in cancer cell motility and invasion [46]. Partial epithelial to mesenchymal transition (EMT) characterized by the expression of both Vimentin and E-cadherin was observed in inflammatory breast cancer, where treatment with the CSF-1R inhibitor BLZ945 reduced the spindle-like phenotype of cancer cells and reversed partial EMT, indicating a role for CSF-1R in breast cancer invasiveness [55]. In canine mammary tumor cells, genetic blockade of CSF-1R negatively affected the migratory and invasive abilities of these cells [51]. Overexpression of CSF-1 in human ovarian cancer cell lines induced the acquisition of invasive and metastatic characteristics observed both in vitro and in vivo. Moreover, the metastatic activity of CSF-1 was mediated by the downstream activation of the urokinase-type plasminogen activator (uPA) pathway, which was linked to motility and invasiveness in several tumor types (Toy et al., 2009). Similarly, the administration of CSF-1 enhanced invasive potential of lung cancer both in vivo and in vitro. Data showed that injection of CSF-1-depleted cancer cells in mice reduced tumor mass and osteolytic bone metastases compared to controls, as well as the percentage of Ki-67-positive tumor cells [57]. In human osteosarcoma cell lines, a pool of CSF-1R+ cells was identified and receptor expression was associated with mesenchymal markers expression and invasive capabilities, as in fact CSF-1R-genetic ablation inhibited EMT and migration of tumor cells. Interestingly, the study identified JAG1 as downstream target gene involved in CSF-1R-dependent cell migration [58]. Shi et al. identified the CSF-1R/STAT3/Mir-34a axis as regulator of pro-tumoral functions in CRC, where CSF-1R expression was positively correlated with the expression of EMT-related genes, like SNAIL and SLUG, and EMT markers such as vimentin and low expression levels of E-cadherin [59]. Similarly, CSF-1R knockdown in glioma cells showed reduced expression of EMT markers such as vimentin [48]. A functional role of CSF-1R in melanoma spread was also demonstrated by Giricz et al., showing a dose-dependent decrease in melanoma invasiveness after treatment with the CSF-1R inhibitor PLX3397 [13]. The clinical importance of CSF-1R was reported in gastric cancer (GC) [60]. According to this study, CSF-1/CSF-1R expression correlated with advanced stage of disease in young patients and tumor metastasis. RT-qPCR and immunohistochemistry indicated CSF-1R expression by GC cells. In vitro assay demonstrated the involvement of CSF-1/CSF-1R axis in tumor migration via anoikis resistance and the induction of pro-angiogenic factor expression, such as VEGFA, in GC tissues [60]. (Table 2).
2.5. CSF-1R in Drug Resistance and Stemness
Vemurafenib resistant melanoma cancer cell lines showed aberrant CSF-1R expression, and a positive link between the transcription factor RUNX1, CSF-1R, and IL-34, which affect the transcriptional regulation of CSF-1R [13]. Notably, CSF-1R and RUNX1 inhibitors reduced aggressiveness of tumor cells and colony size in melanoma [13]. CSF-1R expression was upregulated in a 5-FU chemo-resistant population of CRC cells [59] and the acquisition of the resistant phenotype, as well as the expression of stem-like and EMT-related genes, was linked to downregulation of Mir-34a, thereby suggesting that the CSF-1R/mir-34a pathway could be a potential target against CRC chemoresistance and invasiveness [59]. Mir-34a deficiency correlated with CSF-1R up-regulation and increased expression of the stemness marker Lgr5, as well as tumoroid formation capabilities in intestinal adenoma cells [61]. Studies on mesothelioma primary cultures and cell lines showed that autocrine activation of CSF-1R characterized a pool of malignant cells with stem-like and chemo-resistant phenotypes. The expression of the stemness markers SOX2, NANOG, OCT4, c-MYC and CD44 was observed in CSF-1R+ cells in association with increased ALDH activity and ABCG2 expression [12], suggesting therefore a link between CSF-1R expression and the acquisition of drug-resistant phenotypes. Chemoresistance of CSF-1R+ cells correlated with the activation of AKT signaling pathway, as in fact AKT blockade sensitized cells to cell death following Pemetrexed treatment [12]. Similarly, CSF-1R overexpression induced chemoresistance in ovarian cancer cells via AKT and ERK1/2 pathway activation, while knockdown studies indicated that CSF-1R inhibition increased the apoptotic rate of cisplatin-treated tumor cells [62]. Pass et al. showed a population of Cisplatin resistant CSF-1R+ lung cancer cell lines [63]. Furthermore, the group demonstrated that CSF-1R expression in necessary for chemoresistance in lung cancer. Indeed, the inhibition of the receptor with the tyrosine kinase (TK) inhibitor JNJ-40346527 impaired the sphere-forming capabilities and the expression of both stemness and chemoresistance markers in lung cancer cells. The study also provided evidence of the synergic activity of cisplatin after CSF-1R TK inhibition in vivo [63]. (Table 3).
