
Submitted:
14 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
Glaucoma is a group of ocular diseases that causes irreversible blindness worldwide and is characterized by a multifactorial degeneration of optic nerve and retinal ganglion cells (RGCs), leading to vision loss. One of the significant risk factors in glaucoma pathogenesis is glia-driven neuroinflammation. Impairment of mitochondrial dynamics and bioenergetics is associated with glaucomatous neurodegeneration and is linked to neuroinflammation. Apolipoprotein A-I binding protein (AIBP) binds to toll-like receptor 4 (TLR4) and facilitates the removal of excess cholesterol from the plasma membrane in activated cells, suggesting the regulation of cholesterol-driven inflammation by inhibiting the TLR4-associated lipid rafts. AIBP is localized in mitochondria and regulates mitophagy by ubiquitination of mitofusin 1 and 2. We observed elevated intraocular pressure-induced AIBP deficiency in glaucomatous neurodegeneration. We also observed that AIBP deficiency activated TLR4 in Müller glia, triggered mitochondrial dysfunction in both RGCs and Müller glia and impaired visual function. In mouse models of glaucoma, AIBP treatment protects RGCs by reducing inflammatory responses. Here, our review focuses on investigating the role of AIBP in the retina and examining the underlying mechanism by which AIBP contributes to the pathogenesis of glaucoma, aiming to provide new insight into the therapeutic potential for the treatment of glaucoma.
Keywords:
AIBP
; Glaucoma
; Neuroinflammation
; Cholesterol
; TLR4
; Lipid rafts
; Mitochondria
; Gene therapy
; AAV
1. Introduction
Glaucoma is a leading cause of irreversible blindness globally. It is characterized by a slow, progressive, and irreversible degeneration of retinal ganglion cells (RGCs) and their axons, resulting in a cupping of the optic disc (also called the optic nerve head) and visual field loss in patients with glaucoma [1,2,3]. The global prevalence of glaucoma has been reported, that 3.5% of the population aged 40-80 years are currently being affected worldwide, and that the number of glaucoma patients is expected to reach over 111.8 million by 2040 [4].
Glaucoma is a multifactorial neurodegenerative disorder that is caused by multiple pathogenic stressors, including elevated intraocular pressure (IOP), aging, oxidative/metabolic stress, mitochondrial dysfunction, defects in the retinal vasculature, genetic factors, and neuroinflammation [5,6,7,8]. Despite increasing publications from animal models and human subjects, the underlying pathophysiological mechanisms of glaucoma are not fully understood. To date, IOP is only a proven, modifiable, and treatable risk factor, and lowering IOP pharmaceutically or surgically can reduce glaucoma progression [2,3,9]. However, the reduction of IOP in some patients is often insufficient to restore visual function [10], and the precise mechanisms by which increased IOP leads to axonal degeneration and RGC death remain to be elucidated. The mechanical compression of the axons induced by high IOP at the level of the lamina cribrosa might directly lead to ischemia-hypoxia damage, impairment of axonal transport, deprivation of growth factors, and subsequent RGC loss [11,12,13]. In addition to IOP elevation, accumulating evidence suggested that various mechanisms, such as oxidative stress, mitochondrial dysfunction, and inflammation, contribute to glaucomatous neurodegeneration. Furthermore, crosstalk between mitochondrial dysfunction, inflammasome activation, and inflammatory response exacerbates the progression of glaucoma [11,14,15].
Apolipoprotein A-I (APOA-I) binding protein (AIBP, gene name APOA1BP, also known as NAXE) was identified as a binding protein of human APOA-I, which is the major component of high-density lipoprotein (HDL) particles [16]. The APOA1BP gene is located on chromosome 1q21, and its expression is ubiquitous and constitutive in human and mouse tissues, including the kidney, brain, liver, thyroid/adrenal glands, testis, and retina [16]. AIBP is a secreted protein that promotes cholesterol efflux to HDL/APOA-I in endothelial cells, macrophages, and microglia, resulting in increased cholesterol depletion from the plasma membrane of inflamed cells [17,18,19,20,21]. AIBP protein can be found in the plasma of sepsis patients and healthy subject’s urine and cerebrospinal fluid [16]. In the cell culture system, lipopolysaccharides (LPS) and oxidized LDL increase AIBP expression at the translation level in macrophages [19,22].
In systemic naïve AIBP knockout (Apoa1bp-/-) mice, AIBP deficiency triggered glia-driven neuroinflammation and induced mitochondrial dysfunction in both RGCs and Müller glial cells, leading to visual dysfunction [23]. In addition, administration of recombinant AIBP prevented Müller glia-mediated inflammatory response and promoted RGC survival in a mouse model of acute IOP elevation. Intriguingly, AIBP protein expression is reduced in glaucomatous human and mouse retinas [15,23]. Based on these results, our emerging evidence remarkably indicated that restoring AIBP expression in the retina provides neuroprotection in glaucoma.
Since studies in humans and mouse models have shown that AIBP regulates lipid raft-induced inflammation and cholesterol metabolism in multiple cell types [14,22,24,25,26,27], a comprehensive understanding of AIBP-regulated cholesterol metabolism, mitochondrial function and inflammation involved in glaucoma progression is necessary to develop an attractive therapy. This article will review recent findings on the biological role of AIBP in the context of glaucoma.
2. Potential Role of AIBP on Lipid Rafts and Cholesterol Metabolism in Glaucomatous Retina
Given that lowering IOP is insufficient to prevent the disease progression in some patients with glaucoma, one of the significant potential risk factors influencing glaucoma progression is neuroinflammation associated with toll-like receptor 4 (TLR4) activation, cholesterol dysregulation, inflammasome activation, and inflammatory responses [6]. Cholesterol is a necessary component of cell structure. However, excessive cellular accumulation of cholesterol is toxic and exerts detrimental effects on cellular functions. It is well appreciated that cholesterol accumulation by an increased level of low-density lipoprotein (LDL) in immune cells can promote TLR signaling and inflammasome activations, leading to inflammation in the context of metabolic diseases [28]. In neurodegenerative diseases, e.g., Alzheimer’s disease, a high cholesterol level has been reported as a risk factor for disease progression [29]. Thus, cholesterol has emerged as a regulator of diverse cellular metabolisms and signaling pathways.
A recent review extensively discussed lipid raft-associated lipid metabolism in the pathophysiology of retinal diseases [30]. Lipid rafts are dynamic microdomains in the plasma membrane that act as platforms for intracellular signaling pathways involved in cell migration, membrane trafficking, and inflammation [30]. Cholesterol is a major component of lipid rafts, and its depletion disrupts lipid rafts, leading to the inhibition of receptor-mediated cellular signaling. The new concept of an inflammaraft, a pathological lipid raft, was recently introduced to understand the function of enlarged lipid rafts with concentrated inflammatory receptors and adaptor proteins, such as TLR4, in inflamed cells [31]. Based on previous studies, we characterized the inflammarafts with increased lipid raft abundance in inflammatory cells, increased cholesterol levels, and prolonged inflammatory receptors occupancy in lipid rafts [21,31,32,33]. In macrophages and microglia, which highly express TLR4 on the plasma membrane, LPS leads to activation of the inflammatory response, increasing of lipid raft abundance, and ensuing increase of TLR4 recruitment to lipid rafts via modulation of ABCA1-dependent cholesterol efflux pathway [34,35].
Epidemiology studies has revealed that single nucleotide polymorphisms (SNPs) in lipid metabolism-related genes are linked to the progression of primary open-angle glaucoma (POAG) [36,37,38,39]. In particular, SNPs within the gene responsible for encoding ATP-binding cassette transporter A1 (ABCA1), a membrane transporter involved in phospholipids and cholesterol, have been reported to be linked with human glaucoma [36,40,41,42]. ABCA1 deficiency in the retinal astrocytes induced glaucoma-like optic neuropathy in aged mice and resulted in RGC degeneration [43]. Furthermore, ABCA1 amplification reduced retinal degeneration and prevented RGC death [44]. Of interest, our recent and ongoing studies have revealed that deficiencies of AIBP and ABCA1, crucial regulators of cholesterol metabolism, are notably evident in RGCs and Müller glia endfeet in both glaucomatous human and mouse retinas. These results link to the degeneration of RGCs [15,23]. Importantly, these findings strongly suggest that dysregulation of cholesterol homeostasis plays a critical role in RGC degeneration and glial dysfunction in glaucomatous neuroinflammation and neurodegeneration.
Interestingly, findings on the effect of AIBP on regulating cholesterol efflux show that AIBP increases ABCA1 stability and prevents ubiquitin-mediated degradation via facilitating ABCA1-APOA1 binding in macrophages [45]. Moreover, the combination of AIBP/APOA1 and anti-VEGF treatment showed a beneficial effect on overcoming anti-VEGF resistance via facilitating cholesterol efflux from lipid-laden macrophages in a mouse model of laser-induced choroidal neovascularization [46]. Regarding cancer biology, the anti-tumor effects of AIBP are associated with the regulation of cholesterol efflux and reactive oxygen species (ROS) scavenging in intestinal tumors and hepatocellular carcinoma, respectively [47,48]. Our recent studies demonstrated that AIBP enhanced cholesterol efflux from TLR4-associated lipid rafts and subsequently reduced TLR4-mediated inflammatory response in activated macrophages and microglia [18,19,20,21]. Remarkably, a single intrathecal injection of AIBP in a mouse model of neuropathic pain showed a long-lasting therapeutic effect and induced cholesterol metabolism reprograming in spinal microglia [25], implying that cholesterol-driven TLR4-associated lipid raft formation engages in the development of neuropathic pain.