3. Anti-CSF-1R Therapeutic Strategies and Future Prospectives
Several CSF-1R inhibitors, including small molecules and neutralizing antibodies, have been developed over the last years and many drugs targeting the CSF-1R are currently in clinical development or approved for tumor treatment in many tumor types [64,65]. Studies reported immunotherapeutic activity of anti-CSF-1R molecules due as in fact CSF-1R expression in myeloid cells correlated with the development of an immunosuppressive TME [6,66]. However, few data are currently available on the efficacy of anti-CSF-1R drugs in cancer. Pexidartinib (PLX3397) was tested in phase I clinical trials in several solid tumors, either alone or in combination with paclitaxel, demonstrating good tolerance and low toxicity [67,68]. PLX3397 was approved for treatment of symptomatic tenosynovial giant cell tumor (TGCT), which works through depleting immunosuppressive cells [69]. This FDA-approved CSF-1R inhibitor also interferes with growth and survival of T cell lymphoma cells by acting on both tumoral and microenvironmental cell pools [49]. In BRAF-therapy resistant melanoma, combinatorial treatment with CSF-1R (PLX3397) and BRAF V600E (PLX4720) inhibitors at low dose strongly increased the survival rate of murine xenograft models, as well as decreased proliferation and invasiveness of cancer cell lines [13]. Previous studies already demonstrated the efficacy of the combinatorial strategy in reducing tumor infiltrating myeloid cells in syngeneic mouse model of BRAFV600E-driven melanoma [70]. The efficacy of Pexidartinib was also demonstrated by reducing the proliferation of CSF1R-positive TGCT cell lines. The combined use of Pexidartinib and sotuletinib (BLZ945), another CSF-1R inhibitor, reduced proliferation of tumor cells, their spheroid formation ability as well induced apoptosis in correlation with CSF-1R levels in TGCT [71]. Additionally, PLX3397 administration effectively decreased osteosarcoma tumor growth and metastatic potential of tumor cell line in patient-derived xenograft (PDX)-injected animal models [72]. Another CSF-1R kinase inhibitor, Imatinib, has showed a strong reduction in colony forming ability of breast cancer cells [11]. This potent anti-tumor activity of anti-CSF-1R drugs has been mostly attributed to its action on TAMs and other myeloid-derived cells [64]. A thorough understanding of the significance of CSF-1R expression in tumor cells will greatly help for developing therapeutic drugs that specifically target the neoplastic component, for single use or in combination with other anti-cancer therapies.
4. Conclusions
Aberrant expression of CSF-1R has been observed in tumors, and it is associated with worse prognosis, while high levels of CSF-1R are predictive of an immunosuppressive phenotype and aggressive tumors [5]. Recently, CSF-1R expression was also detected at the cell membrane of cancer cells, where it plays an important role in supporting cancer progression. Thus, the characterization of the mechanisms regulating the expression of a myeloid receptor in cancer cells is an intriguing topic for future studies. Notably, not all tumor cells expressed the receptor but only a subset of cells with aggressive behavior, thereby indicating that CSF-1R could contribute to promoting tumor aggressiveness. As highlighted in this review, CSF-1R expression in tumor cells is indeed associated with increased proliferation, expression of EMT markers, development of invasive capabilities, drug resistance and acquisition of stem-like features. The major CSF-1R-mediated signaling pathways so far described in cancer cells are summarized in Figure 2. Although it was previously thought that anti-CSF-1R therapeutic strategies only targeted M2-TAMs, the evidence demonstrating CSF-1R expression in cancer cells and its contribution to tumor growth makes this receptor an interesting target for therapy. However, still very little is known regarding CSF-1R expression and downstream signaling mechanisms in cancer cells. Different questions are opened regarding CSF-1R activity and regulation in cancer cells: which cells express CSF-1R? In which conditions? Are CSF-1R regulation mechanisms shared between cancer cell of different tumor types? As mentioned above, many studies observed the involvement of CSF-1R signaling in the promotion of stemness and chemoresistance mechanisms, could CSF-1R be considered a negative predictor of outcome in tumor cells? Additionally, its activity in cell cycle progression suggested a specific role and regulation in proliferation. Is CSF-1R differently regulated than other tumor cell membrane receptors? Finally, the possibility that CSF-1R exerts different functions based on its cellular localization is another point to deeply investigate. To conclude, understanding the molecular mechanisms modulating CSF-1R expression and function in cancer cells and whether tumor cells share the same regulatory mechanisms of immune cells, will greatly help to design novel approaches to target CSF-1R in cancer.
Author Contributions
E.G. and F.C. conceived and designed the main content; F.C. and E.G. reviewed all the literature and wrote the manuscript. Methodology was carried out by, S.L. and W.V.; AM, SL, CU, WV, and AG provided valuable critical revisions of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by grants from the Mesothelioma Applied Research Foundation (MARF 2023, E.G.), the Italian Association for Cancer Research (IG-15378; IG-23179, W.V.) and Sbarro Health Research Organization (SHRO).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [PubMed]
- Rojo, R.; Pridans, C.; Langlais, D.; Hume, D.A. Transcriptional mechanisms that control expression of the macrophage colony-stimulating factor receptor locus. Clin. Sci. (Lond.), 2017, 131, 2161–2182. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lin, W.Y.; Chen, Y.; Stawicki, S.; Mukhyala, K.; Wu, Y.; Martin, F.; Bazan, J.F.; Starovasnik, M.A. Structural basis for the dual recognition of helical cytokines IL-34 and CSF-1 by CSF-1R. Structure. 2012, 20, 676–687. [Google Scholar] [CrossRef]
- Muñoz-Garcia, J.; Cochonneau, D.; Télétchéa, S.; Moranton, E.; Lanoe, D.; Brion, R.; Lézot, F.; Heymann, M.F.; Heymann, D. The twin cytokines interleukin-34 and CSF-1: masterful conductors of macrophage homeostasis. Theranostics. 2021, 11, 1568–1593. [Google Scholar] [CrossRef] [PubMed]
- Achkova, D.; Maher, J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem. Soc. Trans. 2016, 44, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Fu, W.; Turley, S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity. 2021, 54, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Sletta, K.Y.; Castells, O.; Gjertsen, B.T. Colony Stimulating Factor 1 Receptor in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 654817. [Google Scholar] [CrossRef]
- Roussel, M.F. Regulation of cell cycle entry and G1 progression by CSF-1. Mol. Reprod. Dev. 1997, 46, 11–18. [Google Scholar] [CrossRef]
- Ridge, S.A.; Worwood, M.; Oscier, D.; Jacobs, A.; Padua, R.A. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 1377–1380. [Google Scholar] [CrossRef]
- Morandi, A.; Barbetti, V.; Riverso, M.; Dello Sbarba, P.; Rovida, E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PloS one. 2011, 6, e27450. [Google Scholar] [CrossRef]
- Cioce, M.; Canino, C.; Goparaju, C.; Yang, H.; Carbone, M.; Pass, H.I. Autocrine CSF-1R signaling drives mesothelioma chemoresistance via AKT activation. Cell Death Dis. 2014, 5, e1167. [Google Scholar] [CrossRef]
- Giricz, O.; Mo, Y.; Dahlman, K.B.; Cotto-Rios, X.M.; Vardabasso, C.; Nguyen, H.; Matusow, B.; Bartenstein, M.; Polishchuk, V.; Johnson, D.B.; Bhagat, T.D.; Shellooe, R.; Burton, E.; Tsai, J.; Zhang, C.; Habets, G.; Greally, J.M.; Yu, Y.; Kenny; P. A., Fields; G., B.; … Verma, A.K. The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma. JCI Insight. 2018, 3, e120422. [Google Scholar] [CrossRef]
- Cho, M.J.; Lee, J.Y.; Shin, M.G.; Kim, H.J.; Choi, Y.J.; Rho, S.B.; Kim, B.R.; Jang, I.S.; Lee, S.H. TSC-22 inhibits CSF-1R function and induces apoptosis in cervical cancer. Oncotarget. 2017, 8, 97990–98003. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; Jones, T.; Jucknischke, U.; Scheiblich, S.; Kaluza, K.; Gorr, I.H.; Walz, A.; Abiraj, K.; Cassier, P.A.; Sica, A.; Gomez-Roca, C.; … Rüttinger, D. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer cell. 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.R.; Pixley, F.J. CSF-1R signaling in health and disease: a focus on the mammary gland. J. Mammary Gland Biol. Neoplasia. 2014, 19, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kacinski, B.M. CSF-1 and its receptor in breast carcinomas and neoplasms of the female reproductive tract. Mol Reprod Dev. 1997, 46, 71–74. [Google Scholar] [CrossRef]
- Espinosa, I.; Beck, A.H.; Lee, C.H.; Zhu, S.; Montgomery, K.D.; Marinelli, R.J.; Ganjoo, K.N.; Nielsen, T.O.; Gilks, C.B.; West, R.B.; van de Rijn, M. Coordinate expression of colony-stimulating factor-1 and colony-stimulating factor-1-related proteins is associated with poor prognosis in gynecological and nongynecological leiomyosarcoma. Am. J. Pathol. 2009, 174, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Toy, E.P.; Azodi, M.; Folk, N.L.; Zito, C.M.; Zeiss, C.J.; Chambers, S.K. Enhanced ovarian cancer tumorigenesis and metastasis by the macrophage colony-stimulating factor. Neoplasia. 2009, 11, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Seligson, D.B.; Memarzadeh, S.; Xin, L.; Horvath, S.; Dubey, P.; Flick, M.B.; Kacinski, B.M.; Palotie, A.; Witte, O.N. Expression of colony-stimulating factor 1 receptor during prostate development and prostate cancer progression. Proc Natl Acad. Sci. U.S.A. 2002, 99, 14404–14409. [Google Scholar] [CrossRef] [PubMed]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.T. The prognostic impact of M-CSF, CSF-1 receptor, CD68 and CD3 in prostatic carcinoma. Histopathology. 