AIBP and ABCA1 deficiency is linked to increased expression of TLR4/IL-1β and MAPKs, which contributes to RGC degeneration in response to elevated IOP in the retina of glaucomatous DBA/2J mice [23]. However, the pathophysiological mechanisms of cholesterol efflux in glaucoma progression and how AIBP regulates retinal cholesterol metabolism and TLR4-associated lipid raft formation remain to be explored. Given that the proposed mechanism for AIBP-mediated RGC protection is associated with cholesterol efflux from inflamed retinal cells, a critical question of great significance is which cholesterol acceptors in the retina serve as a key player in regulating the elimination of excessive cholesterol through the AIBP-ABCA1 pathway. In the brain, which shares many characteristics with the retina and optic nerve, cholesterol fully relies on local biosynthesis, because the blood-brain barrier (BBB) is not permeable to circulating blood lipoproteins [49,50]. APOE-enriched astrocytes are major sources of cholesterol synthesis and provide newly synthesized cholesterol to neurons in the brain [51]. APOE-containing HDL-like lipoproteins produced by astrocytes are involved in cholesterol trafficking to neurons via ABCG1/G4 receptors in the brain [30]. Although APOA1 is the most abundant lipoprotein, mRNA levels of the APOA1 gene are not detected in the brain [51]. Thus, APOA1 in the brain is likely transferred from the periphery [52,53]. Unlike the BBB, the blood-retina barrier is highly permeable to transfer lipoproteins from plasma to retinal pigment epithelium (RPE) cells and plays a critical role in maintaining retinal homeostasis [30,54]. When plasma lipoproteins enter the retina, lipoprotein receptors such as LDL receptor, scavenger receptor class B type I/II, and cluster differentiation 36 in RPE cells are involved in their uptake [55,56,57,58,59]. Like the brain, APOE- or APOA1-containing HDL-like lipoproteins regulate cholesterol trafficking in the retina [58,59]. Moreover, retinal APOA1 not only can be synthesized by RPE, but also is delivered from the periphery [30]. Recent clinical evidence demonstrated higher levels of cholesterol and lower levels of HDL in patients with glaucoma and that statins show a protective effect on glaucoma via other properties such as anti-inflammatory action than lowering LDL levels, suggesting that dysregulation of retinal cholesterol metabolism and inflammation may contribute to glaucoma progression [60]. In addition, ABCA1/G1 deficiency increases TLR4-induced proinflammatory gene expression [61,62,63,64], while cholesterol acceptors such as HDL and APOA1 reduce TLR4-dependent inflammatory responses [65,66,67]. Therefore, the facilitation of cholesterol efflux by AIBP to either HDL or APOA1 may play a role in disrupting TLR4-lipid raft formation. This, in turn, may prevent cellular cholesterol accumulation and inhibit inflammatory response in the retina, ultimately mitigating the progression of glaucomatous neuroinflammation (Figure 1).
3. AIBP in Regulation of Glial-Driven Retinal Neuroinflammation and RGC Degeneration
Neuroinflammation is associated with the activation of glial cells in the central nervous system, resulting in the release of proinflammatory cytokines and chemokines. Initial inflammation, characterized by low levels caused by immune cells in the retina, is essential for maintaining retinal homeostasis. Nevertheless, sustained activation of retinal immune cells contributes to impaired immune function and persistent inflammation, ultimately leading to neuroinflammation in glaucoma. Hence, a comprehensive understanding of the pathophysiological role and therapeutic implication of neuroinflammation is critical to guide the development of potential anti-inflammatory treatments for glaucoma.
AIBP is a secreted protein found in the serum of patients with sepsis and in cerebrospinal fluid and urine from healthy subjects [16]. In addition, cells are stimulated with APOA1/HDL and LPS to induce the secretion of AIBP in vitro [16,19]. Notably, the findings from our work have shown that the secreted AIBP protein selectively binds to TLR4, which is a proinflammatory receptor, in activated cells and augments cholesterol efflux from TLR4-associated lipid rafts, leading to the reduction of TLR4 dimerization, anti-inflammatory response, and cholesterol metabolism reprograming in microglia [20,21]. Moreover, a recent report demonstrated that AIBP attenuates paclitaxel-induced transient receptor potential cation channel subfamily V member 1 (TRPV1) activation by reducing TRPV1-associated lipid raft content and TLR4-TRPV1 interaction in dorsal root ganglion neurons in vivo, resulting in alleviation of lipid raft-associated neuropathic pain [33].
TLR4 is one of the pattern recognition receptors (PRRs) that can be activated by dangerous stimuli, including damaged-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), leading to the secretion of numerous cytokines and chemokines [68,69]. TLR4 can initiate an anti-bacterial response in mammals [70] and is activated in neurodegenerative diseases [71,72,73,74,75]. Studies have shown that activation of TLR4 before exposure to injuries plays a role in neuroprotection [76,77] and increased photoreceptor survival [78]. In contrast, excessive TLR4 activation after injuries induces necroptosis-mediated neuroinflammation and retinal regeneration [79,80,81,82,83]. Proteomic analysis and epidemiological studies of the glaucomatous human retina reveal increased expression levels of TLR2, TLR4, and TLR7, along with their association with SNPs of TLR4 [84,85,86]. Thus, glial activation by TLR4 may contribute to the development of retinal degeneration. Indeed, our recent findings indicate a noteworthy upregulation of TLR4 and IL-1β expression in Müller glia in both glaucomatous human and mouse retinas compared to age/gender-matched control [23]. Activated Müller glia are likely linked to the degeneration of RGCs in patients with glaucoma.
Glial activation in the retina and optic nerve head has been implicated in experimental animal models of glaucoma [87,88,89,90,91,92,93]. In the DBA/2J mice, a extensively studied chronic glaucoma model characterized by the spontaneous development of elevated IOP with age [94,95], convincing evidence indicates a strong correlation between alterations in microglia and retinal degeneration [92,96]. Conversely, microglial inactivation showed a protective effect in experimental glaucoma [97,98,99,100]. In other animal models of glaucoma, including rat and feline, the progression of glaucoma is significantly affected by the induction of retinal inflammation through glial activation, increased production of proinflammatory cytokines, and activation of NF-κB and TLRs [101,102,103,104,105]. Previous studies demonstrated that TLR4-dependent signaling activation induced microglial activation and led to RGC loss in experimental glaucoma [106,107]. Optic nerve crush (ONC), an experimental model of traumatic optic neuropathy, induces axonal degeneration and upregulates the release of proinflammatory cytokines and chemokines, resulting in RGC death [108]. It has been reported that TLR4 activation occurs concurrently with the degeneration of RGCs following ONC. This implies that the inflammation induced by TLR4 activation might contribute to the degeneration of RGC death [109,110,111]. Consistent with these findings, the TLR4 inhibitor TAK242 showed similar effects, diminishing astrocyte activation as evidenced by a reduction of glial fibrillary acidic protein expression, and subsequently suppressing RGC death following ONC [112]. In mouse models of microbead-induced ocular hypertension and retinal ischemia/reperfusion injury, there were significant increases of microglial activation, expression of proinflammatory cytokines and chemokines, and expression of TLR4 and NLR family pyrin domain containing 3 inflammasome [83,113]. Interestingly, a study from Astafurov and coworkers demonstrated that oral bacteria load was higher in glaucomatous patients and that peripheral administration of LPS into two mouse models of glaucoma, DBA/2J and microbead, increased microglial activation and TLR4 signaling, resulting in subsequent enhancement of axonal degeneration and RGC death [114].
Recent findings from our group showed a significant upregulation of TLR4 and IL-1β in the endfeet of Müller glial cells of the ganglion cell layer in glaucomatous DBA/2J and Apoa1bp-/- mice [23]. In addition, MAPK signaling pathways, including ERK1/2 and p38, which lead to inflammation, oxidative stress, and mitochondrial dysfunction [115,116,117,118,119,120], are activated in the retina of Apoa1bp-/- mice and, on the contrary, in vivo recombinant AIBP administration reduces glia-mediated inflammation and RGC death caused by elevated IOP [23]. Considering our findings indicating that AIBP facilitates excess cholesterol removal from the plasma membrane of multiple cell types [25,26,121], a plausible mechanism can be involved in AIBP-mediated augmentation of cholesterol efflux from TLR4-associated lipid rafts in inflamed retinal glial cells, thereby preventing RGC death. However, we have not ruled out the possibility that AIBP may also bind to other receptors in the retinal cells. Collectively, the absence of AIBP could potentially play a role in triggering retinal glial activation and advancing TLR4/IL-1β-induced neuroinflammation, exacerbating glaucoma progression and visual dysfunction.
4. AIBP on Mitochondrial Dynamics and Function in Glaucomatous Neuroinflammation and Neurodegeneration
Mitochondria are dynamic organelles that generate adenosine triphosphate (ATP), which is subsequently used as a major source of energy production in the cell, so they often refer to as the power plants of the cells [122]. While mitochondria are primarily recognized for their role in bioenergy production and metabolite generation, there is increasing attention for their involvement in various other functions such as cell signaling, programmed cell death, ROS generation and elimination, and neuroinflammation [123].
Given that RGCs are highly susceptible to mitochondrial stress caused by glaucomatous insults such as elevated IOP and oxidative stress [5,6,7,8,93,124], mitochondrial dysfunction is considered a critical causing factor in glaucomatous neuroinflammation and neurodegeneration [5,14,15,93]. This could be linked to mitochondrial damage-induced cellular processes, such as aging, excitotoxicity, oxidative stress, inflammasome activation, mitophagy deficiency, and sustained inflammatory response, as downstream pathological pathways contributing to the progression of glaucoma [14,15]. Thus, under glaucomatous conditions, effectively removing damaged mitochondria is an essential step for maintaining mitochondrial quality control and, consequently, protecting RGCs.
In mammalian cells, AIBP is localized in mitochondria [125]. However, its roles in the mitochondria have yet to be extensively studied. Recently, we found for the first time the binding of AIBP to PARKIN (PARK2) and mitofusin (MFN) 1 and 2, which are essential for regulating mitophagy and mitochondrial quality control, resulting in augmentation of mitophagy and protecting macrophages against apoptotic cell death in the context of atherosclerosis [22]. This anti-atherogenic effect of mitochondrial AIBP in regulation of mitophagy was confirmed by others [126]. More importantly, elevated IOP significantly induced a reduction of retinal AIBP expression in mitochondria, suggesting that alterations in retinal AIBP expression are associated with the progression of glaucoma [23]. In parallel, AIBP deficiency decreased mitochondrial dynamics-related proteins, such as optic atrophy type 1 (OPA1) and dynamin-related protein 1 (DRP1), as well as mitochondrial oxidative phosphorylation (OXPHOS) complex proteins in the retina of Apoa1bp-/- mice [23].