2008, 53, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kuropkat, C.; Dünne, A.A.; Plehn, S.; Ossendorf, M.; Herz, U.; Renz, H.; Werner, J.A. Macrophage colony-stimulating factor as a tumor marker for squamous cell carcinoma of the head and neck. Tumour Biol. 2003, 24, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Szmitkowski, M.; Okulczyk, B. Hematopoietic growth factors in colorectal cancer patients. Clin Chem Lab Med. 2003, 41, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Li, N.F.; Kocher, H.M.; Salako, M.A.; Obermueller, E.; Sandle, J.; Balkwill, F. A novel function of colony-stimulating factor 1 receptor in hTERT immortalization of human epithelial cells. Oncogene. 2009, 28, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Hao, Y.; Lv, Y.; Chen, Z.; Shen, J.; Zhou, S.; Yin, M. Overexpression of macrophage-colony stimulating factor-1 receptor as a prognostic factor for survival in cancer: A systematic review and meta-analysis. Medicine. 2021, 100, e25218. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Burugu, S.; Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Nielsen, T.O. (2021). Prognostic Significance of CSF-1R Expression in Early Invasive Breast Cancer. Cancers. 2021, 13, 5769. [Google Scholar] [CrossRef] [PubMed]
- Such, E.; Cervera, J.; Valencia, A.; Garcia-Casado, Z.; Senent, M.L.; Sanz, M.A.; Sanz, G.F. Absence of mutations in the tyrosine kinase and juxtamembrane domains of C-FMS gene in chronic myelomonocytic leukemia (CMML). Leuk. Res. 2009, 33, e162–e163. [Google Scholar] [CrossRef]
- Kirma, N.; Hammes, L.S.; Liu, Y.G.; Nair, H.B.; Valente, P.T.; Kumar, S.; Flowers, L.C.; Tekmal, R.R. Elevated expression of the oncogene c-fms and its ligand, the macrophage colony-stimulating factor-1, in cervical cancer and the role of transforming growth factor-beta1 in inducing c-fms expression. Cancer Res. 2007, 67, 1918–1926. [Google Scholar] [CrossRef]
- Barbetti, V.; Morandi, A.; Tusa, I.; Digiacomo, G.; Riverso, M.; Marzi, I.; Cipolleschi, M.G.; Bessi, S.; Giannini, A.; Di Leo, A.; Dello Sbarba, P.; Rovida, E. Chromatin-associated CSF-1R binds to the promoter of proliferation-related genes in breast cancer cells. Oncogene. 2014, 33, 4359–4364. [Google Scholar] [CrossRef]
- Soares, M.J.; Pinto, M.; Henrique, R.; Vieira, J.; Cerveira, N.; Peixoto, A.; Martins, A.T.; Oliveira, J.; Jerónimo, C.; Teixeira, M.R. CSF1R copy number changes, point mutations, and RNA and protein overexpression in renal cell carcinomas. Mod. Pathol. 2009, 22, 744–752. [Google Scholar] [CrossRef]
- Zwaenepoel, O.; Tzenaki, N. , Vergetaki, A.; Makrigiannakis, A.; Vanhaesebroeck, B.; Papakonstanti, E.A. Functional CSF-1 receptors are located at the nuclear envelope and activated via the p110δ isoform of PI 3-kinase. FASEB J. 2012, 26, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Abiatari, I.; Midelashvili, T.; Motsikulashvili, M.; Tchavtchavadze, A.; Tawfeeq, S.; Amiranashvili, I. OVEREXPRESSED PROGENITOR GENE CSF1R IN PANCREATIC CANCER TISSUES AND NERVE INVASIVE PANCREATIC CANCER CELLS. Georgian Med. News. 2018, (285), 96–100. [Google Scholar]
- Filiberti, S.; Russo, M.; Lonardi, S.; Bugatti, M.; Vermi, W.; Tournier, C.; Giurisato, E. Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways. Biomedicines. 2022, 10, 2709. [Google Scholar] [CrossRef] [PubMed]
- Low, V.; Li, Z.; Blenis, J. Metabolite activation of tumorigenic signaling pathways in the tumor microenvironment. Sci Signal. 2022, 15, eabj4220. [Google Scholar] [CrossRef]
- Cersosimo, F.; Barbarino, M.; Lonardi, S.; Vermi, W.; Giordano, A.; Bellan, C.; Giurisato, E. Mesothelioma Malignancy and the Microenvironment: Molecular Mechanisms. Cancers. 2021, 13, 5664. [Google Scholar] [CrossRef]
- Franzè, E.; Dinallo, V.; Rizzo, A.; Di Giovangiulio, M.; Bevivino, G.; Stolfi, C.; Caprioli, F.; Colantoni, A.; Ortenzi, A.; Grazia, A.D.; Sica, G.; Sileri, P.P.; Rossi, P.; Monteleone, G. Interleukin-34 sustains pro-tumorigenic signals in colon cancer tissue. Oncotarget. 2017, 9, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Hama, N.; Baghdadi, M.; Ishikawa, K.; Otsuka, R.; Wada, H.; Asano, H.; Endo, D.; Konno, Y.; Kato, T.; Watari, H.; Tozawa, A.; Suzuki, N.; Yokose, T.; Takano, A.; Kato, H.; Miyagi, Y.; Daigo, Y.; Seino, K.I. Interleukin-34 expression in ovarian cancer: a possible correlation with disease progression. Int. Immunol. 2020, 32, 175–186. [Google Scholar] [CrossRef]
- Zhou, S.L.; Hu, Z.Q.; Zhou, Z.J.; Dai, Z.; Wang, Z.; Cao, Y.; Fan, J.; Huang, X.W.; Zhou, J. miR-28-5p-IL-34-macrophage feedback loop modulates hepatocellular carcinoma metastasis. Hepatology. 2016, 63, 1560–1575. [Google Scholar] [CrossRef]