Moreover, employing state-of-the-art technology such as serial block-face scanning electron microscopy and three-dimensional electron microscopy tomography, it is well-defined that AIBP deficiency triggers mitochondrial dysfunctions in both Mϋller glia and RGCs in naïve Apoa1bp-/- mice [23]. To provide a detailed insight, mitochondria from Mϋller glia in naïve Apoa1bp-/- mice showed mitochondria fragmentation and ring-shaped mitochondria, which is a hallmark of mitochondrial stress, as well as depletion of mitochondrial cristae, resulting in the reduction of ATP production [23]. In parallel, AIBP deficiency causes mitochondrial abnormalities in RGC somas, such as mitochondrial fragmentation, cristae depletion and ATP reduction. These findings importantly suggested that mitochondrial AIBP is a critical factor in maintaining mitochondrial network and function, and that pathological loss of retinal AIBP expression might exacerbate RGC degeneration and visual dysfunction (Figure 2).
Due to their low cholesterol content, mitochondria highly linked to the progression of various diseases, including Alzheimer’s disease, Niemann-Pick Type C1- deficiency, fatty liver disease. In particular, these conditions are characterized by increasing oxidative stress and compromised OXPHOS system [127,128]. While a novel role of mitochondrial AIBP has been identified in chronic inflammatory diseases [22,23,126], there is currently no evidence of exploring connection between AIBP and cholesterol within mitochondria. Therefore, the precise molecular mechanisms by which AIBP impacts cholesterol metabolism within mitochondria or how AIBP-mediated lipid raft dynamics in the plasma membrane affect mitochondrial quality control under various pathological conditions in glaucomatous neuroinflammation remains to be explored (Figure 2).
Our recent article has reviewed the adverse role of mitochondrial damage-associated molecular patterns (mtDAMPs) in glaucomatous neurodegeneration [89]. Mitochondria play a crucial role in maintaining homeostasis through various pathways, such as quality control protease, unfolded protein response, apoptotic cell death, and mitophagy. Under pathological conditions, cellular defense systems, including mitophagy, are overwhelmed due to the sustained cellular stress and tissue damage. This leads to the release of mtDAMPs into either the intracellular or extracellular compartment [6]. Disrupted mitochondrial membrane potential (MMP) leads to the release of mtDAMPs, such as mtDNA, mtROS, cardiolipin, the mitochondrial transcription factor A, and formyl-peptides [129]. Several studies have demonstrated a link between MMP depolarization and glaucoma. In a rat model of glaucoma induced by a chronic ocular hypertension with the injection of cross-linking hydrogel into the anterior chamber, MMP depolarization was prominent in RGCs [130]. In addition, elevated hydrostatic pressures triggered MMP depolarization in neuronal cell culture system [131].
Release of mtDAMPs activates glial cells through PRRs of the innate immune responses [145], including the NLRP3 inflammasome, TLR9, and cGAS-STING-mediated signaling. Subsequent activation of PRRs increases type 1 interferon (IFN) expression and the levels of proinflammatory cytokines [129]. Specifically, NLRP3 inflammasome activity was upregulated in the retina of glaucomatous DBA/2J mice [132]. TLR9 mRNA expression was significantly increased in trabecular meshwork from patients with POAG and in vitro TM culture system [133]. In addition, cGAS-STING activity was increased in RGCs following ischemia/reperfusion and in poly(dA:dT)-treated BV-2 microglial cells [134]. Given that RGCs are more vulnerable than glial cells to glaucomatous insult-induced mitochondrial stress [135,136] and that a defect in mitophagy in RGCs is linked to AIBP deficiency [22,126], released mtDAMPs from the Apoa1bp-/- RGCs could be the case that AIBP deficiency clearly linked to mitochondrial dysfunction and TLR4-mediated neuroinflammation via glial cell activation in glaucoma. Hence, it would be useful to validate and quantify the levels of mtDAMPs in glaucomatous human and mouse retinas (Figure 2).
5. AIBP-Mediated Neuroprotection in Glaucomatous Neuroinflammation and Neurodegeneration
AIBP protects RGCs against neuroinflammation and mitochondrial dysfunction in elevated IOP-induced retinal neurodegeneration [14]. Specifically, administration of recombinant AIBP enhanced RGC survival against apoptotic cell death and prevented inflammatory responses and cytokine production triggered by the activation of Müller glia, induced by acute IOP elevation in vivo [14]. Because the absence of AIBP remarkably induced the alterations of mitochondrial structure and function in Müller glia and RGCs in vivo [14], these findings importantly indicate that AIBP has a potential to modulate inflammatory mechanisms, protecting against mitochondrial dysfunction that is linked to Müller glia activation and RGC death in retinal neurodegeneration.
Remarkably, our emerging evidence demonstrated that restoring AIBP expression in the retina promotes RGC survival in experimental glaucoma [15]. In the study, we have identified AIBP as the protein regulating several mechanisms of glaucomatous neurodegeneration, including TLR4-mediated inflammatory signaling, excessive cholesterol accumulation, oxidative stress, mitochondrial dysfunction, and metabolic stress. Using a single intravitreal injection of adeno-associated virus (AAV)-AIBP, we found that restoring AIBP expression protected RGCs and their axons in several mouse models of glaucoma, including DBA/2J mice, microbead-induced ocular hypertension, and ONC [15]. In parallel, restoring AIBP expression improved visual function by enhancing RGC function, evident in the increased amplitude of pattern electroretinogram, and enhanced visual acuity through increased spatial frequency against ONC injury. In addition, restoring AIBP expression decreased cholesterol accumulation, TLR4 and IL-1β expression, localization of TLR4 to lipid rafts, and inhibited AMP-activated protein kinase (AMPK) activation in the retina of glaucomatous DBA/2J mice [15]. Hence, our findings suggest for the first time that AIBP attenuates mitochondrial stress and neuroinflammation and protects RGCs by preventing Müller glia activation in glaucoma. Hence, we proposed that the reversing AIBP deficiency by AAV-AIBP delivery has the therapeutic potential to treat glaucoma.
6. Potential Role of AIBP in the NAD(P)HX Repair System of the Retina
To eliminate dangerous toxic metabolites, organisms have developed damage-control systems to remove or recycle damaged metabolites [137]. Nicotinamide adenine dinucleotide (reduced version, NADH; oxidized version, NAD+) and nicotinamide adenine dinucleotide phosphate (reduced version, NADPH; oxidized version, NADP+) are important cofactors for redox equivalents as well as non-redox NAD+-dependent enzymes to maintain metabolic homeostasis [138]. The NAD(P)HX repair system is a highly conserved cellular metabolic reactions from prokaryotes to eukaryotes and two enzymes, the dehydratase and the epimerase, that catalyze the repair of NAD(P)HX are widely expressed in all tissues [139]. Since discovery of AIBP, the APOA1BP gene was recently renamed as NAD(P)HX epimerase (NAXE), due to facilitating the interconversion of R-NAD(P)HX and S-NAD(P)HX [140,141]. NAD(P)H is converted into NAD(P)HX by either glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or spontaneously in an acidic pH or at high temperatures [142,143]. The converted NAD(P)HX exists in the R- and S-forms, which is thought to be toxic. AIBP, as an epimerase converts the R-NAD(P)HX epimers to the S-form. Sequentially, the NAD(P)HX dehydratase converts the S-form back to NAD(P)H, resulting in the inhibition of cyclic NAD(P)HX production, which is an aberrant metabolite that could induce mitochondrial dysfunction. Given that the pathological implications of the NAD(P)HX repair system have recently received attention in human studies, until now, it has been reported a total of 30 clinical cases with 19 rare variants in AIBP that cause lethal neurometabolic disorders, including ataxia, ophthalmoplegia, muscle weakness, and the regression of motor and cognitive functions [139]. Nevertheless, the deficiency of AIBP does not impact growth and viability in mice [18,144], indicating that mouse AIBP may exibit greater tolerance to toxic metabolites or have functions distinct from those in humans [121].
Recent evidence has uncovered the neuroprotective function of nicotinamide (NAM) in both a mouse model of glaucoma and patients with glaucoma [145,146]. However, there is no reported evidence of the NAD(P)HX repair system to the onset of glaucoma. NAM is a precursor form of NAD that is generated from vitamin B-3, or the precursor NAM, nicotinamide riboside, nicotinic acid, or nicotinamide mononucleotide (NMN) defined as the salvage pathway [147]. Therefore, metabolic boosting of NAD+ levels together with the well-conserved or improved NAD(P)HX repair system may have beneficial effect on glaucoma treatment. In both mouse models of glaucoma and the patients with glaucoma, NAD or NAM levels are significantly lower than in control subjects [145,148].
AIBP expression was predominantly reduced in RGCs in both a mouse model of glaucoma, DBA/2J mice, and patients with glaucoma [23](ref). Moreover, AIBP deficiency significantly reduced visual acuity measured by decreasing spatial frequency in male and female naïve Apoa1bp-/- mice [23]. Recent findings demonstrated that the plasma levels of NAD in 3-month-old Apoa1bp-/- mice tended to decrease compared to control mice, although it is not significant [149]. Since the retinal NAD+ or NAM levels have not been tested in Apoa1bp-/- mice and AIBP deficiency did not show morphological distortion [18,144], whether AIBP deficiency confers metabolic alterations in the retina is unclear. Nevertheless, a potential possibility is that, in part, accumulation of toxic metabolites, e.g., cyclic NAD(P)HX, in the retina of Apoa1bp-/- mice may have particularly deleterious effects on metabolic and mitochondrial homeostasis, leading to RGC degeneration and subsequent vision loss (Figure 3).
7. Conclusions and Future Directions
We have reviewed multiple functions of AIBP in glaucoma pathogenesis, from inhibition of TLR4-mediated inflammation via disruption of TLRs by enhancing cholesterol efflux to the regulation of mitochondrial function to NAXE activity. Many clinical findings have shown that NAXE (APOA1BP) gene mutations are associated with neurological disorders [150,151,152,153,154,155]. This suggests that dysregulation of the NAXE-related repair system leads to mitochondrial dysfunction and accumulation of toxic metabolites [150,156]. Nevertheless, no case report presents APOA1BP gene mutation connected with the development of glaucoma in human patients. Because we have revealed a reduction of retinal AIBP expression in both glaucomatous human and mouse retinas, it is crucial to investigate the relevance of AIBP in human glaucoma, particularly considering the absence of genetic variation in the APOA1BP gene.
We have emphasized two main concepts of AIBP action in this article beyond the NAXE-related repair systems. The first concept is that extracellular AIBP selectively reduces pathological lipid raft content from the cholesterol-enriched plasma membrane in TLR4-expressing retinal glial cells, inhibiting of TLR4-mediated inflammation and mitochondrial dysfunction in glaucoma. Here, we focused on the effect of AIBP on TLR4-associated lipid raft formation in glaucoma. However, we do not exclude a potential role of AIBP in regulation of other TLRs-related lipid raft formation, including TLR2 and TLR3, because their expression is increased in human glaucomatous retina [157]. The second concept is the role of AIBP in mitochondria. As a result of reduction of mitochondrial AIBP expression, RGC survival was decreased, and visual acuity was reduced in response to elevated IOP [23]. While most claimed research findings are to explain the role of AIBP in the regulation of cholesterol metabolism, future studies need to be tested the requirement of cholesterol for the function of mitochondrial AIBP.
In the therapeutic point of view, glaucoma is closely associated with neuroinflammation that could be initiated by inflammatory receptor-lipid raft complex, thereby triggering oxidative stress and mitochondrial dysfunction. Indeed, reduced AIBP expression in the retina is linked to glaucomatous neuroinflammation and mitochondrial dysfunction [23]. Therefore, the sustained expression of AIBP which promotes selective removal of excess cholesterol from TLR4-occupied lipid rafts in inflammatory retinal cells could be a possible approach to reduce neuroinflammation and mitochondrial dysfunction in human glaucoma. Clinical development of gene therapy using AAV in ocular disease has drawn attention because of its long-term stable transgene expression at levels that are therapeutic and sustained effects with even a single injection. Hence, restoring AIBP expression delivery by a single intravitreal injection of AAV-AIBP would be a potential treatment for glaucoma.
Here, we highlighted that AIBP is a novel safeguard to protect mechanisms regulating neuroinflammation and mitochondrial dysfunction by controlling cholesterol metabolism in the retina. Notably, the remarkable selectivity of AIBP toward TLR4-associated lipid rafts and regulation of mitochondrial dynamics and function may contribute to developing a new therapeutic strategy for treating glaucoma and other chronic inflammatory diseases.
Author Contributions
S.C., S.-H.C., T.B., Y.P., J.O., K.-Y.K., S.H., Y.I.M., and W.-K.J. were responsible for drafting the manuscript, and S.C., S.-H.C., Y.I.M., and W.-K.J. was responsible for finalizing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health grants EY031697 (W.-K.J.), AG081004 (W.-K.J.), EY034116 (W.-K.J./K.-Y.K/S.-H.C), NS129684 (W.K.J.), HL136275 (Y.I.M.), and AG081037 (Y.I.M./W.-K.J.).
Competing interests
S.-H. Choi, W.-K. Ju and Y.I. Miller are co-inventors named on patents and patent applications by the University of California, San Diego. Y.I. Miller is a scientific co-founder of Raft Pharmaceuticals LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Other authors declare no competing interests exist.
References
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: a review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183–2193. [Google Scholar] [CrossRef]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10. [Google Scholar] [CrossRef]
- Ju, W.K.; Perkins, G.A.; Kim, K.Y.; Bastola, T.; Choi, W.Y.; Choi, S.H. Glaucomatous optic neuropathy: Mitochondrial dynamics, dysfunction and protection in retinal ganglion cells. Prog Retin Eye Res 2022, 101136. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.J.I.o. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. 2008, 49, 4903–4911. [Google Scholar] [CrossRef]
- Kim, K.; Perkins, G.; Shim, M.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.; Weinreb, R.; Ju, W.J.C.d.; disease. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. 2015, 6, e1839–e1839. [Google Scholar] [CrossRef]
- Nemesure, B.; Honkanen, R.; Hennis, A.; Wu, S.Y.; Leske, M.C.; Barbados Eye Studies, G. Incident open-angle glaucoma and intraocular pressure. Ophthalmology 2007, 114, 1810–1815. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Leung, C.K.; Crowston, J.G.; Medeiros, F.A.; Friedman, D.S.; Wiggs, J.L.; Martin, K.R. Primary open-angle glaucoma. Nat Rev Dis Primers 2016, 2, 16067. [Google Scholar] [CrossRef]
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov 2018, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Wang, D.; Grosskreutz, C.L. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp Eye Res 2010, 91, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Nickells, R.W. From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Can J Ophthalmol 2007, 42, 278–287. [Google Scholar] [CrossRef]
- Choi, S.-H.; Kim, K.-Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.J.R.b. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. 2020, 37, 101703. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Ha, Y.; Choi, S.; Kim, K.Y.; Bastola, T.; Kim, J.; Weinreb, R.N.; Zhang, W.; Miller, Y.I.; Choi, S.H. Restoring AIBP deficiency in the retina provides neuroprotection in glaucoma. bioRxiv 2023. [Google Scholar] [CrossRef]
- Ritter, M.; Buechler, C.; Boettcher, A.; Barlage, S.; Schmitz-Madry, A.; Orso, E.; Bared, S.M.; Schmiedeknecht, G.; Baehr, C.H.; Fricker, G.; et al. Cloning and characterization of a novel apolipoprotein A-I binding protein, AI-BP, secreted by cells of the kidney proximal tubules in response to HDL or ApoA-I. Genomics 2002, 79, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Choi, S.H.; Baek, J.S.; Liu, C.; Almazan, F.; Ulrich, F.; Wiesner, P.; Taleb, A.; Deer, E.; Pattison, J.; et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013, 498, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.A.; Choi, S.H.; Agatisa-Boyle, C.; Zhu, L.; Kim, J.; Pattison, J.; Sears, D.D.; Gordts, P.; Fang, L.; Miller, Y.I. AIBP protects against metabolic abnormalities and atherosclerosis. J Lipid Res 2018, 59, 854–863. [Google Scholar] [CrossRef]
- Choi, S.H.; Wallace, A.M.; Schneider, D.A.; Burg, E.; Kim, J.; Alekseeva, E.; Ubags, N.D.; Cool, C.D.; Fang, L.; Suratt, B.T.; et al. AIBP augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Woller, S.A.; Choi, S.H.; An, E.J.; Low, H.; Schneider, D.A.; Ramachandran, R.; Kim, J.; Bae, Y.S.; Sviridov, D.; Corr, M.; et al. Inhibition of Neuroinflammation by AIBP: Spinal Effects upon Facilitated Pain States. Cell Rep 2018, 23, 2667–2677. [Google Scholar] [CrossRef]
- Navia-Pelaez, J.M.; Choi, S.H.; Dos Santos Aggum Capettini, L.; Xia, Y.; Gonen, A.; Agatisa-Boyle, C.; Delay, L.; Goncalves Dos Santos, G.; Catroli, G.F.; Kim, J.; et al. Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. J Exp Med 2021, 218. [Google Scholar] [CrossRef]
- Choi, S.H.; Agatisa-Boyle, C.; Gonen, A.; Kim, A.; Kim, J.; Alekseeva, E.; Tsimikas, S.; Miller, Y.I. Intracellular AIBP (Apolipoprotein A-I Binding Protein) Regulates Oxidized LDL (Low-Density Lipoprotein)-Induced Mitophagy in Macrophages. Arterioscler Thromb Vasc Biol 2021, 41, e82–e96. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, K.Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.; et al. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol 2020, 37, 101703. [Google Scholar] [CrossRef] [PubMed]
- Navia-Pelaez, J.M.; Borges Paes Lemes, J.; Gonzalez, L.; Delay, L.; Dos Santos Aggum Capettini, L.; Lu, J.W.; Goncalves Dos Santos, G.; Gregus, A.M.; Dougherty, P.M.; Yaksh, T.L.; et al. AIBP regulates TRPV1 activation in chemotherapy-induced peripheral neuropathy by controlling lipid raft dynamics and proximity to TLR4 in dorsal root ganglion neurons. Pain 2023, 164, e274–e285. [Google Scholar] [CrossRef]
- Navia-Pelaez, J.M.; Choi, S.-H.; dos Santos Aggum Capettini, L.; Xia, Y.; Gonen, A.; Agatisa-Boyle, C.; Delay, L.; Gonçalves dos Santos, G.; Catroli, G.F.; Kim, J.; et al. Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. Journal of Experimental Medicine 2021, 218. [Google Scholar] [CrossRef]
- Woller, S.A.; Choi, S.-H.; An, E.J.; Low, H.; Schneider, D.A.; Ramachandran, R.; Kim, J.; Bae, Y.S.; Sviridov, D.; Corr, M.J.C.r. Inhibition of neuroinflammation by AIBP: spinal effects upon facilitated pain states. 2018, 23, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, G.-J.; Yin, K.; Xia, X.-D.; Gong, D.; Zhao, Z.-W.; Chen, L.-Y.; Zheng, X.-L.; Tang, X.-E.; Tang, C.-K.J.C.J. Apolipoprotein A-1 binding protein inhibits inflammatory signaling pathways by binding to apolipoprotein A-1 in THP-1 macrophages. 2018, 82, 1396–1404. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer's Disease; From Risk Genes to Pathological Effects. Front Aging Neurosci 2021, 13, 690372. [Google Scholar] [CrossRef]
- Gabrielle, P.H. Lipid metabolism and retinal diseases. Acta Ophthalmol 2022, 100 Suppl 269, 3–43. [Google Scholar] [CrossRef]
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid rafts in glial cells: role in neuroinflammation and pain processing. J Lipid Res 2020, 61, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Navia-Pelaez, J.M.; Agatisa-Boyle, C.; Choi, S.H.; Sak Kim, Y.; Li, S.; Alekseeva, E.; Weldy, K.; Miller, Y.I. Differential Expression of Inflammarafts in Macrophage Foam Cells and in Nonfoamy Macrophages in Atherosclerotic Lesions-Brief Report. Arterioscler Thromb Vasc Biol 2023, 43, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Navia-Pelaez, J.M.; Paes Lemes, J.B.; Gonzalez, L.; Delay, L.; Capettini, L.; Lu, J.W.; Dos Santos, G.G.; Gregus, A.M.; Dougherty, P.M.; Yaksh, T.L.; et al. AIBP regulates TRPV1 activation in CIPN by controlling lipid raft dynamics and proximity to TLR4 in DRG neurons. Pain 2022. [Google Scholar] [CrossRef]