- Blondy, T.; d'Almeida, S.M.; Briolay, T.; Tabiasco, J.; Meiller, C.; Chéné, A.L.; Cellerin, L.; Deshayes, S.; Delneste, Y.; Fonteneau, J.F.; Boisgerault, N.; Bennouna, J.; Grégoire, M.; Jean, D.; Blanquart, C. Involvement of the M-CSF/IL-34/CSF-1R pathway in malignant pleural mesothelioma. J. Immunother. Cancer. 2020, 8, e000182. [Google Scholar] [CrossRef]
- Franzè, E.; Marafini, I.; Troncone, E.; Salvatori, S.; Monteleone, G. Interleukin-34 promotes tumorigenic signals for colon cancer cells. Cell Death Discov. 2021, 7, 245. [Google Scholar] [CrossRef]
- Uemura, Y.; Kobayashi, M.; Nakata, H.; Kubota, T.; Bandobashi, K.; Saito, T.; Taguchi, H. Effects of GM-CSF and M-CSF on tumor progression of lung cancer: roles of MEK1/ERK and AKT/PKB pathways. Int. J. Mol. Med. 2006, 18, 365–373. [Google Scholar] [CrossRef]
- Baghdadi, M.; Endo, H.; Takano, A.; Ishikawa, K.; Kameda, Y.; Wada, H.; Miyagi, Y.; Yokose, T.; Ito, H.; Nakayama, H.; Daigo, Y.; Suzuki, N.; Seino, K.I. High co-expression of IL-34 and M-CSF correlates with tumor progression and poor survival in lung cancers. Sci. Rep. 2018, 8, 418. [Google Scholar] [CrossRef]
- Hua, F.; Tian, Y.; Gao, Y.; Li, C.; Liu, X. Colony-stimulating factor 1 receptor inhibition blocks macrophage infiltration and endometrial cancer cell proliferation. Mol. Med. Rep. 2019, 19, 3139–3147. [Google Scholar] [CrossRef]
- Lee, A.W.; Nambirajan, S.; Moffat, J.G. CSF-1 activates MAPK-dependent and p53-independent pathways to induce growth arrest of hormone-dependent human breast cancer cells. Oncogene. 1999, 18, 7477–7494. [Google Scholar] [CrossRef]
- Patsialou, A.; Wyckoff, J.; Wang, Y.; Goswami, S.; Stanley, E.R.; Condeelis, J.S. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009, 69, 9498–9506. [Google Scholar] [CrossRef]
- Patsialou, A.; Wang, Y.; Pignatelli, J.; Chen, X.; Entenberg, D.; Oktay, M.; Condeelis, J.S. Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFβ in claudin-low breast tumor cells. Oncogene. 2015, 34, 2721–2731. [Google Scholar] [CrossRef]
- Poudel, M.; Kim, G.; Bhattarai, P.Y.; Kim, J.Y.; Choi, H.S. Interleukin-34-CSF1R Signaling Axis Promotes Epithelial Cell Transformation and Breast Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 2711. [Google Scholar] [CrossRef]
- Sun, L.; Liang, H.; Yu, W.; Jin, X. Increased invasive phenotype of CSF-1R expression in glioma cells via the ERK1/2 signaling pathway. Cancer gene Ther. 2019, 26(5-6), 136–144. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.; Rolland, D.C.M.; Polk, A.; Wolfe, A.; Dewar, H.; Chowdhury, P.; Onder, O.; Dewar, R.; Brown, N.A.; Bailey, N.G.; Inamdar, K.; Lim, M.S.; Elenitoba-Johnson, K.S.J.; Wilcox, R.A. Colony-Stimulating Factor 1 Receptor (CSF1R) Activates AKT/mTOR Signaling and Promotes T-Cell Lymphoma Viability. Clin. Cancer Res. 2020, 26, 690–703. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Tipmanee, V.; Tedasen, A.; Thongpanchang, T.; Graidist, P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed. Pharmacother. 2020, 129, 110361. [Google Scholar] [CrossRef]
- Król, M.; Majchrzak, K.; Mucha, J.; Homa, A.; Bulkowska, M.; Jakubowska, A.; Karwicka, M.; Pawłowski, K.M.; Motyl, T. CSF-1R as an inhibitor of apoptosis and promoter of proliferation, migration and invasion of canine mammary cancer cells. BMC Vet. Res. 2013, 9, 65. [Google Scholar] [CrossRef]
- Wrobel, C.N.; Debnath, J.; Lin, E.; Beausoleil, S.; Roussel, M.F.; Brugge, J.S. Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J. Cell Biol. 2004, 165, 263–273. [Google Scholar] [CrossRef]
- Huang, L.; Xu, X.; Hao, Y. The possible mechanisms of tumor progression via CSF-1/CSF-1R pathway activation. Rom. J. Morphol. Embryol. 2014, 55 (2 Suppl), 501–506. [Google Scholar]
- Sapi, E.; Flick, M.B.; Rodov, S.; Kacinski, B.M. Ets-2 transdominant mutant abolishes anchorage-independent growth and macrophage colony-stimulating factor-stimulated invasion by BT20 breast carcinoma cells. Cancer Res. 1998, 58, 1027–1033. [Google Scholar] [PubMed]
- Kai, K.; Iwamoto, T.; Zhang, D.; Shen, L.; Takahashi, Y.; Rao, A.; Thompson, A.; Sen, S.; Ueno, N.T. CSF-1/CSF-1R axis is associated with epithelial/mesenchymal hybrid phenotype in epithelial-like inflammatory breast cancer. Sci. Rep. 2018, 8, 9427. [Google Scholar] [CrossRef] [PubMed]
- Toy, E.P.; Azodi, M.; Folk, N.L.; Zito, C.M.; Zeiss, C.J.; Chambers, S.K. Enhanced ovarian cancer tumorigenesis and metastasis by the macrophage colony-stimulating factor. Neoplasia. 2009, 11, 136–144. [Google Scholar] [CrossRef]
- Hung, J.Y.; Horn, D.; Woodruff, K.; Prihoda, T.; LeSaux, C.; Peters, J.; Tio, F.; Abboud-Werner, S.L. Colony-stimulating factor 1 potentiates lung cancer bone metastasis. Lab. Invest. 2014, 94, 371–381. [Google Scholar] [CrossRef]
- Wen, Z.Q.; Li, X.G.; Zhang, Y.J.; Ling, Z.H.; Lin, X.J. Osteosarcoma cell-intrinsic colony stimulating factor-1 receptor functions to promote tumor cell metastasis through JAG1 signaling. Am. J. Cancer Res. 2017, 7, 801–815. [Google Scholar]