- Zhu, X.; Owen, J.S.; Wilson, M.D.; Li, H.; Griffiths, G.L.; Thomas, M.J.; Hiltbold, E.M.; Fessler, M.B.; Parks, J.S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 2010, 51, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; He, Z.; Wang, J.; Xin, Z.; Wang, J.; Li, F.; Fu, Y. Taraxasterol Inhibits LPS-Induced Inflammatory Response in BV2 Microglia Cells by Activating LXRalpha. Front Pharmacol 2018, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, Y.; Vithana, E.N.; Jia, L.; Zuo, X.; Wong, T.Y.; Chen, L.J.; Zhu, X.; Tam, P.O.; Gong, B.; et al. Common variants near ABCA1 and in PMM2 are associated with primary open-angle glaucoma. Nat Genet 2014, 46, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.N.; Loomis, S.J.; Kang, J.H.; Allingham, R.R.; Gharahkhani, P.; Khor, C.C.; Burdon, K.P.; Aschard, H.; Chasman, D.I.; Igo, R.P., Jr.; et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat Genet 2016, 48, 189–194. [Google Scholar] [CrossRef] [PubMed]
- van Koolwijk, L.M.; Ramdas, W.D.; Ikram, M.K.; Jansonius, N.M.; Pasutto, F.; Hysi, P.G.; Macgregor, S.; Janssen, S.F.; Hewitt, A.W.; Viswanathan, A.C.; et al. Common genetic determinants of intraocular pressure and primary open-angle glaucoma. PLoS Genet 2012, 8, e1002611. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum Mol Genet 2017, 26, R21–R27. [Google Scholar] [CrossRef]
- Gharahkhani, P.; Burdon, K.P.; Fogarty, R.; Sharma, S.; Hewitt, A.W.; Martin, S.; Law, M.H.; Cremin, K.; Bailey, J.N.C.; Loomis, S.J.; et al. Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat Genet 2014, 46, 1120–1125. [Google Scholar] [CrossRef]
- Hysi, P.G.; Cheng, C.Y.; Springelkamp, H.; Macgregor, S.; Bailey, J.N.C.; Wojciechowski, R.; Vitart, V.; Nag, A.; Hewitt, A.W.; Hohn, R.; et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat Genet 2014, 46, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Shiga, Y.; Akiyama, M.; Nishiguchi, K.M.; Sato, K.; Shimozawa, N.; Takahashi, A.; Momozawa, Y.; Hirata, M.; Matsuda, K.; Yamaji, T.; et al. Genome-wide association study identifies seven novel susceptibility loci for primary open-angle glaucoma. Hum Mol Genet 2018, 27, 1486–1496. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Leung, A.; Namekata, K.; Saitoh, S.; Nguyen, H.B.; Takeda, A.; Danjo, Y.; Morizawa, Y.M.; Shigetomi, E.; Sano, F.; et al. Astrocytic dysfunction induced by ABCA1 deficiency causes optic neuropathy. Sci Adv 2022, 8, eabq1081. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, S.; Zhou, Z.; Zhao, Y. Ad- and AAV8-mediated ABCA1 gene therapy in a murine model with retinal ischemia/reperfusion injuries. Mol Ther Methods Clin Dev 2021, 20, 551–558. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, G.J.; Yao, F.; Xia, X.D.; Gong, D.; Zhao, Z.W.; Chen, L.Y.; Zheng, X.L.; Tang, X.E.; Tang, C.K. AIBP reduces atherosclerosis by promoting reverse cholesterol transport and ameliorating inflammation in apoE(-/-) mice. Atherosclerosis 2018, 273, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Parker, M.; Enemchukwu, N.; Shen, M.; Zhang, G.; Yan, Q.; Handa, J.T.; Fang, L.; Fu, Y. Combination of apolipoprotein-A-I/apolipoprotein-A-I binding protein and anti-VEGF treatment overcomes anti-VEGF resistance in choroidal neovascularization in mice. Commun Biol 2020, 3, 386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, Q.; Wang, Y.; Wang, J.; Su, Y.; Wang, F.; Wang, G. AIBP and APOA-I synergistically inhibit intestinal tumor growth and metastasis by promoting cholesterol efflux. J Transl Med 2019, 17, 161. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yu, L.; Xu, C.; Li, Y.M.; Zhao, Y.R.; Cao, M.M.; Yang, L.Y. NAD(P)HX epimerase downregulation promotes tumor progression through ROS/HIF-1alpha signaling in hepatocellular carcinoma. Cancer Sci 2021, 112, 2753–2769. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr Opin Lipidol 2001, 12, 105–112. [Google Scholar] [CrossRef]
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem Rev 2011, 111, 6423–6451. [Google Scholar] [CrossRef]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol 2010, 51, 555–573. [Google Scholar] [CrossRef]
- Stukas, S.; Robert, J.; Lee, M.; Kulic, I.; Carr, M.; Tourigny, K.; Fan, J.; Namjoshi, D.; Lemke, K.; DeValle, N.; et al. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc 2014, 3, e001156. [Google Scholar] [CrossRef]
- Wellington, C.L.; Frikke-Schmidt, R. Relation between plasma and brain lipids. Curr Opin Lipidol 2016, 27, 225–232. [Google Scholar] [CrossRef]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog Retin Eye Res 2019, 69, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.G.; Bailey, K.R.; Kane, J.P.; Schwartz, D.M. Human retinal pigment epithelial cells express scavenger receptors BI and BII. Biochem Biophys Res Commun 2002, 292, 1017–1022. [Google Scholar] [CrossRef]
- Duncan, K.G.; Hosseini, K.; Bailey, K.R.; Yang, H.; Lowe, R.J.; Matthes, M.T.; Kane, J.P.; LaVail, M.M.; Schwartz, D.M.; Duncan, J.L. Expression of reverse cholesterol transport proteins ATP-binding cassette A1 (ABCA1) and scavenger receptor BI (SR-BI) in the retina and retinal pigment epithelium. Br J Ophthalmol 2009, 93, 1116–1120. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Bretillon, L. The ins and outs of cholesterol in the vertebrate retina. J Lipid Res 2010, 51, 3399–3413. [Google Scholar] [CrossRef] [PubMed]
- Tserentsoodol, N.; Gordiyenko, N.V.; Pascual, I.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol Vis 2006, 12, 1319–1333. [Google Scholar] [PubMed]
- Tserentsoodol, N.; Sztein, J.; Campos, M.; Gordiyenko, N.V.; Fariss, R.N.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol Vis 2006, 12, 1306–1318. [Google Scholar]
- Posch-Pertl, L.; Michelitsch, M.; Wagner, G.; Wildner, B.; Silbernagel, G.; Pregartner, G.; Wedrich, A. Cholesterol and glaucoma: a systematic review and meta-analysis. Acta Ophthalmol 2022, 100, 148–158. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ Res 2009, 104, 455–465. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol 2010, 30, 139–143. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, J.Y.; Timmins, J.M.; Brown, J.M.; Boudyguina, E.; Mulya, A.; Gebre, A.K.; Willingham, M.C.; Hiltbold, E.M.; Mishra, N.; et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem 2008, 283, 22930–22941. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Woollard, K.J.; Hoang, A.; Mukhamedova, N.; Stirzaker, R.A.; McCormick, S.P.; Remaley, A.T.; Sviridov, D.; Chin-Dusting, J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol 2008, 28, 2071–2077. [Google Scholar] [CrossRef]
- Chen, R.X.; Jiang, W.J.; Liu, S.C.; Wang, Z.Y.; Wang, Z.B.; Zhou, T.; Chen, Y.A.; Wang, J.F.; Chang, J.; Wang, Y.R.; et al. Apolipoprotein A-1 protected hepatic ischaemia-reperfusion injury through suppressing macrophage pyroptosis via TLR4-NF-kappaB pathway. Liver Int 2023, 43, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Shaul, P.W. Regulation of signal transduction by HDL. J Lipid Res 2013, 54, 2315–2324. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Oda, K.; Kitano, H. A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol 2006, 2, 2006–0015. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Miron, J.; Picard, C.; Frappier, J.; Dea, D.; Theroux, L.; Poirier, J. TLR4 Gene Expression and Pro-Inflammatory Cytokines in Alzheimer's Disease and in Response to Hippocampal Deafferentation in Rodents. J Alzheimers Dis 2018, 63, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Gorecki, A.M.; Anyaegbu, C.C.; Anderton, R.S. TLR2 and TLR4 in Parkinson's disease pathogenesis: the environment takes a toll on the gut. Transl Neurodegener 2021, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh Manjili, F.; Yousefi-Ahmadipour, A.; Kazemi Arababadi, M. The roles played by TLR4 in the pathogenesis of multiple sclerosis; A systematic review article. Immunol Lett 2020, 220, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of Toll-like receptor-4 in the brain. J Neuroimmunol 2014, 268, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, H.L.; Lessov, N.S.; Henshall, D.C.; Minami, M.; Simon, R.P.; Stenzel-Poore, M.P. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004, 35, 2576–2581. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.J.; Fernandez, D.C.; Sande, P.H.; Keller Sarmiento, M.I.; Chianelli, M.; Saenz, D.A.; Rosenstein, R.E. Effect of bacterial lipopolysaccharide on ischemic damage in the rat retina. Invest Ophthalmol Vis Sci 2008, 49, 4604–4612. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Patel, A.K.; Sodhi, C.P.; Hackam, D.J.; Hackam, A.S. Novel role for the innate immune receptor Toll-like receptor 4 (TLR4) in the regulation of the Wnt signaling pathway and photoreceptor apoptosis. PLoS One 2012, 7, e36560. [Google Scholar] [CrossRef] [PubMed]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem 2013, 288, 15326–15341. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ 2018, 25, 180–189. [Google Scholar] [CrossRef]
- Bohm, M.R.; Schallenberg, M.; Brockhaus, K.; Melkonyan, H.; Thanos, S. The pro-inflammatory role of high-mobility group box 1 protein (HMGB-1) in photoreceptors and retinal explants exposed to elevated pressure. Lab Invest 2016, 96, 409–427. [Google Scholar] [CrossRef]
- Han, X.; Chen, X.; Chen, S.; Luo, Q.; Liu, X.; He, A.; He, S.; Qiu, J.; Chen, P.; Wu, Y.; et al. Tetramethylpyrazine attenuates endotoxin-induced retinal inflammation by inhibiting microglial activation via the TLR4/NF-kappaB signalling pathway. Biomed Pharmacother 2020, 128, 110273. [Google Scholar] [CrossRef]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc Natl Acad Sci U S A 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Invest Ophthalmol Vis Sci 2010, 51, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Partida, J.; Martinez-Rizo, A.B.; Ramirez-Barrera, P.; Velazquez-Fernandez, J.B.; Mondragon-Jaimes, V.A.; Santos-Garcia, A.; Benites-Godinez, V. Association of Toll-like receptor 4 single-nucleotide polymorphisms Asp299Gly and Thr399Ile with the risk of primary open angle glaucoma. Graefes Arch Clin Exp Ophthalmol 2017, 255, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, E.; Meguro, A.; Ota, M.; Kashiwagi, K.; Mabuchi, F.; Iijima, H.; Kawase, K.; Yamamoto, T.; Nakamura, M.; Negi, A.; et al. Association of Toll-like receptor 4 gene polymorphisms with normal tension glaucoma. Invest Ophthalmol Vis Sci 2008, 49, 4453–4457. [Google Scholar] [CrossRef]
- Kim, K.Y.; Perkins, G.A.; Shim, M.S.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis 2015, 6, e1839. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Kim, K.Y.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci 2008, 49, 1437–1446. [Google Scholar] [CrossRef]
- Rojas, B.; Gallego, B.I.; Ramirez, A.I.; Salazar, J.J.; de Hoz, R.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Trivino, A.; et al. Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J Neuroinflammation 2014, 11, 133. [Google Scholar] [CrossRef]
- Bosco, A.; Crish, S.D.; Steele, M.R.; Romero, C.O.; Inman, D.M.; Horner, P.J.; Calkins, D.J.; Vetter, M.L. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. 2012.