- Shi, X.; Kaller, M.; Rokavec, M.; Kirchner, T.; Horst, D.; Hermeking, H. Characterization of a p53/miR-34a/CSF1R/STAT3 Feedback Loop in Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 391–418. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Ichikawa, T.; Kawamura, M.; Yasuda, H.; Fujikawa, H.; Saigusa, S.; Ohi, M.; Araki, T.; Tanaka, K.; Inoue, Y.; Tanaka, M.; Miki, C.; Kusunoki, M. Colony-stimulating factor-1 and colony-stimulating factor-1 receptor co-expression is associated with disease progression in gastric cancer. Int. J. Oncol. 2018, 53, 737–749. [Google Scholar] [CrossRef]
- Liu, F.; Bouznad, N.; Kaller, M.; Shi, X.; König, J.; Jaeckel, S.; Hermeking, H. Csf1r mediates enhancement of intestinal tumorigenesis caused by inactivation of Mir34a. Int. J. Biol. Sci. 2022, 18, 5415–5437. [Google Scholar] [CrossRef]
- Yu, R.; Jin, H.; Jin, C.; Huang, X.; Lin, J.; Teng, Y. Inhibition of the CSF-1 receptor sensitizes ovarian cancer cells to cisplatin. Cell. Biochem. Funct. 2018, 36, 80–87. [Google Scholar] [CrossRef]
- Pass, H.I.; Lavilla, C.; Canino, C.; Goparaju, C.; Preiss, J.; Noreen, S.; Blandino, G.; Cioce, M. Inhibition of the colony-stimulating-factor-1 receptor affects the resistance of lung cancer cells to cisplatin. Oncotarget. 2016, 7, 56408–56421. [Google Scholar] [CrossRef]
- Lin, C.C. Clinical Development of Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors. J. Immunother. Precis. Oncol. 2021, 4, 105–114. [Google Scholar] [CrossRef]
- Wen, J.; Wang, S.; Guo, R.; Liu, D. CSF1R inhibitors are emerging immunotherapeutic drugs for cancer treatment. Eur J Med Chem. 2023, 245 Pt 1, 114884. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.S.; You, W.P.; Zhu, F.F.; Wang, J.H.; Guo, F.; Xu, J.J.; Liu, X.L.; Zhong, H.J. Targeted delivery of pexidartinib to tumor-associated macrophages via legumain-sensitive dual-coating nanoparticles for cancer immunotherapy. Colloids Surf. B. Biointerfaces. 2023, 226, 113283. [Google Scholar] [CrossRef]
- Wesolowski, R.; Sharma, N.; Reebel, L.; Rodal, M.B.; Peck, A.; West, B.L.; Marimuthu, A.; Severson, P.; Karlin, D.A.; Dowlati, A.; Le, M.H.; Coussens, L.M.; Rugo, H.S. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors Ther. Adv. Med. Oncol. 2019, 11, 1758835919854238. [Google Scholar] [CrossRef]
- Yin, O.; Wagner, A.J.; Kang, J.; Knebel, W.; Zahir, H.; van de Sande, M.; Tap, W.D.; Gelderblom, H.; Healey, J.H.; Shuster, D.; Stacchiotti, S. Population Pharmacokinetic Analysis of Pexidartinib in Healthy Subjects and Patients With Tenosynovial Giant Cell Tumor or Other Solid Tumors. J. Clin. Pharmacol. 2021, 61, 480–492. [Google Scholar] [CrossRef]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs. 2019, 79, 1805–1812. [Google Scholar] [CrossRef]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Duangkaew, S.; Yotchai, W.; Maungsomboon, S.; Phimolsarnti, R.; Asavamongkolkul, A.; Thuwajit, P.; Thuwajit, C.; Chandhanayingyong, C. Novel CSF1R-positive tenosynovial giant cell tumor cell lines and their pexidartinib (PLX3397) and sotuletinib (BLZ945)-induced apoptosis. Hum. Cell. 2023, 36, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Smeester, B.A.; Slipek, N.J.; Pomeroy, E.J.; Laoharawee, K.; Osum, S.H.; Larsson, A.T.; Williams, K.B.; Stratton, N.; Yamamoto, K.; Peterson, J.J.; Rathe, S.K.; Mills, L.J.; Hudson, W.A.; Crosby, M.R.; Wang, M.; Rahrmann, E.P.; Moriarity, B.S.; Largaespada, D.A. PLX3397 treatment inhibits constitutive CSF1R-induced oncogenic ERK signaling, reduces tumor growth, and metastatic burden in osteosarcoma. Bone. 2020, 136, 115353. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
CSF-1R expression in cancer cells. RNA in situ hybridization (RNA-scope) on paraffin- embedded cell blocks from human cancer cell lines: cervical cancer (HeLa), HER2+ breast cancer (SKBR3), monocytic leukemia (THP1) and biphasic mesothelioma (MSTO-211H). A CSF-1R probe was used following the manufacturer’s (Advanced Cell Diagnostics, Newark, CA, USA) instructions. Signals was detected using RNAscope 2.5 HD Detection Reagent FAST RED (Cat# 322360). Cytoplasmic and perinuclear stains (in red) were considered as specific. Scale bar: 40 microns.
Figure 1.
CSF-1R expression in cancer cells. RNA in situ hybridization (RNA-scope) on paraffin- embedded cell blocks from human cancer cell lines: cervical cancer (HeLa), HER2+ breast cancer (SKBR3), monocytic leukemia (THP1) and biphasic mesothelioma (MSTO-211H). A CSF-1R probe was used following the manufacturer’s (Advanced Cell Diagnostics, Newark, CA, USA) instructions. Signals was detected using RNAscope 2.5 HD Detection Reagent FAST RED (Cat# 322360). Cytoplasmic and perinuclear stains (in red) were considered as specific. Scale bar: 40 microns.
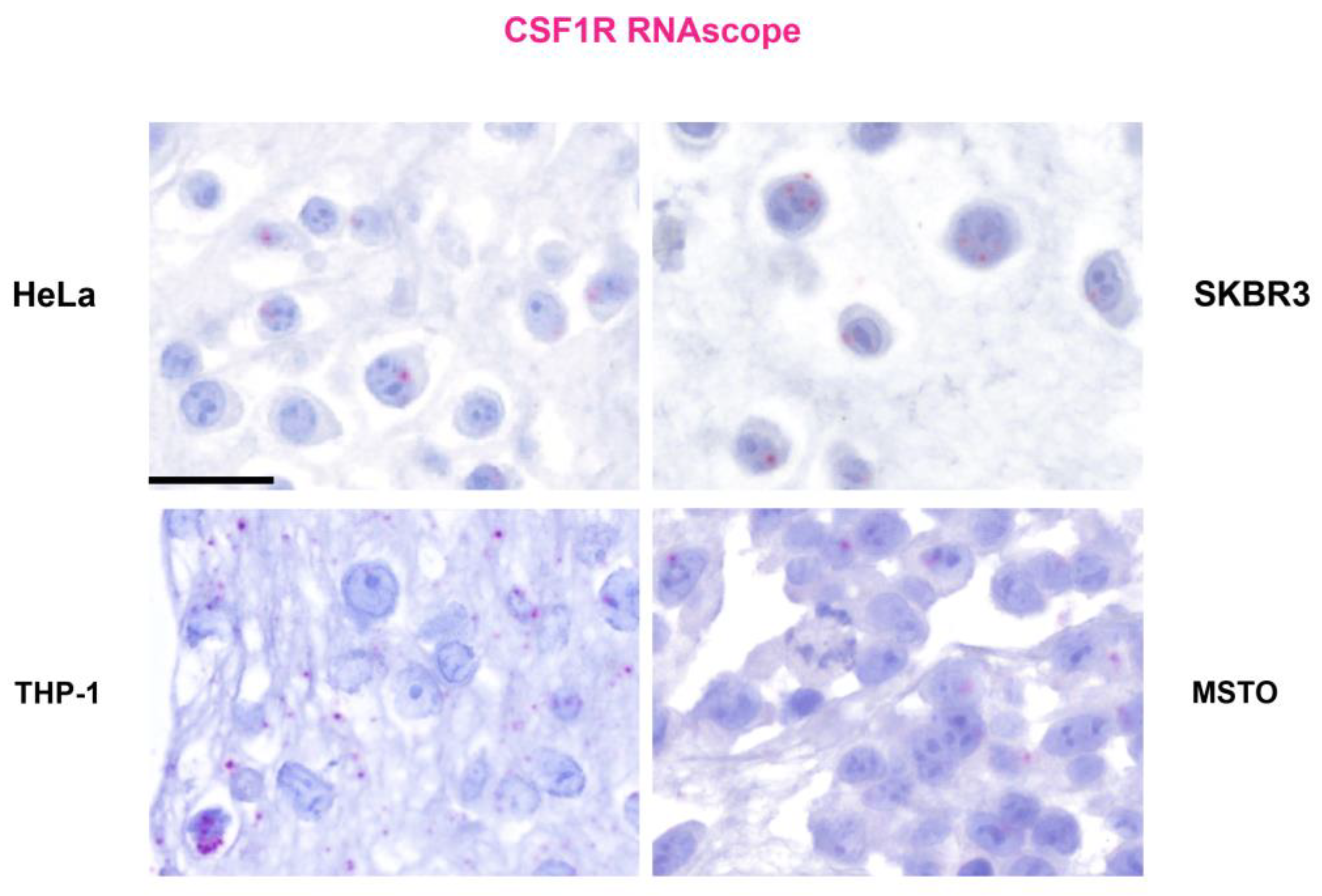
Figure 2.