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol 2011, 519, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog Retin Eye Res 2022, 87, 100998. [Google Scholar] [CrossRef]
- John, S.W.; Smith, R.S.; Savinova, O.V.; Hawes, N.L.; Chang, B.; Turnbull, D.; Davisson, M.; Roderick, T.H.; Heckenlively, J.R. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci 1998, 39, 951–962. [Google Scholar]
- Anderson, M.G.; Smith, R.S.; Hawes, N.L.; Zabaleta, A.; Chang, B.; Wiggs, J.L.; John, S.W. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat Genet 2002, 30, 81–85. [Google Scholar] [CrossRef]
- Bosco, A.; Romero, C.O.; Breen, K.T.; Chagovetz, A.A.; Steele, M.R.; Ambati, B.K.; Vetter, M.L. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis Model Mech 2015, 8, 443–455. [Google Scholar] [CrossRef]
- Wang, K.; Peng, B.; Lin, B. Fractalkine receptor regulates microglial neurotoxicity in an experimental mouse glaucoma model. Glia 2014, 62, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Crish, S.D.; Steele, M.R.; Romero, C.O.; Inman, D.M.; Horner, P.J.; Calkins, D.J.; Vetter, M.L. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. PLoS One 2012, 7, e43602. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One 2012, 7, e40065. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci 2006, 26, 12633–12641. [Google Scholar] [CrossRef]
- Sapienza, A.; Raveu, A.L.; Reboussin, E.; Roubeix, C.; Boucher, C.; Degardin, J.; Godefroy, D.; Rostene, W.; Reaux-Le Goazigo, A.; Baudouin, C.; et al. Bilateral neuroinflammatory processes in visual pathways induced by unilateral ocular hypertension in the rat. J Neuroinflammation 2016, 13, 44. [Google Scholar] [CrossRef]
- Naskar, R.; Wissing, M.; Thanos, S. Detection of early neuron degeneration and accompanying microglial responses in the retina of a rat model of glaucoma. Invest Ophthalmol Vis Sci 2002, 43, 2962–2968. [Google Scholar] [PubMed]
- Johnson, E.C.; Jia, L.; Cepurna, W.O.; Doser, T.A.; Morrison, J.C. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci 2007, 48, 3161–3177. [Google Scholar] [CrossRef]
- Lozano, D.C.; Choe, T.E.; Cepurna, W.O.; Morrison, J.C.; Johnson, E.C. Early Optic Nerve Head Glial Proliferation and Jak-Stat Pathway Activation in Chronic Experimental Glaucoma. Invest Ophthalmol Vis Sci 2019, 60, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, K.; Ver Hoeve, J.N.; Teixeira, L.B.C.; Snyder, K.C.; Kiland, J.A.; Ellinwood, N.M.; McLellan, G.J. Sub-region-Specific Optic Nerve Head Glial Activation in Glaucoma. Mol Neurobiol 2020, 57, 2620–2638. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: a general review. Int J Neurosci 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Niwa, M.; Aoki, H.; Hirata, A.; Tomita, H.; Green, P.G.; Hara, A. Retinal Cell Degeneration in Animal Models. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef]
- Lin, S.; Liang, Y.; Zhang, J.; Bian, C.; Zhou, H.; Guo, Q.; Xiong, Y.; Li, S.; Su, B. Microglial TIR-domain-containing adapter-inducing interferon-beta (TRIF) deficiency promotes retinal ganglion cell survival and axon regeneration via nuclear factor-kappaB. J Neuroinflammation 2012, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Ueda, H. Prothymosin-alpha preconditioning activates TLR4-TRIF signaling to induce protection of ischemic retina. J Neurochem 2015, 135, 1161–1177. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, B.; Hu, Y.; Lu, L.; Lu, X.; Wang, J.; Xu, F.; Yu, S.; Huang, J.; Liang, X. Wogonin prevents TLR4-NF-kappaB-medicated neuro-inflammation and improves retinal ganglion cells survival in retina after optic nerve crush. Oncotarget 2016, 7, 72503–72517. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimazawa, M.; Ojino, K.; Izawa, H.; Takeuchi, H.; Inoue, Y.; Tsuruma, K.; Hara, H. Toll-like receptor 4 inhibitor protects against retinal ganglion cell damage induced by optic nerve crush in mice. J Pharmacol Sci 2017, 133, 176–183. [Google Scholar] [CrossRef]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J Neuroinflammation 2019, 16, 184. [Google Scholar] [CrossRef] [PubMed]
- Astafurov, K.; Elhawy, E.; Ren, L.; Dong, C.Q.; Igboin, C.; Hyman, L.; Griffen, A.; Mittag, T.; Danias, J. Oral microbiome link to neurodegeneration in glaucoma. PLoS One 2014, 9, e104416. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson's Disease. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res 2008, 86, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Du, F.; Douglas, J.T.; Yu, H.; Yan, S.S.; Yan, S.F. Mitochondrial Dysfunction Triggers Synaptic Deficits via Activation of p38 MAP Kinase Signaling in Differentiated Alzheimer's Disease Trans-Mitochondrial Cybrid Cells. J Alzheimers Dis 2017, 59, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson's disease. Cell Death Dis 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Chen, J.; Otsuka, M.; Mols, J.; Ren, S.; Wang, Y.; Han, J. Macrophage deletion of p38alpha partially impairs lipopolysaccharide-induced cellular activation. J Immunol 2008, 180, 5075–5082. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kang, Y.J.; Ren, J.; Jiang, H.; Wang, Y.; Omata, M.; Han, J. Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology 2010, 138, 1255–1265. [Google Scholar] [CrossRef]
- Fang, L.; Miller, Y.I. Regulation of lipid rafts, angiogenesis and inflammation by AIBP. Curr Opin Lipidol 2019, 30, 218–223. [Google Scholar] [CrossRef]
- Lane, N. Mitochondrial disease: powerhouse of disease. Nature 2006, 440, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Skeie, J.M.; Nishimura, D.Y.; Wang, C.L.; Schmidt, G.A.; Aldrich, B.T.; Greiner, M.A. Mitophagy: An Emerging Target in Ocular Pathology. Invest Ophthalmol Vis Sci 2021, 62, 22. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G.J.C.o.i.p. Oxidative stress and mitochondrial dysfunction in glaucoma. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Marbaix, A.Y.; Tyteca, D.; Niehaus, T.D.; Hanson, A.D.; Linster, C.L.; Van Schaftingen, E. Occurrence and subcellular distribution of the NADPHX repair system in mammals. Biochem J 2014, 460, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Chen, H.; Yin, L.; Zhu, X.; Novak, P.; Lv, Y.; Zhao, G.; Yin, K. Mitochondrial apolipoprotein A-I binding protein alleviates atherosclerosis by regulating mitophagy and macrophage polarization. Cell Commun Signal 2022, 20, 60. [Google Scholar] [CrossRef] [PubMed]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nunez, S.; Nuno-Lambarri, N.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol 2019, 24, 101214. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Garcia-Ruiz, C.M.; Fernandez-Checa, J.C. Mitochondrial Cholesterol in Alzheimer's Disease and Niemann-Pick Type C Disease. Front Neurol 2019, 10, 1168. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Huang, S.; Huang, P.; Yu, H.; Chen, J.; Liu, X.; Wang, J.; Shen, X.; Zhong, Y. Hydrogen sulfide supplement preserves mitochondrial function of retinal ganglion cell in a rat glaucoma model. Cell Tissue Res 2022, 389, 171–185. [Google Scholar] [CrossRef]
- Tok, L.; Naziroglu, M.; Uguz, A.C.; Tok, O. Elevated hydrostatic pressures induce apoptosis and oxidative stress through mitochondrial membrane depolarization in PC12 neuronal cells: A cell culture model of glaucoma. J Recept Signal Transduct Res 2014, 34, 410–416. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Inman, D.M. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. J Neuroinflammation 2018, 15, 313. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, N.; Xiong, S.; Xia, X. The correlation between primary open-angle glaucoma (POAG) and gut microbiota: a pilot study towards predictive, preventive, and personalized medicine. EPMA J 2023, 14, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yu, N.; Ye, Z.; Gu, Y.; Zhang, C.; Chen, M.; Wang, K. Inhibition of cGAS-STING pathway alleviates neuroinflammation-induced retinal ganglion cell death after ischemia/reperfusion injury. Cell Death Dis 2023, 14, 615. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog Retin Eye Res 2021, 83, 100916. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; Van Schaftingen, E.; Hanson, A.D. Metabolite damage and its repair or pre-emption. Nat Chem Biol 2013, 9, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Manor, J.; Calame, D.; Gijavanekar, C.; Fisher, K.; Hunter, J.; Mizerik, E.; Bacino, C.; Scaglia, F.; Elsea, S.H. NAXE deficiency: A neurometabolic disorder of NAD(P)HX repair amenable for metabolic correction. Mol Genet Metab 2022, 136, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Marbaix, A.Y.; Noel, G.; Detroux, A.M.; Vertommen, D.; Van Schaftingen, E.; Linster, C.L. Extremely conserved ATP- or ADP-dependent enzymatic system for nicotinamide nucleotide repair. J Biol Chem 2011, 286, 41246–41252. [Google Scholar] [CrossRef]
- Shumilin, I.A.; Cymborowski, M.; Chertihin, O.; Jha, K.N.; Herr, J.C.; Lesley, S.A.; Joachimiak, A.; Minor, W. Identification of unknown protein function using metabolite cocktail screening. Structure 2012, 20, 1715–1725. [Google Scholar] [CrossRef]
- Rafter, G.W.; Chaykin, S.; Krebs, E.G. The action of glyceraldehyde-3-phosphate dehydrogenase on reduced diphosphopyridine nucleotide. J Biol Chem 1954, 208, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Dave, V. Inhibition of NADP-dependent dehydrogenases by modified products of NADPH. Arch Biochem Biophys 1975, 169, 298–303. [Google Scholar] [CrossRef]
- Mao, R.; Meng, S.; Gu, Q.; Araujo-Gutierrez, R.; Kumar, S.; Yan, Q.; Almazan, F.; Youker, K.A.; Fu, Y.; Pownall, H.J.; et al. AIBP Limits Angiogenesis Through gamma-Secretase-Mediated Upregulation of Notch Signaling. Circ Res 2017, 120, 1727–1739. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B(3) modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin Exp Ophthalmol 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab 2018, 27, 1067–1080. [Google Scholar] [CrossRef]
- Kouassi Nzoughet, J.; Chao de la Barca, J.M.; Guehlouz, K.; Leruez, S.; Coulbault, L.; Allouche, S.; Bocca, C.; Muller, J.; Amati-Bonneau, P.; Gohier, P.; et al. Nicotinamide Deficiency in Primary Open-Angle Glaucoma. Invest Ophthalmol Vis Sci 2019, 60, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.D.; Zhu, L.; Sun, Q.; Fang, L. Systemic metabolite profiling reveals sexual dimorphism of AIBP control of metabolism in mice. PLoS One 2021, 16, e0248964. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Danhauser, K.; Herebian, D.; Petkovic Ramadza, D.; Piekutowska-Abramczuk, D.; Seibt, A.; Muller-Felber, W.; Haack, T.B.; Ploski, R.; Lohmeier, K.; et al. NAXE Mutations Disrupt the Cellular NAD(P)HX Repair System and Cause a Lethal Neurometabolic Disorder of Early Childhood. Am J Hum Genet 2016, 99, 894–902. [Google Scholar] [CrossRef]
- Trinh, J.; Imhoff, S.; Dulovic-Mahlow, M.; Kandaswamy, K.K.; Tadic, V.; Schafer, J.; Dobricic, V.; Nolte, A.; Werber, M.; Rolfs, A.; et al. Novel NAXE variants as a cause for neurometabolic disorder: implications for treatment. J Neurol 2020, 267, 770–782. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, T.; Lee, M.; Lee, Y.; Jeon, E.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.H. Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin Genet 2020, 97, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Incecik, F.; Ceylaner, S. Early-onset progressive encephalopathy associated with NAXE gene variants: a case report of a Turkish child. Acta Neurol Belg 2020, 120, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.W.; Lin, S.S.; Chen, C.H.; Lin, C.H.; Lee, N.C.; Hong, S.Y.; Chou, I.C.; Lin, C.L.; Yang, P.Y. NAXE gene mutation-related progressive encephalopathy: A case report and literature review. Medicine (Baltimore) 2021, 100, e27548. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, P.; Heidari, M.; Ashrafi, M.R.; Mahdieh, N.; Garshasbi, M. A novel homozygous missense variant in the NAXE gene in an Iranian family with progressive encephalopathy with brain edema and leukoencephalopathy. Acta Neurol Belg 2022, 122, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, N.J.; Walvekar, A.S.; Patraskaki, M.; Sikora, T.; Linster, C.L.; Christodoulou, J. Clinical and biochemical distinctions for a metabolite repair disorder caused by NAXD or NAXE deficiency. J Inherit Metab Dis 2022, 45, 1028–1038. [Google Scholar] [CrossRef]
- Xu, W.Q.; Wang, Y.S. The role of Toll-like receptors in retinal ischemic diseases. Int J Ophthalmol 2016, 9, 1343–1351. [Google Scholar] [CrossRef]
Figure 1.
AIBP-mediated cholesterol efflux in glaucoma. Schematic overview of AIBP-associated protective effect on glia-driven neuroinflammation in glaucomatous retina. (A) Increased cholesterol accumulation by reducing AIBP and ABCA1 expression leads to TLR4-lipid raft clustering, TLR4 dimerization, TLR4-dependent inflammatory signaling, mitochondrial dysfunction, and cytokine production, resulting in sustained glia-driven neuroinflammation and RGC loss in glaucomatous retina. (B) Enhanced cholesterol efflux by AIBP to HDL or lipid-free APOA1 from TLR4-associated lipid rafts in activated glial cells reduces cholesterol accumulation and the ensuing inhibition of TLR4-dependent inflammation. This results in the inhibition of glia-driven neuroinflammation and protect RGC against cell death in the retina.
Figure 1.
AIBP-mediated cholesterol efflux in glaucoma. Schematic overview of AIBP-associated protective effect on glia-driven neuroinflammation in glaucomatous retina. (A) Increased cholesterol accumulation by reducing AIBP and ABCA1 expression leads to TLR4-lipid raft clustering, TLR4 dimerization, TLR4-dependent inflammatory signaling, mitochondrial dysfunction, and cytokine production, resulting in sustained glia-driven neuroinflammation and RGC loss in glaucomatous retina. (B) Enhanced cholesterol efflux by AIBP to HDL or lipid-free APOA1 from TLR4-associated lipid rafts in activated glial cells reduces cholesterol accumulation and the ensuing inhibition of TLR4-dependent inflammation. This results in the inhibition of glia-driven neuroinflammation and protect RGC against cell death in the retina.
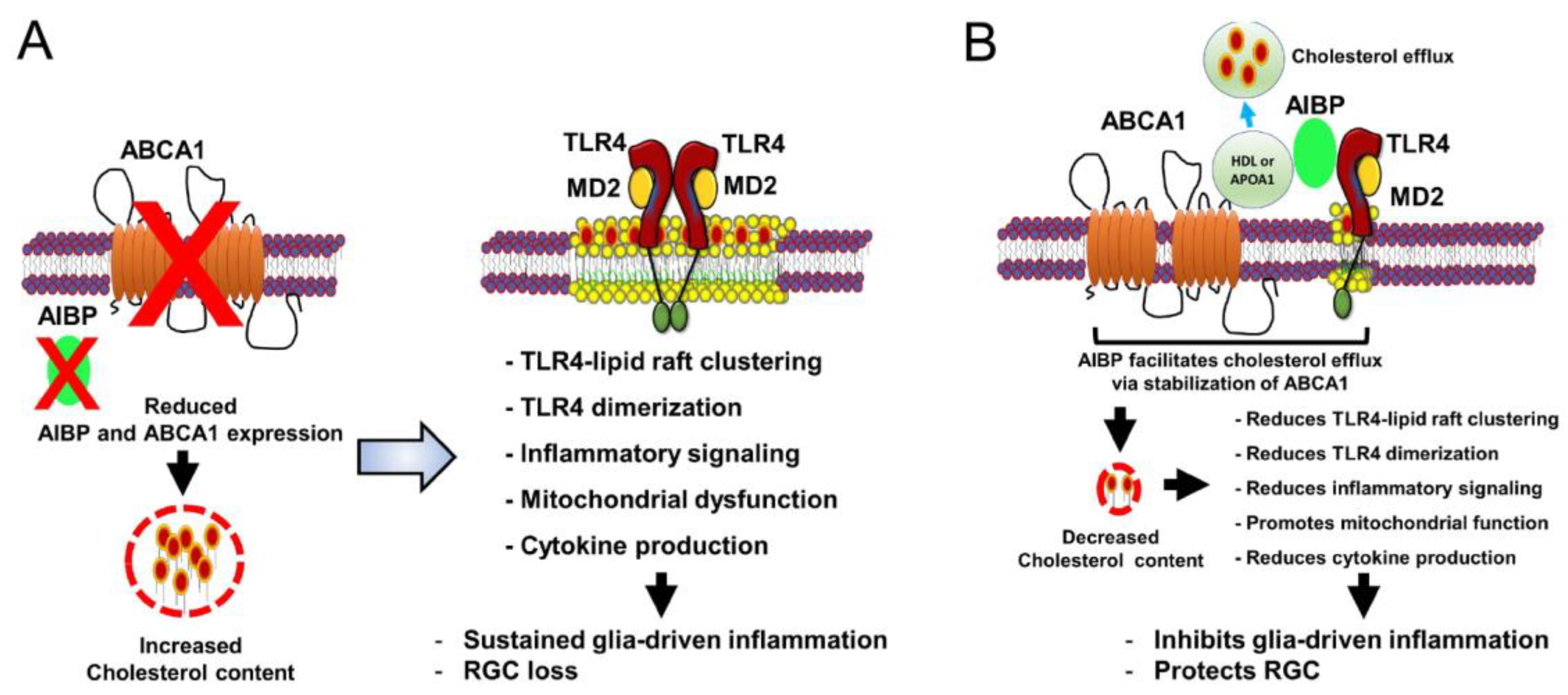
Figure 2.