CSF-1R Signaling in Cancer cells. The image is a schematic representation of the available data regarding CSF-1R signaling and function in different cancer cell types. In mammary epithelial cells, Src signaling correlated with CSF-1R activation [52]. ERK 1/2 pathway activation following CSF-1R induction was reported in lung, breast, and glioma cancer cells, associated with cell proliferation [11,41,48]. In breast cancer, CSF-1R signaling induced the transcription of the proliferative genes CCND1, c-JUN, and c-MYC [29], even though MCF-7 cells overexpressing CSF-1R increased the expression levels of p21 [44]. In different tumor types, CSF-1R promoted proliferation and chemoresistance via the activation of PI3K/AKT signaling [12,49,62]. In colorectal cancer the axis CSF-1R/STAT3/mir-34a was involved in the development of chemoresistant and invasive phenotype [59]. Moreover, mir-34a negatively regulated the expression of stem-like gene Lrg5 and the acquisition of stem-like traits [61]. In mesothelioma, CSF-1R expression correlated with stemness markers expression as SOX2, NANOG, OCT4 and c-MYC and drug resistance markers including ABCG2 transcription [12]. In colorectal cancer, glioma and mesothelioma CSF-1R induced the expression of EMT-related genes, like SNAIL and SLUG, Vimentin (VIM) and matrix metalloproteinase-9 (MMP-9) and CD44[12,48,59]. Moreover, in osteosarcoma cells, CSF-1R expression associated with Twist and N-cadherin expression [58]. According to Giricz et al [13], in a subset of chemoresistant melanoma cells, the main regulator of CSF-1R expression might be RUNX1, which associated with the CSF-1R promoter region [13]. In both cervical and breast cancer the expression levels of CSF-1R linked with TGF-β receptor activity [28,46]. Accumulation of nuclear beta-catenin was also regulated via CSF-1R in intestinal tumor cells [61]. Created in Biorender.com.
Figure 2.
CSF-1R Signaling in Cancer cells. The image is a schematic representation of the available data regarding CSF-1R signaling and function in different cancer cell types. In mammary epithelial cells, Src signaling correlated with CSF-1R activation [52]. ERK 1/2 pathway activation following CSF-1R induction was reported in lung, breast, and glioma cancer cells, associated with cell proliferation [11,41,48]. In breast cancer, CSF-1R signaling induced the transcription of the proliferative genes CCND1, c-JUN, and c-MYC [29], even though MCF-7 cells overexpressing CSF-1R increased the expression levels of p21 [44]. In different tumor types, CSF-1R promoted proliferation and chemoresistance via the activation of PI3K/AKT signaling [12,49,62]. In colorectal cancer the axis CSF-1R/STAT3/mir-34a was involved in the development of chemoresistant and invasive phenotype [59]. Moreover, mir-34a negatively regulated the expression of stem-like gene Lrg5 and the acquisition of stem-like traits [61]. In mesothelioma, CSF-1R expression correlated with stemness markers expression as SOX2, NANOG, OCT4 and c-MYC and drug resistance markers including ABCG2 transcription [12]. In colorectal cancer, glioma and mesothelioma CSF-1R induced the expression of EMT-related genes, like SNAIL and SLUG, Vimentin (VIM) and matrix metalloproteinase-9 (MMP-9) and CD44[12,48,59]. Moreover, in osteosarcoma cells, CSF-1R expression associated with Twist and N-cadherin expression [58]. According to Giricz et al [13], in a subset of chemoresistant melanoma cells, the main regulator of CSF-1R expression might be RUNX1, which associated with the CSF-1R promoter region [13]. In both cervical and breast cancer the expression levels of CSF-1R linked with TGF-β receptor activity [28,46]. Accumulation of nuclear beta-catenin was also regulated via CSF-1R in intestinal tumor cells [61]. Created in Biorender.com.