Mitochondrial AIBP in glaucoma. Role of mitochondrial AIBP in glaucoma. Impairment of mitochondrial function and defective mitophagy in Apoa1bp-/- retina may contribute to glial cell activation via mitochondrial DAMPs (mtDAMPs) that are released from apoptotic cells in glaucoma.
Figure 2.
Mitochondrial AIBP in glaucoma. Role of mitochondrial AIBP in glaucoma. Impairment of mitochondrial function and defective mitophagy in Apoa1bp-/- retina may contribute to glial cell activation via mitochondrial DAMPs (mtDAMPs) that are released from apoptotic cells in glaucoma.
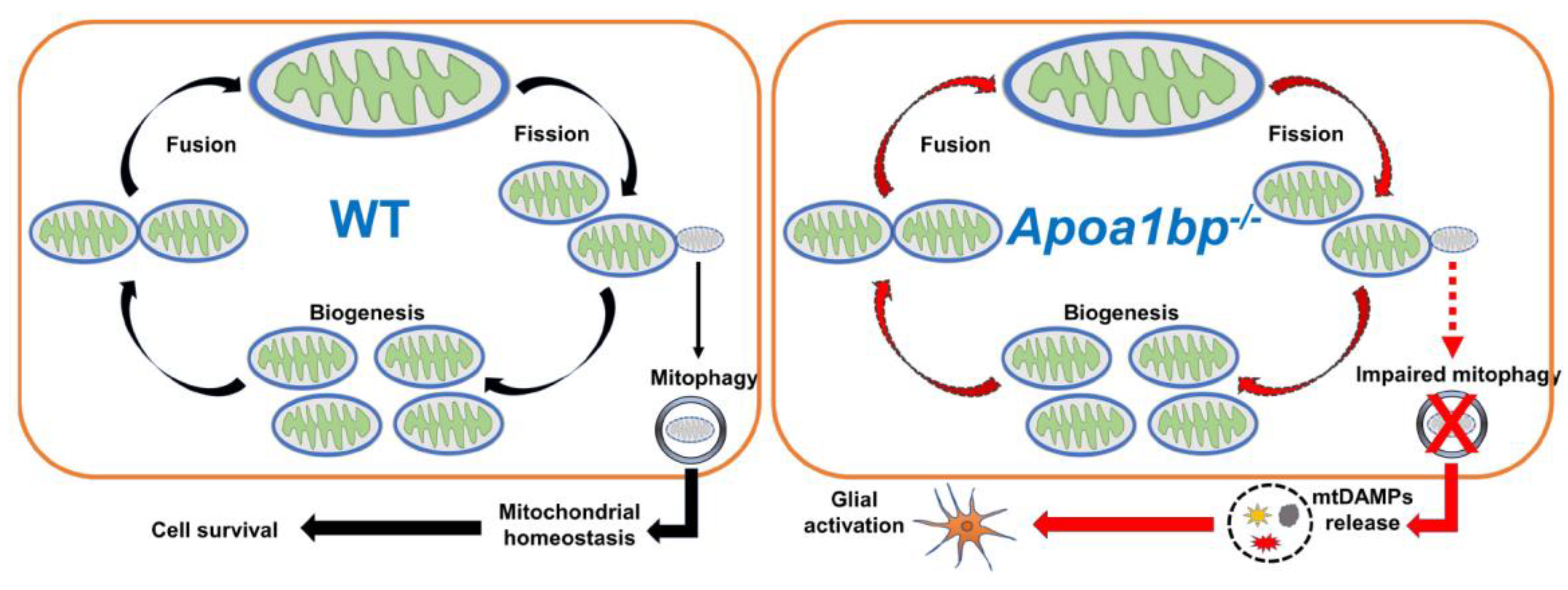
Figure 3.
The NAXE-associated NAD(P)HX repair system. The NAD(P)HX repair system. Nicotinamide adenine dinucleotide (NAD+, oxidized form: NADH) and Nicotinamide adenine dinucleotide phosphate (NADP+, oxidized form: NADPH) are hydrated by the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or spontaneously to form R- and S-epimers. The R-form of NAD(P)HX is converted into the S-form of NAD(P)HX by AIBP (NAXE) and the S-form is then catalyzed by the ATP-dependent dehydratase. AIBP deficiency leads to the accumulation of cyclic NAD(P)HX, a toxic inhibitor of cellular NADH dehydrogenases, in an irreversible path, which in turn decreases the activity of mitochondrial OXPHOS complex I (C-I) and pyruvate dehydrogenase complex (PDHc).
Figure 3.
The NAXE-associated NAD(P)HX repair system. The NAD(P)HX repair system. Nicotinamide adenine dinucleotide (NAD+, oxidized form: NADH) and Nicotinamide adenine dinucleotide phosphate (NADP+, oxidized form: NADPH) are hydrated by the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or spontaneously to form R- and S-epimers. The R-form of NAD(P)HX is converted into the S-form of NAD(P)HX by AIBP (NAXE) and the S-form is then catalyzed by the ATP-dependent dehydratase. AIBP deficiency leads to the accumulation of cyclic NAD(P)HX, a toxic inhibitor of cellular NADH dehydrogenases, in an irreversible path, which in turn decreases the activity of mitochondrial OXPHOS complex I (C-I) and pyruvate dehydrogenase complex (PDHc).
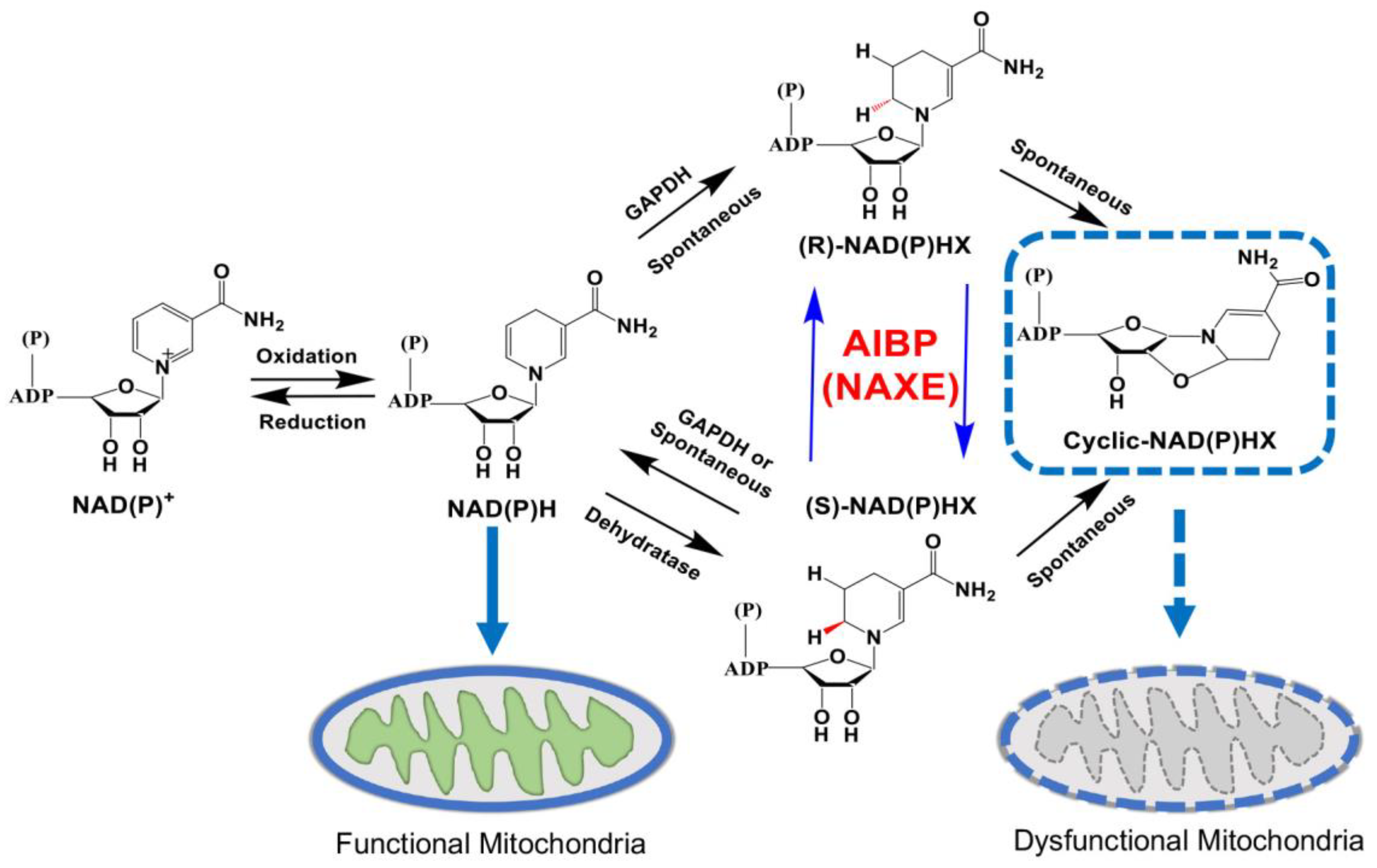
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