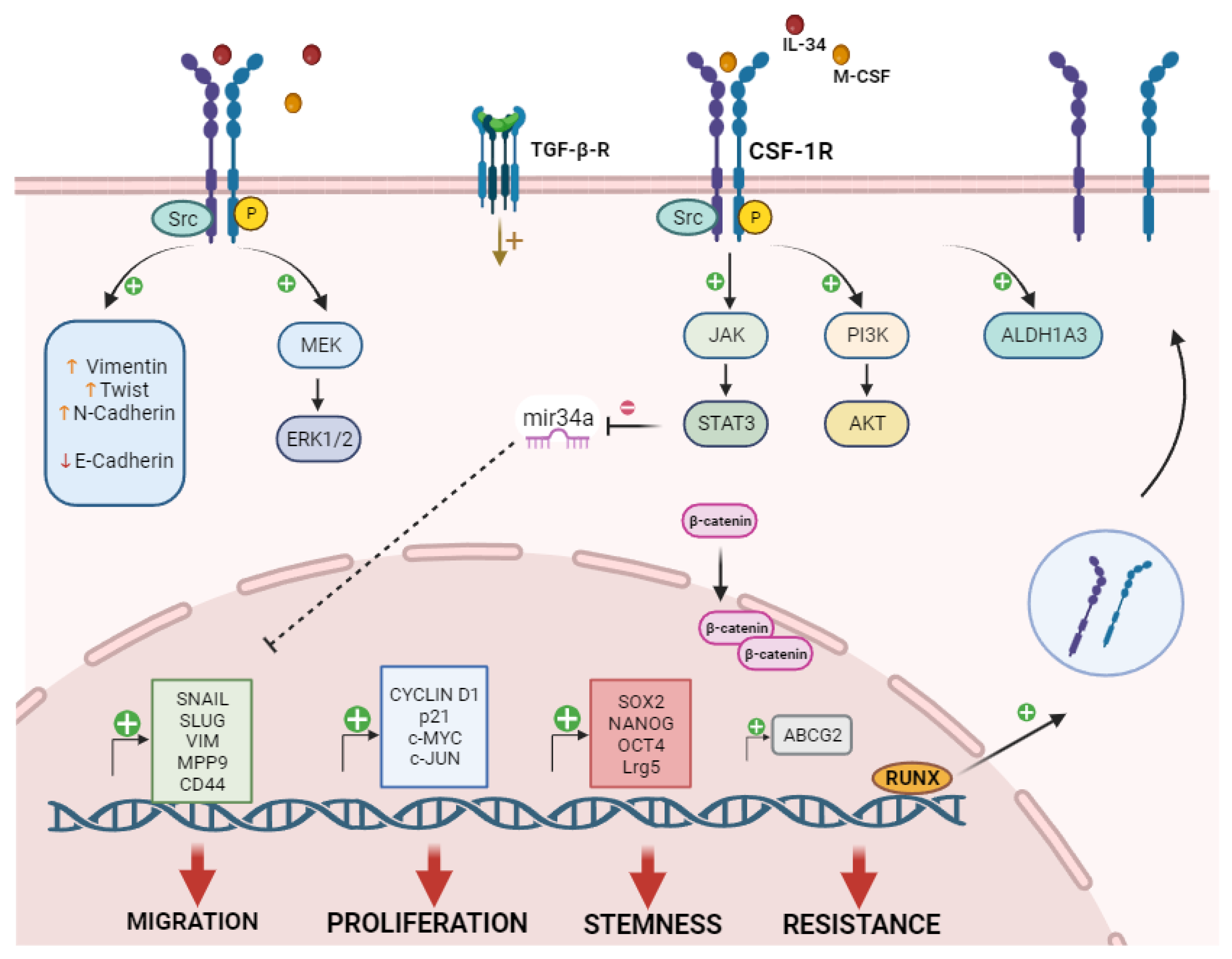

Table 1.
CSF-1R in cancer cell proliferation.
| PROLIFERATION | ||
|---|---|---|
| Tumor type | Function | Reference |
| Breast Cancer | Cell cycle regulation p21 induction |
[44] |
| Breast Cancer | ERK1/2; c-Jun; Cyclin D; c-Myc activation | [11] |
| Breast Cancer | MEK/ERK; JNK/c-Jun c-Fos; c-Jun; AP-1activation |
[47] |
| Cervical Cancer | TSC-22-mediated proliferation | [14] |
| Melanoma | Pro-survival; anti-apoptotic | [13] |
| Glioma | P27 expression Increased cell viability Ki67-+ cells ERK1/2 activation |
[48] |
| T-cell lymphoma | PI3-K/AKT activation | [49] |
| Breast Cancer | G2-M markers expression | [50] |
| Mammary epithelial tissue | Hyperproliferation | [52] |
Table 2.
CSF-1R in cancer cell migration.
| MIGRATION | ||
|---|---|---|
| Tumor type | Function | Reference |
| Mammary epithelial tissue | Destruction of acinar architecture E-Cadherin re-localization Src-kinase activity |
[52] |
| Breast Cancer | Invasiveness and metastases | [46] |
| Breast Cancer | EMT | [55] |
| Ovarian Cancer | Adhesion and motility Tumor metastases Increased Urokinase surface expression |
[19] |
| Lung Cancer | Osteolytic metastases | [57] |
| Osteosarcoma | EMT via JAG-1 pathway activation | [58] |
| Colorectal Cancer | EMT markers expression STAT3 induction Mir-34a downregulation |
[59] |
| Glioma | EMT | [48] |
| Melanoma | Invasiveness | [13] |
| Gastric Cancer | Cell migration and Tumor metastasis | [60] |
Table 3.
CSF-1R in drug resistance and stemness.
| DRUG RESISTANCE & STEMNESS | ||
|---|---|---|
| Tumor type | Function | Reference |
| Melanoma | BRAF resistance | [13] |
| Colorectal cancer | ALDH activity mir-34a downregulation |
[59] |
| Mesothelioma | ALDH activity Stem-like markers expression AKT pathway activation |
[12] |
| Intestinal adenoma | Stem-like gene expression | [61] |
| Ovarian Cancer | Cisplatin resistance AKT; ERK1/2 pathway activation |
[62] |
| Lung Cancer | Cisplatin resistance Stem-like markers expression |
[63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



